

Abstract
In response to myocardial infarction (MI), quiescent cardiac fibroblasts differentiate into myofibroblasts mediating tissue repair. One of the most widely accepted markers of myofibroblast differentiation is the expression of Acta2 which encodes smooth muscle alpha-actin (SMαA) that is assembled into stress fibers. However, the requirement of Acta2/SMαA in the myofibroblast differentiation of cardiac fibroblasts and its role in post-MI cardiac repair remained unknown. To answer these questions, we generated a tamoxifen-inducible cardiac fibroblast-specific Acta2 knockout mouse line. Surprisingly, mice that lacked Acta2 in cardiac fibroblasts had a normal post-MI survival rate. Moreover, Acta2 deletion did not affect the function or histology of infarcted hearts. No difference was detected in the proliferation, migration, or contractility between WT and Acta2-null cardiac myofibroblasts. Acta2-null cardiac myofibroblasts had a normal total filamentous actin level and total actin level. Acta2 deletion caused a significant compensatory increase in the transcription level of non-Acta2 actin isoforms, especially Actg2 and Acta1. Moreover, in myofibroblasts, the transcription levels of cytoplasmic actin isoforms were significantly higher than those of muscle actin isoforms. In addition, we found that myocardin-related transcription factor-A is critical for myofibroblast differentiation but is not required for the compensatory effects of non-Acta2 isoforms. In conclusion, the Acta2 deletion does not prevent the myofibroblast differentiation of cardiac fibroblasts or affect the post-MI cardiac repair, and the increased expression and stress fiber formation of non-SMαA actin isoforms and the functional redundancy between actin isoforms are able to compensate for the loss of Acta2 in cardiac myofibroblasts.
Keywords: cardiac fibroblast, myocardial infarction, stress fiber, actin
1. INTRODUCTION
Among cardiovascular diseases (CVDs), the leading causes of death in developed countries, acute myocardial infarction (MI) is one of the most deadly forms due to its unpredictability, fast disease progression, and poor prognosis [1, 2]. Following MI, cardiomyocytes in the infarcted myocardium are permanently lost, which greatly compromises the integrity of the infarcted ventricle wall [3]. Quiescent cardiac fibroblasts are quickly activated after MI, characterized by massive proliferation and myofibroblast differentiation [4], which is believed to be induced by mechanical stress and cytokine stimulation [5, 6]. Myofibroblasts mediate the formation of infarct scars through expressing high levels of extracellular matrix (ECM) proteins and ECM remodeling enzymes [4, 7]. In addition, myofibroblasts are also known for actin stress fibers mostly composed of smooth muscle alpha-actin (SMαA) encoded by Acta2 [8, 9]. We recently reported that the myofibroblast state of cardiac fibroblasts was largely limited to the first week after MI, after which myofibroblasts further differentiated into matrifibrocytes, a newly identified fibroblast differentiation state lacking the expression of myofibroblast marker genes [4]. The transient myofibroblast state of cardiac fibroblasts suggests that myofibroblasts are especially important in the early stage of post-MI cardiac repair. Indeed, depletion of myofibroblasts after MI increased the risk of cardiac rupture during the first week after MI [7].
Due to the importance of ECM in post-MI tissue healing, the role of cardiac fibroblasts/myofibroblasts in ECM production and remodeling has received special attention [10–13]. However, the function of SMαA, the hallmark of myofibroblasts, has been largely overlooked. A limited number of early in vitro studies manipulating SMαA level or activity found that SMαA promoted fibroblast contraction but inhibited cell migration [14, 15]. A later study showed that in myofibroblasts SMαA stress fibers play an important role in the formation of supermature focal adhesions which are important for the anchorage of myofibroblasts to the surrounding ECM [16, 17]. Several previous studies using mice with global Acta2 deletion to investigate the function of SMαA in myofibroblasts in different disease models suggest that SMαA stress fibers may regulate or affect myofibroblast proliferation, motility, contractility, and ECM remodeling [18–20]. However, some discrepancies are present among the results reported by these studies, which suggests that the role and necessity of SMaA in myofibroblasts of different origins may vary significantly. In addition, the deletion of Acta2 in other cell types, such as the vascular smooth muscle cell, may also affect the interpretation of the function of SMαA in studies using global Acta2 knockout (KO) mice.
Here, we studied the function of SMαA in cardiac myofibroblasts and post-MI cardiac repair using a novel tamoxifen-inducible cardiac fibroblast-specific Acta2 KO mouse line and cardiac fibroblast lineage tracing. It was found that Acta2 deletion in cardiac fibroblasts did not significantly affect the post-MI survival or cardiac function of mice. In response to MI or transforming growth factor β (TGFβ) treatment, Acta2-null cardiac fibroblasts underwent normal myofibroblast differentiation, which was likely due to the compensatory effect of other actin isoforms.
2. MATERIALS AND METHODS
2.1. Mice.
Mouse embryonic stem cells with a “knockout-first” Acta2 allele purchased from European Mouse Mutant Cell Repository were used to generate chimeric mice. Chimeric mice were crossed with WT C57BL/6 mice to obtain offspring carrying the mutant allele which was then crossed with mice expressing FLPe recombinase (Jackson Laboratory, #003946) to delete the LacZ-neo cassette upstream of exon 5 of Acta2 to generate Acta2fl/+ mice with exons 5 to 7 of Acta2 flanked by loxP sites. Tcf21MCM/+ [21] and R26eGFP [22] mice were crossed to generate Tcf21MCM/+;R26eGFP mice. Acta2fl/+ mice were crossed with Tcf2MCM/+;R26eGFP mice to generate Tcf21MCM/+;Acta2fl/fl;R26eGFP mice. R26tdTomato mice were purchased from Jackson Laboratory (#007914). The genotyping for the Acta2fl allele was done using primers targeting the region around 3’ loxP site (Table 2). Experiments include both males and females.
