

Abstract
Lung cancer is the most common cause of cancer‐related death worldwide, accounting for 1.8 million deaths annually. Analysis of The Cancer Genome Atlas data showed that all members of the minichromosome maintenance (MCM) family (hexamers involved in DNA replication: MCM2‐MCM7) were upregulated in lung adenocarcinoma (LUAD) tissues. High expression of MCM4 (P = 0.0032), MCM5 (P = 0.0032), and MCM7 (P = 0.0110) significantly predicted 5‐year survival rates in patients with LUAD. Simurosertib (TAK‐931) significantly suppressed the proliferation of LUAD cells by inhibiting cell division cycle 7‐mediated MCM2 phosphorylation. This finding suggested that MCM2 might be a therapeutic target for LUAD. Moreover, analysis of the epigenetic regulation of MCM2 showed that miR‐139‐3p, miR‐378a‐5p, and miR‐2110 modulated MCM2 expression in LUAD cells. In patients with LUAD, understanding the role of these miRNAs may improve prognoses.
Keywords: lung adenocarcinoma, MCM, miR‐139‐3p, miR‐2110, miR‐378a‐5p, simurosertib
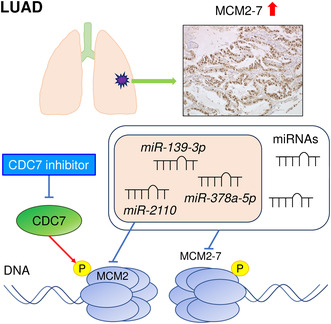
Overexpression of MCM2‐MCM7 is closely associated with the molecular pathogenesis of LUAD. Control of these genes leads to control of lung cancer. We recognized the regulation of miRNAs as an epigenomic factor for MCMs overexpression. The MCM2 were directly regulated by miR‐139‐3p, miR‐378a‐5p, and miR‐2110 in LUAD cells.
Abbreviations
- CDC7
cell division cycle 7
- DBF4
dumbbell former 4 protein
- GAPDH
glyceraldehyde‐3‐phosphate dehydrogenase
- GDC
Genomic Data Commons
- GEO
Gene Expression Omnibus
- GSEA
Gene Set Enrichment Analysis
- IHC
immunohistochemistry
- LUAD
lung adenocarcinoma
- MCM
minichromosome maintenance
- TCGA
The Cancer Genome Atlas
Lung cancer is the most common cause of cancer‐related death worldwide, accounting for 1.8 million deaths annually [1]. Lung cancer is classically divided into two types: small cell lung cancer (SCLC), which accounts for 15% of cases, and non‐small cell lung cancer (NSCLC), which accounts for 85% of cases [2, 3]. The main subtypes of NSCLC are lung adenocarcinoma (LUAD), lung squamous cell carcinoma, and large cell carcinoma. The most common histological subtype is LUAD, which is also the leading cause of cancer‐related deaths [4]. Recent genomic studies have revealed driver mutations that are characteristic of LUAD and molecularly targeted drugs have been developed based on these driver mutations [5, 6]. The survival rate in patients with lung cancer has increased owing to the development of effective treatment strategies, including molecularly targeted drugs and immune checkpoint inhibitors [5, 6, 7].
The prognosis of patients with LUAD is poor (the average 5‐year survival rate is below 20%), despite the availability of many treatment strategies [8]. Moreover, many patients with lung cancer are often diagnosed at an advanced stage, and metastasis to other organs occurs frequently [9, 10]. Unfortunately, effective treatment regimens for patients with metastatic lung cancer are limited [11]. Therefore, additional studies are needed to identify effective targeted molecular therapies for LUAD.
Minichromosome maintenance (MCM) family proteins are composed of six subunits (MCM2‐MCM7), which form a heterohexameric complex, and act as essential players in the initiation and elongation of eukaryotic DNA replication [12]. The MCM complex is also a replication gatekeeper that allows only one DNA replication per cell cycle, and knockdown of these genes induces cell cycle arrest and apoptosis in cancer cells [13]. Aberrant expression of MCM proteins has been reported in several types of cancers, for example, renal cell carcinoma, prostate cancer, and breast cancer [14, 15, 16].
Because overexpression of MCM proteins promotes the malignant transformation of cancer cells, anticancer drugs targeting MCMs are being studied [17, 18, 19]. Previous studies have shown that some antibiotics (e.g., heliquinomycin and ciprofloxacin) inhibit the helicase activity of MCMs and suppress the growth of cancer cells [18, 20]. Some statins, for example, lovastatin and atorvastatin, which inhibit HMG‐CoA reductase to lower blood cholesterol levels, have also been shown to suppress the expression of MCM2 and induce cell cycle arrest and apoptosis [19, 21]. Moreover, biologically active compounds isolated from natural products (e.g., breviscapine and cedrol) suppress the expression of MCMs and inhibit cancer cell proliferation [22, 23]. MCMs are therapeutic targets for a wide range of cancers, and control of these molecules may improve the prognosis of patients with cancers.
Numerous studies have shown that the noncoding RNAs in the human genome are key players in fine‐tuning gene expression [24, 25]. miRNAs are noncoding RNAs that control gene transcription levels by binding to specific sites in target RNAs [26, 27]. Notably, a single miRNA can control many RNA transcripts [25]. Therefore, aberrant expression of miRNAs can trigger the malignant transformation of human cells. Several studies have shown that aberrant expression of miRNAs occurs frequently in cancer cells and that their expression is closely involved in cancer cell progression, metastasis, and drug resistance [28, 29]. Previous studies have shown that miR‐1296 directly regulates MCM2 expression in prostate cancer cells [30]. Furthermore, miR‐214‐3p is downregulated in hepatocellular carcinoma cells, and this miRNA controls the expression of both MCM5 and MCM7 [31].
In this study, we investigated the clinical significance of all MCM family members (hexamers involved in DNA replication: MCM2‐MCM7) in LUAD cells. Analysis of The Cancer Genome Atlas (TCGA) data showed that all MCM family members (MCM2‐MCM7) were upregulated in LUAD tissues. Overexpression of all MCMs in LUAD clinical specimens was confirmed by immunostaining. Notably, high expression of MCM4 (P = 0.0032), MCM5 (P = 0.0032), and MCM7 (P = 0.0110) significantly predicted 5‐year survival rates in patients with LUAD. Functional assays showed that MCM2 was a promising therapeutic target in LUAD cells. Simurosertib significantly suppressed the proliferation of LUAD cells by inhibiting MCM2 phosphorylation.
Finally, we investigated the epigenetic regulation of MCM2 and found that miR‐139‐3p, miR‐378a‐5p, and miR‐2110 controlled MCM2 expression in LUAD cells. Through their functions as DNA helicases, MCM family members were found to be essential for the replication of genomic DNA and were closely involved in the molecular pathogenesis of LUAD. In particular, MCM2 may be a therapeutic target in patients with LUAD, and control of this molecule may improve prognoses in patients with LUAD.
Materials and methods
In silico analysis of gene expression levels and prognosis
The clinical significance of genes in LUAD was evaluated using TCGA datasets (https://www.cancer.gov/tcga). Data on gene expression levels were obtained from FIREBROWSE (http://firebrowse.org/) and the Genomic Data Commons (GDC) Data Portal (https://portal.gdc.cancer.gov/). The overall survival data were obtained from cBioPortal (https://www.cbioportal.org/) [32] and OncoLnc (http://www.oncolnc.org/) (data downloaded on March 26, 2021) [33].
Cell lines and cell culture
Two NSCLC cell lines, A549 and H1299, were used in this study (American Type Culture Collection, Manassas, VA, USA). Both cell lines were grown in RPMI‐1640 medium (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan) supplemented with 10% fetal bovine serum. Both cell lines were cultured in an incubator at 37 °C in an atmosphere containing 5% CO2. Cells were cultured as described previously [34]. Confirmation of negativity for Mycoplasma infection in A549 and H1299 cells was carried out using a LookOut Mycoplasma PCR Detection Kit (catalog no.: MP0035; Sigma‐Aldrich, MO, USA) and JumpStart Taq DNA Polymerase (catalog no.: D9307; Sigma‐Aldrich) according to the manufacturer's protocol.
Transfection with small interfering RNA (siRNA) and miRNA
Opti‐MEM (Gibco, Carlsbad, CA, USA) and Lipofectamine RNAiMax Transfection Reagent (Invitrogen, Carlsbad, CA, USA) were used to transfect siRNA and miRNA into cell lines. The procedure for siRNA and miRNA transfection was described previously [34, 35, 36]. The siRNAs and miRNAs used in this study are listed in Table S1.
RNA extraction and reverse transcription‐quantitative PCR
Total RNA from NSCLC cell lines was extracted using Isogen II (NIPPON GENE Co., Ltd., Tokyo, Japan). A NanoDrop 2000c spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA, USA) was used to check the quantity and quality of total RNA. cDNA was synthesized using a PrimeScript RT Master Mix (catalog no.: RR036A; Takara Bio Inc., Shiga, Japan). TaqMan Real‐Time PCR Assays were conducted to analyze gene expression, as described previously [34, 35, 36]. Reverse transcription‐quantitative PCR (RT‐qPCR) was performed using a StepOnePlus Real‐Time PCR System (Applied Biosystems, Foster City, CA, USA). The reagents used in this study are listed in Table S1.
Western blotting
Non‐small cell lung cancer cells were lysed with RIPA Lysis Buffer (catalog no.: sc‐24948; Santa Cruz Biotechnology Inc., Dallas, TX, USA). When we extract phosphorylated proteins, PhosSTOP (catalog no.: 04906845001; Roche, Mannheim, Germany) was added to the buffer system. A Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Rockford, IL, USA) was used to assay protein concentrations. SuperSep Ace (7.5%, 13‐well; FUJIFILM Wako Pure Chemical Corporation) was used for sodium dodecyl sulfate‐polyacrylamide gel electrophoresis. Precision Plus Protein Dual WesternC Standards (catalog no.: 161‐0376; Bio‐Rad Laboratories, Inc., Herclules, CA, USA) were used as the standard. Proteins were transferred on polyvinylidene fluoride membranes (catalog no.: PPVH00010; Merck KGaA, Darmstadt, Germany). The membranes were blocked with 3% bovine serum albumin (catalog no.: 01863‐77; Nacalai Tesque, Inc., Kyoto, Japan) in TBST. Precision Protein Strep Tactin‐HRP Conjugate (catalog no.: 1610380; Bio‐Rad Laboratories, Inc.) was used to detect the standard. The signal was developed using Amersham ECL Prime Western Blotting Detection Reagent (Cytiva, Marlborough, MA, USA). Chemiluminescence was performed using FluorChem FC2 (Cell Biosciences, Santa Clara, CA, USA) to visualize western blotting. The reagents used in this study are listed in Table S1.
Cell proliferation and cell cycle assays
Cell proliferation was evaluated using a Cell Proliferation Kit (XTT based; Biological Industries, Beit‐Haemek, Israel). BD Cycletest Plus DNA Reagent Kit (BD Biosciences, Franklin Lakes, NJ, USA) was used for the evaluation of the cell cycle distribution on a BD FACSCelesta Flow Cytometer (BD Biosciences). The data were analyzed using FlowJo software (TreeStar, Ashland, OR, USA). The procedures for cell proliferation and cell cycle analyses were described previously [34, 35, 36].
Immunohistochemical staining
Tissue microarray slides (catalog no.: LC811a; US Biomax, Inc., Derwood, MD, USA) were used for immunohistochemical staining. Blocking, the primary antibody reaction, secondary antibody reaction, and binding of avidin to the biotin complex were conducted using a VECTASTAIN Universal Elite ABC Kit (Vector Laboratories, Burlingame, CA, USA) according to the manufacturer's protocol. Dako Real antibody diluent (Agilent, Santa Clara, CA, USA) was used to dilute the primary antibody. The chromogenic reaction was developed using a Dako REAL EnVision Detection System Peroxidase/DAB+, Rabbit/Mouse (Agilent). The primary antibody used in this study is described in Table S1. The characteristics of patients from whom the tissue samples were derived in this tissue array are described in Table S2. The intensity and area of staining were evaluated based on previous reports [37], and immunohistochemical scores were calculated.
