

Abstract
Recombinant adeno-associated viruses (rAAVs) have emerged as one of the most promising gene therapy vectors that have been successfully used in pre-clinical models of heart disease. However, this has not translated well to humans due to species differences in rAAV transduction efficiency. As a result, the search for human cardiotropic capsids is a major contemporary challenge. We used a capsid-shuffled rAAV library to perform directed evolution in human iPSC-derived cardiomyocytes (hiPSC-CMs). Five candidates emerged, with four presenting high sequence identity to AAV6, while a fifth divergent variant was related to AAV3b. Functional analysis of the variants was performed in vitro using hiPSC-CMs, cardiac organoids, human cardiac slices, non-human primate and porcine cardiac slices, as well as mouse heart and liver in vivo. We showed that cell entry was not the best predictor of transgene expression efficiency. The novel variant rAAV.KK04 was the best-performing vector in human-based screening platforms, exceeding the benchmark rAAV6. None of the novel capsids demonstrate a significant transduction of liver in vivo. The range of experimental models used revealed the value of testing for tropism differences under the conditions of human specificity, bona fide, myocardium and cell type of interest.
Keywords: adeno-associated virus, cardiotropic, hiPSC-CM, cardiac organoids, cardiac slices
Graphical abstract
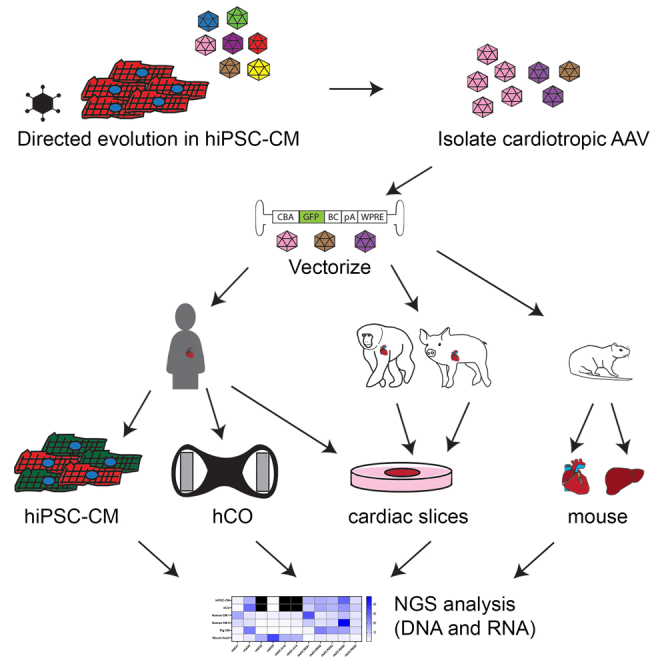
Kizana and colleagues have developed and screened cardiac-specific AAV capsids using clinically relevant models ranging from human iPSC-derived cardiomyocytes and organoids to cardiac slices generated from primary heart tissue (human, non-human primate, and pig). The novel variant rAAV.KK04 showed the most promise for efficient gene delivery to human cardiomyocytes.
Introduction
Recombinant vectors based on adeno-associated virus (rAAV) have gained traction as robust gene delivery systems that can be customized to therapeutically target a range of diseases/cell types. To date, this has resulted in seven clinically approved AAV gene therapies, for the treatment of lipoprotein lipase deficiency (AAV1, Glybera), genetic eye disease (AAV2, Luxturna), spinal muscular atrophy (AAV9, Zolgensma), hemophilia A (AAV5, Roctavian), hemophilia B (AAV5, Hemgenix), aromatic L-amino acid decarboxylase deficiency (AAV2, Upstaza), and Duchenne muscular dystrophy (AAVrh74, Elevidys).1,2,3,4,5,6,7,8,9
The application of the vector system in the context of clinically approved treatment of heart disease was heralded by the CUPID (Calcium Up-Regulation by Percutaneous Administration of Gene Therapy in Cardiac Disease) clinical trials for the treatment of heart failure with rAAV.10 In these trials, despite a promising safety signal, no benefit was observed in patients who were treated with the therapeutic vector. This disappointing result was due to low transduction of the AAV1-based vector, leading to expression levels approximately 1,000× less than achieved in preclinical models.11,12 Several additional gene therapy products using natural AAVs for cardiac disease are on the horizon with the promise of improved performance.13
We and others have reviewed the recent advances in AAV technology and the potential application of improved vectors to cardiac gene therapy.14,15,16,17 Indeed, strategies for identifying the optimal capsid for cardiac-specific delivery have already been tested in small animal models with promising results.18,19 These studies provide an important proof of concept that it is possible to generate novel AAV capsid variants, and subsequently screen for their efficacy in targeting human heart tissue.
There is the risk that vectors screened for clinical potential in animal models may not perform as expected in human patients due to species differences.20 Therefore, it is important to assess vectors in clinically relevant human models, and to undertake capsid screening in the targeted human cells. This dual/combined screening would more effectively identify at an early stage of study whether a candidate AAV capsid was worth the investment of considerable time and resources for development as a clinical vector.
Our study used directed evolution as a strategy to identify novel AAV capsids with optimal potential for gene delivery to human ventricular cardiomyocytes. To achieve this, a shuffled AAV library was generated and screened for the enrichment of cardiotropic variants after six rounds of selection in human induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs).21 The resulting five novel variants obtained were then further characterized for amino acid sequences and parental AAV contributions which may have conferred cardiotropic properties to the candidate capsid variants.
The identified AAV capsid candidates were subsequently functionally tested for gene delivery efficacy in hiPSC-CMs, human cardiac organoids (hCOs), and cardiac slices generated from human, non-human primate (NHP), and porcine left ventricular myocardium. Of note, all five identified capsids outperformed the cardiotropic AAV9. However, the top candidate for cardiac transduction in human-based screening models was rAAV.KK04. Our study confirmed the importance of screening in the most clinically relevant models, as capsids that were efficient vectors in human based models did not show the same performance in non-human screening platforms.
Results
AAV capsids obtained by directed evolution in hiPSC-CMs are related to AAV6 and AAV3b
We sought to identify novel cardiotropic AAV capsids by screening a shuffled AAV library in hiPSC-CMs.21 After six rounds of selection via wild-type (WT) adenovirus-mediated replication, the majority of the enriched cardiotropic AAV variants (AAV.KK01, AAV.KK02, AAV.KK03, and AAV.KK05) were found to be related to AAV6 (Figures 1A and 1C), with a difference of between 6 and 20 amino acid residues (Figure 1B). The most divergent variant was AAV.KK04, which had a sequence most similar to AAV3b (Figures 1A–1C). The propensity of cardiotropic variants to be related to AAV6 may indicate that this parental capsid is particularly efficient at transducing hiPSC-CMs. Indeed, we showed that AAV6 was more efficient than AAV1 and AAV9 for gene delivery to hiPSC-CMs (Figure S1A). AAV1 and AAV6 differ by only six amino acid residues, with five located in the VP3 region (Figure S1B), indicating that these amino acid residues may be critical for transduction as they are in the region most likely to exert structural changes on the capsid surface.22,23,24
Figure 1.

Cardiotropic rAAV variants obtained by directed evolution were closely related to WT AAV6 and AAV3
Directed evolution was performed using the hiPSC-CM line WTCWT. After six rounds of selection, the enriched novel AAVs were sequenced and phylogenetic analysis performed to compare with parental AAVs. (A) Clustering of amino acid sequences from novel variants (red) to WT AAV parental serotypes (black), with relationships depicted as a phylogenetic tree. (B) Heatmap illustrating the percentage identity among select parental AAVs and enriched novel variants. (C) Parental sequence contribution analysis of variants isolated after directed evolution. Black line represents the most probable composition of individual shuffled clones based on the longest sequence of identity to parental variants in a 5′ to 3′ direction. (D) Surface representations of the new capsid variants KK01, KK02, KK03, and KK05. The colored regions (yellow, orange, purple) denote surface residues that differ from the parental AAV6 capsid. Amino acid changes not located on the capsid surface are listed below. (D) Depiction as in (E) for the capsid variant KK04. The colored regions (red and purple) denote surface residues that differ from the parental AAV3b capsid. See also Figure S1.