Table 2.
Primer sequences used in realtime PCR.
| Target gene | Forward primer | Reverse primer |
|---|---|---|
| Actg2 | GACTTCTCACACCCTTGGTGCTC | AAGGGCGGTGGTCTCTTCTTCAC |
| Actg1 | TGAGCAAGAAATGGCTACTGCTG | ACAGGACTCCATGCCCAGGAA |
| Actb | TCCTTCTTGGGTATGGAATCCTGT | TTTACGGATGTCAACGTCACACTTC |
| Acta1 | AAGCCTCACTTCCTACCCTCGG | AGAGCCGTTGTCACACACAAGAG |
| Acta2 | AAGAGCTACGAACTGCCTGACG | GTTTCGTGGATGCCCGCTGA |
| Actc1 | TATAAAGCTGCGCTCCAGGCGA | CTTTGGTGGGTTCTGTAGGCGTG |
| Col1a1 | ACTCTGACTGGAAGAGCGGAGAG | GCTGAGTAGGGAACACACAGGTC |
| Col3a1 | GACCTAAGGGCGAAGATGGCAAA | GGAAGCCACTAGGACCCCTTTCT |
| Postn | ACAGGAGGTGGAGAAACAGGAGA | CCTTGAACCCTTTTGTTGGCTGG |
| 18S | GTAACCCGTTGAACCCCATT | CCATCCAATCGGTAGTAGCG |
| Mrtfa | AAAATGGCTCCTCCAGTCAGCAC | TGGTAGGGATGGTGGCTCTTTGA |
| Acta2 (genotyping) | AGTAGGGATGGTGTCACATTTCCA | CTCTCAGCACTCAGATCGTCTTCC |
2.2. Animal procedures.
To induce the activity of the MerCreMer protein, mice were treated with tamoxifen (MilliporeSigma, T5648) dissolved in corn oil through i.p. injections or gavage at a dosage of 75 mg/kg body weight/day for 5 days starting at 6 weeks of age. Mice were subjected to permanent surgical ligation of the left coronary artery to induce MI at 9 weeks of age [23]. Briefly, mice were anesthetized using isoflurane by a ventilator at 1-3% throughout the entire course of the procedure. Anesthesia was monitored by a combination of movement, stimulus perception, and chest wall movement observation. A left lateral thoracotomy was then performed. The left coronary artery was identified and ligated just below the left atrium. To ensure the efficient deletion of Acta2, mice were given another course of tamoxifen treatment from 1 day before MI to 5 days after MI. Echocardiography was performed in M-mode using a Toshiba Aplio SSA-770a ultrasound system and a 12-MHz transducer as previously described [24]. For pain management related to surgical procedures, mice were given a dose of Carprofen (5 mg/kg body weight) by subcutaneous injection before the surgery followed by a second dose 12 hours after the surgery. Mice were checked every day after MI for survival rate. Mice found dead were subjected to necropsy to identify cardiac rupture. Mice were euthanized by isoflurane overdose followed by cervical dislocation before sample collections.
2.3. Antibodies and biologics.
Antibodies used in this study are listed in Table 1. EdU (sc-284628A) was purchased from Santa Cruz Biotechnology Inc. Collagenase D (11088866001) and Dispase II (4942078001) were purchased from Roche Diagnostics. Collagen, type I (356236) was purchased from Corning. TGF-β (240-B) was purchased from R&D Systems.
Table 1.
Antibody and other staining reagent list.
| Source | Catalog # | Dilution in IHC/ICC | Dilution in WB | |
|---|---|---|---|---|
| Primary antibodies | ||||
| Chicken anti-GFP antibody | Abcam | ab61682 | 1:100 | |
| Rabbit anti-GFP antibody | Rockland | 600-401-215L | 1:100 | |
| Mouse anti-SMαA antibody | Millipore Sigma | 113200-500UL | 1:100 | 1:1000 |
| Rabbit anti-Ki67 antibody | Cell Signaling Technology | 9129S | 1:200 | |
| Goat anti-Pdgfrα antibody | Novus | AF1062 | 1:100 | |
| Mouse anti-β-tubulin antibody | DSHB | E7 | 1:300 | |
| Mouse anti-pan-actin antibody | DSHB | JLA20 | 1:50 | 1:200 |
| Mouse anti-CyβA antibody | Invitrogen | AM4302 | 1:200 | 1:1000 |
| Mouse anti-sarcomeric actin (SkMαA and CMαA) antibody | Millipore Sigma | A2172 | 1:100 | |
| Mouse anti-smooth muscle actin (SMαA and SMγA) antibody | Millipore Sigma | A7607 | 1:100 | |
| Mouse anti-vinculin antibody | NOVUS | NB600-1293 | 1:100 | |
| Mouse anti-CyγA antibody | Millipore Sigma | MABT824 | 1:200 | 1:1000 |
| Secondary antibodies | ||||
| Goat anti-Mouse IgG2a Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Invitrogen | A-21131 | 1:500 | 1:2000 |
| Goat anti-Mouse IgG2b Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Invitrogen | A-21141 | 1:500 | 1:2000 |
| IgG2b Cross-Adsorbed Goat anti-Mouse, Alexa Fluor™ 647 | Invitrogen | A21242 | 1:500 | 1:2000 |
| Goat anti-Mouse IgM (Heavy chain) Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Invitrogen | A-21042 | 1:500 | 1:2000 |
| Goat anti-Mouse IgG2a Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Invitrogen | A-21137 | 1:500 | 1:2000 |
| Goat anti-Mouse IgG1 Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Invitrogen | A-21127 | 1:500 | 1:2000 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Invitrogen | A-21428 | 1:500 | 1:2000 |
| Goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 647 | Invitrogen | A-21235 | 1:500 | 1:2000 |
| Others | ||||
| Alexa Fluor® 647 Phalloidin #8940 | Cell Signaling Technology | 8940S | 1:20 | |
| Click-&-Go Plus EdU 647 Cell Proliferation Assay Kits | Click Chemistry Tools | 1353 | ||
| Click-iT™ Plus EdU Alexa Fluor™ 647 Flow Cytometry Assay Kit | Invitrogen | C10643 A | ||
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 647 | Invitrogen | A31571 | 1:500 | 1:2000 |
| Donkey Anti-Rabbit IgG (H+L), Alexa Fluor® 488 | Jackson ImmunoResearch | 711-547-003 | 1:500 | 1:2000 |
| Donkey anti-Goat IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor™ 555 | Invitrogen | A21432 | 1:500 | 1:2000 |
