

Abstract
瘢痕疙瘩为伤口皮肤结缔组织过度增生引起的良性皮肤肿瘤。精准预测创伤者瘢痕疙瘩患病风险并及时做出早期诊断,对深度管理瘢痕疙瘩和控制其发展具有重大意义。本研究对高通量基因表达综合(GEO)数据库中的4个瘢痕疙瘩数据集进行分析,筛选出瘢痕疙瘩的诊断标志物,并建立列线图预测模型。首先,通过加权基因共表达网络分析(WGCNA)、差异表达分析和蛋白质互作网络中心性算法,筛选出37个核心蛋白质编码基因。随后,利用最小绝对值收敛和选择算子(LASSO)以及支持向量机−递归特征消除(SVM-RFE)两种机器学习算法,从中筛选出4个最具预测能力的瘢痕疙瘩诊断标志物,分别为肝细胞生长因子(HGF)、多配体蛋白聚糖4(SDC4)、外核苷酸焦磷酸酶/磷酸二酯酶2(ENPP2)和Rho家族三磷酸鸟苷酶3(RND3),并通过单基因的基因集富集分析(GSEA)探索可能涉及的生物途径。最后,对诊断标志物进行单因素与多因素逻辑回归分析,并构建列线图预测模型。经内外部验证发现,该模型校准曲线贴近理想曲线,决策曲线优于其他策略,接受者操作特征曲线下面积高于对照模型(最佳截断值为0.588),表明该模型具有较高的校准度、临床收益率以及预测能力,有望为临床诊断提供有效先期手段。
Keywords: 瘢痕疙瘩, 加权基因共表达网络分析, 最小绝对值收敛和选择算子, 支持向量机−递归特征消除, 列线图预测模型
Abstract
Keloids are benign skin tumors resulting from the excessive proliferation of connective tissue in wound skin. Precise prediction of keloid risk in trauma patients and timely early diagnosis are of paramount importance for in-depth keloid management and control of its progression. This study analyzed four keloid datasets in the high-throughput gene expression omnibus (GEO) database, identified diagnostic markers for keloids, and established a nomogram prediction model. Initially, 37 core protein-encoding genes were selected through weighted gene co-expression network analysis (WGCNA), differential expression analysis, and the centrality algorithm of the protein-protein interaction network. Subsequently, two machine learning algorithms including the least absolute shrinkage and selection operator (LASSO) and the support vector machine-recursive feature elimination (SVM-RFE) were used to further screen out four diagnostic markers with the highest predictive power for keloids, which included hepatocyte growth factor (HGF), syndecan-4 (SDC4), ectonucleotide pyrophosphatase/phosphodiesterase 2 (ENPP2), and Rho family guanosine triphophatase 3 (RND3). Potential biological pathways involved were explored through gene set enrichment analysis (GSEA) of single-gene. Finally, univariate and multivariate logistic regression analyses of diagnostic markers were performed, and a nomogram prediction model was constructed. Internal and external validations revealed that the calibration curve of this model closely approximates the ideal curve, the decision curve is superior to other strategies, and the area under the receiver operating characteristic curve is higher than the control model (with optimal cutoff value of 0.588). This indicates that the model possesses high calibration, clinical benefit rate, and predictive power, and is promising to provide effective early means for clinical diagnosis.