The novel AAV variants were therefore further characterized with a focus on VP3 amino acid residues. For AAV.KK01, AAV.KK02, and AAV.KK05, there were minor predicted changes to the capsid surface relative to the parental AAV6 (Figure 1D). However, AAV.KK04 was compared with parental AAV3 and predicted to have significantly more changes to the capsid surface (Figure 1E). All the novel variants had differences in amino acid residues within VP1 and VP2, which are not depicted. However, in contrast to changes in VP3, these were not in regions that would be expected to induce structural changes on the capsid surface. (Figures S1B–S1D).
Since HSPG binding can alter capsid performance in vivo, we tested whether the novel variants were likely to bind to heparin. rAAV vectors ± soluble heparin (400 IU/mL) were used to transduce hiPSC-CMs.19,25 Both the proportion of transduced cardiomyocytes (GFP+ cTnT+ cells) and the GFP fluorescence of the cTnT population were significantly reduced in the heparin-treated groups (Figure S2). Despite differences in the capsid surface residues (Figures 1D and 1E), there has been no change in the heparin binding affinity of the novel capsids relative to parental AAV6.
We next assessed what impact the capsid sequence changes in the novel variants had on vector packaging efficiency. When the five variants and AAV6 were used to package rAAV.CBA.GFP, AAV.KK01, rAAV.KK02, and AAV.KK05 were shown to yield significantly more genome-containing particles than that of AAV6 (Figure S3). This may indicate that changes in capsid surface-exposed amino acids improved vector manufacturability of the AAV variants, which is an advantageous characteristic for translational application. The vector titer for rAAV.KK04 was the same as AAV6.
Efficient cell entry and gene expression in hiPSC-CMs transduced with rAAV.KK04 vectors
The novel capsids were vectorized and functionally compared with WT AAV1, AAV6, and AAV9 capsids using a competitive transduction assay. Each vectorized capsid was separately used to package a rAAV.CMV.GFP-BC cassette identified by a unique barcode, which allowed the pool of vectors to be mixed at equimolar ratio and used to simultaneously transduce hiPSC-CMs from three different lines obtained from Stanford Cardiovascular Institute (SCVI). These lines are designated SCVI 8, 100, and 480. To account for potential saturation of receptors at high vector dose, cells were transduced with multiplicity of transduction (MOT) 100, 1,000, and 10,000.
Testing was performed in cardiomyocytes differentiated from three hiPSC lines (Figures 2 and S4) and transduction efficiency was determined at the level of cell entry (DNA) and transgene expression (mRNA/cDNA). At all tested MOTs, rAAV.KK04 was the most efficient at cell entry and transgene expression in human cardiomyocytes. In contrast, rAAV9 was consistently most inefficient at transduction at all MOTs and in cardiomyocytes derived from all three iPSC lines, whereas rAAV6 was the best among the three WT variants tested (Figures 2A, S4A, and S4B).
Figure 2.

rAAV.KK04 was most efficient at gene delivery to hiPSC-CMs
(A) hiPSC-CMs competitively transduced with a barcoded library of rAAV.CMV.GFP vectors, then harvested at D5 post transduction. Extraction of DNA/RNA was performed, followed by analysis using next-generation sequencing (n = 3). The relative proportions of barcode reads post NGS analysis are given at the level of cell entry (gDNA) and gene expression (mRNA) for MOT 100, 1,000, and 10,000. Results are expressed as percentage of total reads for each cell line. (B) hiPSC-CMs were transduced with unbarcoded rAAV.CBA.GFP vectors at MOT 1,000, followed by analysis using microscopy and flow cytometry to quantify GFP (n = 3 per group for SCVI 8 and 100, n = 4 per group for SCVI 480) on day 5 post transduction. Fluorescence images showing GFP auto-fluorescence in live cells. Scale bars, 100 μM. (C) Flow cytometry dot plots quantifying proportions of GFP-positive cardiomyocytes (cTnT+ cells) in three hiPSC lines. Data are mean ± SEM. Statistical significance of differences was calculated using an ordinary one-way ANOVA, and the difference between the mean of the variants and AAV6 was calculated with Dunnett’s multiple comparison test (∗ p ≤ 0.05, ∗∗ p ≤ 0.01, ∗∗∗ p ≤ 0.001, ∗∗∗∗ p ≤ 0.0001).
To confirm this result, we then individually tested the five novel capsids. All vectors were used to package a non-barcoded transgene (rAAV.CBA.GFP). We observed that rAAV.KK04 induced the strongest GFP expression in cardiomyocytes across all hiPSC lines (Figure 2B). This was further quantified by flow cytometry, which showed a significantly higher proportion of transduced cardiomyocytes (Figure 2C).
Interestingly, when the promoter was changed from the ubiquitous CMV or CBA to the cardiac restricted cTnT, rAAV.KK02 showed improved transduction efficiency, which was equivalent to rAAV.KK04 (Figures S5A and S5B). Despite this unexpected result, consistent with the competitive transduction assay, these data indicate that rAAV.KK04 generally outperforms the other AAV6-based variants in hiPSC-CMs.
Efficient cell entry and gene expression observed in rAAV.KK04-transduced cardiac organoids
The maturity of hiPSC-CMs can be enhanced by culturing into hCOs.26 This system is more clinically relevant for AAV application in mature myocardium as hiPSC-CMs have a relatively fetal phenotype compared with primary cardiomyocytes. Therefore, hCOs were transduced with AAV vectors to evaluate the effect of cell maturity on transduction.
We used the barcoded rAAV.CMV.GFP-BC vectors in a competitive transduction assay (MOT = 100, 1,000, and 10,000). For these assays we focused on bulk mRNA reads, as AAV could potentially bind to the HSPGs in the matrix-rich hCO environment without cellular entry, as seems to be the result for AAV1. Consistent with our 2D hiPSC-CM data, both rAAV1 and rAAV9 had limited expression, while rAAV6 had robust expression in hCO (Figure 3A). Also consistent with 2D hiPSC-CM, the transduction efficiency rAAV.KK04 was the highest 30% and 30% at MOT 100 and 1,000, respectively (Figure 3A). However, it became outcompeted with rAAV6 at the highest MOT of 10,000 (Figure 3A).
Figure 3.

Competitive transduction assay and functional validation of novel AAV variants in hCO
(A) Human cardiac organoids were transduced with a barcoded library of rAAV.CMV.GFP vectors at MOT 100, 1,000, and 10,000. This was followed by analysis using next-generation sequencing (n = 8). The relative proportions of barcode reads post NGS analysis were given for all cell lines at the level of cell entry (gDNA) and gene expression (cDNA). Results are expressed as percentage of total reads for each cell line. (B) Human cardiac organoids were then transduced with unbarcoded rAAV vectors at MOT 10,000, followed by analysis by microscopy to quantify GFP (n = 6 or 7 per group). Fluorescence images showing GFP auto-fluorescence in live organoids. (C) GFP fluorescence intensity was quantified from microscopy acquired imaging by MATLAB. Data are mean ± SEM. Statistical significance of differences was calculated using an ordinary one-way ANOVA, and the difference between the mean of the variants and AAV6 was calculated with Dunnett’s multiple comparison test (∗ p ≤ 0.05, ∗∗ p ≤ 0.01, ∗∗∗ p ≤ 0.001, ∗∗∗∗ p ≤ 0.0001).
We also confirmed these findings using individual unbarcoded rAAV.CBA.GFP. While GFP expression was challenging to image in the 3D hCO at the lower MOT of 100 and 1,000 (data not shown), we observed robust hCOs transduced with MOT of 10,000 for rAAV6 and the new rAAV variants (Figures 3B and 3C). This experiment revealed a discrepancy between the DNA/RNA-based ranking of functional performance and that of GFP protein at high doses of each individual vector in competitive transduction, which may be explained by AAV binding in the hCO matrix. However, ranking of gene expression from the barcoded vectors is consistent with that observed in the 2D hiPSC-CM experiments.