Cytotoxicity assay
For cytotoxicity assays, 100 μL of cell suspension (approximately 2 × 103 cells·mL−1) was seeded in each well of a 96‐well plate, and cells were allowed to attach overnight. The next day, culture media were discarded, and the cells were treated with different concentrations of TAK‐931 (catalog no.: HY‐100888/CS‐0020550; MedChemExpress, NJ, USA) ranging from 1 to 1000 nm. The plates were incubated for 5 days, and media containing TAK‐931 were replaced every other day. Cells treated with 0.1% DMSO served as a negative control. To investigate cell toxicity, XTT assays were performed using a Cell Proliferation Kit (Biological Industries) according to the manufacturer's protocol. The signals were measured using a microplate reader (Bio‐Rad Laboratories, Inc.).
Colony formation assay
For colony formation assays, 1 mL medium containing 300 cells was added to each well of a 12‐well plate, and the cells were treated with different concentrations of TAK‐931 the next day. TAK‐931 was replaced every other day, and the cells were incubated for 10 consecutive days. Cells treated with 0.1% DMSO were used as a control. At the end of the incubation, the colonies were washed with phosphate‐buffered saline and subsequently fixed using 4% paraformaldehyde for 15 min at room temperature. After fixation, the colonies were dyed with 0.5% crystal violet (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan) for 25 min. The wells were washed with water, and the plates were air‐dried.
Identification of putative miRNAs binding to MCM2‐MCM7 and analysis of miRNA expression
Data for putative miRNAs binding to MCM2‐MCM7 were obtained from the targetscanhuman database (release 8.0) (https://www.targetscan.org/vert_80/) [38]. Data for miRNA expression levels were obtained from FIREBROWSE (http://firebrowse.org/) and the GDC Data Portal (https://portal.gdc.cancer.gov/) (data downloaded on August 4, 2022).
The construction of plasmid and dual‐luciferase reporter assays
The wild‐type sequences containing miRNAs binding sites in the 3′‐untranslated regions (UTRs) of MCM2 and the deletion‐type sequences lacking them were cloned into the psiCHECK‐2 vector (C8021; Promega, Madison, WI, USA). The procedures for transfection and dual‐luciferase reporter assays were provided in previous studies [34, 35].
MCM family alteration analysis based GSEA analysis
The data were obtained from cBioPortal (http://cbioportal.org, accessed on May 19, 2023 [32]). We executed queries to specify MCM2‐MCM7 mRNA expression changes (Z‐score ≥0) and analyzed survival. Based on mRNA expression levels compared between altered and non‐altered groups, the ranked gene list for Gene Set Enrichment Analysis (GSEA) was obtained and uploaded into WEB‐based GEne SeT AnaLysis Toolkit, ‘WebGestalt’ (http://www.webgestalt.org), accessed on May, 19 2023 [39]. We applied ‘Wikipathway cancer’ dataset for GSEA (https://www.wikipathways.org/index.php/WikiPathways, accessed on May 19, 2023 [40]).
Statistical analysis
Statistical analyses were performed using graphpad prism 8 (GraphPad Software, La Jolla, CA, USA) and jmp pro 14 (SAS Institute Inc., Cary, NC, USA). Differences between the two groups were analyzed using Student's t‐tests or Mann–Whitney U‐tests. Multiple group comparisons were performed using one‐way ANOVA and Tukey tests for post hoc analyses. Survival rates were analyzed using Kaplan–Meier survival curves and log‐rank tests.
Results
Expression and clinical significance of MCM2‐MCM7 in patients with LUAD by TCGA analysis
First, we analyzed the Gene Expression Omnibus (GEO) database (GEO accession no: GSE19188) to identify genes exhibiting oncogenic effects in NSCLC. The upregulated genes in GSE19188 were narrowed down based on the criteria of log2 (fold change) greater than 1 and P‐value less than 0.05; using these parameters, 613 genes were identified. These candidate genes were classified according to the Kyoto Encyclopedia of Genes and Genomes pathways by GeneCodis 4 database (https://genecodis.genyo.es/) (data downloaded on March 17, 2021) [41]. The top 10 annotations based on relative enrichment score were mucin type O‐glycan biosynthesis, DNA replication, glycosaminoglycan biosynthesis—heparan sulfate/heparin, RNA degradation, homologous recombination, protein digestion and absorption, Fanconi anemia pathway, cell cycle, ether lipid metabolism, and glycosphingolipid biosynthesis—lacto and neolacto series (Table 1). In our previous reports, we found that cell cycle and DNA replication genes regulated by miRNAs affected lung cancer progression. Twenty‐four and eight genes were classified as cell cycle‐ and DNA replication‐related genes, respectively (Tables 2 and 3). MCM2, MCM4, MCM6, and MCM7 were common in these two annotations.
Table 1.
Enriched annotations of the upregulated genes identified by GSE19188.
| Annotation | Annotation ID | Number of genes | Relative enrichment score | P‐value | Adjusted P‐value | Genes |
|---|---|---|---|---|---|---|
| Mucin‐type O‐glycan biosynthesis | hsa00512 | 3 | 50.31 | < 0.001 | < 0.001 | GCNT3, GALNT14, GALNT7 |
| DNA replication | hsa03030 | 8 | 47.36 | < 0.001 | < 0.001 | RNASEH2A, RFC4, POLE2, MCM7, MCM6, MCM4, MCM2, FEN1 |
| Glycosaminoglycan biosynthesis—heparan sulfate/heparin | hsa00534 | 2 | 40.25 | < 0.001 | 0.003 | HS3ST1, HS6ST2 |
| RNA degradation | hsa03018 | 4 | 33.54 | < 0.001 | < 0.001 | PFKP, HSPD1, ENO1, PABPC1L |
| Homologous recombination | hsa03440 | 5 | 29.60 | < 0.001 | < 0.001 | RAD54B, BLM, BARD1, RAD51, BRIP1 |
| Protein digestion and absorption | hsa04974 | 9 | 29.22 | < 0.001 | < 0.001 | KCNN4, COL12A1, COL11A1, COL10A1, COL5A2, COL5A1, COL3A1, COL1A2, COL1A1 |
| Fanconi anemia pathway | hsa03460 | 6 | 28.75 | < 0.001 | < 0.001 | BLM, RAD51, RMI2, BRIP1, FANCI, UBE2T |
| Cell cycle | hsa04110 | 24 | 27.44 | < 0.001 | < 0.001 | CHEK1, CDKN2A, CDC25C, CDC20, CDC6, CDK1, ESPL1, CCNE1, CCNB1, CCNA2, PTTG1, CCNE2, CCNB2, CDC45, TTK, BUB1B, BUB1, TFDP2, PLK1, MCM7, MCM6, MCM4, MCM2, MAD2L1 |
| Ether lipid metabolism | hsa00565 | 5 | 26.48 | < 0.001 | < 0.001 | PLPP2, UGT8, PLA2G4A, PAFAH1B3, EPT1 |
| Glycosphingolipid biosynthesis—lacto and neolacto series | hsa00601 | 3 | 25.16 | < 0.001 | 0.001 | B3GNT3, FUT3, FUT2 |
Table 2.
Upregulated genes associated with cell cycle.
| Entrez GeneID | Gene symbol | Gene name |
Log2 fold change |
P‐value |
|---|---|---|---|---|
| 991 | CDC20 | Cell division cycle 20 | 2.73 | < 0.001 |
| 891 | CCNB1 | Cyclin B1 | 2.64 | < 0.001 |
| 7272 | TTK | TTK protein kinase | 2.49 | < 0.001 |
| 9133 | CCNB2 | Cyclin B2 | 2.36 | < 0.001 |
| 4085 | MAD2L1 | Mitotic arrest deficient 2 like 1 | 2.30 | < 0.001 |
| 701 | BUB1B | BUB1 mitotic checkpoint serine/threonine kinase B | 2.25 | < 0.001 |
| 983 | CDK1 | Cyclin‐dependent kinase 1 | 2.10 | < 0.001 |
| 699 | BUB1 | BUB1 mitotic checkpoint serine/threonine kinase | 2.06 | < 0.001 |
| 890 | CCNA2 | Cyclin A2 | 1.92 | < 0.001 |
| 4173 | MCM4 | Minichromosome maintenance complex component 4 | 1.78 | < 0.001 |
| 1111 | CHEK1 | checkpoint kinase 1 | 1.75 | < 0.001 |
| 4171 | MCM2 | Minichromosome maintenance complex component 2 | 1.71 | < 0.001 |
| 898 | CCNE1 | Cyclin E1 | 1.71 | < 0.001 |
| 1029 | CDKN2A | Cyclin‐dependent kinase inhibitor 2A | 1.67 | < 0.001 |
| 990 | CDC6 | Cell division cycle 6 | 1.63 | < 0.001 |
| 9232 | PTTG1 | PTTG1 regulator of sister chromatid separation, securin | 1.59 | < 0.001 |
| 9134 | CCNE2 | Cyclin E2 | 1.57 | < 0.001 |
| 8318 | CDC45 | Cell division cycle 45 | 1.47 | < 0.001 |
| 4176 | MCM7 | Minichromosome maintenance complex component 7 | 1.24 | < 0.001 |
| 995 | CDC25C | Cell division cycle 25C | 1.11 | < 0.001 |
| 5347 | PLK1 | Polo‐like kinase 1 | 1.09 | < 0.001 |
| 4175 | MCM6 | Minichromosome maintenance complex component 6 | 1.09 | < 0.001 |
| 9700 | ESPL1 | Extra spindle pole bodies like 1, separase | 1.06 | < 0.001 |
| 7029 | TFDP2 | Transcription factor Dp‐2 | 1.02 | < 0.001 |
Table 3.
Upregulated genes associated with DNA replication.
| Entrez GeneID | Gene symbol | Gene name |
Log2 fold change |
P‐value |
|---|---|---|---|---|
| 4173 | MCM4 | Minichromosome maintenance complex component 4 | 1.78 | < 0.001 |
| 4171 | MCM2 | Minichromosome maintenance complex component 2 | 1.71 | < 0.001 |
| 5984 | RFC4 | Replication factor C subunit 4 | 1.59 | < 0.001 |
| 10,535 | RNASEH2A | Ribonuclease H2 subunit A | 1.28 | < 0.001 |
| 2237 | FEN1 | Flap structure‐specific endonuclease 1 | 1.28 | < 0.001 |
| 5427 | POLE2 | DNA polymerase epsilon 2, accessory subunit | 1.24 | < 0.001 |
| 4176 | MCM7 | Minichromosome maintenance complex component 7 | 1.24 | < 0.001 |
| 4175 | MCM6 | Minichromosome maintenance complex component 6 | 1.09 | < 0.001 |
MCM2‐MCM7 form a hexamer and act as a DNA helicase essential for genomic DNA replication. Notably, MCM2‐MCM7 were significantly upregulated in LUAD tissues compared with normal tissues, and the 5‐year overall survival rates tended to be poor in patients with high MCM2‐MCM7 expression (Fig. 1). In this study, we focused on MCM2‐MCM7.
Fig. 1.