2.4. EdU in vivo fibroblast proliferation assay.
Mice were treated with EdU at a dosage of 50 mg/kg body weight through i.p. injections. Four hours after EdU injections, mice were sacrificed, and heart samples were collected. EdU detection was carried out after IHC staining using the Click-iT Plus Alexa Fluor 647 Picolyl Azide Toolkit (C10643) from Thermo Fisher Scientific.
2.5. Cell isolation and culture.
Cardiac fibroblasts were isolated as previously described [4]. Briefly, heart tissue was minced and digested in DMEM containing 0.75 U/ml collagenase D (Roche, 11088866001), 1.0 U/ml Dispase II (Roche, 10165859001), and 1 mM CaCl2 at 37°C for 40 minutes. The slurry was then passed through a 100 μm cell strainer and then a 40 μm cell strainer. Cells were collected by centrifugation at 350 g for 10 minutes. The cell pellets were resuspended in a growth medium composed of DMEM with 10% bovine growth serum (BGS) and 1% of an antibiotic mixture containing 10,000 U/ml penicillin, 10 mg/ml streptomycin, and 25 μg/ml amphotericin B and seeded on cell culture plates. To induce Acta2 deletion, WT and Acta2fl/fl cardiac fibroblasts were treated with adenoviruses expressing Cre (Adeno-Cre) at 60-80 multiplicity of infection (MOI). Myofibroblast differentiation was induced by TGFβ (10 ng/ml) treatment for 2 days. For Mrtfa knock down (KD) experiments, cardiac fibroblasts were treated with lentiviruses expressing shRNA targeting mouse Mrtfa (Lenti-shMrtfa) or control lentiviruses expressing GFP (Lenti-GFP) at 20 MOI.
2.6. Adenoviral and lentiviral particle production.
Adeno-Cre were produced and purified as previously published [25–27]. To produce Lenti-shMrtfa and Lenti-GFP, HEK293T cells were co-transfected with psPAX2, pMD2.G, and pLKO.1 carrying shRNA targeting Mrtfa (Sigma #TRCN0000095961) or pLKO.1-puro-CMV-TurboGFP (Sigma # SHC003). Lentiviral particles were purified as previously described [28].
2.7. Gel contraction assay.
Gel contraction assay was performed as previously described with some modifications [26]. Briefly, cardiac fibroblasts transduced with Adeno-Cre were treated with TGFβ (10 ng/ml) for 2 days to induced myofibroblast differentiation. 60,000 treated cells were resuspended in 0.4 ml of growth medium and mixed with 0.2 ml of collagen solution that contains 0.1% acetic acid. 4 μl of 1 M NaOH was added to neutralize the acetic acid. 0.5 ml of the mixture was added to each well of 24 well plates and allowed to solidify at room temperature for 20 min, followed by the addition of 0.6 ml of growth medium supplemented with TGFβ. 12 hours after the incubation at 37 °C, gels were carefully released from the well to allow the contraction.
2.8. Rheology.
Cell-free and cell-laden collagen gels were made and cultured as described above. After 24 hours of incubation, collagen gels were released and trimmed using a 5 mm biopsy punch. Using a TA Discovery HR-2 rheometer and a 5 mm crosshatched plate, the storage moduli of gels were determined by frequency sweeping from 6.28 to 25.13 (rad/s) at 2% strain and 10% compression.
2.9. Cell motility assay.
Cell motility assay was performed using Oris Cell Migration Assembly Kit (Platypus Technologies) following the manufacturer’s protocol with some modifications. Briefly, R26tdTomato and Acta2fl/fl;R26eGFP cardiac fibroblasts transduced with Adeno-Cre were treated with TGFβ (10 ng/ml) for 2 days to induce myofibroblast differentiation. Treated cells of the 2 groups were mixed at a 1:1 ratio. A total of 20,000 cells in a maintenance medium containing DMEM with 0.5% BGS and 1% of the antibiotic mixture were seeded into each well of 96 well plates with a stopper placed in the center of the well. The stoppers were removed overnight and then the wells were gently washed with PBS, followed by the addition of a fresh maintenance medium with or without TGFβ. For the 3D migration test, after the rinse, 0.5 ml neutralized collagen solution (3 mg/ml) was added to each well and allowed to solidify for 20 minutes before adding a fresh maintenance medium with or without TGFβ. The migration of cells into the center of the well was monitored using an Echo Revolve microscope. Cells in the area previously occupied by the stoppers were quantified and used to calculate the ratio between WT and Acta2-null cardiac myofibroblasts. Images randomly taken in the areas with attached cells were used to verify the 1:1 initial ratio before removing the stoppers.