Keywords: Keloids, Weighted gene co-expression network analysis, Least absolute shrinkage and selection operator, Support vector machine-recursive feature elimination, Nomogram prediction model
0. 引言
瘢痕疙瘩是一种良性皮肤纤维增生性肿瘤[1-2],其主要特征是损伤愈合后,局部皮肤过度生长,形成瘢痕疙瘩[3]。瘢痕疙瘩的发生率在4.5%~16.0%之间,且可持续增长多年,很少会自然消退[4]。尽管研究人员对瘢痕疙瘩的发病机制进行了一些探索,但目前仍缺乏明确的病理诊断标准和根治方案[5]。目前,较为有效的治疗方式是尽早判断瘢痕疙瘩的发病可能,在创伤后尽快干预,并进行深度的瘢痕疙瘩管理[6],可有效延缓或控制瘢痕疙瘩的无限发展。在临床实践中,瘢痕疙瘩的诊断主要依赖于医生的主观判断[7],若患者错误进行手术切除治疗,可能会导致更大范围的瘢痕疙瘩形成[8]。因此,为了能尽早诊断瘢痕疙瘩,降低误诊率,避免瘢痕疙瘩的过度发展,急需建立一种有效的预测模型。
列线图预测模型的设计初衷是为了对数学函数或公式进行近似的图形化计算,通过图形方式表达多因素逻辑回归结果[9]。列线图预测模型已广泛应用于各种疾病的临床风险预测,例如前列腺癌和糖尿病患者术中皮肤压力性损伤的风险预测[10-11]。目前,关于瘢痕疙瘩发病预测的研究在国内外都相对有限,相关的预测模型也尚未建立。近年来,许多研究开始探索基因作为临床预测因素的可能性[12-13]。同时,生物信息学技术也在飞速发展,众多研究者尝试运用各种方法,如差异表达分析、最小绝对值收敛和选择算子(least absolute shrinkage and selection operator,LASSO)、支持向量机−递归特征消除(support vector machine-recursive feature elimination,SVM-RFE)[14-16] 、加权基因共表达网络分析(weighted gene co-expression network analysis,WGCNA)以及蛋白质互作网络中心性算法等[17],尽可能地挖掘疾病的潜在生物标志物。在此过程中,Bi等[18]通过差异表达分析与蛋白质互作网络寻找到关键的蛋白质集群。随后,Li等[19]依据WGCNA、差异表达分析与蛋白质互作网络中心性算法筛选出重要蛋白质的编码基因。同时,Yin等[20]利用差异表达分析结合机器学习算法(SVM-RFE和LASSO)挑选出瘢痕疙瘩的特征基因。尽管上述方法已展开相关应用,但遗憾的是,瘢痕疙瘩的预测特征均未与瘢痕疙瘩疾病进行关联,也未建立基于上述方法的列线图预测模型。
本文借助上述方法,为瘢痕疙瘩的预测因素提出了一种创新的深层基因信息挖掘模式。首先,结合WGCNA、差异表达分析和蛋白质互作网络中心性算法,分析基因的瘢痕疙瘩关联性、组间表达水平以及它们在蛋白质互作网络中的重要性,从而确定核心蛋白质编码基因。随后,利用机器学习算法(SVM-RFE和LASSO),识别出最具预测能力的诊断标志物,并对其进行单基因的基因集富集分析(gene set enrichment analysis,GSEA)。然后,通过多因素逻辑回归分析,建立了列线图预测模型。从内部和外部两方面对模型进行验证,评估它的准确性、校准度以及临床效用,并将其与缺失单项算法的对照模型进行比对。如图1研究流程图所示,其成果或将为临床上瘢痕疙瘩的诊断和治疗提供新的思路。
图 1.

Flowchart of the research
研究流程图
1. 方法
1.1. 数据获取和差异表达分析
美国国立生物技术信息中心的高通量基因表达综合(gene expression omnibus,GEO)数据库(网址为:http: //www.ncbi.nlm.nih.gov/geo/)是一个全球性公开资源库。该库收录并整理了世界各地研究人员上传的高通量基因组数据,并向公众提供免费下载和使用的服务。在GEO数据库中检索并下载了瘢痕疙瘩相关的GEO系列(GEO series,GSE)数据,包括GSE121618[21](5例瘢痕疙瘩和6例正常皮肤)和GSE145725[22](9例瘢痕疙瘩和10例正常皮肤)。首先,利用数据处理可视化软件R 4.2.0(Lucent Inc,美国)的替代变量分析(sva)R程序包消除数据集的批次效应。然后,使用微阵列数据线性模型(limma)R程序包[23],以|log2倍数变化| >1和检验水准为0.05(即P < 0.05表示差异具有统计学意义)为标准,对数据集基因进行筛选。最后,利用 绘图语法(ggplot2)R程序包,将这些基因展现为火山图。
