

Standfirst:
The Tuberculosis Drug Accelerator, an experiment designed to facilitate collaboration in TB drug discovery by breaking down barriers among competing labs and institutions, has reached the 10-year landmark. We review the consortium’s achievements, advantages and limitations and advocate for application of similar models to other diseases.
Tuberculosis (TB) claimed 1.4 million lives in 2019, ranking it among the leading causes of death and one of the most lethal caused by a single infectious agent. That year, 10 million people fell ill with TB, 2% of whom had multidrug resistant (MDR) disease. As the cornerstone of TB control, “short-course” combination chemotherapy has two interrelated limitations: its 6-month duration, which reduces adherence, and its diminishing efficacy, reflecting the emergence and spread of drug-resistant strains of the etiologic agent, Mycobacterium tuberculosis (Mtb). Encouragingly, recently developed regimens comprising new and repurposed drugs simplify and shorten the treatment of MDR TB1. However, the major objective in TB chemotherapy is to develop dramatically shortened, less toxic, universal drug regimens that can achieve rapid, durable cure irrespective of resistance to existing drugs.
This goal underpinned the formation of the TB Drug Accelerator (TBDA), an innovative multi-sector, multi-partner, multi-disciplinary, virtual collaborative network. In addition to fueling the TB drug pipeline with new targets, leads, clinical candidates and regimens, the TBDA has enabled technological advances of broad relevance to anti-infective drug discovery and regimen design. Here, we review the first decade of the TBDA, highlight some of its achievements, and reflect on how the learnings from this experiment might inform approaches to drug discovery for other diseases.
A bold mission
At its inception in 2012, the bold mission of the TBDA was to discover drugs that in combination would create shorter, safer and simpler regimens to cure all TB patients. The TBDA is a unique collaboration among academic, pharmaceutical, governmental and other organizations operating within a cooperation and sharing agreement that pledges to make medicines affordable to those in need, builds knowledge and seeks to identify and pursue the most promising biological targets and chemical series for breakthrough drug candidates.
At the outset, a campaign of unprecedented scale phenotypically screened over 10 million compounds from the TBDA partners under conditions designed to capture various metabolic states of Mtb in human disease. The members triaged the data and disclosed unencumbered hits to the consortium for evaluation. Sharing datasets reduced duplicative efforts and allowed members to identify common mechanisms of action, focus on the most promising compounds, efficiently analyze structure-activity relationships and prioritize the best leads for optimization. As leads emerged, the introduction of a portfolio management team with rotating membership and review by an external advisory panel of drug discovery experts maximized scientific quality and applied consistent, rigorous standards for compound progression.
The consortium membership has evolved dynamically to bring in teams with distinctive assets and cutting-edge scientific capabilities. Members form cross-organizational sub-teams to define and address high-value drug targets and ensure that high priority projects are optimally resourced. The Bill & Melinda Gates Foundation (BMGF) coordinates strategic and operational aspects and supplements the in-kind support provided by the pharmaceutical industry partners with grants to other participating institutions and engagement of contract research organizations. Drug candidates emerging from formal hit assessment enter a collective portfolio and are typically taken into clinical development by the TBDA members with product development capabilities.
Promoting the collaborative culture are twice-yearly, two-day meetings of all members in person, when possible; bimonthly virtual meetings on select scientific topics; sharing of assays, compounds and expertise among members; and consensus-building for target product profiles. The development of friendships, trust and respect sustains a candid exchange of views.
Scientific contributions
Development of a fast-acting, universal drug regimen for TB is hindered by incomplete understanding of TB pathogenesis. The TBDA has responded on nine fronts: (i) bringing increased chemical diversity to bear on (ii) phenotypic screens against Mtb in diverse metabolic states (e.g.2–4), while also (iii) conducting screens against recombinant protein targets prioritized by essentiality and vulnerability5; (iv) determining if the whole-cell activity of a given enzyme inhibitor is on-target via use of Mtb strains that are conditionally hypomorphic for the target (e.g.6) and (v) by studying the inhibitor’s metabolomic impact on Mtb (e.g.6–8); (vi) testing compounds in diverse animal models7–9 and (vii) under conditions found in pulmonary lesions10; (viii) conducting studies of pharmacokinetics and pharmacodynamics not just in blood but within TB lesions7,11,12 and within Mtb itself6,13,14 and (ix) exploring how to prioritize specific combinations15. Determining the mechanism of action of a new hit series in Mtb can be a daunting task; one reward can be to highlight critical pathways that are subject to inhibition in other ways. Figure 1 illustrates some of these approaches and Table 1 illustrates some of the findings.
