

Abstract
With the growing worldwide prevalence of antibiotic-resistant strains of tuberculosis (TB), new targets are urgently required for the development of treatments with novel modes of action. Fumarate hydratase (fumarase), a vulnerable component of the citric acid cycle in Mycobacterium tuberculosis (Mtb), is a metabolic target that could satisfy this unmet demand. A key challenge in the targeting of Mtb fumarase is its similarity to the human homolog, which shares an identical active site. A potential solution to this selectivity problem was previously found in a high-throughput screening hit that binds in a non-conserved allosteric site. In this work, a structure-activity relationship study was carried out with the determination of further structural biology on the lead series, affording derivatives with sub-micromolar inhibition. Further, the screening of this series against Mtb in vitro identified compounds with potent minimum inhibitory concentrations (MIC).
Keywords: Mycobacterium tuberculosis, Mtb, tricarboxylic acid cycle, citric acid cycle, Krebs cycle, fumarate hydratase, fumarase, fumarate, L-malate, high-throughput screening, allosteric, deconstruction-reconstruction, defragmentation, differential scanning fluorimetry, DSF
INTRODUCTION
Mycobacterium tuberculosis (Mtb), the causative agent of the disease tuberculosis (TB), infects 10 million people per year and is responsible for the deaths of 1.6 million annually, of which 300,000 are HIV-positive.1 Further, a quarter of the global population, 1.7 billion individuals, are thought to be infected with asymptomatic latent TB and hence at risk of active infection.2 Despite the scope of the disease, treatment remains intensive and long-term with multi- and extensively-drug resistant TB strains a growing problem, hence there is a continuing urgent need for novel improved anti-TB therapeutics.3
Mtb fumarate hydratase (fumarase) is a component of the citric acid cycle in Mtb,4 where it catalyzes the reversible interconversion of fumarate and L-malate. When normoxic cells are rapidly dividing fumarase operates in the forward direction (fumarate to L-malate) and is involved in energy generation and the synthesis of precursors for biosynthetic pathways. Under conditions of lowered oxygen availability fumarase operates in the reverse direction allowing fermentative production and secretion of succinate.5,6 During pathogenesis of tuberculosis anoxia develops in the center of caseous lesions prior to the development of cavitary lesions, where sudden reaeration allows rapid bacterial growth to promote bacterial transmission from host-to-host.7 Mtb fumarase is a non-redundant and therefore vulnerable component of the citric acid cycle,8 a situation that is not guaranteed in bacteria.9 The regulation of fumarase is therefore central to the realignment of bacterial metabolism that occurs during normal disease progression in Mtb, which makes it a very high value target for drugs that might allow faster bacterial clearance. Mtb fumarase has been shown to be essential for Mtb survival with depletion linked to impaired growth due to accumulation of fumarate, which can react with cysteine thiols.10 This modification adversely impacts the antioxidants catalase and mycothiol, inducing hypersensitivity to oxidative stress and ultimately Mtb death both in vitro and in vivo.
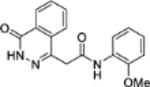
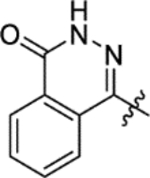
Mtb fumarase is a 210 kDa class II fumarase,11 characterized by thermostability, iron-independence and a homotetrameric structure.9,12 Each subunit is defined by an N-terminal domain (residues 1–137), a central domain (residues 138–393) whose α-helices pack with those from other subunits to form a central 20-helix bundle that holds together the quaternary structure, and a conformationally flexible C-terminal domain (residues 394–474). The protein possesses four symmetry-related active sites, each located in clefts formed by three subunits and covered by an ‘SS’ loop (residues 316–325) that can switch between ‘open’ and ‘closed’ states.11 The selective targeting of Mtb fumarase is challenging due to its 53% overall sequence identity with the human homolog, which was recently the focus of a study on cancer cell lines.13 In our previous work, the conservation of active site residues between Mtb and human fumarase was circumvented through the discovery by high-throughput screening (HTS) of an allosteric inhibitor 1 of Mtb fumarase (IC50 2.5 ± 1.1 μM) (Figure 1a).14
Figure 1.
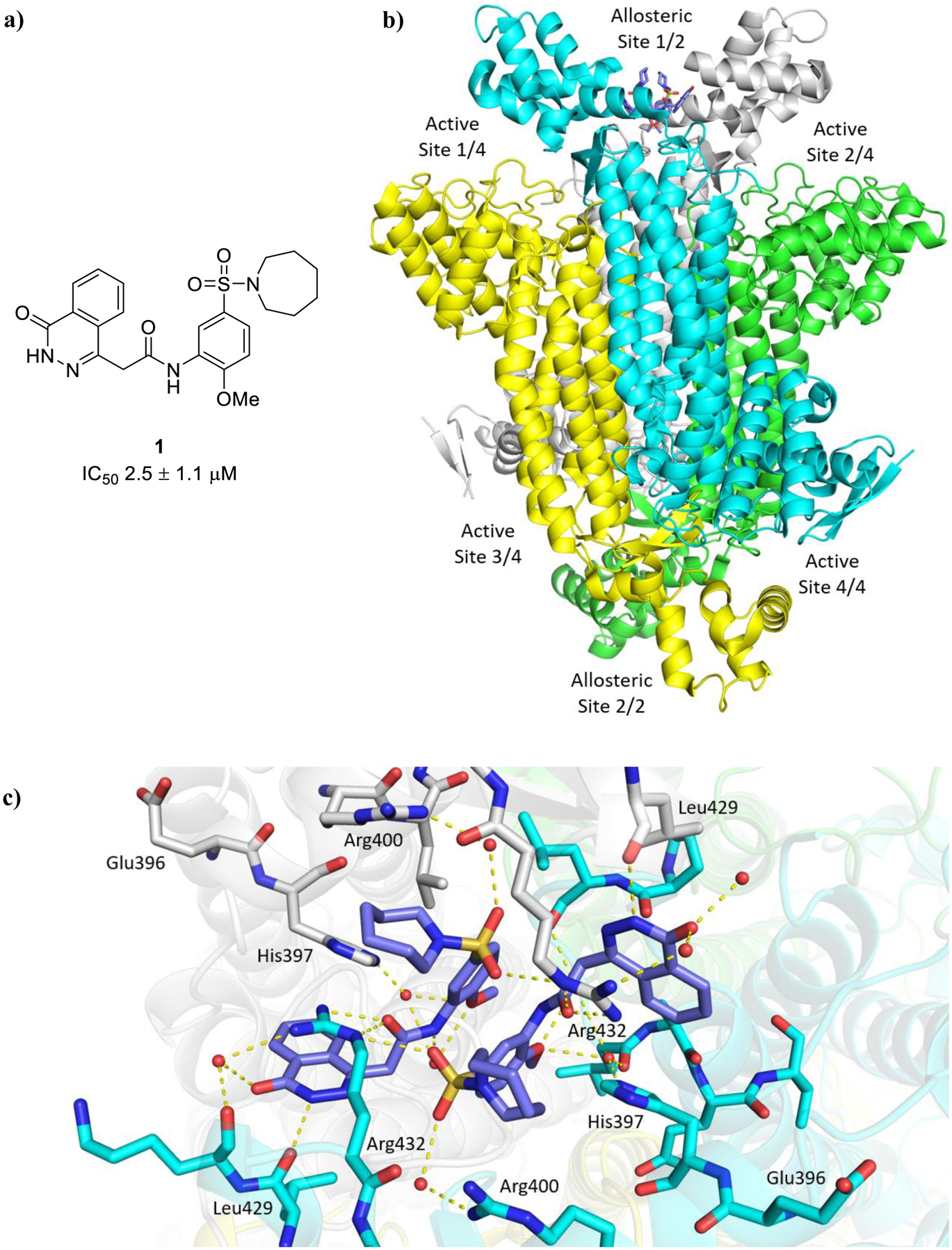
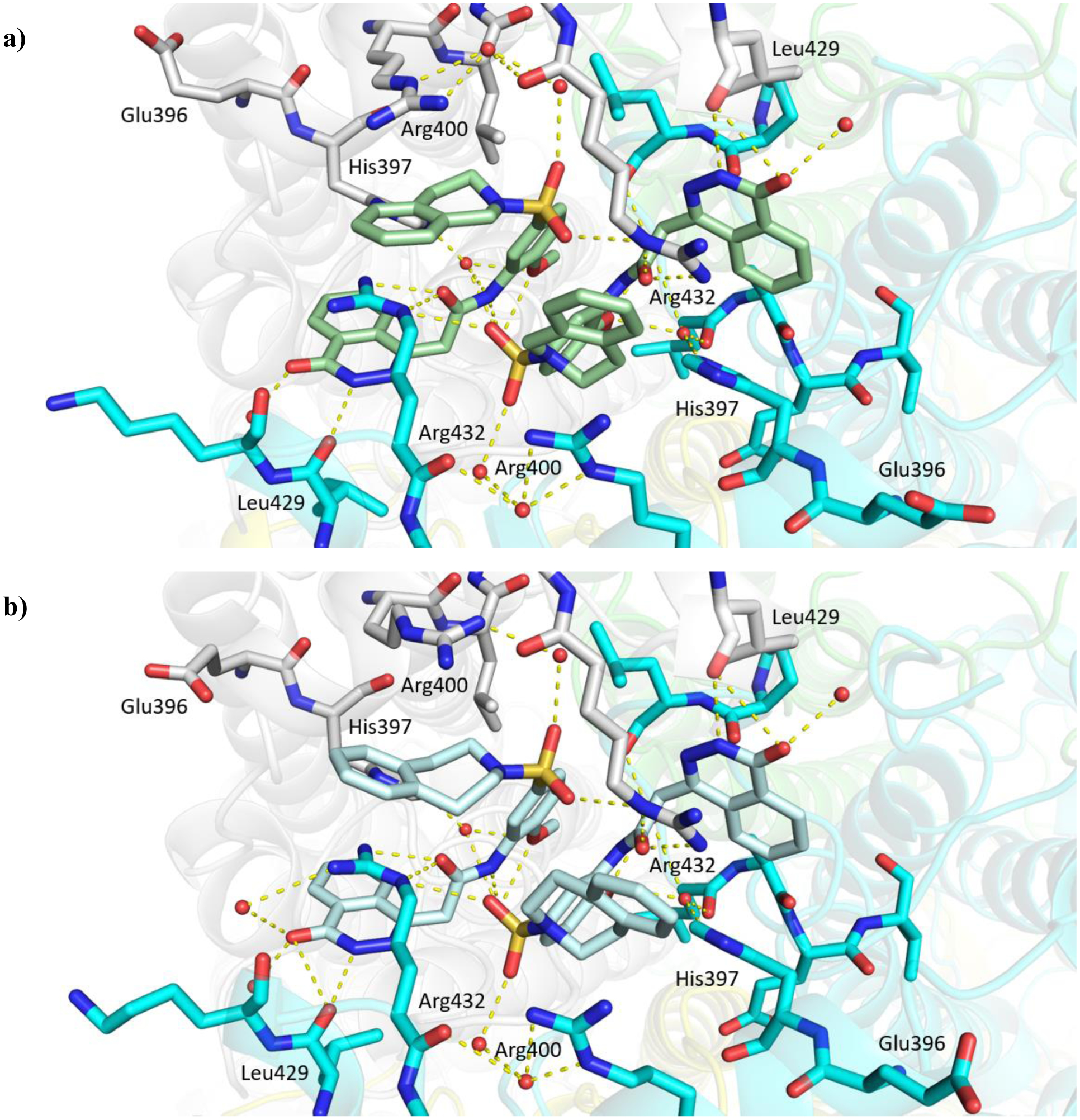
a) Allosteric inhibitor 1 of Mtb fumarase.14 b) X-ray crystal structure of Mtb fumarase in complex with 1 (PDB code 5F91, subunit A = white, subunit B = green, subunit C = cyan, subunit D = yellow),14 illustrating relative locations of the allosteric and active sites in the homotetramer. c) The interactions (yellow dashed lines) of 1 (lilac) in the allosteric site
X-ray crystallography showed that inhibitor 1 binds dimerically to Mtb fumarase at the interface between two adjacent C-terminal domains in an induced pocket (Figure 1b), locking the nearest active site in an ‘open’ conformation, with the bound molecules interacting with each other through π-stacking of their central phenyl rings (Figures 1c and S8a).14 A second symmetry-related allosteric site exists in the homotetramer however this is unoccupied in the X-ray crystal structure (Figure 1b), likely due to its participation in a crystal contact. Whilst 1 did not exhibit bactericidal properties it was able to exert inhibition of Mtb growth in vitro (65% inhibition at 250 μM) that, in combination with the demonstrated essentiality of Mtb fumarase,8,10 warranted additional investigation into inhibitor 1. Due to the dimeric-binding mode of 1, a possible strategy would be to link the molecules together into one inhibitor, however the geometry of the π-stacking interaction makes this route challenging. One way of circumventing this problem could be the use of a deconstruction-reconstruction approach,15 where the HTS hit is defragmented into fragment-like molecules allowing the identification of key binding motifs. This would allow the use of fragment-based methods to more effectively sample the ligand-binding site and develop a sub-micromolar inhibitor with activity against Mtb in vitro.16
RESULTS AND DISCUSSION
Defragmentation of HTS hit 1.
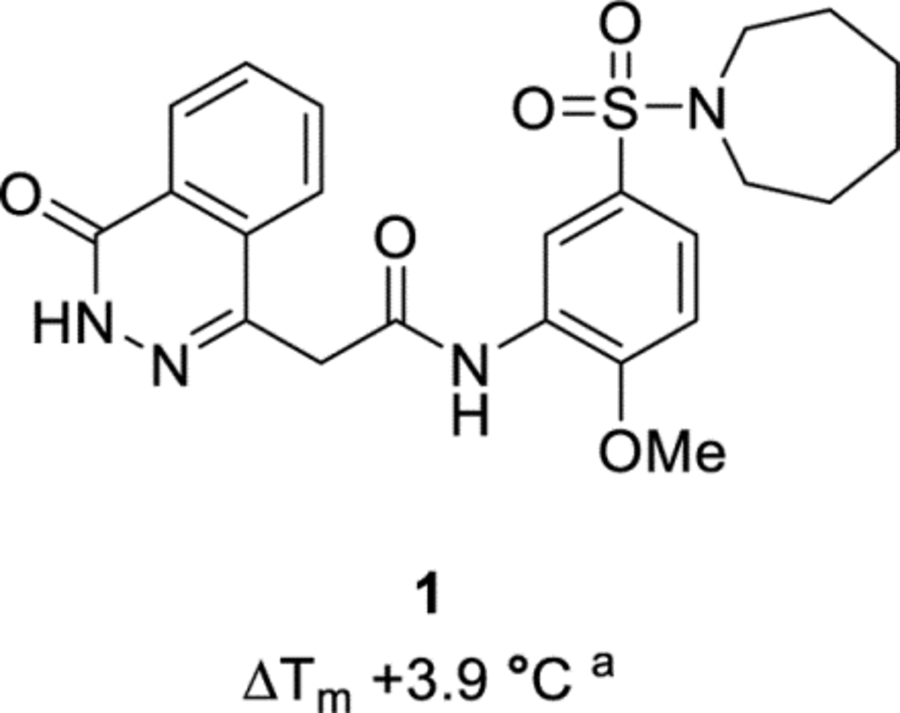
The defragmentation of HTS hit 1 into ‘fragment-like’ molecules resulted in the assembly of a focused library (Table 1), with members spanning a range of molecular weights (204 to 326 Da) and incorporating various binding motifs of 1. The screening of the library with a biochemical assay afforded no measurable inhibition at concentrations of 100 μM or 1 mM (data not shown). Similarly, the use of differential scanning fluorimetry (DSF) showed either negligible change in the melting temperature (ΔTm +0.1 °C for 10) or negative thermal shifts (ΔTm −1.6 °C for 7) at a ligand concentration of 5 mM (Table 1), in contrast to the thermal shift of +3.9 °C afforded by 1 at 0.625 mM. This includes the higher molecular weight compounds in the library 4 (ΔTm −0.4 °C) and 6 (ΔTm −0.6 °C), which retain the substituted phenyl ring of 1 in addition to either of the sulfonamide or phthalazinone motifs. These results suggest that the inhibitory capability of 1 for Mtb fumarase is dependent upon all its binding motifs, with high sensitivity to truncation. The negative impact of defragmentation is not surprising due to the large number of interactions that doubly-binding 1 makes in the allosteric site with the two C-terminal domains of Mtb fumarase (in addition to itself through the π-stacking interaction of its phenyl ring), as well as the induced nature of the site with cryptic pockets not easily targeted by weak fragments.
Table 1.
The change in the melting temperatures (ΔTm) of compounds 2-11.
| |||||
|---|---|---|---|---|---|
| Compound | ΔTmb (°C) | Compound | ΔTmb (°C) | ||
| 2 |
|
−0.6 | 7 |
|
−1.6 |
| 3 |
|
−0.4 | 8 |
|
−1.2 |
| 4 |
|
−0.4 | 9 |
|
−0.4 |
| 5 |
|
−0.7 | 10 |
|
+0.1 |
| 6 |
|
−0.6 | 11 |
|
−0.4 |
0.625 mM ligand and 2.5 μM Mtb fumarase.
5 mM ligand and 2.5 μM Mtb fumarase.
Structure-activity Relationship Study of HTS hit 1.
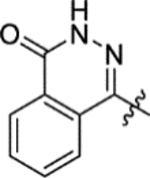
Due to lack of success in defragmentation of the HTS hit 1, attention was shifted to a structure-activity relationship study. Initial modification of the central phenyl ring of 1 through the removal of its methoxy group, which indirectly engages Leu303, Gly305, Leu306 and His397 through hydrogen bonds with two ordered water molecules (Figures 2 and S8b), was not tolerated in 12 (16% inhibition at 50 μM) (Table 2). A similar result was seen upon replacement of the methoxy group with a methyl group in 13 (<10% inhibition at 50 μM), illustrating the sensitivity of the central phenyl ring of 1 to alteration. The replacement of the sulfonamide group with an amide, and the resultant rigidification and rotation of the azepanyl ring was also not tolerated in 14 (14% inhibition at 50 μM).
Figure 2.
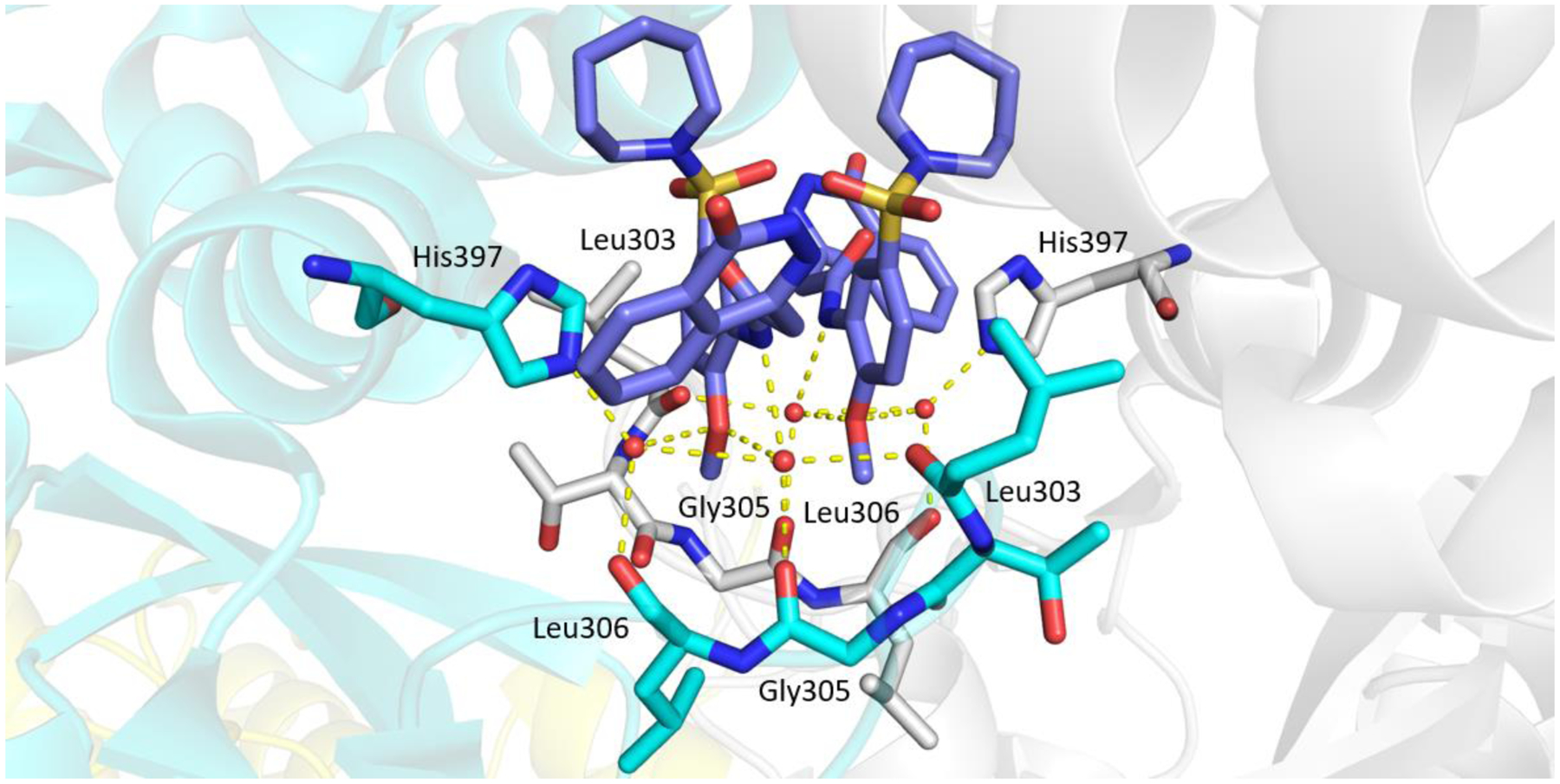
X-ray crystal structure of Mtb fumarase in complex with 1 (lilac) (PDB code 5F91, subunit A = white, subunit B = green, subunit C = cyan, subunit D = yellow),14 illustrating the interactions (yellow dashed lines) of the methoxy group of 1 in the allosteric site.