Performance of novel variants in the myocytes of human cardiac slices
The screening platforms tested thus far were based on hiPSCs and hiPSC-derived hCOs, which are known to be relatively immature and some aspects may not be reflective of primary myocardial tissue. As such, the AAV variants were subsequently tested in cardiac slices taken from the left ventricular myocardium of two human donor hearts (Table 1). Slices were transduced with the barcoded rAAV.CMV.GFP-BC mix on the day of slicing, then collected after 48 h for analysis of transduction efficiency.
Table 1.
Demographic information for each human donor heart
| Donor heart ID no. | Heart 1 | Heart 2 |
|---|---|---|
| Donor sex | male | female |
| Donor age (years) | 51 | 51 |
| Ethnicity | Hispanic/Latino | white |
| Weight (kg) | 80.4 | 98.4 |
| Cause of death | head trauma | anoxia/cardiovascular |
Cardiac slices contain a heterogeneous population of cells. To determine the best capsid for efficiently targeting cardiomyocytes, PCM1+ nuclear sorting was used. In the nuclei from the human cardiac slices, we observed that rAAV1, rAAV6, and rAAV.KK02-05 were lowly expressed in cardiomyocytes and non-myocytes, with robust expression of rAAV.KK04 in both nuclear populations (Figure 4).
Figure 4.

Competitive transduction assay to compare efficiency of barcoded AAV variants in human myocardial slices
Cardiac slices were generated from the left ventricular myocardium of fresh human heart tissue. Slices were transduced with a barcoded library of rAAV.CMV.GFP vectors at MOT 10,000 (n = 6 from two human hearts). Two days post transduction, DNA and RNA were extracted from the slices and analyzed by next-generation sequencing. The relative proportions of barcode reads post NGS analysis are given at the level of cell entry and gene expression. Results are expressed as percentage of total reads for each condition. Only five data points are shown for PCM1+ gene expression as the AAV amplicon could not be amplified from one replicate of heart 2. Data are mean ± SEM. Statistical significance of differences was calculated using an ordinary one-way ANOVA, and the difference between the mean of the variants and AAV6 was calculated with Dunnett’s multiple comparison test (∗∗ p ≤ 0.01, ∗∗∗ p ≤ 0.001, ∗∗∗∗ p ≤ 0.0001).
Performance of novel variants in the myocytes of pig and NHP cardiac slices
To assess whether the novel AAV variants show robust cardiotropism in pre-clinical animal models, we also assessed the rAAV.KK variants in cardiac slices from NHP and pig hearts. Cardiac slices were generated and transduced with the barcoded rAAV.CMV.GFP-BC vector mix on the day of slicing.
Consistent with the human cardiac slices, rAAV1, rAAV6, and rAAV.KK02-05 drove transgene expression in pig cardiomyocytes and non-myocytes (Figure 5). However, in contrast to the human cardiac slices rAAV6, rAAV.KK02, and rAAV.KK03 gave similar expression to rAAV.KK04 (Figure 5). This was not a pig vs. primate difference as NHP sections expressed rAAV1, rAAV6, and all variants rAAV.KK01-05 with rAAV6, rAAV.KK01, and rAAV.KK02 being the highest (Figure S6).
Figure 5.

Competitive transduction assay to compare efficiency of barcoded AAV variants in pig myocardial slices
Cardiac slices were generated from the left ventricular myocardium of fresh pig heart tissue. Slices were transduced with a barcoded library of rAAV.CMV.GFP vectors at MOT 10,000 (n = 4). Two days post transduction, DNA and RNA were extracted from the slices and analyzed by next-generation sequencing. The relative proportions of barcode reads post NGS analysis are given at the level of cell entry and gene expression. Results are expressed as percentage of total reads for each condition. Data are mean ± SEM. Statistical significance of differences was calculated using an ordinary one-way ANOVA, and the difference between the mean of the variants and AAV6 was calculated with Dunnett’s multiple comparison test (∗∗ p ≤ 0.01, ∗∗∗ p ≤ 0.001, ∗∗∗∗ p ≤ 0.0001).
Novel variants do not show increased risk of liver transduction in vivo
The liver is a site of significant off-target transduction due to the propensity of AAV variants to target this organ. Therefore, it is important to determine whether our novel capsids have increased risk of off-target transduction to the liver after systemic delivery.
Male mice injected via tail vein with the barcoded rAAV vector mix were evaluated for cardiac and hepatic transduction analysis by next-generation sequencing. In the heart, cell entry was highest for rAAV.KK03, while the other four rAAV.KK variants showed reduced cell entry compared with other WT control variants (Figure 6). When gene expression at the RNA level was analyzed, rAAV9 showed the highest gene expression in the mouse heart. All five novel variants showed relatively inefficient transduction of the mouse heart.
Figure 6.

AAV variants exhibiting human cardiotropism did not show increased risk of liver transduction in vivo, although efficient cardiac expression was also not seen in the mouse heart
C57Bl6 mice (6–8 week males, n = 6) were injected with a barcoded library of rAAV.CMV.GFP vectors at 1011 vg per animal via the tail vein. Four weeks post transduction, mice were sacrificed. Heart and liver tissues were collected for DNA and RNA extraction for next-generation sequencing. The relative proportions of barcode reads post NGS analysis are given at the level of cell entry and gene expression. Results are expressed as percentage of total reads for each condition. Data are mean ± SEM. Statistical significance of differences was calculated using an ordinary one-way ANOVA, and the difference between the mean of the variants and AAV9 (for heart) or AAV8 (for liver) was calculated with Dunnett’s multiple comparison test (∗∗ p ≤ 0.01, ∗∗∗ p ≤ 0.001, ∗∗∗∗ p ≤ 0.0001).
Analysis of the liver showed that rAAV8, rAAV.rh74, rAAV.KK03, and rAAV.KK04 had more efficient cell entry compared with the other variants. The strongest gene expression was from rAAV8, with intermediate levels seen from rAAV9, rAAV.rh10, and rAAV.rh74. The four novel variants most similar to rAAV6 (rAAV.KK01-03, rAAV.KK05) showed relatively minimal gene expression in the liver. While rAAV.KK04 alone showed slightly increased gene expression, this was not higher than the WT rAAV capsids tested.
Comparison of the screening platforms tested
Despite the higher transduction efficiency of rAAV9 in the mouse heart in vivo, other capsids showed better utility in human, NHP, and pig models tested (Figure 7). It was also observed that while all novel variants showed successful transduction in human-based models, rAAV.KK04 showed the most promise overall as a cardiac-specific therapeutic vector.
Figure 7.

Summary of rAAV transduction efficiencies across all cardiac models tested
Relative proportion of barcode reads showing rAAV performance at cell entry (gDNA) and gene expression (mRNA). With the exception of the hiPSC-CM, hCO and mouse heart, all other models show the barcode reads from the myocyte fraction (identified by PCM1+ staining for myocyte nuclei in the human, NHP, and pig cardiac slices). Higher proportions are indicated by both larger dot size and darker color. Lower proportions are indicated by smaller dot size and lighter color. NA shows variants which were not tested in that particular model.
Discussion
Development of human- and organ-specific AAV vectors is critical to optimizing the safety and efficacy of gene therapy strategies targeting the heart. We have taken the approach of screening and selecting for cardiotropic AAVs from a shuffled-capsid library-derived AAV in hiPSC-CMs using directed evolution. Most of the isolated cardiotropic variants were related to AAV6, indicating that AAV6 based vectors have potential to be cardiotropic in human cells. The outstanding variant AAV.KK04 was most related to AAV.3b and either equaled or out-performed the other selected variants or naturally occurring cardiotropic AAVs in a spectrum of human cardiac models and other relevant preclinical models.