Figure 1. Overview of TBDA activities.
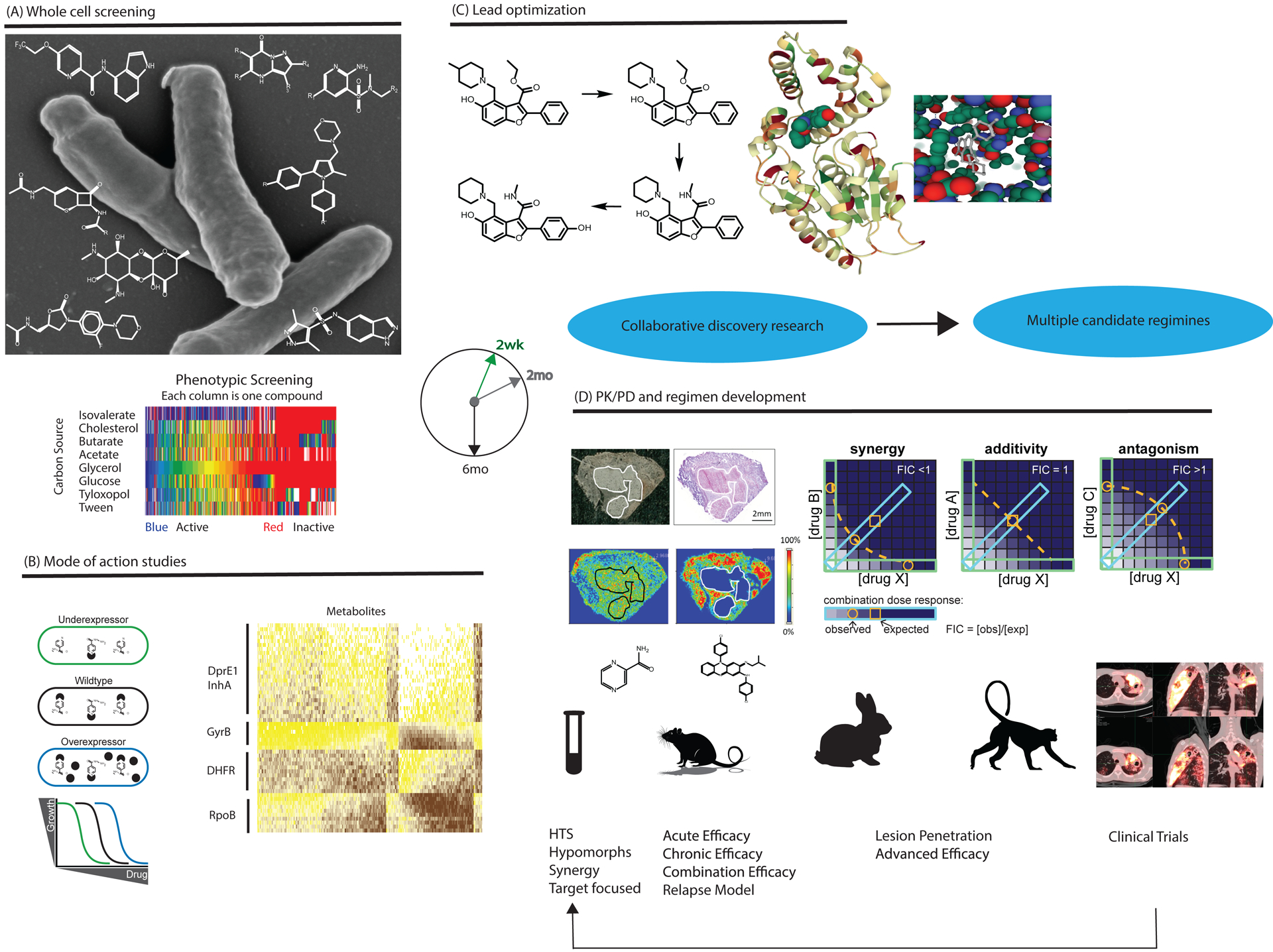
A. Phenotypic whole cell screening of Mtb against substantial chemical diversity. Examples of hit series from TBDA screening programs are shown against a scanning electron microscopy image of the pathogen. TBDA screens use diverse biological conditions illustrated by the heat map showing divergent results from screening the same compound deck against Mtb grown with different nutrients. B. Genetics and metabolomics facilitate studies of mechanism of action. For example, strains under- and over-expressing the target show selectively differential responses to an inhibitor of that target (left), while novel inhibitors of a pathway often elicit metabolomic responses similar to those seen with known inhibitors of the same pathway (right). C. Advanced series enter lead optimization with crystallography support, as illustrated for PKS 13 inhibitors. D. Advances in preclinical tools for evaluating new regimens include imaging mass spectrometry to quantify drug penetration into TB lesions and new approaches to measure synergies and antagonisms among multiple agents. Higher animal models of TB are studied with the same tools applied in early human studies, helping to develop better preclinical tools to predict treatment responses in humans. Lessons from clinical trials are applied to the ongoing process of drug discovery. (All studies involving animals were ethically reviewed and conducted in accordance with the relevant institution’s policy on the care, welfare and treatment of laboratory animals.)
Table 1.
Examples of scientific contributions from the TBDA
| Approach | Illustrative learnings |
|---|---|
| Metabolomics can help reveal mechanism of action of antibacterial compounds, quantify their uptake and detect their intrabacterial transformation | The second-line antitubercular, p-aminosalicylic acid, is a prodrug. It gets incorporated into an analog of dihydrofolate that subsequently poisons the cell.13 |
| Bedaquiline, a newly approved ATP synthase inhibitor used for the treatment of drug-resistant TB, kills Mtb through a complex response that involves glutamine synthetase as a collateral target, suggesting the possibility of rationally engineering drug synergy14. | |