Table 2.
The inhibition at 50 μM ligand concentration afforded by compounds 1 and 12–14.
| Compound | Structure | Inhibition (%) at 50 μM |
|---|---|---|
| 1 |
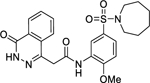
|
> 90 |
| 12 |
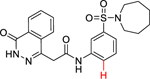
|
16 ± 1 |
| 13 |
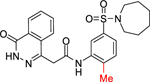
|
< 10 |
| 14 |
|
14 ± 1 |
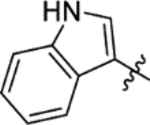
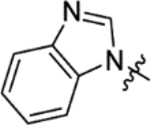
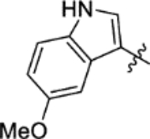
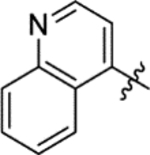
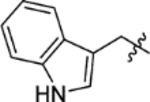
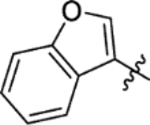
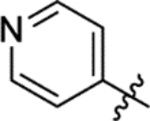
The replacement of the phthalazinone ring of HTS hit 1 with an indole in 15a (27% inhibition at 50 μM) resulted in a significant drop in inhibition (Table 3), despite X-ray crystallography showing the maintenance of the cation-π interaction with Arg432 and a hydrogen-bond with the backbone carbonyl of Leu429 (Figure 3a). The substitution of the ring in 15b (38% inhibition at 50 μM) or extension of the linker in 15c (12% inhibition at 50 μM) did not result in a significant improvement on 15a. The use of alternative 5–6 fused rings including benzofuran (15d), benzisoxazole (15e) and benzimidazole (15f) derivatives failed to provide inhibition in the biochemical assay at 50 μM. In contrast, the screening of quinoline derivative 15g (IC50 4.1 μM) resulted in a comparable IC50 value to 1 (IC50 2.0 μM), with the X-ray crystal structure of the Mtb fumarase-15g complex showing the quinoline ring hydrogen-bonding to a water molecule as opposed to the protein backbone (Figure 3b). Truncation of the quinoline ring of 15g to a pyridine in 15i eliminated inhibitory activity at 50 μM, in a similar manner to the defragmentation of 1.
Table 3.
The inhibition at 50 μM ligand concentration or half-maximal inhibitory concentrations (IC50) afforded by compounds 1 and 15a-i.
| |||||
|---|---|---|---|---|---|
| Compound | R | Inhibition (%) at 50 μM | Compound | R | Inhibition (%) at 50 μM |
| 1 |
|
> 90a | 15e |
|
< 10 |
| 15a |
|
27 ± 6 | 15f |
|
< 10 |
| 15b |
|
38 ± 5 | 15g |
|
86 ± 2b |
| 15c |
|
12 ± 9 | 15h |
|
21 ± 17 |
| 15d |
|
< 10 | 15i |
|
< 10 |
IC50 2.0 ± 0.1 μM, consistent with previously reported value.14
IC50 4.1 ± 0.3 μM.
Figure 3.
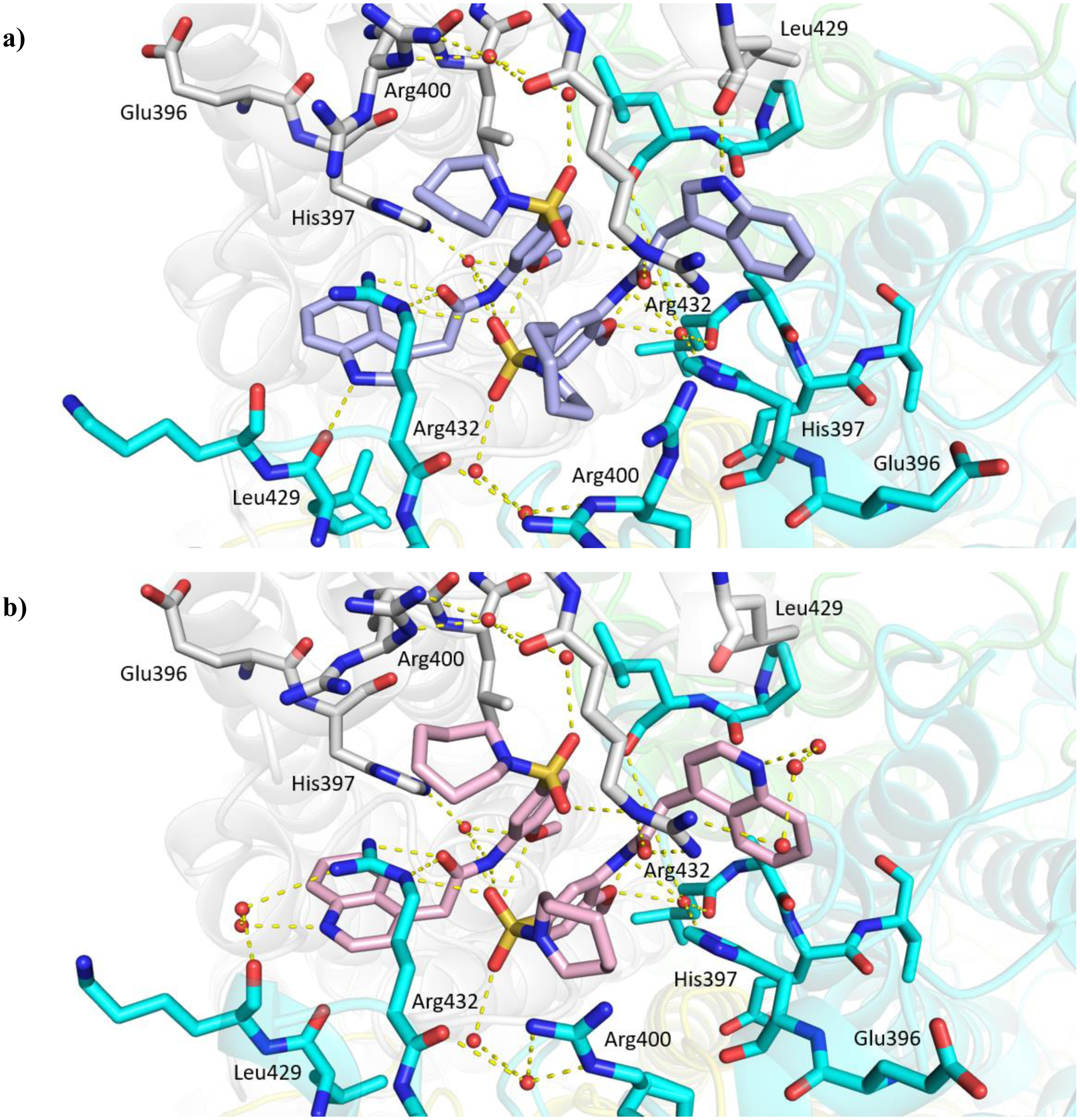
X-ray crystal structures of Mtb fumarase (subunit A = white, subunit B = green, subunit C = cyan, subunit D = yellow) in complex with a) 15a (blue) (PDB code 6S7U) and b) 15g (pink) (PDB code 6S7W), illustrating the interactions (yellow dashed lines) of the ligands in the allosteric site.
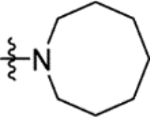
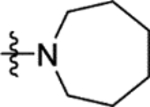
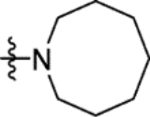
The truncation of HTS hit 1 to the N-methyl sulfonamide 16a (IC50 57 μM) afforded the lowest molecular weight derivative (402 Da) to give a measurable IC50 value (Table 4). An X-ray crystal structure was obtained of the complex of 16a with Mtb fumarase, illustrating a 5 Å movement in the Arg400 side chain (at Cε) relative to the complex with 1 to form a salt-bridge with Glu396, significantly reducing the apparent volume for growth around the sulfonamide (Figures 4a and S9b). Heterocyclic derivatives of 1 were also produced to explore the volume defined by His397, Arg400 and Arg432, beginning with the azocanyl and piperidinyl analogues 16b (IC50 4.0 μM) and 16c (IC50 4.4 μM). These 8- and 6-membered derivatives possessed slightly attenuated inhibition in comparison to 1 (IC50 2.0 μM), however were not sufficiently different to preclude testing of alternative heterocycles of similar size. X-ray crystallography showed no significant difference in the binding mode of 16b in Mtb fumarase in comparison to 1 except for Arg400 located above the larger 8-membered ring, which showed evidence of a second alternative conformation (Figures 4b and S10a). The N-methyl piperazinyl derivative 16d (IC50 38 μM), along with morpholino and thiomorpholine dioxide analogues 16e (IC50 12 μM) and 16g (IC50 13 μM) afforded IC50 values weaker than 1 or the thiomorpholino derivative 16f (IC50 4.7 μM).
Table 4.
The inhibition at 50 μM ligand concentration or half-maximal inhibitory concentrations (IC50) afforded by compounds 16a-n
| |||||
|---|---|---|---|---|---|
| Compound | R | IC50 (μM) | Compound | R | IC50 (μM) |
| 16a |

|
57 ± 3 | 16h |
|
17 ± 3 |
| 16b |
|
4.0 ± 0.1 | 16i |
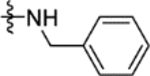
|
NDa |
| 16c |
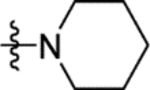
|
4.4 ± 0.1 | 16j |
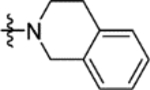
|
2.2 ± 0.2 |
| 16d |
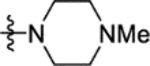
|
38 ± 2 | 16k |
|
3.4 ± 0.2 |
| 16e |
|
12 ± 1 | 16l |
|
0.67 ± 0.03 |
| 16f |
|
4.7 ± 0.2 | 16m |
|
0.67 ± 0.01 |
| 16g |
|
13 ± 1 | 16n |
|
NDb |
53 ± 3% inhibition at 50 μM concentration.
44 ± 5% inhibition at 50 μM concentration.
Figure 4.
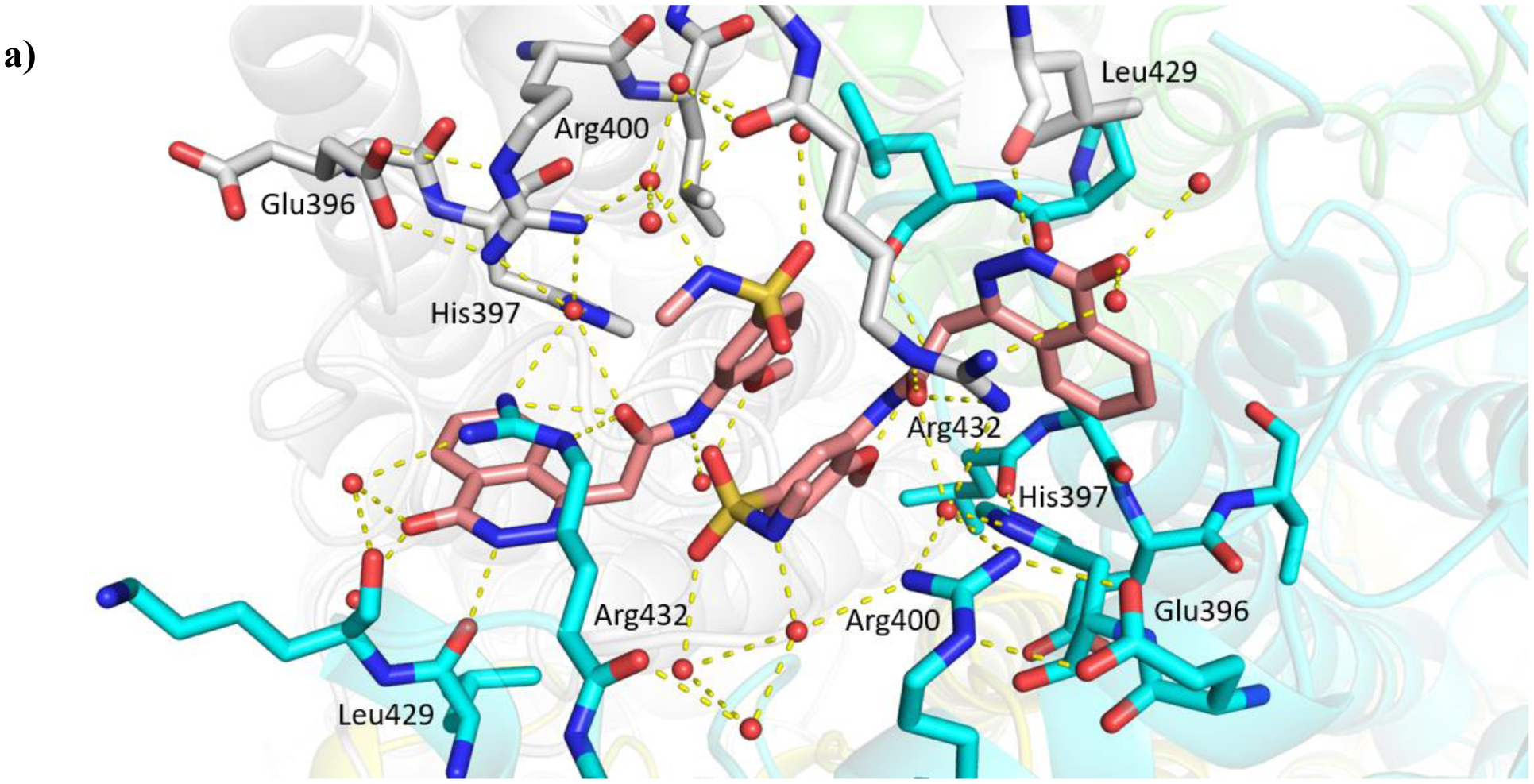
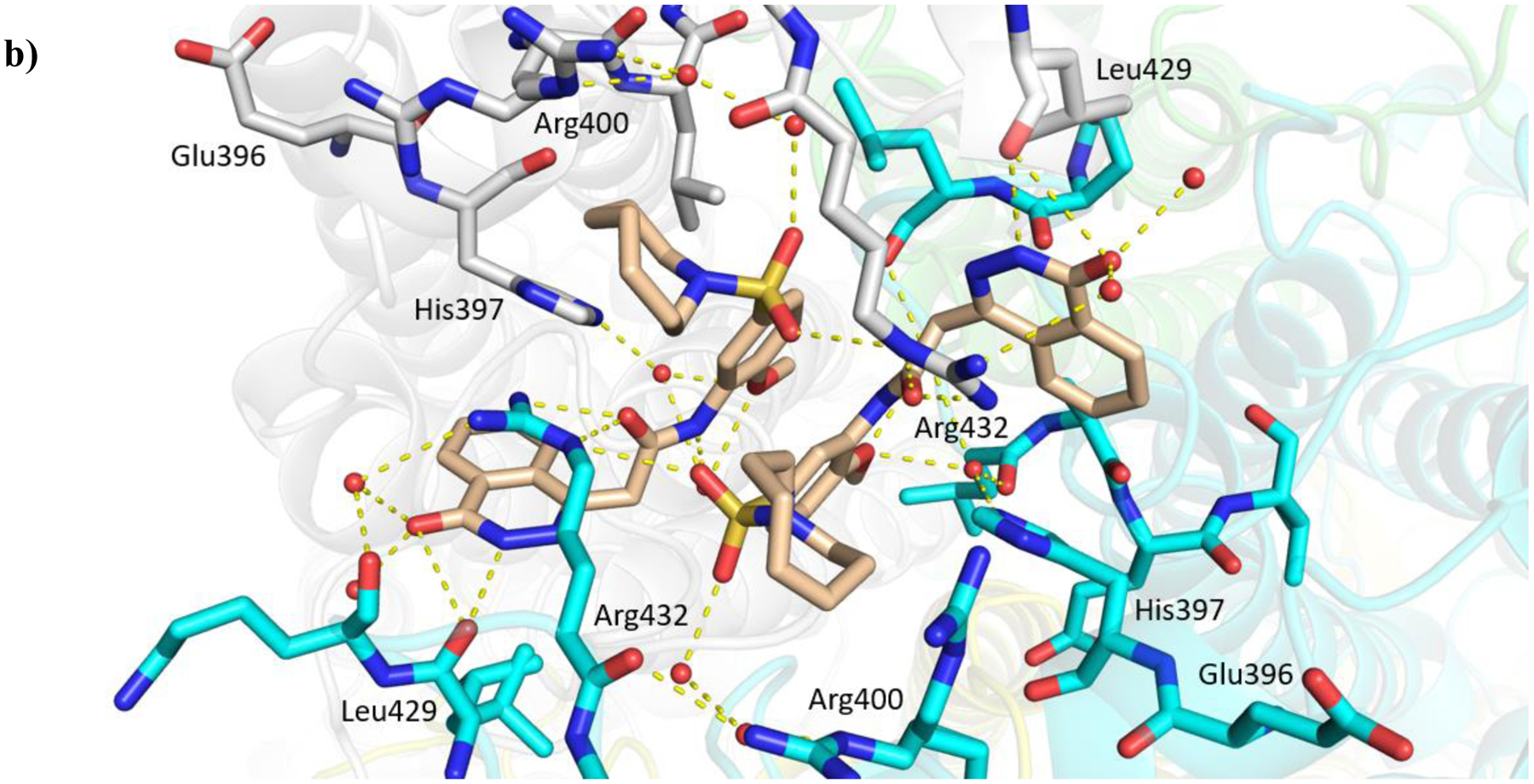
X-ray crystal structure of Mtb fumarase (subunit A = white, subunit B = green, subunit C = cyan, subunit D = yellow) in complex with a) 16a (light pink) (PDB code 6S7K) and b) 16b (beige) (PDB code 6S43), illustrating the interactions (yellow dashed lines) of the ligands in the allosteric site.
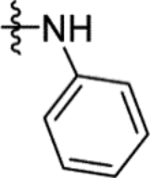
The replacement of the methyl group of 16a with a phenyl ring in 16h (IC50 17 μM) (Table 4) partially restored the conformation of the Arg400 side chain, with observation of a cation-π interaction by X-ray crystallography (Figures S9a and S10b). The addition of more flexibility to 16h in 16i through insertion of a methylene linker had a detrimental impact on inhibition (53% inhibition at 50 μM). The rigidification of 16i through incorporation of its benzyl group into a tetrahydroisoquinolyl ring system in 16j (IC50 2.2 μM) was more successful, with comparable inhibition to 1 (IC50 2.0 μM). X-ray crystallography showed the tetrahydroisoquinolyl ring of 16j lying between Arg400 and Arg432, with the side chain of Arg400 maintaining the conformation exhibited in the Mtb fumarase-16h structure through a cation-π interaction with the phenyl portion of the ring (Figure 5a). Ring expansion of 16j to improve the interaction with Arg432 in 16l (IC50 0.67 μM) resulted in a sub-micromolar IC50 value, 3-fold lower than that of 1 (IC50 2.0 μM) (Figure 5b). Addition of a methoxy group in 16m (IC50 0.67 μM) maintained the sub-micromolar inhibitory profile, however further modification of 16l through addition of a methylene bridge to the tetrahydrobenzoazepanyl ring in 16n was not tolerated (44% inhibition at 50 μM), revealing the limits of occupancy of this pocket in the allosteric site.
