

Abstract
Introduction:
Accumulating evidence supports the evaluation of glucagon-like peptide-1 (GLP-1) receptor (R) agonists for the treatment of the underlying pathology causing Parkinson’s Disease (PD). Not only are these effects evident in models of PD and other neurodegenerative disorders but recently in a randomized, double-blind, placebo-controlled clinical trial, a GLP-1R agonist has provided improved cognition motor functions in humans with moderate PD.
Areas covered:
In this mini-review, we describe the development of GLP-1R agonists and their potential therapeutic value in treating PD. Many GLP-1R agonists are FDA approved for the treatment of metabolic disorders, and hence can be rapidly repositioned for PD. Furthermore, we present preclinical data offering insights into the use of monomeric dual- and tri-agonist incretin-based mimetics for neurodegenerative disorders. These drugs combine active regions of GLP-1 with those of glucose-dependent insulinotropic peptide (GIP) and/or glucagon (Gcg).
Expert opinion:
GLP-1Ragonists offer a complementary and enhanced therapeutic value to other drugs used to treat PD. Moreover, the use of the dual- or tri-agonist GLP-1-based mimetics may provide combinatory effects that are even more powerful than GLP-1R agonism alone. We advocate for further investigations into the repurposing of GLP-1R agonists and the development of classes of multiagonists for PD treatment.
Keywords: Glucagon-like peptide-1 (GLP-1), glucose-dependent insulinotropic peptide (GIP), glucagon (Gcg), Parkinson’s disease, incretin mimetics, neurodegeneration, microglia, brain trauma
1. Introduction to GLP-1R agonist drug development
The gut-derived incretin hormones glucagon-like peptide 1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP) are produced and released from enteroendocrine L and K cells within the small intestine in response to food intake and act on their corresponding receptors in the islet β-cells of the pancreas to promote insulin secretion. These two hormones are responsible for the majority of insulin production, with GLP-1 showing more profound effects than GIP in Type 2 diabetes mellitus (T2DM) [1]. Glucagon (Gcg), another member of the secretin protein family, is produced in pancreatic α-cells as well as in the gut but, conversely, acts to raise glucose levels in the bloodstream. Aberrant production of these signaling hormones is a hallmark of several metabolic disorders, including T2DM. Counteracting impairments in their generation and/or activity with commercial monomeric GLP-1-based mimetics has proven effective for treatment [2,3]. Endogenous GLP-1, GIP, and Gcg have short half-lives and are susceptible to cleavage and inactivation by the enzyme dipeptidyl peptidase-4 (DPP-IV) (Figure 1(a)). Overcoming the action of DPP-IV has been achieved following the discovery of a naturally occurring analog of GLP-1 found in the venom of the Gila monster lizard (Heloderma suspectum) of the southwest United States [4]. Known as Exendin-4 (Ex-4) and commercially as Exenatide (Figure 1(b)), this GLP-1 analog does not break down in the presence of DPP-IV. Since the discovery of Exenatide and its FDA approval in 2005 for the treatment of T2DM, a suite of GLP-1R agonists has been developed and approved by government regulatory agencies for use as treatments for metabolic disorders (Table 1). There are further single GLP-1R agonists in preclinical development and clinical trials that seek to improve potency, duration of action, and compliance of users by reducing the number of subcutaneous injections, for example. Broad classes of monomeric dual- and tri-agonist formulations of GLP-1 combined with active regions of GIP, Gcg, or both are also advancing treatment options for a range of disorders including T2DM, nonalcoholic steatohepatitis (NASH), nonalcoholic fatty liver disease (NAFLD), and obesity (for comprehensive reviews see [5,6]).
Figure 1.
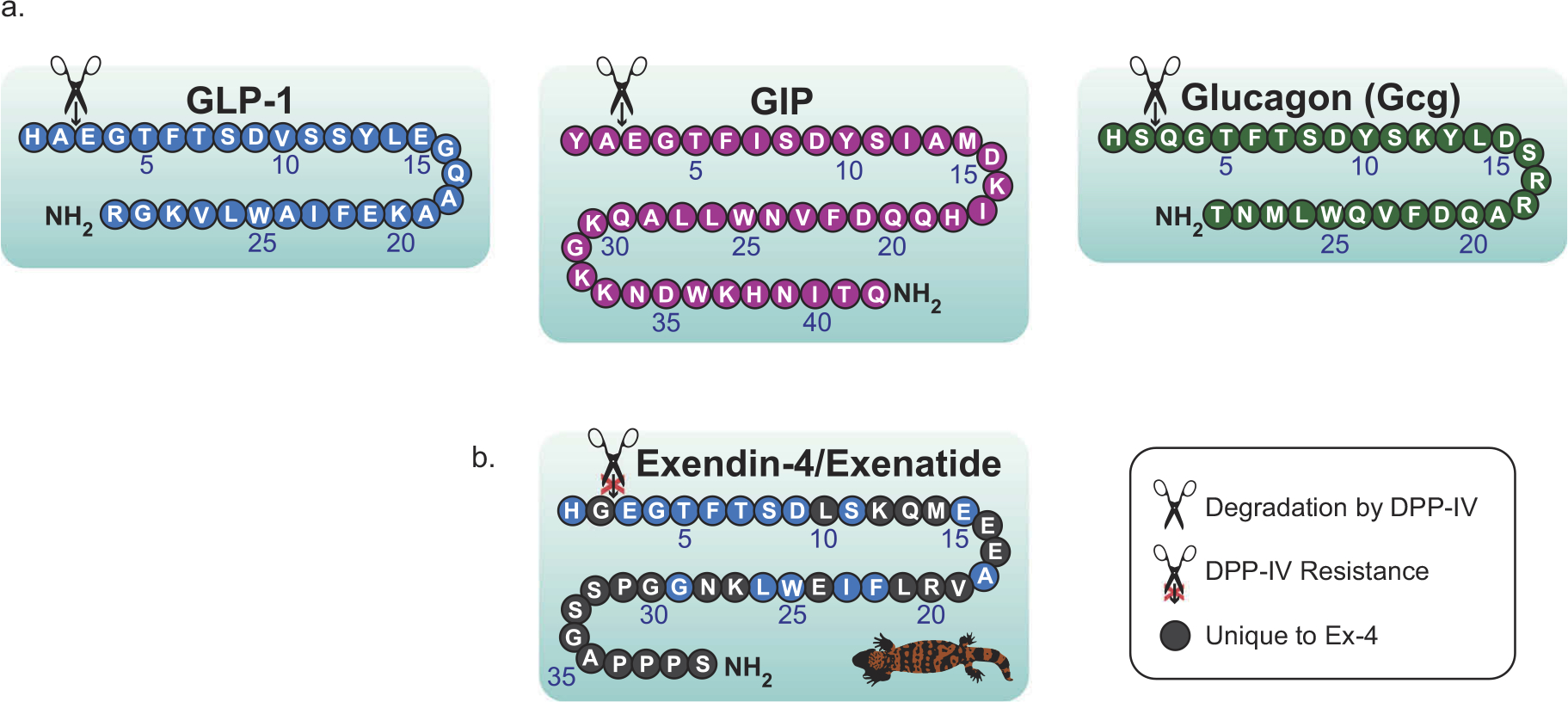
Amino acid architecture of endogenous secretin proteins and of Exendin-4/Exenatide. (a) The incretins GLP-1 and GIP along with the related secretin protein Gcg, are structurally similar proteins produced in the gut and pancreas. These proteins act in a glucose-dependent manner and remain short-lived following their production due to rapid degradation by dipeptidyl peptidase-4 (DPP-IV). (b) A naturally occurring analog of GLP-1, known as Exendin-4 (Ex-4) and commercially as Exenatide was discovered in the venom of the Gila monster and has allowed for the production of a wide variety of long-acting drugs for the treatment of metabolic diseases. Figure adapted from Glotfelty et al., 2019.