| Genetically engineered variation of target protein levels can help identify targets and determine if cidality of inhibitors is due to on-target action | The enzyme protein-biotin ligase is a target that has potential to shorten chemotherapy22. |
| Inhibition of a step in the coenzyme A biosynthesis pathway suffices to kill Mtb23. | |
| An inhibitor of phosphopantetheinyl transferase kills Mtb by inhibiting the enzyme. Mtb’s vulnerability is amplified by the action of a hydrolase that undoes the transferase reaction6. | |
| Given the efficacy of bedaquiline, inhibiting other targets in the bacterial respiration machinery offers potential synergies24–26. | |
| Spatially resolved mass spectrophotometry and transcriptomics of TB lesions can provide reality-checks for prospective therapeutics | TB granulomas contain metabolites that can enable Mtb to bypass the need for de novo biosynthesis of certain critical molecules by Mtb enzymes that might otherwise be prioritized as targets27. |
| TB granulomas are highly structured with respect to expression of host inflammatory molecules28 | |
| TB drugs distribute differentially within lesions7,10 | |
| Pursuit of mechanism of action of phenotypically active compounds can reveal new biology | An essential enoyl CoA hydratase is catalytically inactive but plays a crucial role in enabling lipid transport during mycolic acid biosynthesis29. |
| Hundreds of repetitive proteins in a particular family play an essential role as solute-specific transporters of various small molecules across the mycobacterial outer membrane30. |
Where possible, TBDA members have made sets of hit compounds from such efforts available to the TB research community either directly16 or by participating in collation of sets for wider usage within and beyond TB (https://www.mmv.org/mmv-open/pathogen-box/about-pathogen-box).
The phenotypic screening campaigns refocused attention on known scaffolds such as carbapenems, which were further energized by early clinical indications of activity in a Phase 2 clinical study17. Certain targets – two of which have been actively pursued in the TBDA18,19–were discovered with high frequency. To enrich target opportunities, TBDA members developed ways to find compounds active against critical biosynthetic pathways in the bacterium20.