Figure 5.
X-ray crystal structure of Mtb fumarase (subunit A = white, subunit B = green, subunit C = cyan, subunit D = yellow) in complex with a) 16j (light green) (PDB code 6S7Z) and b) 16l (light blue) (PDB code 6S88), illustrating the interactions (yellow dashed lines) of the ligands in the allosteric site.
Screening against H37Rv Mycobacterium tuberculosis.
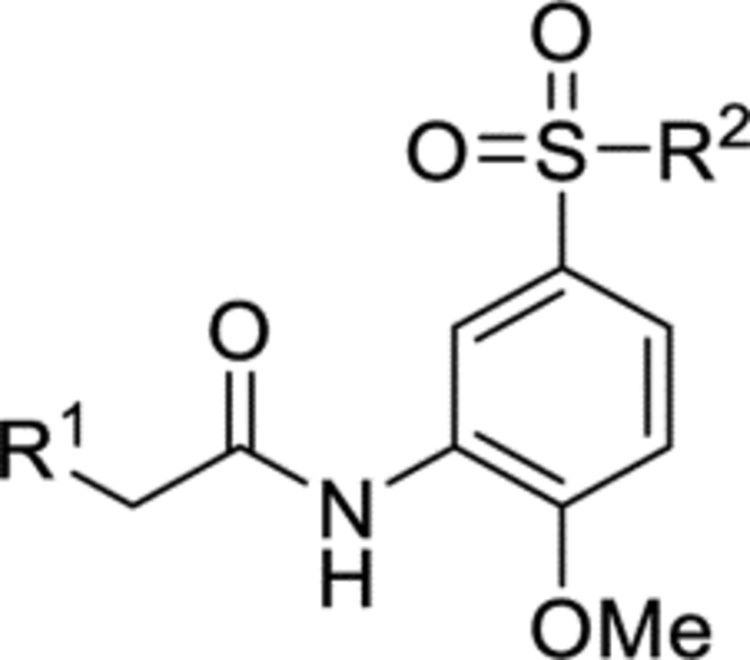
As previously reported,14 no bactericidal activity was observed for HTS hit 1 (Table 5). This was also found with derivatives 15g-i, 16c-h and 16j-m (data not shown). However, activity was observed with 15a-d and 16b (Table 5). The balance of metabolic intermediates in Mtb can be programmed by feeding the bacterium various carbon sources. As such, Mtb growing under glucose as the sole carbon source relies on glycolysis while bacteria that grow on dipalmitoyl phosphatidylcholine (DPPC) will rely heavily on β-oxidation to supply intermediates for the central carbon metabolism. Both glucose and DPPC are carbon sources predicted to be relevant during the in vivo pathogenesis of Mtb.17 Because pyruvate from glycolysis will go on to feed several other anabolic pathways, on balance bacteria get more acetyl-CoA equivalence under growth in DPPC. These acetyl-CoA feed directly into the citric acid cycle (among other pathways) in which fumarase lies. Therefore, the flux of intermediates in the citric acid cycle is greater under growth in DPPC and Mtb are more reliant on the activity of enzymes in this pathway for growth. Hence, it is reassuring that compounds 15a-d and 16b were highly active in DPPC-containing medium. In particular, the benzofuran analogue 15d gave the lowest MIC value against Mtb at 6.3 μM in DPPC-containing medium. Of these active compounds 16b (IC50 4.0 μM, MIC 19 μM in 7H9/DPPC) was the most potent against purified Mtb fumarase, and all were more lipophilic than 1 (cLogP 1.6) with the majority possessing a cLogP between 3.6 and 4.1. The poor activity in 7H9 medium supplemented with ADC was likely due to high protein binding since this medium contains 0.4% bovine serum albumin. This suggests overcoming protein binding will be an important component of future optimization.
Table 5.
The minimum inhibitory concentrations (MIC) against H37Rv Mycobacterium tuberculosis (Mtb) afforded by compounds 1, 151-d and 16b.
| |||||||
|---|---|---|---|---|---|---|---|
| Compound | R1 | R2 | cLogPa | H37Rv Mtb MIC (μM) | |||
| GAST-Fe | 7H9/ADC | 7H9/glucose | 7H9/DPPC | ||||
| 1 |
|
|
1.6 | > 100 | > 100 | > 100 | > 100 |
| 15a |
|
|
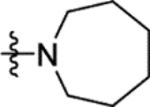
3.6 | > 100 | > 100 | 100 | 9.4 |
| 15b |
|
|
3.6 | > 100 | > 100 | > 100 | 12.5 |
| 15c |
|
|
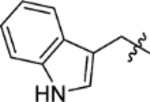
4.1 | > 100 | > 100 | 25 | 9.4 |
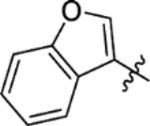
| 15d |
|
|
4.1 | > 100 | > 100 | 9.4 | 6.3 |
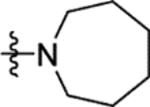
| 16b |
|
|
2.1 | > 100 | > 100 | 25 | 19 |
Calculated using ChemDraw (PerkinElmer, Waltham MA).
Synthetic Chemistry.
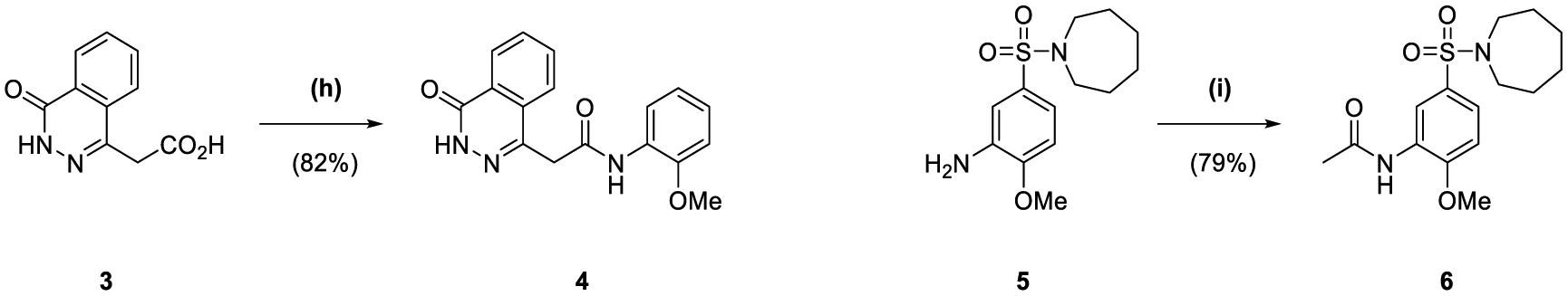
The synthesis of 1 was achieved in a convergent manner with the T3P®-mediated amide coupling of carboxylic acid 3 and aniline 5 (60% yield) (Scheme 1), each produced as previously reported.14 Synthesis of aniline 5 involved the trifluoroacetyl-protection of the amino group of o-anisidine 19 as 20 (99% yield), followed by SEAr of a sulfonyl chloride group by treatment with chlorosulfonic acid (77% yield). Reaction of 21 with hexamethyleneimine and sodium hydride converted the sulfonyl chloride to a sulfonamide, with the resultant intermediate heated under reflux in an aqueous mixture of ethanol and HCl to remove the trifluoroacetyl protecting group, affording 5 (83% yield). Carboxylic acid 3 was obtained by a route beginning with the Horner-Wadsworth-Emmons reaction of phthalic anhydride 17 and (carbethoxymethylene)triphenylphosphorane to give 18 (67% yield), followed by heating under reflux with hydrazine in ethanol to afford the phthalazinone ester 2 (99% yield). Hydrolysis of 2 produced acid 3 (44% yield), which was also used in the synthesis of 4 through T3P®-mediated amide coupling with o-anisidine 19 (82% yield) (Scheme 1). The application of acetic anhydride and pyridine to aniline 5 also allowed the production of 6 (79% yield).
Scheme 1.
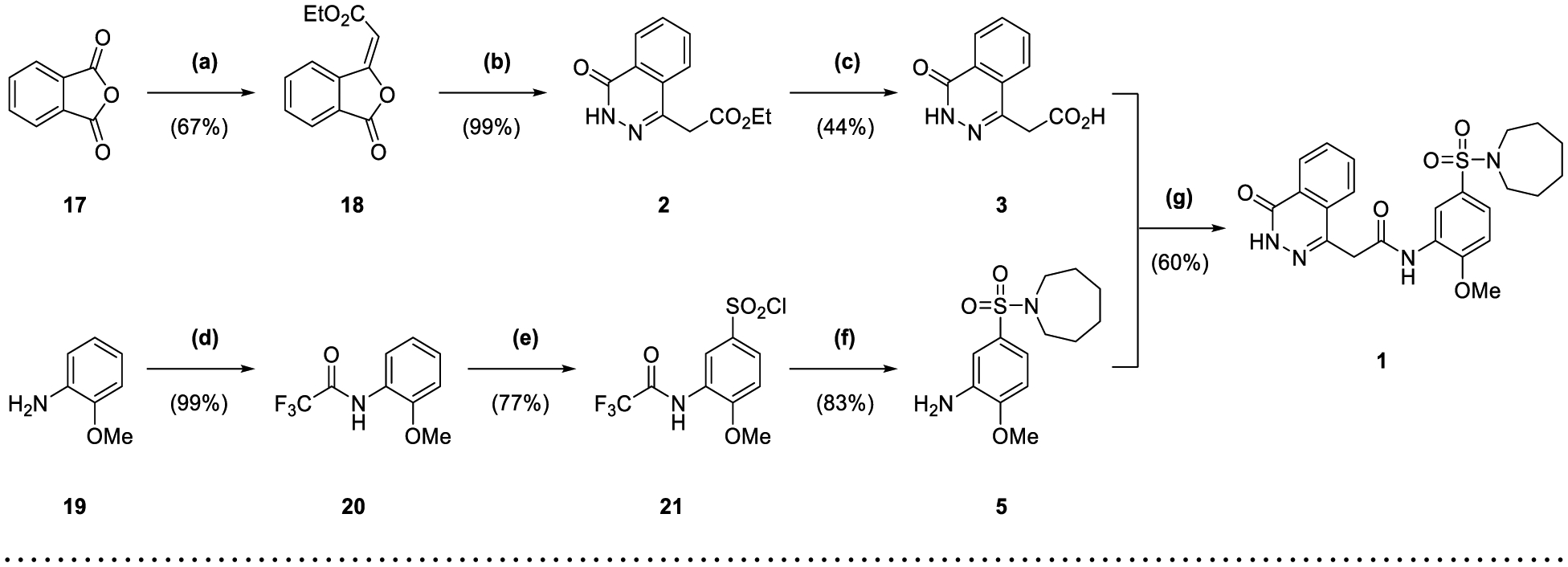
Synthesis of Compounds 1-6 and 21.
Reagents and Conditions: (a) (carbethoxymethylene)triphenylphosphorane, CHCl3, reflux, 3 h; (b) N2H4·H2O, EtOH, 50 °C, 2 h; (c) NaOH (10% w/v), THF, reflux, 1 h; (d) TFAA, pyridine, DCM, 0 °C to rt, 3 d; (e) HSO3Cl, DCM, 0 °C to rt, 16 h; (f) (i) hexamethyleneimine, NaH, DMF, 0 °C to rt, 3 h (ii) EtOH, HCl (37.5% w/v), H2O, reflux, 20 h; (g) T3P® (50 wt. % in DMF), DIPEA, DMF, 70 °C, 1 h; (h) 19, T3P® (50 wt. % in DMF), DIPEA, DMF, 40 °C, 2 h; (i) Ac2O, pyridine, DCM, 90 min.
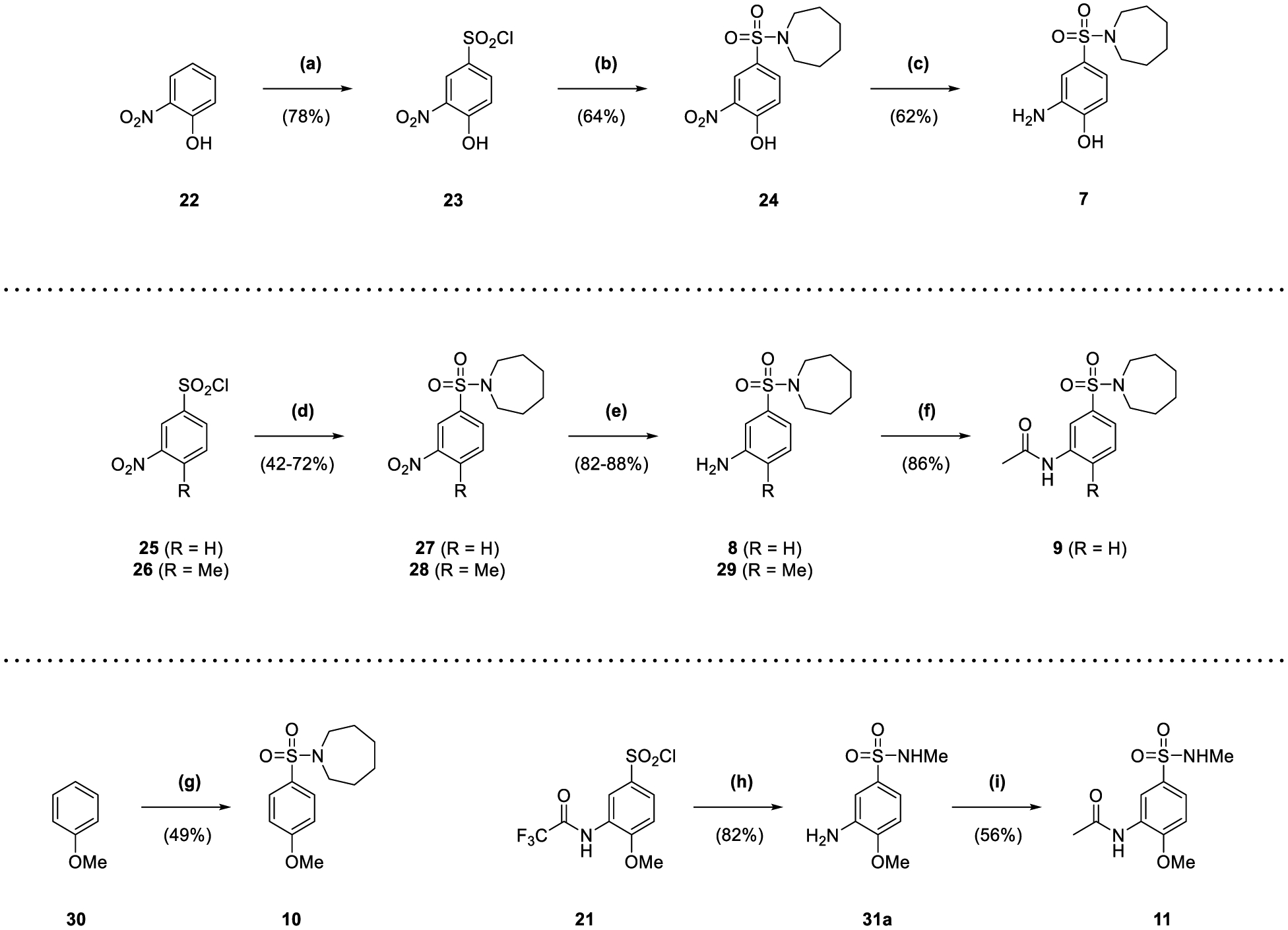
Fragment 7 was synthesized in three steps, beginning with the treatment of phenol 22 with chlorosulfonic acid in a similar manner to 20 (78% yield) (Scheme 2). The resultant sulfonyl chloride 23 was reacted with hexamethyleneimine in the presence of DIPEA to afford sulfonamide 24 (64% yield), whose nitro group was reduced by a mixture of sodium borohydride and nickel(II) chloride (62% yield). The production of fragment 8 and the corresponding methyl analogue 29 involved the functionalization of commercial sulfonyl chlorides 25 and 26 with hexamethyleneimine and sodium hydride, affording sulfonamides 27 and 28 (42–72% yield) (Scheme 2). The nitro groups of 27 and 28 were reduced in the same manner as 24 to the corresponding anilines 8 and 29 (82–88% yield). Aniline 8 was also acetylated to fragment 9 in the same manner as 5 (86% yield). Anisole 30 was used to make fragment 10 through stirring with chlorosulfonic acid, with the resultant sulfonyl chloride taken forwards crude for reaction with hexamethyleneimine and sodium hydride (49% yield overall) (Scheme 2). The sulfonyl chloride 21 was converted to N-methyl sulfonamide 31a by heating under reflux with methylamine in THF (82% yield), with 31a also acetylated in the same manner as 5 to afford fragment 11 (56% yield).
Scheme 2.
Synthesis of Compounds 7-11, 29 and 31a.
Reagents and Conditions: (a) HSO3Cl, CHCl3, 0 °C to reflux, 90 min; (b) hexamethyleneimine, DIPEA, DCM, 15 h; (c) NaBH4, NiCl2·6H2O, MeOH, 0 °C to rt, 2 h; (d) hexamethyleneimine, NaH, DMF, 0 °C to rt, 1 h to 90 min; (e) NaBH4, NiCl2, MeOH, 0 °C to rt, 45 min; (f) Ac2O, pyridine, DCM, 5 h; (g) (i) HSO3Cl, CHCl3, 0 °C to rt, 30 min (ii) hexamethyleneimine, NaH, DMF, 0 °C to rt, 1 h; (h) (i) methylamine (2 M in THF), THF, reflux, 90 min (ii) EtOH, HCl (37.5% w/v), H2O, reflux, 3 h 30 min; (i) Ac2O, pyridine, DCM, 2 d.
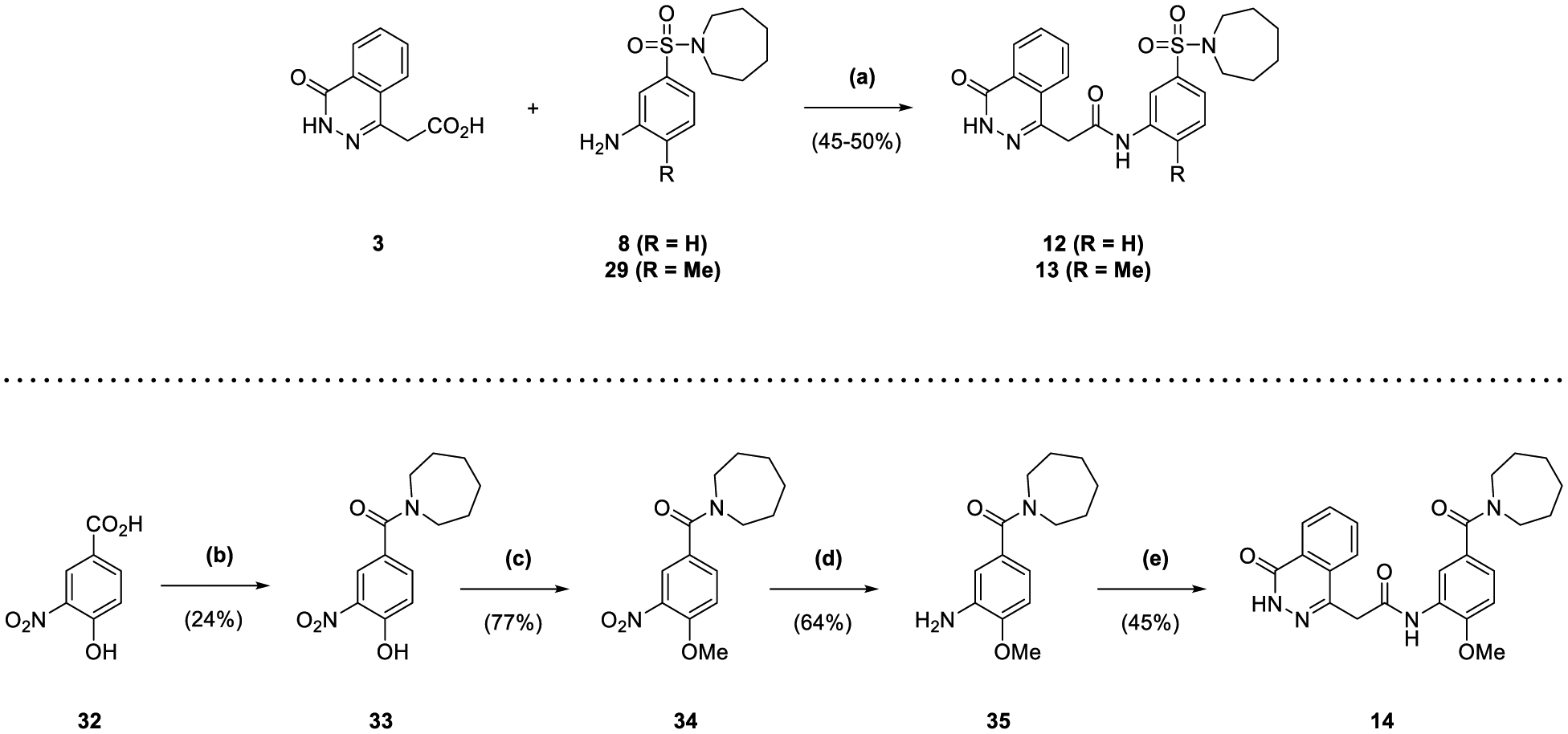
Compounds 12 and 13 were synthesized using anilines 8 and 29 (Scheme 2), which were coupled with carboxylic acid 3 using T3P® as with the synthesis of 1 (45–50% yield) (Scheme 3). In contrast, the route for 14 required the initial T3P®-mediated amide coupling of carboxylic acid 32 with hexamethyleneimine, affording phenol 33 (24% yield) that was methylated with methyl 21 iodide (77% yield) (Scheme 3). The nitro group of 34 was reduced in the same manner as 27 and 28 (64% yield), with the resultant aniline 35 coupled with acid 3 to afford 14 (45% yield).