One of the factors known to affect capsid tropism is the heparin sulfate proteoglycan receptor (HSPG), which is widely distributed in the body. Variants such as AAV2 that utilize HSPG to facilitate transduction show reduced tissue specificity.27,28 AAV6 is known to bind to heparan, with this ability being attributed to a unique lysine residue at position 531 of the viral capsid protein. The novel variants AAV.KK01, AAV.KK02, AAV.KK03, and AAV.KK05 all retain lysine at this key position, which supports HSPG binding. In AAV1 and AAV9, this lysine reside is substituted with glutamate, leading to attenuated heparin binding.29 AAV.KK04 has a glutamate at this position, indicating that it is not as likely to be a heparin binding variant. However, we have shown that all novel variants, including AAV.KK04, have binding affinity to soluble heparin. While AAV9 is used in pre-clinical studies and approved for clinical application for cardiac gene therapy, we have shown that AAV9 does not efficiently transduce the human, pig, or NHP cardiomyocytes. Considering that AAV1 and AAV9 performed poorly across all human models tested, despite being identical to AAV.KK04 at position 531, our results may indicate that the transduction efficacy of rAAV.KK04 in human-based models is via a mechanism independent of HSPG binding.
The variable regions (VRs) are important to consider when comparing the structure and subsequent tropism of AAV variants. Overall, the AAV capsid structure is highly conserved, with a core eight-stranded antiparallel β barrel (βB-βI) region which gives rise to the classic icosahedral shape homologous for all serotypes. However, the VRs (I-IX), loops between the β strands that form the capsid surface are highly diverse and have a direct effect on the tropism of AAV variants.30 Among the five AAV variants studied, rAAV.KK04 was the outlier, as the overall protein sequence was most similar to AAV3b rather than AAV6. Despite this, we observed that many of the amino acid differences resulted in sequence swaps that rendered the VR I region of rAAV.KK04 identical to AAV1 and AAV6. This may have resulted in enhanced muscle tropism, as a similar mechanism was described for the engineered capsid variant AAV2.5, and may underpin the performance of rAAV.KK04 as the better performing capsid at cell entry and gene expression in hiPSC-CM and hCO.23
To broadly test the transduction efficiency of the novel capsids, we compared cassettes with three different promoters in hiPSC-CMs. The two ubiquitous promoters, CBA and CMV, showed similar results. Interestingly, when we tested a cardiac restricted promoter (cTnT), rAAV.KK04 still outperformed rAAV6, but we were surprised to see that rAAV.KK02 performance was improved beyond initial results using the CBA or CMV promoter. This may be due to the phenomenon of AAV capsids influencing the epigenetic marking of the rAAV-delivered episomal genomes. Histone epigenetic modifications related to active transcription (such as H3K4me3) may have been increased in the case of the cTnT promoter construct packaged in rAAV.KK02 due to capsid sequence-specific interaction with host proteins during vector uncoating.31
Human iPSC-CMs are relatively immature and do not fully recapitulate the primary adult cardiomyocyte. Even though AAV.KK04 was highly expressed in all cardiac models, the greatest performance was seen in hiPSCs. The variability in performance are highly likely to be due to the method in which the five novel variants were derived. Directed evolution was initially performed in hiPSC-CMs from one donor cell line. While we have shown that the high transduction efficiency of rAAV.KK04 in cardiomyocytes is consistent across the 2D and 3D cultures from multiple iPSC lines, we have also observed variation in performance across the two human hearts tested. This may be due to biological variation, leading to differences in AAV receptor usage within human cardiomyocytes. However, further validation with larger sample size is required to answer this definitively.
We also observed that rAAV.KK02 may also potentially be a promising candidate in pig and NHP heart tissue. Species differences as well as tissue maturity may explain the disparity in the performance the variants between models. DNA damage response (DDR) has been shown to regulate AAV vector expression, with DDR proteins physically binding AAV genomes and restricting transduction.32,33 The DDR is increased in hCOs, and further in fully mature cardiomyocytes, which may explain the difference in expression of the novel variants in cardiomyocytes across the different models.34
The cardiac slice experiments also show the discrepancy between cell entry and gene expression. For example, AAV.KK01 shows relatively high cell entry in human myocytes, but low transgene expression. Given the short period of time available for transduction, early differences in onset of transgene expression could reflect differences among capsid variants in the speed of uncoating and formation of double-stranded forms. It would have been ideal to perform longer-term experiments of novel AAVs in cardiac slices to assess the effect of time on transduction and vector expression.
Finally, we observed that our novel capsids could transduce human cells, but had limited utility when tested in vivo using mice. This result contrasts with previous studies that show that cardiotropic capsids selected in mouse heart can also transduce the hiPSC-CMs.18,19 These differences are most likely due to the initial selection platform used to isolate the capsids with desired specificity restricted to human cardiac cells. This therefore rendered it difficult to fully address the potential propensity of our variants to target the human liver, an important consideration as it affects the ability of AAVs to reach the heart. While we showed that our novel variants did not have increased risk of liver transduction in mice in vivo, we cannot conclude that there is no risk in the context of the human liver. There is currently no optimal in vivo model of clinical relevance as use of mice, rats, pigs, etc., would then introduce the confounding factor of species difference. As such, it would also be important to develop humanized models that would be able to overcome this challenge (dual humanized heart and liver in vivo model).
Collectively, these data suggest that rAAV.KK04 is effective and optimal in human cardiomyocytes and can also be used in large animal pig/NHP studies for pre-clinical models. However, caution may be required when other animal models are used as expression may not be robust across all animal models.
Conclusion
In conclusion, we have obtained five AAV variants via directed evolution in hiPSC-CMs. These variants showed different patterns of expression depending on whether the model for testing was based on hiPSC-CMs or primary heart tissue (human, NHP, or pig). We have demonstrated the utility of functionally validating the AAV variants at the DNA/RNA level, as capsids that show high efficiency at cell entry may not perform as the best vectors in terms of gene expression. We have also successfully applied the AAVs in a cardiac slice model and shown that nuclear separation of cardiac cell subpopulations can allow for focused analysis of vector performance specifically in cardiomyocytes. Our study indicates that rAAV.KK04 may be useful as a cardiotropic variant, and merits further validation in additional human hearts.
Materials and methods
AAV capsid library construction and directed evolution
A library of AAV capsids was constructed as described previously.21 Differentiated hiPSC-CMs in Geltrex (Gibco, Waltham, MA, no. A14133-02)-coated 24-well plates from the WTC iPSC line were obtained at >90% purity based on cardiac troponin (cTnT) expression. Cardiomyocytes were infected with four 10-fold dilutions of AAV library from 3.6 × 1010 vg. The following day, cells were washed with 1× PBS, then co-infected with WT human adenovirus 5 (Ad5) (ATCC VR-5, Manassas, VA), propagated as described previously.35 After 4 days, the cells were harvested and lysed by three freeze-thaw cycles. The clarified supernatant was then analyzed for AAV amplification by qPCR as described previously.21 Primers specific to AAV rep2 were used after each round of selection to verify AAV amplification and to select the appropriate dilution to be used for subsequent rounds of selection. The library dilution selected for the next round of selection was heated to 65°C for 30 min to inactivate the Ad5, before a dilution series was applied to the next batch of cells. In total, six rounds of selection were performed on fresh batches of hiPSC-CMs.
Phylogenetic analysis and vectorization of enriched shuffled AAV variants
After selection, AAV capsid sequences were recovered by PCR as described previously.21 Twenty clones were sequenced after each round to track the progress of selection. The amino acid sequences of enriched AAV variants were aligned to known parental AAVs using Geneious Prime 2022.2.1 (https://www.geneious.com). This software was used to align sequences shown in Figure S1. A phylogenetic tree was drawn to scale, with branch lengths measured in the number of substitutions per site. Amino acid substitutions were also displayed using a heatmap generated from GraphPad Prism v.8.2.1 for Windows (GraphPad Software, Boston, MA). To visualize parental contribution in the capsids rAAV.KK01-05, the Xover tool was used (http://qpmf.rx.umaryland.edu/xover.html) for Figure 1C. Vectorization was then performed as described previously.21 The novel capsid variants AAV.KK01-KK05 were then used to package one set of non-barcoded pAAV.CBA.GFP-expressing vectors, and another set of barcoded pAAV.CMV.GFP-BC-expressing vectors. AAV1, AAV6, and AAV9 were also included as control vectors. Vector packaging and titration was done as described previously.35 The capsids AAV.KK02, AAV.KK04, and AAV6 were also used to package pAAV.cTnT.GFP, which contains a cardiac specific promoter.