1.2. 加权基因共表达网络分析筛选关键基因
根据数据准确性和样本数量的需求,选择GSE145725数据集进行WGCNA,并设置标准差阈值为0.1,剔除波动过小基因。使用WGCNA R程序包对样本进行聚类,并检测数据集是否存在离群样本[24]。在建立无尺度网络的过程中,将无尺度网络模型指数R2>0.85作为标准,计算出软阈值,并构建拓扑重叠矩阵。用模块的特征向量来计算模块关联度,并用层次聚类方法将相似模块合并,设置剪切线高度为0.65。然后,绘制了模块−特征关联图,以分析模块特征向量与疾病的相关性以及它们之间差异是否具有统计学意义。通过比较基因显著性(gene significance,GS)和模块成员关系(module membership,MM)的中位数,筛选出了高GS与MM模块基因。最后,将这些基因与差异表达基因相交,得到瘢痕疙瘩的关键基因。
1.3. 构建蛋白互作网络并筛选核心蛋白质编码基因
使用预测蛋白质−蛋白质相互作用的相邻基因重复实例搜索工具(search tool for recurring instances of neighboring genes,STRING)数据库(网址为:https://string-db.org/)[25]构建关键基因的蛋白质互作网络。接着,利用可视化分子相互作用网络软件Cytoscape 3.8.2(国际开源开发者联盟,美国)[26]的网络中心性分析插件(cytoNCA)对蛋白质互作网络的介数中心性和度中心性进行计算,选择两种算法排名前50的交集作为核心蛋白质编码基因。
1.4. 诊断标志物的鉴定与使用单基因基因集富集分析探索其生物途经
利用广义线性模型(glmnet)R程序包[27]对数据集进行LASSO回归分析,包括模型拟合、数据预测、10折交叉验证和提取模型系数。通过选取使误差最小的非负调节参数λ,提取出模型系数不为0的预测特征。接下来,使用分类与回归训练(caret)R程序包 [28]进行SVM−RFE分析,通过反向特征选择辅助函数、10折交叉验证以及带有径向基核函数的支持向量机识别最优特征集。两种机器算法的结果相交,即得到瘢痕疙瘩的诊断标志物。接着,进行单基因GSEA。根据单基因表达量的中位数,将疾病组分为低表达和高表达两个亚组,并对两个亚组进行差异表达分析。然后,基于倍数变化对数据集基因进行排序,利用生物信息学富集分析(clusterProfiler)R程序包基于京都基因和基因组百科全书(kyoto encyclopedia of genes and genomes,KEGG)数据库进行GSEA,并绘制山脊图来展示诊断标志物可能涉及的生物途经。
1.5. 列线图预测模型的构建与验证
为了增加样本数量,整合GSE121618、GSE145725、GSE92566[29]和GSE44270[30](共35例瘢痕疙瘩和33例正常皮肤)四个数据集。对这些数据集消除批次效应和进行标准化处理。利用caret R程序包进行分层比例抽样,将其分为训练组和验证组。在训练集数据上,进行诊断标志物的单因素和多因素逻辑回归分析。接着,使用回归模型策略(rms)R程序包建立列线图预测模型,并在训练组数据上进行了内部验证。绘制校准图,以评价模型的校准度。接下来,使用风险模型决策分析(rmda)R程序包,对模型进行决策曲线分析(decision curve analysis,DCA),从而评估模型的临床收益。该过程需绘制接受者操作特征(receiver operating characteristic curve,ROC)曲线,故使用部分ROC(partial ROC,pROC)R程序包,比较该模型与缺失单项算法对照模型的优劣。然后,利用报告ROC(reportROC)R程序包,进行约登指数计算,确定模型的最佳截断值。最后,利用DCA和ROC曲线,对验证组数据进行分析,以便评估模型在外部验证中的表现。
1.6. 统计分析
采用R软件 4.2.0和统计处理软件SPSS 13.0(IBM Corp.,美国)进行统计分析。首先,进行了正态分布检验,结果显示所得数据符合正态分布。对于瘢痕疙瘩和正常皮肤的组间差异,采用独立样本t检验进行比较。使用皮尔逊相关系数构建了加权基因共表达网络。对诊断标志物进行了单因素与多因素回归分析,并使用χ2检验计算了优势比和95%置信区间。为了验证模型的性能,绘制了校准曲线以观察预测结果与实际情况的拟合程度,并使用霍斯默·莱梅肖(Hosmer-Lemeshow)拟合优度检验评价模型的校准度。最后,使用约登指数确定模型的最佳截断值,并通过绘制ROC曲线和DCA评价模型的准确性和临床效用。P<0.05,表示差异具有统计学意义。
2. 实验结果与讨论
2.1. 差异表达分析和加权基因共表达网络分析筛选的瘢痕疙瘩关键基因
首先,将GSE121618和GSE145725两个数据集整合,并进行差异表达分析。如图2差异表达基因的火山图所示,在正常皮肤和瘢痕疙瘩之间存在247个差异表达基因,其中包括109个上调(红点)和138个下调基因(蓝点)。
图 2.