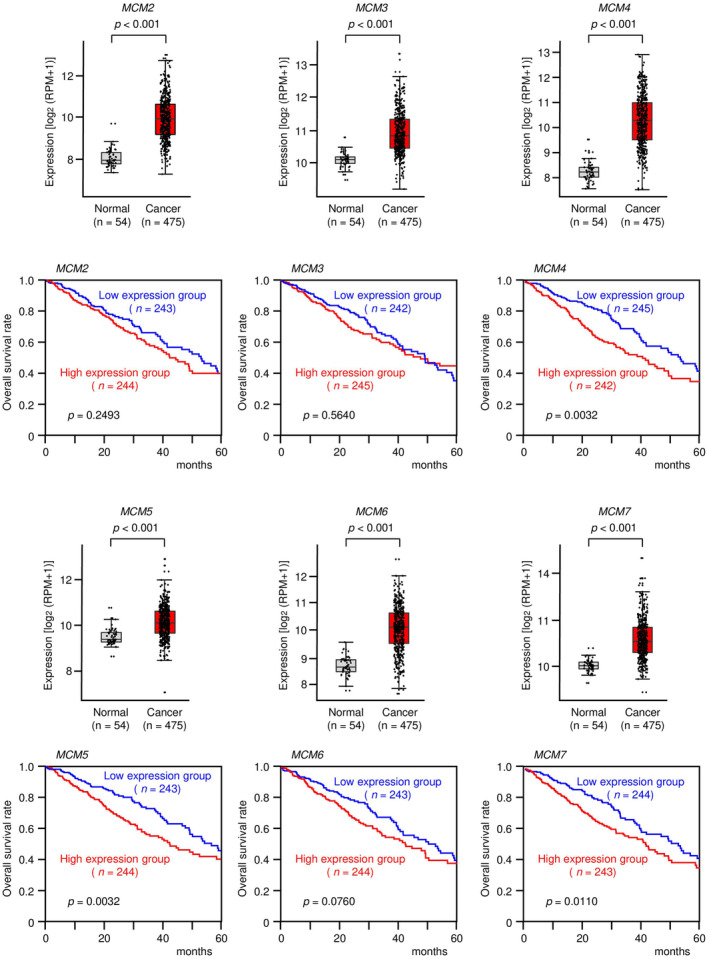
Expression levels of MCM2, MCM3, MCM4, MCM5, MCM6, and MCM7 and association with overall survival in patients with LUAD. The expression levels of six genes (MCM2, MCM3, MCM4, MCM5, MCM6, and MCM7) in normal and cancer tissues were evaluated using data from TCGA database. In total, 475 LUAD tissues and 54 normal lung tissues were analyzed. The lower and upper hinges represent the 25th and 75th percentiles, respectively. The lower/upper whiskers represent the lowest/largest points inside the range defined by the first/third quartiles ± 1.5 times the IQR, respectively. Dots represent individual samples. Student's t‐test. Kaplan–Meier 5‐year overall survival curves were created from TCGA data. Patients were divided into high‐ and low‐expression groups according to miRNA expression (based on median expression). The red line shows the high‐expression group, and the blue line shows the low‐expression group. Log‐rank test. RPM, reads per million.
Overexpression of MCM2‐MCM7 in LUAD clinical specimens
We performed immunohistochemistry (IHC) to investigate MCM2–MCM7 protein expression in LUAD. The nuclear expression of MCM2‐MCM7 was high in LUAD but low in normal lung tissues. MCM2‐MCM7 were significantly overexpressed in LUAD compared with normal lung tissues (Fig. 2). The characteristics of patients are described in Table S2.
Fig. 2.
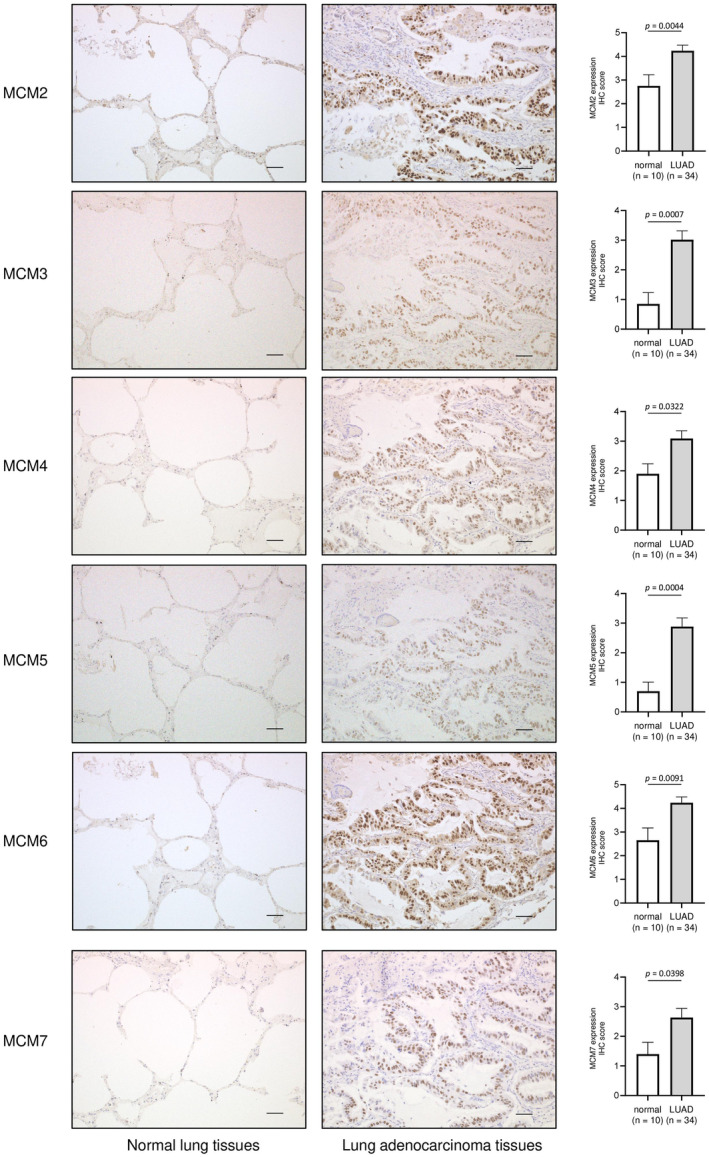
MCM2, MCM3, MCM4, MCM5, MCM6, and MCM7 expression levels in LUAD clinical specimens. Expression levels of MCM2, MCM3, MCM4, MCM5, MCM6, and MCM7 in LUAD and normal lung tissues were evaluated by IHC using a tissue microarray. Representative LUAD tissues (patient no. 9) and normal lung tissues (patient no. 75) are shown. The data are means and standard errors of the means. Mann–Whitney U‐tests. Scale bars = 50 μm.
Search for miRNAs that negatively regulate MCMs (MCM2–MCM7) expression in LUAD cells
We hypothesized that one of the reasons for the overexpression of MCMs in LUAD cells is that some miRNAs expression is repressed. We recently created the RNA‐sequencing‐based miRNA expression signature using LUAD clinical specimens (GEO accession no: GSE230229). Based on this signature, we selected miRNAs whose expression is repressed in LUAD tissues. Next, we integrated these downregulated miRNAs with TargetScanHuman database (release 8.0) (https://www.targetscan.org/vert_80/), and selected miRNAs that may negatively regulate MCMs (MCM2–MCM7) in LUAD cells. Our strategy for miRNA selection is shown in Fig. 3A and Fig. S1. Candidate miRNAs that negatively regulated MCM2–MCM7 are listed in Table 4 and Tables [Link], [Link].
Fig. 3.
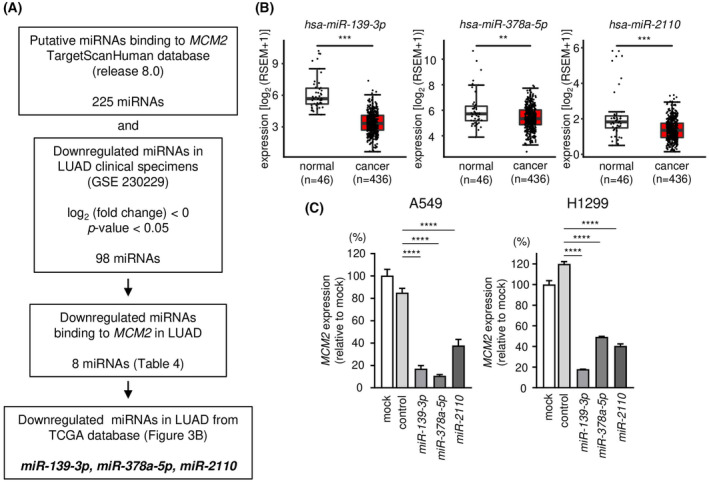
Selection of miRNAs regulating MCM2 in LUAD cells. (A) This shows a flowchart for the strategy to identify the candidate of miRNAs regulating MCM2 in LUAD cells. (B) The expression levels of miR‐139‐3p, miR‐378a‐5p, and miR‐2110 were evaluated in a LUAD dataset from TCGA. A total of 436 LUAD tissues and 46 normal lung tissues were analyzed. The lower and upper hinges represent the 25th and 75th percentiles, respectively. The lower/upper whiskers represent the lowest/largest points inside the range defined by first/third quartile ± 1.5 times IQR, respectively. Dots mean each sample. Student's t‐test. (C) mRNA expressions of MCM2 were evaluated by RT‐qPCR. Internal control was the expression of GAPDH. The time for transfection is 72 h. RT‐qPCR was measured in triplicate. The data are mean and SD. One‐way ANOVA and Tukey tests for post hoc analysis. **P < 0.01, ***P < 0.001, ****P < 0.001. RSEM, RNA‐seq by Expectation‐Maximization.
Table 4.
Candidate tumor‐suppressive miRNAs binding to MCM2. FDR, false discovery rate.
| miRNA | miRBase accession No. | Log2 fold change GSE230229 | Normalized read count GSE230229 |
FDR |
P‐value |
|
|---|---|---|---|---|---|---|
| LUAD tissues | Normal lung tissues | |||||
| hsa‐miR‐603 | MIMAT0003271 | −2.90 | 0.00 | 2.90 | < 0.001 | < 0.001 |
| hsa‐miR‐373‐3p | MIMAT0000726 | −2.09 | 0.00 | 2.09 | 0.152 | 0.038 |
| hsa‐miR‐139‐3p | MIMAT0004552 | −1.83 | 5.04 | 6.87 | 0.047 | 0.009 |
| hsa‐miR‐4732‐3p | MIMAT0019856 | −1.72 | 3.70 | 5.42 | 0.014 | 0.002 |
| hsa‐miR‐4516 | MIMAT0019053 | −1.38 | 3.56 | 4.94 | 0.164 | 0.042 |
| hsa‐miR‐378a‐5p | MIMAT0000731 | −0.80 | 8.58 | 9.38 | 0.010 | 0.002 |
| hsa‐miR‐2110 | MIMAT0010133 | −0.69 | 6.91 | 7.59 | 0.141 | 0.034 |
| hsa‐miR‐6513‐3p | MIMAT0025483 | −0.58 | 5.51 | 6.10 | 0.186 | 0.049 |
To verify the effectiveness of our list, we performed further analysis using MCM2 as a model case. Our strategy identified a candidate of eight miRNAs that negatively controlled the expression of MCM2 in LUAD cells (Fig. 3A and Table 4). Among these miRNAs, three miRNAs (miR‐139‐3p, miR‐378a‐5p, and miR‐2110) were significantly downregulated in LUAD specimens based on TCGA‐LUAD database analysis (Fig. 3B). MCM2 mRNA expressions were significantly suppressed by ectopic expression of these miRNAs (miR‐139‐3p, miR‐378a‐5p, and miR‐2110) in LUAD cells (Fig. 3C).
