

Abstract
We describe a role of CD44-mediated signaling during host-defense against tuberculosis (TB) using a mouse model of TB and studies in M. tuberculosis (Mtb) infected human macrophage (MΦ). Liposomes targeting CD44 using thioaptamers (CD44TA-LIP) were designed and tested as new vaccines to boost host immunity in TB. CD44TA-LIP enhanced killing of Mtb in human MΦ, which correlated with an increased production of pro-inflammatory cytokines IL-1β, TNF-α and IL-12. CD44TA-LIP activated MΦ showed an enhanced MHC-II dependent antigen presentation to CD4 T-cells. Inhibition of cellular proliferation and cytoskeleton rearrangement pathways downstream of CD44 signaling abrogated CD44TA-LIP-induced antimicrobial effects. Blockade of inflammatory pathways also reduced antigen presentation by MΦ and activation of CD4 T-cells. Mtb infected MΦ treated with CD44TA-LIP exhibited increased nitric oxide and HβD2 defensin peptide production. Among Mtb infected mice with increased lung and spleen loads of organisms, intranasal administration of CD44TA-LIP led to a ten-fold reduction of colony forming units of Mtb and elevated IFN-γ+CD4, effector, central and resident memory T-cells. Biodistribution studies demonstrated that CD44TA-LIP preferentially accumulated in the lungs and were associated with CD11b+ cells. CD44TA-LIP treated mice showed no weight loss or increased liver LDH levels. This study highlights the importance of CD44-mediated signaling in host-defense during TB and the therapeutic potential of CD44TA-LIP.
Keywords: CD44, host-defense, Tuberculosis, Macrophages, Immunity, Thioaptamers, Liposomes, Mycobacterium tuberculosis, CD4 T cells, host-directed therapy
Graphical Abstract
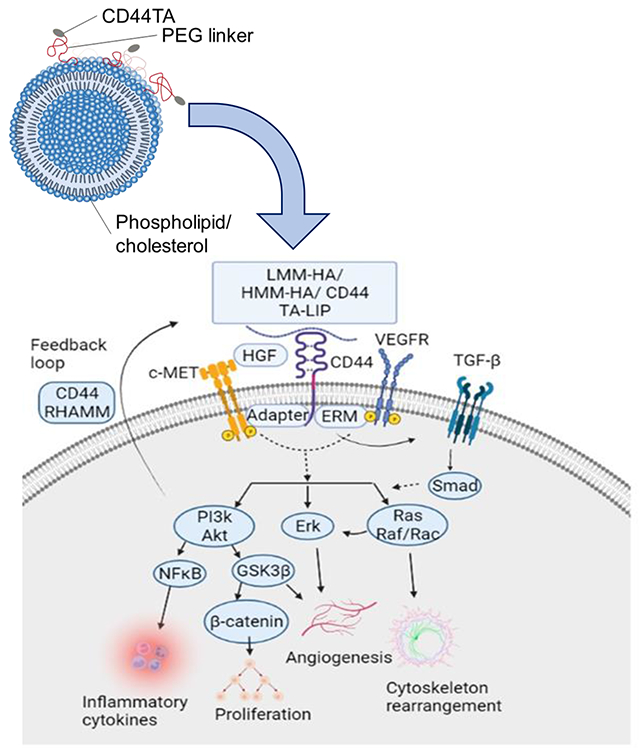
INTRODUCTION
Tuberculosis (TB), due to Mycobacterium tuberculosis (Mtb) infection remains a major health concern worldwide as it causes more than one million deaths per year (1). Treatment with multiple antibiotics and vaccination with BCG are currently the two clinically used major TB control strategies. However, BCG vaccine has poor efficacy in preventing pulmonary tuberculosis, and standard treatments with multiple antibiotics are lengthy (6-9 months) and associated with many complications, contributing to patient non-compliance and the development of drug-resistant M. tuberculosis (MDR and XDR) strains (2). The comorbidity conditions, such as HIV infection, diabetes, malnutrition and other immune suppressive disorders further complicate potential TB treatment, increasing the patients’ mortality rate (3). These facts underscore the need for the development of new and improved TB prevention/treatment strategies. Alternative treatments aimed at re-educating the host immune system are currently being actively pursued as Host-directed therapeutic (HDT) methods to find a more resilient TB control strategy (4). Thus, identifying therapeutic targets for HDT is critical. Most humans exposed to the pathogen develop natural immunity without progressing to an active disease, indicating the existence of immune mechanisms that can provide protection against TB (5). However, finding the key factors of the host immune system protecting against TB remains a major challenge.
Traditionally, protective immunity to Mtb has been suggested to be primarily mediated by T-cells, with CD4+ T cells playing a central role (6,7). However, many population-based immunologic and genetic studies demonstrate that innate immunity is equally important in generating protective response against the pathogen (8,9). Recent reports from both clinical and in vivo studies, accentuating the role of innate cells in host defense against TB focused on re-examining the innate immune system as a target for HDT (HDT) (8,9). Macrophages (MΦ), one of the critical components of the innate immune system are a primary intracellular niche for the growth and survival of Mtb. Thus, boosting the immune response of MΦ could not only help in augmenting protective adaptive immune response, but also enhance their intrinsic intracellular antimicrobial activity against Mtb. Several surface receptors, including complement receptors, C-type lectin receptors, FC gamma receptors, scavenger receptor and TLRs, known to bind the pathogen and facilitate its intracellular uptake, have been identified as possible HDT targets on MΦ to alter the intracellular fate of Mtb (10). Nonetheless, little success has been achieved in elucidating the mechanisms and identifying therapeutic molecules that ligate with these receptors to devise HDT strategies. CD44 is a surface receptor that has been reported to be a binding site for Mtb and to mediate MΦ recruitment in TB (11). Although activation of CD44 signaling pathways have been known to affect multiple cellular processes, such as cell proliferation, adhesion, inflammation, migration, and invasion (12,13), the mechanisms through which it contributes to protective immunity against TB remain unknown.
Molecules that can bind to CD44 receptor also remain unexplored for their therapeutic effect in context of Mtb infection. In this study we designed and characterized CD44 thioaptamer conjugated liposomes (CD44TA-LIP) that can specifically target CD44 receptors. CD44 family comprises proteoglycan transmembrane glycoproteins, which are universally expressed in physiological states and various pathological conditions. Being the primary membranal cell receptor for hyaluronic acid (HA), CD44 binds HA via a lectin-like fold, a Link module. The HA binding domain (HABD), is located near the N-terminal Link module(14) encompassing Link module and a few additional basic residues. The Link module and HABD are highly conserved among CD44 family members and the crystal structures of the HABD have been previously published (15,16). The thioaptamers used in this study were preselected for HABD binding with binding affinities for the HABD in the range of ~200 nM, a few orders of magnitude higher than that of hyaluronic acid (Kd > μM range) (17).
We characterized specific immune response induced by CD44TA-LIP in human MΦ and examined their downstream mechanisms along with evaluation of its therapeutic potential in vivo. We identified cellular proliferation and cytoskeleton rearrangement as key cellular pathways that mediated antimicrobial activity against Mtb after activation of CD44 signaling via CD44TA-LIP. CD44TA-LIP enhanced the priming of T cells and antigen presentation capabilities of MΦ independently on cellular proliferation and cytoskeleton rearrangement pathways, which translated in vivo in an experimental mouse model of TB. In summary, the study demonstrates the immunostimulatory effect of CD44TA-LIP on MΦ based immune responses against Mtb infection and identifies the cellular pathways that are critical for antitubercular immunity.
RESULTS
Synthesis and characterization of CD44TA-LIP
CD44TA-LIP were prepared as described in methods section. Fig. 1 shows the design and characterization of the CD44TA-LIP system. CD44TA- LIP were found to have the average size of 204.9 ± 6.3 nm with narrow size distribution and a negative zeta potential of −22.8 ± 1.9 mV. The targeted liposomes are very uniform in size and have narrow size distribution and zeta potential. The fabrication process that involves microfluidic technique is GMP compatible and allows for production of reproducible systems in scaled larger quantities.
Fig. 1: Design (A) and characterization (B-D) of CD44TA-LIP.
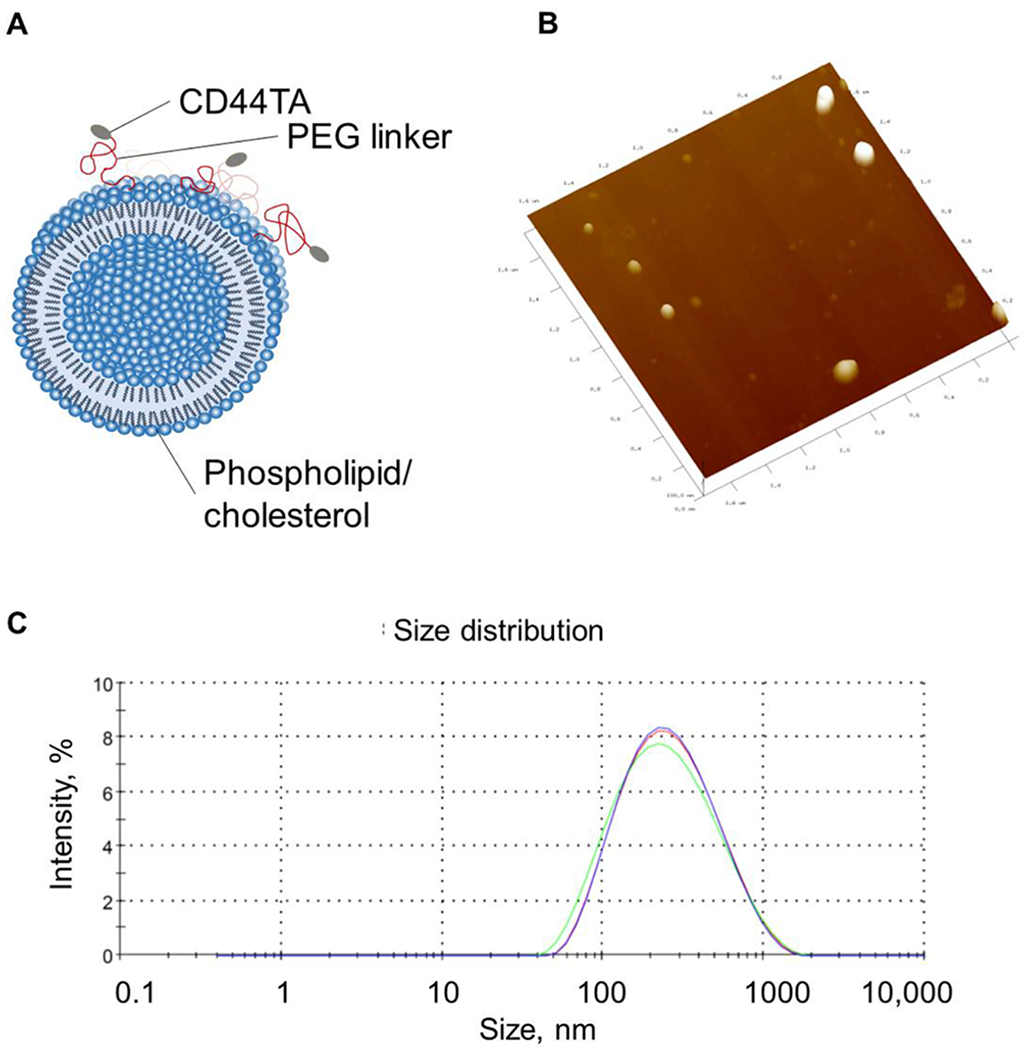
A) Schematics of LIP composition and conjugation with CD44-TA. (B) Atomic force microscopy (AFM) image shows CD44TA-LIP of ~200 nm, confirmed by dynamic light scattering measurement (C) which showed the diameter of 204.9 ± 6.3 nm.