Results from differential expression analysis and WGCNA, and key genes in keloids
差异表达分析和WGCNA的结果以及瘢痕疙瘩的关键基因
随后,针对GSE145725数据集进行了WGCNA,以识别出与瘢痕疙瘩疾病相关的基因。依据最佳软阈值β=12构建了无尺度网络,并进行了基因的层次聚类。如图2基因树图和动态树剪切图所示,共识别出了60个模块。在合并相似模块并保留剪切树高度低于0.65的模块后,最终得到了5个模块。如图2模块−特征关联图所示,深绿松石和绿色模块的模块特征基因与瘢痕疙瘩疾病相关(相关性分别为0.88、−0.59,P<0.01)。深绿松石模块包含3 191个基因,绿色模块则包含1 926个基因。此外,深绿松石和绿色模块的GS与MM之间也存在相关性(相关性系数r分别为0.96、−0.75,P<0.01),如图2深绿松石模块和绿色模块的GS与MM图所示。在疾病相关模块中,共1 840个基因的GS和MM值大于模块的中位数,包括深绿松石模块的1 323个基因和绿色模块的517个基因。最后,通过将差异表达基因和高GS与MM模块基因取交集,如图2瘢痕疙瘩关键基因的韦恩图所示,得出了136个与瘢痕疙瘩疾病密切相关的关键基因。
2.2. 诊断标志物的鉴定及单基因基因集富集分析结果
为了筛选出蛋白质互作网络中的核心蛋白质编码基因,本文基于STRING数据库构建了关键基因的蛋白质互作网络(节点数:136,边数:779,平均节点度:11.5,平均局部聚类系数:0.305),如图3关键基因的蛋白质互作网络图所示。使用度中心性和介数中心性作为筛选指标,取两者的交集,以鉴定在蛋白质互作网络中具有信息传递能力且处于中心地位的核心蛋白质。通过筛选,得到了介数中心性和度中心性排名前50的基因的交集,共有37个核心蛋白质编码基因,如图3 核心蛋白质编码基因的韦恩图所示。接着,采用了两种机器学习算法,即SVM-RFE和LASSO,来计算对瘢痕疙瘩最具预测能力的诊断标志物。如图3 SVM-RFE交叉验证图和LASSO回归变量筛选图所示,以最小化均方根误差为目标进行交叉验证,在SVM-RFE算法中筛选出了10个基因,分别是肝细胞生长因子(hepatocyte growth factor,HGF)、多配体蛋白聚糖4(syndecan-4,SDC4)、外核苷酸焦磷酸酶/磷酸二酯酶2(ectonucleotide pyrophosphatase/phosphodiesterase 2,ENPP2)、Rho家族三磷酸鸟苷酶3(Rho family guanosine triphosphatase 3,RND3)、第1类D型肌动蛋白、C3肉毒杆菌毒素底物2、成对框基因9、肌肉结节蛋白同源序列1、前列腺素D2合成酶、肾上腺素受体β2。选择最小化10折交叉验证误差的最佳非负调节参数λ,LASSO算法筛选出了6个基因,包括ENPP2、HGF、SDC4、RND3、同源框B7、易洛魁族同源框蛋白5。最后,综合LASSO和SVM-RFE算法的结果,成功地筛选出了4个最具预测能力的基因,分别为HGF、SDC4、ENPP2和RND3,如图3瘢痕疙瘩诊断标志物的韦恩图所示。
图 3.

Screening for diagnostic markers of keloids
瘢痕疙瘩诊断标志物筛选
为了更深入地理解诊断标志物在瘢痕疙瘩中的作用,依据诊断标志物的表达量中位数将瘢痕疙瘩样本分为两组进行单基因GSEA。如图4所示,在ENPP2高表达亚组中,发现核苷酸结合寡聚化结构域(nucleotide-binding oligomerization domain,NOD)样信号通路、过氧化物酶体增殖物激活受体(peroxisome proliferator-activated receptor,PPAR)信号通路、两面神激酶−信号传导及转录激活蛋白(Janus kinase-signal transducer and activator of transcription,JAK−STAT)信号通路和核因子活化B细胞κ轻链增强子(nuclear factor kappa-light-chain-enhancer of activated B cells,NF-kappa B)信号通路等相关通路的富集。而在ENPP2低表达亚组中,N−聚糖的生物合成过程更为活跃。ENPP2可以产生磷脂酸,它通过同源G蛋白偶联受体,影响多种生物学功能,包括应激上皮细胞产生促炎信号、激活转化生长因子信号和积累成纤维细胞[31]。通过核糖核酸干扰(RNA interference,RNAi)的小鼠实验,研究人员发现ENPP2受到无翼/整合(wingless/integrated,Wnt)信号蛋白/β−连环蛋白信号通路的调控,这是调节肌细胞分化的核心机制之一[32]。此外,ENPP2还可通过N−聚糖的生物合成通路,改变细胞外基质的组成和结构[33]。
图 4.