Furthermore, we examined whether these miRNAs (miR‐139‐3p, miR‐378a‐5p, and miR‐2110) directly bind to 3′UTR of MCM2 by dual‐luciferase reporter assays. TargetScanHuman database shows the putative miR‐139‐3p‐binding site in the 3′‐UTR of MCM2 (Fig. 4A upper). Luminescence intensity was significantly reduced following co‐transfected with miR‐139‐3p and a wild‐type vector (containing the miR‐139‐3p binding site in the 3′UTR of MCM2) (Fig. 4A lower). By contrast, no change in luminescence intensity following co‐transfected with miR‐139‐3p and a deletion‐type vector (without the miR‐139‐3p binding site in the 3′UTR of MCM2) (Fig. 4A lower). These results indicated that miR‐139‐3p directly bound to 3′UTR of MCM2 and controlled MCM2 expression in LUAD cells. Similar analyses were performed for the other miRNAs (miR‐378a‐5p and miR‐2110). The results revealed that two miRNAs also directly bound to the 3′UTR of MCM2 and negatively regulate its expression in LUAD cells (Fig. 4B,C).
Fig. 4.
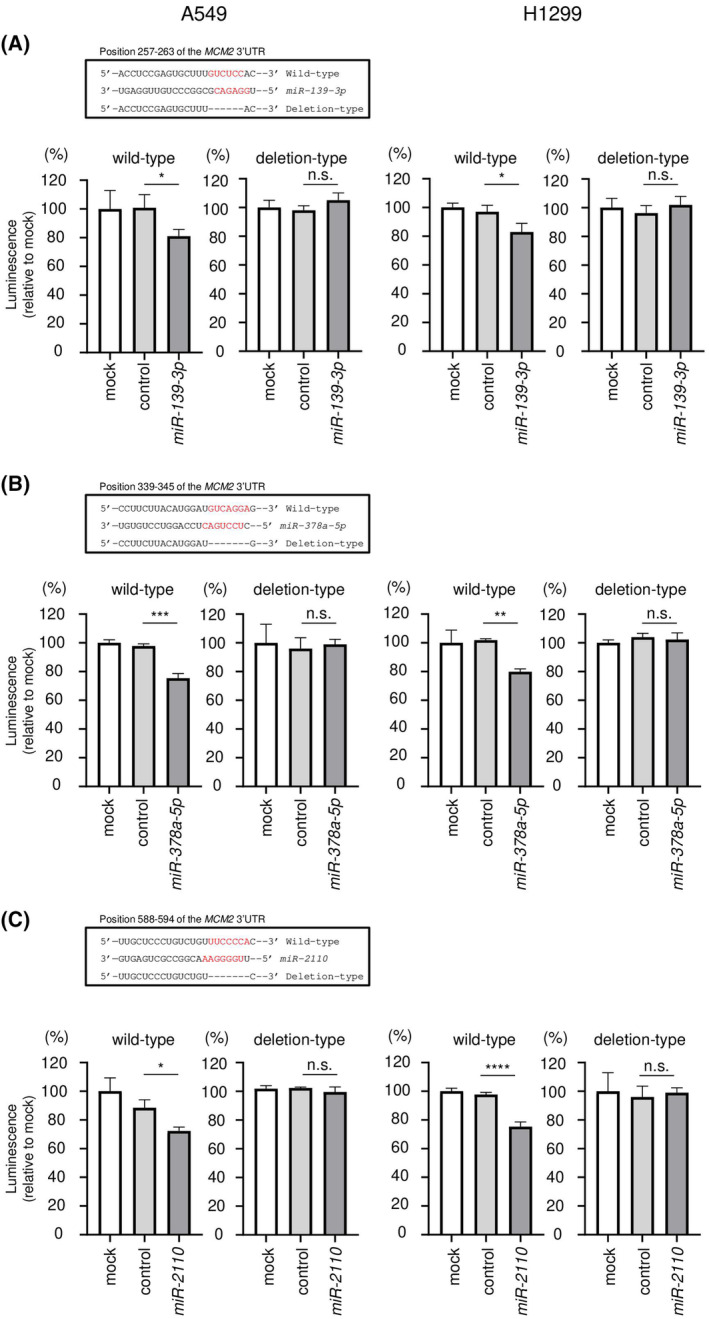
Direct regulation of MCM2 expression by miR‐139‐3p, miR‐378a‐5p, and miR‐2110 in LUAD cells. This figure shows dual‐luciferase reporter assay results following transfection with (A) miR‐139‐3p, (B) miR‐378a‐5p, and (C) miR‐2110. The upper panels are putative miR‐139‐3p, miR‐378a‐5p, and miR‐2110 binding sites predicted within the 3′‐UTR of MCM2 by TargetScanHuman database analysis. After 24 h co‐transfection of miRNA and wild‐type or deletion‐type psiCHECK‐2 vector, dual‐luciferase reporter assay was conducted (lower panel). Wild‐type vector contains the miRNA binding site of MCM2 3′‐UTR sequence, and deletion‐type one does not. Normalized data were calculated as the Renilla/firefly luciferase activity ratio. Luminescence activity was measured in triplicate. The data are mean and SD. One‐way ANOVA and Tukey tests for post hoc analysis. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, n.s., not significant. RSEM, RNA‐seq by Expectation‐Maximization.
Alteration of mRNA expression of MCMs (MCM2–MCM7) in LUAD patients based on TCGA‐LUAD analysis
We investigated the clinical impact of alteration of MCMs (MCM2–MCM7) expression in patients with LUAD using TCGA‐LUAD cohort dataset.
In 586 LUAD clinical samples, a large number of LUAD cohort data showed high mRNA expression for MCM2 at 49%, MCM3 at 44%, MCM4 at 49%, MCM5 at 48%, MCM6 at 53%, and MCM7 at 45% (Z‐score >0) (Fig. 5A). Interestingly, we found that there were many cases in which the expression of all 6 MCM genes was simultaneously increased (Fig. 5A).
Fig. 5.
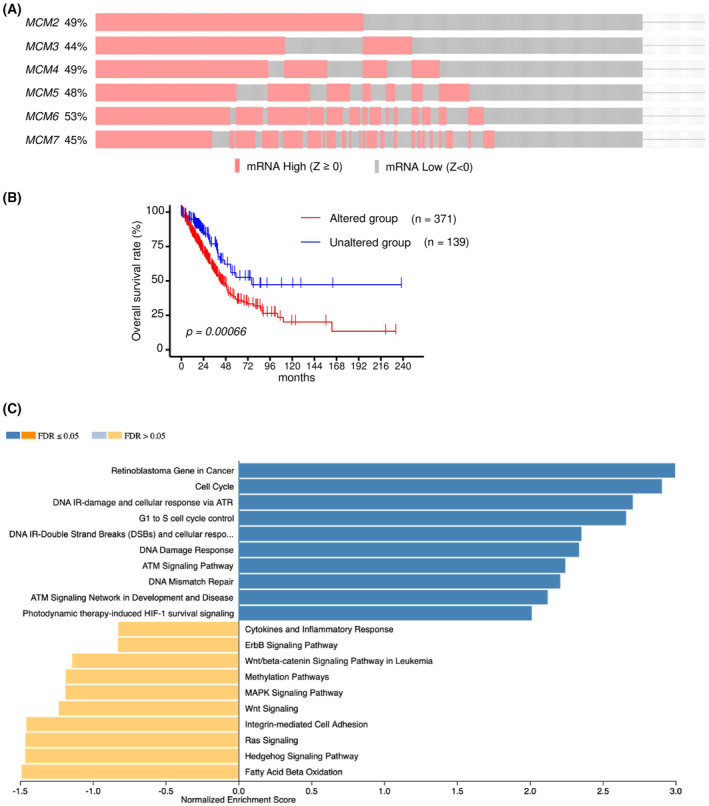
MCM family mRNA expression alternations in LUAD clinical specimens from TCGA‐LUAD analyses. (A) Oncoprint of TCGA‐LUAD on cBioPortal filtered by the mRNA expression (Z‐score ≥0) from the query for genes. (B) Kaplan–Meier curves of overall survivals between altered and unaltered groups. (C) Bar chart and enrichment plots of GSEA of alteration of MCM family. FDR, false discovery rate.
Patients with LUAD who had high expression in at least one of the MCM genes (MCM2–MCM7) showed a worse survival outcome compared with patients with no MCM gene alteration (Fig. 5B). Gene set enrichment analysis using TCGA‐LUAD dataset revealed that ‘Retinoblastoma Gene in Cancer’, ‘Cell Cycle’, ‘DNA IR‐damage and cellular response via ATR’, and ‘G1 to S cell cycle control’ were identified as enrichment pathways in high expression of MCM genes group (Fig. 5C).