Scheme 3.
Synthesis of Compounds 12-14.
Reagents and Conditions: (a) T3P® (50 wt. % in EtOAc), DIPEA, DMF, 70 °C, 2 h; (b) hexamethyleneimine, T3P® (50% in DMF), DIPEA, DMF, 1 d; (c) Me2SO4, K2CO3, acetone, reflux, 2 h; (d) NaBH4, NiCl2·6H2O, MeOH, 0 °C to rt, 90 min; (e) 3, T3P® (50 wt. % in EtOAc), DIPEA, DMF, 70 °C, 2 h.
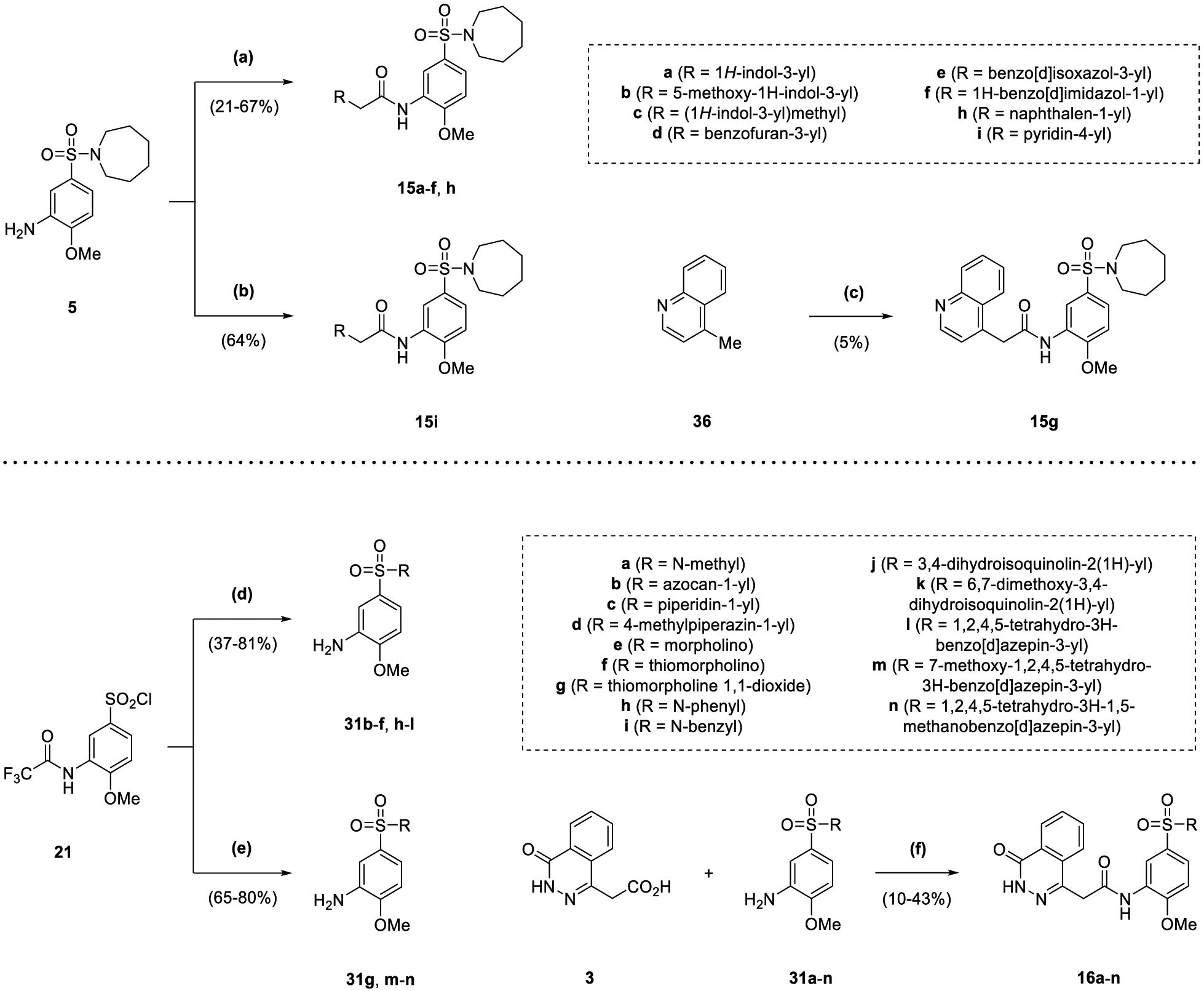
Compounds 15a-f and 15h were synthesized in one step from aniline 5 via T3P®-mediated amide coupling with commercially available carboxylic acids (21–67% yield), whilst 15i required alternative amide-coupling conditions with the use of EDC (64% yield) (Scheme 4). In the production of 15g, 4-methylquinoline 36 was converted using LDA and carbon dioxide to 4-quinolylacetic acid, which was taken forwards crude for reaction with aniline 5 (5% yield overall). The synthesis of compounds 16a-n involved the T3P®-mediated amide coupling of anilines 31a-n with carboxylic acid 3 as with the synthesis of 1 (10–43% yield) (Scheme 4). Anilines 31b-f and 31h-l were produced by the reaction of sulfonyl chloride 21 with commercially available amines in the presence of sodium hydride, followed by trifluoroacetyl deprotection via heating under reflux with ethanol and HCl as in 5 (37–81% yield). In comparison, whilst the trifluoroacetyl deprotection step in the synthesis of anilines 31g and 31m-n was the same as 31b-f and 31h-l, the prior reaction of sulfonyl chloride 21 with an amine was carried out with triethylamine and DMAP (65–80% yield overall).
Scheme 4.
Synthesis of Compounds 15-16.
Reagents and Conditions: (a) RCH2CO2H, T3P® (50 wt. % in EtOAc), DIPEA, DMF, 70 °C, 1 to 4 h; (b) 4-pyridylacetic acid HCl, EDC.HCl, DIPEA, DMAP, DCM, 90 min; (c) (i) CO2 (s), LDA (2 M in THF/heptane/ethylbenzene), THF, −78 °C to rt, 1 h (ii) 5, T3P® (50 wt. % in EtOAc), DIPEA, DMF, 70 °C, 40 min; (d) (i) RH, NaH, DMF, 0 °C to rt, 30 min to 20 h (ii) EtOH, HCl (37.5% w/v), H2O, reflux, 9 h to 2 d; (e) (i) RH, NEt3, DMAP, DCM, 30 min to 1 h (ii) EtOH, HCl (37.5% w/v), H2O, reflux, 3 to 16 h; (f) T3P® (50 wt. % in EtOAc), DIPEA, DMF, 70 °C, 45 min to 5 h.
CONCLUSIONS
The application of a deconstruction-reconstruction approach with the previously identified HTS hit 1 did not lead to the development of improved inhibitors, with defragmentation failing to afford fragments with either measurable inhibition in the biochemical assay or a positive result by DSF. This points to the essentiality of all components of 1 for its unique dimeric, induced-fit binding mode in the allosteric site of Mtb fumarase. A subsequent SAR study of 1 showed that modifications including replacement of both the phthalazinone and azepanyl ring systems were possible, with benzoazepanyl derivatives 16l-m affording sub-micromolar IC50 values (0.67 μM) three-fold stronger than that of 1. Derivatives of 1 maintained the dimeric-binding mode in the allosteric site of Mtb fumarase under X-ray crystallography soaking conditions, including in the case of attenuated inhibition as in 16a (IC50 57 μM). In contrast to 1, a subset of the derivatives afforded measurable MIC values against Mtb in vitro. These derivatives ranged from conservative expansion of the azepanyl ring of 1 with a subsequent rise in lipophilicity in 16b (MIC 19 μM in 7H9/DPPC) to complete replacement of the phthalazinone ring with alternative heterocyclic systems in 15a-d (MIC 6.3–12.5 μM in 7H9/DPPC). These results represent a significant improvement on the original HTS hit 1 and encourage further work on Mtb fumarase as a target for the development of compounds with bactericidal activity.
EXPERIMENTAL SECTION
General Chemistry.
All reactions were carried out in oven-dried glassware under a positive pressure of dry nitrogen atmosphere. Temperatures of 0 and −78 °C were obtained by submerging the reaction vessel in a bath containing either ice or a mixture of solid CO2 pellets and acetone respectively. The solvents DCM, ethyl acetate, acetonitrile, methanol, petroleum ether and toluene were distilled over calcium hydride under a dry nitrogen atmosphere prior to use, with THF distilled over a mixture of lithium aluminium hydride, calcium hydride and triphenylphosphine. DMF was purchased as anhydrous from commercial suppliers, with ethanol obtained in the absolute form. All purchased chemicals were used as received. Solutions of Na2CO3 and NaCl (brine) were aqueous and saturated.
Flash column chromatography was performed using automated Biotage® Isolera™ Spektra purification systems with appropriately sized Biotage® SNAP cartridges, containing either KP 50 μm silica in ‘normal phase’ purification or HP-sphere 25 μm C18 silica in ‘reverse phase’ purification. Analytical thin layer chromatography (TLC) was performed using Merck glass-backed silica plates, with visualization by 254 or 365 nm ultraviolet light.
Liquid chromatography mass spectrometry (LCMS) was carried out using a Waters® Acquity UPLC® H-Class system, with samples run on a solvent gradient from 0 to 95% acetonitrile in water (+ 0.1% formic acid) over 4 minutes. Peaks corresponding to desired product are described, including the retention time (rt) and % purity by integration. High-resolution mass spectrometry (HRMS) was mainly performed using ThermoFinnigan Orbitrap Classic, Waters® LCT Premier™ or Waters® Vion™ IMS QTof systems. A Perkin-Elmer® Spectrum One FT-IR spectrometer fitted with a universal attenuated total reflectance accessory was used to record infrared spectra, with wavelengths of maximum absorbance (νmax) quoted in wavenumbers (cm−1) for signals outside of the fingerprint region (br = broad). Only peaks corresponding to key functional groups were characterized. Nuclear magnetic resonance (NMR) spectra were recorded in the indicated deuterated solvents with Avance™ III HD (400 MHz), QNP Cryoprobe (400 MHz) or DCH Cryoprobe (500 MHz) Bruker spectrometers. 1H NMR data are presented in the following order: chemical shift (in ppm on a δ scale relative to the residual solvent resonance peak), integration, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, quin = quintet, sep = septet, m = multiplet) and coupling constant (J, in Hz). 13C NMR spectra were proton-decoupled, with chemical shifts presented.
A combination of TLC and LCMS analysis was used to monitor reactions. All tested compounds possessed a purity of at least 95% as determined by LCMS analysis.
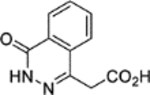
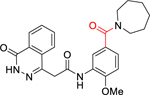
N-(5-(Azepan-1-ylsulfonyl)-2-methoxyphenyl)-2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetamide (1).
T3P® (50 wt. % in DMF, 1.3 mL, 2.2 mmol) and N,N-diisopropylethylamine (0.64 mL, 3.7 mmol) were added to a solution of 2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetic acid 3 (0.150 g, 0.735 mmol) and 5-(azepan-1-ylsulfonyl)-2-methoxyaniline 5 (0.209 g, 0.735 mmol) in DMF (2 mL). The reaction mixture was heated to 70 °C over 1 hour. The reaction mixture was diluted with water (15 mL), adjusted to pH 1 and extracted into ethyl acetate (3 × 20 mL). The combined organic extracts were dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (0 – 20% methanol in DCM) afforded 1 (0.206 g, 60% yield). LCMS (ESI+): m/z 471.3 [M + H]+, (ESI-): m/z 469.1 [M - H]−, rt 1.92 minutes, >99%; 1H NMR (400 MHz, (CD3)2SO) 12.62 (1H, s), 9.86 (1H, s), 8.45 (1H, d, J = 2.1 Hz), 8.27 (1H, d, J = 7.9 Hz), 7.99–7.91 (2H, m), 7.90–7.82 (1H, m), 7.49 (1H, dd, J = 8.5, 2.2 Hz), 7.24 (1H, d, J = 8.8 Hz), 4.21 (2H, s), 3.96 (3H, s), 3.12 (4H, t, J = 5.9 Hz), 1.64–1.51 (4H, m), 1.50–1.39 (4H, m); 13C NMR (125 MHz, (CD3)2SO) 168.4, 159.5, 152.0, 142.1, 133.5, 131.6, 130.4, 129.8, 127.7, 127.6, 125.9, 125.5, 123.4, 119.1, 111.2, 56.3, 47.6, 28.5, 26.3 (1 peak missing); spectroscopic data consistent with literature.14
Ethyl 2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetate (2).14,18
Hydrazine monohydrate (0.47 mL, 9.0 mmol) was added dropwise to a solution of ethyl (E)-2-(3-oxoisobenzofuran-1(3H)-ylidene)acetate 18 (1.97 g, 9.03 mmol) in ethanol (10 mL). The reaction mixture was stirred at 50 °C over 2 hours. The reaction mixture was cooled to room temperature and left to stand for 12 hours. The resulting precipitate was obtained by vacuum filtration and washed with ethanol (10 mL) to afford 2 (2.21 g, 99% yield). LCMS (ESI+): m/z 233.2 [M + H]+, rt 1.53 minutes, >99%; 1H NMR (400 MHz, (CD3)2SO) 12.63 (1H, s), 8.30–8.24 (1H, m), 7.98–7.91 (1H, m), 7.90–7.83 (2H, m), 4.11 (2H, q, J = 7.1 Hz), 4.05 (2H, s), 1.17 (3H, t, J = 7.1 Hz); 13C NMR (100 MHz, (CD3)2SO) 169.8, 159.4, 140.9, 133.6, 131.7, 129.4, 127.5, 125.9, 125.5, 60.7, 37.9, 14.0; 1H NMR spectroscopic data consistent with literature.18
2-(4-Oxo-3,4-dihydrophthalazin-1-yl)acetic acid (3).14
Ethyl 2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetate 2 (0.260 g, 1.12 mmol) was dissolved in a mixture of aqueous NaOH (10% w/v, 10 mL) and THF (10 mL). The reaction mixture was heated under reflux for 1 hour. The reaction mixture was adjusted to pH 1 by the addition of aqueous HCl (2 M) at 0 °C, then extracted into diethyl ether (3 × 100 mL). The combined organic extracts were washed (brine), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (0 – 10% methanol in DCM) afforded 3 (0.101 g, 44% yield). 1H NMR (400 MHz, (CD3)2SO) 12.61 (1H, s), 8.26 (1H, d, J = 7.7 Hz), 7.94 (1H, ddd, J = 8.3, 6.9, 1.4 Hz), 7.90–7.82 (2H, m), 3.95 (2H, s); 13C NMR (100 MHz, (CD3)2SO) 171.4, 159.5, 141.5, 133.5, 131.6, 129.6, 127.5, 125.9, 125.6, 38.2; 1H NMR spectroscopic data consistent with literature.18
N-(2-Methoxyphenyl)-2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetamide (4).
T3P® (50 wt. % in DMF, 0.35 mL, 0.59 mmol) and N,N-diisopropylethylamine (0.10 mL, 0.59 mmol) were added to a solution of 2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetic acid 3 (40 mg, 0.20 mmol) and o-anisidine 19 (33 μL, 0.29 mmol) in DMF (1 mL). The reaction mixture was heated to 40 °C over 2 hours. The reaction mixture was diluted with water (15 mL) and extracted into DCM (3 × 20 mL). The combined organic extracts were dried (MgSO4) and concentrated in vacuo. The residue was dissolved in toluene (10 mL) and concentrated in vacuo. Purification by flash column chromatography (50 – 100% ethyl acetate in petroleum ether, 0 – 20% methanol in DCM) afforded 4 (50 mg, 82% yield). LCMS (ESI+): m/z 332.1 [M + Na]+, rt 1.64 minutes, >99%; 1H NMR (500 MHz, (CD3)2SO) 12.61 (1H, s), 9.56 (1H, s), 8.27 (1H, d, J = 7.9 Hz), 8.01–7.79 (4H, m), 7.11–7.00 (2H, m), 6.92–6.83 (1H, m), 4.16 (2H, s), 3.86 (3H, s); 13C NMR (125 MHz, (CD3)2SO) 167.6, 159.5, 149.6, 142.3, 133.5, 131.5, 129.8, 127.6, 127.2, 125.8, 125.6, 124.6, 121.8, 120.2, 111.2, 55.7 (1 peak missing); νmax/cm−1 2905, 1665 (C=O), 1646, 1597, 1539; HRMS (ESI)+: m/z calculated for [C17H15N3O3 + H]+ = 310.1186, observed 310.1175.
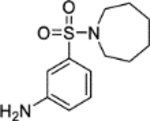
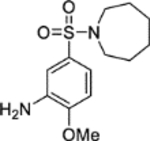
5-(Azepan-1-ylsulfonyl)-2-methoxyaniline (5).14
Hexamethyleneimine (0.212 mL, 1.89 mmol) was added dropwise at 0 °C to a suspension of sodium hydride (60% in mineral oil, 0.189 g, 4.72 mmol) in DMF (2 mL). The reaction mixture was stirred at 0 °C over 30 minutes. A solution of 4-methoxy-3-(2,2,2-trifluoroacetamido)benzenesulfonyl chloride 21 (0.500 g, 1.57 mmol) in DMF (3 mL) was added dropwise at 0 °C to the reaction mixture. The reaction mixture was warmed to room temperature and stirred over 3 hours. Ethanol (10 mL) was added dropwise at 0 °C to the reaction mixture, followed by water (10 mL) and aqueous HCl (37.5% w/v, 10 mL). The reaction mixture was heated under reflux for 20 hours. The reaction mixture was concentrated in vacuo to remove ethanol, then adjusted to pH 9 by the dropwise addition of aqueous NaOH (10% w/v). The mixture was diluted with ethyl acetate (50 mL), and the resultant aqueous layer discarded. The organic layer was washed with water (3 × 50 mL) and brine (50 mL), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (20 – 50% ethyl acetate in petroleum ether) afforded 5 (0.373 g, 83% yield). LCMS (ESI+): m/z 285.2 [M + H]+, rt 1.85 minutes, >99%; 1H NMR (400 MHz, CDCl3) 7.17 (1H, dd, J = 8.4, 2.3 Hz), 7.09 (1H, d, J = 2.2 Hz), 6.81 (1H, d, J = 8.4 Hz), 3.98 (2H, br s), 3.90 (3H, s), 3.23 (4H, t, J = 5.9 Hz), 1.75–1.64 (4H, m), 1.62–1.53 (4H, m); 13C NMR (100 MHz, CDCl3) 150.1, 136.7, 131.6, 118.1, 112.7, 109.7, 55.8, 48.3, 29.3, 27.1; νmax/cm−1 3484 (N-H), 3379 (N-H), 2932, 2849, 1610, 1577, 1513; HRMS (ESI)+: m/z calculated for [C13H20N2O3S + K]+ = 323.0826, observed 323.0835.
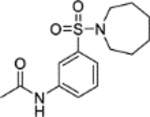
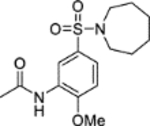
N-(5-(Azepan-1-ylsulfonyl)-2-methoxyphenyl)acetamide (6).