In silico capsid structure prediction
3D models of the VP monomers for the new capsid variants were generated from the protein sequence in the online tool SWISS-MODEL using the structure of AAV6 (PDB: 3OAH) or AAV3 (PDB: 3KIC) as templates, respectively.36 These reference monomer models were used to generate 60mer capsids (based on 60 copies of the VP3 protein) with the VIPERdb2 oligomer generator.37 The resulting 60mer models were imported into PyMol to generate surface maps and the amino acids changed compared with the parental AAV capsids highlighted.
Maintenance of hiPSCs and differentiation into cardiomyocytes
Four different hiPSC lines were used in this study: the WTCWT iPSC line obtained from Prof. Bruce Conklin (The J. David Gladstone Institutes) and SCVI 8, 100 and 480 iPSC lines from Prof. Joseph Wu (Stanford Cardiovascular Institute). All lines were maintained on Matrigel (Corning, New York, no. 354277) coated 60 mm cell culture dishes using the mTeSR Plus kit (STEMCELL Technologies, Vancouver, Canada, no. 05825). Upon confluence, cells were passaged as colonies using gentle cell dissociation reagent (STEMCELL Technologies, no. 07174) every 6–7 days.
For cardiomyocyte differentiation, WTCWT cells were maintained and differentiated as described previously.38,39 The SCVI 8, 100, and 480 lines were differentiated as follows. Cells were dissociated from confluent dishes on D2 (2 days prior to differentiation start on D0) using TrypLE Express Enzyme (Thermo Fisher Scientific, Waltham, MA, 12604-021), then seeded into Matrigel-coated 12-well plates at 700,000 cells/well using mTeSR Plus supplemented with Y-27632 (STEMCELL Technologies, no. 72304). On D1, a medium change was performed to remove the Y-27632. On D0, the differentiation was then commenced using the STEMdiff Cardiomyocyte Differentiation Kit according to the manufacturer’s instructions until the point of maintenance in STEMdiff Cardiomyocyte Maintenance Medium (CMM). On D11, beating cells were subjected to 2 days of metabolic selection using lactate medium composed of glucose free DMEM (Thermo Fisher Scientific, no. A14430-01) supplemented with L-(+)-lactic acid (Sigma-Aldrich, St. Louis, MO, no. L1750-10G) to final concentration of 4 mM and bovine serum albumin (Sigma, no. A9418-10G) to 15 μM. On D13, cells were then returned to CMM and maintained until differentiation completion on D15.
After 1 h of incubation with 10 μM Y-27632, differentiated cardiomyocytes were dissociated using TrypLE Express supplemented with 2 μg/mL DNase I (STEMCELL Technologies, no. 07900). Dissociated cells were then replated into Geltrex-coated 24-well plates at 500,000 cells/well using RPMI 1640 medium (Thermo Fisher Scientific, 21870076) and B27 supplement (Thermo Fisher Scientific, 17504-001) accompanied with Y-27632.
Transduction of hiPSC-CM
One well of cells was used for counting to determine viability at D2 post replating. Subsequently, cells were transduced with rAAV.GFP vectors in RPMI 1640 + B27 at MOT 1,000. For competitive transduction assays, barcoded AAV variants were mixed at equimolar ratio to total MOT of 100, 1,000, or 10,000. On D5 post transduction, cells were imaged using the Zeiss Axiovert 200M Live Cell Imaging Microscope (Zeiss, Oberkochen, Germany), then harvested for nucleic acid extraction using the AllPrep DNA/RNA Micro Kit (QIAGEN, Hilden, Germany, no. 80284) or flow cytometry to quantify transduced cardiomyocytes.
Flow cytometry
Cells were dissociated using TrypLE Express, then washed with Dulbecco’s phosphate-buffered saline without calcium and magnesium (Lonza, Basel, Switzerland, no. 12001-664). Cells were then stained using the Zombie NIR Fixable Viability Kit (BioLegend, San Diego, no. 423105 CA). After washing, cells were fixed in 4% PFA (w/v) for 30 min, then washed and further stained with BV421-conjugated mouse anti-cTnT antibody (BD Biosciences, San Diego, CA, no. 565618) for 2 h. Upon further washing, cells were analyzed on the FACSCanto II Cell Analyzer or LSRFortessa and data recorded using FACSDiva Software (BD Biosciences, Franklin Lakes, NJ). Analysis was subsequently performed using FlowJo (FlowJo, Ashland, OR) version 10.
Heparin competition assay
AAV vectors were diluted into RPMI 1640 + B27 (±400 IU/mL soluble heparin) at MOT 1,000.25 Diluted vector mix was incubated at 37°C for 15 min, then used to transduce hiPSC-CMs. Five days post transduction, cells were harvested and stained with cTnT as described above. Cells were then analyzed by flow cytometry.
Embryonic stem cell culture
Ethical approval for the use of human embryonic stem cells was obtained from QIMR Berghofer’s Ethics Committee and RCH Human Research Ethics Committee Approval (P2385) and was carried out in accordance with the National Health and Medical Research Council of Australia (NHMRC) regulations. Female HES3 (WiCell) were maintained in mTeSR PLUS (STEMCELL Technologies)/Matrigel (Millipore) and passaged using ReLeSR (STEMCELL Technologies). Quality control was performed with karyotyping and mycoplasma testing.
Cardiomyocyte and stromal cell differentiation was achieved using previously described protocols.26,40 hPSCs were seeded on Matrigel-coated flasks at 2 × 104 cells/cm2 and cultured in mTeSR PLUS for 3 days and then mTeSR-1 for 1 day prior to differentiation. To induce cardiac mesoderm, hPSCs were cultured in RPMI B27 medium (RPMI 1640 GlutaMAX + 2% B27 supplement without insulin, 200 μM L-ascorbic acid 2-phosphate sesquimagnesium salt hydrate) (Sigma) and 1% penicillin/streptomycin (Thermo Fisher Scientific), supplemented with 5 ng/mL BMP-4 (R&D Systems), 9 ng/mL activin A (R&D Systems), 5 ng/mL FGF-2 (R&D Systems), and 1 μM CHIR99021 (STEMCELL Technologies), with daily medium exchanges for 3 days. Cardiac specification was performed using RPMI B27– containing 5 μM IWP-4 (STEMCELL Technologies) for another 3 days, and then a further 7 days using 5 μM IWP-4 RPMI B27+ (RPMI1640 GlutaMAX + 2% B27 supplement with insulin, 200 μM L-ascorbic acid 2-phosphate sesquimagnesium salt hydrate and 1% penicillin/streptomycin) with medium changes every 2–3 days. The cells were then cultured another 2 days in RPMI B27+ before harvesting using 0.2% collagenase type I (Sigma) in 20% fetal bovine serum (FBS) in PBS (with Ca2+ and Mg2+) at 37°C for 1 h, and then washed before incubation in 0.25% trypsin-EDTA at 37°C for 10 min. Cells were neutralized in organoid formation medium (10% FBS, 200 μM L-ascorbic acid 2-phosphate sesquimagnesium salt hydrate and 1% penicillin/streptomycin), filtered through a 100 μm mesh cell strainer (BD Biosciences), centrifuged at 300 × g for 3 min, and re-suspended in organoid formation medium.