CD44TA-LIP enhance antimicrobial and proinflammatory immune response of MΦ against tuberculosis
Our previous study found that CD44 targeting thioaptamers (TA), conjugated to discoidal silicon mesoporous microparticles (CD44TA-SMP), accumulated in Mtb infected MΦ and enhanced the intracellular killing of the pathogen ex vivo (18). SMP characteristics (material and geometry) used in this previous work were chosen based on the preferential lung accumulation of discoidal SMP after intravenous injections (19–21). Furthermore, CD44TA-SMP treated mice through intravenous route exhibited diminished bacterial load in the lungs and caused recruitment of T lymphocytes at the site of infection. While these results indicated the role of CD44 signaling in generating protective immune response via MΦ activation, they also presented the usefulness of TA based targeting of CD44 as a potential HDT strategy. Nonetheless, SMP conjugated TA may be a limiting factor for their utility as a therapeutic agent, as no silicon-based technologies have yet reached clinical applications. Moreover, the proposed administration route, intravenous injection, for CD44TA-SMP is not applicable in most rural settings largely affected by TB. Thus, in the current work we have changed the delivery strategy to liposomes conjugated to CD44 thioaptamers (CD44TA-LIP). Liposomes are phospholipid nanovectors that have been used in the clinic as antibacterial and anticancer drug carriers for more than 25 years (22,23). Liposomes can be designed for non-invasive pulmonary administration, which is more compatible in rural settings and will target better alveolar MΦ affected by Mtb (24). First, to compare between CD44TA-SMP and CD44TA-LIP anti-Mtb properties, MΦ were treated with both systems simultaneously with Mtb infection. The dose was adjusted to 10μg CD44TA/106 MΦ and the intracellular survival of the pathogen was determined through CFU assays. A time killing kinetics of Mtb in MΦ revealed that both CD44TA-SMP and CD44TA-LIP induced a significantly increased killing of intracellular bacilli until day 3 post treatment (Fig 2A). However, compared to CD44TA-SMP, CD44TA-LIP-induced killing of Mtb in MΦ persisted longer with a significant difference of more than 0.5 log at day 7 post infection, indicating that CD44TA-LIP have a more profound and sustained effect on CD44 activation mediated intracellular killing of Mtb. To further understand if pretreatment of MΦ with CD44TA-LIP prior to Mtb infection (prophylactic) could induce CD44 activation-mediated antibacterial effects, cells were treated 24 h prior to infection and intracellular survival of the pathogen was subsequently determined in a time-dependent manner. While untreated MΦ exhibited significant intracellular growth of Mtb over time, cell pretreated with CD44TA-LIP, prevented the growth inducing a significant killing of the pathogen over a period of 7 days (Fig 2B). These results indicate that not only concurrent treatment but pretreatment of MΦ with CD44TA-LIP can augment intracellular antimicrobial defense to enhance the killing of intracellular Mtb bacilli. These findings are important for TB prophylaxis since active disease can be prevented when an exposure to the pathogen is anticipated. Since CD44TA-LIP treated MΦ were able to efficiently control Mtb infection, it was intriguing to examine if these cells had a distinct immune response in terms of the cytokines they produced. In untreated and CD44TA-LIP treated Mtb infected MΦ (concurrently with infection as well as pre-treated prior to the infection), a panel of pro-inflammatory and anti-inflammatory cytokines was examined at 24h post infection (Fig 2C). Cytokines released by uninfected MΦ untreated, pretreated and treated with CD44TA-LIP were also assessed. During Mtb infection, proinflammatory cytokines, TNF-α and IL-1β, secreted by CD44TA-LIP treated and pre-treated MΦ were significantly higher as compared to untreated MΦ. This indicates that CD44TA-LIP caused generation of proinflammatory immune responses. Secreted levels of IL-6, IL-12, IL-4 and IL-10 were not found to be significantly different in CD44TA-LIP treated vs untreated Mtb infected MΦ. Uninfected MΦ secreted significantly lower levels of all the tested cytokines and did not exhibit any significant difference in untreated vs CD44TA-LIP treated group, pointing towards the importance of Mtb infection for CD44TA-LIP response and biocompatibility of CD44TA-LIP in uninfected (normal) cells. Individual components of the system, namely CD44TA and LIP had no effect on infected and uninfected cells (Supplementary Fig SF1).
Figure 2: Effect of CD44TA-LIP treatment on antimicrobial activity and cytokine response of human MΦ during Mtb infection.
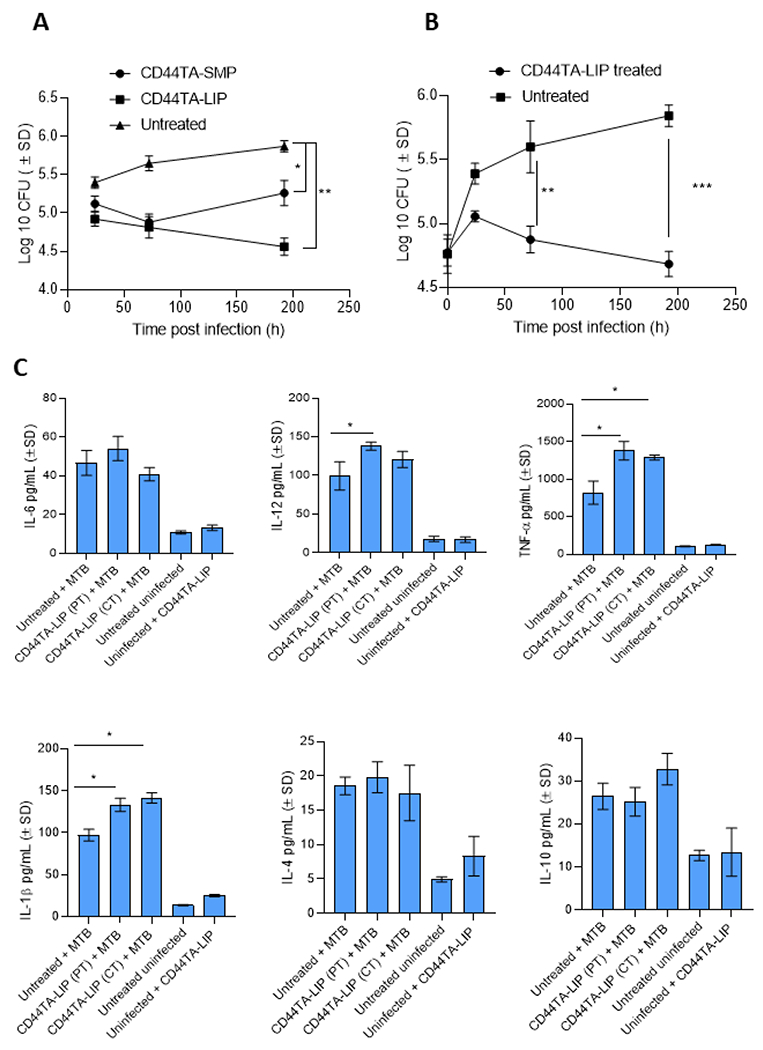
Mtb burden in CD44TA-LIP treated (Panel A: Concurrent treatment; Panel B: pre-treatment) vs untreated human MΦ over seven days of infection as measured by CFU assay. During concurrent treatment MΦ were treated with CD44TA-LIP at the beginning of infection (4 h after the infection) whereas during pre-treatment MΦ were treated 24 h prior to infection. B. Secreted levels of pro- and anti-inflammatory cytokines by CD44TA-LIP treated, untreated and uninfected human MΦ at 24 h post-infection/treatment. CD44TA-LIP concentration used: 10 μg/106 MΦ. PT: pre-treatment; CT: concurrent treatment. pg = picograms; MOI used for the experiments=1. Number of MΦ used per replicate: 1x106. h= h. Representative data shown from three independent experiments carried out in duplicate. Bars and error bars represent mean and SD, respectively. *p≤0.05, **p≤0.005, ***p≤0.0005, ****p≤0.0001.
CD44TA-LIP induce autophagy, nitric oxide and beta defensin-2 (HβD2) peptide production in MΦ during Mtb infection
In order to identify possible mechanism/s through which CD44TA-LIP triggered increased killing of Mtb in MΦ, we examined different antimicrobial components that are known to restrict the intracellular growth of the pathogen. Since overexpression of CD44 is known to enhance autophagy flux (25,26), which is a critical innate component for the containment of intracellular pathogens, we first examined the expression of autophagy related genes in untreated vs CD44TA-LIP treated MΦ during Mtb infection. Strikingly, increased autophagy flux (as examined through the LC3B antibody staining) in CD44TA-LIP treated MΦ was observed as compared to untreated MΦ during Mtb infection (Fig 3A). However, we did not observe a significant co-localization of LC3B protein (red) with GFPMtb (green) containing phagosomes in CD44TA-LIP treated vs untreated MΦ. An increased expression of autophagy related genes Atg5 and Atg7 both at transcription and translation levels was observed in CD44TA-LIP treated MΦ (Fig 3B and 3C). Increased LC3B expression indicated autophagy mediated endosomal maturation, and corroborating the same, Rab7 which is a marker of endosome maturation, found to be overexpressed in CD44TA-LIP treated MΦ (Fig 2B). Expression of lysosomal marker Lamp1 was also enhanced (both at transcription and translation level) in CD44TA-LIP treated MΦ, indicating an increased distribution of lysosomes in MΦ (Fig 3B and 3C) (Quantitation of protein levels shown in Supplementary Fig SF2). To determine if elevated autophagy flux could have caused the increased killing of Mtb in MΦ after CD44TA-LIP treatment, autophagy associated gene beclin1 (Atg6) was knocked down using siRNA vs beclin1 and Mtb survival was subsequently monitored (Fig 3D). CD44TA-LIP mediated intracellular killing of Mtb was observed in beclin1 siRNA treated MΦ as well as scrambled siRNA treated MΦ. Interestingly, upon beclin1 siRNA knockdown, Mtb growth in untreated MΦ increased indicating the critical role of autophagy in containment of Mtb growth in MΦ. Nonetheless, the data clearly indicate that despite upregulation of autophagy flux, CD44TA- LIP mediated killing of Mtb in MΦ was independent of autophagy.
Figure 3: Effect of CD44TA-LIP treatment on autophagy, reactive species and antimicrobial peptide production by human MΦ during Mtb infection.
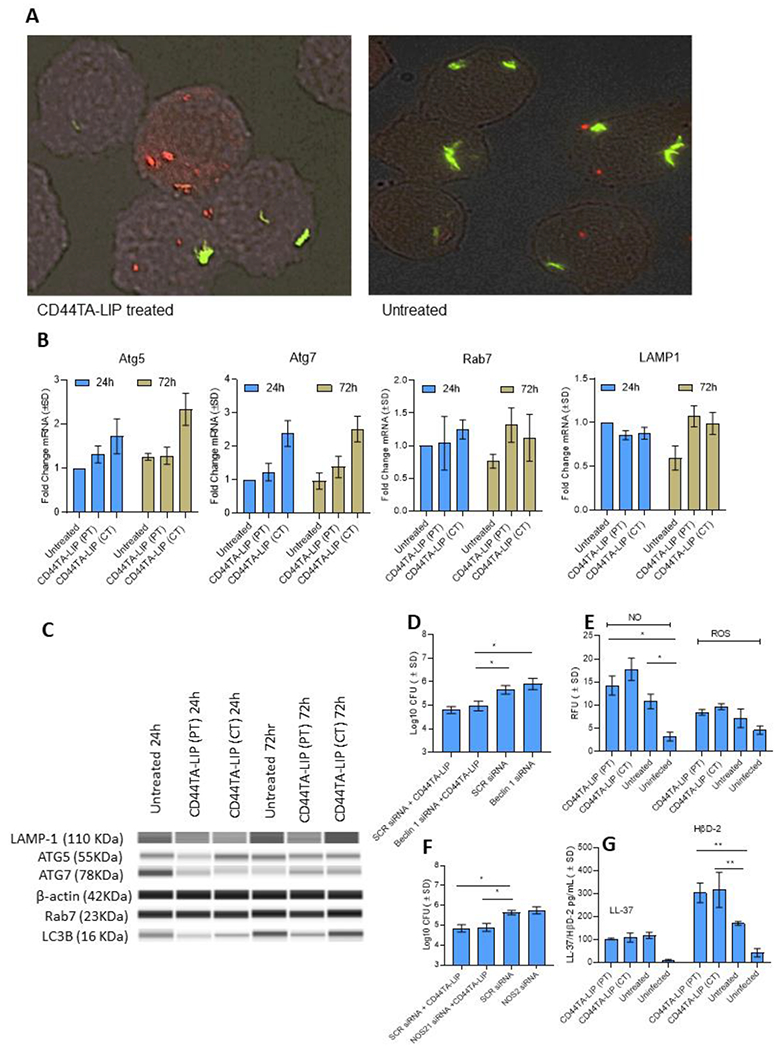
A: Localization of Mtb (green) phagosomes and autophagy related LC3B+ protein (red) within CD44TA-LIP treated vs untreated MΦ after 24 h of infection. Phagosomes were labeled using GFP Mtb and LC3B was labeled using specific antibody. Images were acquired at 60x magnification with an inverted fluorescent microscope using MΦ of a single random healthy donor and were analyzed using confocal microscopy and NIKON NIS element software. Transcriptional (B) and translational (C) level expression of autophagy and phagosome maturation associated genes Atg5, Atg7, Rab7 and Lamp in CD44TA-LIP treated and untreated MΦ at 24 and 72 h post Mtb infection. Mtb burden (at 72 h post infection) in CD44TA-LIP treated (Concurrent treatment) and untreated human MΦ after siRNA mediated knockdown of beclin1 (D) and NOS2 (F) as measured by CFU assay. Production of ROS/ NO (E) and antimicrobial peptides LL-37/HβD2 (G) by CD44TA-LIP treated and untreated MΦ at 24 h post-Mtb infection, as measured through fluorescent probes, and ELISA assays respectively, using a fluorimeter/plate reader. CD44TA-LIP concentration used: 10 μg/106 MΦ. PT: pre-treatment; CT: concurrent treatment. pg = picograms; MOI used for the experiments = 1. Number of MΦ used per replicate: 1x106. h= h. Representative data shown from three independent experiments carried out in duplicate. Bars and error bars represent mean and SD, respectively. *p≤0.05, **p≤0.005, ***p≤0.0005, ****p≤0.0001.