Table 1.
GLP-1R agonists approved by the United States FDA and international regulatory agencies.
| Company | Drug name | Administration | |
|---|---|---|---|
|
| |||
| AstraZeneca | Exenatide | Bydureon Bcise® | Once-weekly SC injection |
| Bydureon® | Once-weekly SC injection | ||
| Byetta® | 2x/day SC injection | ||
| Sanofi-Aventis | Lixisenatide | Adlyxin®/aLyxumia® | Once-daily SC injection |
| GlaxoSmithKline | Albiglutide | Tanzeum® | Once-weekly SC injection |
| Novo Nordisk | Liraglutide | Victoza® | Once-daily SC injection |
| Saxenda® | Once-daily SC injection | ||
| Semaglutide | Ozempic® | Once-weekly SC injection | |
| Rybelsus® | Oral once daily | ||
| Eli Lilly | Dulaglutide | Trulicity® | Once-weekly SC |
Internationally approved for use; SC = subcutaneous.
Evidence from epidemiological studies has identified T2DM and related insulin resistance conditions as a risk factor for PD and other age-related neurological disorders, suggesting a possible link between dysregulated communication of the gut-brain axis and progression of neurodegeneration [7–10]. Moreover, metabolic and neurodegenerative disorders share common pathophysiological features that, in addition to insulin resistance, include inflammation, oxidative stress, abnormal protein aggregation, and disrupted cognition [11]. Targeting disrupted gut-brain axis communication as an avenue to mitigate and possibly halt neurodegenerative disease progression provides an opening for PD treatment. Importantly, GLP-1R agonists and related incretin-based mimetics have extrapancreatic actions and are able to enter the brain where they act on a variety of cell types [12,13]. Repositioning the widely available FDA-approved GLP-1-based antidiabetic drugs and other incretin-based therapeutics in development may be an effective strategy for modifying the progression of PD and other neurodegenerative disorders.
2. GLP-1R agonism and PD pathology
PD is a progressive neurodegenerative disorder of the dopaminergic system that may arise from a variety of genetic predispositions or possibly environmental factors; however, the single highest risk factor for disease development is age. In the PD brain, the structure and function of the basal ganglia are predominantly affected, causing motor and coordination impairments. In addition, cognitive and gastrointestinal issues are associated with the development of the disease. Damage to the dopaminergic neurons of the substantia nigra is visible via positron emission tomography (PET), among other methods, in postmortem mid-brain tissue visibly by the lack of a pigmented substantia nigra, and microscopically exemplified by reduced immunohistochemical labeling of tyrosine hydroxylase, the rate-limiting precursor to dopamine (DA) (Figure 2A) [14].
Figure 2.
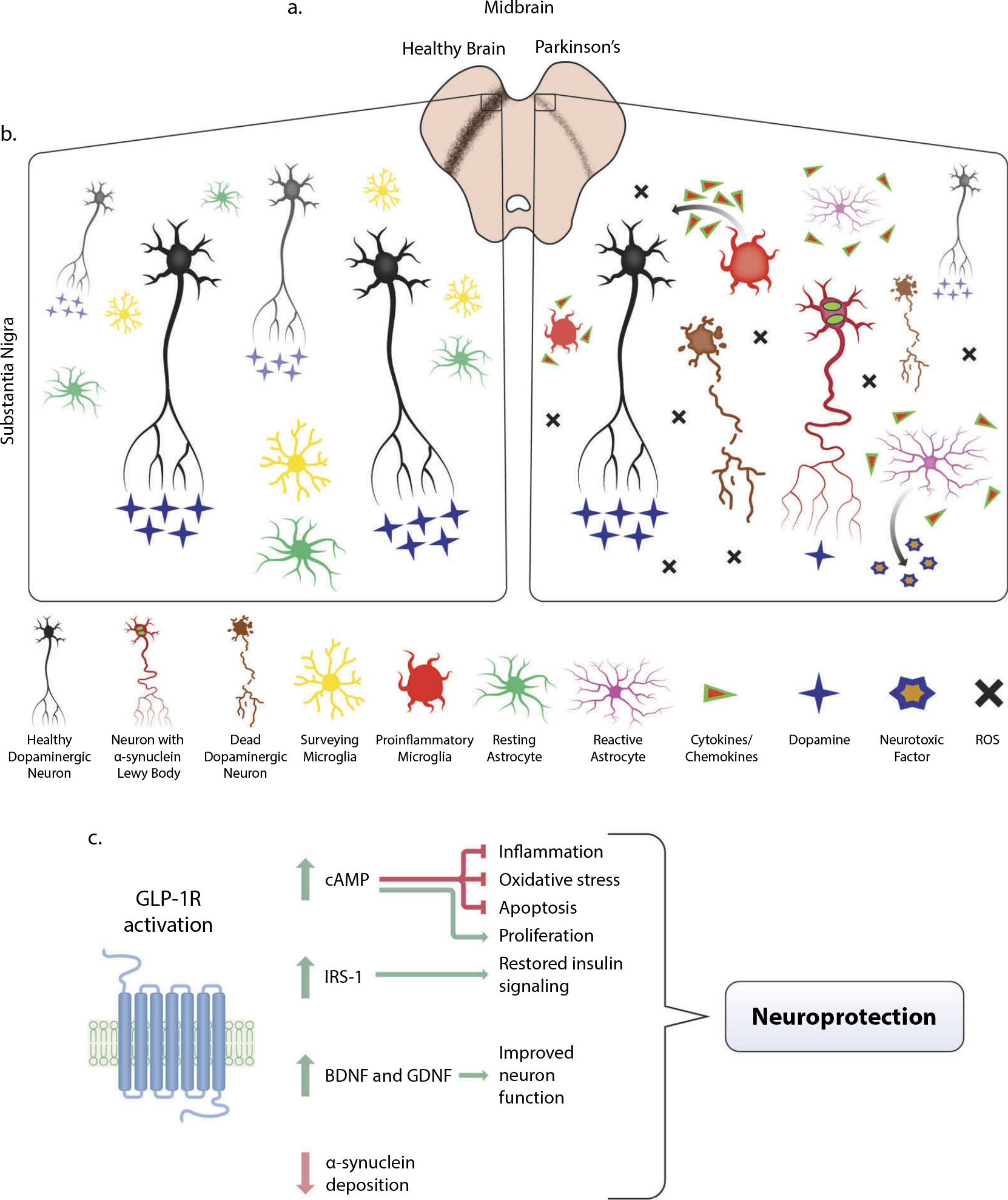
Pathology of Parkinson’s Disease (PD) in the midbrain and mitigation through GLP-1R activation. (a) In the midbrain of PD patients, loss of dopaminergic neurons is visible via decreased immunostaining of tyrosine hydroxylase (TH) (brown), the precursor to dopamine. In animal models of PD, including the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and 6-hydroxydopamine (6-OHDA) lesion methods, similar loss of dopaminergic neurons in the midbrain is observed. (b) A microenvironment comparison of a healthy and PD afflicted midbrain. Dopaminergic neuron dysfunction, reduced dopamine transmission, α-synuclein deposition within neurites, and eventual death may arise from a variety of genetic or environmental factors that cause mitochondrial dysfunction and high amounts of oxidative stress. The accumulation of reactive oxygen species (ROS), neuronal α-synuclein, and dying cells are some of the components that contribute to a proinflammatory environment in the PD midbrain. Highly dynamic surveying microglial cells respond to this milieu by altering their activation state and producing proinflammatory