

SUMMARY
The immunopathogenesis of inflammatory bowel disease (IBD) has been attributed to a combination of host genetics and intestinal dysbiosis. Previous work in a small cohort of IBD patients suggested that pro-inflammatory bacterial taxa are highly coated with secretory immunoglobulin IgA. Using bacterial fluorescence-activated cell sorting coupled with 16S rRNA gene sequencing (IgA-SEQ), we profiled IgA coating of intestinal microbiota in a large cohort of IBD patients and identified bacteria associated with disease and treatment. Forty-three bacterial taxa displayed significantly higher IgA coating in IBD compared with controls, including 8 taxa exhibiting differential IgA coating but similar relative abundance. Patients treated with anti-TNF-α therapies exhibited dramatically altered microbiota-specific IgA responses compared with controls. Furthermore, increased IgA coating of Oscillospira was associated with a delay in time to surgery. These results demonstrate that investigating IgA responses to microbiota can uncover potential disease-modifying taxa and reveal improved biomarkers of clinical course in IBD.
Graphical Abstract
In Brief
Shapiro et al. explored patterns of secretory IgA-coated bacteria in a cohort of patients with inflammatory bowel disease (IBD). Medications, specifically TNFα inhibitors, had a notable impact on microbiota-specific IgA responses. IgA coating of Oscillospira was associated with a delay in surgery. IgA-SEQ may serve as a biomarker in IBD.
INTRODUCTION
The pathogenesis of inflammatory bowel disease (IBD), including Crohn’s disease (CD) and ulcerative colitis (UC), has been related to a combination of factors including defects in the innate and adaptive immune system, various environmental exposures, and alterations in the gut microbiome (Khor et al., 2011; Ng et al., 2013; Manichanh et al., 2012). Genome-wide association studies (GWAS) have identified over 200 IBD susceptibility loci to date, a large fraction of which are associated with microbial defense pathways (de Lange et al., 2017). Genetic predisposition alone, however, is insufficient to explain disease onset and environmental factors thus also play critical roles in disease etiology (Ananthakrishnan et al., 2018). Despite ongoing debates regarding the relative importance of various genetic and environmental contributors to disease, current models have coalesced around a core mechanism of disease that is characterized by a dysregulated immune response to the commensal microbiota in genetically susceptible individuals (Knights et al., 2014).
Alterations in gut microbiota composition, which are often referred to as dysbiosis, have been proposed to play a critical role in IBD pathogenesis (Knights et al., 2014; Kostic et al., 2014). Dysbiosis in IBD can take various forms, including overall decreases in microbial diversity, increases in the abundance of deleterious taxa, or decreases in abundance of protective taxa. Whether these changes are a root cause of disease or are secondary effects of (chronic) mucosal inflammation and/or treatment exposures is difficult to assess. We and others have previously proposed that, similar to mouse models of IBD, individual pathogenic members of the microbiota (“pathobionts”) may directly drive the development of IBD in a subset of patients by triggering chronic inflammatory responses in the intestine. To test this theory, we developed a technology called IgA-SEQ that combines bacterial cell sorting with 16S rRNA gene sequencing to determine taxa-specific levels of coating with the secreted mucosal immunoglobulin IgA (Palm et al., 2014). We and others have shown that coating with IgA can be used as a proxy to identify putative immunostimulatory or immunoregulatory members of the microbiota, including specific members of the human microbiota that confer profound susceptibility to mouse models of IBD upon transplantation into germ-free mice (Palm et al., 2014; Viladomiu et al., 2017). Nonetheless, the full spectrum of commensal microbes that interact intimately with the mucosal immune system and potentially drive intestinal inflammation and IBD progression, remains to be defined (Shapiro et al., 2015).
Here, we report a large-scale evaluation of IgA coating of the gut microbiota in a clinically well-defined cohort of IBD patients, the Ocean State Crohn’s and Colitis Area Registry (OSCCAR) (Sands et al., 2009). We evaluated patterns of IgA coating, as well as traditional relative abundance measurements in 184 IBD patients and 32 healthy controls and integrated these data with the rich clinical datasets available from the OSCCAR cohort, including IBD phenotype, inflammatory burden, medication exposures, and clinical outcomes, including the risk of surgical resection. We found that IgA-SEQ revealed multiple potential bacterial contributors to IBD that were identified exclusively based on IgA coating and that low IgA coating of one specific taxon, Oscillospira, was predictive of progression to surgical resection. We also noted a significant change in bacterial IgA-coating patterns in anti-TNFα-treated patients, suggesting that these medications may ameliorate disease and reduce IgA coating by addressing a core molecular mediator of commensal-driven inflammation in IBD. These studies thus expand our understanding of immune-microbiota interactions in IBD, suggest potential targets for the treatment or prevention of IBD, and reveal unique biomarkers that may predict disease progression.
RESULTS
The OSCCAR Patient Cohort
We performed 16S rRNA gene sequencing and IgA-SEQ on 184 baseline fecal samples from the OSCCAR cohort. We determined the microbial composition of both IgA-coated and non-coated fractions, as well as unsorted microbial composition via sequencing and analysis of the V4 region of the 16S rRNA gene. To control for read coverage and to maximize concordance between microbial composition and clinical phenotypes, we selected samples that were collected within 200 days of IBD diagnosis and with sequencing coverage over 1,000 reads per sample for all sorting conditions for further analysis (n = 184). Table 1 summarizes this cohort relative to disease phenotype at the time of study enrollment, as expressed by the Montreal Classification System (Silverberg et al., 2005). C-reactive protein (CRP) and fecal calprotectin were measured as surrogate markers of inflammation in all patients. In CD patients, we observed a significant correlation between CRP and calprotectin (Figure S1A, left). In UC patients, however, CRP levels did not correlate with calprotectin levels (Figure S1A, right). There were no significant differences in calprotectin or CRP levels according to disease type (CD vs UC, Figure S1B), disease location (Figure S1C), or disease behavior or extension in CD patients or UC patients, respectively (Figures S1D and S1E). The majority of patients in OSCCAR were enrolled within 3 months of IBD diagnosis and these markers thus reflect early IBD disease course after initiation of treatment.
Table 1.
Clinical Characteristics at Time of Baseline Stool Collection
| OSCCAR N = 184 |
|
|---|---|
| Age, Mean (Range) | 31.6 (4–83) |
| Sex, N (%) | |
| Male | 76 (41) |
| Female | 108 (59) |
| Diagnosis | |
| CD, N (%) | 115 (62) |
| Disease Location | |
| L1 – Ileal | 25 (22) |
| L2 – Colonic | 53 (46) |
| L3 - Ileocolonic | 34 (30) |
| L4a- Upper GI tract | 31 (27) |
| Disease Behavior | |
| B1 – Inflammatory | 94 (82) |
| B2 – Stricturing | 15 (13) |
| B3 – Penetrating | 6 (5) |
| Pb – Perianal | 10 (9) |
| UC, N (%) | |
| 69 (38) | |
| E1 – Proctitis | 13 (19) |
| E2 – Left sided | 22 (32) |
| E3 - Pancolitis | 34 (49) |
| Medications | |
| Corticosteroids | 81 (44) |
| Antibioticsc | 45 (25) |
| Immune Modulatorsd | 11 (6) |
| Biologics (anti-TNF α) | 6 (3) |
Concomitant upper tract (L4)
or perianal disease (P),which can be present in addition to the luminal disease distributions summarized above.
Antibiotics limited to metronidazole and ciprofloxacin.
Immune modulators include 6-mercaptopurine, azathioprine, and methotrexate.
Microbiota Composition in OSCCAR Patients
IBD patients exhibit characteristic alterations in gut microbiota composition (Knights et al., 2014; Gevers et al., 2014). As expected, when we evaluated unsorted (i.e., total) microbiota composition, we observed that IBD patients and healthy controls exhibited distinct microbial community compositions. Principal coordinate analysis (PCoA) based on unweighted UniFrac revealed significant differences in microbiota composition between healthy controls and IBD patients (Figure 1A). However, microbial alpha diversity was not significantly different between healthy, CD, and UC (Figure 1B), although CD and UC patients both had higher variance than healthy subjects (F test; p = 0.0004 and p = 0.0001, respectively). In CD, disease behavior and location were not significantly associated with alpha diversity (ANOVA and Tukey’s HSD test, p > 0.05). In UC, pancolitis had significantly lower diversity than proctitis (ANOVA and Tukey’s HSD, p = 0.001). As previously reported, several taxa displayed differential relative abundance in IBD patients versus controls (Figure 1C). For example, we observed a significant enrichment of the phyla Actinobacteria and Bacteroidetes and a moderate enrichment of Proteobacteria in IBD, whereas Firmicutes and Verrucomicrobia were depleted compared with controls (Figure 1D). A random forest classifier trained on the unsorted microbial composition (Figure 1E) distinguished between controls and IBD patients with high accuracy (area under the curve AUC = 0.997). Within the group of IBD patients, CD and UC patients were indistinguishable using this approach (AUC = 0.454).