hCO fabrication
The hCO culture inserts were fabricated using SU-8 photolithography and PDMS molding (Mills et al., 201734). Acid-solubilized bovine collagen 1 (Devro) was salt balanced using 10× DMEM and pH neutralized using 0.1M NaOH before combining with Matrigel and then the cell suspension was placed on ice. Each hCO contained 5 × 104 cells, at a final concentration of 2.6 mg/mL collagen I and 9% Matrigel. Suspension (3.5 μL) was pipetted into the hCO culture insert and incubated at 37°C with 5% CO2 for 45–60 min to gel. After gelling, organoid formation medium was added and hCO were cultured for 2 days. The hCO were subsequently cultured for 5 days in maturation medium: 4% B27– insulin (Thermo Fisher Scientific), 1 mM glucose, 200 μM L-ascorbic acid 2-phosphate sesquimagnesium salt hydrate (Sigma), 1% penicillin/streptomycin (Thermo Fisher Scientific), 1% GlutaMAX (100×) (Thermo Fisher Scientific), 33 μg/mL aprotinin (Sigma), and 100 μM palmitate (conjugated to bovine serum albumin in B27, Sigma) in DMEM without glucose, glutamine, and phenol red (Thermo Fisher Scientific) with a medium change after 2 days.34 hCOs were then cultured in weaning medium: 4% B27– insulin (Thermo Fisher Scientific), 5.5 mM glucose, 1 nM insulin (Sigma), 200 μM L-ascorbic acid 2-phosphate sesquimagnesium salt hydrate (Sigma), 1% penicillin/streptomycin (Thermo Fisher Scientific), 1% GlutaMAX (100×) (Thermo Fisher Scientific), 33 μg/mL aprotinin (Sigma), and 100 μM palmitate (conjugated to bovine serum albumin in B27, Sigma) in DMEM without glucose, glutamine, and phenol red (Thermo Fisher Scientific).41
hCO AAV infection and analysis
After 7 days of hCO culture they were infected with AAV at the specified MOT. The hCOs were imaged using a Leica Thunder microscope after 2 days and then again after 5 days when substantial GFP was observed. Fluorescence images were captured at 5 days and intensity quantified using custom MATLAB files.34 The hCOs were subsequently snap frozen for other downstream analyses.
Generation of cardiac slices and transduction with AAV
The human hearts were acquired from deceased donors according to IRB approval obtained from the Institutional Review Boards committee at the University of Louisville. All human tissue processing had ethical approval (Western Sydney Local Health District HREC 2020/ETH02191) and were performed in accordance with the National Health and Medical Research Council (NHMRC) Code for the care and use of animals for scientific purposes. Human cardiac slices were generated from two healthy donor hearts by researchers at the University of Louisville as described previously.42 In brief, hearts were stored in cardioplegia solution. For each heart, three 1–2 cm3 blocks were cut. Each block was mounted onto a 4% agar bed with the endocardium facing up. The block was then cut in cold (4°C) oxygenated modified Tyrode’s solution using a vibrating microtome 700SMZ (Campden Instruments). Slices were cut to 300 μm thickness and transferred to modified 6 well plates for stimulated culture (10 V, 1.2 Hz continuously).43 Slices were transduced by incubating with barcoded rAAV mix at MOT 10,000 in a 2 mL volume of medium for 15 min at room temperature. Slice cultures were then supplemented with an additional 3–4 mL of medium. After 18 h incubation at 37°C, the virus-containing medium was removed and cultures replenished with fresh medium. Slices were maintained with regular change of oxygenated medium every 8 h until day 2 post slicing. At the endpoint, the slices were snap frozen and stored at –80°C.
Porcine and NHP left ventricular myocardium was obtained from tissues discarded by researchers at the Westmead Institute for Medical Research. All animal procedures had ethical approval (Western Sydney Local Health District animal ethics protocol 5155) and were performed in accordance with the National Health and Medical Research Council (NHMRC) Code for the care and use of animals for scientific purposes. Cardiac slices were generated as described previously.44 In brief, LV tissue was obtained and stored in cold cardioplegic solution within 30 min of euthanasia. Slices of 300 μm thickness were cut using a Leica vibratome (Leica VT1000S or VT1200S, Biosystems, Germany) within 4–6 h of tissue collection. Slices were maintained in transwell plates using the culture-air-liquid interface method and transduced with rAAV vectors at MOT 10,000 on the day of slicing as described previously.45 Slices were maintained until day 2 post slicing. Viability was measured as described previously.45 All slices were then imaged using a Zeiss Axiovert 200M Live Cell Imaging Microscope (Zeiss), then snap frozen and stored at – 80°C.
Nuclear extraction from heart slices
For nuclear isolation, tissues were lysed and processed as described previously with minor modifications.46 Once tissues were lysed in the dounce homogenizer, the lysate was filtered through a 70 μm filter, followed by a 40 μm filter. The lysate was centrifuged at 700 × g for 10 min. Supernatant was removed and the pellet resuspended in 1 mL of NSB. The nuclear pellet was then stained for PCM1+, washed, stained with goat anti-rabbit IgG (Sigma-Aldrich, no. HPA023370-100UL), and washed again in NSB. The nuclear pellet was stained in DAPI, then sorted using the imaging cytometer to separate the PCM1+ cardiomyocyte fraction and the PCM1− non-myocyte fraction. The sorted nuclei were immediately used for nucleic acid extraction using the AllPrep DNA/RNA Micro Kit.
Next-generation sequencing analysis
For next-generation sequencing analysis, DNA/RNA samples from hiPSC-CMs, organoids, and cardiac slices were prepared and analyzed. In brief, each variant was used to package the pAAV.CMV.GFP-BC construct. The uniquely labeled variants (two barcodes per construct) were then mixed at equimolar ratio to create a custom barcoded AAV kit. This was then used to transduce the various screening platforms described in the study. DNA and RNA were extracted and processed for next-generation sequencing. Detailed workflow for analysis using Snakemake to process reads and count barcodes, and Python script to identify barcodes corresponding to AAV variants were as described previously.35 Next-generation sequencing reads from DNA and RNA populations were normalized to the reads from the pre-transduction mix.
Transduction of mouse heart and liver
All animal procedures had ethical approval (Western Sydney Local Health District animal ethics protocol 4348) and were performed in accordance with the National Health and Medical Research Council (NHMRC) Code for the care and use of animals for scientific purposes. C57Bl6 mice (6–8 week males) were injected with 1011 vg of rAAV via the tail vein. After 4 weeks, mice were sacrificed. Livers and hearts were collected and used for DNA/RNA extraction, which were then used for subsequent next-generation sequencing analysis as described above.
Statistical analysis and summary of models
Depending on the data, different statistical analyses detailed in the figure legends were performed. For all used tests, significance was represented as follows: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. The summary in Figure 7 was generated using ggplot in Rstudio (see Tables S1 and S2 for script and data used, respectively).
Acknowledgments
We thank CMRI Vector and Genome Engineering Facility for help in vector preparation. Flow cytometry, live cell imaging using the Zeiss microscope, and PCR were performed at the Westmead Scientific Platforms, which are supported by the Westmead Research Hub, the Westmead Institute for Medical Research, the Cancer Institute New South Wales, the National Health and Medical Research Council, and the Ian Potter Foundation. This work was supported by NHMRC Project grant GNT1128864. C.Y.K. was supported by NHMRC Ideas grant APP1184929. L.L. was supported by NHMRC Project grants APP1156431 and APP1161583, and a grant from the National Science Centre, Republic of Poland (OPUS 13) (UMO-2017/25/B/NZ1/02790). J.E.H. is supported by a Snow Medical Fellowship. E.K. was funded by NHMRC Ideas grants APP1184929 and APP1188348 and NSW Health – Cardiovascular Research Capacity Program. R.M. and M.M were funded by the NIH grant R01 NIH GM082946.
Author contributions
Conceptualization, C.Y.K., M.C.-C., R.M., I.E.A., L.L., and E.K.; methodology, C.Y.K., M.C.-C., J.E.H., L.L., and E.K.; software, M.C.-C., M.M., R.F., and S.S.; validation, C.Y.K., S.T., S.I., and R.R.; investigation, C.Y.K., S.T., M.C.-C., S.I., R.S., S.H.Y.L., S.L.G., M.M., R.F., J.M., and M.K.; formal analysis, C.Y.K., S.T., M.C.-C., S.I., M.M., R.F., and L.L.; resources, L.L., R.M., J.J.H.C., A.P.H., J.E.H., T.M.A.M., L.L., and E.K.; data curation, C.Y.K., S.T., M.M., and R.F.; writing – original draft, C.Y.K., M.C.-C., and E.K.; writing – review & editing, C.Y.K., S.T., M.C.-C., S.I., R.R., R.S., S.H.Y.L., S.L.G., M.M., M.K., S.S., R.F., J.M., T.M.A.M., R.M., J.J.H.C., A.P.H., J.E.H., I.E.A., L.L., and E.K.; funding acquisition, L.L. and E.K.; visualization, C.Y.K., M.M., R.F., and S.C.; supervision, C.Y.K., R.M., J.J.H.C., A.P.H., J.E.H., I.E.A., L.L., and E.K.