Effects of MCM2‐MCM7 knockdown on cell proliferation and the cell cycle
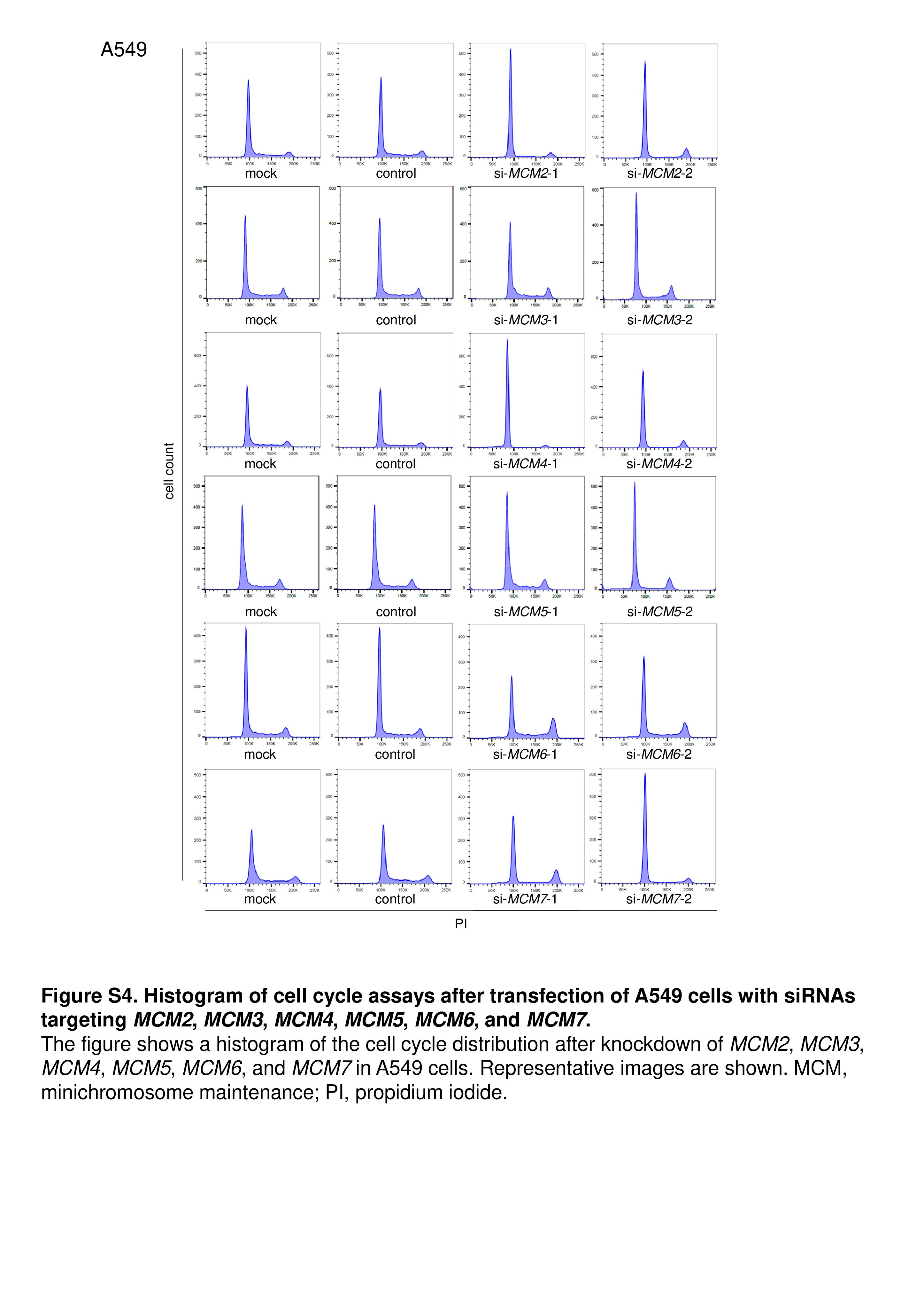
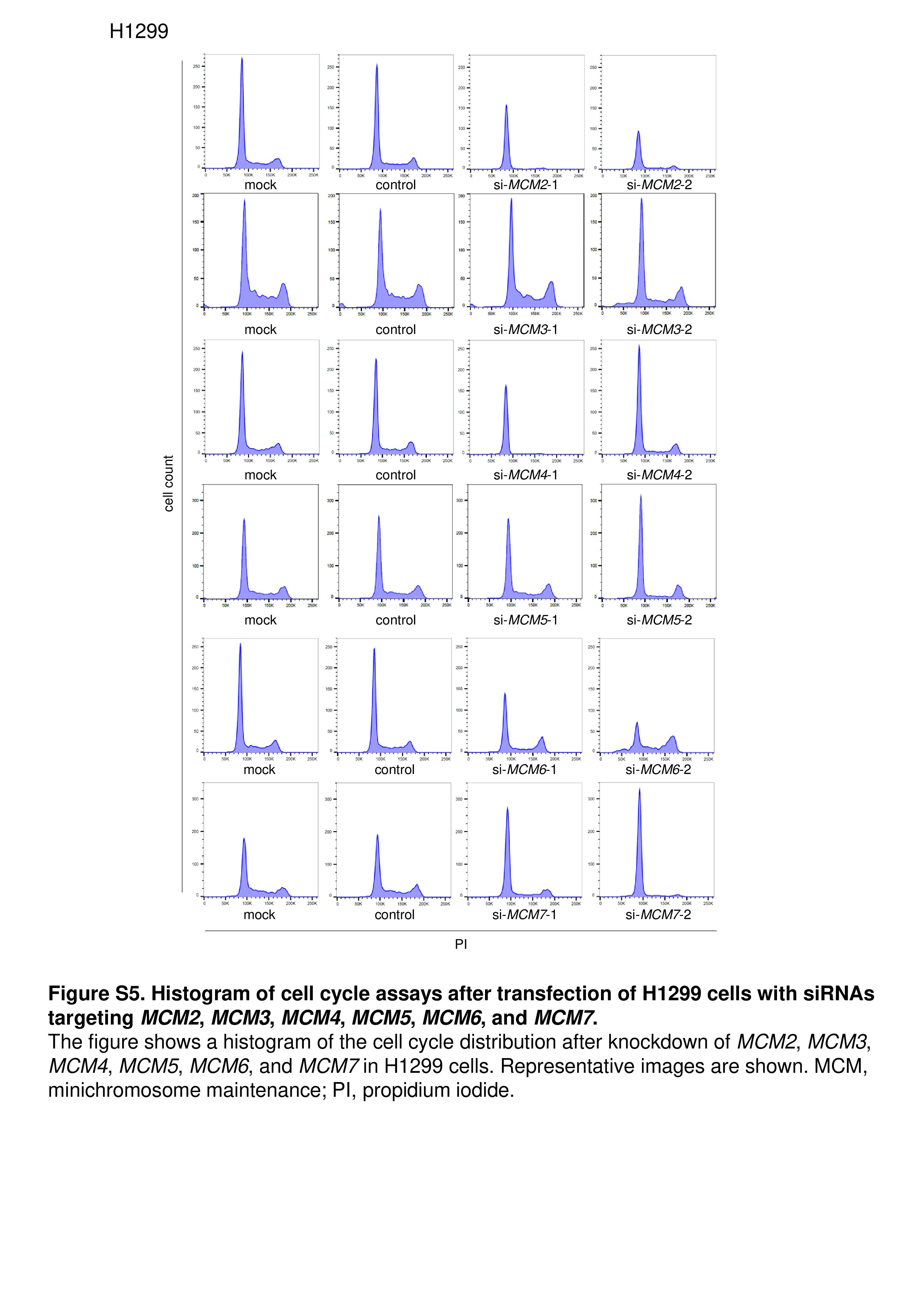
Next, we investigated the effects of MCM2‐MCM7 knockdown to evaluate the functions of MCM2‐MCM7 in A549 and H1299 cells. First, we confirmed the knockdown efficiencies of MCM2‐MCM7 siRNAs using RT‐qPCR and western blotting. Transfection with two siRNAs reduced the mRNA and protein expression of MCM2‐MCM7 in A549 and H1299 cells (Figs S2 and S3). Next, we conducted cell proliferation assays. Most siRNAs targeting MCM2‐MCM7 suppressed cell proliferation (Fig. 6). Lastly, cell cycle assays were performed after transfection with siRNAs targeting MCM2‐MCM7. siRNAs targeting MCM2‐MCM5 and MCM7 led to G0/G1 arrest, and those targeting MCM6 induced G2/M arrest (Fig. 7; Figs S4 and S5). These results suggested that knockdown of MCM2‐MCM7 inhibited cell proliferation and cell cycle progression in A549 and H1299 cells.
Fig. 6.
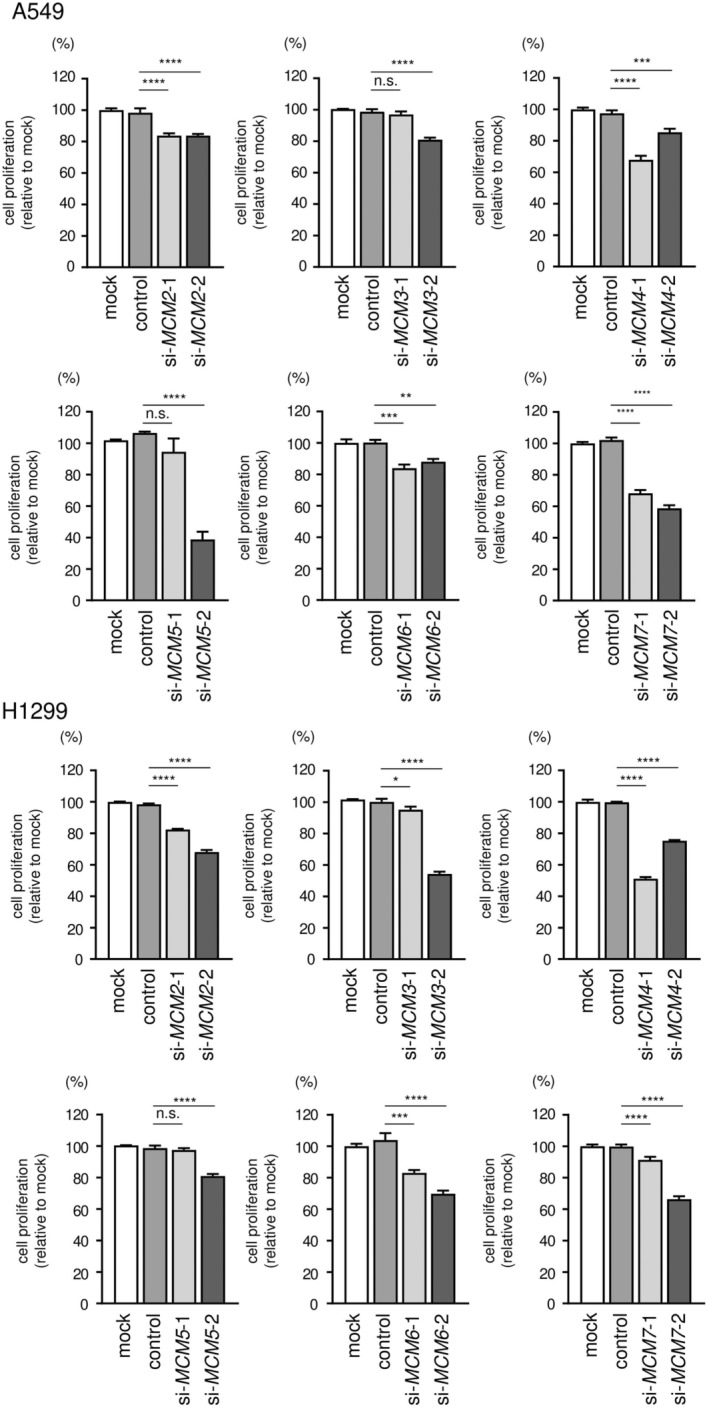
Cell proliferation assays after knockdown of MCM2, MCM3, MCM4, MCM5, MCM6, and MCM7. Cell proliferation after knockdown of MCM2, MCM3, MCM4, MCM5, MCM6, and MCM7 was assessed using XTT assays. The transfection time was 96 h. Cell proliferation was measured in triplicate. The data are means and standard deviations. One‐way ANOVA and Tukey tests were used for post hoc analysis. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. n.s., not significant.
Fig. 7.
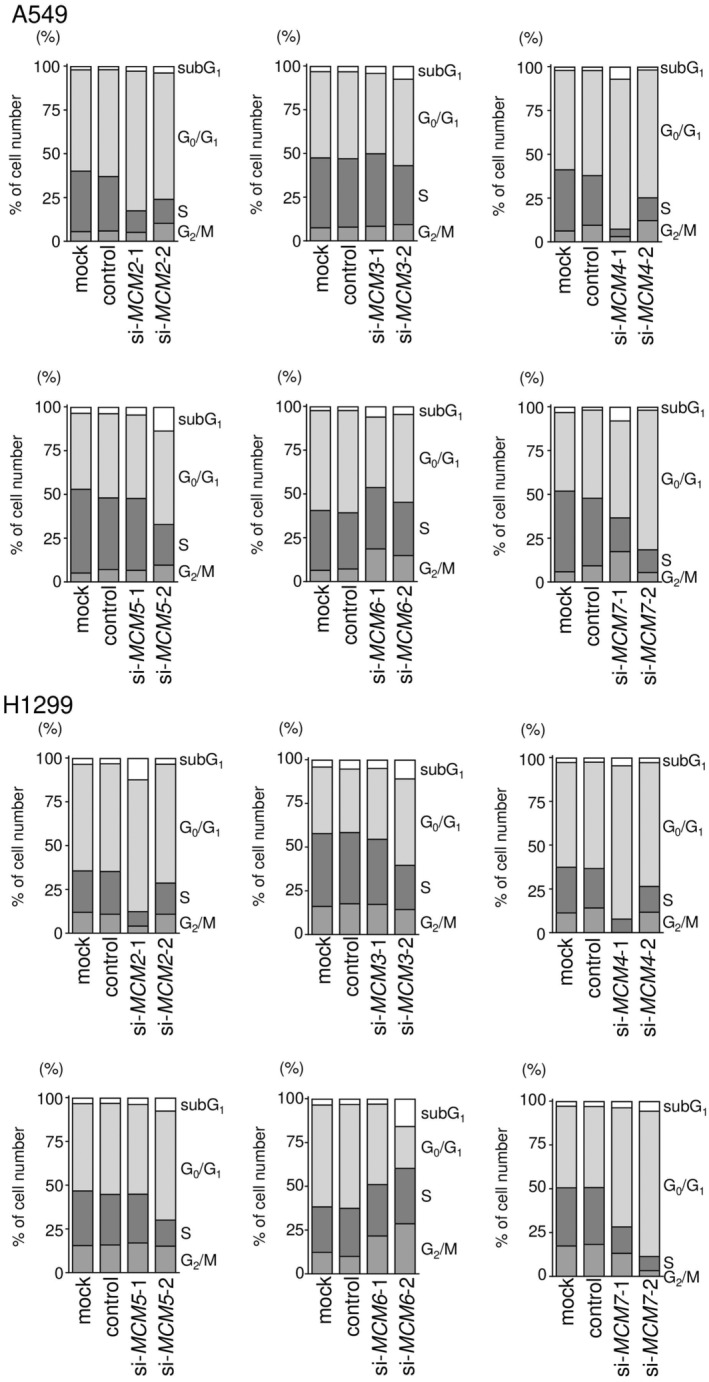
Cell cycle assays after knockdown of MCM2, MCM3, MCM4, MCM5, MCM6, and MCM7. Cell cycle distribution was tested by flow cytometry. The samples were evaluated 72 h after transfection with siRNAs targeting MCM2, MCM3, MCM4, MCM5, MCM6, and MCM7. Cell cycle distribution was measured in triplicate. Data in the stacked bar graphs are means.