cytokines and chemokines that cascade to further evoke reactive astrocyte activation from their resting quiescent state. These astrocytes secrete a neurotoxic factor that selectively ablates subsets of neurons and oligodendrocytes and further exacerbates the already chronically inflamed region. Though inflammation is an immune response necessary for repair, chronic inflammation is especially detrimental. (c) Upon activation of GLP-1 receptor (GLP-1R), a multitude of downstream pathways are activated that mitigate the effects of PD pathology, most notably the major upstream secondary messenger cyclic adenosine monophosphate (cAMP). Upregulation of cAMP induces downstream effector proteins that ameliorate inflammation, oxidative stress, and apoptosis, which provides neuroprotection and proliferative capabilities for neurite outgrowth (see Athauda and Foltynie, 2016b and Glotfelty et al., 2019 for more detailed signaling pathways). Restoration of insulin signaling through the upregulation of active insulin receptor substrate-1 (IRS-1) and downstream proteins provide additional neuroprotection (see Athauda and Foltynie, 2016a and Hölscher, 2020). This, coupled with increased production of brain-derived neurotrophic factor (BDNF) and glial cell line-derived neurotrophic factor (GDNF), contributes to the amelioration of deficits associated with PD.
Much of what causes the death of dopaminergic neurons in the substantia nigra of the midbrain remains a mystery, although hints toward an understanding of the development of PD pathology comes from a subset of drug users in the 1980s. These individuals mistakenly used heroin contaminated with the toxic chemical MPTP (1-methyl-4-phenyl-1,2,3,6-tetra-hydropyridine) and shortly thereafter developed PD-like symptoms, including dopaminergic neuron death. The MPTP metabolite, MPP+, accumulates in the mitochondria of DA producing neurons, leading to dysfunction and an array of oxidation products that further contribute to damage. It has been established that key genetic mutations associated with PD (LRRK2, parkin, DJ1, and PINK) likewise cause mitochondrial dysfunction of DA neurons and later cell death [15]. Aggregation of α-synuclein in Lewy bodies has been suggested to be caused by a presynaptic neurotransmitter deficiency that disrupts the surviving DA neuron function [16]. However, it remains unclear whether Lewy bodies, located in nerve cell bodies, are derived from presynaptic or cell body located α-synuclein. An array of reactive oxygen species, cell debris, other damage-associated molecular patterns (DAMPS), and α-synuclein deposits are sensed by surveying microglia, the innate immune cells of the brain, causing their phenotypic diversification to perform reparative functions as well as to recruit other microglia to control the local damage. Some microglia become chronically pro-inflammatory in this process, and secretion of a cascade of cytokines and chemokines can induce phenotypic shifts in astrocytes, another glial cell-type involved in metabolic support of neurons among other homeostatic functions. Recently, these reactive astrocytes have been shown to produce a soluble neurotoxic factor that exacerbates chronic inflammatory damage in the brain [17]. Clearly, there is an inflammatory component to PD pathology including myeloperoxidase immunoreactive cells (Gellhaar et al. 2017) (summarized in Figure 2(b)). Actions of GLP-1R agonists on neurons as well as microglia and astrocytes have been directly linked to providing potent anti-inflammatory, neurotrophic, and neuroprotective actions [5], even blocking the development of pro-inflammatory microglia and reactive astrocytes in genetic animal models of PD [13,18]. A recent study [13] provides strong evidence for focus on inflammation as a main contributor of PD, with effects of a GLP-1R agonist increasing lifespan and decreasing pathology in genetic animal models of the disease.