Given that non-replicating Mtb shows phenotypic resistance to most TB drugs in vitro, one approach to treatment shortening has been to develop genetic tools that allow tightly controlled protein depletion in non-replicating cells to evaluate candidate targets. Another approach to the treatment duration problem is to appreciate and overcome the challenge of drug distribution throughout the fibrous capsule and lipidic core of the pulmonary lesions within which many of the Mtb reside that spread the disease. A clinical study that monitored the lesion-penetrating ability of known antituberculosis medicines10 guided the development of mouse, marmoset and rabbit models that allow assessment of penetration of agents during development7,9. Recognition of the phenotypic drug tolerance of the bacteria in necrotic lesions11 allowed development of more predictive pharmacokinetic/pharmacodynamic (PK/PD) models for regimen and dose prediction12.
Medical contributions
The TBDA model allows compounds under development by one group to be tested in animal models developed by another group, enriching the preclinical data package and facilitating compound prioritization. Groups developing inhibitors of the same target can share their specialized assays for off-target activities that contribute to toxicity. Below are two of many examples of these efficiency-enhancing collaborative actions enabled by the TBDA model.
TBDA members used a relapse mouse model to determine the relative potential of various combinations of novel drugs given for various lengths of time to improve relapse-free cure relative to the standard of care (rifampicin, isoniazid, pyrazinamide, and ethambutol). A study of bedaquiline, pretomanid and linezolid8 indicated that the addition of linezolid significantly contributed to sterilization and shortening of treatment, motivating clinical efforts that led to relapse-free cure of 90% of subjects with extensively drug-resistant (XDR) TB in the Nix-TB trial1, leading in turn to FDA and EMA approval of pretomanid as part of the regimen with bedaquiline and linezolid. The mouse study also demonstrated that similar sterilization was achieved in this regimen with only 1–2 months of linezolid, providing the rationale for the ongoing ZeNix trial.
Simultaneously, the TBDA undertook discovery of oxazolidinones safer than linezolid for use in drug-sensitive as well as MDR and XDR TB, along with second-generation diarylquinolines with higher therapeutic indices than bedaquiline. TB Alliance and MSD in partnership with the NIH have each discovered oxazolidinones with sterilizing activity equivalent to linezolid but significantly less potential for toxicity from inhibition of mammalian mitochondrial protein synthesis. The new oxazolidinone TBI-223 (TB Alliance) is entering a Phase 1 multiple ascending dose trial and the MSD compound is undergoing IND enabling studies. Efforts on diarylquinolines led by the TB Alliance produced two compounds with >10-fold higher in vitro potency than bedaquiline, which results in shorter treatment times when combined with pretomanid and linezolid. Both analogs demonstrate superior in vitro activity against bedaquiline-resistant Mtb strains with mutations in the Rv0678 regulator that de-repress expression of the MmpL5/MmpS5 efflux system, and sterilize Mtb in mice better than bedaquiline21. Both compounds are or soon will be in Phase 1 clinical trials.
The TBDA structure enables pre-publication knowledge-sharing among members. This has promoted phenotypic screening under conditions relevant to human infection. Moreover, it allows pharmaceutical companies to conduct preclinical tests in combination with drugs being developed by other members. For example, a screen by GSK and partners for inhibitors of Mtb growth in macrophages identified a compound, GSK2556286 (www.newtbdrugs.org/pipeline/compound/gsk-286), that operates via a novel, complex pathway related to cholesterol catabolism that has been reported by other TBDA partners3. This mechanism of action gives moderate efficacy in vivo in standard infection models, making further development challenging. Extensive efforts within the TBDA and with additional partners demonstrated that GSK286 contributes important efficacy when combined with compounds with other mechanisms of action. With the scientific endorsement and practical support of the TBDA and help from consortia that contribute farther downstream in drug development, such as ERA4TB, this exciting candidate has entered Phase 1 clinical development.