2.10. Relative proliferation assay.
R26tdTomato and Acta2fl/fl;R26eGFP cardiac fibroblasts were treated with Adeno-Cre and TGFβ as described in the cell motility assay. Treated cells of the 2 genotypes were mixed at a 1:1 ratio. A total of 25,000 cells were seeded into each well of 24 well plates and cultured in a growth medium with 10% BGS and 10 ng/ml TGFβ. Images captured after 2 days of incubation were used for quantification to calculate the relative abundance of cells of the 2 genotypes.
2.11. FACS
After tissue digestion, Tcf21 lineage-traced eGFP+ cardiac fibroblasts were sorted on a FACSaria II flow cytometer (BD Biosciences).
2.12. Realtime PCR.
Realtime PCR was performed as previously described [29]. Briefly, RNA was extracted using a Direct-zol RNA Microprep Kit (Zymo). cDNA synthesis of RNA was performed using an iScript cDNA Synthesis Kit (Bio-Rad). Realtime PCR was carried out using a CFX RT-PCR detection system (Bio-Rad) with SsoAdvanced Universal SYBR Green Supermix (Bio-Rad). Relative mRNA content was normalized to 18S rRNA content. The primers used are listed in Table 2.
2.13. Western blot.
Western blot was performed as previously described [30] with some modifications. Briefly, cells were lysed in RIPA buffer with Halt Protease Inhibitor Cocktail (Thermo Scientific) to extract the protein. SDS-PAGE and blotting were performed using Bio-Rad Mini-PROTEAN Electrophoresis System. After the immunostaining, images were taken using a ProteinSimple FluorChem R System. Band density was normalized to β-tubulin content.
2.14. Immunocytochemical staining (ICC).
Cells grown on multiple-chamber slides or multiple well plates were fixed in 4% paraformaldehyde (PFA) for 10 minutes, rinsed 3 times in TBS with 0.1% Triton X-100, incubated in blocking buffer (TBS, 0.1% Triton X-100, and 3% BSA), and then incubated with primary antibodies diluted in blocking buffer overnight at 4°C. Cells were then rinsed in TBS with 0.1% Triton X-100 3 times and stained with corresponding secondary antibodies diluted in blocking buffer for 1 hour at room temperature. Stained slides were then rinsed and mounted in a mounting medium containing Dapi (Vector Laboratories). Images were captured using an Echo Revolve microscope. Signal intensity was determined using ImageJ. The signal intensity of each image was normalized to the number of cells indicated by Dapi staining.
2.15. Immunohistochemical staining (IHC).
IHC staining was performed as previously described [4]. Briefly, mouse heart samples were fixed in 4% PFA, incubated in 30% sucrose dissolved in PBS, and embedded in OCT (Tissue-Tek) for cryosectioning. Cryosections (5 μm thick) were blocked with 5% goat serum and 0.2% Triton X-100 diluted in TBS, incubated in primary antibodies diluted in blocking buffer overnight at 4°C, and then incubated in appropriate fluorophore-conjugated secondary antibodies diluted in blocking buffer for 1 hour at room temperature. Stained sections were then mounted in a mounting medium containing Dapi (Vector Laboratories). Images were captured using a Leica SP8X confocal microscope.
2.16. Trichrome staining.
Trichrome staining was performed using reagents for Masson’s Trichrome for Connective Tissue (Electron Microscopy Sciences) following the manufacturer’s protocol. Percentage of the fibrotic area in the left ventricle and average left ventricle thickness at 3 different segments were determined using ImageJ.
2.17. Picrosirius red staining.
Picrosirius red staining was carried out as previously described [4]. Briefly, longitudinal heart sections were collected from frozen samples and incubated in Bouin’s fixative (Electron Microscopy Sciences, 26367-01) for 1 hour at 55°C and then stained with picrosirius red (Electron Microscopy Science, 26357-02) for 1 hour at room temperature. Sections were subsequently dehydrated and cleared with xylene. Pictures were captured in bright-field mode or under polarized light to capture the birefringence of collagen fibers using a Leica light microscope. ImageJ was used to analyze the birefringence images to quantify the signal intensities of type I collagen (yellow-red) and type III collagen (green) and their ratio.
2.18. RNAseq
RNAseq was performed as previously described [31]. Briefly, cardiac fibroblasts isolated from WT and Acta2fl/fl mice were transduced with Adeno-Cre, treated with TGFβ for 2 days, and then subjected to RNA isolation using a Direct-zol RNA Microprep Kit (Zymo). cDNA libraries were constructed using the NEBNext single cell/low input RNA library prep kit for Illumina (E6420) from New England BioLabs. The cDNA libraries were sequenced on the Illumina Hi-seq platform using 150 bp paired-end sequencing. RNA-seq reads that passed filters were trimmed to remove low-quality reads and adaptors by TrimGalore-0.6. The quality of reads after filtering was assessed by fastQC, followed by alignment to the mouse genome (MM10) by STAR (2.5.3a) with default parameters. Individual mapped reads were adjusted to provide TPM (transcripts per million) values with mouse genome as the reference.