Acetic anhydride (10 μL, 0.11 mmol) was added to a solution of 5-(azepan-1-ylsulfonyl)-2-methoxyaniline 5 (15 mg, 0.053 mmol) and pyridine (8.5 μL, 0.11 mmol) in DCM (2 mL). The reaction mixture was stirred over 90 minutes. The reaction mixture was diluted with water (10 mL) and extracted into DCM (3 × 15 mL). The combined organic extracts were dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (50 – 80% ethyl acetate in petroleum ether) afforded 6 (14 mg, 79% yield). LCMS (ESI+): m/z 327.3 [M + H]+, (ESI-): m/z 325.2 [M - H]−, rt 1.90 minutes, 96%; 1H NMR (400 MHz, CDCl3) 8.78 (1H, d, J = 1.8 Hz), 7.76 (1H, br s), 7.53 (1H, dd, J = 8.5, 2.3 Hz), 6.91 (1H, d, J = 8.6 Hz), 3.94 (3H, s), 3.30 (4H, t, J = 6.0 Hz), 2.22 (3H, s), 1.80–1.67 (4H, m), 1.65–1.53 (4H, m); 13C NMR (125 MHz, CDCl3) 168.4, 150.4, 132.1, 128.2, 123.5, 118.0, 109.5, 56.2, 48.5, 29.3, 27.1, 25.0; νmax/cm−1 3349, 2930, 2855, 1673 (C=O), 1592, 1518; HRMS (ESI)+: m/z calculated for [C15H22N2O4S + H]+ = 327.1373, observed 327.1375.
2-Amino-4-(azepan-1-ylsulfonyl)phenol (7).
Sodium borohydride (28 mg, 0.75 mmol) was added portionwise at 0 °C to a suspension of NiCl2·6H2O (59 mg, 0.25 mmol) in methanol (2 mL). The reaction mixture was warmed to room temperature and stirred over 20 minutes. A solution of 4-(azepan-1-ylsulfonyl)-2-nitrophenol 24 (0.170 g, 0.498 mmol) in methanol (2 mL) was added at 0 °C to the reaction mixture, followed by further sodium borohydride (94 mg, 2.5 mmol). The reaction mixture was warmed to room temperature and stirred over 2 hours. Water (15 mL) was added at 0 °C and the reaction mixture filtered through celite. The product was extracted into DCM (3 × 25 mL). The combined organic extracts were washed (brine), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (20 – 50% ethyl acetate in petroleum ether) afforded 7 (83 mg, 62% yield). LCMS (ESI+): m/z 271.2 [M + H]+, (ESI-): m/z 269.2 [M - H]−, rt 1.71 minutes, >99%; 1H NMR (400 MHz, CD3CN) 7.45 (1H, br s), 7.04 (1H, d, J = 2.3 Hz), 6.94 (1H, dd, J = 8.2, 2.2 Hz), 6.80 (1H, d, J = 8.3 Hz), 4.26 (2H, br s), 3.18 (4H, t, J = 5.9 Hz), 1.74–1.60 (4H, m), 1.59–1.50 (4H, m); 13C NMR (100 MHz, CD3CN) 148.0, 137.6, 131.9, 117.8, 114.8, 113.6, 48.9, 29.8, 27.6; νmax/cm−1 3397, 3350, 3320, 3285, 2927, 2855, 1593, 1510; HRMS (ESI)-: m/z calculated for [C12H18N2O3S - H]− = 269.0965, observed 269.0965.
3-(Azepan-1-ylsulfonyl)aniline (8).
Sodium borohydride (51 mg, 1.4 mmol) was added portionwise at 0 °C to a suspension of NiCl2 (59 mg, 0.45 mmol) in methanol (2 mL). The reaction mixture was warmed to room temperature and stirred over 30 minutes. 1-((3-Nitrophenyl)sulfonyl)azepane 27 (0.257 g, 0.904 mmol) was added at 0 °C to the reaction mixture, followed by further methanol (4 mL) and sodium borohydride (0.171 g, 4.52 mmol). The reaction mixture was warmed to room temperature and stirred over 45 minutes. Water (10 mL) was added at 0 °C and the mixture filtered through celite, eluted with methanol (10 mL) and water (15 mL). The filtrate was concentrated in vacuo to remove methanol, then extracted into ethyl acetate (3 × 25 mL). The combined organic extracts were washed (brine), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (20 – 50% ethyl acetate in petroleum ether) afforded 8 (0.188 g, 82% yield). LCMS (ESI+): m/z 255.2 [M + H]+, rt 1.77 minutes, >99%; 1H NMR (400 MHz, CDCl3) 7.24 (1H, t, J = 7.8 Hz), 7.15–7.10 (1H, m), 7.08 (1H, t, J = 2.0 Hz), 6.81 (1H, ddd, J = 7.9, 2.3, 0.7 Hz), 3.89 (2H, br s), 3.26 (4H, t, J = 5.9 Hz), 1.77–1.65 (4H, m), 1.63–1.54 (4H, m); 13C NMR (100 MHz, CDCl3) 147.2, 140.4, 130.0, 118.6, 116.7, 113.0, 48.4, 29.3, 27.0; νmax/cm−1 3391 (N-H), 3326 (N-H), 2928, 2851, 1639, 1596; HRMS (ESI)+: m/z calculated for [C12H18N2O2S + H]+ = 255.1162, observed 255.1166.
N-(3-(Azepan-1-ylsulfonyl)phenyl)acetamide (9).
Acetic anhydride (11 μL, 0.12 mmol) was added to a solution of 3-(azepan-1-ylsulfonyl)aniline 8 (15 mg, 0.059 mmol) and pyridine (10 μL, 0.12 mmol) in DCM (2 mL). The reaction mixture was stirred over 5 hours. The reaction mixture was diluted with water (10 mL) and extracted into DCM (3 × 15 mL). The combined organic extracts were dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (35 – 70% ethyl acetate in petroleum ether) afforded 9 (15 mg, 86% yield). LCMS (ESI+): m/z 297.2 [M + H]+, (ESI-): m/z 295.2 [M - H]−, rt 1.87 minutes, >99%; 1H NMR (500 MHz, CDCl3) 7.98 (1H, d, J = 7.9 Hz), 7.91 (1H, br s), 7.83–7.78 (1H, m), 7.52–7.47 (1H, m), 7.45 (1H, t, J = 7.9 Hz), 3.27 (4H, t, J = 6.0 Hz), 2.21 (3H, s), 1.76–1.66 (4H, m), 1.65–1.53 (4H, m); 13C NMR (125 MHz, CDCl3) 169.0, 139.9, 139.1, 130.0, 123.7, 122.2, 117.9, 48.5, 29.3, 27.0, 24.7; νmax/cm−1 3305, 3257, 3192, 3117, 2929, 2851, 1670 (C=O), 1592, 1545; HRMS (ESI)+: m/z calculated for [C14H20N2O3S + H]+ = 297.1267, observed 297.1268.
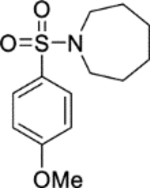
1-((4-Methoxyphenyl)sulfonyl)azepane (10).
Chlorosulfonic acid (0.25 mL, 3.7 mmol) was added dropwise at 0 °C to a mixture of anisole 30 (0.201 mL, 1.85 mmol) and chloroform (5 mL). The reaction mixture was warmed to room temperature and stirred over 30 minutes. Water (15 mL) was added dropwise at 0 °C to the reaction mixture. The mixture was extracted into DCM (3 × 20 mL). The combined organic extracts were washed (brine), dried (MgSO4) and concentrated in vacuo to afford a crude residue. Hexamethyleneimine (0.141 mL, 1.26 mmol) was added dropwise at 0 °C to a suspension of sodium hydride (60% in mineral oil, 0.137 g, 3.43 mmol) in DMF (1 mL). The reaction mixture was stirred at 0 °C over 20 minutes. A solution of the crude residue in DMF (2 mL) was added dropwise at 0 °C to the reaction mixture. The reaction mixture was warmed to room temperature and stirred over 1 hour. Water (15 mL) was added dropwise at 0 °C to the reaction mixture. The product was extracted into DCM (3 × 20 mL). The combined organic extracts were washed (brine), dried (MgSO4) and concentrated in vacuo. The residue was dissolved in toluene (5 mL) and concentrated in vacuo. Purification by flash column chromatography (0 – 35% ethyl acetate in petroleum ether) afforded 10 (0.243 g, 49% yield). LCMS (ESI+): m/z 270.2 [M + H]+, rt 2.15 minutes, >99%; 1H NMR (400 MHz, CDCl3) 7.75–7.70 (2H, m), 6.99–6.93 (2H, m), 3.86 (3H, s), 3.25 (4H, t, J = 5.9 Hz), 1.75–1.66 (4H, m), 1.62–1.53 (4H, m); 13C NMR (100 MHz, CDCl3) 162.6, 131.5, 129.1, 114.2, 55.7, 48.3, 29.2, 27.1; νmax/cm−1 2929, 2848, 1595, 1579, 1501; HRMS (ESI)+: m/z calculated for [C13H19NO3S + H]+ = 270.1158, observed 270.1153.
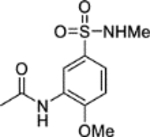
N-(2-Methoxy-5-(N-methylsulfamoyl)phenyl)acetamide (11).
Acetic anhydride (18 μL, 0.19 mmol) was added to a solution of 3-amino-4-methoxy-N-methylbenzenesulfonamide 31a (42 mg, 0.19 mmol) and pyridine (16 μL, 0.19 mmol) in DCM (2 mL). The reaction mixture was stirred over 2 days. The reaction mixture was diluted with water (15 mL) and extracted into DCM (3 × 20 mL). The combined organic extracts were dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (50 – 100% ethyl acetate in petroleum ether) afforded 11 (28 mg, 56% yield). LCMS (ESI+): m/z 259.2 [M + H]+, (ESI-): m/z 257.2 [M - H]−, rt 1.31 minutes, >99%; 1H NMR (400 MHz, CD3CN) 8.74 (1H, d, J = 2.0 Hz), 8.30 (1H, br s), 7.51 (1H, dd, J = 8.5, 2.2 Hz), 7.10 (1H, d, J = 8.6 Hz), 5.43–5.27 (1H, m), 3.94 (3H, s), 2.47 (3H, d, J = 5.3 Hz), 2.14 (3H, s); 13C NMR (100 MHz, CD3CN) 170.0, 152.2, 131.6, 129.5, 124.0, 118.7, 111.2, 56.9, 29.5, 24.7; νmax/cm−1 3422 (N-H), 3168, 1672 (C=O), 1594, 1530; HRMS (ESI)+: m/z calculated for [C10H14N2O4S + Na]+ = 281.0566, observed 281.0569.
N-(3-(Azepan-1-ylsulfonyl)phenyl)-2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetamide (12).
T3P® (50 wt. % in ethyl acetate, 0.35 mL, 0.59 mmol) and N,N-diisopropylethylamine (0.17 mL, 0.98 mmol) were added to a solution of 2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetic acid 3 (47 mg, 0.20 mmol) and 3-(azepan-1-ylsulfonyl)aniline 8 (50 mg, 0.20 mmol) in DMF (2 mL). The reaction mixture was heated to 70 °C over 2 hours. The reaction mixture was diluted with ethyl acetate (25 mL), washed with water (3 × 25 mL) and brine (25 mL), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (50 – 90% ethyl acetate in petroleum ether) afforded 12 (40 mg, 45% yield). LCMS (ESI+): m/z 441.3 [M + H]+, (ESI-): m/z 439.2 [M - H]−, rt 1.82 minutes, 98%; 1H NMR (500 MHz, (CD3)2SO) 12.63 (1H, s), 10.67 (1H, s), 8.30–8.25 (1H, m), 8.13 (1H, t, J = 1.8 Hz), 7.98–7.92 (2H, m), 7.86 (1H, ddd, J = 8.1, 5.7, 2.4 Hz), 7.78 (1H, dq, J = 8.2, 1.0 Hz), 7.54 (1H, t, J = 8.0 Hz), 7.44 (1H, dq, J = 7.8, 0.9 Hz), 4.10 (2H, s), 3.18 (4H, t, J = 5.9 Hz), 1.66–1.56 (4H, m), 1.53–1.44 (4H, m); 13C NMR (125 MHz, (CD3)2SO) 168.1, 159.5, 141.8, 139.7, 139.5, 133.6, 131.6, 130.0, 129.8, 127.6, 125.8, 125.6, 122.6, 121.2, 116.7, 47.7, 28.5, 26.3 (1 peak missing); νmax/cm−1 2929, 2852, 1644 (C=O), 1591, 1541; HRMS (ESI)+: m/z calculated for [C22H24N4O4S + H]+ = 441.1591, observed 441.1613.
N-(5-(Azepan-1-ylsulfonyl)-2-methylphenyl)-2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetamide (13).
T3P® (50 wt. % in ethyl acetate, 0.33 mL, 0.56 mmol) and N,N-diisopropylethylamine (0.16 mL, 0.93 mmol) were added to a solution of 2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetic acid 3 (44 mg, 0.19 mmol) and 5-(azepan-1-ylsulfonyl)-2-methylaniline 29 (50 mg, 0.19 mmol) in DMF (2 mL). The reaction mixture was heated to 70 °C over 2 hours. The reaction mixture was diluted with ethyl acetate (25 mL), washed with water (3 × 25 mL) and brine (25 mL), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (50 – 90% ethyl acetate in petroleum ether, 0 – 5% methanol in DCM) afforded 13 (42 mg, 50% yield). LCMS (ESI+): m/z 455.3 [M + H]+, (ESI-): m/z 453.2 [M - H]−, rt 1.82 minutes, >99%; 1H NMR (500 MHz, (CD3)2SO) 12.64 (1H, s), 9.84 (1H, s), 8.28 (1H, d, J = 7.7 Hz), 8.02–7.92 (3H, m), 7.87 (1H, ddd, J = 8.0, 6.7, 1.5 Hz), 7.49–7.41 (2H, m), 4.15 (2H, s), 3.14 (4H, t, J = 6.0 Hz), 2.33 (3H, s), 1.65–1.53 (4H, m), 1.51–.1.42 (4H, m); 13C NMR (125 MHz, (CD3)2SO) 168.0, 159.5, 142.0, 136.8, 136.5, 135.8, 133.5, 131.6, 131.3, 129.8, 127.6, 125.9, 125.5, 122.8, 122.1, 47.7, 28.5, 26.3, 18.0 (1 peak missing); νmax/cm−1 3177, 3045, 2915, 2853, 1651 (C=O), 1612, 1600, 1582, 1553; HRMS (ESI)+: m/z calculated for [C23H26N4O4S + H]+ = 455.1748, observed 455.1769.
N-(5-(Azepane-1-carbonyl)-2-methoxyphenyl)-2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetamide (14).
T3P® (50 wt. % in ethyl acetate, 0.12 mL, 0.20 mmol) and N,N-diisopropylethylamine (58 μL, 0.33 mmol) were added to a solution of 2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetic acid 3 (14 mg, 0.067 mmol) and (3-amino-4-methoxyphenyl)(azepan-1-yl)methanone 35 (17 mg, 0.067 mmol) in DMF (1 mL). The reaction mixture was heated to 70 °C over 2 hours. The reaction mixture was diluted with ethyl acetate (25 mL), washed with water (3 × 10 mL) and brine (25 mL), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (50 – 100% ethyl acetate in petroleum ether, 0 – 10% methanol in DCM) afforded 14 (13 mg, 45% yield). LCMS (ESI+): m/z 435.3 [M + H]+, (ESI-): m/z 433.3 [M - H]−, rt 1.68 minutes, >99%; 1H NMR (400 MHz, (CD3)2CO) 11.69 (1H, br s), 9.01 (1H, br s), 8.39–8.32 (2H, m), 8.04 (1H, d, J = 8.1 Hz), 7.94 (1H, td, J = 7.7, 1.5 Hz), 7.89–7.82 (1H, m), 7.09 (1H, dd, J = 8.4, 2.0 Hz), 7.04 (1H, d, J = 8.4 Hz), 4.21 (2H, s), 3.91 (3H, s), 3.56 (2H, br s), 3.40 (2H, br s), 1.81–1.47 (8H, m); 13C NMR (125 MHz, (CD3)2SO) 169.9, 167.9, 159.5, 149.7, 142.2, 133.5, 131.5, 129.8, 129.1, 127.6, 126.8, 125.8, 125.5, 122.9, 119.9, 110.8, 55.9, 49.2, 45.5, 28.9, 27.2, 26.8, 25.8 (1 peak missing); νmax/cm−1 3191, 2929, 2856, 1674, 1648, 1618, 1548; HRMS (ESI)+: m/z calculated for [C24H26N4O4 + H]+ = 435.2027, observed 435.2025.
N-(5-(Azepan-1-ylsulfonyl)-2-methoxyphenyl)-2-(1H-indol-3-yl)acetamide (15a).
T3P® (50 wt. % in ethyl acetate, 0.15 mL, 0.26 mmol) and N,N-diisopropylethylamine (75 μL, 0.43 mmol) were added to a solution of 3-indoleacetic acid (15 mg, 0.086 mmol) and 5-(azepan-1-ylsulfonyl)-2-methoxyaniline 5 (24 mg, 0.086 mmol) in DMF (1 mL). The reaction mixture was heated to 70 °C over 4 hours. The reaction mixture was diluted with ethyl acetate (25 mL), washed with water (3 × 10 mL) and brine (25 mL), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (40 – 80% ethyl acetate in petroleum ether) afforded 15a (8 mg, 21% yield). LCMS (ESI+): m/z 442.3 [M + H]+, (ESI-): m/z 440.2 [M - H]−, rt 2.13 minutes, 97%; 1H NMR (400 MHz, (CD3)2SO) 10.98 (1H, s), 9.34 (1H, s), 8.50 (1H, d, J = 2.2 Hz), 7.61 (1H, d, J = 7.9 Hz), 7.45 (1H, dd, J = 8.6, 2.4 Hz), 7.37 (1H, d, J = 8.1 Hz), 7.30 (1H, d, J = 2.3 Hz), 7.18 (1H, d, J = 8.8 Hz), 7.09 (1H, ddd, J = 8.1, 7.0, 1.1 Hz), 6.99 (1H, ddd, J = 7.9, 7.0, 1.0 Hz), 3.87 (2H, s), 3.85 (3H, s), 3.12 (4H, t, J = 5.9 Hz), 1.65–1.52 (4H, m), 1.51–1.38 (4H, m); 13C NMR (100 MHz, (CD3)2SO) 170.4, 151.7, 136.2, 130.4, 127.9, 127.2, 124.3, 123.1, 121.2, 118.7, 118.6, 118.5, 111.5, 111.1, 108.2, 56.3, 47.7, 33.5, 28.5, 26.4; νmax/cm−1 3345 (br, N-H), 2928, 2856, 1673 (C=O), 1594, 1524; HRMS (ESI)+: m/z calculated for [C23H27N3O4S + Na]+ = 464.1614, observed 464.1615.
N-(5-(Azepan-1-ylsulfonyl)-2-methoxyphenyl)-2-(5-methoxy-1H-indol-3-yl)acetamide (15b).
T3P® (50 wt. % in ethyl acetate, 0.31 mL, 0.53 mmol) and N,N-diisopropylethylamine (0.15 mL, 0.88 mmol) were added to a solution of 5-methoxy-3-indoleacetic acid (36 mg, 0.18 mmol) and 5-(azepan-1-ylsulfonyl)-2-methoxyaniline 5 (50 mg, 0.18 mmol) in DMF (2 mL). The reaction mixture was heated to 70 °C over 2 hours. The reaction mixture was diluted with ethyl acetate (100 mL), washed with water (3 × 100 mL) and brine (100 mL), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (30 – 70% ethyl acetate in petroleum ether) afforded 15b (32 mg, 38% yield). LCMS (ESI+): m/z 472.3 [M + H]+, (ESI-): m/z 470.2 [M - H]−, rt 2.09 minutes, 98%; 1H NMR (500 MHz, (CD3)2SO) 10.81 (1H, d, J = 1.6 Hz), 9.29 (1H, s), 8.51 (1H, d, J = 2.1 Hz), 7.45 (1H, dd, J = 8.7, 2.3 Hz), 7.29–7.24 (2H, m), 7.17 (1H, d, J = 8.8 Hz), 7.13 (1H, d, J = 2.4 Hz), 6.74 (1H, dd, J = 8.7, 2.4 Hz), 3.84 (3H, s), 3.83 (2H, s), 3.74 (3H, s), 3.13 (4H, t, J = 6.0 Hz), 1.64–1.54 (4H, m), 1.51–1.42 (4H, m); 13C NMR (125 MHz, (CD3)2SO) 170.4, 153.2, 151.6, 131.3, 130.4, 127.8, 127.4, 124.9, 123.0, 118.5, 112.1, 111.2, 111.0, 107.9, 100.5, 56.2, 55.3, 47.7, 33.6, 28.4, 26.3; νmax/cm−1 3350 (br, N-H), 2933, 1719, 1675 (C=O), 1593, 1522; HRMS (ESI)+: m/z calculated for [C24H29N3O5S + H]+ = 472.1901, observed 472.1890.