Ridge plot of single-gene GSEA results
单基因GSEA结果的山脊图
对于HGF高表达亚组,观察到JAK−STAT信号通路、奇异(Toll)样受体信号通路、叉头盒O蛋白(forkhead box O,FoxO)信号通路、NOD样信号通路以及NF-kappa B信号通路等相关通路的富集。HGF的通路主要集中在TGF-β、肿瘤坏死因子(tumor necrosis factor,TNF)和趋化因子等信号通路,失调可能导致成纤维细胞的活化和细胞外基质蛋白的过量产生[34]。HGF在瘢痕疙瘩的发展过程中主要通过抑制胶原合成、调控细胞外基质的周转以及增强基质金属蛋白酶的表达来发挥作用[35-36]。
在RND3高表达亚组中,发现白细胞介素17信号通路、NF-kappa B信号通路、转化生长因子−β信号通路等相关通路活性增强。而在RND3低表达亚组中,单磷酸腺苷酸活化蛋白激酶(adenosine monophosphate-activated protein kinase,AMPK)信号通路、环磷酸鸟苷(cyclic guanosine monophosphate,cGMP)−蛋白激酶G(protein kinase G,PKG)信号通路和血管平滑肌收缩显得更为活跃。RND3能够调节张力纤维的收缩和肌动蛋白丝的伸长,这些过程是控制细胞骨架的重组以及细胞迁移的关键环节[37-38]。此外,RND3被发现能够降低特发性肺纤维化中纤连蛋白、I型胶原蛋白和α-平滑肌肌动蛋白的表达[39]。
在SDC4高表达亚组中,黏液素型O−聚糖生物合成、细胞周期、视黄酸诱导基因蛋白I(retinoic acid-inducible gene-I,RIG−I)样受体信号通路、细胞黏附分子、细胞外基质−受体相互作用、黏着斑等生物学功能更为活跃。而在SDC4的低表达亚组中发现,TNF信号通路和白细胞介素17信号通路的富集。SDC4是一种细胞表面糖蛋白,它通过与多个配基结合,以共受体的形式来调节细胞黏附、迁移、信号转导以及细胞增殖等生物过程[40-41]。SDC4也可能间接影响细胞周期进程,从而调控细胞增殖[42]。这些诊断标志物在免疫反应、细胞增殖、分化、迁移以及细胞外基质的组成和结构调控等多个生物过程和信号通路中发挥作用。
2.3. 基于诊断标志物的列线图预测模型
瘢痕疙瘩是皮肤损伤后伤口异常愈合的极端表现[43],无论接受何种形式的治疗,都面临极高的复发率[1]。目前尚未发现针对瘢痕疙瘩易感人群的生物标志物,这导致手术后发病风险增大[44]。研究发现,瘢痕疙瘩患者比普通人更容易患上癌症,尤其是皮肤癌[45]。此外,具有瘢痕疙瘩家族史的人群,其患病风险明显增加且病情更严重[46]。不同瘢痕疙瘩患者的易感位点各不相同,这表明瘢痕疙瘩可能符合寡基因遗传模式[47]。尽管对瘢痕疙瘩的临床诊断主要依赖病理检查,但手术操作可能加速瘢痕疙瘩的进展[48]。因此,构建基于诊断标志物的列线图预测模型可以为医生的诊断提供辅助依据,减少侵入性诊断技术带来的危害,具有较高的临床应用价值。同时,不同人种或不同地区的瘢痕疙瘩发生率存在差异,甚至同一种族在不同地区也存在发病率的差异[4, 6, 49]。因此,制定适应各个区域人群的早期瘢痕疙瘩诊断和治疗策略将对管理瘢痕疙瘩起到积极的作用。
在本研究中,采用训练组数据,以诊断标志物为自变量,病理检查结果是否为瘢痕疙瘩为因变量,进行了单因素和多因素逻辑回归分析。通过分析,得到了四个变量的回归分析参数,包括回归系数、优势比、置信区间和P值,详细参数如表1所示。单因素回归分析结果表明,ENPP2是瘢痕疙瘩发生的危险因素,而HGF、RND3和SDC4则表现为保护因素,这一发现与多因素回归分析的结果保持一致。依据多因素回归分析结果,构建了一个线性逻辑模型,如式(1)所示:
表 1. Single factor and multi-factor logistic regression analysis of each variable in the training group.