Figure 1. Microbiome Characterization of OSCCAR Patients and Healthy Controls.
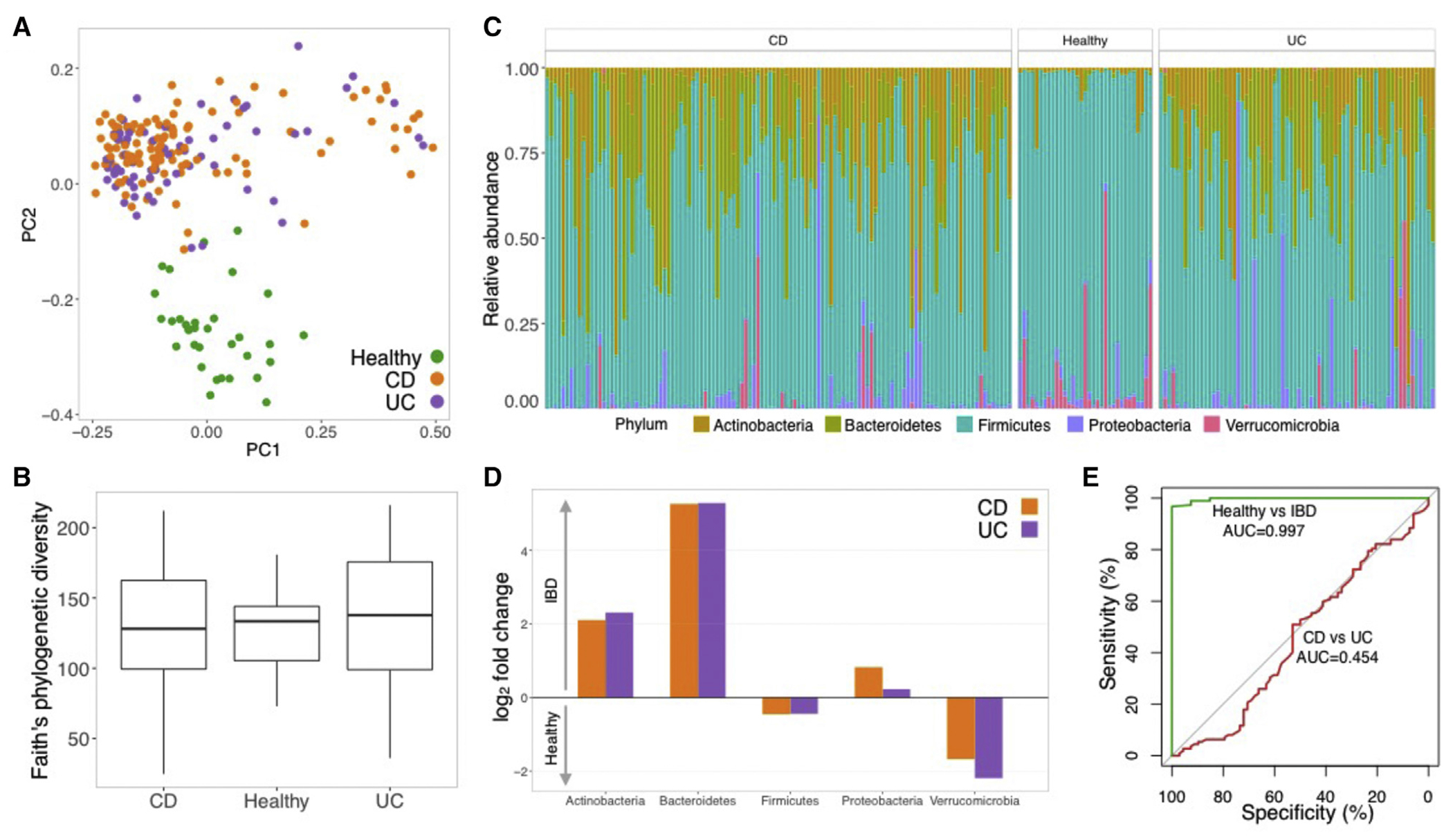
(A) PCoA based on unweighted UniFrac distances. Samples colored by disease status: healthy (green, N = 33); CD (orange, N = 114); UC (purple; N = 68).
(B) Boxplot with alpha diversity estimated using Faith’s phylogenetic diversity in healthy, CD, and UC subjects.
(C) Relative abundance of different phyla in each subject.
(D) Bar plot with fold change in the mean relative abundance of different phyla in IBD (top) versus healthy (bottom). Orange bars indicate abundance in CD; purple indicates relative abundance in UC.
(E) ROC curve constructed from a random forest model trained to distinguish healthy versus IBD patients (green curve; AUC = 0.997) and CD versus UC patients (red curve; AUC = 0.454).
IgA-SEQ Highlights a Unique Subset of Putative Immunostimulatory Human Gut Bacteria in Patients with IBD
To evaluate the full spectrum of potentially immunostimulatory gut microbes that may contribute to IBD, we performed IgA-SEQ on baseline fecal samples from OSCCAR and an established cohort of geographically similar healthy controls. After filtering for reading coverage and fecal collection time points, we evaluated IgA coating patterns in 184 IBD patients and 32 healthy controls. We compared IgA coating (as measured using an IgA-coating Index (Palm et al., 2014), ICI; see STAR Methods) and relative abundance of taxa between IBD patients and controls (Figure 2). We identified a subset of 43 bacterial taxa that displayed significantly higher IgA coating in IBD patients than healthy controls (Figure 2). Of these 43 species, 35 also showed significant differences in relative abundance between IBD patients and controls, including Bacteroides fragilis, Haemophilus parainfluenzae, Clostridium methylpentosum, Bifidobacterium animalis, and Bilophila sp. In contrast, eight taxa showed differential IgA coating but similar relative abundance in controls and patients, including Anaerococcus sp., Gemellaceae sp., Collinsella sp., and Clostridium perfringens. Thirty-three species showed differential relative abundance in IBD patients versus controls but did not differ in terms of IgA coating. This group included multiple putative “beneficial” taxa that are reduced in patients with IBD, such as Faecalibacterium prausnitzii.
Figure 2. Differential Enrichment of IgA Coating and Relative Abundance in IBD-associated Taxa.