N-(5-(Azepan-1-ylsulfonyl)-2-methoxyphenyl)-3-(1H-indol-3-yl)propanamide (15c).
T3P® (50 wt. % in ethyl acetate, 0.25 mL, 0.42 mmol) and N,N-diisopropylethylamine (0.12 mL, 0.70 mmol) were added to a solution of 3-indolepropionic acid (27 mg, 0.14 mmol) and 5-(azepan-1-ylsulfonyl)-2-methoxyaniline 5 (40 mg, 0.14 mmol) in DMF (2 mL). The reaction mixture was heated to 70 °C over 1 hour. The reaction mixture was diluted with ethyl acetate (25 mL), washed with water (3 × 25 mL) and brine (25 mL), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (30 – 70% ethyl acetate in petroleum ether) afforded 15c (26 mg, 41% yield). LCMS (ESI+): m/z 456.3 [M + H]+, (ESI-): m/z 454.3 [M - H], rt 2.17 minutes, >99%; 1H NMR (400 MHz, (CD3)2SO) 10.78 (1H, s), 9.37 (1H, s), 8.51 (1H, d, J = 1.9 Hz), 7.57 (1H, d, J = 7.8 Hz), 7.47 (1H, dd, J = 8.7, 2.4 Hz), 7.33 (1H, d, J = 8.0 Hz), 7.19 (1H, d, J = 8.7 Hz), 7.14 (1H, d, J = 2.0 Hz), 7.06 (1H, t, J = 7.5 Hz), 6.97 (1H, t, J = 7.4 Hz), 3.89 (3H, s), 3.16 (4H, t, J = 5.9 Hz), 3.01 (2H, t, J = 7.5 Hz), 2.82 (2H, t, J = 7.5 Hz), 1.70–1.56 (4H, m), 1.55–1.41 (4H, m); 13C NMR (100 MHz, (CD3)2SO) 171.8, 152.0, 136.2, 130.3, 127.8, 127.0, 123.1, 122.3, 120.9, 119.4, 118.5, 118.2, 113.6, 111.3, 111.0, 56.2, 47.7, 36.8, 28.5, 26.4, 20.7; νmax/cm−1 3360 (br, N-H), 2927, 2856, 1674 (C=O), 1594, 1524; HRMS (ESI)+: m/z calculated for [C24H29N3O4S + H]+ = 456.1952, observed 456.1970.
N-(5-(Azepan-1-ylsulfonyl)-2-methoxyphenyl)-2-(benzofuran-3-yl)acetamide (15d).
T3P® (50 wt. % in ethyl acetate, 0.31 mL, 0.53 mmol) and N,N-diisopropylethylamine (0.15 mL, 0.88 mmol) were added to a solution of benzo[b]furan-3-ylacetic acid (31 mg, 0.18 mmol) and 5-(azepan-1-ylsulfonyl)-2-methoxyaniline 5 (50 mg, 0.18 mmol) in DMF (2 mL). The reaction mixture was heated to 70 °C over 90 minutes. The reaction mixture was diluted with ethyl acetate (25 mL), washed with water (3 × 25 mL) and brine (25 mL), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (30 – 60% ethyl acetate in petroleum ether) afforded 15d (50 mg, 64% yield). LCMS (ESI+): m/z 443.3 [M + H]+, (ESI-): m/z 441.3 [M - H]−, rt 2.24 minutes, >99%; 1H NMR (500 MHz, (CD3)2SO) 9.70 (1H, s), 8.46 (1H, d, J = 2.0 Hz), 7.92 (1H, s), 7.69 (1H, d, J = 7.6 Hz), 7.57 (1H, d, J = 8.2 Hz), 7.49 (1H, dd, J = 8.5, 2.3 Hz), 7.32 (1H, td, J = 7.7, 1.1 Hz), 7.27 (1H, t, J = 7.5 Hz), 7.22 (1H, d, J = 8.7 Hz), 3.94 (3H, s), 3.92 (2H, s), 3.13 (4H, t, J = 5.9 Hz), 1.64–1.53 (4H, m), 1.51–1.42 (4H, m); 13C NMR (125 MHz, (CD3)2SO) 169.0, 154.5, 152.1, 143.5, 130.4, 127.8, 127.6, 124.4, 123.4, 122.6, 120.2, 119.3, 114.6, 111.3, 111.2, 56.3, 47.6, 31.2, 28.4, 26.3; νmax/cm−1 3353, 2929, 2855, 1680 (C=O), 1593, 1523; HRMS (ESI)+: m/z calculated for [C23H26N2O5S + H]+ = 443.1635, observed 443.1652.
N-(5-(Azepan-1-ylsulfonyl)-2-methoxyphenyl)-2-(benzo[d]isoxazol-3-yl)acetamide (15e).
T3P® (50 wt. % in ethyl acetate, 81 μL, 0.14 mmol) and N,N-diisopropylethylamine (39 μL, 0.23 mmol) were added to a solution of 2-(1,2-benzisoxazol-3-yl)acetic acid (8 mg, 0.05 mmol) and 5-(azepan-1-ylsulfonyl)-2-methoxyaniline 5 (13 mg, 0.045 mmol) in DMF (1 mL). The reaction mixture was heated to 70 °C over 3 hours. The reaction mixture was diluted with ethyl acetate (25 mL), washed with water (3 × 15 mL) and brine (25 mL), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (30 – 70% ethyl acetate in petroleum ether) followed by reverse phase column chromatography (0 – 100% acetonitrile in water (+ 0.1% NH3)) afforded 15e (11 mg, 55% yield). LCMS (ESI+): m/z 444.3 [M + H]+, (ESI-): m/z 442.2 [M - H]−, rt 2.15 minutes, >99%; 1H NMR (400 MHz, (CD3)2SO) 10.03 (1H, s), 8.44 (1H, d, J = 1.9 Hz), 7.91 (1H, d, J = 8.0 Hz), 7.75 (1H, d, J = 8.5 Hz), 7.66 (1H, t, J = 7.7 Hz), 7.52 (1H, dd, J = 8.6, 2.2 Hz), 7.41 (1H, t, J = 7.4 Hz), 7.25 (1H, d, J = 8.7 Hz), 4.34 (2H, s), 3.97 (3H, s), 3.13 (4H, t, J = 5.9 Hz), 1.67–1.52 (4H, m), 1.51–1.39 (4H, m); 13C NMR (100 MHz, (CD3)2SO) 166.8, 162.4, 154.2, 152.2, 130.4, 127.4, 123.7, 122.6, 121.5, 119.5, 111.3, 109.7, 56.3, 47.6, 32.8, 28.5, 26.3 (2 peaks missing); νmax/cm−1 2923, 2856, 1689 (C=O), 1595, 1527; HRMS (ESI)+: m/z calculated for [C22H25N3O5S + H]+ = 444.1588, observed 444.1592.
N-(5-(Azepan-1-ylsulfonyl)-2-methoxyphenyl)-2-(1H-benzo[d]imidazol-1-yl)acetamide (15f).
T3P® (50 wt. % in ethyl acetate, 0.25 mL, 0.42 mmol) and N,N-diisopropylethylamine (0.12 mL, 0.70 mmol) were added to a solution of 2-(1H-benzimidazol-1-yl)acetic acid (25 mg, 0.14 mmol) and 5-(azepan-1-ylsulfonyl)-2-methoxyaniline 5 (40 mg, 0.14 mmol) in DMF (2 mL). The reaction mixture was heated to 70 °C over 1 hour. The reaction mixture was diluted with ethyl acetate (25 mL), washed with water (3 × 25 mL) and brine (25 mL), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (50 – 100% ethyl acetate in petroleum ether, 0 – 5% methanol in DCM) afforded 15f (33 mg, 53% yield). LCMS (ESI+): m/z 443.3 [M + H]+, (ESI-): m/z 441.2 [M - H]−, rt 1.62 minutes, >99%; 1H NMR (500 MHz, (CD3)2SO) 10.00 (1H, s), 8.45 (1H, d, J = 2.2 Hz), 8.23 (1H, s), 7.70–7.65 (1H, m), 7.54 (1H, d, J = 7.8 Hz), 7.51 (1H, dd, J = 8.7, 2.3 Hz), 7.29–7.19 (3H, m), 5.31 (2H, s), 3.98 (3H, s), 3.11 (4H, t, J = 5.9 Hz), 1.62–1.51 (4H, m), 1.49–1.40 (4H, m); 13C NMR (125 MHz, (CD3)2SO) 166.5, 152.0, 145.0, 143.2, 134.4, 130.4, 127.3, 123.7, 122.4, 121.6, 119.4, 119.1, 111.3, 110.3, 56.4, 47.6, 47.3, 28.4, 26.3; νmax/cm−1 2930, 2853, 1694 (C=O), 1596, 1533; HRMS (ESI)+: m/z calculated for [C22H26N4O4S + H]+ = 443.1748, observed 443.1769.
N-(5-(Azepan-1-ylsulfonyl)-2-methoxyphenyl)-2-(quinolin-4-yl)acetamide (15g).
Lithium diisopropylamide (2 M in THF/heptane/ethylbenzene, 1.5 mL, 3.0 mmol) was added dropwise over 30 minutes at −78 °C to a mixture of 4-methylquinoline 36 (0.33 mL, 2.5 mmol) and THF (5 mL). The reaction mixture was stirred at −78 °C over 1 hour, then solid CO2 pellets (1 g) were added. The reaction mixture was stirred at −78 °C over 10 minutes, then warmed to room temperature and stirred over 1 hour. Water (20 mL) was added dropwise at 0 °C, followed by aqueous NaOH (10% w/v, 5 mL). The mixture was washed with DCM (3 × 25 mL), then adjusted to pH 6 and washed with DCM/methanol (9:1, 3 × 50 mL). The aqueous phase was concentrated in vacuo to afford a crude residue. 5-(azepan-1-ylsulfonyl)-2-methoxyaniline 5 (0.140 g, 0.492 mmol) and DMF (10 mL) were added to the crude residue, followed by T3P® (50 wt. % in ethyl acetate, 3.5 mL, 5.9 mmol) and N,N-diisopropylethylamine (1.7 mL, 9.9 mmol). The reaction mixture was heated to 70 °C over 40 minutes. The reaction mixture was diluted with ethyl acetate (100 mL), washed with water (3 × 100 mL) and brine (100 mL), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (50 – 100% ethyl acetate in petroleum ether, 0 – 10% methanol in DCM) followed by reverse phase column chromatography (0 – 50% acetonitrile in water (+ 0.1% NH3)) afforded 15g (11 mg, 5% yield). LCMS (ESI+): m/z 454.3 [M + H]+, (ESI-): m/z 452.3 [M - H]−, rt 1.70 minutes, >99%; 1H NMR (400 MHz, (CD3)2SO) 9.90 (1H, s), 8.85 (1H, d, J = 4.3 Hz), 8.42 (1H, d, J = 2.2 Hz), 8.19 (1H, d, J = 8.4 Hz), 8.05 (1H, dd, J = 8.4, 0.7 Hz), 7.77 (1H, ddd, J = 8.4, 6.9, 1.4 Hz), 7.65 (1H, ddd, J = 8.4, 6.9, 1.3 Hz), 7.53–7.46 (2H, m), 7.24 (1H, d, J = 8.8 Hz), 4.39 (2H, s), 3.97 (3H, s), 3.11 (4H, t, J = 5.9 Hz), 1.63–1.51 (4H, m), 1.50–1.39 (4H, m); 13C NMR (125 MHz, CDCl3) 167.1, 150.6, 150.5, 148.8, 140.2, 132.2, 130.6, 130.0, 127.63, 127.58, 127.4, 124.0, 123.7, 122.8, 118.1, 109.6, 56.2, 48.5, 42.1, 29.3, 27.0; νmax/cm−1 3295, 2923, 1663 (C=O), 1593, 1537; HRMS (ESI)+: m/z calculated for [C24H27N3O4S + H]+ = 454.1795, observed 454.1781.
N-(5-(Azepan-1-ylsulfonyl)-2-methoxyphenyl)-2-(naphthalen-1-yl)acetamide (15h).
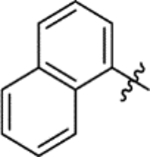
T3P® (50 wt. % in ethyl acetate, 0.31 mL, 0.53 mmol) and N,N-diisopropylethylamine (0.15 mL, 0.88 mmol) were added to a solution of 1-naphthaleneacetic acid (33 mg, 0.18 mmol) and 5-(azepan-1-ylsulfonyl)-2-methoxyaniline 5 (50 mg, 0.18 mmol) in DMF (2 mL). The reaction mixture was heated to 70 °C over 2 hours. The reaction mixture was diluted with ethyl acetate (25 mL), washed with water (3 × 25 mL) and brine (25 mL), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (0 – 50% ethyl acetate in petroleum ether) afforded 15h (53 mg, 67% yield). LCMS (ESI+): m/z 453.3 [M + H]+, (ESI-): m/z 451.3 [M - H]−, rt 2.37 minutes, >99%; 1H NMR (400 MHz, (CD3)2SO) 9.69 (1H, s), 8.44 (1H, d, J = 2.3 Hz), 8.12 (1H, d, J = 7.9 Hz), 7.98–7.91 (1H, m), 7.86 (1H, dd, J = 7.2, 2.2 Hz), 7.59–7.44 (5H, m), 7.22 (1H, d, J = 8.8 Hz), 4.30 (2H, s), 3.94 (3H, s), 3.11 (4H, t, J = 5.9 Hz), 1.62–1.51 (4H, m), 1.49–1.40 (4H, m); 13C NMR (125 MHz, CDCl3) 169.0, 150.6, 134.2, 132.2, 132.1, 130.6, 129.0, 128.9, 128.6, 127.9, 127.1, 126.4, 125.8, 123.9, 123.6, 117.9, 109.4, 56.0, 48.5, 43.1, 29.4, 27.0; νmax/cm−1 3360, 2925, 2862, 1675 (C=O), 1592, 1519; HRMS (ESI)+: m/z calculated for [C25H28N2O4S + Na]+ = 475.1662, observed 475.1652.
N-(5-(Azepan-1-ylsulfonyl)-2-methoxyphenyl)-2-(pyridin-4-yl)acetamide (15i).
EDC.HCl (83 mg, 0.43 mmol), N,N-diisopropylethylamine (0.15 mL, 0.86 mmol) and DMAP (5 mg, 0.04 mmol) were added to a solution of 4-pyridylacetic acid hydrochloride (50 mg, 0.29 mmol) and 5-(azepan-1-ylsulfonyl)-2-methoxyaniline 5 (98 mg, 0.35 mmol) in DCM (2 mL). The reaction mixture was stirred over 90 minutes, then diluted with water (25 mL) and extracted into DCM (3 × 25 mL). The combined organic extracts were washed (brine), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (50 – 100% ethyl acetate in petroleum ether, 0 – 5% methanol in DCM) afforded 15i (74 mg, 64% yield). LCMS (ESI+): m/z 404.3 [M + H]+, (ESI-): m/z 402.2 [M - H]−, rt 1.54 minutes, >99%; 1H NMR (400 MHz, (CD3)2SO) 9.73 (1H, s), 8.54–8.48 (2H, m), 8.45 (1H, d, J = 2.2 Hz), 7.49 (1H, dd, J = 8.6, 2.3 Hz), 7.38–7.31 (2H, m), 7.23 (1H, d, J = 8.8 Hz), 3.94 (3H, s), 3.86 (2H, s), 3.14 (4H, t, J = 5.9 Hz), 1.66–1.54 (4H, m), 1.52–1.41 (4H, m); 13C NMR (125 MHz, CDCl3) 167.2, 150.6, 150.5, 143.1, 132.3, 127.6, 124.7, 124.0, 118.2, 109.6, 56.3, 48.5, 44.2, 29.3, 27.0; νmax/cm−1 3300, 2931, 2858, 1658 (C=O), 1596, 1536; HRMS (ESI)+: m/z calculated for [C20H25N3O4S + Na]+ = 426.1458, observed 426.1447.
N-(2-Methoxy-5-(N-methylsulfamoyl)phenyl)-2-(4-oxo-3,4-dihydrophthalazin-1yl)acetamide (16a).
T3P® (50 wt. % in ethyl acetate, 93 μL, 0.16 mmol) and N,N-diisopropylethylamine (46 μL, 0.26 mmol) were added to a solution of 2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetic acid 3 (11 mg, 0.052 mmol) and 3-amino-4-methoxy-N-methylbenzenesulfonamide 31a (11 mg, 0.052 mmol) in DMF (1 mL). The reaction mixture was heated to 70 °C over 5 hours. The reaction mixture was diluted with ethyl acetate (25 mL), washed with water (3 × 15 mL) and brine (15 mL), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (50 – 100% ethyl acetate in petroleum ether, 0 – 10% methanol in DCM) afforded 16a (6 mg, 29% yield). LCMS (ESI+): m/z 403.2 [M + H]+, (ESI-): m/z 401.1 [M - H]−, rt 1.44 minutes, >99%; 1H NMR (500 MHz, (CD3)2SO) 12.61 (1H, s), 9.83 (1H, s), 8.47 (1H, d, J = 2.2 Hz), 8.27 (1H, d, J = 7.5 Hz), 8.00–7.92 (2H, m), 7.86 (1H, ddd, J = 8.0, 6.3, 1.9 Hz), 7.50 (1H, dd, J = 8.7, 2.3 Hz), 7.31–7.22 (2H, m), 4.21 (2H, s), 3.96 (3H, s), 2.35 (3H, d, J = 5.0 Hz); 13C NMR (125 MHz, (CD3)2SO) 168.2, 159.5, 152.0, 142.2, 133.5, 131.6, 130.7, 129.8, 127.6, 127.5, 125.8, 125.6, 123.5, 119.5, 111.1, 56.3, 28.6 (1 peak missing); νmax/cm−1 3277, 3173, 3016, 2904, 1660 (C=O), 1594, 1537; HRMS (ESI)+: m/z calculated for [C18H18N4O5S + H]+ = 403.1071, observed 403.1064.
N-(5-(Azocan-1-ylsulfonyl)-2-methoxyphenyl)-2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetamide (16b).
T3P® (50 wt. % in ethyl acetate, 0.24 mL, 0.40 mmol) and N,N-diisopropylethylamine (0.12 mL, 0.67 mmol) were added to a solution of 2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetic acid 3 (32 mg, 0.13 mmol) and 5-(azocan-1-ylsulfonyl)-2-methoxyaniline 31b (40 mg, 0.13 mmol) in DMF (2 mL). The reaction mixture was heated to 70 °C over 2 hours. The reaction mixture was diluted with ethyl acetate (25 mL), washed with water (3 × 25 mL) and brine (25 mL), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (50 – 100% ethyl acetate in petroleum ether, 0 – 5% methanol in DCM) afforded 16b (24 mg, 37% yield). LCMS (ESI+): m/z 485.3 [M + H]+, (ESI-): m/z 483.2 [M - H]−, rt 1.94 minutes, >99%; 1H NMR (500 MHz, (CD3)2SO) 12.62 (1H, s), 9.86 (1H, s), 8.45 (1H, d, J = 2.2 Hz), 8.30–8.25 (1H, m), 7.98–7.92 (2H, m), 7.89–7.83 (1H, m), 7.48 (1H, dd, J = 8.6, 2.3 Hz), 7.24 (1H, d, J = 8.8 Hz), 4.21 (2H, s), 3.97 (3H, s), 3.01 (4H, t, J = 5.8 Hz), 1.67–1.44 (10H, m); 13C NMR (125 MHz, (CD3)2SO) 168.4, 159.5, 152.1, 142.1, 133.5, 131.6, 129.8, 129.5, 127.7, 127.6, 125.9, 125.5, 123.5, 119.2, 111.2, 56.3, 48.0, 27.2, 26.2, 24.6 (1 peak missing); νmax/cm−1 3307, 2923, 1693, 1644 (C=O), 1596, 1530; HRMS (ESI)+: m/z calculated for [C24H28N4O5S + H]+ = 485.1853, observed 485.1848.
N-(2-Methoxy-5-(piperidin-1-ylsulfonyl)phenyl)-2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetamide (16c).