Effects of the CDC7 inhibitor TAK‐931 on A549 and H1299 cells
Phosphorylation of MCM2 by cell division cycle 7 (CDC7) at the G1 phase to S phase initiates DNA synthesis [42]. CDC7 is a dumbbell former 4 protein (DBF4)‐dependent serine/threonine kinase and plays important roles in DNA replication [43]. CDC7 kinase activity is essential for MCM2‐MCM7 hexamer‐induced DNA replication. We hypothesized that indirect suppression of MCM2‐MCM7 hexamer function caused by CDC7 inhibition may decrease tumor cell progression (Fig. 8A). TAK‐931 (simurosertib) was used as a CDC7 inhibitor in this study.
Fig. 8.
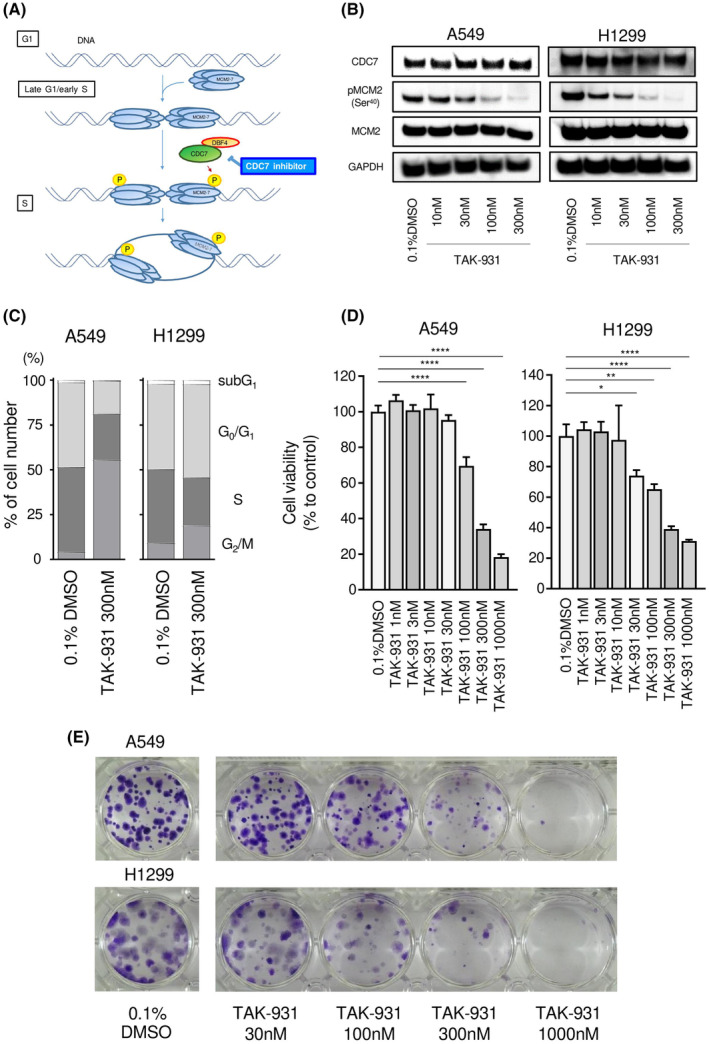
Effects of CDC7 inhibition on the phosphorylation of MCM2 and tumor progression in NSCLC. (A) The scheme shows the function of the CDC7 inhibitor. (B) Protein levels of CDC7, phospho‐MCM2, and MCM2 were evaluated by western blot analysis. Glyceraldehyde‐3‐phosphate dehydrogenase was used as a loading control. (C) Cell cycle distribution after treatment with TAK‐931 was tested by flow cytometry. The samples were evaluated 24 h after TAK‐931 administration. Cell cycle distribution was measured in triplicate. (D) Cytotoxicity was evaluated using XTT assays. The treatment duration was 5 days. Cytotoxicity assays were performed in triplicate. Data are means and standard deviations. One‐way ANOVA and Tukey tests were performed for post hoc analysis. *P < 0.05, **P < 0.01, ****P < 0.0001. (E) Colony formation assays were conducted. The TAK‐931 treatment duration was 10 days. Representative images are shown. Cells treated with 0.1% DMSO were used as the negative control.
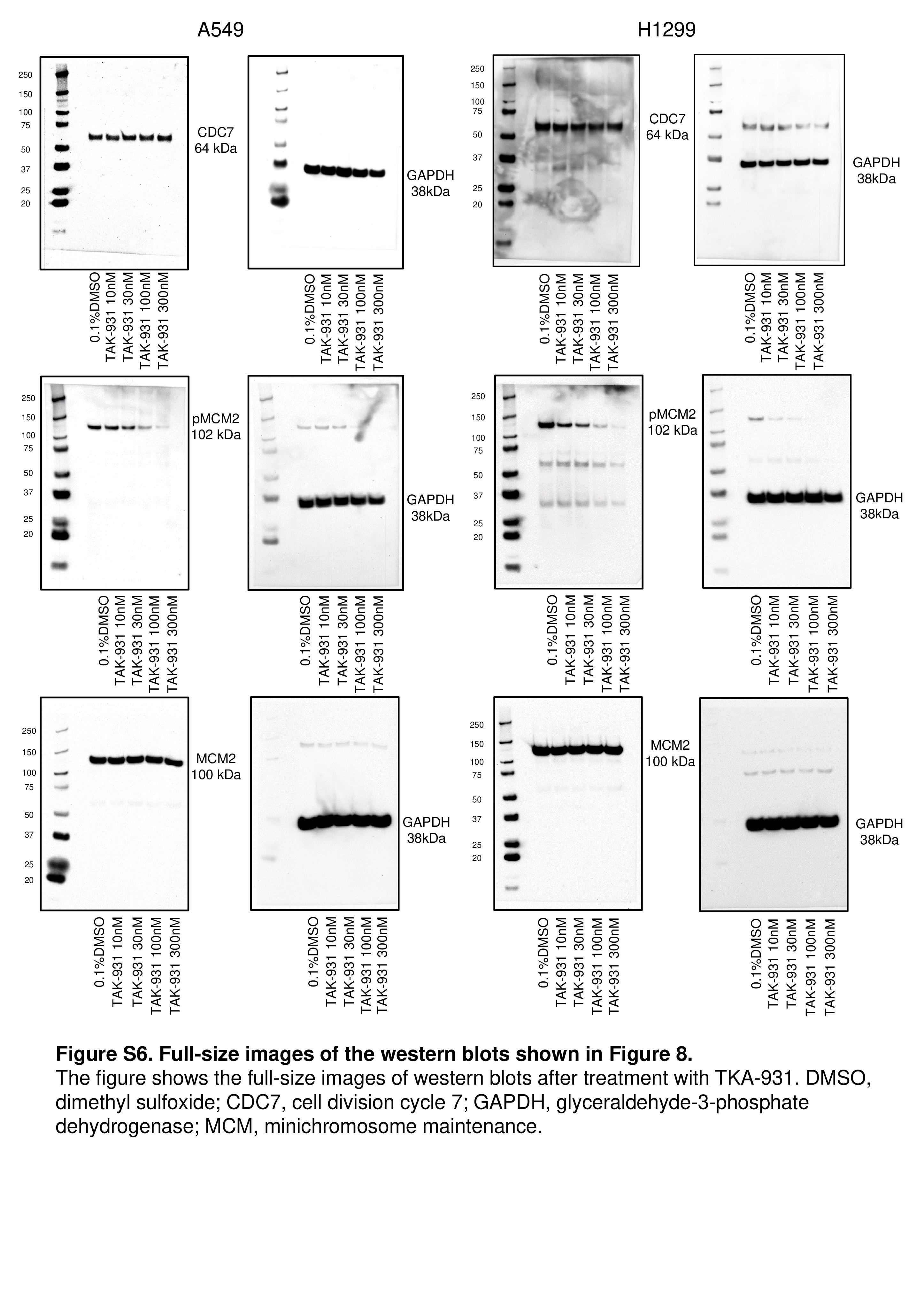
In A549 and H1299 cells, TAK‐931 blocked the phosphorylation of MCM2 (Ser40) in a concentration‐dependent manner (Fig. 8B; Fig. S6). Moreover, cell cycle assays showed that cells treated with TAK‐931 exhibited late S to late G2/M arrest in a concentration‐ and time‐dependent manner (Fig. 8C; Fig. S7). This result suggested that TAK‐931 suppressed the functions of MCM2‐MCM7 and that DNA replication was delayed in A549 and H1299 cells.
Cytotoxicity was evaluated using XTT assays. After treatment with TAK‐931, cell viability decreased significantly when the concentration of TAK‐931 was greater than 100 nm (Fig. 8D). In 10‐day colony formation assays, TAK‐931 inhibited colony formation in a concentration‐dependent manner (Fig. 8E). We found that inhibition of CDC7 suppressed tumor cell progression. CDC7 inhibitors may be another treatment strategy for NSCLC based on these findings.
Discussion
DNA replication is the most critical issue for the normal maintenance of cells, and its regulation is extremely strict. In cancer cells, however, DNA replication proceeds chaotically, which triggers abnormal cell proliferation. Aberrant expression of various genes involved in DNA replication has been reported in cancer cells [44, 45].
The hexameric protein complex formed by MCM proteins (MCM2–MCM7) is a key component of the prereplication complex and recruits other DNA replication‐associated proteins to form replication forks [42, 46]. Aberrant expression of MCM proteins has been reported in several types of cancers, for example, renal cell carcinoma, prostate cancer, and breast cancer, and these events promote genome instability and uncontrolled cell cycle progression [23, 47]. Several sets of cohort data in TCGA for LUAD revealed that all MCM family members were significantly upregulated in LUAD tissues. Moreover, MCM4, MCM5, and MCM7 expression levels may be used as prognostic molecular markers for patients with LUAD. In the future, these molecules may have applications in predicting the efficacy of treatment regimens and in the selection of therapeutic drugs for patients with LUAD.
Minichromosome maintenance family members have been reported to be phosphorylated by various kinases and activated in human cells [44, 48]. Aberrant activation of the MCM family by excess phosphorylation causes disruption of DNA replication and the cell cycle and is involved in the development and progression of cancer cells [49, 50]. MCM7 forms a complex with MCM2, MCM4, and MCM6 and has been shown to regulate the helicase activity of the complex [51]. Aberrant expression of MCM7 has been reported in several types of cancers, and its expression leads to a poor prognosis in these patients [52, 53]. MCM7 phosphorylation (Y600) through the epidermal growth factor/Lyn kinase cascade enhances MCM complex assembly and cancer cell proliferation [49]. RACK1 is a highly conserved WD40 repeat scaffold protein that is involved in several cellular processes [50]. Overexpression of RACK1 promotes AKT‐dependent phosphorylation of MCM7 and is involved in DNA replication and proliferation in NSCLC cells [50].