2.19. ATACseq
ATACseq was performed as previously described [31]. Briefly, cardiac fibroblasts isolated from WT and Acta2fl/fl mice were transduced with Adeno-Cre, treated with TGFβ for 2 days, and then 10,000 cells were lysed on ice for 20 minutes to isolate the nuclei. Isolated nuclei were then incubated with the Tn5 transposase (TDE1, Illumina) and tagmentation buffer at 37°C for 30 minutes with shaking on a thermomixer at 700 rpm. Tagmentated DNA was purified using MinElute Reaction Cleanup Kit (Qiagen). PCR was performed to amplify the ATACseq libraries using Illumina TrueSeq primers and multiplex by indexes primers. After the PCR reaction, libraries were purified with the 1.1X AMPure beads (Beckman). The ATACseq libraries were sequenced on the Illumina Hi-seq platform using 150 bp paired-end sequencing. Sequencing reads of all samples underwent adapter removal using TrimGalore-0.6, followed by quality assessment using FastQC. Reads were then aligned to the mouse reference genome MM10 using Bowtie 2.3 with the following options: – very-sensitive -X 2000 – no-mixed – no-discordant. Only unique alignments within each sample were retained in subsequent analysis. Moreover, alignments resulting from PCR duplicates or located in mitochondria were excluded. The mouse genome was tiled with consecutive non-overlapping 300bp bins. The accessibility of each of the 300bp bins was assessed by the number of fragments per million mapped (FPM) that was aligned to the bin. ATAC-seq peaks were called separately for each sample by MACS2 [32] with the following options: – keep-dup all – nolambda – nomodel. The identification of transcriptional factor motifs in peaks was evaluated using HOMER (http://homer.ucsd.edu/homer/motif/).
2.20. Statistics.
All data are expressed as mean ± SEM unless otherwise stated. Data were analyzed using GraphPad Prism 9 (GraphPad Software, Inc.). Two-tailed t test was used to determine the significance of differences between the 2 groups. One-way ANOVA with post hoc Tukey’s test was used to determine the significance of difference when more than 2 groups were compared. Gehan–Breslow–Wilcoxon test was used to determine the significance of difference between the survival rates of Tcf21MCM/+;R26eGFP and Tcf21MCM/+;Acta2fl/fl;R26eGFP mice. All data are shown as mean with SEM. P < 0.05 was considered significant.
2.21. Data availability.
RNAseq data of cardiac fibroblasts isolated from uninjured hearts and infarct scars were from our previous study [31] (GEO #GSE186079). All new raw sequencing data have been deposited into the public database at NCBI (GEO #GSE193163).
2.22. Study approval.
All experiments involving mice were approved by the IACUC at LSU (approval numbers IACUC 18-024 and 21-034).
3. RESULTS
3.1. Cardiac fibroblast-specific deletion of Acta2 does not affect post-MI survival, cardiac function, or remodeling.
To generate a mouse line with cardiac fibroblast-specific tamoxifen-inducible Acta2 deletion, mice with Acta2 exons 5-7 flanked by loxP sites (Acta2fl/fl) were crossed with Tcf21-MerCreMer (Tcf21MCM/+) mice that also carry a Cre-dependent eGFP cassette in the Rosa26 locus (R26eGFP) (Figure 1A). In these mice (Tcf21MCM/+;Acta2fl/fl;R26eGFP), the deletion of Acta2 exons 5-7 and expression of eGFP in Tcf21-lineage traced cardiac fibroblasts can be induced by tamoxifen treatment. Tcf21MCM/+;Acta2fl/fl;R26eGFP and Tcf21MCM/+;R26eGFP (WT control) mice were treated with tamoxifen for 5 days to induce Cre activity starting at 6 weeks of age and then subjected to surgeries to induce MI at 9 weeks of age (Figure 1B). To maximize the efficiency of Acta2 deletion and reduce the negative effect of prolonged continuous tamoxifen treatment on the viability of mice, another course of tamoxifen treatment was applied to mice from 1 day before MI to 5 days after MI. No significant difference in the post-MI survival rate between Tcf21MCM/+;Acta2fl/fl;R26eGFP and Tcf21MCM/+;R26PeGFP mice was observed (Figure 1C). Moreover, the cardiac function of the two groups was not significantly different from each other without injury and on days 7 and 28 after MI (Figures 1DE). To study the potential impact of Acta2 deletion on the post-MI cardiac repair and remodeling, heart samples were collected from Tcf21MCM/+;Acta2fl/fl;R26eGFP and Tcf21MCM/+;R26eGFP mice on days 7 and 28 after MI and subjected to IHC for type I collagen, trichrome staining, and picrosirius red staining. No difference in the histology and fibrotic response of the heart was observed between Tcf21MCM/+;Acta2fl/fl;R26eGFP and Tcf21MCM/+;R26eGFP mice (Figures 1F–J), suggesting relatively normal tissue remodeling in Tcf21MCM/+;Acta2fl/fl;R26eGFP mice after MI. The infiltration of Tcf21-lineage traced cardiac fibroblasts into the infarct scar was also not affected by the loss of Acta2 (Figure 1F), suggesting that the motility of cardiac fibroblasts is not affected by the lack of SMαA stress fibers. Together, these results suggest that the loss of SMαA in cardiac myofibroblasts does not affect post-MI cardiac repair or tissue remodeling.
Figure 1.