T3P® (50 wt. % in ethyl acetate, 0.14 mL, 0.23 mmol) and N,N-diisopropylethylamine (66 μL, 0.38 mmol) were added to a solution of 2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetic acid 3 (16 mg, 0.076 mmol) and 2-methoxy-5-(piperidin-1-ylsulfonyl)aniline 31c (21 mg, 0.076 mmol) in DMF (1 mL). The reaction mixture was heated to 70 °C over 2 hours. The reaction mixture was diluted with ethyl acetate (25 mL), washed with water (3 × 10 mL) and brine (25 mL), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (50 – 100% ethyl acetate in petroleum ether, 0 – 10% methanol in DCM) afforded 16c (14 mg, 40% yield). LCMS (ESI+): m/z 457.3 [M + H]+, (ESI-): m/z 455.2 [M - H]−, rt 1.76 minutes, 98%; 1H NMR (500 MHz, (CD3)2SO) 12.62 (1H, s), 9.88 (1H, s), 8.39 (1H, d, J = 2.2 Hz), 8.28 (1H, d, J = 7.9 Hz), 7.99–7.92 (2H, m), 7.86 (1H, ddd, J = 8.0, 5.7, 2.5 Hz), 7.45 (1H, dd, J = 8.6, 2.3 Hz), 7.28 (1H, d, J = 8.7 Hz), 4.22 (2H, s), 3.98 (3H, s), 2.81 (4H, t, J = 5.2 Hz), 1.50 (4H, quin, J = 5.7 Hz), 1.39–1.28 (2H, m); 13C NMR (125 MHz, (CD3)2SO) 168.4, 159.5, 152.4, 142.1, 133.5, 131.6, 129.8, 127.59, 127.56, 126.7, 125.9, 125.5, 124.3, 119.9, 111.2, 56.4, 46.5, 24.6, 22.8 (1 peak missing); νmax/cm−1 3176, 3057, 2923, 1674, 1646 (C=O), 1594, 1542; HRMS (ESI)+: m/z calculated for [C22H24N4O5S + H]+ = 457.1540, observed 457.1537.
N-(2-Methoxy-5-((4-methylpiperazin-1-yl)sulfonyl)phenyl)-2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetamide (16d).
T3P® (50 wt. % in ethyl acetate, 0.24 mL, 0.41 mmol) and N,N-diisopropylethylamine (0.12 mL, 0.68 mmol) were added to a solution of 2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetic acid 3 (32 mg, 0.14 mmol) and 2-methoxy-5-((4-methylpiperazin-1-yl)sulfonyl)aniline 31d (40 mg, 0.14 mmol) in DMF (2 mL). The reaction mixture was heated to 70 °C over 2 hours. The reaction mixture was diluted with ethyl acetate (25 mL), washed with water (3 × 25 mL) and brine (25 mL), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (50 – 100% ethyl acetate in petroleum ether, 0 – 10% methanol in DCM) followed by reverse phase column chromatography (0 – 30% acetonitrile in water (+ 0.1% NH3)) afforded 16d (10 mg, 16% yield). LCMS (ESI+): m/z 472.3 [M + H]+, (ESI-): m/z 470.2 [M - H]−, rt 1.23 minutes, >99%; 1H NMR (500 MHz, (CD3)2SO) 12.62 (1H, s), 9.91 (1H, s), 8.40 (1H, d, J = 2.2 Hz), 8.30–8.25 (1H, m), 7.99–7.92 (2H, m), 7.86 (1H, ddd, J = 8.0, 5.8, 2.4 Hz), 7.46 (1H, dd, J = 8.6, 2.3 Hz), 7.30 (1H, d, J = 8.7 Hz), 4.22 (2H, s), 3.99 (3H, s), 2.81 (4H, br s), 2.38–2.28 (4H, m), 2.10 (3H, s); 13C NMR (125 MHz, (CD3)2SO) 168.4, 159.4, 152.6, 142.1, 133.5, 131.6, 129.8, 127.62, 127.56, 126.0, 125.8, 125.5, 124.5, 120.0, 111.3, 56.4, 53.5, 45.7, 45.2 (1 peak missing); νmax/cm−1 3376, 2922, 1684, 1655 (C=O), 1594, 1533; HRMS (ESI)+: m/z calculated for [C22H25N5O5S + H]+ = 472.1649, observed 472.1662.
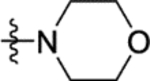
N-(2-Methoxy-5-(morpholinosulfonyl)phenyl)-2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetamide (16e).
T3P® (50 wt. % in ethyl acetate, 0.26 mL, 0.44 mmol) and N,N-diisopropylethylamine (0.13 mL, 0.73 mmol) were added to a solution of 2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetic acid 3 (35 mg, 0.15 mmol) and 2-methoxy-5-(morpholinosulfonyl)aniline 31e (40 mg, 0.15 mmol) in DMF (2 mL). The reaction mixture was heated to 70 °C over 2 hours. The reaction mixture was diluted with ethyl acetate (25 mL), washed with water (3 × 25 mL) and brine (25 mL), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (50 – 100% ethyl acetate in petroleum ether, 0 – 10% methanol in DCM) followed by reverse phase column chromatography (0 – 60% acetonitrile in water (+ 0.1% NH3)) afforded 16e (7 mg, 10% yield). LCMS (ESI+): m/z 459.3 [M + H]+, (ESI-): m/z 457.3 [M - H]−, rt 1.55 minutes, >99%; 1H NMR (400 MHz, (CD3)2SO) 12.62 (1H, s), 9.93 (1H, s), 8.41 (1H, d, J = 2.0 Hz), 8.27 (1H, d, J = 7.8 Hz), 8.01–7.91 (2H, m), 7.86 (1H, ddd, J = 8.0, 5.7, 2.5 Hz), 7.47 (1H, dd, J = 8.7, 2.2 Hz), 7.31 (1H, d, J = 8.7 Hz), 4.23 (2H, s), 3.99 (3H, s), 3.59 (4H, t, J = 4.5 Hz), 2.79 (4H, t, J = 4.4 Hz); 13C NMR (100 MHz, (CD3)2SO) 168.5, 159.5, 152.7, 142.1, 133.6, 131.6, 129.8, 127.7, 127.6, 125.9, 125.6, 125.5, 124.6, 120.1, 111.4, 65.2, 56.4, 45.9 (1 peak missing); νmax/cm−1 3175, 3027, 2924, 2854, 1672, 1646 (C=O), 1593, 1538; HRMS (ESI)+: m/z calculated for [C21H22N4O6S + Na]+ = 481.1152, observed 481.1143.
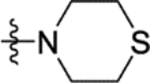
N-(2-Methoxy-5-(thiomorpholinosulfonyl)phenyl)-2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetamide (16f).
T3P® (50 wt. % in ethyl acetate, 0.24 mL, 0.40 mmol) and N,N-diisopropylethylamine (0.12 mL, 0.67 mmol) were added to a solution of 2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetic acid 3 (32 mg, 0.13 mmol) and 2-methoxy-5-(thiomorpholinosulfonyl)aniline 31f (40 mg, 0.13 mmol) in DMF (2 mL). The reaction mixture was heated to 70 °C over 2 hours. The reaction mixture was diluted with ethyl acetate (25 mL), washed with water (3 × 25 mL) and brine (25 mL), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (50 – 100% ethyl acetate in petroleum ether, 0 – 10% methanol in DCM) afforded 16f (23 mg, 36% yield). LCMS (ESI+): m/z 475.2 [M + H]+, (ESI-): m/z 473.2 [M - H]−, rt 1.72 minutes, >99%; 1H NMR (500 MHz, (CD3)2SO) 12.62 (1H, s), 9.92 (1H, s), 8.41 (1H, d, J = 2.3 Hz), 8.27 (1H, d, J = 7.9 Hz), 8.00–7.92 (2H, m), 7.86 (1H, ddd, J = 8.0, 5.8, 2.3 Hz), 7.47 (1H, dd, J = 8.7, 2.3 Hz), 7.29 (1H, d, J = 8.7 Hz), 4.23 (2H, s), 3.99 (3H, s), 3.13 (4H, t, J = 4.7 Hz), 2.63 (4H, t, J = 5.1 Hz); 13C NMR (125 MHz, (CD3)2SO) 168.4, 159.5, 152.6, 142.1, 133.6, 131.6, 129.8, 127.8, 127.6, 127.2, 125.9, 125.6, 124.2, 119.6, 111.4, 56.4, 47.8, 26.3 (1 peak missing); νmax/cm−1 3391, 2929, 2849, 1674, 1645, 1607, 1516; HRMS (ESI)+: m/z calculated for [C21H22N4O5S2 + H]+ = 475.1104, observed 475.1104.
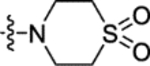
N-(5-((1,1-Dioxidothiomorpholino)sulfonyl)-2-methoxyphenyl)-2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetamide (16g).
T3P® (50 wt. % in ethyl acetate, 0.67 mL, 1.1 mmol) and N,N-diisopropylethylamine (0.33 mL, 1.9 mmol) were added to a solution of 2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetic acid 3 (89 mg, 0.38 mmol) and 4-((3-amino-4-methoxyphenyl)sulfonyl)thiomorpholine 1,1-dioxide 31g (0.120 g, 0.375 mmol) in DMF (2 mL). The reaction mixture was heated to 70 °C over 1 hour. The reaction mixture was diluted with ethyl acetate (25 mL), washed with water (3 × 25 mL) and brine (25 mL), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (50 – 100% ethyl acetate in petroleum ether, 0 – 20% methanol in DCM) followed by reverse phase column chromatography (0 – 60% acetonitrile in water (+ 0.1% NH3)) afforded 16g (55 mg, 28% yield). LCMS (ESI+): m/z 507.2 [M + H]+, (ESI-): m/z 505.2 [M - H]−, rt 1.61 minutes, 97%; 1H NMR (500 MHz, (CD3)2SO) 12.62 (1H, s), 9.96 (1H, s), 8.47 (1H, s), 8.27 (1H, d, J = 7.8 Hz), 8.02–7.82 (3H, m), 7.55 (1H, d, J = 8.8 Hz), 7.32 (1H, d, J = 8.7 Hz), 4.23 (2H, s), 4.00 (3H, s), 3.27–3.19 (4H, m) (1 peak suspected to be obscured by H2O signal); 13C NMR (125 MHz, (CD3)2SO) 168.6, 159.5, 152.9, 142.1, 133.6, 131.6, 129.8, 128.0, 127.6, 126.8, 125.9, 125.6, 124.2, 119.5, 111.7, 56.5, 49.9, 45.1 (1 peak missing); νmax/cm−1 3403, 3001, 2933, 1663 (C=O), 1597, 1525; HRMS (ESI)-: m/z calculated for [C21H22N4O7S2 - H]− = 505.0857, observed 505.0851.
N-(2-Methoxy-5-(N-phenylsulfamoyl)phenyl)-2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetamide (16h).
T3P® (50 wt. % in ethyl acetate, 0.16 mL, 0.27 mmol) and N,N-diisopropylethylamine (79 μL, 0.45 mmol) were added to a solution of 2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetic acid 3 (19 mg, 0.090 mmol) and 3-amino-4-methoxy-N-phenylbenzenesulfonamide 31h (25 mg, 0.090 mmol) in DMF (1 mL). The reaction mixture was heated to 70 °C over 2 hours. The reaction mixture was diluted with ethyl acetate (25 mL), washed with water (3 × 10 mL) and brine (25 mL), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (50 – 100% ethyl acetate in petroleum ether, 0 – 10% methanol in DCM) afforded 16h (14 mg, 33% yield). LCMS (ESI+): m/z 465.2 [M + H]+, (ESI-): m/z 463.2 [M - H]−, rt 1.71 minutes, 98%; 1H NMR (500 MHz, (CD3)2SO) 12.61 (1H, s), 10.17 (1H, s), 9.78 (1H, s), 8.55 (1H, d, J = 2.2 Hz), 8.31–8.25 (1H, m), 7.98–7.91 (2H, m), 7.90–7.84 (1H, m), 7.47 (1H, dd, J = 8.7, 2.4 Hz), 7.22–7.14 (3H, m), 7.07–7.03 (2H, m), 7.00–6.94 (1H, m), 4.20 (2H, s), 3.91 (3H, s); 13C NMR (125 MHz, (CD3)2SO) 168.2, 159.4, 152.2, 142.1, 137.9, 133.5, 131.6, 131.0, 129.8, 129.1, 127.6, 127.5, 125.8, 125.6, 123.7, 123.6, 119.6, 119.3, 111.0, 56.3 (1 peak missing); νmax/cm−1 3175, 3016, 1674, 1647 (C=O), 1592, 1538; HRMS (ESI)+: m/z calculated for [C23H20N4O5S + H]+ = 465.1227, observed 465.1223.
N-(5-(N-Benzylsulfamoyl)-2-methoxyphenyl)-2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetamide (16i).
T3P® (50 wt. % in ethyl acetate, 0.26 mL, 0.43 mmol) and N,N-diisopropylethylamine (0.12 mL, 0.71 mmol) were added to a solution of 2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetic acid 3 (30 mg, 0.14 mmol) and 3-amino-N-benzyl-4-methoxybenzenesulfonamide 31i (45 mg, 0.14 mmol) in DMF (2 mL). The reaction mixture was heated to 70 °C over 2 hours. The reaction mixture was diluted with ethyl acetate (25 mL), washed with water (3 × 25 mL) and brine (25 mL), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (50 – 100% ethyl acetate in petroleum ether, 0 – 8% methanol in DCM) afforded 16i (25 mg, 37% yield). LCMS (ESI+): m/z 479.3 [M + H]+, (ESI-): m/z 477.2 [M - H]−, rt 1.74 minutes, >99%; 1H NMR (500 MHz, (CD3)2SO) 12.62 (1H, s), 9.81 (1H, s), 8.51 (1H, d, J = 2.2 Hz), 8.27 (1H, d, J = 7.8 Hz), 8.03–7.92 (3H, m), 7.86 (1H, ddd, J = 8.0, 6.6, 1.6 Hz), 7.53 (1H, dd, J = 8.6, 2.3 Hz), 7.28–7.17 (6H, m), 4.22 (2H, s), 3.96 (3H, s), 3.89 (2H, d, J = 6.5 Hz); 13C NMR (125 MHz, (CD3)2SO) 168.1, 159.5, 152.0, 142.2, 137.7, 133.5, 132.1, 131.6, 129.8, 128.2, 127.6, 127.53, 127.49, 127.1, 125.8, 125.6, 123.4, 119.4, 111.1, 56.3, 46.1 (1 peak missing); νmax/cm−1 3287, 3176, 3026, 1672, 1647 (C=O), 1593, 1538; HRMS (ESI)+: m/z calculated for [C24H22N4O5S + H]+ = 479.1384, observed 479.1381.
N-(5-((3,4-Dihydroisoquinolin-2(1H)-yl)sulfonyl)-2-methoxyphenyl)-2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetamide (16j).
T3P® (50 wt. % in ethyl acetate, 0.28 mL, 0.47 mmol) and N,N-diisopropylethylamine (0.14 mL, 0.79 mmol) were added to a solution of 2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetic acid 3 (37 mg, 0.16 mmol) and 5-((3,4-dihydroisoquinolin-2(1H)-yl)sulfonyl)-2-methoxyaniline 31j (50 mg, 0.16 mmol) in DMF (2 mL). The reaction mixture was heated to 70 °C over 1 hour. The reaction mixture was diluted with ethyl acetate (25 mL), washed with water (3 × 25 mL) and brine (25 mL), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (50 – 100% ethyl acetate in petroleum ether, 0 – 5% methanol in DCM) afforded 16j (35 mg, 43% yield). LCMS (ESI-): m/z 503.3 [M - H]−, rt 1.90 minutes, 98%; 1H NMR (500 MHz, (CD3)2SO) 12.62 (1H, s), 9.89 (1H, s), 8.50 (1H, d, J = 2.2 Hz), 8.28 (1H, d, J = 7.9 Hz), 7.99–7.92 (2H, m), 7.86 (1H, ddd, J = 8.0, 5.9, 2.3 Hz), 7.56 (1H, dd, J = 8.6, 2.3 Hz), 7.28 (1H, d, J = 8.7 Hz), 7.17–7.05 (4H, m), 4.22 (2H, s), 4.11 (2H, s), 3.97 (3H, s), 3.22 (2H, t, J = 6.0 Hz), 2.83 (2H, t, J = 5.8 Hz); 13C NMR (125 MHz, (CD3)2SO) 168.4, 159.5, 152.5, 142.1, 133.5, 133.0, 131.6, 131.5, 129.8, 128.6, 127.7, 127.6, 126.9, 126.6, 126.4, 126.1, 125.8, 125.6, 124.4, 119.9, 111.4, 56.4, 47.2, 43.6, 28.0 (1 peak missing); νmax/cm−1 3354, 2929, 2853, 1679 (C=O), 1592, 1523; HRMS (ESI)+: m/z calculated for [C26H24N4O5S + H]+ = 505.1540, observed 505.1534.
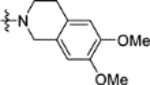
N-(5-((6,7-Dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)sulfonyl)-2-methoxyphenyl)-2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetamide (16k).
T3P® (50 wt. % in ethyl acetate, 0.38 mL, 0.63 mmol) and N,N-diisopropylethylamine (0.18 mL, 1.1 mmol) were added to a solution of 2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetic acid 3 (50 mg, 0.21 mmol) and 5-((6,7-dimethoxy-3,4-dihydroisoquinolin-2(1H)-yl)sulfonyl)-2-methoxyaniline 31k (80 mg, 0.21 mmol) in DMF (2 mL). The reaction mixture was heated to 70 °C over 1 hour. The reaction mixture was diluted with ethyl acetate (25 mL), washed with water (3 × 25 mL) and brine (25 mL), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (50 – 100% ethyl acetate in petroleum ether, 0 – 7% methanol in DCM) followed by reverse phase column chromatography (0 – 50% acetonitrile in water (+ 0.1% NH3)) afforded 16k (48 mg, 40% yield). LCMS (ESI+): m/z 565.4 [M + H]+, (ESI-): m/z 563.2 [M - H]−, rt 1.78 minutes, >99%; 1H NMR (500 MHz, (CD3)2SO) 12.62 (1H, s), 9.89 (1H, s), 8.50 (1H, d, J = 2.2 Hz), 8.28 (1H, d, J = 7.7 Hz), 7.99–7.92 (2H, m), 7.86 (1H, ddd, J = 8.0, 6.0, 2.1 Hz), 7.54 (1H, dd, J = 8.6, 2.3 Hz), 7.28 (1H, d, J = 8.9 Hz), 6.71 (1H, s), 6.65 (1H, s), 4.22 (2H, s), 4.01 (2H, s), 3.97 (3H, s), 3.67 (3H, s), 3.65 (3H, s), 3.18 (2H, t, J = 5.9 Hz), 2.74 (2H, t, J = 5.8 Hz); 13C NMR (125 MHz, (CD3)2SO) 168.4, 159.5, 152.5, 147.5, 147.3, 142.1, 133.5, 131.6, 129.8, 127.7, 127.6, 127.0, 125.8, 125.6, 124.7, 124.4, 123.1, 119.8, 111.7, 111.3, 109.8, 56.4, 55.5, 55.4, 46.9, 43.7, 27.7 (1 peak missing); νmax/cm−1 3299, 3010, 2938, 2837, 1695, 1641 (C=O), 1596, 1520; HRMS (ESI)+: m/z calculated for [C28H28N4O7S + Na]+ = 587.1571, observed 587.1543.
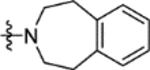
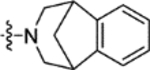
N-(2-Methoxy-5-((1,2,4,5-tetrahydro-3H-benzo[d]azepin-3-yl)sulfonyl)phenyl)-2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetamide (16l).