训练组各变量的单因素与多因素回归分析
| 变量 | 单因素分析 | 多因素分析 | |||
| 优势比(95%置信区间) | P值 | 优势比(95%置信区间) | 回归系数 | ||
| ENPP2 | 3.719(1.486~11.717) | 0.011 | 9.053(1.448~249.898) | 2.203 | |
| HGF | 0.599(0.244~1.318) | 0.183 | 0.272(0.034~1.148) | − 1.301 | |
| RND3 | 0.121(0.012~0.639) | 0.031 | 0.806(0.027~17.398) | − 0.339 | |
| SDC4 | 0.102(0.013~0.425) | 0.012 | 0.189(0.014~1.379) | − 1.667 | |
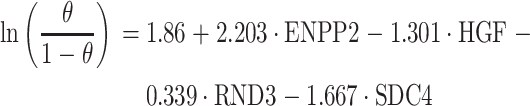 |
1 |
其中,式(1)是逻辑回归模型的对数形式,即对数几率(logit)形式,主要用于计算事件发生和不发生的比值的自然对数。该式包含了截距和各个预测变量的回归系数(截距:1.86,ENPP2:2.203,HGF:−1.301,RND3:−0.339,SDC4:−1.667)。
将式(1)的左式表示为变量y,采用logit变换将线性逻辑模型y转化为概率值θ,如式(2)所示:
 |
2 |
其中,e 是自然对数的底数,约为2.71828。概率值θ表示瘢痕疙瘩发生的概率,线性逻辑模型y是预测变量(在本例中,这些变量为ENPP2、HGF、RND3和 SDC4)的加权总和。当预测变量的值已知,可以通过模型计算出疾病发生的概率θ。
如图5训练组和验证组的ROC图所示,比较了四个模型(模型1:本文模型;模型2:WGCNA + 蛋白质互作网络中心性算法+机器学习;模型3:差异表达分析 + WGCNA + 机器学习;模型4:差异表达分析 + 蛋白质互作网络中心性算法 + 机器学习)的曲线下面积。对照实验结果表明,任何单个环节的缺失都会降低模型的准确性。相比之下,本文模型展现出较高的诊断能力。如图5列线图预测模型所示,它不仅提供了预测事件发生的概率,还阐明了预测变量影响事件发生概率的方式和程度。例如,某创伤初诊患者,其样本数据中诊断标志物RND3、SDC4、ENPP2和HGF的表达量log2值分别为5.16、5.19、4.85和1.12。通过将这些值投射到顶部标尺,得到每个变量的对应分数,将它们相加得到总分数167。然后再将总分数向下投射,即可预测该患者病理诊断为瘢痕疙瘩的概率为66.7%。通过计算约登指数,发现模型的最佳截断值为0.588;在此阈值下,模型的特异度为58.8%,敏感度为100.0%。这意味着当预测疾病概率大于58.8%时,建议进行病理检查;反之,应进行主动监测。此后,对列线图预测模型的校准曲线进行了Hosmer-Lemeshow拟合优度检验,如图5列线图预测模型的校准图所示,结果表明校准曲线与理想曲线的差异不具有统计学意义(χ2 = 5.368,P = 0.718),该模型的预测概率与实际概率接近,具有较高的校准度。此外,如图5训练组和验证组的DCA图所示,不论是在训练组还是验证组中,列线图预测模型的临床净收益都优于“单诊断标志物预测”、“全部处理”以及“不作处理”策略,显示出良好的临床收益率。经过内部和外部两方面的验证,该模型展现了较强的适应性,可用于瘢痕疙瘩的风险预测。
图 5.

Nomogram model establishment and validation
列线图预测模型建立与验证
3. 结论
本文提出了一种新颖的深层基因信息挖掘模式,旨在寻找瘢痕疙瘩的预测因素。该模式运用了WGCNA、差异表达分析、蛋白质互作网络中心性算法以及机器学习,并基于诊断标志物建立了瘢痕疙瘩的列线图预测模型。与传统依赖于临床因素的列线图预测模型相比,本文的方法结合了新兴的生物信息学技术,顺应精准医疗的发展趋势,并为瘢痕疙瘩预测研究提供了新的思路。然而,该方法仍存在改进空间,未来可考虑结合本地医院的临床数据以提高瘢痕疙瘩风险预测的准确性,并通过扩大样本规模来进一步验证其效用。此外,还需要进一步探究本文鉴定的诊断标志物在瘢痕疙瘩的分子机制和生物功能方面的作用。
重要声明
利益冲突声明:本文全体作者均声明不存在利益冲突。
作者贡献声明:李政宇主要负责数据处理、数据分析、论文撰写与修改;田保华主要负责数据分析指导、协助论文撰写、修改文章关键内容;梁海霞主要负责协助论文撰写、论文审阅修订并提供基金支持。
Funding Statement
国家自然科学基金青年项目(31501124)
National Natural Science Foundation of China
References
- 1.Tan S, Khumalo N, Bayat A Understanding keloid pathobiology from a quasi-neoplastic perspective: less of a scar and more of a chronic inflammatory disease with cancer-like tendencies. Front Immunol. 2019;10:1810. doi: 10.3389/fimmu.2019.01810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ogawa R Keloid and hypertrophic scars are the result of chronic inflammation in the reticular dermis. Int J Mol Sci. 2017;18(3):606. doi: 10.3390/ijms18030606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jfri A, O'Brien E, Alavi A, et al Association of hidradenitis suppurativa and keloid formation: a therapeutic challenge. JAAD Case Rep. 2019;5(8):675–678. doi: 10.1016/j.jdcr.2019.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Alhady S M, Sivanantharajah K Keloids in various races. a review of 175 cases. Plast Reconstr Surg. 1969;44(6):564–566. doi: 10.1097/00006534-196912000-00006. [DOI] [PubMed] [Google Scholar]
- 5.Lee H J, Jang Y J Recent understandings of biology, prophylaxis and treatment strategies for hypertrophic scars and keloids. Int J Mol Sci. 2018;19(3):711. doi: 10.3390/ijms19030711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ekstein S F, Wyles S P, Moran S L, et al Keloids: a review of therapeutic management. Int J Dermatol. 2021;60(6):661–671. doi: 10.1111/ijd.15159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wong T W, Lee J Y Should excised keloid scars be sent for routine histologic analysis? Ann Plas Surg. 2008;60(6):724. doi: 10.1097/SAP.0b013e318178d9f1. [DOI] [PubMed] [Google Scholar]
- 8.Alexandrescu D, Fabi S, Yeh L C, et al Comparative results in treatment of keloids with intralesional 5-FU/kenalog, 5-FU/verapamil, enalapril alone, verapamil alone, and laser: a case report and review of the literature. J Drugs Dermatol. 2016;15(11):1442–1447. [PubMed] [Google Scholar]