ADVANTAGES AND DISADVANTAGES
The TBDA addresses two daunting challenges—to bring rapid adaptability to TB drug discovery through ongoing identification of progress-blocking and rate-limiting obstacles, and to overcome those obstacles by mobilizing resources across three interfaces: academia-industry; competitor-competitor; and basic and applied research. Gifts in kind from pharmaceutical companies, grants to academics, and engagement of contract research organizations put cutting edge technologies to work, often while they are being developed. Sustaining all this is a non-modifiable collaboration agreement that spells out requirements for data sharing, public disclosure and global access; defines the rights of signatories regarding changes in membership; introduces a standard material transfer agreement; and leaves intellectual property agreements to the collaborating institutions. The TBDA’s approach complements much larger and broader investments by the US National Institutes of Health and the European Research Council that involve a broader and lengthier process of proposal review.
Pharmaceutical companies are attracted to the TBDA model because it provides an opportunity to contribute know-how and resources to a global health drug discovery program. In the few cases where the companies have internal TB programs, they can access leading academic expertise and save costs in developing drugs that will face a poor return on investment. Benefits of success include incurring goodwill in major emerging markets, improving the safety of employees in endemic areas, and gratifying the altruism of scientific staff and corporate leaders.
Similarly, academics are gratified by addressing major unmet medical needs. They are privileged to see their ideas tested in real-world translational and clinical settings and benefit from early access to tool compounds. They are gladdened by rapid, realistic funding decisions. Their mutual progress is accelerated by collaborating rather than working in isolation and by hearing early about each other’s latest advances.
Of course, there are negatives: Some companies fear risk of IP diffusion. Resentment of ‘free riders’ can challenge the trust on which the enterprise depends. Companies can abruptly change course. The TBDA began with 6 pharmaceutical company partners. Since then, 4 more joined and 4 left, with 3 of the originals staying the course. Contributing to such shifts are the trade-offs that companies face between other investments and the financial, personnel, and technical contributions required by the TBDA, whose extensive interactions draw effort and resources from other tasks.
For academics who are not developing compositions of matter, putting IP at risk is of less concern. However, the academics await a foundation’s invitation and evaluation rather than initiating an application that will undergo peer review. Provision of funding is agile, but so is its termination.
CONCLUSIONS
We believe that the advantages of the TBDA model of multi-sector, multi-party, multi-disciplinary collaboration greatly outweigh its limitations. Sharing that assessment, the BMGF has launched drug accelerators for malaria (http://winzeler.ucsd.edu/malda/), helminths and Cyptosporidium. However, as an engine for agility, cooperativity and efficiency that complements traditional approaches to disease-oriented research, the model deserves wider application. The global community would do well to set in place the organizational and legal structures and build the culture for similar collaborations to address additional WHO type 3 diseases (e.g., Ebola, Chikungunya, dengue), and to tackle WHO type 2 diseases, such as pandemic viral infections.
Footnotes
Ethics Statement: The following had or have conflicting interests by virtue of the indicated employment: David Barros-Aguirre, Robert H. Bates, Fabian Gusovsky, Philip A. Hipskind, Dale J. Kempf, Joel Lelievre, David B. Olsen, H. Michael Petrassi, Alexander Pym, Michael Schrimpf, Anna M. Upton. The other authors declare no conflicting interests.