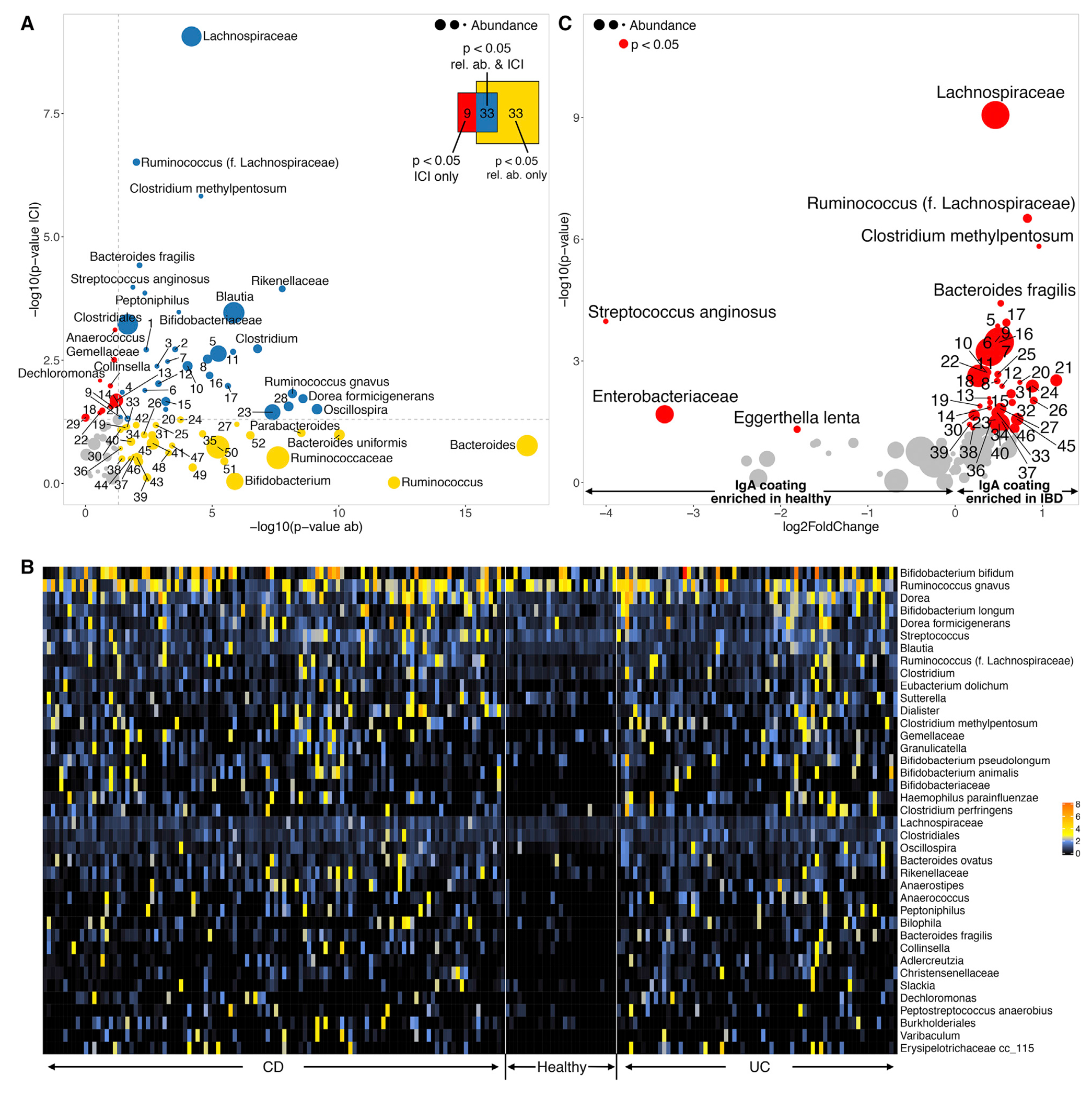
(A) Taxa differentially enriched in healthy (N = 27) or IBD (N = 180) subjects estimated using their relative abundance (horizontal axis: −log10 p value ofthe test) or IgA coating (vertical axis). Mean relative abundance is indicated by the size of the circles representing each taxa. Colors represent significantly differential taxa based on relative abundance (yellow), IgA coating (red), or both (blue). Venn diagram indicating the total number of taxa enriched as measured by abundance, IgA coating, or both. Numbers are exact, the area of the diagrams is not necessarily proportional.
(B) Heatmap of IgA Coating Index (ICI) scores in the 43 taxa with differential IgA coating between healthy (N=27) and IBD (N = 180) subjects.
(C) Volcano plot depicting taxa with differential IgA coating between healthy (N = 27) and IBD (N = 180) subjects. Horizontal axis indicates the fold change in ICI score (negative, enriched in healthy; positive, enriched in IBD). Vertical axis reflects −log10 p value of the test. The size of the circles indicates mean relative abundance, significant taxa (p < 0.05) colored in red. To improve legibility, some taxa have been replaced by numeric IDs (Table S4).
We also observed specific bacterial taxa that were enriched in the IBD population (as measured by relative abundance) and were highly coated in select individual patients but were not significantly more coated in IBD patients versus controls as a group, including Anaerostipes sp., Ruminococcus gnavus, Sutterella sp., and various species from the genus Bifidobacterium (Figure S2). These IgA-coated “outliers” may represent unique strains that are highly immunogenic or may result from additional patient-specific factors (e.g., genetics or environment). Patterns of IgA coating between CD and UC were nearly identical–only three taxa displayed significantly higher IgA coating in CD, and even these differences were relatively modest (Figure S3).
We observed that the relationship between IgA coating and relative abundance was taxa-specific (Table S1). The vast majority of taxa (70/81) showed no significant correlation (positive or negative) between relative abundance and ICI scores. However, about 9% of all taxa (8/81), including Enterobacteriaceae, Coprobacillus, Sutterella and E. biforme, exhibited a negative correlation between relative abundance and ICI score, which is consistent with a model where IgA coating drives bacterial depletion. By contrast, a small minority of taxa (3/81), including B. plebeius and R. torques, displayed a significant positive correlation between relative abundance and ICI scores. Thus, in select taxa, IgA coating may enhance bacterial colonization, growth, or persistence, for example via antibody-enhanced biofilm formation or adhesion to the host mucosa.
IBD Treatments Differentially Affect IgA Coating of the Gut Microbiota
We next took advantage of the robust clinical information available for the OSCCAR cohort to determine the effect of different IBD treatment regimens on disease activity, microbiota composition, and IgA coating of the gut microbiota. While most medications had modest effects on disease activity as measured by fecal calprotectin, biologics significantly reduced measureable inflammation in CD patients (Figure S4A). In contrast, we did not observe any significant associations between treatment regimens and inflammatory markers in UC patients (Figure S4B).
Biologic agents (limited to anti-tumor necrosis factor α [TNF-α] inhibitors), immune modulators (6-mercaptopurine and methotrexate), and antibiotics (ciprofloxacin and metronidazole) had the most noticeable effects on microbiota composition, while systemic corticosteroids had a lesser effect. Biologics, in particular, had a notable impact on bacterial IgA coating: thirty taxa were differentially IgA coated in patients treated with biologics compared with naive patients but showed similar relative abundances, fifteen taxa displayed differential relative abundance but had similar IgA coating, and nine taxa were altered based on both IgA coating and relative abundance (Figure 3A). Faecalibacterium prausnitzii was the most dramatically impacted taxon as measured by relative abundance. Immune modulators also affected IgA coating or relative abundance of twelve and fifteen taxa, respectively, with seven taxa shared (Figure 3B). The relative abundances of Bacteroides uniformis and Ruminococcus sp. were both highly affected by immune modulators. Unsurprisingly, antibiotics had a large impact on microbiome composition (24 taxa showed differential relative abundance), while the effects on IgA coating were less marked (9 taxa), and 11 taxa were differential both in relative abundance and IgA coating (Figure 3C). The relative abundances of Blautia obeum and Eggerthella lenta were dramatically reduced by antibiotics. Finally, steroids had minimal impact on either relative abundance (8 differential taxa) or IgA coating (3 taxa), and no taxa impacted both in their abundance and IgA coating (Figure 3D). Ruminococcaceae sp. was the taxa most affected by steroids in its relative abundance.
Figure 3. Impact of Medications on Relative Abundance and IgA Coating.
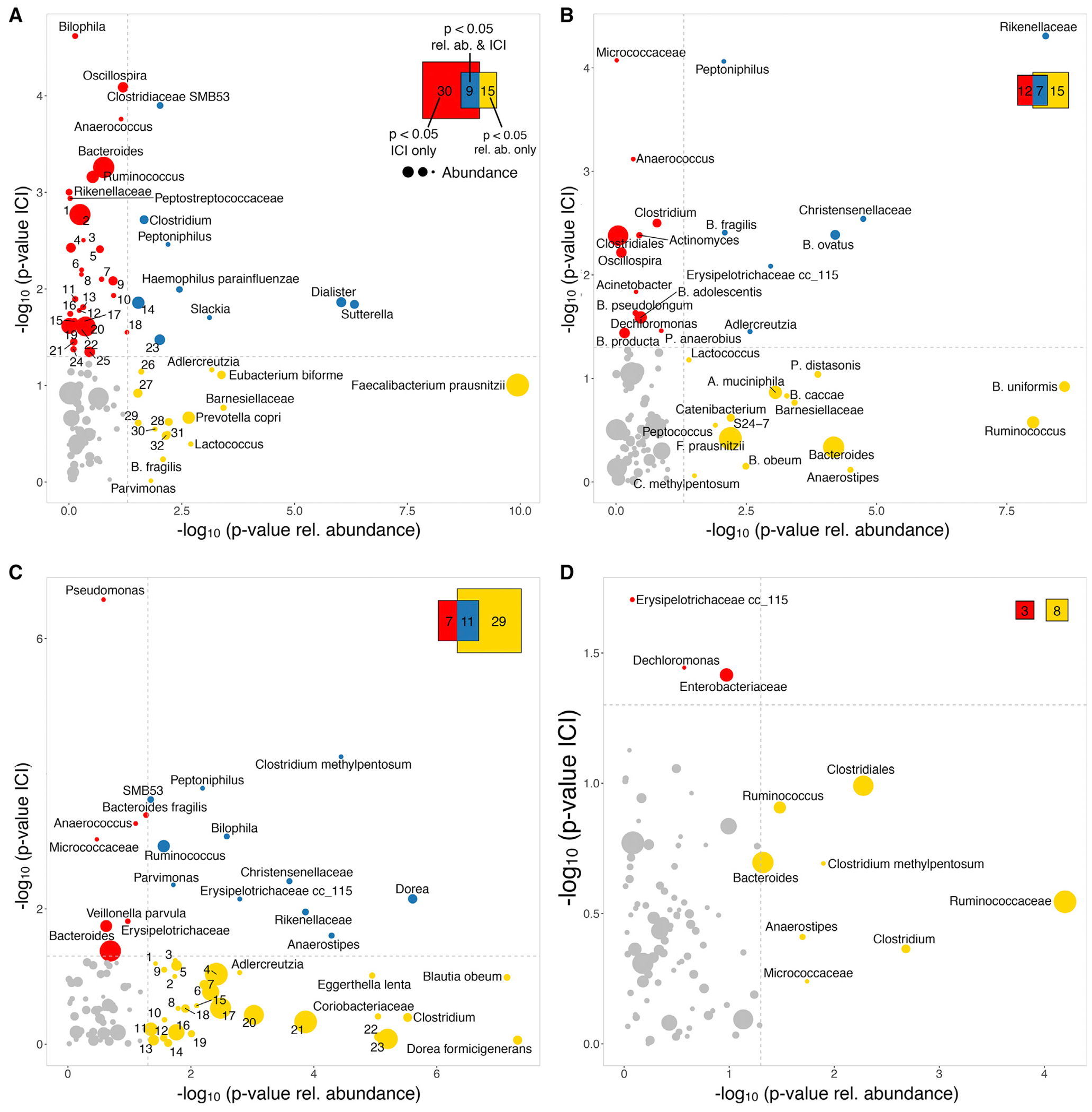
Taxa differentially enriched in subjects depending on whether they received a specific medication or not. Significance was estimated using their relative abundance (horizontal axis: −log10 p value ofthe test) or IgA coating (vertical axis). Mean relative abundance is indicated by the size of the circles representing each taxa. Colors represent significantly differential taxa based on relative abundance (yellow), IgA coating (red), or both (blue). Venn diagram shows the total number of taxa enriched as measured by abundance, IgA coating, or both.