CDC7, a serine/threonine kinase, regulates DNA replication, cell cycle, DNA repair, and gene expression by phosphorylating many intracellular target molecules [42, 54]. Notably, MCM is a key target of CDC7, and the CDC7/DBF4 complex selectively phosphorylates MCM2 (Ser40 and Ser53) and is involved in regulating the initiation of DNA replication [55, 56]. Therefore, CDC7 is a therapeutic target molecule for cancer therapy, and various CDC7 inhibitors have been developed [42]. TAK‐931 is a novel CDC7‐selective inhibitor that has been shown to exhibit significant antitumor effects both in vitro and in vivo [57]. Our data showed that treatment of LUAD cells with TAK‐931 markedly suppressed cell proliferation by targeting MCM2 phosphorylation. The development of therapeutic agents that target the CDC7/MCM2 cascade will contribute to improving the prognosis of patients with LUAD.
Based on the epigenomic perspective, we further investigated the cause of MCM2 upregulation in LUAD cells. Numerous studies have clarified that noncoding RNAs are involved in the regulation of gene expression. In particular, miRNAs play pivotal roles as regulators of gene expression [24, 25]. Previous studies have shown that miR‐186‐3p is downregulated in cervical cancer tissues and cell lines and that the expression of this miRNA directly controls MCM2 expression [58]. In colon cancer, MCM2 expression is regulated by miR‐195‐5p and miR‐497‐5p, which are downregulated in cancer tissues and cultured cancer stem cells. Overexpression of MCM2 is involved in the maintenance of stemness [59].
Our current findings showed that miR‐139‐3p, miR‐378a‐5p, and miR‐2110 were downregulated in LUAD tissues and controlled the expression of MCM2 in LUAD cells. Previous studies have demonstrated that miR‐139‐3p was downregulated in lung squamous cell carcinoma tissues and that checkpoint kinase 1 (CHEK1) could be targeted by miR‐139‐3p [60]. Notably, CHEK1 is closely involved in various biological processes, for example, cell cycle arrest, DNA repair, and cell death [61, 62].
The Cancer Genome Atlas analysis revealed that aberrant expression of MCM genes (MCM2–MCM7) has a negative impact on prognosis with LUAD patients. In this study, we showed candidate miRNAs that negatively regulate MCM genes. For example, miR‐144‐3p, miR‐2110, miR‐30c‐2‐3p, etc. may regulate multiple MCMs from our data (Table 4 and Tables [Link], [Link]), and some of them are also downregulated in TCGA‐LUAD cohort dataset. Functional analyses of these miRNAs will be a future challenge. Elucidation of the molecular networks regulated by tumor‐suppressive miRNAs will reveal the novel oncogenic pathways underlying the malignant transformation of LUAD.
Conclusions
Analysis of TGCA‐LUAD datasets and immunostaining revealed that all MCM family members (MCM2–MCM7) were overexpressed in cancer tissues. Notably, high expression of MCM4, MCM5, and MCM7 significantly predicted 5‐year survival rates in patients with LUAD. Functional assays showed that knockdown of each MCM gene significantly suppressed the proliferation of LUAD cells. The CDC7‐selective inhibitor TAK‐931 attenuated the proliferation of LUAD cells by suppressing MCM2 phosphorylation. Moreover, MCM2 expression was controlled by miR‐139‐3p, miR‐378a‐5p, and miR‐2110. MCM family members have DNA helicase activity that is essential for the replication of genomic DNA and are closely involved in the molecular pathogenesis of LUAD. In particular, regulation of MCM2 phosphorylation may be a promising target when developing novel treatments for this disease.
Conflict of interest
The authors declare no conflict of interest.
Peer review
The peer review history for this article is available at https://www.webofscience.com/api/gateway/wos/peer‐review/10.1002/2211‐5463.13681.
Author contributions
KT, KM, HI, and NS conceived the study and designed the experiments. KT, TS, and NS wrote the manuscript. KT, YT, and SM performed the experiments. KT, YT, NK, SA, and YH analyzed the data. All authors read and approved the manuscript.
Supporting information
Fig. S1. Selection of miRNAs regulating MCM3‐MCM7 in LUAD cells.
{kind=link}
Fig. S2. mRNA and protein expression of MCM2, MCM3, MCM4, MCM5, MCM6, and MCM7 after transfection with siRNAs into NSCLC cells.
{kind=link}
Fig. S3. Full‐size images of the western blots shown in Fig. S2.
{kind=link}
Fig. S4. Histogram of cell cycle assays after transfection of A549 cells with siRNAs targeting MCM2, MCM3, MCM4, MCM5, MCM6, and MCM7.
{kind=link}
Fig. S5. Histogram of cell cycle assays after transfection of H1299 cells with siRNAs targeting MCM2, MCM3, MCM4, MCM5, MCM6, and MCM7.
{kind=link}
Fig. S6. Full‐size images of the western blots shown in Fig. 8.
{kind=link}
Fig. S7. Effects of CDC7 inhibition on MCM2 phosphorylation and late S to early G2/M cell cycle arrest in NSCLC cell lines.
{kind=link}
Table S1. Reagents used in this study.
Table S2. Characteristics of the patients used for IHC.
Table S3. Candidate tumor‐suppressive miRNAs binding to MCM3.
Table S4. Candidate tumor‐suppressive miRNAs binding to MCM4.
Table S5. Candidate tumor‐suppressive miRNAs binding to MCM5.
Table S6. Candidate tumor‐suppressive miRNAs binding to MCM6.
Table S7. Candidate tumor‐suppressive miRNAs binding to MCM7.
Acknowledgments
The current study was supported by JSPS KAKENHI Grant Number JP22K08260, JP22K16196.
Contributor Information
Keiko Mizuno, Email: keim@m.kufm.kagoshima-u.ac.jp.
Naohiko Seki, Email: naoseki@faculty.chiba-u.jp.
Data accessibility
The data that support the findings of this study are openly available in NCBI's Gene Expression Omnibus and are accessible through https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE19188, GEO Series accession number GSE19188; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE230229, GEO Series accession number GSE230229.
References
- 1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A and Bray F (2021) Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 71, 209–249. [DOI] [PubMed] [Google Scholar]
- 2. Alvarado‐Luna G and Morales‐Espinosa D (2016) Treatment for small cell lung cancer, where are we now?–A review. Transl Lung Cancer Res 5, 26–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Travis WD, Brambilla E, Nicholson AG, Yatabe Y, Austin JHM, Beasley MB, Chirieac LR, Dacic S, Duhig E, Flieder DB et al. (2015) The 2015 World Health Organization classification of lung tumors: impact of genetic, clinical and radiologic advances since the 2004 classification. J Thorac Oncol 10, 1243–1260. [DOI] [PubMed] [Google Scholar]
- 4. Cheng TY, Cramb SM, Baade PD, Youlden DR, Nwogu C and Reid ME (2016) The International Epidemiology of Lung Cancer: latest trends, disparities, and tumor characteristics. J Thorac Oncol 11, 1653–1671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hida T, Nokihara H, Kondo M, Kim YH, Azuma K, Seto T, Takiguchi Y, Nishio M, Yoshioka H, Imamura F et al. (2017) Alectinib versus crizotinib in patients with ALK‐positive non‐small‐cell lung cancer (J‐ALEX): an open‐label, randomised phase 3 trial. Lancet 390, 29–39. [DOI] [PubMed] [Google Scholar]
- 6. Ramalingam SS, Vansteenkiste J, Planchard D, Cho BC, Gray JE, Ohe Y, Zhou C, Reungwetwattana T, Cheng Y, Chewaskulyong B et al. (2020) Overall survival with osimertinib in untreated, EGFR‐mutated advanced NSCLC. N Engl J Med 382, 41–50. [DOI] [PubMed] [Google Scholar]
- 7. Gandhi L, Rodríguez‐Abreu D, Gadgeel S, Esteban E, Felip E, De Angelis F, Domine M, Clingan P, Hochmair MJ, Powell SF et al. (2018) Pembrolizumab plus chemotherapy in metastatic non‐small‐cell lung cancer. N Engl J Med 378, 2078–2092. [DOI] [PubMed] [Google Scholar]
- 8. Goldstraw P, Chansky K, Crowley J, Rami‐Porta R, Asamura H, Eberhardt WE, Nicholson AG, Groome P, Mitchell A, Bolejack V et al. (2016) The IASLC lung cancer staging project: proposals for revision of the TNM stage groupings in the forthcoming (eighth) edition of the TNM classification for lung cancer. J Thorac Oncol 11, 39–51. [DOI] [PubMed] [Google Scholar]
- 9. Heyneman LE, Herndon JE, Goodman PC and Patz EF Jr (2001) Stage distribution in patients with a small (< or = 3 cm) primary nonsmall cell lung carcinoma. Implication for lung carcinoma screening. Cancer 92, 3051–3055. [DOI] [PubMed] [Google Scholar]
- 10. Lin PY, Chang YC, Chen HY, Chen CH, Tsui HC and Yang PC (2010) Tumor size matters differently in pulmonary adenocarcinoma and squamous cell carcinoma. Lung Cancer 67, 296–300. [DOI] [PubMed] [Google Scholar]
- 11. Joo JW, Hong MH and Shim HS (2018) Clinical characteristics of T790M‐positive lung adenocarcinoma after resistance to epidermal growth factor receptor‐tyrosine kinase inhibitors with an emphasis on brain metastasis and survival. Lung Cancer 121, 12–17. [DOI] [PubMed] [Google Scholar]
- 12. Labib K and Diffley JF (2001) Is the MCM2‐7 complex the eukaryotic DNA replication fork helicase? Curr Opin Genet Dev 11, 64–70. [DOI] [PubMed] [Google Scholar]
- 13. Yu S, Wang G, Shi Y, Xu H, Zheng Y and Chen Y (2020) MCMs in cancer: prognostic potential and mechanisms. Anal Cell Pathol 2020, 3750294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhong H, Chen B, Neves H, Xing J, Ye Y, Lin Y, Zhuang G, Zhang SD, Huang J and Kwok HF (2017) Expression of minichromosome maintenance genes in renal cell carcinoma. Cancer Manag Res 9, 637–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Stewart PA, Khamis ZI, Zhau HE, Duan P, Li Q, Chung LWK and Sang QA (2017) Upregulation of minichromosome maintenance complex component 3 during epithelial‐to‐mesenchymal transition in human prostate cancer. Oncotarget 8, 39209–39217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wojnar A, Pula B, Piotrowska A, Jethon A, Kujawa K, Kobierzycki C, Rys J, Podhorska‐Okolow M and Dziegiel P (2011) Correlation of intensity of MT‐I/II expression with Ki‐67 and MCM‐2 proteins in invasive ductal breast carcinoma. Anticancer Res 31, 3027–3033. [PubMed] [Google Scholar]
- 17. Simon NE and Schwacha A (2014) The Mcm2‐7 replicative helicase: a promising chemotherapeutic target. Biomed Res Int 2014, 549719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ishimi Y, Sugiyama T, Nakaya R, Kanamori M, Kohno T, Enomoto T and Chino M (2009) Effect of heliquinomycin on the activity of human minichromosome maintenance 4/6/7 helicase. FEBS J 276, 3382–3391. [DOI] [PubMed] [Google Scholar]
- 19. Zhang X, Teng Y, Yang F, Wang M, Hong X, Ye LG, Gao YN and Chen GY (2015) MCM2 is a therapeutic target of lovastatin in human non‐small cell lung carcinomas. Oncol Rep 33, 2599–2605. [DOI] [PubMed] [Google Scholar]