Declaration of interests
C.Y.K., S.L.G., M.C.-C., I.E.A., L.L., and E.K. are inventors on patent applications filed by Children’s Medical Research Institute related to AAV capsid sequences and in vivo function of novel AAV variants. L.L. is a cofounder and a scientific advisor at LogicBio Therapeutics. L.L. and I.E.A. are co-founders of Exigen Biotherapeutics. L.L. has a sponsored research agreement with LogicBio Therapeutics. M.C.-C., I.E.A., and L.L. have IP licensed to biopharmaceutical companies. L.L. and I.A.E. have consulted on technologies addressed in this paper. L.L. and I.A.E. have stock and/or equity in companies with technology broadly related to this paper. J.E.H. is a co-founder, scientific advisor, and stockholder in Dynomics. All other authors declare no conflicts of interest.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2023.08.010.
Supplemental information
Data and code availability
The data that support the findings of this study are available from the corresponding author upon request.
References
- 1.Oh J.G., Ishikawa K. Recent highlights and advances in cardiac gene therapy. Discov. Med. 2019;28:229–235. [PMC free article] [PubMed] [Google Scholar]
- 2.Büning H. Gene therapy enters the pharma market: The short story of a long journey. EMBO Mol. Med. 2013;5:1–3. doi: 10.1002/emmm.201202291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoy S.M. Onasemnogene Abeparvovec: First Global Approval. Drugs. 2019;79:1255–1262. doi: 10.1007/s40265-019-01162-5. [DOI] [PubMed] [Google Scholar]
- 4.First systemic CRISPR agent in humans. Nat. Biotechnol. 2020;38:1364. doi: 10.1038/s41587-020-00773-8. [DOI] [PubMed] [Google Scholar]
- 5.Stroes E.S., Nierman M.C., Meulenberg J.J., Franssen R., Twisk J., Henny C.P., Maas M.M., Zwinderman A.H., Ross C., Aronica E., et al. Intramuscular administration of AAV1-lipoprotein lipase S447X lowers triglycerides in lipoprotein lipase-deficient patients. Arterioscler. Thromb. Vasc. Biol. 2008;28:2303–2304. doi: 10.1161/ATVBAHA.108.175620. [DOI] [PubMed] [Google Scholar]
- 6.Ozelo M.C., Mahlangu J., Pasi K.J., Giermasz A., Leavitt A.D., Laffan M., Symington E., Quon D.V., Wang J.-D., Peerlinck K., et al. Valoctocogene Roxaparvovec Gene Therapy for Hemophilia A. N. Engl. J. Med. 2022;386:1013–1025. doi: 10.1056/NEJMoa2113708. [DOI] [PubMed] [Google Scholar]
- 7.Pipe S.W., Leebeek F.W.G., Recht M., Key N.S., Castaman G., Miesbach W., Lattimore S., Peerlinck K., Van der Valk P., Coppens M., et al. Gene Therapy with Etranacogene Dezaparvovec for Hemophilia B. N. Engl. J. Med. 2023;388:706–718. doi: 10.1056/NEJMoa2211644. [DOI] [PubMed] [Google Scholar]
- 8.Tai C.-H., Lee N.-C., Chien Y.-H., Byrne B.J., Muramatsu S.-I., Tseng S.-H., Hwu W.-L. Long-term efficacy and safety of eladocagene exuparvovec in patients with AADC deficiency. Mol. Ther. 2022;30:509–518. doi: 10.1016/j.ymthe.2021.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mendell J.R., Sahenk Z., Lehman K., Nease C., Lowes L.P., Miller N.F., Iammarino M.A., Alfano L.N., Nicholl A., Al-Zaidy S., et al. Assessment of Systemic Delivery of rAAVrh74.MHCK7.micro-dystrophin in Children With Duchenne Muscular Dystrophy: A Nonrandomized Controlled Trial. JAMA Neurol. 2020;77:1122–1131. doi: 10.1001/jamaneurol.2020.1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gabisonia K., Recchia F.A. Gene Therapy for Heart Failure: New Perspectives. Curr. Heart Fail. Rep. 2018;15:340–349. doi: 10.1007/s11897-018-0410-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jessup M., Greenberg B., Mancini D., Cappola T., Pauly D.F., Jaski B., Yaroshinsky A., Zsebo K.M., Dittrich H., Hajjar R.J., Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease CUPID Investigators Calcium Upregulation by Percutaneous Administration of Gene Therapy in Cardiac Disease (CUPID): a phase 2 trial of intracoronary gene therapy of sarcoplasmic reticulum Ca2+-ATPase in patients with advanced heart failure. Circulation. 2011;124:304–313. doi: 10.1161/CIRCULATIONAHA.111.022889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greenberg B., Butler J., Felker G.M., Ponikowski P., Voors A.A., Desai A.S., Barnard D., Bouchard A., Jaski B., Lyon A.R., et al. Calcium upregulation by percutaneous administration of gene therapy in patients with cardiac disease (CUPID 2): a randomised, multinational, double-blind, placebo-controlled, phase 2b trial. Lancet. 2016;387:1178–1186. doi: 10.1016/S0140-6736(16)00082-9. [DOI] [PubMed] [Google Scholar]
- 13.Sheridan C. Genetic medicines aim straight for the heart. Nat. Biotechnol. 2023;41:435–437. doi: 10.1038/s41587-023-01745-4. [DOI] [PubMed] [Google Scholar]
- 14.Kok C.Y., MacLean L.M., Ho J.C., Lisowski L., Kizana E. Potential Applications for Targeted Gene Therapy to Protect Against Anthracycline Cardiotoxicity. JACC. CardioOncol. 2021;3:650–662. doi: 10.1016/j.jaccao.2021.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ravindran D., Kok C., Farraha M., Selvakumar D., Clayton Z.E., Kumar S., Chong J., Kizana E. Gene and Cell Therapy for Cardiac Arrhythmias. Clin. Therapeut. 2020;42:1911–1922. doi: 10.1016/j.clinthera.2020.09.001. [DOI] [PubMed] [Google Scholar]
- 16.Liu Y., Zhang X., Yang L. Genetic Engineering of AAV Capsid Gene for Gene Therapy Application. Curr. Gene Ther. 2020;20:321–332. doi: 10.2174/1566523220666200930105521. [DOI] [PubMed] [Google Scholar]
- 17.Sasaki N., Kok C.Y., Westhaus A., Alexander I.E., Lisowski L., Kizana E. Heart Lung Circ; 2023. In Search of Adeno-Associated Virus Vectors with Enhanced Cardiac Tropism for Gene Therapy. [DOI] [PubMed] [Google Scholar]
- 18.Weinmann J., Weis S., Sippel J., Tulalamba W., Remes A., El Andari J., Herrmann A.-K., Pham Q.H., Borowski C., Hille S., et al. Identification of a myotropic AAV by massively parallel in vivo evaluation of barcoded capsid variants. Nat. Commun. 2020;11:5432. doi: 10.1038/s41467-020-19230-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rode L., Bär C., Groß S., Rossi A., Meumann N., Viereck J., Abbas N., Xiao K., Riedel I., Gietz A., et al. AAV capsid engineering identified two novel variants with improved in vivo tropism for cardiomyocytes. Mol. Ther. 2022;30:3601–3618. doi: 10.1016/j.ymthe.2022.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lisowski L., Dane A.P., Chu K., Zhang Y., Cunningham S.C., Wilson E.M., Nygaard S., Grompe M., Alexander I.E., Kay M.A. Selection and evaluation of clinically relevant AAV variants in a xenograft liver model. Nature. 2014;506:382–386. doi: 10.1038/nature12875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cabanes-Creus M., Ginn S.L., Amaya A.K., Liao S.H.Y., Westhaus A., Hallwirth C.V., Wilmott P., Ward J., Dilworth K.L., Santilli G., et al. Codon-Optimization of Wild-Type Adeno-Associated Virus Capsid Sequences Enhances DNA Family Shuffling while Conserving Functionality. Molecular therapy. Mol. Ther. Methods Clin. Dev. 2019;12:71–84. doi: 10.1016/j.omtm.2018.