Reactive molecules such as nitric oxide (NO) and reactive oxygen species (ROS), are also known to be critical for intracellular host defense against Mtb (27). Therefore, we next examined the production of NO and ROS in infected MΦ and found significantly increased production NO after CD44TA-LIP treatment (Fig 3E). However, blockade of NO synthesis in MΦ through NOS2 siRNA did not significantly abrogate the CD44TA-LIP mediated killing of Mtb (Fig 3F). These results indicated that although stimulation of CD44 signaling pathway in MΦ increased production of NO, this was not the main factor contributing to Mtb killing. Since antimicrobial peptides (AMPs), are important elements of the innate immune defense of MΦ, we further examined the expression of LL-37 and HβD2 peptide in CD44TA-LIP treated and untreated Mtb infected MΦ. We observed significantly increased production of HβD2 in CD44TA-LIP treated and pretreated MΦ as compared to untreated cells, while levels of LL-37 did not change significantly (Fig 3G). Thus, it is possible that the expression of HβD2 in CD44TA-LIP treated/pretreated MΦ plays an important role in enhanced Mtb killing.
CD44TA-LIP mediated increased killing of Mtb in MΦ is associated with CD44 signaling mediated cellular proliferation and cytoskeleton rearrangement pathways
Although CD44TA-LIP treatment induced autophagy, and increased NO and HβD2 expression in MΦ (Fig 3), the mechanism/s through which it induced killing of intracellular Mtb still needed to be further elucidated. Thus, to identify the components of CD44 signaling pathway that could have contributed to intra- MΦ killing of Mtb, we examined the CD44 receptor mediated signal transduction pathways that are induced after treatment with CD44TA-LIP. Ligand binding to CD44 cell surface receptor is known to trigger a variety of signaling events, including a complex formation between CD44 and co-receptors such as ERM, VEGFR, and TGF-β receptors, and activation of downstream effectors such as Src, c-Met, Akt, MAPK, PP2A, PI3K, ERK1/2, and Ras/Raf/Rac (12,13) (Fig 4A). These signaling events lead to modulation of various downstream effectors that drive cellular proliferation, cellular invasion, cytoskeleton rearrangement, angiogenesis and inflammation. Using specific pharmacological inhibitors, we blocked each downstream signaling pathway and examined its effect on CD44TA-mediated killing of Mtb in MΦ. Inhibition of Akt kinase and PI3K with afuresertib and alpelisib, respectively, did not affect the CD44TA-LIP mediated killing of Mtb in MΦ (Fig 4B). On the other hand, inhibition of B-Raf and GSK3β via GDC-0879 and TWS119, respectively, nullified the antimicrobial effect induced by CD44TA-LIP. These inhibitors did not significantly affect the antimicrobial activity in MΦ that were not treated with CD44TA-LIP. These results indicate that intra- MΦ antitubercular activity induced by CD44TA-LIP is mediated through CD44 signaling dependent cellular proliferation/survival and cytoskeleton rearrangement pathways.
Figure 4: CD44 signal transduction pathways and effect of specific pharmacological inhibitors of its downstream effectors on antimicrobial effect of CD44TA-LIP on survival of Mtb in human MΦ.
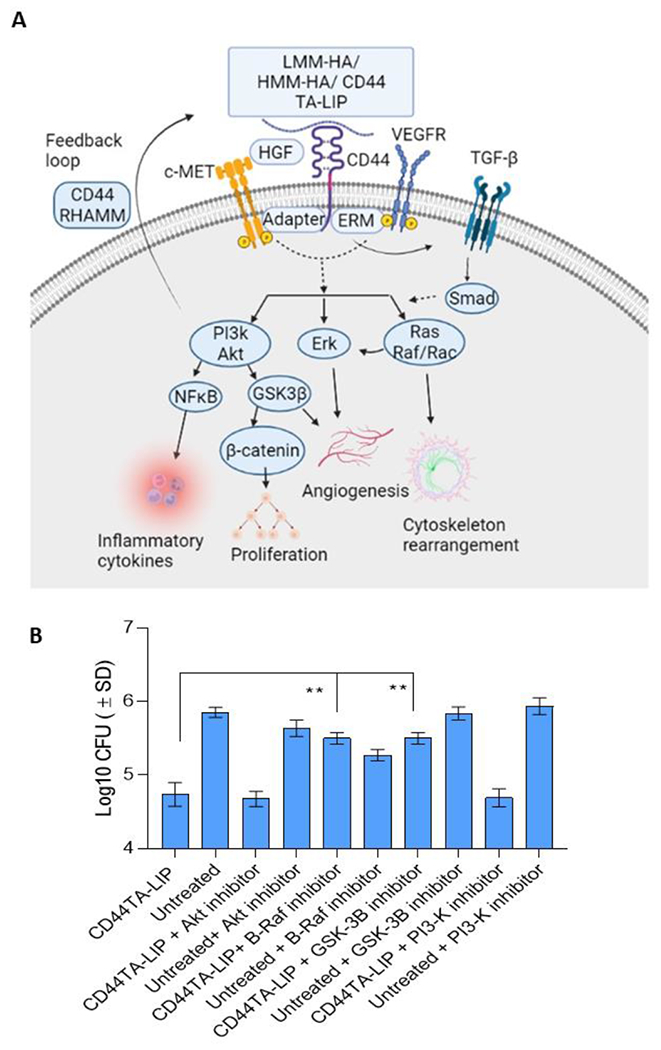
A: Ligand binding to cell surface receptor CD44 triggers a complex formation between CD44 and co-receptors such as c-Met, VEGFR, and TGF-β receptors leading to activation of downstream effectors such as Akt, PI3K, PP2A, ERK1/2, and Ras/Raf/Rac. By inducing these signaling events and downstream effectors, CD44 signaling can modulate various cellular processes including proliferation, invasion, cytoskeleton rearrangement and inflammation. B: Effect of pharmacological inhibitors of Akt, PI3K, GSK3β and B-Raf on Mtb burden in CD44TA-LIP treated (Concurrent treatment) vs untreated human MΦ at 72 h post infection as measured by CFU assay. CD44TA-LIP concentration used: 10 μg/106 MΦ. Respective concentration used for Akt, PI3K, GSK3β and B-Raf inhibitors/106 MΦ= 10 nM, 50 nM, 5 nM and 100 nM. Pharmacological inhibitors of Akt, PI3K, GSK3β and B-Raf were added at the beginning of infection and removed after 24 h post infection. pg= picograms; MOI used for the experiments = 1. Representative data shown from three independent experiments carried out in duplicate. Bars and error bars represent mean and SD, respectively. *p≤0.05, **p≤0.005, ***p≤0.0005, ****p≤0.0001.
CD44TA-LIP boosted the antigen processing and presentation in MΦ to induce more efficient priming of CD4 T cells ex vivo
While CD44 signaling mediated inflammation was not found to be involved in intra- MΦ killing of Mtb (Fig 3B), induction of pro-inflammatory cytokines after CD44TA-LIP treatment in infected MΦ (Fig 2C) indicated that inflammatory pathways could also be involved in generating an immune response against Mtb infection. Since inflammatory cytokines IL-1β, IL-12 and TNF-α, induced by CD44TA-LIP, are also linked to MΦ -mediated adaptive immune response, we further examined if priming of CD4 T cells was affected by MΦ after CD44TA-LIP treatment. Ex vivo antigen presentation assay used here has been described in details in our recent work (28). Briefly, the assay quantitates the antigen presentation capabilities of MΦ based on the IL-2 levels secreted by hybridoma T cells F9A6 upon co-culture with Mtb infected human MΦ. Upon recognition of one of the specific epitopes of Mtb Ag85B, when presented through HLA-DR1/4, F9A6 cells start secreting IL-2 which can be used as a quantitative marker of total antigen presentation capabilities of MΦ. An increased production of IL-2 by Mtb Ag85B specific hybridoma T cells (F9A6) was observed upon overlay to CD44TA-LIP treated MΦ as compared to untreated MΦ 24 h after the infection (Fig 5A). This was observed during pre-treatment as well as concurrent treatment of MΦ with CD44TA-LIP during Mtb infection indicating that CD44 signaling induced enhanced the MHC-II mediated antigen processing and presentation capabilities of MΦ. To further understand the CD44 signaling pathway/s that could have contributed to a more efficient priming of CD4 T cells, each signaling pathway was blocked downstream via specific pharmacological inhibitor as mentioned above (Fig 4A) and their effect on antigen presentation capabilities of MΦ was examined using the ex vivo antigen presentation assay. Inhibition of inflammatory pathways of CD44 signaling via pharmacological inhibitors of Akt and PI3K significantly reduced the CD44TA-LIP mediated IL-2 secretion by CD4 T cells (F9A6) upon overlay to Mtb infected MΦ treated with CD44TA-LIP (Fig 5B). Interestingly, pharmacological inhibition of cellular proliferation and cytoskeleton rearrangement pathways that are induced by CD44TA-LIP, did not significantly alter the IL-2 levels produced by CD4 T cells upon overlay to CD44TA-LIP treated and Mtb infected MΦ. On the other hand, PI3K and Akt inhibitors significantly reduced the IL-2 levels produced by CD4 T cells upon overlay to CD44TA-LIP treated and Mtb infected MΦ. None of the specific pharmacological inhibitors affected the antigen presentation capabilities of untreated MΦ. These results, thus, indicate that CD44TA-LIP treatment (preventative and concurrent) enhanced the antigen presentation abilities of MΦ via induction of PI3K and Akt dependent inflammatory pathways.
Figure 5: Effect of CD44 signaling on antigen presentation capabilities of CD44TA-LIP treated vs untreated human MΦ during Mtb infection.
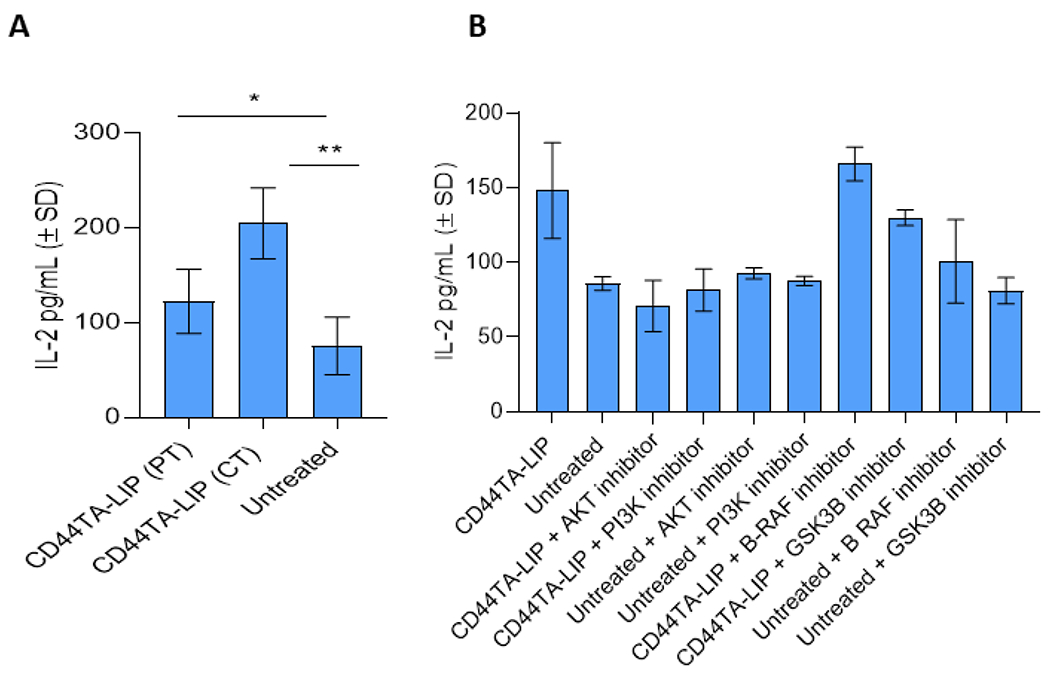
A: Secreted levels of IL-2 by Mtb Ag85B-specific T cells (F9A6) during co-culture with CD44TA-LIP treated and untreated MΦ at 72 h post-infection. B: Effect of pharmacological inhibitors of Akt, PI3K, GSK3β and B-Raf on antigen presentation capabilities of CD44TA-LIP treated (Concurrent treatment) vs untreated human MΦ at 72 h post infection as measured by CFU assay. CD44TA-LIP concentration used: 10 μg/106 MΦ. Respective concentration used for Akt, PI3K, GSK3β and B-Raf inhibitors /106 MΦ= 10 nM, 50 nM, 5 nM and 100 nM. Pharmacological inhibitors of Akt, PI3K, GSK3β and B-Raf were added at the beginning of infection and removed after 24 h post infection. pg= picograms; MOI used for the experiments = 1. Representative data shown from three independent experiments carried out in duplicate. Bars and error bars represent mean and SD, respectively. *p≤0.05, **p≤0.005, ***p≤0.0005, ****p≤0.0001.