(A) Biologics (untreated N = 173; treated N = 6). To improve legibility, some taxa have been replaced by numeric IDs (Table S4).
(B) Immune modulators (untreated N = 169; treated N = 10).
(C) Antibiotics (N = 154, N = 26). To improve legibility, some taxa have been replaced by numeric IDs (Table S4).
(D) Steroids (untreated N = 105; treated N = 74).
Out of the 39 taxa that displayed significantly altered IgA coating in patients on biologics, Bilophila sp., Oscillospira sp., Clostridiaceae SMB53, and Anaerococcus sp. showed the greatest difference (Figure 4A). In contrast, Faecalibacterium prausnitzii, which was significantly altered in its relative abundance in patients receiving antibiotics, displayed equivalent IgA coating regardless of treatment. Immune modulators also resulted in significant changes in IgA coating of several taxa, particularly Micrococcaceae sp. and Anaerococcus sp., while those highly impacted in their relative abundance (Bacteroides uniformis, Ruminococcus sp.) again displayed similar IgA coating (Figure 4B). Among the few taxa that showed differential IgA coating in antibiotic-treated patients, Pseudomonas sp., Micrococcaceae sp. and Peptoniphilus sp. were the most significant (Figure 4C). Steroids had a minimal effect on IgA coating, and only Erysipelotrichaceae sp. and Dechloromonas sp. displayed moderate reduced coating (Figure 4D). Interestingly, treatment with biologics, immune modulators, antibiotics, and steroids all led to decreased IgA coating of bacterial species in IBD patients, with one exception: treatment with steroids resulted in an increased IgA coating of Enterobacteraceae sp (Figure 4D). Although multivariate analysis limits the power to detect significant associations due to the addition of confounders, we confirmed that IgA coating of several of these taxa were independently associated with medication, including Pseudomonas, Burkholderiales, Varibaculum, and L. zeae, as well as multiple members of the genera Peptostreptococcus, Enterobacter and Bifidobacterium (Table S2).
Figure 4. Medications Induce Differential Enrichment of IgA Coating.
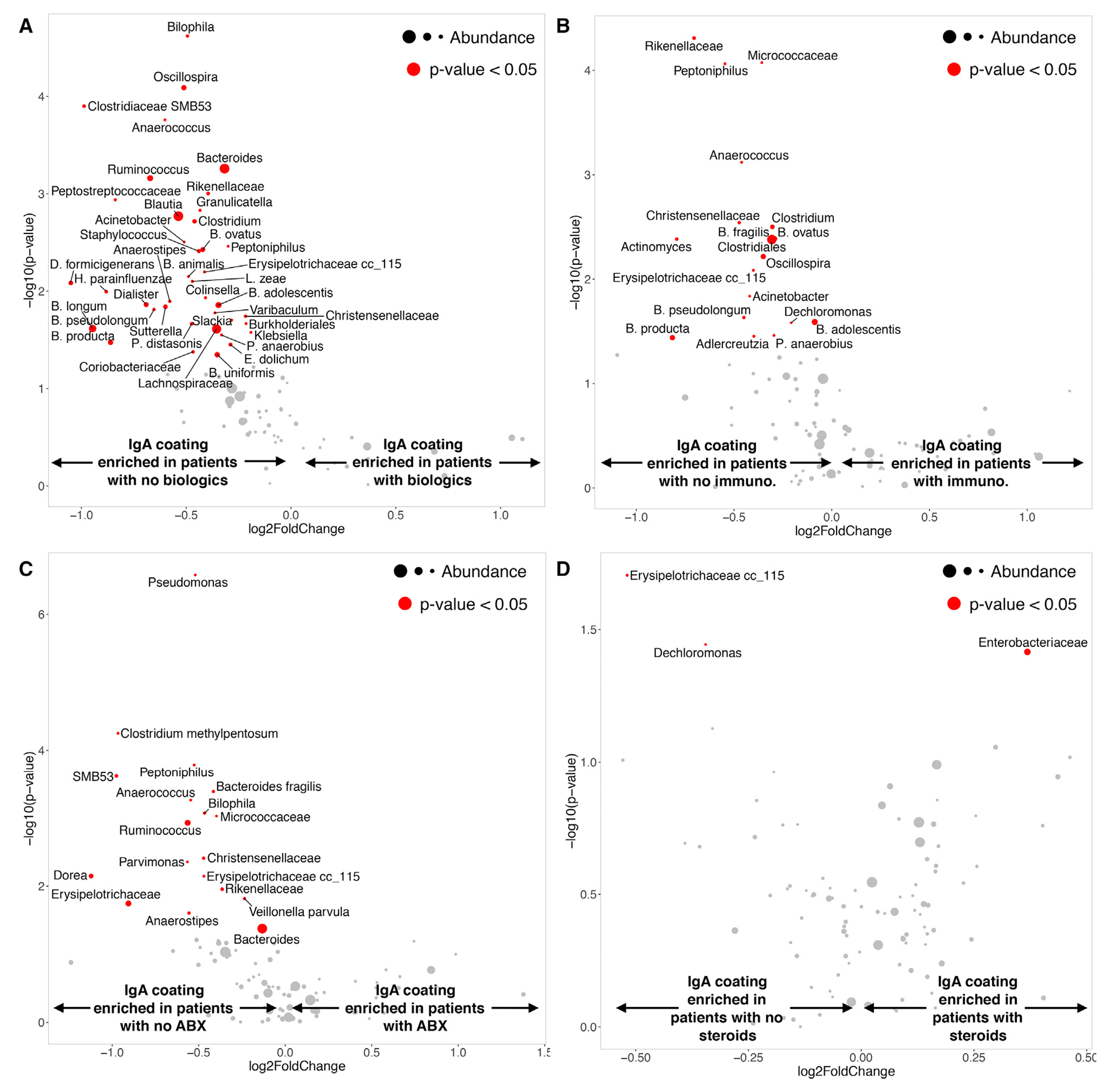
Volcano plot depicting taxa with differential IgA coating between subjects receiving or not receiving specific medication. Horizontal axis indicates the fold change in ICI score (negative: enriched in healthy; positive: enriched in IBD). The vertical axis reflects −log10 p value of the test. Size of the circles indicate mean relative abundance, significant taxa (p < 0.05) colored in red.
(A) Biologics (untreated N = 173; treated N = 6).
(B) Immune modulators (untreated N = 169; treated N = 10).
(C) Antibiotics (N = 154, N = 26).
(D) Steroids (untreated N = 105; treated N = 74).