Activation of GLP-1R in multiple cell types induces the upregulation of neuroprotective intracellular pathways [5,19], production of trophic factors [20–23], and restored brain insulin sensitivity/signaling [9] which are all beneficial for combating PD (Figure 2(c)). Further evidence of GLP-1 mediated restoration of neuronal insulin signaling in humans comes from a recent clinical trial of Exenatide in patients with PD. Individuals treated with Exenatide had significant improvements in PD symptoms following the 48-week regimen that persisted 12-weeks off medication. Neuronal derived exosomes were isolated from these patients’ blood, and significant upregulation of active insulin receptor substrate-1 (IRS-1) and other insulin receptor substrate proteins including mammalian target of rapamycin (mTOR) and protein kinase B (Akt) was observed [24]. During the past 10 years, a multitude of studies demonstrating potent anti-PD effects of GLP-1R agonists (Figure 2(c)) have been published (extensively reviewed in [19,25]), and promising research indicates enhanced efficacy with monomeric combinations of GLP-1 with either GIP, Gcg, or both (Figure 3(a)) [5,26].
Figure 3.
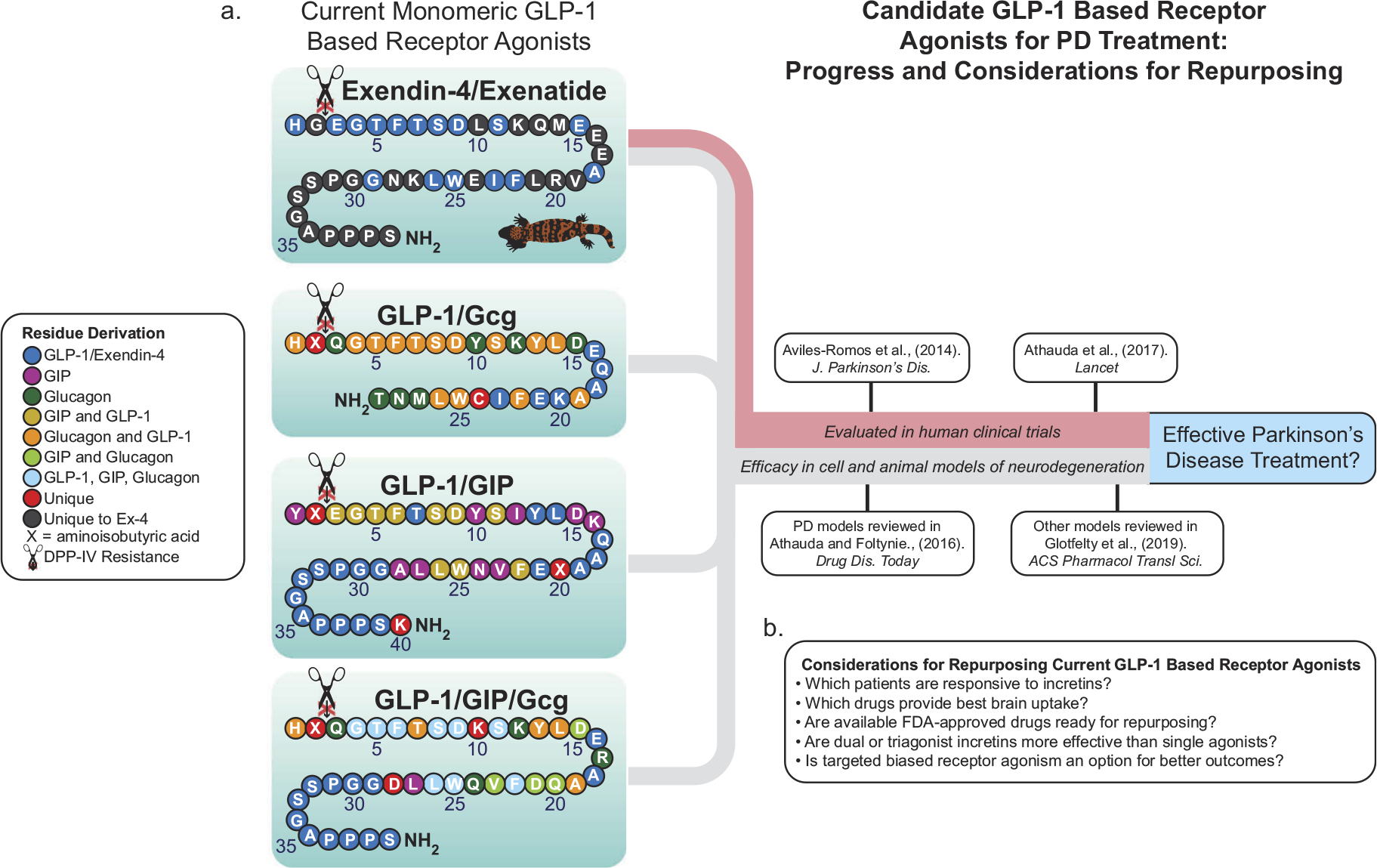
Incretin-based mimetics as possible PD treatments. (a) Many FDA-approved GLP-1R agonists are available on the market and several have been tested in human clinical trials for PD (red line), while multi-agonists incorporating elements of GLP-1, GIP, and Gcg have proven efficacious in preclinical models of PD among other neurodegenerative injuries and diseases (gray line). (B) Several questions remain as to which of these to repurpose or develop further into a treatment for PD. Amino acid structures adapted from Glotfelty et al., 2019.
Dual GLP-1R/GcgR (oxyntomodulin) and GLP-1R/GIPR agonists are safe for use in humans [27,28], with the former showing superior effectiveness for weight loss and positive metabolic changes in obese mice and non-human primates than comparable doses of GLP-1R agonists alone [29]. Similar findings were demonstrated by Finan and colleagues [30] using a dual GLP-1R/GIPR agonist versus a single GLP-1R agonist in a variety of mouse models and in non-human primates. In this same study, improved insulinotropic and antihyperglycemic properties with this dual agonist were shown in humans versus a GLP-1R agonist alone. Supraphysiological effects in reducing body weight, enhancing glycemic control, and reversing hepatic steatosis have been exhibited using a balanced GLP-1R/GIPR/GcgR triagonist compared to dual agonists and FDA-approved single GLP-1R agonists [31]. Notably, recent studies in neuroblastoma cell cultures show that GLP-1R/GIPR, a dual-incretin agonist known as ‘twincretin’ [32], and the GLP-1R/GIPR/GcgR triagonist [33] significantly enhance cyclic adenosine monophosphate (cAMP) production compared to single GLP-1R agonists alone. Furthermore, the triagonist shows superior neuroprotective effects against glutamate toxicity versus twincretin [33]. Neurodegenerative animal models are also revealing improved outcomes using monomeric multi-agonists versus single agonists. These effects include enhanced reductions in inflammation and increased production of neurotrophic factors using a dual GLP-1R/GIPR agonist in an MPTP model of PD [34]. More studies in neurodegeneration models are needed to verify these functional improvements using the multi-agonists and to identify specific contributions of individual receptor activation. It is important to consider that genetically modified mice that knock out certain receptors may compensate for these absences and exhibit atypical physiological responses to the monomeric multi-agonists [35]. Regardless, more studies will likely find improvements using the multi-agonist approach. As GLP-1R agonists are the most established class of incretin mimetics, they will continue to drive new research and clinical trials in humans for neurodegenerative disorders. Multi-agonist incretin-based mimetics are beginning to be evaluated in human clinical trials for metabolic diseases and will likely emerge as a major class of drugs in the near future [5].