- 9.Park S Y Nomogram: an analogue tool to deliver digital knowledge. J Thorac Cardiovasc Surg. 2018;155(4):1793. doi: 10.1016/j.jtcvs.2017.12.107. [DOI] [PubMed] [Google Scholar]
- 10.南力宾, 李茹, 霍红沙, 等 基于多参数建立前列腺癌列线图预测模型及验证的研究. 大连医科大学学报. 2021;43(2):139–145. [Google Scholar]
- 11.付佳, 田甜. 糖尿病患者术中皮肤压力性损伤风险列线图预测模型的构建. 中国医科大学学报, 2021, 50(11): 1014-1019, 1025.
- 12.Zhao Z, He S, Yu X, et al Analysis and experimental validation of rheumatoid arthritis innate immunity gene CYFIP2 and pan-cancer. Front Immunol. 2022;13:954848. doi: 10.3389/fimmu.2022.954848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.武杰, 李岚, 张惠博, 等 结肠癌淋巴结转移的风险基因及列线图预测模型的构建. 肿瘤防治研究. 2020;47(12):947–952. [Google Scholar]
- 14.周嫱, 柏娜, 刘生刚, 等 基于生物信息学和机器学习方法探索缺血性脑卒中关键风险基因. 中国神经精神疾病杂志. 2022;48(9):525–532. [Google Scholar]
- 15.王玉潇, 姜威, 刘治, 等 基于共空间模式算法和支持向量机二重分类的癫痫发病预测. 生物医学工程学杂志. 2021;38(1):39–46. doi: 10.7507/1001-5515.201911042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.高智勇, 龚健雅, 秦前清, 等 支持向量机在早期癌症检测中的应用. 生物医学工程学杂志. 2005;22(5):1045–1048. [PubMed] [Google Scholar]
- 17.程丽珍, 郭起浩, 李蔚, 等 基于WGCNA分析和SVM建模对轻度认知功能障碍患者血液基因生物标志物的筛选研究. 重庆医科大学学报. 2021;46(11):1334–1341. [Google Scholar]
- 18.Bi S, Liu R, Wu B, et al Bioinformatic analysis of key genes and pathways related to keloids. Biomed Res Int. 2021;2021:5897907. doi: 10.1155/2021/5897907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li X, Jiang R, Jin H, et al Identification of hub genes of keloid fibroblasts by coexpression network analysis and degree algorithm. J Healthc Eng. 2022;2022:1272338. doi: 10.1155/2022/1272338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yin X, Bu W, Fang F, et al Keloid biomarkers and their correlation with immune infiltration. Front Genet. 2022;13:784073. doi: 10.3389/fgene.2022.784073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsumoto N M, Aoki M, Okubo Y, et al Gene expression profile of isolated dermal vascular endothelial cells in keloids. Front Cell Dev Biol. 2020;8:658. doi: 10.3389/fcell.2020.00658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kang Y, Roh M R, Rajadurai S, et al Hypoxia and HIF-1α regulate collagen production in keloids. J Invest Dermatol. 2020;140(11):2157–2165. doi: 10.1016/j.jid.2020.01.036. [DOI] [PubMed] [Google Scholar]
- 23.Ritchie M E, Phipson B, Wu D, et al limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Langfelder P, Horvath S WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szklarczyk D, Gable A L, Nastou K C, et al The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021;49(D1):D605–D612. doi: 10.1093/nar/gkaa1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shannon P, Markiel A, Ozier O, et al Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Friedman J, Hastie T, Tibshirani R Regularization paths for generalized linear models via coordinate descent. J Stat Softw. 2010;33(1):1–22. [PMC free article] [PubMed] [Google Scholar]
- 28.Van Essen D C Cortical cartography and caret software. NeuroImage. 2012;62(2):757–764. doi: 10.1016/j.neuroimage.2011.10.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fuentes-Duculan J, Bonifacio K M, Suárez-Fariñas M, et al Aberrant connective tissue differentiation towards cartilage and bone underlies human keloids in African Americans. Exp Dermatol. 2017;26(8):721–727. doi: 10.1111/exd.13271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hahn J M, Glaser K, Mcfarland K L, et al Keloid-derived keratinocytes exhibit an abnormal gene expression profile consistent with a distinct causal role in keloid pathology. Wound Repair Regen. 2013;21(4):530–544. doi: 10.1111/wrr.12060. [DOI] [PubMed] [Google Scholar]
- 31.Ninou I, Magkrioti C, Aidinis V. Autotaxin in pathophysiology and pulmonary fibrosis. Front Med(Lausanne). 2018, 5: 180.