IgA-SEQ Identifies Unique Bacterial Predictors of Disease Progression
Because of the proposed central importance of immune-microbiota interactions in the progression of IBD, we hypothesized that patterns of IgA coating of the microbiota might correlate with disease progression. Since our cohort only included cross-sectional samples from patients at the time of enrollment, we used a number of years to surgical resection (CD, N = 16) or colectomy (UC, N = 3) as a proxy for disease progression, with shorter time to surgery indicating a more rapid disease progression. We therefore, investigated whether the IgA coating of different bacteria might predict this outcome. Among the taxa that were significantly associated with treatment (Figure 4), Oscillospira sp. was the only taxon where IgA-coating levels and time to resection were significantly correlated, with the decreased coating being associated with earlier resection (Figure 5A, rightmost panel); interestingly, its relative abundance was not correlated (Figure 5B). On the other hand, the relative abundances of Erysipelotrichaceae sp. and Faecalibacterium prausnitzii were significantly correlated with number of years to surgery (Figure 5B, left and middle panels), while this effect was not observed based on their IgA coating levels (Figure 5A). These patterns again emphasize the complementary nature of IgA coating and relative abundance and may serve as microbiome-based biomarkers for the prediction of disease progression.
Figure 5. Relative Abundance and IgA Coating-based Bacterial Predictors of Disease Progression Correlation between Erysipelotrichaceae, Faecaliabcterium prausnitzii, and Oscillospira and number of years to intestinal resection or colectomy (N = 19).
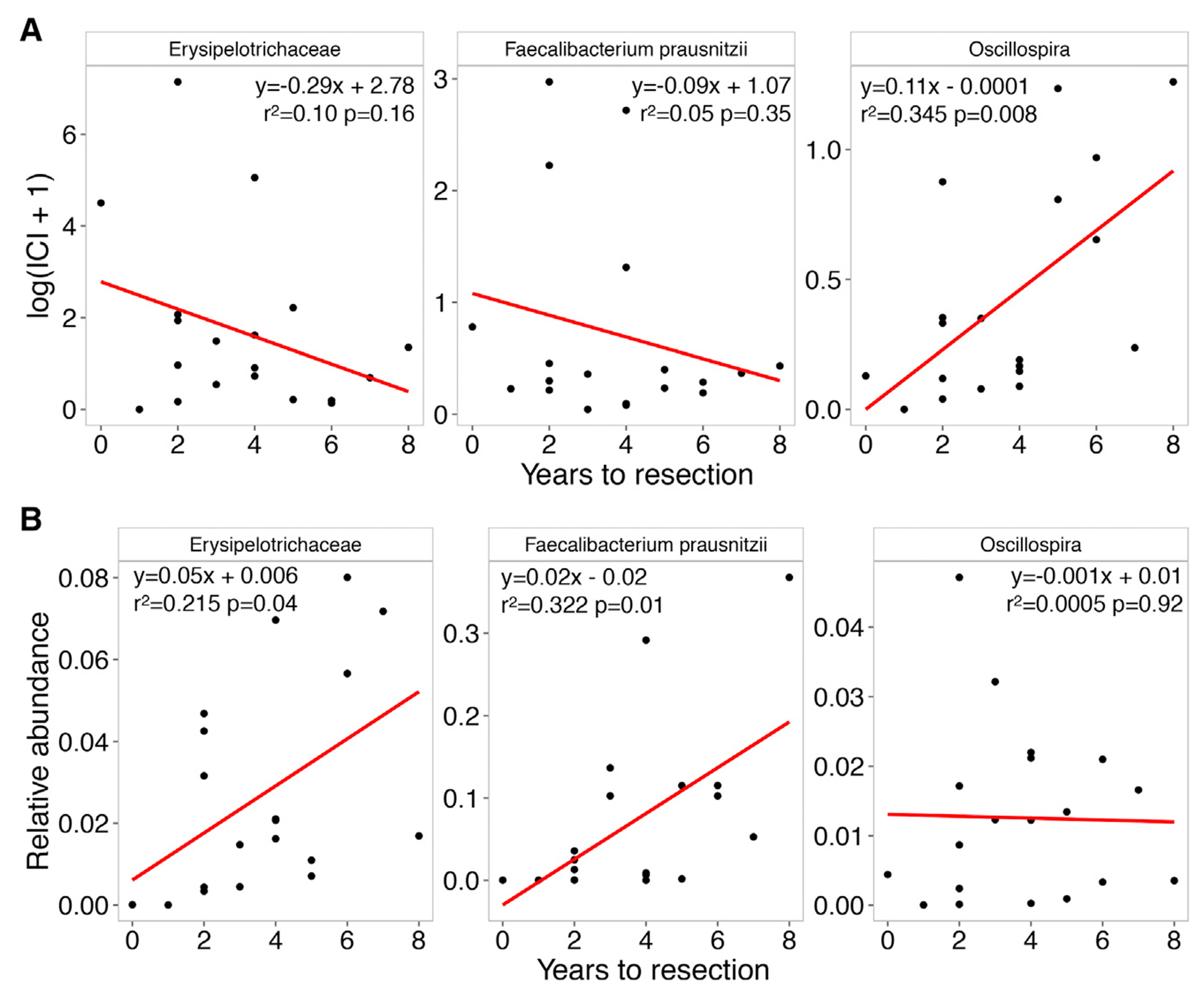
(A) Scatterplot of ICI scores and years to resection. Red line shows linear regression, strength, and significance of the correlation indicated in each panel.
(B) Scatterplot of relative abundance and years to resection.
DISCUSSION
We report here a large-cohort examination of the IgA response to the intestinal microbiota in IBD. In line with prior reports that examine complete gut microbial communities in IBD using 16S rRNA gene sequencing, we observed significant differences in relative abundances between IBD patients and healthy controls with increases in Actinobacteria, Bacteroidetes, and Proteobacteria noted in IBD (Kostic et al., 2014; Gevers et al., 2014). Neither 16S rRNA gene sequencing nor IgA-SEQ could reliably discriminate between CD and UC. Disease activity and inflammatory burden, as measured by fecal calprotectin and CRP, did not exert a noticeable effect on microbial composition or IgA-SEQ results. The lack of association between inflammatory state and microbial composition is similar to prior reports and, in our cohort, may be at least partially attributed to the fact that fecal samples were obtained within 3 months of IBD diagnosis at which point the majority of patients had initiated treatment (Gevers et al., 2014).
Examining microbial signatures in a large cohort of treatment-naive pediatric CD patients enrolled in the Pediatric RISK Stratification Study, Gevers et al. similarly noted that large-scale analysis of 16S rRNA from stool samples did not accurately reflect IBD phenotype (Gevers et al., 2014). Considering both luminal and mucosa-associated bacteria, they observed that increased abundance of Enterobacteriaceae, Pasturellacaea, Veillonellaceae, and Fusobacteriaceae and a decreased abundance of Erysipelotrichales, Bacteriodales, and Clostridiales correlated with disease status and severity. We observed differential IgA coating of many of these same taxa including Enterobacteriaceae, Clostridiales, and Bacteriodales.
Lewis and colleagues explored longitudinal changes in the gut microbiome in pediatric patients with CD treated with either a defined formula diet or anti-TNF agents (Lewis et al., 2015). Among patients treated with anti-TNF’s, they observed 11 taxa that differed in abundance in patients responsive to anti-TNF versus healthy controls at baseline; six of these taxa also remained significantly different from controls after treatment. In our study, the relative abundances of six bacterial taxa within the clades detected in Lewis’ results were also significantly different between healthy and untreated patients (Table S3). It should be noted, however, that differences in study design (longitudinal in Lewis et al. versus cross-sectional in our study) makes a direct comparison of results difficult to interpret, and future studies will be required to assess the longitudinal impact of medications on IgA coating.
Treatment exposures within the OSCCAR cohort were noted to exert a variable effect on pre- and post-IgA-sorted 16S rRNA datasets. Similar to other studies, antibiotics resulted in a large change in relative abundances across various taxa (Gevers et al., 2014; Lewis et al., 2015; Morgan et al., 2012); however, they did not appear to have a significant impact on patterns of IgA coating. This underscores previous observations that relative abundance does not necessarily correlate with IgA coating (Palm et al., 2014; Viladomiu et al., 2017). Systemic corticosteroids appeared to have minimal effect on both pre-IgA-sorted 16S rRNA and IgA-SEQ results. The lack of corticosteroid effect on IgA coating is likely related to the superficial level at which these anti-inflammatory medications mitigate the immune reaction in IBD. Corticosteroids are widely used in IBD for symptom control but are not successful in obtaining deeper levels of remission via mucosal healing like biologic agents that block TNFα (Modigliani et al., 1990; Cholapranee et al., 2017). In line with this, exposure to anti-TNFα agents and, to a lesser extent, immune modulators, appeared to have the most dramatic effect on IgA coating.