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Limberis M.P., Vandenberghe L.H., Zhang L., Pickles R.J., Wilson J.M. Transduction efficiencies of novel AAV vectors in mouse airway epithelium in vivo and human ciliated airway epithelium in vitro. Mol. Ther. 2009;17:294–301. doi: 10.1038/mt.2008.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yan Z., Lei-Butters D.C.M., Keiser N.W., Engelhardt J.F. Distinct transduction difference between adeno-associated virus type 1 and type 6 vectors in human polarized airway epithelia. Gene Ther. 2013;20:328–337. doi: 10.1038/gt.2012.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang L.Y., Patel A., Ng R., Miller E.B., Halder S., McKenna R., Asokan A., Agbandje-McKenna M. Characterization of the Adeno-Associated Virus 1 and 6 Sialic Acid Binding Site. J. Virol. 2016;90:5219–5230. doi: 10.1128/JVI.00161-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cabanes-Creus M., Westhaus A., Navarro R.G., Baltazar G., Zhu E., Amaya A.K., Liao S.H.Y., Scott S., Sallard E., Dilworth K.L., et al. Attenuation of Heparan Sulfate Proteoglycan Binding Enhances In Vivo Transduction of Human Primary Hepatocytes with AAV2. Mol. Ther. Methods Clin. Dev. 2020;17:1139–1154. doi: 10.1016/j.omtm.2020.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Voges H.K., Mills R.J., Elliott D.A., Parton R.G., Porrello E.R., Hudson J.E. Development of a human cardiac organoid injury model reveals innate regenerative potential. Development. 2017;144:1118–1127. doi: 10.1242/dev.143966. [DOI] [PubMed] [Google Scholar]
- 27.Summerford C., Samulski R.J. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J. Virol. 1998;72:1438–1445. doi: 10.1128/jvi.72.2.1438-1445.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kern A., Schmidt K., Leder C., Müller O.J., Wobus C.E., Bettinger K., Von der Lieth C.W., King J.A., Kleinschmidt J.A. Identification of a heparin-binding motif on adeno-associated virus type 2 capsids. J. Virol. 2003;77:11072–11081. doi: 10.1128/JVI.77.20.11072-11081.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arnett A.L.H., Beutler L.R., Quintana A., Allen J., Finn E., Palmiter R.D., Chamberlain J.S. Heparin-binding correlates with increased efficiency of AAV1- and AAV6-mediated transduction of striated muscle, but negatively impacts CNS transduction. Gene Ther. 2013;20:497–503. doi: 10.1038/gt.2012.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Govindasamy L., Padron E., McKenna R., Muzyczka N., Kaludov N., Chiorini J.A., Agbandje-McKenna M. Structurally mapping the diverse phenotype of adeno-associated virus serotype 4. J. Virol. 2006;80:11556–11570. doi: 10.1128/JVI.01536-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gonzalez-Sandoval A., Pekrun K., Tsuji S., Zhang F., Hung K.L., Chang H.Y., Kay M.A. The AAV capsid can influence the epigenetic marking of rAAV delivered episomal genomes in a species dependent manner. Nat. Commun. 2023;14:2448. doi: 10.1038/s41467-023-38106-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lovric J., Mano M., Zentilin L., Eulalio A., Zacchigna S., Giacca M. Terminal differentiation of cardiac and skeletal myocytes induces permissivity to AAV transduction by relieving inhibition imposed by DNA damage response proteins. Mol. Ther. 2012;20:2087–2097. doi: 10.1038/mt.2012.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zentilin L., Marcello A., Giacca M. Involvement of cellular double-stranded DNA break binding proteins in processing of the recombinant adeno-associated virus genome. J. Virol. 2001;75:12279–12287. doi: 10.1128/JVI.75.24.12279-12287.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mills R.J., Titmarsh D.M., Koenig X., Parker B.L., Ryall J.G., Quaife-Ryan G.A., Voges H.K., Hodson M.P., Ferguson C., Drowley L., et al. Functional screening in human cardiac organoids reveals a metabolic mechanism for cardiomyocyte cell cycle arrest. Proc. Natl. Acad. Sci. USA. 2017;114:E8372–E8381. doi: 10.1073/pnas.1707316114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cabanes-Creus M., Navarro R.G., Zhu E., Baltazar G., Liao S.H.Y., Drouyer M., Amaya A.K., Scott S., Nguyen L.H., Westhaus A., et al. Novel human liver-tropic AAV variants define transferable domains that markedly enhance the human tropism of AAV7 and AAV8. Mol. Ther. Methods Clin. Dev. 2022;24:88–101. doi: 10.1016/j.omtm.2021.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwede T., Kopp J., Guex N., Peitsch M.C. SWISS-MODEL: An automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ho P.T., Montiel-Garcia D.J., Wong J.J., Carrillo-Tripp M., Brooks C.L., 3rd, Johnson J.E., Reddy V.S. VIPERdb: A Tool for Virus Research. Annu. Rev. Virol. 2018;5:477–488. doi: 10.1146/annurev-virology-092917-043405. [DOI] [PubMed] [Google Scholar]
- 38.Lian X., Hsiao C., Wilson G., Zhu K., Hazeltine L.B., Azarin S.M., Raval K.K., Zhang J., Kamp T.J., Palecek S.P. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc. Natl. Acad. Sci. USA. 2012;109:E1848–E1857. doi: 10.1073/pnas.1200250109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burridge P.W., Matsa E., Shukla P., Lin Z.C., Churko J.M., Ebert A.D., Lan F., Diecke S., Huber B., Mordwinkin N.M., et al. Chemically defined generation of human cardiomyocytes. Nat. Methods. 2014;11:855–860. doi: 10.1038/nmeth.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hudson J., Titmarsh D., Hidalgo A., Wolvetang E., Cooper-White J. Primitive cardiac cells from human embryonic stem cells. Stem Cell. Dev. 2012;21:1513–1523. doi: 10.1089/scd.2011.0254. [DOI] [PubMed] [Google Scholar]
- 41.Mills R.J., Humphrey S.J., Fortuna P.R.J., Lor M., Foster S.R., Quaife-Ryan G.A., Johnston R.L., Dumenil T., Bishop C., Rudraraju R., et al. BET inhibition blocks inflammation-induced cardiac dysfunction and SARS-CoV-2 infection. Cell. 2021;184:2167–2182.e22. doi: 10.1016/j.cell.2021.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ou Q., Jacobson Z., Abouleisa R.R.E., Tang X.L., Hindi S.M., Kumar A., Ivey K.N., Giridharan G., El-Baz A., Brittian K., et al. Physiological Biomimetic Culture System for Pig and Human Heart Slices. Circ. Res. 2019;125:628–642. doi: 10.1161/CIRCRESAHA.119.314996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ou Q., Abouleisa R.R.E., Tang X.L., Juhardeen H.R., Meki M.H., Miller J.M., Giridharan G., El-Baz A., Bolli R., Mohamed T.M.A. Slicing and Culturing Pig Hearts under Physiological Conditions. J. Vis. Exp. 2020 doi: 10.3791/60913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Watson S.A., Scigliano M., Bardi I., Ascione R., Terracciano C.M., Perbellini F. Preparation of viable adult ventricular myocardial slices from large and small mammals. Nat. Protoc. 2017;12:2623–2639. doi: 10.1038/nprot.2017.139. [DOI] [PubMed] [Google Scholar]
- 45.Liu Z., Klose K., Neuber S., Jiang M., Gossen M., Stamm C. Comparative analysis of adeno-associated virus serotypes for gene transfer in organotypic heart slices. J. Transl. Med. 2020;18:437. doi: 10.1186/s12967-020-02605-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bergmann O., Jovinge S. Isolation of Cardiomyocyte Nuclei from Post-mortem Tissue. JoVE. 2012:e4205. doi: 10.3791/4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request.