3. The need for new Parkinson’s disease treatments
Despite numerous advances in modern medicine, neurodegenerative disorders remain some of the most elusive to tackle. In 2020, a void still remains in establishing effective treatments to cure these diseases, and PD is no exception. Arvid Carlssońs groundbreaking discovery of DA’s marked effects on movement control in 1957, along with subsequent discoveries of DA deficiencies in the striata of PD patients, led to the development of levodopa (L-3,4-dihydroxyphenylalanine) as the primary treatment for symptomatic relief, a legacy that continues to this day. Although levodopa provides relief of akinesia and other Parkinsońs related motor symptoms, it, along with other PD treatments do nothing to halt the debilitating disease progression. In addition to aberrant motor control experienced by many PD patients, non-motor symptoms, including gastrointestinal tract and metabolic issues, are quite common early in the disease onset and remain as the disease advances.
In recognition of the lack of disease-modifying therapies, the Parkinson’s Disease Linked Clinical Trials (LCT) initiative was formed in 2012. This brought together world PD experts from industry, academia, government, and major non-profit/other funding agencies to prioritize existing drugs that presented the most promise for repositioning as a PD treatment. The group surveyed many candidate drugs and settled on 26 to prioritize for clinical trials. GLP-1R agonists built the highest consensus scores for having the most potential and were selected for pre-prioritization in clinical trials [36]. The LCT gave GLP-1R agonists priority, in part, due to the multiple biochemical pathways they activate. The upregulation of cAMP by GLP-1 was identified as an ideal target outcome. This results from activation of its class B G-protein coupled receptor (GPCR) and, via several cascades, leads to the upregulation of multiple key downstream proteins that may blunt a host of common problems associated with PD. GLP-1-based dual- and tri-receptor agonists (incorporating GIP, Gcg, or both) were in the infant stage of their development during the initial LCT gathering and thus were not considered in the initiative. However, it is notable that these act on multiple class B GPCRs simultaneously, possibly activating similar pathways as single GLP-1R agonists with increased potency or synergy. Of the 26 drug candidates selected by the LCT in 2012, most provided single modes of action to target PD. These included PARP inhibitors (Veliparib and Olaparib), drugs with anti-oxidant effects (Deferiprone, Cysteamine, Deferirpone, Resveratrol, Trehalose, and Genistein), and those with anti-inflammatory potency (Resveratrol and Sativex) [36]. The synthetic GLP-1R agonists, including those combined with GIPR and GcgR agonists, differ in that they are potentially disease modifying in all of the aforementioned categories. Since the initial LCT 2012 meeting, several FDA-approved GLP-1R agonists and others in development have successfully moved through or been initiated in human clinical trials for PD (summarized in Table 2).
Table 2.
GLP-1R agonists in clinical trials for PD. A variety of FDA-approved and newly developed (*) GLP-1R agonists either have completed trials or are currently in progress. SC = subcutaneous; # clinical trials can be located at www.clinicaltrials.gov.
| Company | Drug name | PD clinical trial information | |
|---|---|---|---|
|
| |||
| AstraZeneca | Exenatide | Byetta® | Phase 2 clinical trial completed (#NCT01174810) (See Aviles-Olmos et al., 2013) |
| Bydureon® | Phase 2 clinical trial completed (#NCT01971242) (See Athauda et al., 2017) | ||
| Phase 1 clinical trial recruiting (#NCT0346687) | |||
| Phase 2 clinical trial recruiting (#NCT04305002) | |||
| Phase 3 clinical trial not yet recruiting (#NCT04232969) | |||
| Peptron Inc. | Exenatide SR | *PT-302 | Phase I clinical trial completed (#NCT00964262) |
| Phase 2 clinical trial recruiting (#NCT04269642) | |||
| Neuraly Inc. | Exenatide | *NLY01 | Phase 1 clinical trial completed (#NCT03672604) (See Yun et al., 2018) |
| Phase 2 clinical trial recruiting (#NCT04154072) | |||
| Novo Nordisk | Liraglutide | Victoza® | Phase 2 clinical trial recruiting (#NCT02953665) |
| Semiglutide | Ozempic® | Phase 2 clinical trial not yet recruiting (#NCT03659682) | |
| Sanofi-Aventis | Lixesentatide | Adlyxin® | Phase 2 clinical trial recruiting (#NCT03439943) |
Findings from completed human PD clinical trials with GLP-1R agonists indicate positive preliminary results. The initial open-label proof-of-concept clinical trial (NCT01174810) notes persistent cognitive and motor function improvements up to 12-months post-treatment [37] with a later randomized, double-blind, placebo-controlled trial (NCT01971242) similarly showing improvements in motor scores in treated patients [38], thereby, cross-validating them. Results from the later clinical trial described in Athauda et al. [2017, 38] have provided additional insight into which patients might best respond to GLP-1R agonists as treatments, which may clarify future recruitment into clinical trials. Tremor-dominant phenotypes and lower Movement Disorder Society-Sponsored Revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS) Part-2 scores predicted the greatest motor response to treatment, indicating PD patients at earlier stages of disease progression may be best suited for a GLP-1R drug regimen [39]. This is reasonable because, in later stages of the disease, there are fewer remaining DA neurons for the drug to positively impact. As mentioned above, further analysis of blood-derived neuronal exosomes from the patients indicates modified insulin, Akt, and mTOR signaling as contextual foundations for improvements seen in this study [24]. Although it is unknown how other cell types in the brains of the patients may have responded pathologically, the studies present profound results supporting the potential for GLP-1R agonists as a new class of PD disease-modifying drugs. Future clinical trials, including those in Table 2, will provide more insight into the benefits of using different GLP-1R agonists for PD treatment. Although no human clinical trials or epidemiological studies have measured the PD modifying effects of multi-agonist incretin-based mimetics, a recent Swedish nation-wide study found significantly reduced incidences of PD in diabetic patients with a history of using DPP-IV inhibitors but not GLP-1R agonists [40]. The GLP-1R agonist patient cohort may have been underpowered as indicated by the authors, although the results related to the DPP-IV compounds are profound and provide evidence that improved signaling of GLP-1, GIP, and Gcg may have a prophylactic effect on PD development. Further studies are needed to confirm these results, but they provide context for future studies analyzing how incretin-based therapies affect the development of PD.