Recent studies have explored IgA-SEQ’s ability to identify bacteria with distinct immunomodulatory properties capable of inducing intestinal pathology (Palm et al., 2014; Viladomiu et al., 2017; Planer et al., 2016). Evaluating the fecal microbiome of IBD patients with and without spondyloarthritis (SpA), Viladomiu et al. demonstrated selective enrichment of IgA-coated Escherichia coli in patients with Crohn’s disease-associated SpA (Viladomiu et al., 2017). While only Escherichia coli was differentially enriched in SpA, they observed differential IgA coating of Akkermansia, Bacteroidetes, Blautia, Lachnoclostridium, Lactobacillus, Pseudomonas, Staphylococcus, and Streptococcus in CD patients. These results are consistent with our observation that Lachnospiraceae, B. fragilis, Blautia, and Streptococcus are differentially coated with IgA in IBD patients compared with healthy controls (Figure 3), indicating that the IgA response to selected individual bacterial species is relatively consistent across multiple IBD patient cohorts. The OSCCAR cohort also revealed a number of identified IBD-associated taxa to be coated with IgA, including Clostridium methylpentosum and Haemophilus parainfluenza. Thus, distinct taxa are identified when taking the IgA response into account and can reveal insight into or predict specific clinical phenotypes.
While IgA coating in IBD patients marks bacteria that can initiate or exacerbate inflammatory diseases in mouse models (Palm et al., 2014; Viladomiu et al., 2017), IgA coating also plays a critical role in limiting the full pathogenic potential of IgA-targeted strains (Cullender et al., 2013; Kubinak et al., 2015; Suzuki et al., 2004). Thus, IgA itself does not appear to contribute to the inflammatory response in IBD (at least in mouse models) and instead may constrain further detrimental impacts of IgA-targeted taxa. Furthermore, in contrast to IBD patients and dysbiotic mice, IgA-coated strains from healthy controls may support immunoregulatory responses in the gut and protect against intestinal inflammation (Kau et al., 2015; Cong et al., 2009). The opposing impacts of distinct IgA-inducing strains on intestinal inflammation may reflect two distinct mechanisms of T-cell-dependent IgA production: an inflammatory pathway that proceeds through T helper type 17 cells and an immunoregulatory pathway that relies upon regulatory T cells (Viladomiu et al., 2017; Cong et al., 2009; Kawamoto et al., 2014; Hirota et al., 2013).
The clinical course of IBD is highly variable and there is a lack of reliable translational biomarkers that can accurately predict which patients will go on to develop complicated disease or require surgery (Jess et al., 2007; Guizzetti et al., 2018). Kugathasan et al. observed distinct microbial signatures to be associated with certain CD-related outcomes (Kugathasan et al., 2017). For instance, Rothia and Ruminococcus were associated with stricture development whereas Veillonella was increased in ileal biopsies of patients who developed penetrating disease. No microbiota-specific associations with future surgical risk were reported within this cohort. A number of factors, including age at diagnosis, medication exposures, anti-microbial serologies, and various disease activity metrics, have also been considered in predictive models of surgical resection in IBD (Guizzetti et al., 2018). Specific microbial signatures have thus far not been implicated or associated with future surgical risk.
Our results reveal three potential biomarkers for IBD disease progression and “years-to-resection:” low relative abundance (based on 16S rRNA) of Erysipelotrichaceae sp. and Faecalibacterium prausnitzii, as well as low IgA coating of Oscillospira. While prior studies have noted a decreased relative abundance of Erysipelotrichaceae and Faecalibacterium prausnitzii in IBD, no direct relationship with clinical outcomes was reported (Gevers et al., 2014; Sokol et al., 2008). The potential clinical importance of Oscillospira in the IBD microbiome was similarly observed in a recent report from the multicenter pediatric UC study, PROTECT (Predicting Response to Standardized Pediatric Colitis Therapy). Examining 16S rRNA from an inception cohort of 428 pediatric UC patients, a decrease in Oscillospira abundance in patient stool samples was independently associated with escalation to anti-TNFα therapy (Hyams et al., 2019). Likewise, a decreased abundance of Oscillospira has previously been reported in CD and pediatric non-alcoholic fatty liver disease (Santoru et al., 2017; Del Chierico et al., 2017). While we did not observe a role for Oscillospira based on 16S rRNA analysis, consideration of the host immune response to this organism correlates with future surgical risk in our cohort suggesting that failure to mount a sufficient protective IgA response against a potential pathobiont (in this case, Oscillospira) may lead to more rapid disease progression. This highlights the potential importance of evaluating IgA coating when identifying predictive biomarkers.
The immunopathogenesis of IBD has been attributed to a combination of genetic susceptibility, microbial contributors, and environmental factors. Our results demonstrate that consideration of host immune response to specific bacteria reveals unique, potentially disease-modifying taxa, as well as potential biomarkers for disease progression, that would have otherwise not been identified via examination of unsorted 16S rRNA relative abundance. The ability to further discriminate clinically significant bacterial taxa based on their interactions with and effects on the host immune response may thus provide a framework for the development of more refined biomarkers of disease course and direct future microbial-based therapeutic approaches.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jose C. Clemente (jose.clemente@mssm.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
Raw sequences and associated metadata for this study are available at the National Center for Biotechnology Information Sequence Read Archive under the BioProject accession number PRJNA674841. This study used pre-existing software and did not generate new custom code or algorithms.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patient Cohort
A total of 408 adult and pediatric patients were enrolled in the Ocean State Crohn’s and Colitis Area Registry (OSCCAR) between 2008-2013. OSCCAR is an observational cohort of patients with newly diagnosed IBD in the state of the Rhode Island (Sands et al., 2009). All patients were managed at the discretion of their gastroenterologist. As an observational study, the use of medications or decision to complete diagnostic procedures was not dictated by study enrollment. Consent for participation and specimen collection was obtained prior to enrollment and the study was approved by the Institutional Review Board at Rhode Island Hospital and Mount Sinai School of Medicine. In addition to detailed clinical data, biological specimens such as blood, urine, and stool were collected at baseline and annually, thereafter. Only baseline stool samples collected within 200 days from IBD diagnosis with a sequencing coverage over 1,000 reads per sample for all sorting conditions were sequenced and analyzed (n=184). Of the 184 patients included in this study the mean age was 31.6 with a range from 4 to 83 years old. Over half (59%) of these patients were female.
METHOD DETAILS
IgA-SEQ and 16S rRNA Gene Sequencing
Fecal material was incubated for 15 min in 1ml PBS per 100 mg fecal material in Fast Prep Lysing Matrix D tubes containing ceramic beads (MP Biomedicals). To remove large particles, fecal material was homogenized via bead beating for 5 s (Minibeadbeater; Biospec) and then centrifuged (50 x g, 15 min, 4°C). Bacteria in the supernatants were removed (100 μl/sample), washed with 1 ml PBS containing 1% (w/v) Bovine Serum Albumin (BSA, American Bioanalytical; staining buffer), and centrifuged for 5 min (8,000 x g, 4°C) before resuspension in 1 ml staining buffer (PBS containing 1% (w/v) Bovine Serum Albumin). A 20 μl fraction of this suspension was saved for 16S rRNA gene sequencing analysis of the pre-sort fraction before centrifugation. Bacterial pellets were resuspended in 100 μl blocking buffer (20% Normal Mouse Serum, Jackson ImmunoResearch) and incubated for 20 min on ice before adding 100 μl staining buffer containing PE-conjugated Anti-Human IgA (1:10; Miltenyi Biotec clone IS11-8E10) and incubating for an additional 30 min on ice. Samples were washed 3 times with 1ml staining buffer before FACS of 500.000 IgA-positive and IgA-negative bacteria. After centrifugation, bacterial pellets were frozen at −80°C prior to DNA extraction.