CD44TA-LIP exerted therapeutic effect against experimental aerosol-induced tuberculosis in mice through modulation of T cell functions
In our previous studies we found that intravenously injected CD44TA-SMP accumulated in the TB infected MΦs in the lungs, resulting in bacterial killing in vivo in an experimental mouse model (18). Since, as compared to CD44TA-SMP, CD44TA-LIP can be administered non-invasively and were found more effective in generating protective immune response against Mtb infection in vitro (Fig 1), we further evaluated the therapeutic effect of CD44TA-LIP in a C57Bl/6 mouse based experimental model of TB. Both prophylactic (pre-treatment) as well as therapeutic effect of CD44TA-LIP were evaluated using pulmonary intranasal administration of CD44TA-LIP. Bacterial burden within lung and spleen was analyzed and correlated with the T cell functional phenotypes to evaluate the effect of CD44TA-LIP and understand its possible mechanism of action. With the NIH mouse TB vaccine evaluation model using two different treatment regimens (as described in methods), in comparison to untreated mice, treatment with CD44TA-LIP post Mtb infection reduced the bacterial burden in lungs and spleens of mice by more than 1 order of magnitude when measured at 30 days post infection (Fig 6A and 6B). However, pre-treatment with CD44TA-LIP did not have any significant effect on bacterial burden in lungs and spleens of infected mice. These data, thus, suggested that therapeutic regimen of CD44TA-LIP could be more efficacious than its prophylactic regimen in vivo. Tissue-resident memory (TRM) T cells have been implicated in protective immune responses against Mtb infection in recent studies (29) and, therefore, we first examined the CD4 and CD8 TRM population in lungs and spleens of mice of CD44TA-LIP treated vs untreated groups. In lungs, frequencies of CD4 TRMs were not found to be significantly different in mice treated with CD44TA-LIP post- Mtb infection when compared with untreated mice (Fig 6C). However, higher frequencies of CD8 TRMs were found in the lungs of mice treated with CD44TA-LIP post Mtb infection. Mice treated with CD44TA-LIP prior to Mtb infection, demonstrated increased percentage of CD4 as well as CD8 TRMs in lungs. In spleens on the other hand, mice treated with CD44TA-LIP post Mtb infection, exhibited significantly higher number of CD4 as well as CD8 TRMs as compared to untreated mice (Fig 7A). Mice treated with CD44TA-LIP prior to Mtb infection, also demonstrated increased frequencies of CD4 as well as CD8 TRMs, though to a relatively lower degree as compared to mice treated with CD44TA-LIP post Mtb infection. Analysis of T cells in spleens and lungs via flow cytometry for the presence of IFN-γ +/IL-2+ CD4 and CD8 T cells was also done in the above-mentioned treatment regimens to find correlates of protection with multifunctional T cells. In lungs, mice pre-treated with CD44TA-LIP, demonstrated a higher % of IFN-γ+ CD4 T cells as compared to untreated mice (Fig 6D). In spleens, however, mice treated post infection exhibited an increase in IFN-γ + as well as IL-2+ CD4 T cells (Fig 7B, gating information is provided in Supplementary SF3). Analysis of CD8 T cells, on the other hand, revealed an increased number of IFN-γ+ as well as IL-2+ CD8 T cells in both lungs and spleens of treated mice as compared to untreated mice (Fig 6E and 7C). Interestingly, in mice treated with CD44TA-LIP prior to Mtb infection, there was no difference in IFN-γ+ and IL-2+ CD8 T cells in both lungs and spleens when compared with untreated mice, indicating that pre-treatment with CD44TA-LIP may not induce long lasting effect on multifunctional CD8 T cells (Fig 6E and 7C).
Figure 6: Effect of CD44TA-LIP on bacterial burden and various T cell populations in vivo during experimental murine tuberculosis.
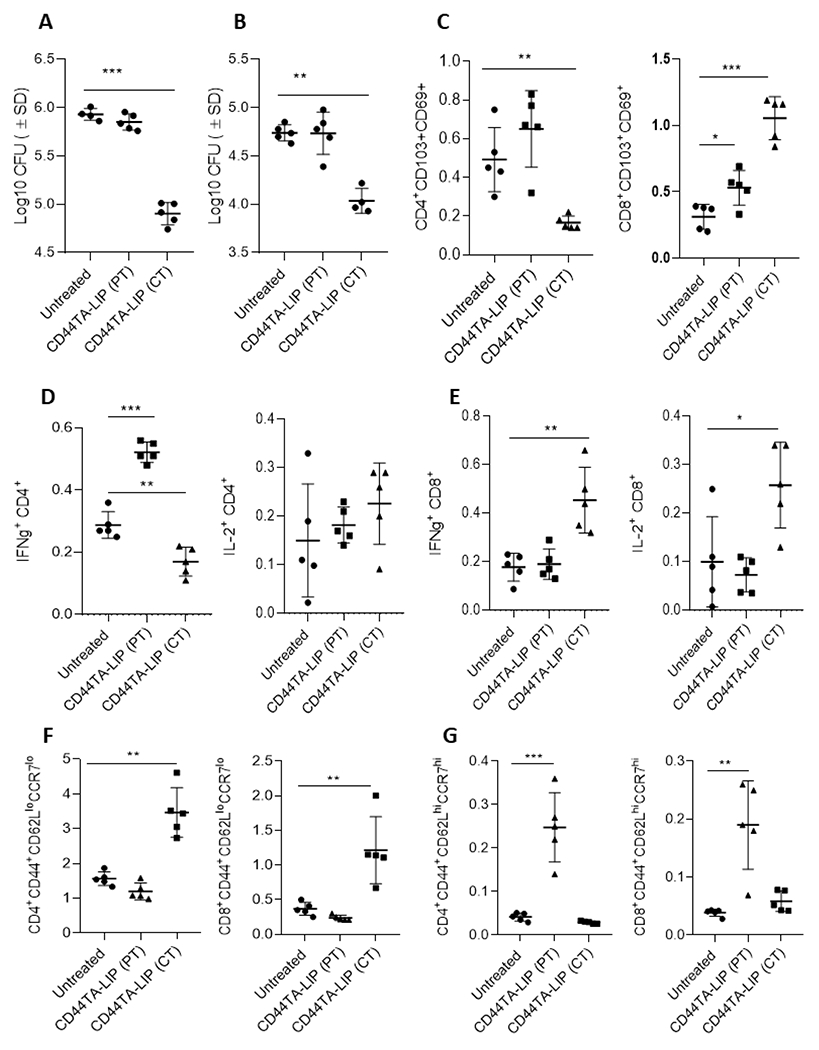
Mtb CFU in Lungs (A) and spleens (B) of mice treated intranasally with CD44TA-LIP. Immune cell analysis was done by flow cytometry. Frequencies of resident memory CD4 and CD8 T cells (CD103+CD69+) in lungs (C) of untreated, and CD44TA-LIP treated mice. Percentages of IFN-γ and IL-2 expressing CD4 (D) and CD8 (E) T cells in lungs of untreated, and CD44TA-LIP treated mice. Frequencies of effector memory (F) and central memory (G) CD4 and CD8 T cells in lungs of untreated, and CD44TA-LIP treated mice. Treatment with CD44TA-LIP prior to infection is depicted as CD44TA-LIP (PT) whereas treatment with CD44TA-LIP after infection is depicted as CD44TA-LIP (CT) in all panels. Flow panels show the % of live leukocytes. CD44+CD62LloCCR7lo= Effector memory population; CD44+CD62LhiCCR7hi= Central memory population. *p < 0.05, **p < 0.001, ***p < 0.0001, ***p < 0.0001; t-test.
Figure 7: Effect of CD44TA-LIP treatment on various T cell populations of mice during tuberculosis infection.
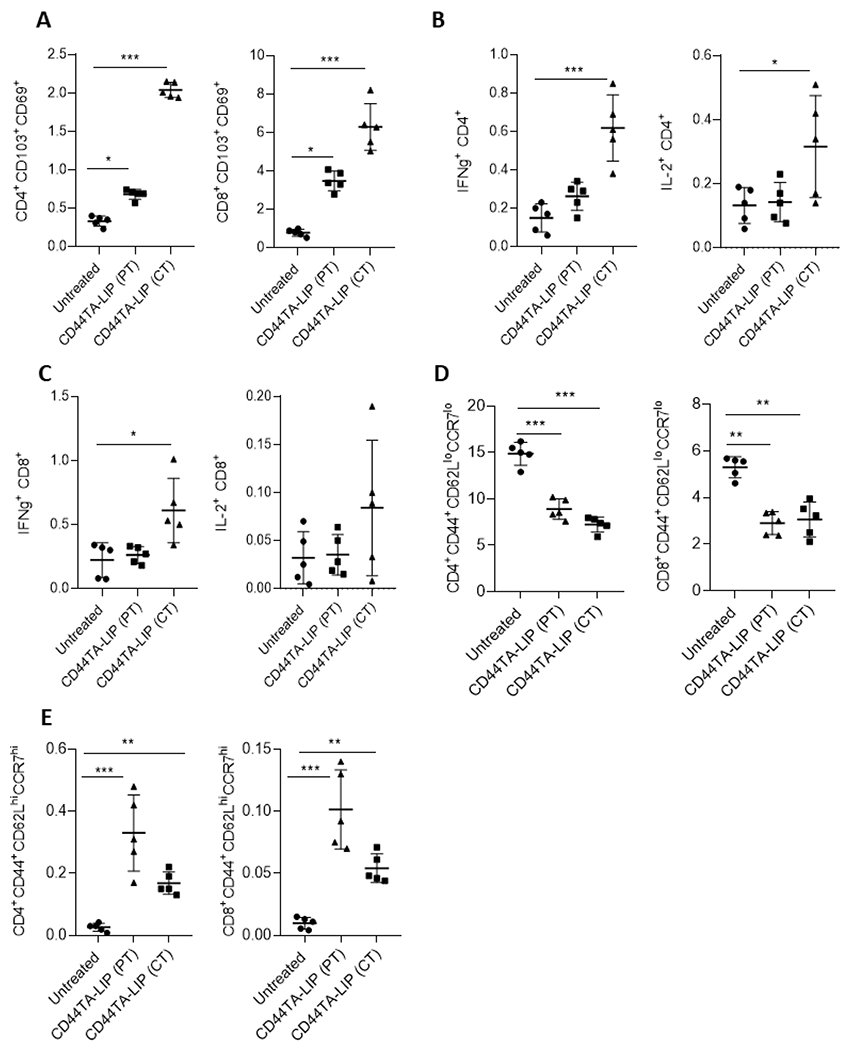
Frequencies of resident memory CD4 and CD8 T cells (CD103+CD69+) in spleens (A) of untreated, and CD44TA-LIP treated mice. Percentages of IFN-γ and IL-2 expressing CD4 (B) and CD8 (C) T cells in spleens of untreated, and CD44TA-LIP treated mice. Frequencies of effector memory (D) and central memory (E) CD4 and CD8 T cells in spleens of untreated, and CD44TA-LIP treated mice. Treatment with CD44TA-LIP prior to infection is depicted as CD44TA-LIP (PT) whereas treatment with CD44TA-LIP after infection is depicted as CD44TA-LIP (CT) in all panels. CD44+CD62LloCCR7lo= Effector memory population; CD44+CD62LhiCCR7hi= Central memory population. *p < 0.05, **p < 0.001, ***p < 0.0001, ***p < 0.0001; t-test.
We further examined if treatment with CD44TA-LIP affects generation of memory T cell. In lungs, CD4 and CD8 effector memory T cell populations increased in mice treated with CD44TA-LIP post infection, but not prior to infection (Fig 6F). On the other hand, both CD4 as well CD8 effector memory T cell population decreased in spleens of mice treated with CD44TA-LIP prior to infection or after infection (Fig 7D). On the other hand, central memory CD4 and CD8 T cell population were found to be significantly increased in lungs of mice pretreated with CD44TA-LIP as compared to untreated mice (Fig 6G). However, in spleens, mice treated with CD44TA-LIP post Mtb infection exhibited significant increase in both central memory CD4 and CD8 T cell population, as compared to untreated mice (Fig 7E). Since bacterial burden in both lungs and spleen of mice treated with CD44TA-LIP prior to infection did not decrease, these results suggest that effector memory T cell response rather than central memory T cell population could have contributed to the reduction of Mtb burden in mice. Nonetheless, analysis of various T cell population as a whole suggests that therapeutic effect of CD44TA- LIP promoted the expansion of TRMs, IFN-γ+ and IL-2+ multifunctional T cells as well as effector and central memory T cells both locally in lungs and systemically in spleens, which could have all contributed in generating protective immune response against Mtb infection in mice.
Analysis of CD44TA-LIP biodistribution and liver functions
We have assessed the biodistribution of CD44TA-LIP in lung inflammation model. CD44TA-LIP were tagged with Cy 5.5 labeled phospholipid. The data presented in Fig. 8 show that the liposomes concentrated in the lungs of the animals at 24h post-treatment. No signal was observed in hearts or spleens of the animals. Cy 5.5 signal in the kidneys increased between 2 and 4 h following CD44TA-LIP administration, indicating possible detachment of the fluorescent probe and clearance through renal route. A signal in the liver also increased with time, which can be linked to the clearance of the Cy 5.5 conjugated phospholipid.
Figure 8: Organ distribution of CD44TA-LIP in lung inflammation model.