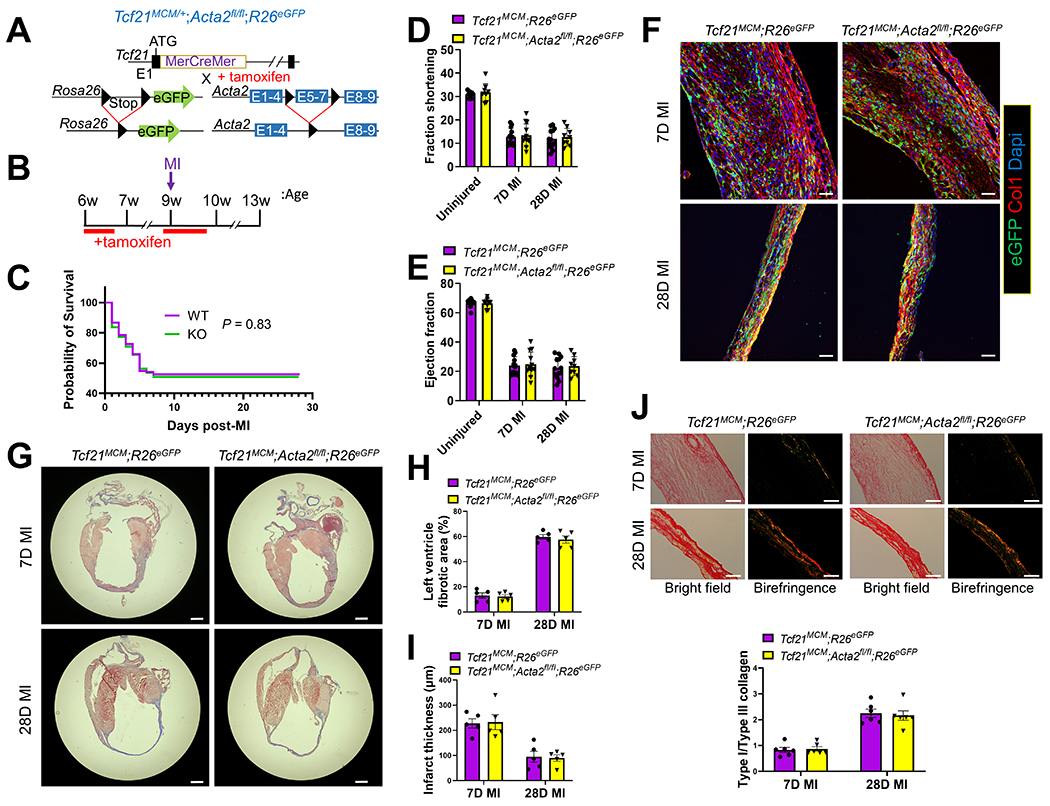
Cardiac fibroblast-specific deletion of Acta2 does not affect the post-MI survival or cardiac function of mice.
(A) Schematic of the generation of Tcf21MCM/+;Acta2fl/fl;R26eGFP mice. In these mice, a tamoxifen-regulated MerCreMer cDNA cassette inserted into exon 1 (E1) enables the tamoxifen-induced deletion of the loxP site-flanked stop cassette upstream of eGFP inserted into the R26 locus and the deletion of the loxP site-flanked exons 5-7 (E5-7) of Acta2 only in cells expressing Tcf21. (B) Experimental scheme of tamoxifen treatment and surgical procedure to induce MI. (C) Survival curves showing the survival rate of Tcf21MCM/+;R26eGFP and Tcf21MCM/+;Acta2fl/fl;R26eGFP mice after MI. n=135 (Tcf21MCM/+;R26eGFP); n=110 (Tcf21MCM/+;Acta2fl/fl;R26eGFP). (D-E) Fraction shortening (D) and ejection fraction (E) of Tcf21MCM/+;R26eGFP and Tcf21MCM/+;Acta2fl/fl;R26eGFP mice without MI and at 7 and 28 days after MI. n=9 for uninjured mice; n=13 for Tcf21MCM/+;R26eGFP; mice at 7 and 28 days after MI; n=12 for for Tcf21MCM/+;Acta2fl/fl;R26eGFP mice 7 days after MI; n=9 for Tcf21MCM/+;Acta2fl/fl;R26eGFP mice 28 days after MI. (F-I) Heart samples were collected from tamoxifen-treated Tcf21MCM/+;R26eGFP and Tcf21MCM/+;Acta2fl/fl;R26eGFP mice at 7 and 28 days after MI. (F) Representative IHC images from 5 analyzed hearts per group show the presence of eGFP+ Tcf21 lineage-traced cardiac fibroblasts and the expression of type I collagen (Col1). Nuclei are shown with Dapi. Scale bar: 50 μm. (G) Representative trichrome staining images from 5 analyzed hearts per group showing the dilation of the left ventricle after MI and collagen deposition in the infarct area. Scale bar: 1 mm. (H-I) Percentage of the fibrotic area in the left ventricle (H) and average left ventricle thickness (I) determined by analysis of trichrome staining images using ImageJ. n=5 for each group. (J) Representative picrosirius red staining images from 6 analyzed hearts per group showing the collagen maturation (type I/III collagen ratio) in the infarct area. n=6. Scale bar: 50 μm.
3.2. Cardiac fibroblast-specific deletion of Acta2 does not affect the expansion of cardiac fibroblasts in the infarct after MI or their proliferation in vitro.
It was reported that global Acta2 KO led to increased renal fibroblast expansion [18]. To study if specific deletion of Acta2 in cardiac fibroblasts also affects their post-MI expansion, a 5-Ethynyl-2’-deoxyuridine (EdU)-based in vivo proliferation assay was performed. Using this method, we previously identified that the proliferation of cardiac fibroblasts after MI peaked at day 3 after MI and was largely diminished beyond day 7 post-MI [4]. Thus, a single EdU injection was given to mice on days 3 or 7 after MI, followed by sample collection 4 hours after the EdU injection. Efficient Acta2 deletion in Tcf21 lineage-traced cardiac fibroblasts of Tcf21MCM/+;Acta2fl/fl;R26eGFP mice was observed, indicated by the absence of SMαA expression in GFP+ cardiac fibroblasts (Figure 2AB). However, in these mice, the expression of SMαA in vascular smooth muscle cells and non-Tcf21 lineage-traced cardiac fibroblasts was not affected, showing the fidelity of our cell type-specific KO system (Figure S1). EdU and Ki67 staining showed that the post-MI proliferation of Tcf21-lineage traced cardiac fibroblasts was not different between Tcf21MCM/+;Acta2fl/fl;R26eGFP and Tcf21MCM/+;R26eGFP mice (Figure 2AB). To verify these findings in vitro, cardiac fibroblasts were isolated from Acta2fl/fl and WT mice, treated with Adeno-Cre, and induced for myofibroblast differentiation by TGFβ treatment. No difference in the percentage of cells expressing Ki67 was observed between the 2 groups (Figure 2C). A proliferation assay was also performed by co-culturing TGFβ- and Adeno-Cre-treated cardiac fibroblasts isolated from Acta2fl/fl;R26eGFP mice and R26tdTomato mice (tdTomato is expressed in response to Adeno-Cre) mixed at a 1:1 ratio. The ratio remained the same after 48 hours of incubation, further suggesting that Acta2 deletion does not affect the proliferation of cardiac fibroblasts (Figure 2D).