4. Conclusion
Although originally developed for metabolic disorders, GLP-1R agonists present a promising treatment for PD as well as other neurodegenerative disorders. Utilization and repurposing of already FDA-approved drugs, such as GLP-1R agonists, will accelerate the treatment options which have remained stagnant over the past 50 years, importantly addressing the growing population diagnosed with PD [36,41]. Preclinical in vitro and in vivo PD modeling shows that long-acting incretin-based mimetics evoke strong neuroprotective and reparative processes that contribute to the amelioration of PD. Building on the successes of GLP-1R agonism in these models, new classes of long-acting dual- and tri-agonist incretin-based mimetics that combine active regions of GLP-1 with GIP and/or Gcg are emerging as safe and possibly more efficacious than single GLP-1R agonists. As the single GLP1R agonists are the most developed class of incretin mimetics, human clinical trials are currently focused on their use and are revealing significant benefits in treating PD, especially during the early onset of the disease [39].
5. Expert opinion
The past 10 years have marked a paradigm shift in the potential to treat and modify PD outcomes. With the already available FDA-approved GLP-1R agonists, additional funding is needed to assess which compounds are best suited to treat PD. Several considerations need to be made to optimally move forward with GLP-1R agonists for PD treatment (Figure 3(b)) and can potentially be followed by probing the contents of blood-derived neuronal and/or astrocytic exosomes and brain imaging for evidence of target engagement. These clinical trials will be key to understanding mechanisms incretin-based mimetics employ to affect the human brain under pathological conditions.
5.1. Dual- and tri-agonist options
Attention to monomeric dual- and tri-agonists incorporating combinations of GLP-1 with GIP and/or Gcg (shown in Figure 3 (a)), present even further potential for more potent classes of incretin-based mimetics (for a comprehensive list of preclinical multi-agonists see Glotfelty et al., 2019). Dual GLP-1R/GIPR and GLP-1R/GcgR agonists have been shown to be safe in humans [27,42], while the GLP-1R/GIPR/GcgR triagonist is in its infancy and has not yet been tested in humans. Preliminary studies in PD cellular and animal models of neurodegeneration show efficacy in using these multi-agonists for enhanced brain function, including improvements over single GLP-1R agonism [5,19,26,33,43,44]. Although these monomeric multi-agonists are currently in development for metabolic disorders [6,45], the horizon for future PD drugs appears promising. As the need for PD treatments is time sensitive, repurposing already available approved drugs is the best short-term solution for bringing new treatments to the public. It is crucial that those with the greatest brain uptake are identified and considered in future trials. With our available knowledge from previous clinical trials, we suggest that enrolling those who may respond best to GLP-1R agonists, specifically those in the early onset of disease, should be further explored and tested with more rigor.
5.2. Biased agonism and improved blood-brain barrier penetration
In addition to multi-agonists, other forms of GLP-1R agonists that can selectively activate downstream GPCR pathways are currently being explored. These include biased agonists which can preferentially upregulate cAMP and other secondary messengers [46]. Identifying crucial pathways, such as improved cAMP activation, to target in PD treatment can perhaps provide enhanced outcomes as compared to the current classes of GLP-1R agonists. As all FDA-approved GLP-1R agonists are designed for metabolic disorders, blood-brain barrier (BBB) penetration was of little consideration in their design. Brain concentrations of Ex-4 in humans [38] and rats [12] are around 2–3% of levels in the plasma, and similar findings have been observed from the administration of a non-peptidic GLP-1R agonist designed to treat obesity [47]. With the advent of designer, non-peptidic GLP-1R agonists such as TTP273 [48] and TT-OAD2 [49], improved BBB penetration and potency may be possible, adding to the therapeutic benefits of currently available incretin-based mimetics. Further neurological-related research into non-peptidic designer GLP-1R agonists compared to those currently available should be a priority in the coming years.
5.3. The current and potential landscape
GLP-1R agonists are a fruitful area for repositioning across a variety of neurodegenerative and some neuropsychiatric disorders, with the greatest inroads having been made for treating PD thus far. Current widespread prescribed use and studies in humans indicate that these compounds are well tolerated and clinical trials demonstrate promising results in treating PD. Because of their widespread long-term use for diseases such as T2DM, there should be a wealth of epidemiological data available to examine population-wide effects of GLP-1R agonists and PD prevalence (see 40). These agents were not developed for neurodegenerative disorders, but their repurposing should be prioritized. Furthermore, the new multi-agonists and long-term delivery techniques will likely further optimize the use of these drugs not only in T2DM but also for neurodegenerative disorders that affect wide portions of the population.
Article Highlights.
FDA-approved glucagon-like peptide-1 receptor (GLP-1R) agonists are well positioned for repurposing for the treatment of Parkinson’s disease (PD). Although originally developed for metabolic disorders, GLP-1R agonists offer a promising treatment for PD and other neurodegenerative disorders
GLP-1R agonists are broadly neuroprotective in models of PD. Numerous clinical trials using GLP-1R agonists as PD treatments have been completed or are underway.
Monomeric GLP-1-based peptide analogs in combination with glucose-dependent insulinotropic peptide (GIP) and/or glucagon (Gcg) are emerging therapeutics and may provide improved efficacy.
Single GLP-1R agonists are the most developed class of incretin mimetics; hence, human clinical trials are currently focused on their use and are revealing significant benefits in treating PD, especially during the early onset of the disease. Enrolling patients who may respond best to GLP-1R agonists, specifically those in the early onset of disease, should be further explored and tested with more rigor.
Because the need for PD treatments is time sensitive, repurposing already available approved drugs is the best short-term solution for bringing new treatments to the market
It is crucial that those therapies with the greatest brain uptake are identified and considered in future trials.
Acknowledgments
The authors thank Lauren Brick, Visual Media core, National Institute on Drug Abuse, National Institutes of Health, USA, in relation to the figures.