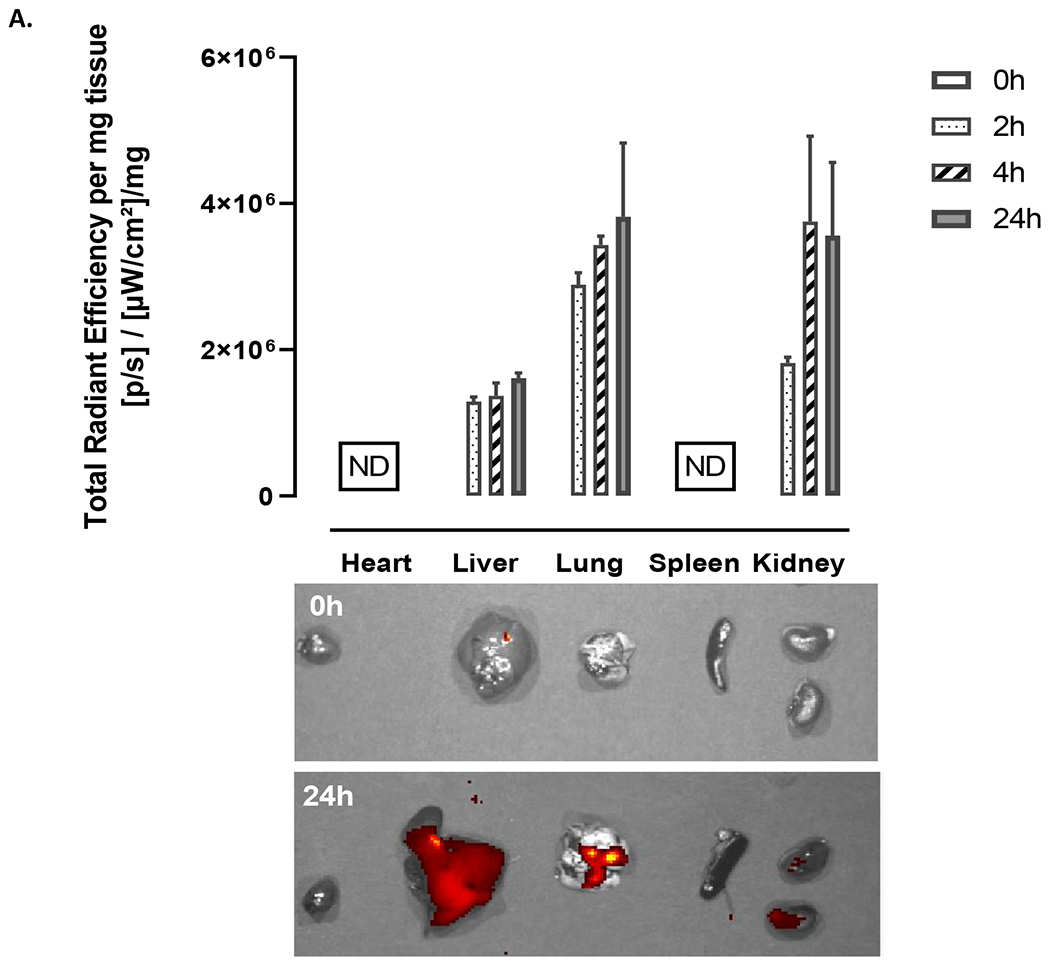
CD44TA-LIP were labeled with Cy 5.5. At pre-determined times, animals were sacrificed and IVIS images were taken at 0 (prior to administration), 2, 4 and 24h. (A) The signal (total radiant efficiency) was assessed using IVIS Spectrum Imaging System (Perkin Elmer, MA, USA) at excitation and emissions wavelengths of 675nm and 720nm for Cy 5.5 fluorescence intensity detection. The data was quantified using Living Image® Software and normalized to the organ weight.
We have further analyzed the cells that were associated with CD44TA-LIP in the lungs (Fig. 9). The data show that Cy 5.5 labeled CD44TA-LIP were associated mainly with CD11b cells and CD 80 positive cells. At 24h 11.33% CD11b and 0.62% CD80 positive cells were associated with Cy 5.5 signal. Since CD11b and CD80 markers are expressed primarily by lung interstitial MΦ and dendritic cells in mice, the data indicate that these were the major cell population associated with CD44TA-LIP.
Figure 9:
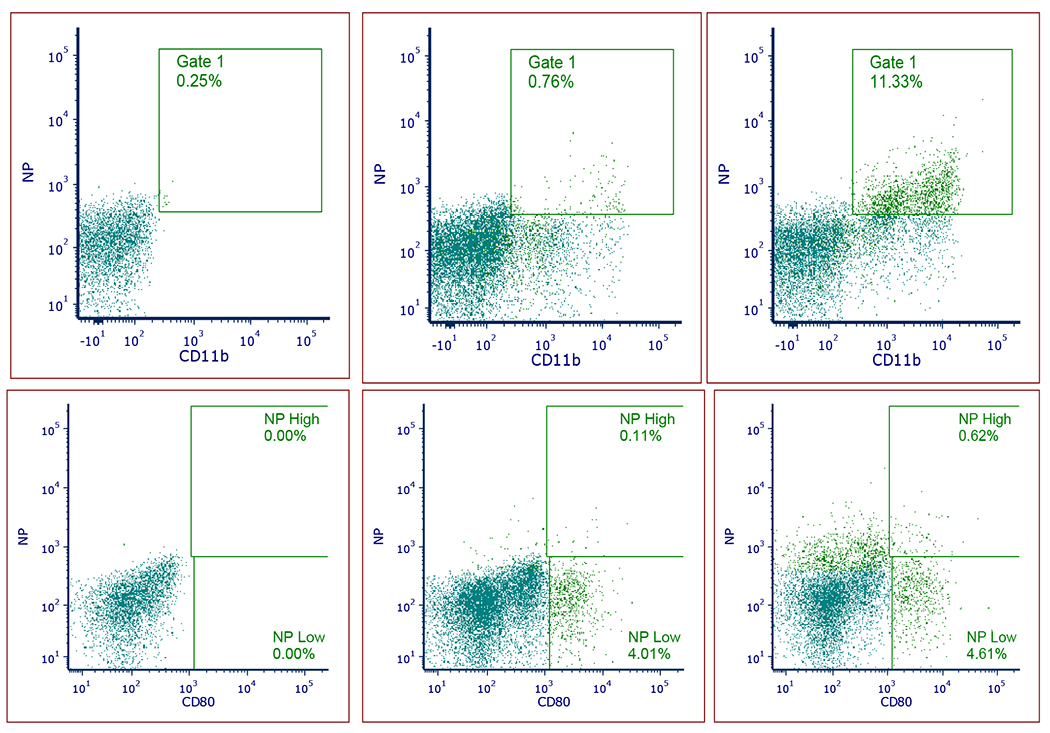
Flow cytometry analysis of CD11b (A, B, C) and CD80 (D, E. F) positive cell populations associated with CD44TA-LIP (NP on the scatters). No treatment control (A and D); 2h (B and E) and 24h (C and D). CD44TA-LIP were Cy 5.5 labeled and the samples were stained with CD11b-Pacific Blue (117322, Biolegend) and CD80-PE (104708, Biolegend). Samples were analyzed using a Becton-Dickenson FACS Fortessa (5 laser 17 channel) using Diva 9.1. Gates were based on single and unstained controls.
The liver function was tested using LDH assay (Fig. 10). There were no significant changes in the LDH levels in the liver 2, 4 and 24h following the administration of the CD44TA-LIP as compared to untreated (PBS) control. Additionally, no changes in the weights of the animals were observed for up to one week following the administration of CD44TA-LIP.
Figure 10:
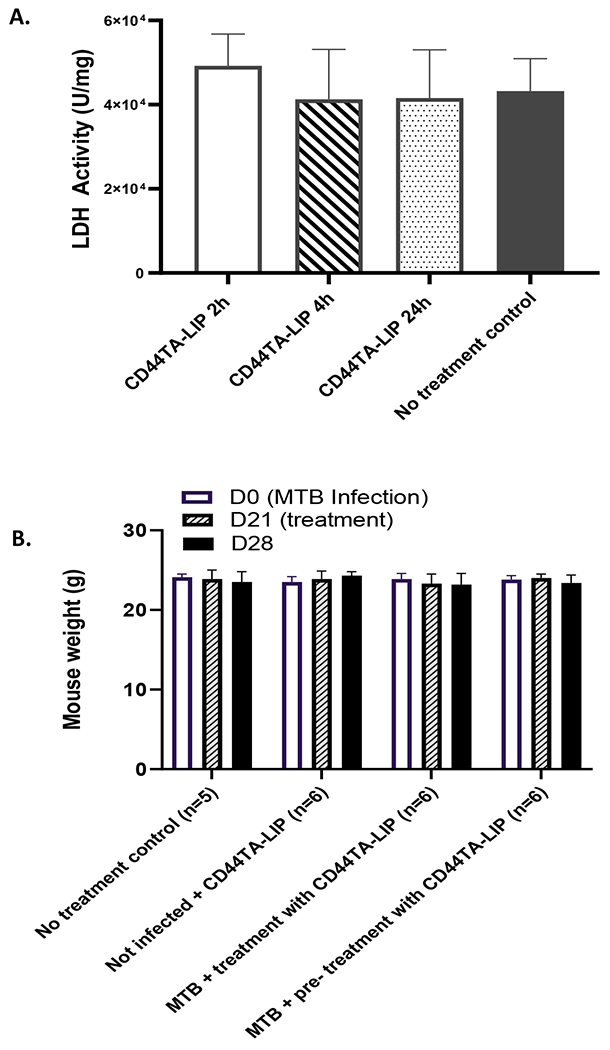
Safety of CD44TA-LIP: (A) LDH levels in the liver 2, 4, and 24h following CD44TA-LIP administration. (B) Weights of the mice in therapy experiments. No changes were observed up to 7 days following CD44TA-LIP administration.
DISCUSSION
During infection, CD44 receptor-mediated signaling plays important role in host defense against invading pathogens, specifically via activation and migration of lymphocytes (30). Apart from promoting activation, homing, rolling, and extravasation of lymphocytes into inflammatory/infection sites, CD44 signaling is also known to modulate MΦ mediated host defenses against various infections (31,32). Role of CD44 receptor mediated signaling in TB immunopathogenesis remains undefined at large, though some evidence suggests that CD44 signaling assists in mediating host resistance against mycobacterial infection. CD44 surface receptors on MΦ can directly bind to Mtb bacilli, facilitating the cellular uptake of the pathogen (11). CD44−/− mice exhibited increased Mtb growth in lungs and liver during pulmonary tuberculosis, clearly indicating the protective role of CD44 receptor against TB (11). However, the cell population/s that contributed to CD44 mediated protective immunity against TB remained to be determined. The results presented in this study demonstrate that MΦ could be contributing to host defense against TB in a CD44 receptor dependent manner. Time dependent increased killing of Mtb bacilli in MΦ after treatment with CD44TA-LIP clearly indicates that activation of CD44 signaling enhances the intracellular antimicrobial defense mechanisms of MΦ (Fig 2A). While previous investigations suggest that CD44 receptors are important for recruitment of MΦ to the site of infection and phagocytosis of Mtb bacilli (11), our data demonstrates that CD44 signaling is critical for intracellular killing of the pathogen (Fig 2A and 2B). Although, binding of Mtb to CD44 during phagocytosis may activate CD44 receptor mediated downstream signaling, many other receptors such as mannose receptors, scavenger receptors, complement receptors, Fcγ receptors are also involved in uptake of the pathogen (33,34), which may lead to less than optimal activation of CD44 signaling mediated cellular defense pathways in MΦ. Further enhancement of CD44 signaling through CD44 targeting aptamers and subsequent killing of intra-macrophage Mtb indicated that CD44 receptor could be a viable target of TB HDT. Both concurrent treatment and pretreatment with CD44TA-LIP resulted in killing of Mtb in MΦ (Fig 2B), indicating the potential of CD44TA-LIP as prophylactic and post infection therapeutic options.
Although different variants of CD44 receptor are distributed spatially on the cell surface, hyaluronic acid (HA) is the main natural ligand for these isoforms (12,35). The size of the HA is known to be critical with low molecular mass HA (LMW-HA) having more specific effect on inducing pro-inflammatory immune response against pathogens (36). Thioaptamers used in this study mimic LMW-HA and their ability to induce production of proinflammatory cytokines TNF-α, IL-1β and IL-12 confirmed that their effect was very similar to LMW-HA ligands (Fig 2C).