Figure 2.

Cardiac fibroblast-specific deletion of Acta2 does not alter cardiac fibroblast proliferation after MI.
(A-B) Tamoxifen-treated Tcf21MCM/+;R26eGFP and Tcf21MCM/+;Acta2fl/fl;R26eGFP mice were subjected to MI and EdU based proliferation assays at 3 days and 7 days post-MI. IHC was performed to identify and quantify Tcf21 lineage-traced (eGFP+) cardiac fibroblasts that are positive for SMαA, Ki67, or EdU in the infarct region at 3 days (A) and 7 days (B) after MI. n=3 for each group. Scale bar: 20 μm. (C) ICC was performed to identify the expression of SMαA and Ki67 in WT and Acta2-null cardiac myofibroblasts. Nuclei are shown with Dapi. n=4 for each group. Scale bar: 100 μm. (D) Cardiac fibroblasts isolated from R26tdTomato (WT) and Acta2fl/fl;R26eGFP mice were transduced with Adeno-Cre and co-cultured at a 1:1 ratio. The ratio between WT and Acta2-null cardiac fibroblasts was calculated again after 48 hours of co-culture in the presence of TGFβ. n=3 for each group. Scale bar: 250 nm. **P < 0.01; ***P < 0.0001.
3.3. Cardiac myofibroblasts lacking Acta2 have normal motility, contractility, and matrix stabilization ability.
Besides the unaffected cardiac fibroblast proliferation, in vivo experiments using Tcf21MCM/+;R26eGFP and Tcf21MCM/+;Acta2fl/fl;R26eGFP mice also suggest that the deletion of Acta2 in cardiac fibroblasts does not affect their motility or contractility after MI (Figures 1F–I). To verify these results in more controlled environments, the effects of Acta2 deletion on the motility and contraction of cardiac myofibroblasts were tested in vitro. For a direct comparison of motility, Adeno-Cre-treated Acta2fl/fl;R26eGFP cardiac fibroblasts and R26tdTomato cardiac fibroblasts co-cultured at 1:1 ratio were monitored for their migration toward the center of the well. No difference was observed between the 2 groups in the presence or absence of TGFβ (Figure 3A). To better simulate the 3D matrix environment in the infarct, the same experiment was repeated with the addition of a layer of collagen gel cast on top of the cocultured cardiac myofibroblasts, which however also failed to show any significant difference (Figure 3A). To test the effect of Acta2 deletion on the contractility of cardiac myofibroblasts, a gel contraction assay was performed using cardiac fibroblasts isolated from Acta2fl/fl and WT mice and treated with Adeno-Cre and TGFβ, which showed no difference between the 2 groups (Figure 3B). It has been shown that cells can function as a crosslinker to strengthen the mechanical property of the matrix [33], a possible mechanism through which cardiac fibroblasts/myofibroblasts maintain the structural integrity of the infarct. Such a process likely involves the connection between actin stress fibers and ECM through focal adhesion. Thus, we employed rheology to test the impact of Acta2 deletion on the effect of cardiac fibroblasts on the stability of the collagen gel they reside in. In rheology, the storage modulus indicates the ability of the material to store deformation energy in an elastic manner while maintaining structural integrity and has been used to measure the strength of biomaterials [34–36]. Hydrogels with a greater level of crosslinking have a higher storage modulus [37]. Cardiac fibroblasts isolated from Acta2fl/fl and WT mice were treated with Adeno-Cre and TGFβ, mixed with collagen gel, and cultured for 24 hours to allow for the development of tension, followed by tests using a rheometer. The presence of cardiac fibroblasts increased the storage modulus of collagen gels (Figure 3C). However, no significant difference was observed in the storage modulus between collagen gels laden with WT myofibroblasts and those laden with Acta2-null myofibroblasts. This suggests that the loss of Acta2 does not affect the matrix stabilization capacity of myofibroblasts, which is in line with the similar post-MI survival rates between Tcf21MCM/+;R26eGFP and Tcf21MCM/+;Acta2fl/fl;R26eGFP mice.
Figure 3.

Deletion of Acta2 does not affect the motility, contractility, and matrix stabilization ability of cardiac fibroblasts.
(A) WT (tdTomato+) and Acta2-null (GFP+) cardiac myofibroblasts were seeded onto the same wells of 96-well plates at a 1:1 ratio. The migration of cells into the center of wells was quantified every day. n=3 for each group. Scale bar: 300 μm. (B-C) WT and Acta2-null cardiac myofibroblasts were mixed with collagen gel and poured onto 24 well plates. (B) Gels were released from wells after 12 hours of incubation. Images and quantification show the degree of contraction of gels 48 hours after the release of gels. n=3 for each group. (C) The storage moduli of cell-free gels and cell-laden gels 24 hours after incubation were measured using a rheometer. n=5 for each group.