Funding
The research of the authors was supported in part by (i) the Intramural Research Program of the National Institute on Aging, National Institutes of Health, USA, (ii) the Swedish Research Council K2012–62X-03185–42-4, and (iii) the Swedish Brain Foundation. E Glotfelty is supported by the National Institutes of Health—Karolinska Institute Graduate Partnership Program.
Footnotes
Declaration of interest
The NH Greig is a co-inventor on patents related to the use of GLP-1R agonists for the treatment of neurodegenerative disorders and has assigned all rights to the National Institutes of Health, USA.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as either of interest (•) or of considerable interest (••) to readers.
- 1.Nauck MA, Heimesaat MM, Orskov C, et al. Preserved incretin activity of glucagon-like peptide 1 [7–36 amide] but not of synthetic human gastric inhibitory polypeptide in patients with type- 2 diabetes mellitus. J. Clin. Invest. 1993;91:301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drucker DJ. Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metab. 2018;27:740–756. [DOI] [PubMed] [Google Scholar]
- 3.Müller TD, Finan B, Bloom SR, et al. Glucagon-like peptide 1 (GLP-1). Mol. Metab. 2019;30:72–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Eng J, Kleinman WA, Singh L, et al. Isolation and characterization of exendin-4, an exendin-3 analogue, from Heloderma suspectum venom: further evidence for an exendin receptor on dispersed acini from guinea pig pancreas. J Biol Chem. 1992;267:7402–7405. •• This publication outlines the discovery of Exendin-4, a DPP-IV-resistant analog of human GLP-1. This discovery led to the new class of long-acting T2DM drugs and is the foundation for the structures of emerging multiagonists.
- 5. Glotfelty EJ, Delgado TE, Tovar-y-Romo LB, et al. Incretin mimetics as rational candidates for the treatment of traumatic brain injury. ACS Pharmacol. Transl. Sci. 2019;2:66–91. • This is a recent comprehensive review that outlines much of the research and use of incretin mimetics for neurodegenerative disorders. It covers major signalling mechanisms common among these disorders.
- 6.Quiñones M, Fernø J, Diéguez C, et al. Exciting advances in GPCR-based drugs discovery for treating metabolic disease and future perspectives. Expert Opin Drug Discov. 2019;14:421–431. [DOI] [PubMed] [Google Scholar]
- 7.Salcedo I, Tweedie D, Li Y, et al. Neuroprotective and neurotrophic actions of glucagon-like peptide-1: an emerging opportunity to treat neurodegenerative and cerebrovascular disorders. Br J Pharmacol. 2012;166:1586–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hogg E, Athreya K, Basile C, et al. High prevalence of undiagnosed insulin resistance in non-diabetic subjects with Parkinson’s disease. J. Parkinsons. Dis. 2018;8:259–265. [DOI] [PubMed] [Google Scholar]
- 9.Hölscher C Brain insulin resistance: role in neurodegenerative disease and potential for targeting. Expert Opin Investig Drugs. 2020;29(4):333–348. [DOI] [PubMed] [Google Scholar]
- 10.Athauda D, Foltynie T. Insulin resistance and Parkinson’s disease: a new target for disease modification? Prog Neurobiol. 2016;145–146:98–120. [DOI] [PubMed] [Google Scholar]
- 11.Procaccini C, Santopaolo M, Faicchia D, et al. Role of metabolism in neurodegenerative disorders. Metabolism. 2016;65(9):1376–1390. [DOI] [PubMed] [Google Scholar]
- 12.Bader M, Li Y, Lecca D, et al. Pharmacokinetics and efficacy of PT302, a sustained-release Exenatide formulation, in a murine model of mild traumatic brain injury. Neurobiol. Dis. 2018;124:439–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yun SP, Kam T-I, Panicker N, et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018;24:931–938. • This recent publication is important for establishing the role of antiinflammatory mechanisms of GLP-1R agonists for PD treatment. It establishes a recently discovered inflammatory mechanism for glial cell activation as a major influence on developing PD pathology and comprehensively shows its mitigation through the use of a GLP-1R agonist.
- 14.Chen S, Yu S-J, Li Y, et al. Post-treatment with PT302, a long-acting Exendin-4 sustained release formulation, reduces dopaminergic neurodegeneration in a 6-Hydroxydopamine rat model of Parkinson’s disease. Sci. Rep. 2018;8:10722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Langston JW. The MPTP story. J Parkinsons Dis. 2017;7:S11–S19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schulz-Schaeffer WJ. The synaptic pathology of α-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 2010;120:131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541 (7638):481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hinkle JT, Dawson VL, Dawson TM. The A1 astrocyte paradigm: New avenues for pharmacological intervention in neurodegeneration. Mov. Disord. 34;mds.27718. DOI: 10.1002/mds.27718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Athauda D, Foltynie T. The glucagon-like peptide 1 (GLP) receptor as a therapeutic target in Parkinson’s disease: mechanisms of action. Drug Discov Today. 2016;21:802–818. [DOI] [PubMed] [Google Scholar]
- 20.Jalewa J, Sharma MK, Gengler S, et al. A novel GLP-1/GIP dual receptor agonist protects from 6-OHDA lesion in a rat model of Parkinson’s disease. Neuropharmacology. 2017;117:238–248. [DOI] [PubMed] [Google Scholar]
- 21.Spielman LJ, Gibson DL, Klegeris A. Incretin hormones regulate microglia oxidative stress, survival and expression of trophic factors. Eur J Cell Biol. 2017;96:240–253. [DOI] [PubMed] [Google Scholar]
- 22.Zhang L, Zhang L, Li L, et al. Semaglutide is neuroprotective and reduces α-synuclein levels in the chronic MPTP mouse model of Parkinson’s disease. J Parkinsons Dis. 2019;9:157–171. [DOI] [PubMed] [Google Scholar]
- 23.Ji C, Xue G-F, Lijun C, et al. A novel dual GLP-1 and GIP receptor agonist is neuroprotective in the MPTP mouse model of Parkinson′s disease by increasing expression of BNDF. Brain Res. 2016;1634:1–11. [DOI] [PubMed] [Google Scholar]
- 24. Athauda D, Gulyani S, Karnati HK, et al. Utility of neuronal-derived exosomes to examine molecular mechanisms that affect motor function in patients with Parkinson disease. JAMA Neurol. 2019;76(4):420. •• This study is of particular importance as it (1) examines a novel and accessible mechanism for evaluating treatment outcomes (blood-derived neuronal exosomes) for PD and (2) provides a contextual basis for the mechanisms by which GLP-1R agonism is affecting PD patients.