We believe that since MΦ are professional phagocytes, the presence of nanoparticles that are engulfed by Mtb infected MΦ enables activation of intracellular mechanisms that include induction of NO and autophagy along with production of antimicrobial peptides (Fig 3). CD44 receptors interaction with its ligand leads to intracellular recruitment of host autophagy proteins that help in forming phagosomes after the internalization of Mtb (37). Thus, induction of autophagy by CD44TA-LIP is likely due to a combinatorial effect of CD44 receptor ligation and internalization of Mtb by MΦ. On the other hand, ligation of LMW hyaluronan fragments with CD44 have been shown to upregulate the expression of iNOS through nuclear factor Kappa B dependent mechanisms (38). Nonetheless, induction of NO by CD44TA-LIP did not contribute to killing of intra- MΦ bacilli indicating that pro-inflammatory signaling alone may not result in antimicrobial activity of MΦ towards Mtb (Fig 3F). Induction of HβD2 by CD44TA-LIP in Mtb infected MΦ (Fig 3G) suggested CD44 receptor activation dependent antimicrobial response, though it was not clear if it also contributed to the killing of the intracellular organism. Previous studies have demonstrated the association of upregulation of HβD2 and inhibition of intracellular growth of Mtb in human MΦ suggesting that HβD2 could be critical for containment of infection (39). In the intestinal epithelium, HA dependent CD44 signaling is known to induce HβD2 production which enhances antimicrobial defense against invading enteric pathogens during early infancy (40). Similar behavior was also reported in skin epithelium (41). It has also been demonstrated that LMW-HA can strongly upregulate HβD2 expression as compared to high molecular weight hyaluronan (HMW-HA) (42). Interestingly, HA-CD44 signaling mediated up-regulation of HβD2 was found to be co-dependent on toll-like receptor 2 and 4 (TLR4) (41). It is, thus, possible that CD44TA-LIP may require co-activation of TLR2/4 along with CD44 to induce HβD2 expression and antimicrobial activity against Mtb infection. Very low levels of HβD2 production by uninfected cells (Fig 3G) indicated that TLR4 co-activation may be required for CD44TA-LIP dependent HβD2 production.
Interestingly, CD44 receptor mediated signaling induced by CD44TA-LIP associated with cytoskeleton rearrangement and cellular proliferation, was critical for the antimicrobial activity against Mtb (Fig 4A and 4B). B-Raf, which regulates actin cytoskeleton to control cellular movement, morphology and growth in a CD44-dependent manner, was found to be a restriction factor for intracellular Mtb survival in MΦ (Fig 4B). Similarly, cellular proliferation pathways mediated by GSK3β dependent CD44 signaling were also vital for intracellular killing of Mtb. Local proliferation of interstitial MΦ has been observed during Mtb infection though it remains to be determined whether it also promotes their antimicrobial immune effectors pathways(43). In this context, the mechanism through which CD44 signaling dependent cytoskeleton and cellular proliferation pathways affect intracellular survival of Mtb will be worth exploring in future studies to identify new targets for HDT for TB.
Similarly, CD44 dependent inflammatory pathways, though did not contribute to the innate antimicrobial effector functions of MΦ, were critical for their antigen processing and presentation (Fig 5), suggesting that CD44 signaling may also be important for augmentation of adaptive immunity against intracellular pathogens. Both Akt and PI3 kinase dependent pathways of MΦ inflammatory immune response are known to induce maturation of pathogen containing phagosomes leading to a better processing and presentation of antigen to effectively prime T cells during infection (44). Since Mtb infection is known to suppress the PI3K-Akt-mTOR signaling pathway to reduce the optimal activation of T cell dependent immunity (45), enhancement of this signaling by CD44 dependent signaling through CD44TA-LIP may help in overcoming this evasion mechanism of the pathogen. More importantly, these findings were translated in vivo reducing bacterial burden in lungs and spleens of Mtb infected mice treated with CD44TA-LIP (Fig 6 and 7). Remarkably, treatment with CD44TA-LIP enhanced the % of TRMs and IFN-γ +/IL-2+ CD4/CD8 T cells known to be associated with protective immunity against TB (Fig 6 and 7). Although more efficient presentation of antigen in MΦ is known to enhance the priming of CD4T cells which could lead to induction of IFN- γ producing helper T cells, it is possible that CD44TA-LIP could directly affect the CD4 T cells as suggested by previous studies (46,47). CD44 is a known prominent activation marker activated helper T cells which differentiates them from their naïve counterparts. Activation of CD44 receptors plays a role in enhancing T cell receptor signaling and thereby production of IFN- γ and other Th1 cytokines. CD44 receptor on T cells has indeed been demonstrated to participate in the regulation of Th1 differentiation and in that context, it is likely that Th1 bias of T cells could have been enhanced due to the direct interaction of T cell CD44 receptor with CD44TA-LIP (48). Local resident memory (TRM) as well as effector/central memory T cells increased in lungs and spleens of mice treated with CD44TA-LIP, though differences were observed in pre vs post infection treatment groups (Fig 6 and 7). Specifically, correlating with reduction in bacterial burden, mice treated post infection demonstrated significantly increased number of effector memory T cells as well as resident memory T cells. Increased % of central memory T cell population though did not correlate with reduction in bacterial burden in lungs and spleens suggesting that central memory T cells may not be directly involved in CD44TA-LIP mediated killing of Mtb in mice.
In the current work, we have used a C57Bl/6 mouse based experimental model of tuberculosis to test the efficacy of CD44TA-LIP. This mouse model is widely used and is considered to be the most suitable model for testing candidate vaccines and HDT as it mimics the immune response generated and the protection provided against M. tuberculosis aerosol challenge (49). Measurement of survival of animals is not feasible in this animal model due to slow progression of disease in C57Bl/6 mice. This model relies on measurement of specific cell mediated immune response and corresponding bacterial burden in various infected organs to determine the measurement of protection generated by a candidate vaccine or therapeutic molecule. Multifunctional CD4 and CD8 T cells that produce IL-2 and IFN- γ have been shown to correlate with the protection provided by various candidate vaccines in earlier studies and hence used as a measure of efficacy of therapeutic molecules(50,51). As compared to untreated mice, we found increased number of resident memory as well as IFN-γ +/IL-2+ CD4/CD8 T cells in lungs and spleens of the mice that were treated with CD44TA-LIP hence indicating their efficacy (Fig 6 and 7). Although other mouse-based animal models of TB have been reported where survival studies can be carried out, they do not resemble the pathology, immune response and the disease manifestation as it occurs during human TB and hence are not suitable to determine the efficacy of prophylactic or therapeutic vaccines.
In summary, the identified mechanisms through which CD44TA-LIP promoted both innate and adaptive immunity show the important role that CD44 receptor can potentially have in regulating the MΦ and T cell functions for protective immune responses against TB. Evidences present here also emphasize the potential of CD44TA-LIP as a therapeutic vaccine against TB.
MATERIALS AND METHODS
CD44TA-LIP design and characterization
CD44TA-LIP were prepared using a microfluidic device NanoAssemblr (Precision NanoSystems). This instrument has been optimized and proven to be able to improve LIP production, enabling reproducibility and scalability, as well as ability to work in accordance to GLP as well as GMP practice. The LIP were prepared by mixing ethanol (organic solvent) containing 12.5 μmole S100 soybean phosphatidylcholine (Lipoid, Ludwigshafen, Germany), 2.0 μmole cholesterol (Sigma, St. Louis, MO), and 0.4 μmole 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[carboxy(polyethylene glycol)-2000] (DSPE-PEG(2000) Carboxylic Acid) (Avanti Polar Lipids, Alabaster, AL). One volume of the solvent was mixed with one volume of 1X PBS (Thermo Fisher Scientific, USA) using dual syringe pump (model S200, kD Scientific, Holliston, MA) to drive the solutions through the micro-mixer at a combined flow rate of 4mL/min. The produced LIP centrifugal filtered (100 kDa Amicon Ultra 0.5mL, Sigma, St. Louis, MO) at 14k x g for 5mins to remove ethanol. The resulting LIP were analyzed for size and zeta potential by dynamic light scattering (Malvern Zetasizer, Malvern Instruments, Malvern, UK) measurement. The conjugation of LIP with CD44TA (IDT, USA) was conducted following the LIP production using 0.4-10 μmole EDC (1-ethyl-3-[3-dimethylaminopropyl]carbodiimide) (Life technologies, Carlsbad, CA) and 15 μmole NHS (N-hydroxysuccinimide) (Life technologies, Carlsbad, CA) in 0.1M MES buffer (PolyLink Protein Coupling Kit, Polysciences, Warrington, PA) as catalyst. 5.0 nM CD44TA (IDT, Coralville, IA) was dissolved in the catalyst mixture and LIP were added to the solution and let to react in rotatory at room temperature (RT) for at least 15 min. After conjugation, the LIP were centrifuge- filtered (100 kDa Amicon Ultra 0.5mL, Sigma, St. Louis, MO) at 14k x g for 5mins to remove MES buffer. The CD44TA-LIP were resuspended in PBS under aseptic conditions and analyzed for size and zeta potential by dynamic light scattering (DLS, Malvern Zetasizer, Malvern Instruments, Malvern, UK), for concentration (NanoSight, Malvern Instruments, UK), and visualized by Atomic Force Microscopy (AFM, Bio-Catalyst AFM, Bruker).
Monocytes/ MΦ from healthy human subjects
All blood samples were collected per approved institutional review board protocols. CD14 magnetic beads (Miltenyi Inc., USA) were used to purify monocytes from PBMCs that were plated in 6 or 24 tissue culture well plates at a density of 5x106 and 1x106 cells per well, respectively. Eight-well slide chambers or coverslips received 104 cells per chamber for confocal/immunofluorescent imaging studies. CD14 bead purified monocytes were grown in Iscove’s medium (IMDM) with 10% fetal bovine serum (FBS) and 10 μg/mL penicillin and gentamycin for 24 h and then plated in antibiotics-free medium until 7 days for before differentiation into MΦ.
Infection and Colony forming unit (CFU) counts of Mtb in human MΦ
MΦ were plated at a density of 106 per well in 24 well adherent plates. Mtb H37Rv (ATCC 27294), was grown in Middlebrook 7H9 broth supplemented with 0.05% (v/v) Tween 80 and Middlebrook AODC enrichment (Difco, Becton Dickinson) to mid-log phase (OD 600 nm 0.6±0.8). Mid log phase cultures of Mtb suspensions were sonicated at 4 watts for 60 seconds using a sonicator to prepare a uniform single cell suspension, diluted in serum-free medium for addition to MΦ at a multiplicity of infection (MOI) of 1 in 24 well plates. After a 4-hr. mixing, cells were washed and added with CD44TA-LIP/CD44TA-SNP at a concentration of 10μg/106 MΦ and then further incubated for determination of CFU at different time post infection. In the pre-treatment protocol, MΦ were treated with CD44TA-LIP for 24 hrs., at the same concentration and incubated for 24 hr., followed by washing and infection (MOI=1) with Mtb for 4 hr. After infection, MΦ were washed to remove extracellular Mtb and incubated further for determination of CFUs at different time post infection. An untreated set of MΦ was infected with Mtb as a control. Medium was replaced every alternate day. At different time points post infection Mtb counts within MΦ were determined by lysing the cells with 0.05% SDS and plating cell lysates at tenfold dilutions on 7H11 agar plates and reading the CFU after incubation at 37° C for 21 days. Mtb counts were expressed as log10 CFU counts averaged for triplicate wells of MΦ per group or combination from 2 independent experiments. P value for differences in CFU between different groups was determined using 1-way ANOVA using Tukey’s posttest.
Cytokine and LL-37/HβD2 ELISA assays
Cell supernatants were tested for cytokines using sandwich ELISA kits (Biolegend and R&D systems, USA) for various cytokines (TNF-α, IL-1β, IL-4, IL-6, IL-10 and IL-12) secreted by CD44TA-LIP treated and untreated MΦ after infection with Mtb. For the quantification of Cathilicidin/HβD2 through ELISA (Novus Biologicals), cell lysates of GM-CSF competent and GM-CSF depleted MΦ after infection with Mtb were used.
Immunofluorescence microscopy for localization and labeling of LC3B and Mtb containing phagosomes
Naive MΦ were infected using GFP expressing Mtb (MOI=1) for 4 h, washed and treated with either CD44TA-LIP or left untreated and incubated until 18 h post infection. The cells were then washed, plated onto coverslips, fixed in 3.7% paraformaldehyde, permeabilized using tween80-digitonin--BSA buffer. A specific mAb to LC3 (Cell signaling #3868) was used to stain autophagosomes using a Texas red conjugated secondary antibody (ThermoFisher Scientific, T-2767). IF images were acquired using a NIS element deconvolution software and N90 Nikon microscope. Colocalization was determined by scoring the localization of LC3B and GFP Mtb phagosomes of 25 MΦ in triplicate chambers per treatment group.
Quantitative PCR (qPCR) analysis
Total RNA from CD4TA-LIP treated/ untreated MΦ was extracted using RNAeasy mini kit (Qiagen, Germany). RNA concentration and purity ratios (OD260/280, OD260/230) were measured using the NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, USA). cDNA synthesis was performed on a CFX96 Real-Time PCR System (BioRad, USA) using the 2X OneStep qRT-PCR Mastermix Kit (Applied Biosystems, USA) according to manufacturer’s instruction. Quantitative PCR (qPCR) was performed using SYBR green probe and gene specific primers (Supplementary Table ST1). Threshold cycle numbers were transformed to ΔΔCt values, and the results were expressed relative to the reference gene, GAPDH. Gene expression data was performed using GraphPad Prism ver. 6.0 suite (GraphPad Software). Student’s t-test was used for mean comparison between GM-CSF competent and depleted MΦ. Significance was set at the 0.05 level.