REFERENCES
- 1.Conradie F, et al. Treatment of highly drug-resistant pulmonary tuberculosis. N Engl J Med 382, 893–902 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Early J, et al. Identification of compounds with pH-dependent bactericidal activity against Mycobacterium tuberculosis. ACS Infect Dis 5, 272–280 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.VanderVen BC, et al. Novel inhibitors of cholesterol degradation in Mycobacterium tuberculosis reveal how the bacterium’s metabolism is constrained by the intracellular environment. PLoS Pathog 11, e1004679 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Warrier T, et al. Identification of novel anti-mycobacterial compounds by screening a pharmaceutical small-molecule library against nonreplicating Mycobacterium tuberculosis. ACS Infect Dis 1, 580–585 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Kim JH, et al. A genetic strategy to identify targets for the development of drugs that prevent bacterial persistence. Proc Natl Acad Sci U S A 110, 19095–19100 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ballinger E, et al. Opposing reactions in coenzyme A metabolism sensitize Mycobacterium tuberculosis to enzyme inhibition. Science 363(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blanc L, et al. High-resolution mapping of fluoroquinolones in TB rabbit lesions reveals specific distribution in immune cell types. Elife 7(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tasneen R, et al. Contribution of oxazolidinones to the efficacy of novel regimens containing bedaquiline and pretomanid in a mouse model of tuberculosis. Antimicrob Agents Chemother 60, 270–277 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Via LE, et al. Host-mediated bioactivation of pyrazinamide: Implications for efficacy, resistance, and therapeutic alternatives. ACS Infect Dis 1, 203–214 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Prideaux B, et al. The association between sterilizing activity and drug distribution into tuberculosis lesions. Nat Med 21, 1223–1227 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sarathy JP, et al. Prediction of drug penetration in tuberculosis lesions. ACS Infect Dis 2, 552–563 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Strydom N, et al. Tuberculosis drugs’ distribution and emergence of resistance in patient’s lung lesions: A mechanistic model and tool for regimen and dose optimization. PLoS Med 16, e1002773 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chakraborty S, Gruber T, Barry CE 3rd, Boshoff HI & Rhee KY Para-aminosalicylic acid acts as an alternative substrate of folate metabolism in Mycobacterium tuberculosis. Science 339, 88–91 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang Z, et al. Mode-of-action profiling reveals glutamine synthetase as a collateral metabolic vulnerability of M. tuberculosis to bedaquiline. Proc Natl Acad Sci U S A 116, 19646–19651 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cokol M, Kuru N, Bicak E, Larkins-Ford J & Aldridge BB Efficient measurement and factorization of high-order drug interactions in Mycobacterium tuberculosis. Sci Adv 3, e1701881 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ballell L, et al. Fueling open-source drug discovery: 177 small-molecule leads against tuberculosis. ChemMedChem 8, 313–321 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diacon AH, et al. beta-Lactams against tuberculosis--new trick for an old dog? N Engl J Med 375, 393–394 (2016). [DOI] [PubMed] [Google Scholar]
- 18.Aggarwal A, et al. Development of a novel lead that targets M. tuberculosis polyketide synthase 13. Cell 170, 249–259 e225 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ray PC, et al. Spirocycle MmpL3 inhibitors with improved hERG and cytotoxicity profiles as Inhibitors of Mycobacterium tuberculosis growth. ACS Omega 6, 2284–2311 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chengalroyen MD, et al. Biological profiling enables rapid mechanistic classification of phenotypic screening hits and identification of KatG activation-dependent pyridine carboxamide prodrugs with activity against Mycobacterium tuberculosis. Front Cell Infect Microbiol 10, 582416 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu J, et al. Comparative efficacy of the novel diarylquinoline TBAJ-587 and bedaquiline against a resistant Rv0678 mutant in a mouse model of tuberculosis. bioRxiv (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tiwari D, et al. Targeting protein biotinylation enhances tuberculosis chemotherapy. Sci Transl Med 10(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Evans JC, et al. Validation of CoaBC as a bactericidal target in the coenzyme A pathway of Mycobacterium tuberculosis. ACS Infect Dis 2, 958–968 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beites T, et al. Plasticity of the Mycobacterium tuberculosis respiratory chain and its impact on tuberculosis drug development. Nat Commun 10, 4970 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berube BJ, et al. Novel MenA inhibitors are bactericidal against Mycobacterium tuberculosis and synergize with electron transport chain inhibitors. Antimicrob Agents Chemother 63(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Smith J, et al. Anthranilic amide and imidazobenzothiadiazole compounds disrupt Mycobacterium tuberculosis membrane potential. Medchemcomm 10, 934–945 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Park Y, et al. Essential but not vulnerable: Indazole sulfonamides targeting inosine monophosphate dehydrogenase as potential leads against Mycobacterium tuberculosis. ACS Infect Dis 3, 18–33 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marakalala MJ, et al. Inflammatory signaling in human tuberculosis granulomas is spatially organized. Nat Med 22, 531–538 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cox JA, et al. THPP target assignment reveals EchA6 as an essential fatty acid shuttle in mycobacteria. Nat Microbiol 1, 15006 (2016). [DOI] [PubMed] [Google Scholar]
- 30.Wang Q, et al. PE/PPE proteins mediate nutrient transport across the outer membrane of Mycobacterium tuberculosis. Science 367, 1147–1151 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]