T3P® (50 wt. % in ethyl acetate, 0.38 mL, 0.63 mmol) and N,N-diisopropylethylamine (0.18 mL, 1.1 mmol) were added to a solution of 2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetic acid 3 (50 mg, 0.21 mmol) and 2-methoxy-5-((1,2,4,5-tetrahydro-3H-benzo[d]azepin-3-yl)sulfonyl)aniline 31l (70 mg, 0.21 mmol) in DMF (2 mL). The reaction mixture was heated to 70 °C over 2 hours. The reaction mixture was diluted with ethyl acetate (25 mL), washed with water (3 × 25 mL) and brine (25 mL), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (50 – 80% ethyl acetate in petroleum ether, 0 – 5% methanol in DCM) followed by reverse phase column chromatography (0 – 100% acetonitrile in water (+ 0.1% NH3)) afforded 16l (20 mg, 18% yield). LCMS (ESI+): m/z 519.3 [M + H]+, (ESI-): m/z 517.2 [M - H]−, rt 2.06 minutes, >99%; 1H NMR (500 MHz, (CD3)2SO) 12.61 (1H, s), 9.85 (1H, s), 8.42 (1H, d, J = 2.1 Hz), 8.27 (1H, d, J = 7.8 Hz), 7.97–7.90 (2H, m), 7.89–7.83 (1H, m), 7.48 (1H, dd, J = 8.7, 2.3 Hz), 7.23 (1H, d, J = 8.8 Hz), 7.12–7.04 (4H, m), 4.19 (2H, s), 3.94 (3H, s), 3.18–3.04 (4H, m), 2.96–2.83 (4H, m); 13C NMR (125 MHz, (CD3)2SO) 168.4, 159.4, 152.3, 142.1, 140.4, 133.5, 131.6, 129.8, 129.1, 128.7, 127.7, 127.6, 126.5, 125.9, 125.5, 123.9, 119.3, 111.3, 56.3, 48.1, 35.5 (1 peak missing); νmax/cm−1 3302, 3173, 3011, 2906, 1687, 1650 (C=O), 1595, 1530; HRMS (ESI)-: m/z calculated for [C27H26N4O5S - H]− = 517.1551, observed 517.1549.
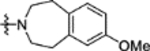
N-(2-Methoxy-5-((7-methoxy-1,2,4,5-tetrahydro-3H-benzo[d]azepin-3-yl)sulfonyl)phenyl)-2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetamide (16m).
T3P® (50 wt. % in ethyl acetate, 0.50 mL, 0.84 mmol) and N,N-diisopropylethylamine (0.24 mL, 1.4 mmol) were added to a solution of 2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetic acid 3 (66 mg, 0.28 mmol) and 2-methoxy-5-((7-methoxy-1,2,4,5-tetrahydro-3H-benzo[d]azepin-3-yl)sulfonyl)aniline 31m (0.101 g, 0.279 mmol) in DMF (2 mL). The reaction mixture was heated to 70 °C over 1 hour. The reaction mixture was diluted with ethyl acetate (25 mL), washed with water (3 × 25 mL) and brine (25 mL), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (50 – 100% ethyl acetate in petroleum ether, 0 – 7% methanol in DCM) afforded 16m (48 mg, 30% yield). LCMS (ESI+): m/z 549.4 [M + H]+, (ESI-): m/z 547.3 [M - H]−, rt 1.96 minutes, 96%; 1H NMR (500 MHz, (CD3)2SO) 12.61 (1H, s), 9.85 (1H, s), 8.41 (1H, d, J = 2.1 Hz), 8.27 (1H, d, J = 7.9 Hz), 7.97–7.90 (2H, m), 7.89–7.83 (1H, m), 7.47 (1H, dd, J = 8.7, 2.3 Hz), 7.22 (1H, d, J = 8.8 Hz), 6.98 (1H, d, J = 8.2 Hz), 6.67 (1H, d, J = 2.6 Hz), 6.62 (1H, dd, J = 8.2, 2.8 Hz), 4.20 (2H, s), 3.94 (3H, s), 3.66 (3H, s), 3.15–3.02 (4H, m), 2.91–2.76 (4H, m); 13C NMR (125 MHz, (CD3)2SO) 168.4, 159.5, 157.8, 152.3, 142.1, 141.7, 133.6, 132.4, 131.6, 130.2, 129.8, 128.7, 127.7, 127.6, 125.9, 125.5, 123.9, 119.3, 115.0, 111.3, 111.2, 56.3, 55.0, 48.6, 48.1, 35.7, 34.7 (1 peak missing); νmax/cm−1 3308, 2913, 1691, 1642 (C=O), 1595, 1529; HRMS (ESI)+: m/z calculated for [C28H28N4O6S + Na]+ = 571.1622, observed 571.1615.
N-(2-Methoxy-5-((1,2,4,5-tetrahydro-3H-1,5-methanobenzo[d]azepin-3yl)sulfonyl)phenyl)-2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetamide (16n).
T3P® (50 wt. % in ethyl acetate, 0.26 mL, 0.43 mmol) and N,N-diisopropylethylamine (0.13 mL, 0.72 mmol) were added to a solution of 2-(4-oxo-3,4-dihydrophthalazin-1-yl)acetic acid 3 (34 mg, 0.14 mmol) and 2-methoxy-5-((1,2,4,5-tetrahydro-3H-1,5-methanobenzo[d]azepin-3-yl)sulfonyl)aniline 31n (55 mg, 0.14 mmol) in DMF (2 mL). The reaction mixture was heated to 70 °C over 45 minutes. The reaction mixture was diluted with ethyl acetate (25 mL), washed with water (3 × 25 mL) and brine (25 mL), dried (MgSO4) and concentrated in vacuo. Purification by flash column chromatography (50 – 80% ethyl acetate in petroleum ether, 0 – 5% methanol in DCM) followed by reverse phase column chromatography (0 – 50% acetonitrile in water (+ 0.1% NH3)) afforded 16n (26 mg, 33% yield). LCMS (ESI+): m/z 531.3 [M + H]+, (ESI-): m/z 529.3 [M - H]−, rt 1.98 minutes, 96%; 1H NMR (500 MHz, (CD3)2SO) 12.64 (1H, s), 9.82 (1H, s), 8.28 (1H, d, J = 7.9 Hz), 8.22 (1H, d, J = 1.6 Hz), 8.01–7.92 (2H, m), 7.87 (1H, ddd, J = 8.0, 6.3, 1.9 Hz), 7.22–7.06 (6H, m), 4.23 (2H, s), 3.97 (3H, s), 3.40 (2H, d, J = 10.6 Hz), 3.18–3.12 (2H, m), 2.80 (2H, d, J = 10.8 Hz), 2.11–2.01 (1H, m), 1.51 (1H, d, J = 10.9 Hz); 13C NMR (125 MHz, (CD3)2SO) 168.2, 159.5, 152.1, 144.3, 142.1, 133.5, 131.6, 129.8, 128.1, 127.6, 127.5, 126.9, 125.9, 125.6, 123.6, 122.4, 119.6, 111.0, 56.3, 49.2, 41.2 (2 peaks missing); νmax/cm−1 3177, 3020, 2937, 2908, 2851, 1673, 1646 (C=O), 1597, 1540; HRMS (ESI)+: m/z calculated for [C28H26N4O5S + Na]+ = 553.1516, observed 553.1517.
Expression and Purification of Mycobacterium tuberculosis fumarate hydratase.
A single freshly transformed Escherichia coli strain BL21 (DE3) colony was transferred to LB media (20 mL) with ampicillin (100 μg mL−1) and incubated overnight (37 °C, 200 rpm). The starter culture was used to inoculate 2 flasks, each containing TB media (1 L) with ampicillin (100 μg mL−1), with incubation (37 °C, 200 rpm) until an optical density (A600nm) of 1.1 was reached. Protein expression was induced by the addition of isopropyl β-D-1-thiogalactopyranoside (0.5 mM), followed by overnight incubation (15 °C, 200 rpm). Cells were harvested by centrifugation (4 °C, 4000 g, 20 minutes), then frozen.
The cells were resuspended in 50 mL lysis buffer (50 mM Tris pH 8.0, 150 mM NaCl, 20 mM imidazole) with a tablet of cOmplete™ Protease Inhibitor Cocktail (Roche). The suspension was sonicated (15 minutes, 10 seconds on/ 20 seconds off) and centrifuged (4 °C, 30000 g, 20 minutes). The lysate was loaded onto a 7.5 mL nickel Sepharose™ fast flow column (GE Healthcare), pre-equilibrated with lysis buffer. The column was washed with 5 column volumes of lysis buffer and eluted with buffer B (50 mM Tris pH 8.0, 150 mM NaCl, 300 mM imidazole) in 8 × 5 mL aliquots. Protein-containing aliquots, as determined by SDS-PAGE, were combined and concentrated (30 kDa cutoff), then loaded onto a Superdex 200 Hiload™ 26/60 column (GE Healthcare) pre-equilibrated with filtration buffer (10 mM Tris pH 8.0, 150 mM NaCl, 0.5 mM TCEP). Chromatography was conducted using an ÄKTA™ FPLC system (GE Healthcare), with protein-containing fractions, as determined by SDS-PAGE, combined and concentrated to 27.9 mg mL−1 (24.3 mg L−1 yield), then flash-frozen in liquid nitrogen and stored at −80 °C. The identity of the protein was confirmed by LCMS analysis.
Differential Scanning Fluorimetry.
DSF was performed using a BioRad CFX Connect™ system, from 25 to 95 °C in 0.5 °C increments of 30 seconds duration. Samples were run in 96-well plates, with each well containing a final volume of 50 μL. Screening was conducted with 100 mM Tris pH 7.5, 50 mM NaCl, 2.5x SyproOrange®, 2.5 μM Mtb fumarase and either 5% DMSO (control) or 5% ligand stock solution in DMSO.
Biochemical Assay.
The enzymatic activity of Mtb fumarase was followed by a CLARIOstar® microplate spectrometer (BMG Labtech) in a 96-well UV plate (Greiner). 2 μL of either DMSO (control) or ligand stock solution in DMSO was pipetted per well. 150 μL of a solution containing acetyl coenzyme A sodium salt (267 μM), β-nicotinamide adenine dinucleotide hydrate (200 μM), malate dehydrogenase (13.3 units mL−1), citrate synthase (1.33 units mL−1) and Mtb fumarase (33.3 nM) in buffer (250 mM Tris pH 8.0, 5 mM MgCl2) was pipetted per well. The plate was left to equilibrate for 5 minutes at 25 °C, then 48 μL of either buffer (negative control) or fumaric acid (1.67 mM) (positive control or ligand) was pipetted per well. After 2 minutes the plate was read at a wavelength of 340 nm at intervals of 24 seconds over 10 minutes.
Inhibition values for each ligand at a particular concentration were calculated from the gradient of the assay read over the 10 minutes, with correction by the negative control and normalization by the positive control. Inhibition experiments at 50 μM ligand concentration were performed at least twice (n ≥ 2), with % inhibition and standard error of the mean reported. IC50 experiments (n = 6) were performed with 10 ligand concentrations, obtained through serial dilution. Dose-response curves were calculated using Origin software (OriginLab, Northampton, MA, USA), with IC50 and standard error reported.
Soaking of Mycobacterium tuberculosis fumarate hydratase Crystals.
Crystals were grown in Intelli-Plate® 24–4 well sitting drop plates (Art Robbins Instruments), incubated at 19 °C in a ROCK IMAGER® 1000 system (FORMULATRIX®). A sitting drop was set up with 2 μL protein solution (14 mg mL−1 Mtb fumarase, 150 mM NaCl, 10 mM Tris pH 8.0 and 0.5 mM TCEP) and 1 μL reservoir solution (10% w/v PEG3350, 5% DMSO and 0.30 M magnesium formate), equilibrated against 400 μL reservoir solution. After 3 weeks the resultant crystals were used to make seed stocks to produce reliable crystals for ligand screening. Sitting drops were set up with 3 μL protein solution (14 mg mL−1 Mtb fumarase, 150 mM NaCl, 10 mM Tris pH 8.0 and 0.5 mM TCEP), 1 μL reservoir solution (10 – 11.4% w/v PEG3350, 5% DMSO and 0.30 M magnesium formate) and 0.5 μL seed stock, equilibrated against 400 μL reservoir solution. After 1 – 2 weeks drops were treated with 2 μL soaking solution (0.5 – 3 mM ligand, 7.5% DMSO, 26.25% w/v PEG3350 and 0.20 M magnesium formate) and incubated overnight. Crystals were mounted into loops and flash frozen in liquid nitrogen.
X-ray Data Collection and Processing.
X-ray data were collected on beamlines I03 and I04 at the Diamond Light Source synchrotron. Data was processed using autoPROC.19 Molecular replacement for the structure of Mtb fumarase in complex with 16b was carried out using PHASER,20 accessed through the CCP4 software suite,21 with the previously published 1-bound Mtb fumarase structure (PDB code 5F91) used as a search model.14 For structures of Mtb fumarase in complex with 15a, 15g, 16a, 16h, 16j or 16l, the solved 16b-bound structure was used as a search model. Models were manually rebuilt using the COOT molecular graphics software package,22 and refined with REFMAC5,23 accessed through CCP4.21 All coordinates have been deposited to the Protein Data Bank and the accession numbers, with data collection statistics compiled using PHENIX,24 are presented in Table S1.
Screening against H37Rv Mycobacterium tuberculosis.
Antimicrobial susceptibility testing against Mycobacterium tuberculosis H37Rv was performed in various media including: GAST-Fe, 7H9/ADC, 7H9/DPPC (4.7 g/L 7H9 base, 14 mg/L DPPC, 0.81 g/L NaCl, 0.3 g/L Casitone, and 0.05% Tyloxapol) and 7H9/Glucose (4.7 g/L 7H9 base, 4 g/L glucose, 0.81 g/L NaCl, 0.3 g/L Casitone, and 0.05% Tyloxapol). H37Rv was grown in the corresponding media to OD 0.2–0.4 prior to use. 50 μL of a two-fold serial dilution series was placed in each well of a sterile 96-well round bottom plate, then 50 μL of H37Rv diluted to OD 0.0002 was added. Plates were incubated at 37 °C for two weeks prior to determining MIC. The MIC is defined here as the drug concentration that completely inhibits growth of cells.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank Dr Sitthivut Charoensutthivarakul for the collection of HRMS data, Moriam Masha for the LCMS analysis of purified Mtb fumarase and Dr Marko Hyvӧnen for the provision of equipment and facilities for the production and handling of crystals for X-ray crystallography. The authors would like to thank Diamond Light Source for beam-time (proposal mx18548) and the staff of beamlines I03 and I04 for assistance with data collection.
Funding Sources
A.J.W was funded by the EPSRC. M.K. was supported by the National Institutes of Health–Oxford–Cambridge Scholars Program and the Washington University in St. Louis Medical Scientist Training Program. This work was funded in part by the Intramural Research Program of NIH, NIAID (AI000693-25).
ABBREVIATIONS
- ΔTm
change in melting temperature
- cLogP
partition coefficient
- DSF
differential scanning fluorimetry
- IC50
half-maximal inhibitory concentration
- MIC
minimum inhibitory concentration
- Mtb
Mycobacterium tuberculosis
- TB
tuberculosis
Footnotes
Supporting Information
The following files are available free of charge.
Synthesis of intermediates, Biochemical Assay Dose-response Curves (Figures S1 to S7), stereo views of the X-ray crystal structure of Mtb fumarase in complex with 1 (Figure S8), X-ray crystal structure of Mtb fumarase in complex with 16h (Figure S9a), overlays of the X-ray crystal structure of Mtb fumarase in complex with 1 with structures in complex with 16a, 16b and 16h (Figures S9b and S10), omit Fo-Fc electron density maps (Figures S11 to S14), X-ray crystallographic data collection and refinement statistics (Table S1) (PDF)
Molecular formula strings (CSV)
Accession Codes
Atomic coordinates for the X-ray structures of compounds 1 (PDB code 5F91), 15a (PDB code 6S74), 15g (PDB code 6S7W), 16a (PDB code 6S7K), 16b (PDB code 6S43), 16j (PDB code 6S7Z), 16l (PDB code 6S88) and 16h (PDB code 6S7S) are available from the RCPB Protein Data Bank (www.rcpb.org). Authors will release the atomic coordinates and experimental data upon article publication.
REFERENCES
- 1.Global tuberculosis report 2018; World Health Organization: Geneva, 2018, pp 27–64. [Google Scholar]
- 2.Houben RMGJ; Dodd PJ The global burden of latent tuberculosis: a re-estimation using mathematical modelling. PLoS Med. 2016, 13, e1002152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Libardo MDJ; Boshoff HIM; Barry CE The present state of the tuberculosis drug development pipeline. Curr. Opin. Pharmacol 2018, 42, 81–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cole ST; Brosch R; Parkhill J; Garnier T; Churcher C; Harris D; Gordon SV; Eiglmeier K; Gas S; Barry CE; Tekaia F; Badcock K; Basham D; Brown D; Chillingworth T; Connor R; Davies R; Devlin K; Feltwell T; Gentles S; Hamlin N; Holroyd S; Hornsby T; Jagels K; Krogh A; McLean J; Moule S; Murphy L; Oliver K; Osborne J; Quail MA; Rajandream M-A; Rogers J; Rutter S; Seeger K; Skelton J; Squares R; Squares S; Sulston JE; Taylor K; Whitehead S; Barrell BG Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature 1998, 393, 537–544. [DOI] [PubMed] [Google Scholar]
- 5.Watanabe S; Zimmermann M; Goodwin MB; Sauer U; Barry CE; Boshoff HI Fumarate reductase activity maintains an energized membrane in anaerobic Mycobacterium tuberculosis. PLoS Pathog. 2011, 7, e1002287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eoh H; Rhee KY Multifunctional essentiality of succinate metabolism in adaptation to hypoxia in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 6554–6559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Prosser G; Brandenburg J; Reiling N; Barry CE; Wilkinson RJ; Wilkinson KA The bacillary and macrophage response to hypoxia in tuberculosis and the consequences for T cell antigen recognition. Microbes Infect. 2017, 19, 177–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sassetti CM; Boyd DH; Rubin EJ Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol 2003, 48, 77–84. [DOI] [PubMed] [Google Scholar]
- 9.Woods SA; Schwartzbach SD; Guest JR Two biochemically distinct classes of fumarase in Escherichia coli. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol 1988, 954, 14–26. [DOI] [PubMed] [Google Scholar]
- 10.Ruecker N; Jansen R; Trujillo C; Puckett S; Jayachandran P; Piroli GG; Frizzell N; Molina H; Rhee KY; Ehrt S Fumarase deficiency causes protein and metabolite succination and intoxicates Mycobacterium tuberculosis. Cell Chem. Biol 2017, 24, 306–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mechaly AE; Haouz A; Miras I; Barilone N; Weber P; Shepard W; Alzari PM; Bellinzoni M Conformational changes upon ligand binding in the essential class II fumarase Rv1098c from Mycobacterium tuberculosis. FEBS Lett. 2012, 586, 1606–1611. [DOI] [PubMed] [Google Scholar]
- 12.Weaver TM; Levitt DG; Donnelly MI; Wilkens Stevens PP; Banaszak LJ The multisubunit active site of fumarase C from Escherichia coli. Nat. Struct. Biol 1995, 2, 654–662. [DOI] [PubMed] [Google Scholar]
- 13.Takeuchi T; Schumacker PT; Kozmin SA Identification of fumarate hydratase inhibitors with nutrient-dependent cytotoxicity. J. Am. Chem. Soc 2015, 137, 564–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kasbekar M; Fischer G; Mott BT; Yasgar A; Hyvönen M; Boshoff HIM; Abell C; Barry CE; Thomas CJ Selective small molecule inhibitor of the Mycobacterium tuberculosis fumarate hydratase reveals an allosteric regulatory site. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 7503–7508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen H; Zhou X; Wang A; Zheng Y; Gao Y; Zhou J Evolutions in fragment-based drug design: the deconstruction–reconstruction approach. Drug Discovery Today 2015, 20, 105–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scott DE; Coyne AG; Hudson SA; Abell C Fragment-based approaches in drug discovery and chemical biology. Biochemistry 2012, 51, 4990–5003. [DOI] [PubMed] [Google Scholar]
- 17.Ehrt S; Rhee K Mycobacterium tuberculosis metabolism and host interaction: mysteries and paradoxes. Curr. Top. Microbiol. Immunol 2013, 374, 163–188. [DOI] [PubMed] [Google Scholar]
- 18.Agrawal M; Kharkar P; Moghe S; Mahajan T; Deka V; Thakkar C; Nair A; Mehta C; Bose J; Kulkarni-Almeida A; Bhedi D; Vishwakarma RA Discovery of thiazolyl-phthalazinone acetamides as potent glucose uptake activators via high-throughput screening. Bioorg. Med. Chem. Lett 2013, 23, 5740–5743. [DOI] [PubMed] [Google Scholar]
- 19.Vonrhein C; Flensburg C; Keller P; Sharff A; Smart O; Paciorek W; Womack T; Bricogne G Data processing and analysis with the autoPROC toolbox. Acta Crystallogr., Sect. D: Biol. Crystallogr 2011, 67, 293–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser crystallographic software. J. Appl. Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AGW; McCoy A; McNicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr 2011, 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Emsley P; Cowtan K Coot: Model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 23.Murshudov GN; Skubák P; Lebedev AA; Pannu NS; Steiner RA; Nicholls RA; Winn MD; Long F; Vagin AA REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr 2011, 67, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adams PD; Afonine PV; Bunkóczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung L-W; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.