Western blot analysis
Six-well tissue culture plates were seeded with MΦ. They were infected with Mtb for 4 h at MOI of 1 and then washed three times with PBS and re-plated in the medium. At different time points post infection/treatment with CD44TA-LIP, MΦ were washed three times with 1x PBS, and 50 μL RIPA buffer containing APM (anti-protease mix) was added to each well and incubated for 15 minutes. Lysates were then collected, and protein quantification (Bradford assay, Pierce Coomassie Plus cat no. 23238; Thermo Fisher Scientific) was performed. The quantitative Wes capillary immunoassay was used for the Western blot, in which the lysates were separated and detected using Wes separation capillary cartridge 12-230 kDa along with Wes Anti-Rabbit Detection Module (Simple Western system and Compass Software, Protein Simple). In brief, glass microcapillaries were loaded with stacking and separation matrices followed by sample loading. During capillary electrophoresis, proteins were separated by size and then immobilized to the capillary wall (28). Samples were loaded at 1 mg/mL dilution and the primary rabbit antibody ATG 5, ATG7, Rab7 and Lamp1 were used at a dilution of 1:50 (ATG5: cat no.2360;cell signaling; ATG7: cat no.8558; Rab7: cat no.9367; Lamp1: cat no.9091) and β-actin (Rabbit monoclonal, Sigma-Aldrich # SAB5600204) were used at 1:50 dilution. Data were analyzed with the Compass software (version 2.6.7). The area under the curve (AUC), which represents the signal intensity of the chemiluminescent reaction was analyzed for all the antibodies and β-actin. Values given for protein expression were normalized to β-actin. Quantitation of protein levels (area under each peak; arbitrary units [A.U.]) were performed using the Compass software (version 2.6.7).
Nitric Oxide (NO) and Reactive Oxygen Species (ROS) Estimation
MΦ were plated in 96-well plates at a density of 1x104 cells per well in triplicate and treated with CD44TA-LIP, followed by infection with Mtb (MOI=1). Cells were then treated with fluorescent probes for the quantification of NO using diaminofluorescein diacetate (DAF-2 DA), per manufacturer’s instructions (Enzo Life Sciences, USA); 2’,7’-dichlorodihydrofluorescein diacetate (DCFDA) was used to detect ROS. Quantification of NO/ROS release was determined by calculating the fluorescence emitted over time using an ex485 nm/em515 nm and plotting AFUs (± SD) against time using Ascent fluoroscan software version 2.6.
siRNA blockade assay
MΦ were subjected to siRNA knockdown using manufacturer’s instructions (Origene technologies, USA). The kits for human siRNAs (mixture of duplexes), were purchased from Origene technologies (Beclin-1 siRNA: SR322490; iNOS siRNA: SR303202). MDSCs were treated with siRNA and the scrambled control, washed and added with Mtb (H37Rv) for 4 h. infection (MOI of 1). Cells were washed added with CD44TA-LIP (10 μg/mL) or left untreated and incubated further for CFU assays at 72 h post infection.
Ex vivo Mtb Ag85B antigen presentation to CD4 T cells
Ex vivo Mtb Ag858 antigen presentation to CD4 T cells was performed as previously described. The original method described by the Harding lab has been extensively used by us and others for in vitro antigen presentation by MΦ (52). Briefly, Mtb -infected MΦ (+/− CD44TA-LIP/pharmacological inhibitors of CD44 signaling pathway) were washed after a 4-h infection and overlaid with the F9A6-CD4 T cell hybridoma (gift from Dr. David Canaday lab) that recognizes an Ag85B epitope in the context of human HLA-DR1. IL-2 secreted from hybridoma T cells were determined using a sandwich ELISA kit (Biolegend).
CD44TA-LIP efficacy in Mtb mouse model: analysis of bacterial burden and immune cells
The main mouse vaccine validation protocol is identical to the NIH mouse model that has been described by us previously (53). Age and sex matched mice (4-6 weeks) were tested with a combination of treatment regimen with CD44TA-LIP. Regimen 1: In post infection treatment regimen, mice were first infected with Mtb through aerosol route using Glas-Col inhalation exposure system, implanting ~ 100 CFU in the lungs of each mice. 3 weeks post infection, they were treated intranasally with CD44TA-LIP twice a week for one week at a dose of 20 μg/20 μL. One week after treatment, organs were harvested for analysis of bacterial burden and immune cells. Regimen 2: In pre-treatment regimen, mice were first treated intranasally with CD44TA-LIP twice a week for one week at the same dose. One week later they were infected with Mtb through aerosol route using Glas-Col inhalation exposure system, implanting ~ 100 CFU in the lungs of each mice. Three weeks post infection, organs were harvested for analysis of bacterial burden and immune cells. Control experimental groups of untreated, unvaccinated and uninfected mice were also used for comparison and analysis purpose. 5 mice per experimental group per time point were used. For analysis of bacterial burden via CFU assay, lungs and spleens were homogenized in 5 mL saline with 0.05% Tween 80 and 100 μL aliquots were then plated in replicates on Middlebrook 7H11 agar plates.
For analysis of immune cell via flow cytometry, lungs perfused with sterile phosphate-buffered saline were teased in buffer with 1 mg/mL collagenase and 1 mg/mL elastase (Sigma Biologicals, USA) to break down the fibrous tissue material. Single-cell suspensions from lungs and red blood cell lysed spleens were prepared in PBS with 10% FCS and 2 mM EDTA (Sigma-Aldrich) by straining through 40-μm cell strainer using a plunger of 5 mL syringe (BD Biosciences). Lung- and splenocytes were stained for CD4 and CD8 T cells using antibodies to surface markers and intracellular cytokines as described before (53). Results were reported as absolute numbers of T cells per organ after performing an initial viable cell count using trypan blue exclusion followed by immuno-staining and acquiring a fixed number of cells. One million splenocytes or lung cells were stained using antibodies to CD3 (564380), CD8 (551162), CD4 (561025), CD62L (553150), CD44 (562464), CCR7 (12-1971-80), CD103 (562772), CD69 (740220), CD11a (740849), Live/Dead Fixable Aqua Dead Cell Stain Kit (L34957). All the antibodies were purchased by BD Biosciences. CCR7 antibody was purchased from eBiosciences and Live/Dead staining kit was purchased from Thermo Fisher Scientific. After fixation and storage at 4°C overnight, the cells were analyzed by flow cytometry. For intracellular staining, cells were fixed and permeabilized using Fixation/Permeabilization Solution Kit and stained for IFN- γ (564336: BD Biosciences) and IL-2 (560547: BD Biosciences). Cells were incubated under culture conditions with BD GolgiStop (554715: BD Biosciences) for several h to enhance the detection of intracellular cytokines. All staining antibodies were used at 1:200 or 1:300 or 1:500 dilution depending on the experiment. Sample acquisition was performed on a BD Fortessa flow cytometer using FACS Diva software (BD Biosciences) followed by data analysis with FlowJo software (Tree Star Inc., Stanford, CA).
CD44TA-LIP organ distribution and liver LDH analysis
C57BL/6 mice (10 weeks) were purchased from Charles Rivers Laboratories and were housed with controlled temperature (25oC), 12h lighting cycles, and access to standard diet and water.
In Vivo Lung Inflammation and CD44-NP Treatment Biodistribution
Lung inflammation was induced in mice, intranasally, to mimic the inflammation associated with TB burden, for NP biodistribution studies with 10μg of Lipopolysaccharides, from Pseudomonas aeruginosa 10 (L9143, Sigma, MA, USA) in 20μL solution; 10 μL per nostril. Control mice (no inflammation) were intranasally introduced 20μL of 1X PBS (Gibco, MA, USA); 10μL per nostril. Twenty four h following the procedure, mice were randomly assigned to the groups of 4 and treated with CD44TA-LIP labeled with 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000]-N-(Cy 5.5) (Avanti Polar lipids) at the dose of 5μg in 20μL per mouse for 2, 4 and 24h. At the above time points, mice were sacrificed and the main organs including lungs, heart, liver, spleen, kidneys, excised for fluorescence imagining of CD44TA-LIP distribution with IVIS Spectrum Imaging System (Perkin Elmer, MA, USA) at excitation and emissions wavelengths of 675nm and 720nm for Cy 5.5 fluorescence intensity detection. The data was quantified using Living Image® Software.
Following the study, the left lateral liver lobes of the mice were flash frozen in liquid nitrogen, homogenized with TissueLyser (Qiagen, MD, USA) and normalized to 1mg/mL protein. Lactate dehydrogenase (LDH) activity was quantified using the LDH Fluorometric Assay Kit (ab197000, Abcam, MA, USA) according to manufacturer’s protocol.
Additionally, the lungs (processed as above) were analyzed for the cell populations associated with CD44TA-LIP. CD44TA-LIP were Cy 5.5 labeled and the samples were stained with CD11b-Pacific Blue (117322, Biolegend) and CD80-PE (104708, Biolegend). Samples were analyzed using a Becton-Dickenson FACS Fortessa (5 laser 17 channel) using Diva 9.1. Compensation was accomplished using Diva auto-compensation with single color controls. Acquisition in Diva and analysis in FCS Express V7 (DeNovo Software) was accomplished by excluding debris with an FSC/SSC gate, singlet gate using FSC-A/FSC-H was used to isolate single cells, and Live/Dead was used to isolate on viable cells. Gates were based on single and unstained controls.
Statistics
For all statistical analyses, PRISM (Version 8, GraphPad, San Diego) software was used. Statistical analysis was performed with Paired two-tailed Student’s t-test/one-way ANOVA with post-hoc analysis. P values <0.05 were considered significant.
Study Approval
All animal experiments in this study were performed as per animal protocols verified and approved by the Institutional Animal Care and Use Committee of Houston Methodist Research Institute, Houston. The animal study was carried out in accordance with the US government PHS policy on Humane Care and Use of Laboratory Animals. All animal experiments were conducted in accordance with approved protocols. With a written informed consent, all blood samples were collected per approved institutional review board approved protocols for the isolation of PBMCs and MΦ.
Supplementary Material
Acknowledgments
The authors wish to acknowledge funding support from NIH (R21 7R21AI137533-02) and intramural funds from HMRI. We wish to gratefully acknowledge Drs. Sasha Pejerrey and Heather McConnell, Lead Scientific writer, Houston Methodist Research Institute, for assistance with editing of the manuscript.