V4 16S rRNA gene sequencing was performed as described previously using 2×250 sequencing on an Illumina MiSeq (Palm et al., 2014; Caporaso et al., 2010). Briefly, samples were suspended in 400 μl staining buffer before adding 250 ml 0.1 mm zirconia/silica beads (Biospec), 300 ml Lysis buffer (200 mM NaCl, 200 mM Tris, 20 mM EDTA, pH 8), 200 μl 20% SDS and 500 μl phenol:chloroform:isoamylalcohol (25:24:1, pH 7.9; Sigma). Samples were chilled on ice for 4 min and then homogenized by beat beating (2 min bead beating, 2 min on ice, 2 min bead beating). After centrifugation (6000 x g, 4°C), the aqueous phase was transferred to a Phase Lock Gel tube (Light; 5 PRIME), an equal volume of phenol:chloroform:isoamylalcohol was added, and samples were mixed by inversion and then centrifuged for 3 min (16,100 x g, room temperature). The DNA was precipitated by adding 1/10 volume of 3 M NaOAc (pH5.5) and 1 volume Isopropanol to the aqueous phase before incubation at −20°C overnight. Precipitated DNA was pelleted (20 min, 16,100 x g, 4°C), washed with 500 μl 100% EtOH (3 min, 16,100 x g, 4°C), dried (miVac GeneVac 15 min, no heat, Auto Run setting), and resuspended in 100 μl TE buffer (pH 7; 50°C for 30 min). The DNA was then treated with 35 U/ml RNase A (QIAGEN) before purification (QIAquick PCR purification; QIAGEN), and elution in 40 μl Elution Buffer. The V4 region of 16S ribosomal RNA was then PCR amplified (28 cycles; primer pair F515/R806) in triplicate (10 μl purified DNA per reaction; Phusion polymerase, New England Bioscience). After amplification, PCR triplicates were pooled, purified (MinElute, QIAGEN), and resuspended in 20 μl H2O. PCR products were then quantified with Picogreen (Invitrogen) and pooled at a final concentration of 10 nM before sequencing on a miSeq sequencer (Illumina, 2x250bp paired-end reads, up to 200 samples per sequencing run).
CRP and Fecal Calprotectin
Serum C-reactive protein (CRP) levels were quantified by Prometheus Laboratories (San Diego, California). Fecal calprotectin extraction was performed using the Buhlmann Calprotectin ELISA kit according to the manufacturer’s instructions (Alpco Immunoassays, Salem, New Hampshire).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data Analysis
Analyses of 16S rRNA data were performed using the Quantitative Insights into Microbial Ecology (QIIME) 1.9.1 (http://qiime.org), an open-source bioinformatics pipeline for performing analysis of microbiome sequence data. Analyses were performed as previously described (Dominguez-Bello et al., 2016; Clemente et al., 2015). Briefly, raw sequencing data were demultiplexed using unique barcodes assigned to each sample, and low quality reads discarded from downstream analyses. Remaining reads were then clustered into Operational Taxonomic Units (OTUs) with a 97% similarity threshold, and using Greengenes v13-8 as a reference set to assign taxonomy to each OUT (McDonald et al., 2012). OTU tables were rarified at the sequencing depth of 1,000 sequences/sample. There were no significant differences in sequencing depth per group or disease status. For IgA-sorted samples, an IgA coating index (ICI) was calculated for each taxa as the fraction between the relative abundance of the taxa in the IgA-positive sample and the abundance in the IgA-negative sample, as previously described (Palm et al., 2014). Taxa with ICI lower than 1 were discarded from additional analyses. Alpha diversity (bacterial richness of a sample expressed as a function of the number of OTUs identified in it) was estimated using Faith’s phylogenetic diversity (Faith, 2007). Beta diversity (distance between samples based on differences in OTUs present in each sample) was measured using unweighted UniFrac (Lozupone and Knight, 2005) calculated from the rarefied OTU tables. Principal coordinate analysis (PCoA) was used to visualize clustering patterns between samples based on beta diversity distances. Linear discriminant analysis effect size (LEfSe), a method for biomarker discovery, was used to identify bacterial taxa differentially enriched in each population of interest (Segata et al., 2011). Association between microbiome composition and covariates were tested using PERMANOVA, a nonparametric test similar to ANOVA but that does not require the data to be normally distributed (Anderson, 2001). Significance of PERMANOVA tests were determined using 999 permutations with adjustment for multiple testing. Examination of bacterial abundance/ICI scores of all taxa relative to continuous variables such as fecal calprotectin and serum CRP was completed via Spearman’s rank correlation or Pearson’s correlation, depending on the distribution of the data. Unpaired t-tests were used to evaluate differences in ICI scores of taxa differentially enriched or depleted of IgA. Multivariate analysis was performed using the ”glm” function in R v3.4.1. For each taxa, we calculated a linear regression model examining time to surgical resection using the “lm” function in R v3.4.1. P-values and coefficients of determination were directly obtained from the models.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PE-conjugated Anti-Human IgA | Miltenyi Biotec | clone IS11-8E10 |
| Biological Samples | ||
| Human fecal samples from the OSCCAR cohort | (Sands et al., 2009) | N/A |
| Blood samples from the OSCCAR cohort | (Sands et al., 2009) | N/A |
| Critical Commercial Assays | ||
| Buhlmann Calprotectin ELISA kit | Alpco Immunoassays | 30-CALPHU-E01 |
| C-Reactive Protein | Prometheus Laboratories | N/A |
| Deposited Data | ||
| Raw data | This paper | 16S rRNA gene sequence data (NCBI) BioProject: PRJNA674841 |
| Oligonucleotides | ||
| 16S rRNA Forward Primer for V4 region 515F: GTGCCAGCMGCCGCGGTAA | (Caporaso et al., 2012) | N/A |
| 16S rRNA Reverse Primer for V4 region 806R: GGACTACHVGGGTWTCTAAT | (Caporaso et al., 2012) | N/A |
| Software and Algorithms | ||
| QIIME v1.9.1 | (Caporaso et al., 2010) | http://qiime.org |
| R v3.6.3 | (R Core Team, 2017) | https://www.r-project.org/ |
| ggplot2 | (Wickham, 2016) | N/A |
Highlights.
IgA-coated bacteria were evaluated in a large cohort of IBD patients using IgA-SEQ
IgA-SEQ identified distinct IBD-associated bacteria compared with the relative abundance
Anti-TNFα treated patients exhibited altered microbiota-specific IgA responses
IgA coating of Oscillospira was associated with a delay in time to surgery in IBD
ACKNOWLEDGMENTS
J.M.S. was supported by a COBRE Immune Based Interventions Against Infectious Diseases Pilot grant (P20 GM104317). M.R.dZ. was supported by a VIDI grant from the Netherlands Organization for Scientific Research (NWO, grant 91715377) and the Utrecht exposome Hub of Utrecht Life Sciences (www.uu.nl/exposome), funded by the Executive Board of Utrecht University. J.H.C. was supported by a grant from NIH NIDDK (U01 DK062422). J.C.C. was partially supported by a grant from NIH NIDDK (5R01DK114038).
The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.chom.2020.12.003.
DECLARATION OF INTERESTS
N.W.P., M.R.d.Z., and R.A.F. are co-founders of, consultants for, and hold equity in Artizan Biosciences. N.W.P., M.R.d.Z., and R.A.F. are inventors on a patent application for IgA-SEQ.