- 25.Kim DS, Choi H-I, Wang Y, et al. A new treatment strategy for Parkinson’s disease through the gut-brain axis: the glucagon-like peptide-1 receptor pathway. Cell Transplant. 2017;26:1560–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hölscher C Novel dual GLP-1/GIP receptor agonists show neuroprotective effects in Alzheimer’s and Parkinson’s disease models. Neuropharmacology. 2018;136:251–259. [DOI] [PubMed] [Google Scholar]
- 27.Wynne K, Park AJ, Small CJ, et al. Subcutaneous oxyntomodulin reduces body weight in overweight and obese subjects: a double-blind, randomized, controlled trial. Diabetes. 2005;54(8):2390–2395. [DOI] [PubMed] [Google Scholar]
- 28.Frias JP, Bastyr EJ, Vignati L, et al. The sustained effects of a dual GIP/GLP-1 receptor agonist, NNC0090–2746, in patients with type 2 diabetes. Cell Metab. 2017;26(2):343–352.e2. [DOI] [PubMed] [Google Scholar]
- 29.Elvert R, Herling AW, Bossart M, et al. Running on mixed fuel-dual agonistic approach of GLP-1 and GCG receptors leads to beneficial impact on body weight and blood glucose control: a comparative study between mice and non-human primates. Diabetes, Obes. Metab. 2018;20:1836–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Finan B, Ma T, Ottaway N, et al. Unimolecular dual incretins maximize metabolic benefits in rodents, monkeys, and humans. Sci Transl Med. 2013;5. DOI: 10.1126/scitranslmed.3007218. • This publication has been foundational in supporting use of a dual incretin approach for treating diabetes. Demonstration of the safety and efficacy of a monomeric dual GLP-1R/GIPR agonist has been important moving forward neurological research associated with the compound.
- 31.Finan B, Yang B, Ottaway N, et al. A rationally designed monomeric peptide triagonist corrects obesity and diabetes in rodents. Nat. Med. 2015;21:27–36. [DOI] [PubMed] [Google Scholar]
- 32.Tamargo IA, Bader M, Li Y, et al. Novel GLP-1R/GIPR co-agonist “twincretin” is neuroprotective in cell and rodent models of mild traumatic brain injury. Exp Neurol. 2017;288:176–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Y, Glotfelty EJ, Namdar I, et al. Neurotrophic and neuroprotective effects of a monomeric GLP-1/GIP/Gcg receptor triagonist in cellular and rodent models of mild traumatic brain injury. Exp. Neurol. 2020;324:113113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yuan Z, Li D, Feng P, et al. A novel GLP-1/GIP dual agonist is more effective than liraglutide in reducing inflammation and enhancing GDNF release in the MPTP mouse model of Parkinson’s disease. Eur. J. Pharmacol. 2017;812:82–90. [DOI] [PubMed] [Google Scholar]
- 35.Capozzi ME, DiMarchi RD, Tschöp MH, et al. Targeting the incretin/glucagon system with triagonists to treat diabetes. Endocr Rev. 2018;39:719–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brundin P, Barker RA, Conn PJ, et al. Linked clinical trials-the development of new clinical learning studies in Parkinson’s disease using screening of multiple prospective new treatments. J. Parkinsons. Dis. 2013;3:231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aviles-Olmos I, Dickson J, Kefalopoulou Z, et al. Motor and cognitive advantages persist 12 months after exenatide exposure in Parkinson’s disease. J. Parkinsons. Dis. 2014;4:337–344. [DOI] [PubMed] [Google Scholar]
- 38. Athauda D, Maclagan K, Skene SS, et al. Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet. 2017;390:1664–1675. •• This is the first randomized, double-blind, placebo-controlled clinical trial of a GLP-1R agonist for the treatment of PD.
- 39.Athauda D, Maclagan K, Budnik N, et al. Post hoc analysis of the Exenatide-PD trial – factors that predict response. Eur. J. Neurosci. 2019;49:410–421. [DOI] [PubMed] [Google Scholar]
- 40.Svenningsson P, Wirdefeldt K, Yin L, et al. Reduced incidence of Parkinson’s disease after dipeptidyl peptidase-4 inhibitors – a nationwide case-control study. Mov. Disord. 2016;31:1422–1423. [DOI] [PubMed] [Google Scholar]
- 41.Goetz CG. The history of Parkinson’s disease: early clinical descriptions and neurological therapies. Cold Spring Harb Perspect Med. 2011;1:a008862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Frias JP, Nauck MA, Van J, et al. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet. 2018;392(10160):2180–2193. [DOI] [PubMed] [Google Scholar]
- 43.Li T, Jiao J-J, Hölscher C, et al. A novel GLP-1/GIP/Gcg triagonist reduces cognitive deficits and pathology in the 3xTg mouse model of Alzheimer’s disease. Hippocampus. 2018;28:358–372. [DOI] [PubMed] [Google Scholar]
- 44.Li Y, Wu K-J, Yu S-J, et al. Neurotrophic and neuroprotective effects of oxyntomodulin in neuronal cells and a rat model of stroke. Exp. Neurol. 2017;288:104–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tschöp MH, Finan B, Clemmensen C, et al. Unimolecular polypharmacy for treatment of diabetes and obesity. Cell Metab. 2016;24(1):51–62. [DOI] [PubMed] [Google Scholar]
- 46.Hager MV, Johnson LM, Wootten D, et al. β-arrestin-biased agonists of the GLP-1 receptor from β-amino acid residue incorporation into GLP-1 analogues. J Am Chem Soc. 2016;138:14970–14979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Freeman JL, Agolory J, Valcarce C. Preclinical findings with GLP-1 receptor agonist TTP273 reinforce importance of neuro-enteroendocrine signaling, In: diabetes. New Orleans:A309–A309. DOI: 10.2337/db16-861-1374. [DOI] [Google Scholar]
- 48.Tomlinson B, Hu M, Zhang Y, et al. Investigational glucagon-like peptide-1 agonists for the treatment of obesity. Expert Opin Investig Drugs. 2016;25:1167–1179. [DOI] [PubMed] [Google Scholar]
- 49.Zhao P, Liang Y-L, Belousoff MJ, et al. Activation of the GLP-1 receptor by a non-peptidic agonist. Nature. 2020;577(7790):432–436. [DOI] [PubMed] [Google Scholar]