REFERENCES
- 1.Chakaya J, Khan M, Ntoumi F, Aklillu E, Fatima R, Mwaba P, Kapata N, Mfinanga S, Hasnain SE, Katoto PDMC, et al. Global Tuberculosis Report 2020 – Reflections on the Global TB burden, treatment and prevention efforts. Int J Infect Dis (2021) doi: 10.1016/j.ijid.2021.02.107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Seung KJ, Keshavjee S, Rich ML. Multidrug-Resistant Tuberculosis and Extensively Drug-Resistant Tuberculosis. Cold Spring Harb Perspect Med (2015) 5:a017863. doi: 10.1101/cshperspect.a017863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bates M, Marais BJ, Zumla A. Tuberculosis Comorbidity with Communicable and Noncommunicable Diseases. Cold Spring Harb Perspect Med (2015) 5:a017889. doi: 10.1101/cshperspect.a017889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Palucci I, Delogu G. Host Directed Therapies for Tuberculosis: Futures Strategies for an Ancient Disease. Chemotherapy (2018) 63:172–180. doi: 10.1159/000490478 [DOI] [PubMed] [Google Scholar]
- 5.O’Garra A, Redford PS, McNab FW, Bloom CI, Wilkinson RJ, Berry MPR. The Immune Response in Tuberculosis. Annu Rev Immunol (2013) 31:475–527. doi: 10.1146/annurev-immunol-032712-095939 [DOI] [PubMed] [Google Scholar]
- 6.Zeng G, Zhang G, Chen X. Th1 cytokines, true functional signatures for protective immunity against TB? Cell Mol Immunol (2018) 15:206–215. doi: 10.1038/cmi.2017.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sakai S, Mayer-Barber KD, Barber DL. Defining features of protective CD4 T cell responses to Mycobacterium tuberculosis. Curr Opin Immunol (2014) 29:137–142. doi: 10.1016/j.coi.2014.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu CH, Liu H, Ge B. Innate immunity in tuberculosis: host defense vs pathogen evasion. Cell Mol Immunol (2017) 14:963–975. doi: 10.1038/cmi.2017.88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Crevel R, Ottenhoff THM, van der Meer JWM. Innate Immunity to Mycobacterium tuberculosis. Clin Microbiol Rev (2002) 15:294–309. doi: 10.1128/CMR.15.2.294-309.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hawn TR, Matheson AI, Maley SN, Vandal O. Host-Directed Therapeutics for Tuberculosis: Can We Harness the Host? Microbiol Mol Biol Rev (2013) 77:608–627. doi: 10.1128/MMBR.00032-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leemans JC, Florquin S, Heikens M, Pals ST, Neut R van der, van der Poll T. CD44 is a macrophage binding site for Mycobacterium tuberculosis that mediates macrophage recruitment and protective immunity against tuberculosis. J Clin Invest (2003) 111:681–689. doi: 10.1172/JCI16936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen C, Zhao S, Karnad A, Freeman JW. The biology and role of CD44 in cancer progression: therapeutic implications. J Hematol Oncol (2018) 11:64. doi: 10.1186/s13045-018-0605-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jordan AR, Racine RR, Hennig MJP, Lokeshwar VB. The Role of CD44 in Disease Pathophysiology and Targeted Treatment. Front Immunol (2015) 6: doi: 10.3389/fimmu.2015.00182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kohda D, Morton CJ, Parkar AA, Hatanaka H, Inagaki FM, Campbell ID, Day AJ. Solution structure of the link module: a hyaluronan-binding domain involved in extracellular matrix stability and cell migration. Cell (1996) 86:767–75. doi: 10.1016/s0092-8674(00)80151-8 [DOI] [PubMed] [Google Scholar]
- 15.Takeda M, Ogino S, Umemoto R, Sakakura M, Kajiwara M, Sugahara KN, Hayasaka H, Miyasaka M, Terasawa H, Shimada I. Ligand-induced structural changes of the CD44 hyaluronan-binding domain revealed by NMR. J Biol Chem (2006) 281:40089–95. doi: 10.1074/jbc.M608425200 [DOI] [PubMed] [Google Scholar]
- 16.Banerji S, Wright AJ, Noble M, Mahoney DJ, Campbell ID, Day AJ, Jackson DG. Structures of the Cd44-hyaluronan complex provide insight into a fundamental carbohydrate-protein interaction. Nat Struct Mol Biol (2007) 14:234–9. doi: 10.1038/nsmb1201 [DOI] [PubMed] [Google Scholar]
- 17.Somasunderam A, Thiviyanathan V, Tanaka T, Li X, Neerathilingam M, Lokesh GLR, Mann A, Peng Y, Ferrari M, Klostergaard J, et al. Combinatorial selection of DNA thioaptamers targeted to the HA binding domain of human CD44. Biochemistry (2010) 49:9106–12. doi: 10.1021/bi1009503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leonard F, Ha NP, Sule P, Alexander JF, Volk DE, Lokesh GLR, Liu X, Cirillo JD, Gorenstein DG, Yuan J, et al. Thioaptamer targeted discoidal microparticles increase self immunity and reduce Mycobacterium tuberculosis burden in mice. J Control Release (2017) 266:238–247. doi: 10.1016/j.jconrel.2017.09.038 [DOI] [PubMed] [Google Scholar]
- 19.Decuzzi P, Godin B, Tanaka T, Lee S-Y, Chiappini C, Liu X, Ferrari M. Size and shape effects in the biodistribution of intravascularly injected particles. J Control Release (2010) 141:320–7. doi: 10.1016/j.jconrel.2009.10.014 [DOI] [PubMed] [Google Scholar]
- 20.Godin B, Chiappini C, Srinivasan S, Alexander JF, Yokoi K, Ferrari M, Decuzzi P, Liu X. Discoidal Porous Silicon Particles: Fabrication and Biodistribution in Breast Cancer Bearing Mice. Adv Funct Mater (2012) 22:4225–4235. doi: 10.1002/adfm.201200869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu R, Zhang G, Mai J, Deng X, Segura-Ibarra V, Wu S, Shen J, Liu H, Hu Z, Chen L, et al. An injectable nanoparticle generator enhances delivery of cancer therapeutics. Nat Biotechnol (2016) 34:414–8. doi: 10.1038/nbt.3506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stone NRH, Bicanic T, Salim R, Hope W. Liposomal Amphotericin B (AmBisome(®)): A Review of the Pharmacokinetics, Pharmacodynamics, Clinical Experience and Future Directions. Drugs (2016) 76:485–500. doi: 10.1007/s40265-016-0538-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barenholz Y Doxil®--the first FDA-approved nano-drug: lessons learned. J Control Release (2012) 160:117–34. doi: 10.1016/j.jconrel.2012.03.020 [DOI] [PubMed] [Google Scholar]
- 24.Cipolla D, Gonda I, Chan H-K. Liposomal formulations for inhalation. Ther Deliv (2013) 4:1047–72. doi: 10.4155/tde.13.71 [DOI] [PubMed] [Google Scholar]
- 25.Ryoo I, Choi B, Ku S-K, Kwak M-K. High CD44 expression mediates p62-associated NFE2L2/NRF2 activation in breast cancer stem cell-like cells: Implications for cancer stem cell resistance. Redox Biol (2018) 17:246–258. doi: 10.1016/j.redox.2018.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poluzzi C, Nastase M-V, Zeng-Brouwers J, Roedig H, Hsieh LT-H, Michaelis JB, Buhl EM, Rezende F, Manavski Y, Bleich A, et al. Biglycan evokes autophagy in macrophages via a novel CD44/Toll-like receptor 4 signaling axis in ischemia/reperfusion injury. Kidney Int (2019) 95:540–562. doi: 10.1016/j.kint.2018.10.037 [DOI] [PubMed] [Google Scholar]
- 27.Shastri MD, Shukla SD, Chong WC, Dua K, Peterson GM, Patel RP, Hansbro PM, Eri R, O’Toole RF. Role of Oxidative Stress in the Pathology and Management of Human Tuberculosis. Oxid Med Cell Longev (2018) 2018:1–10. doi: 10.1155/2018/7695364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khan A, Sayedahmed EE, Singh VK, Mishra A, Dorta-Estremera S, Nookala S, Canaday DH, Chen M, Wang J, Sastry KJ, et al. A recombinant bovine adenoviral mucosal vaccine expressing mycobacterial antigen-85B generates robust protection against tuberculosis in mice. Cell Reports Med (2021) 2:100372. doi: 10.1016/j.xcrm.2021.100372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ogongo P, Porterfield JZ, Leslie A. Lung Tissue Resident Memory T-Cells in the Immune Response to Mycobacterium tuberculosis. Front Immunol (2019) 10: doi: 10.3389/fimmu.2019.00992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Naor D, Sionov RV, Ish-Shalom D. “CD44: Structure, Function and Association with the Malignant Process,” in, 241–319. doi: 10.1016/S0065-230X(08)60101-3 [DOI] [PubMed] [Google Scholar]
- 31.Hodge-Dufour J, Noble PW, Horton MR, Bao C, Wysoka M, Burdick MD, Strieter RM, Trinchieri G, Puré E. Induction of IL-12 and chemokines by hyaluronan requires adhesion-dependent priming of resident but not elicited macrophages. J Immunol (1997) 159:2492–500. [PubMed] [Google Scholar]
- 32.Vachon E, Martin R, Kwok V, Cherepanov V, Chow C-W, Doerschuk CM, Plumb J, Grinstein S, Downey GP. CD44-mediated phagocytosis induces inside-out activation of complement receptor-3 in murine macrophages. Blood (2007) 110:4492–4502. doi: 10.1182/blood-2007-02-076539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Queval CJ, Brosch R, Simeone R. The Macrophage: A Disputed Fortress in the Battle against Mycobacterium tuberculosis. Front Microbiol (2017) 8: doi: 10.3389/fmicb.2017.02284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ernst JD. Macrophage receptors for Mycobacterium tuberculosis. Infect Immun (1998) 66:1277–81. doi: 10.1128/IAI.66.4.1277-1281.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mackay C, Terpe H, Stauder R, Marston W, Stark H, Günthert U. Expression and modulation of CD44 variant isoforms in humans. J Cell Biol (1994) 124:71–82. doi: 10.1083/jcb.124.1.71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rayahin JE, Buhrman JS, Zhang Y, Koh TJ, Gemeinhart RA. High and Low Molecular Weight Hyaluronic Acid Differentially Influence Macrophage Activation. ACS Biomater Sci Eng (2015) 1:481–493. doi: 10.1021/acsbiomaterials.5b00181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ding S, Yang J, Feng X, Pandey A, Barhoumi R, Zhang D, Bell SL, Liu Y, da Costa LF, Rice-Ficht A, et al. Interactions between fungal hyaluronic acid and host CD44 promote internalization by recruiting host autophagy proteins to forming phagosomes. iScience (2021) 24:102192. doi: 10.1016/j.isci.2021.102192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McKee CM, Lowenstein CJ, Horton MR, Wu J, Bao C, Chin BY, Choi AMK, Noble PW. Hyaluronan Fragments Induce Nitric-oxide Synthase in Murine Macrophages through a Nuclear Factor κB-dependent Mechanism. J Biol Chem (1997) 272:8013–8018. doi: 10.1074/jbc.272.12.8013 [DOI] [PubMed] [Google Scholar]
- 39.Nickel D, Busch M, Mayer D, Hagemann B, Knoll V, Stenger S. Hypoxia Triggers the Expression of Human β Defensin 2 and Antimicrobial Activity against Mycobacterium tuberculosis in Human Macrophages. J Immunol (2012) 188:4001–4007. doi: 10.4049/jimmunol.1100976 [DOI] [PubMed] [Google Scholar]
- 40.Hill DR, Rho HK, Kessler SP, Amin R, Homer CR, McDonald C, Cowman MK, de la Motte CA. Human Milk Hyaluronan Enhances Innate Defense of the Intestinal Epithelium. J Biol Chem (2013) 288:29090–29104. doi: 10.1074/jbc.M113.468629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gariboldi S, Palazzo M, Zanobbio L, Selleri S, Sommariva M, Sfondrini L, Cavicchini S, Balsari A, Rumio C. Low Molecular Weight Hyaluronic Acid Increases the Self-Defense of Skin Epithelium by Induction of β-Defensin 2 via TLR2 and TLR4. J Immunol (2008) 181:2103–2110. doi: 10.4049/jimmunol.181.3.2103 [DOI] [PubMed] [Google Scholar]
- 42.Hill DR, Kessler SP, Rho HK, Cowman MK, de la Motte CA. Specific-sized Hyaluronan Fragments Promote Expression of Human β-Defensin 2 in Intestinal Epithelium. J Biol Chem (2012) 287:30610–30624. doi: 10.1074/jbc.M112.356238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang L, Nazarova EV, Tan S, Liu Y, Russell DG. Growth of Mycobacterium tuberculosis in vivo segregates with host macrophage metabolism and ontogeny. J Exp Med (2018) 215:1135–1152. doi: 10.1084/jem.20172020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee H-J, Ko H-J, Song D-K, Jung Y-J. Lysophosphatidylcholine Promotes Phagosome Maturation and Regulates Inflammatory Mediator Production Through the Protein Kinase A–Phosphatidylinositol 3 Kinase–p38 Mitogen-Activated Protein Kinase Signaling Pathway During Mycobacterium tuberculosis Infect. Front Immunol (2018) 9: doi: 10.3389/fimmu.2018.00920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang X, Huang T, Wu Y, Peng W, Xie H, Pan M, Zhou H, Cai B, Wu Y. Inhibition of the PI3K-Akt-mTOR signaling pathway in T lymphocytes in patients with active tuberculosis. Int J Infect Dis (2017) 59:110–117. doi: 10.1016/j.ijid.2017.04.004 [DOI] [PubMed] [Google Scholar]
- 46.Schumann J, Stanko K, Schliesser U, Appelt C, Sawitzki B. Differences in CD44 Surface Expression Levels and Function Discriminates IL-17 and IFN-γ Producing Helper T Cells. PLoS One (2015) 10:e0132479. doi: 10.1371/journal.pone.0132479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baaten BJG, Li C-R, Bradley LM. Multifaceted regulation of T cells by CD44. Commun Integr Biol (2010) 3:508–512. doi: 10.4161/cib.3.6.13495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guan H, Nagarkatti PS, Nagarkatti M. Role of CD44 in the Differentiation of Th1 and Th2 Cells: CD44-Deficiency Enhances the Development of Th2 Effectors in Response to Sheep RBC and Chicken Ovalbumin. J Immunol (2009) 183:172–180. doi: 10.4049/jimmunol.0802325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kramnik I, Beamer G. Mouse models of human TB pathology: roles in the analysis of necrosis and the development of host-directed therapies. Semin Immunopathol (2016) 38:221–237. doi: 10.1007/s00281-015-0538-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cooper AM. T cells in mycobacterial infection and disease. Curr Opin Immunol (2009) 21:378–384. doi: 10.1016/j.coi.2009.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Forbes EK, Sander C, Ronan EO, McShane H, Hill AVS, Beverley PCL, Tchilian EZ. Multifunctional, High-Level Cytokine-Producing Th1 Cells in the Lung, but Not Spleen, Correlate with Protection against Mycobacterium tuberculosis Aerosol Challenge in Mice. J Immunol (2008) 181:4955–4964. doi: 10.4049/jimmunol.181.7.4955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Torres M, Ramachandra L, Rojas RE, Bobadilla K, Thomas J, Canaday DH, Harding CV, Boom WH. Role of Phagosomes and Major Histocompatibility Complex Class II (MHC-II) Compartment in MHC-II Antigen Processing of Mycobacterium tuberculosis in Human Macrophages. Infect Immun (2006) 74:1621–1630. doi: 10.1128/IAI.74.3.1621-1630.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Khan A, Singh VK, Mishra A, Soudani E, Bakhru P, Singh CR, Zhang D, Canaday DH, Sheri A, Padmanabhan S, et al. NOD2/RIG-I Activating Inarigivir Adjuvant Enhances the Efficacy of BCG Vaccine Against Tuberculosis in Mice. Front Immunol (2020) 11: doi: 10.3389/fimmu.2020.592333 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.