REFERENCES
- Ananthakrishnan AN, Bernstein CN, Iliopoulos D, Macpherson A, Neurath MF, Ali RAR, Vavricka SR, and Fiocchi C (2018). Environmental triggers in IBD: a review of progress and evidence. Nat. Rev. Gastroenterol. Hepatol 15, 39–49. [DOI] [PubMed] [Google Scholar]
- Anderson MJ (2001). A new method for non-parametric multivariate analysis of variance. Austral Ecol 26, 32–46. [Google Scholar]
- Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, et al. (2012). Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J 6, 1621–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholapranee A, Hazlewood GS, Kaplan GG, Peyrin-Biroulet L, and Ananthakrishnan AN (2017). Systematic review with meta-analysis: comparative efficacy of biologics for induction and maintenance of mucosal healing in Crohn’s disease and ulcerative colitis controlled trials. Aliment. Pharmacol. Ther 45, 1291–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente JC, Pehrsson EC, Blaser MJ, Sandhu K, Gao Z, Wang B, Magris M, Hidalgo G, Contreras M, Noya-Alarcón Ó, et al. (2015). The microbiome of uncontacted Amerindians. Sci. Adv 1, e1500183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong Y, Feng T, Fujihashi K, Schoeb TR, and Elson CO (2009). A dominant, coordinated T regulatory cell-IgA response to the intestinal microbiota. Proc. Natl. Acad. Sci. USA 106, 19256–19261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullender TC, Chassaing B, Janzon A, Kumar K, Muller CE, Werner JJ, Angenent LT, Bell ME, Hay AG, Peterson DA, et al. (2013). Innate and adaptive immunity interact to quench microbiome flagellar motility in the gut. Cell Host Microbe 14, 571–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange KM, Moutsianas L, Lee JC, Lamb CA, Luo Y, Kennedy NA, Jostins L, Rice DL, Gutierrez-Achury J, Ji S-G, et al. (2017). Genome-wide association study implicates immune activation of multiple integrin genes in inflammatory bowel disease. Nat. Genet 49, 256–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Chierico F, Nobili V, Vernocchi P, Russo A, De Stefanis C, Gnani D, Furlanello C, Zandonà A, Paci P, Capuani G, et al. (2017). Gut microbiota profiling of pediatric nonalcoholic fatty liver disease and obese patients unveiled by an integrated meta-omics-based approach. Hepatology 65, 451–464. [DOI] [PubMed] [Google Scholar]
- Dominguez-Bello MG, De Jesus-Laboy KM, Shen N, Cox LM, Amir A, Gonzalez A, Bokulich NA, Song SJ, Hoashi M, Rivera-Vinas JI, et al. (2016). Partial restoration of the microbiota of cesarean-born infants via vaginal microbial transfer. Nat. Med 22, 250–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith DP (2007). The role of the phylogenetic diversity measure, PD, in bioinformatics: getting the definition right. Evol. Bioinform. Online 2, 277–283. [PMC free article] [PubMed] [Google Scholar]
- Gevers D, Kugathasan S, Denson LA, Vázquez-Baeza Y, Van Treuren W, Ren B, Schwager E, Knights D, Song SJ, Yassour M, et al. (2014). The treatment-naïve microbiome in new-onset Crohn’s disease. Cell Host Microbe 15, 382–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guizzetti L, Zou G, Khanna R, Dulai PS, Sandborn WJ, Jairath V, and Feagan BG (2018). Development of clinical prediction models for surgery and complications in Crohn’s disease. J. Crohns Colitis 12, 167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota K, Turner JE, Villa M, Duarte JH, Demengeot J, Steinmetz OM, and Stockinger B (2013). Plasticity of Th17 cells in Peyer’s patches is responsible for the induction of T cell-dependent IgA responses. Nat. Immunol 14, 372–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyams JS, Davis Thomas SD, Gotman N, Haberman Y, Karns R, Schirmer M, Mo A, Mack DR, Boyle B, Griffiths AM, et al. (2019). Clinical and biological predictors of response to standardised paediatric colitis therapy (PROTECT): a multicentre inception cohort study. Lancet 393, 1708–1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jess T, Riis L, Vind I, Winther KV, Borg S, Binder V, Langholz E, Thomsen OØ, and Munkholm P (2007). Changes in clinical characteristics, course, and prognosis of inflammatory bowel disease during the last 5 decades: a population-based study from Copenhagen, Denmark. Inflam. Bowel Dis 13, 481–489. [DOI] [PubMed] [Google Scholar]
- Kau AL, Planer JD, Liu J, Rao S, Yatsunenko T, Trehan I, Manary MJ, Liu T-C, Stappenbeck TS, Maleta KM, et al. (2015). Functional characterization of IgA-targeted bacterial taxa from undernourished Malawian children that produce diet-dependent enteropathy. Sci. Transl. Med 7, 276ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamoto S, Maruya M, Kato LM, Suda W, Atarashi K, Doi Y, Tsutsui Y, Qin H, Honda K, Okada T, et al. (2014). Foxp3(+) T cells regulate immunoglobulin A selection and facilitate diversification of bacterial species responsible for immune homeostasis. Immunity 41, 152–165. [DOI] [PubMed] [Google Scholar]
- Khor B, Gardet A, and Xavier RJ (2011). Genetics and pathogenesis of inflammatory bowel disease. Nature 474, 307–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knights D, Silverberg MS, Weersma RK, Gevers D, Dijkstra G, Huang H, Tyler AD, van Sommere S, Imhann F, Stempak JM, et al. (2014). Complex host genetics influence microbiome in inflammatory bowel disease. Genome Med 6, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostic AD, Xavier RJ, and Gevers D (2014). The microbiome in inflammatory bowel diseases: current status and the future ahead. Gastroenterology 146, 1489–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubinak JL, Petersen C, Stephens WZ, Soto R, Bake E, O’Connell RM, and Round JL (2015). MyD88 signaling in T cells directs IgA-mediated control of the microbiota to promote health. Cell Host Microbe 17, 153–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kugathasan S, Denson LA, Walters TD, Kim MO, Marigorta UM, Schirmer M, Mondal K, Liu C, Griffiths A, Noe JD, et al. (2017). Prediction of complicated disease course for children newly diagnosed with Crohn’s disease: a multicentre inception cohort study. Lancet 389, 1710–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JD, Chen EZ, Baldassano RN, Otley AR, Griffiths AM, Lee D, Bittinger K, Bailey A, Friedman ES, Hoffmann C, et al. (2015). Inflammation, antibiotics, and diet as environmental stressors of the gut microbiome in pediatric Crohn’s disease. Cell Host Microbe 18, 489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozupone C, and Knight R (2005). UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol 71, 8228–8235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manichanh C, Borruel N, Casellas F, and Guarner F (2012). The gut microbiota in IBD. Nat. Rev. Gastroenterol. Hepatol 9, 599–608. [DOI] [PubMed] [Google Scholar]
- McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, and Hugenholtz P (2012). An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6, 610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modigliani R, Mary JY, Simon JF, Cortot A, Soule JC, Gendre JP, and Rene E (1990). Clinical, biological, and endoscopic picture of attacks of Crohn’s disease. Evolution on prednisolone. Groupe d’Etude thérapeutique des affections inflammatoires digestives. Gastroenterology 98, 811–818. [DOI] [PubMed] [Google Scholar]
- Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, Reyes JA, Shah SA, LeLeiko NS, Snapper SB, et al. (2012). Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol 13, R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng SC, Bernstein CN, Vatn MH, Lakatos PL, Loftus EV, Tysk C, O’Morain C, Moum B, and Colombel JF; Epidemiology and Natural History Task Force of the International Organization of Inflammatory Bowel Disease (IOIBD) (2013). Geographical variability and environmental risk factors in inflammatory bowel disease. Gut 62, 630–649. [DOI] [PubMed] [Google Scholar]
- Palm NW, de Zoete MR, Cullen TW, Barry NA, Stefanowski J, Hao L, Degnan PH, Hu J, Peter I, Zhang W, et al. (2014). Immunoglobulin A coating identifies colitogenic bacteria in inflammatory bowel disease. Cell 158, 1000–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planer JD, Peng Y, Kau AL, Blanton LV, Ndao IM, Tarr PI, Warner BB, and Gordon JI (2016). Development of the gut microbiota and mucosal IgA responses in twins and gnotobiotic mice. Nature 534, 263–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2017). R: A Language and Environment for (Statistical Computing). https://www.R-project.org/.
- Sands BE, LeLeiko N, Shah SA, Bright R, and Grabert S (2009). OSCCAR: ocean state Crohn’s and Colitis area registry. Med. Health RI 92, 82–85, 88. [PubMed] [Google Scholar]
- Santoru ML, Piras C, Murgia A, Palmas V, Camboni T, Liggi S, Ibba I, Lai MA, Orrù S, Blois S, et al. (2017). Cross sectional evaluation ofthe gut-microbiome metabolome axis in an Italian cohort of IBD patients. Sci. Rep 7, 9523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, and Huttenhower C (2011). Metagenomic biomarker discovery and explanation. Genome Biol 12, R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro JM, Cho JH, Sands BE, and LeLeiko NS (2015). Bridging the gap between host immune response and intestinal dysbiosis in inflammatory bowel disease: does immunoglobulin A mark the spot? Clin. Gastroenterol. Hepatol 13, 842–846. [DOI] [PubMed] [Google Scholar]
- Silverberg MS, Satsangi J, Ahmad T, Arnott ID, Bernstein CN, Brant SR, Caprilli R, Colombel JF, Gasche C, Geboes K, et al. (2005). Toward an integrated clinical, molecular and serological classification of inflammatory bowel disease: report of a Working Party of the 2005 Montreal world congress of gastroenterology. Can. J. Gastroenterol 19, 5A–36A. [DOI] [PubMed] [Google Scholar]
- Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, et al. (2008). Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 105, 16731–16736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Meek B, Doi Y, Muramatsu M, Chiba T, Honjo T, and Fagarasan S (2004). Aberrant expansion of segmented filamentous bacteria in IgA-deficient gut. Proc. Natl. Acad. Sci. USA 101, 1981–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viladomiu M, Kivolowitz C, Abdulhamid A, Dogan B, Victorio D, Castellanos JG, Woo V, Teng F, Tran NL, Sczesnak A, et al. (2017). IgA-coated E. coli enriched in Crohn’s disease spondyloarthritis promote TH17-dependent inflammation. Sci. Transl. Med 9, 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2016). Ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag; ). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequences and associated metadata for this study are available at the National Center for Biotechnology Information Sequence Read Archive under the BioProject accession number PRJNA674841. This study used pre-existing software and did not generate new custom code or algorithms.