- 32.Sah J P, Hao N T T, Han X, et al Ectonucleotide pyrophosphatase 2 (ENPP2) plays a crucial role in myogenic differentiation through the regulation by WNT/β-Catenin signaling. Int J Biochem Cell Biol. 2020;118:105661. doi: 10.1016/j.biocel.2019.105661. [DOI] [PubMed] [Google Scholar]
- 33.Ohtsubo K, Marth J D Glycosylation in cellular mechanisms of health and disease. Cell. 2006;126(5):855–867. doi: 10.1016/j.cell.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 34.Wynn T A Cellular and molecular mechanisms of fibrosis. J Pathol. 2008;214(2):199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lee W J, Park S E, Rah D K Effects of hepatocyte growth factor on collagen synthesis and matrix metalloproteinase production in keloids. J Korean Med Sci. 2011;26(8):1081–1086. doi: 10.3346/jkms.2011.26.8.1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jeon Y R, Ahn H M, Choi I K, et al Hepatocyte growth factor-expressing adenovirus upregulates matrix metalloproteinase-1 expression in keloid fibroblasts. Int J Dermatol. 2016;55(3):356–361. doi: 10.1111/ijd.12965. [DOI] [PubMed] [Google Scholar]
- 37.Guasch R M, Scambler P, Jones G E, et al RhoE regulates actin cytoskeleton organization and cell migration. Mol Cell Biol. 1998;18(8):4761–4771. doi: 10.1128/MCB.18.8.4761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhou J, Li K, Gu Y, et al Transcriptional up-regulation of RhoE by hypoxia-inducible factor (HIF)-1 promotes epithelial to mesenchymal transition of gastric cancer cells during hypoxia. Biochem Biophys Res Commun. 2011;415(2):348–354. doi: 10.1016/j.bbrc.2011.10.065. [DOI] [PubMed] [Google Scholar]
- 39.Jiang C, Huang H, Liu J, et al. Fasudil, a Rho-kinase inhibitor, attenuates bleomycin-induced pulmonary fibrosis in mice. Int J Mol Sci. 2012,13(7): 8293-8307.
- 40.Keller-Pinter A, Gyulai-Nagy S, Becsky D, et al Syndecan-4 in tumor cell motility. Cancers (Basel) 2021;13(13):3322. doi: 10.3390/cancers13133322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gattazzo F, Urciuolo A, Bonaldo P Extracellular matrix: a dynamic microenvironment for stem cell niche. Biochim Biophys Acta. 2014;1840(8):2506–2519. doi: 10.1016/j.bbagen.2014.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Massagué, J G1 cell-cycle control and cancer. Nature. 2004;432(7015):298–306. doi: 10.1038/nature03094. [DOI] [PubMed] [Google Scholar]
- 43.Jumper N, Paus R, Bayat A. Functional histopathology of keloid disease. Histol Histopathol. 2015, 30(9): 1033-1057.
- 44.Jiao H, Fan J, Cai J, et al. Analysis of characteristics similar to autoimmune disease in keloid patients. Aesthetic Plast Surg. 2015, 39(5): 818-825.
- 45.Lu Y Y, Tu H P, Wu C H, et al Risk of cancer development in patients with keloids. Sci Rep. 2021;11(1):9390. doi: 10.1038/s41598-021-88789-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lu W S, Zheng X D, Yao X H, et al. Clinical and epidemiological analysis of keloids in Chinese patients. Arch Dermatol Res. 2015, 307(2): 109-114.
- 47.Marneros A G, Norris J E, Watanabe S, et al. Genome scans provide evidence for keloid susceptibility loci on chromosomes 2q23 and 7p11. J Invest Dermatol. 2004, 122(5): 1126-1132.
- 48.Butler P D, Longaker M T, Yang G P Current progress in keloid research and treatment. J Am Coll Surg. 2008;206(4):731–741. doi: 10.1016/j.jamcollsurg.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 49.Chen Y, Gao J H, Liu X J, et al Characteristics of occurrence for Han Chinese familial keloids. Burns. 2006;32(8):1052–1059. doi: 10.1016/j.burns.2006.04.014. [DOI] [PubMed] [Google Scholar]