- 20. Simon N, Bochman ML, Seguin S, Brodsky JL, Seibel WL and Schwacha A (2013) Ciprofloxacin is an inhibitor of the Mcm2‐7 replicative helicase. Biosci Rep 33, e00072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bruemmer D, Yin F, Liu J, Kiyono T, Fleck E, Van Herle A, Graf K and Law RE (2003) Atorvastatin inhibits expression of minichromosome maintenance proteins in vascular smooth muscle cells. Eur J Pharmacol 462, 15–23. [DOI] [PubMed] [Google Scholar]
- 22. Guan YB, Yang DR, Nong SJ, Ni J, Hu CH, Li J, Zhu J and Shan YX (2017) Breviscapine (BVP) inhibits prostate cancer progression through damaging DNA by minichromosome maintenance protein‐7 (MCM‐7) modulation. Biomed Pharmacother 93, 103–116. [DOI] [PubMed] [Google Scholar]
- 23. Yun HJ, Jeoung DJ, Jin S, Park JH, Lee EW, Lee HT, Choi YH, Kim BW and Kwon HJ (2022) Induction of cell cycle arrest, apoptosis, and reducing the expression of MCM proteins in human lung carcinoma A549 cells by Cedrol, isolated from Juniperus chinensis . J Microbiol Biotechnol 32, 918–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116, 281–297. [DOI] [PubMed] [Google Scholar]
- 25. Bartel DP (2009) MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Filipowicz W, Bhattacharyya SN and Sonenberg N (2008) Mechanisms of post‐transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet 9, 102–114. [DOI] [PubMed] [Google Scholar]
- 27. Singh SK, Pal Bhadra M, Girschick HJ and Bhadra U (2008) MicroRNAs – micro in size but macro in function. FEBS J 275, 4929–4944. [DOI] [PubMed] [Google Scholar]
- 28. Ma L (2016) MicroRNA and metastasis. Adv Cancer Res 132, 165–207. [DOI] [PubMed] [Google Scholar]
- 29. Bayraktar R and Van Roosbroeck K (2018) miR‐155 in cancer drug resistance and as target for miRNA‐based therapeutics. Cancer Metastasis Rev 37, 33–44. [DOI] [PubMed] [Google Scholar]
- 30. Majid S, Dar AA, Saini S, Chen Y, Shahryari V, Liu J, Zaman MS, Hirata H, Yamamura S, Ueno K et al. (2010) Regulation of minichromosome maintenance gene family by microRNA‐1296 and genistein in prostate cancer. Cancer Res 70, 2809–2818. [DOI] [PubMed] [Google Scholar]
- 31. Wang J, Amin A, Cheung MH, Shi L and Liang C (2022) Targeted inhibition of the expression of both MCM5 and MCM7 by miRNA‐214 impedes DNA replication and tumorigenesis in hepatocellular carcinoma cells. Cancer Lett 539, 215677. [DOI] [PubMed] [Google Scholar]
- 32. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E et al. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Anaya J (2016) OncoLnc: linking TCGA survival data to mRNAs, miRNAs, and lncRNAs. PeerJ Comput Sci 2, e67. [Google Scholar]
- 34. Mizuno K, Tanigawa K, Nohata N, Misono S, Okada R, Asai S, Moriya S, Suetsugu T, Inoue H and Seki N (2020) FAM64A: a novel oncogenic target of lung adenocarcinoma regulated by both strands of miR‐99a (miR‐99a‐5p and miR‐99a‐3p). Cells 9, 2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sanada H, Seki N, Mizuno K, Misono S, Uchida A, Yamada Y, Moriya S, Kikkawa N, Machida K, Kumamoto T et al. (2019) Involvement of dual strands of miR‐143 (miR‐143‐5p and miR‐143‐3p) and their target oncogenes in the molecular pathogenesis of lung adenocarcinoma. Int J Mol Sci 20, 4482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Misono S, Mizuno K, Suetsugu T, Tanigawa K, Nohata N, Uchida A, Sanada H, Okada R, Moriya S, Inoue H et al. (2021) Molecular signature of small cell lung cancer after treatment failure: the MCM complex as therapeutic target. Cancers 13, 1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cheng AL, Huang WG, Chen ZC, Peng F, Zhang PF, Li MY, Li F, Li JL, Li C, Yi H et al. (2008) Identification of novel nasopharyngeal carcinoma biomarkers by laser capture microdissection and proteomic analysis. Clin Cancer Res 14, 435–445. [DOI] [PubMed] [Google Scholar]
- 38. Agarwal V, Bell GW, Nam JW and Bartel DP (2015) Predicting effective microRNA target sites in mammalian mRNAs. Elife 4, e05005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang B, Kirov S and Snoddy J (2005) WebGestalt: an integrated system for exploring gene sets in various biological contexts. Nucleic Acids Res 33, W741–W748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Martens M, Ammar A, Riutta A, Waagmeester A, Slenter DN, Hanspers K, Miller RA, Digles D, Lopes EN, Ehrhart F et al. (2021) WikiPathways: connecting communities. Nucleic Acids Res 49, D613–d621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Garcia‐Moreno A, López‐Domínguez R, Villatoro‐García JA, Ramirez‐Mena A, Aparicio‐Puerta E, Hackenberg M, Pascual‐Montano A and Carmona‐Saez P (2022) Functional enrichment analysis of regulatory elements. Biomedicine 10, 590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Montagnoli A, Moll J and Colotta F (2010) Targeting cell division cycle 7 kinase: a new approach for cancer therapy. Clin Cancer Res 16, 4503–4508. [DOI] [PubMed] [Google Scholar]
- 43. Yamada M, Masai H and Bartek J (2014) Regulation and roles of Cdc7 kinase under replication stress. Cell Cycle 13, 1859–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Fei L and Xu H (2018) Role of MCM2‐7 protein phosphorylation in human cancer cells. Cell Biosci 8, 43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lei M (2005) The MCM complex: its role in DNA replication and implications for cancer therapy. Curr Cancer Drug Targets 5, 365–380. [DOI] [PubMed] [Google Scholar]
- 46. Méndez J and Stillman B (2003) Perpetuating the double helix: molecular machines at eukaryotic DNA replication origins. Bioessays 25, 1158–1167. [DOI] [PubMed] [Google Scholar]
- 47. Ishimi Y (2018) Regulation of MCM2‐7 function. Genes Genet Syst 93, 125–133. [DOI] [PubMed] [Google Scholar]
- 48. Francis LI, Randell JC, Takara TJ, Uchima L and Bell SP (2009) Incorporation into the prereplicative complex activates the Mcm2‐7 helicase for Cdc7‐Dbf4 phosphorylation. Genes Dev 23, 643–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Huang TH, Huo L, Wang YN, Xia W, Wei Y, Chang SS, Chang WC, Fang YF, Chen CT, Lang JY et al. (2013) Epidermal growth factor receptor potentiates MCM7‐mediated DNA replication through tyrosine phosphorylation of Lyn kinase in human cancers. Cancer Cell 23, 796–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fei L, Ma Y, Zhang M, Liu X, Luo Y, Wang C, Zhang H, Zhang W and Han Y (2017) RACK1 promotes lung cancer cell growth via an MCM7/RACK1/ Akt signaling complex. Oncotarget 8, 40501–40513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yabuta N, Kajimura N, Mayanagi K, Sato M, Gotow T, Uchiyama Y, Ishimi Y and Nojima H (2003) Mammalian Mcm2/4/6/7 complex forms a toroidal structure. Genes Cells 8, 413–421. [DOI] [PubMed] [Google Scholar]
- 52. Zhang J, Zhang H, Wang Y and Wang Q (2021) MCM2‐7 in clear cell renal cell carcinoma: MCM7 promotes tumor cell proliferation. Front Oncol 11, 782755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Marnerides A, Vassilakopoulos TP, Boltetsou E, Levidou G, Angelopoulou MK, Thymara I, Kyrtsonis MC, Pappi V, Tsopra O, Panayiotidis P et al. (2011) Immunohistochemical expression and prognostic significance of CCND3, MCM2 and MCM7 in Hodgkin lymhoma. Anticancer Res 31, 3585–3594. [PubMed] [Google Scholar]
- 54. Cheng AN, Fan CC, Lo YK, Kuo CL, Wang HC, Lien IH, Lin SY, Chen CH, Jiang SS, Chang IS et al. (2017) Cdc7‐Dbf4‐mediated phosphorylation of HSP90‐S164 stabilizes HSP90‐HCLK2‐MRN complex to enhance ATR/ATM signaling that overcomes replication stress in cancer. Sci Rep 7, 17024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Jiang W, McDonald D, Hope TJ and Hunter T (1999) Mammalian Cdc7‐Dbf4 protein kinase complex is essential for initiation of DNA replication. EMBO J 18, 5703–5713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cho WH, Lee YJ, Kong SI, Hurwitz J and Lee JK (2006) CDC7 kinase phosphorylates serine residues adjacent to acidic amino acids in the minichromosome maintenance 2 protein. Proc Natl Acad Sci U S A 103, 11521–11526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Iwai K, Nambu T, Dairiki R, Ohori M, Yu J, Burke K, Gotou M, Yamamoto Y, Ebara S, Shibata S et al. (2019) Molecular mechanism and potential target indication of TAK‐931, a novel CDC7‐selective inhibitor. Sci Adv 5, eaav3660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lu X, Song X, Hao X, Liu X, Zhang X, Yuan N, Ma H and Zhang Z (2021) MicroRNA‐186‐3p attenuates tumorigenesis of cervical cancer by targeting MCM2. Oncol Lett 22, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wang L, Guo J, Zhou J, Wang D, Kang X and Zhou L (2020) NF‐κB maintains the stemness of colon cancer cells by downregulating miR‐195‐5p/497‐5p and upregulating MCM2. J Exp Clin Cancer Res 39, 225. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 60. Zheng X, Zhang Y, Wu S, Jiang B and Liu Y (2022) MiR‐139‐3p targets CHEK1 modulating DNA repair and cell viability in lung squamous carcinoma cells. Mol Biotechnol 64, 832–840. [DOI] [PubMed] [Google Scholar]
- 61. Gupta N, Huang TT, Horibata S and Lee JM (2022) Cell cycle checkpoints and beyond: exploiting the ATR/CHK1/WEE1 pathway for the treatment of PARP inhibitor‐resistant cancer. Pharmacol Res 178, 106162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Matt S and Hofmann TG (2016) The DNA damage‐induced cell death response: a roadmap to kill cancer cells. Cell Mol Life Sci 73, 2829–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Selection of miRNAs regulating MCM3‐MCM7 in LUAD cells.
Fig. S2. mRNA and protein expression of MCM2, MCM3, MCM4, MCM5, MCM6, and MCM7 after transfection with siRNAs into NSCLC cells.
Fig. S3. Full‐size images of the western blots shown in Fig. S2.
Fig. S4. Histogram of cell cycle assays after transfection of A549 cells with siRNAs targeting MCM2, MCM3, MCM4, MCM5, MCM6, and MCM7.
Fig. S5. Histogram of cell cycle assays after transfection of H1299 cells with siRNAs targeting MCM2, MCM3, MCM4, MCM5, MCM6, and MCM7.
Fig. S6. Full‐size images of the western blots shown in Fig. 8.
Fig. S7. Effects of CDC7 inhibition on MCM2 phosphorylation and late S to early G2/M cell cycle arrest in NSCLC cell lines.
Table S1. Reagents used in this study.
Table S2. Characteristics of the patients used for IHC.
Table S3. Candidate tumor‐suppressive miRNAs binding to MCM3.
Table S4. Candidate tumor‐suppressive miRNAs binding to MCM4.
Table S5. Candidate tumor‐suppressive miRNAs binding to MCM5.
Table S6. Candidate tumor‐suppressive miRNAs binding to MCM6.
Table S7. Candidate tumor‐suppressive miRNAs binding to MCM7.
Data Availability Statement
The data that support the findings of this study are openly available in NCBI's Gene Expression Omnibus and are accessible through https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE19188, GEO Series accession number GSE19188; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE230229, GEO Series accession number GSE230229.