3.4. Acta2 deletion in cardiac fibroblasts does not prevent myofibroblast differentiation.
Studies using global Acta2 KO mice showed that the deletion of Acta2 did not seem to prevent the formation of filamentous actin (F-actin) stress fiber in myofibroblasts derived from fibroblasts of several different organs [18–20]. To test if this was also the case in cardiac fibroblasts, cardiac fibroblasts were isolated from Acta2fl/fl and WT mice, treated with Adeno-Cre, and induced for myofibroblast differentiation using TGFβ. ICC using an antibody against SMαA combined with phalloidin staining showed that the amount of F-actin stress fibers was not significantly different between WT and Acta2-null cardiac myofibroblasts (Figure 4A). To specifically study the effect of Acta2 deletion on the myofibroblast differentiation of Tcf21 lineage-traced cardiac fibroblasts, cardiac fibroblasts were isolated from tamoxifen-treated Tcf21MCM/+;Acta2fl/fl;R26eGFP and Tcf21MCM/+;R26eGFP mice and cultured in a medium supplemented with TGFβ. ICC showed the lack of SMαA expression in most Tcf21 lineage-traced cardiac myofibroblasts from Tcf21MCM/+;Acta2fl/fl;R26eGFP mice but not in Tcf21 lineage-traced cardiac myofibroblasts from Tcf21MCM/+;R26eGFP mice (Figure 4B). However, no significant difference was observed in the total F-actin level between Tcf21 lineage-traced cardiac myofibroblasts of the 2 groups of mice (Figure 4B). Similarly, on day 7 after MI the amount of F-actin stress fibers in Tcf21 lineage-traced cardiac fibroblasts in the infarct scar of Tcf21MCM/+;Acta2fl/fl;R26eGFP mice was similar compared to that in Tcf21MCM/+;R26eGFP mice (Figure 4C). Stress fibers guide the maturation of focal adhesion [38]. We then questioned if the lack of SMαA stress fiber in cardiac fibroblasts had an effect on the formation of focal adhesion. ICC using an anti-vinculin antibody showed that Acta2 deletion did not significantly affect the abundance of mature focal adhesion (Figure 4D). These results suggest that the deletion of Acta2 in cardiac fibroblasts does not prevent myofibroblast differentiation.
Figure 4.

Deletion of Acta2 does not prevent the myofibroblast differentiation of cardiac fibroblasts.
(A) WT and Acta2-null cardiac myofibroblasts were subjected to ICC to identify the expression of SMαA and presence of F-actin using an anti-SMαA antibody and phalloidin, respectively. Nuclei are shown with Dapi. Scale bar: 100 μm. The fluorescence intensity was determined using ImageJ. n=3 for each group. **P < 0.01. (B) Cardiac fibroblasts isolated from tamoxifen-treated Tcf21MCM/+;R26eGFP and Tcf21MCM/+;Acta2fl/fl;R26eGFP mice were treated with TGFβ for 2 days and subjected to ICC to identify the expression of SMαA and presence of F-actin. Nuclei are shown with Dapi. Images represent 3 independent replications. Scale bar: 40 μm. (C) IHC was performed to identify Tcf21 lineage-traced (eGFP+) cardiac fibroblasts that are positive for SMαA or/and phalloidin in Tcf21MCM/+;R26eGFP and Tcf21MCM/+;Acta2fl/fl;R26eGFP mice at post-MI day 7. Nuclei are shown with Dapi. Images represent 3 analyzed hearts per group. Scale bar: 20 μm. (D) WT and Acta2-null cardiac myofibroblasts were subjected to ICC to identify the expression of SMαA and vinculin using specific antibodies. Nuclei are shown with Dapi. n=4 for each group. Scale bar: 40 μm. The average area of focal adhesion was measured using ImageJ. (E-F) Western blot (E) and ICC (F) were performed to quantify SMαA and total actin protein levels in WT and Acta2-null cardiac myofibroblasts using an anti-SMαA antibody and a pan-actin antibody, respectively. Scale bar: 100 μm. n=3 (western blot) or n=4 (ICC) for each group; **P < 0.01.
3.5. Compensatory effects of non-SMαA actin isoforms in Acta2-null cardiac myofibroblasts.
Besides Acta2/SMαA, 5 other actin isoforms are also expressed in mice, including Acta1 (skeletal muscle alpha-actin, SkMαA), Actb (cytoplasmic beta-actin, CyβA), Actc1 (cardiac muscle alpha-actin, CMαA), Actg1 (cytoplasmic gamma-actin, CyγA), and Actg2 (smooth muscle gamma-actin, SMγA). To study the impact of Acta2 deletion on the total actin level in cardiac fibroblasts, Acta2fl/fl and WT cardiac fibroblasts treated with Adeno-Cre and induced for myofibroblast differentiation were subjected to western blot and ICC using a pan-actin antibody that recognizes all actin isoforms (Figures 4EF). It was found that the deletion of Acta2 did not affect the total actin protein level in cardiac myofibroblasts. The unaffected F-actin and total actin levels in Acta2-null cardiac fibroblasts suggest some compensatory effects mediated by other actin isoforms. To explore this possibility, we first examined the relative expression levels of different actin isoforms in cardiac fibroblasts after MI using our recently published RNAseq data obtained using Tcf21 lineage-traced cardiac fibroblasts isolated from uninjured hearts and infarct scars at different days after MI [31]. The result showed that even though Acta2 was the most upregulated actin isoform in cardiac fibroblasts after MI by fold change, the expression of Actb, Actg1, and Actg2 were also upregulated (Figure 5A). Moreover, in cardiac myofibroblasts, Actg1 was expressed at a level comparable to Acta2 and the expression level of Actb was about 2.5 folds of that of Acta2 (Figure 5A). Adeno-Cre- and TGFβ-treated WT and Acta2fl/fl cardiac fibroblasts were then compared to study the impact of Acta2 KO on the expression of other actin isoforms by realtime PCR. A unique upregulation in the expression of Acta1 and Actg2 was identified in Acta2-null cardiac myofibroblasts as compared to the WT control (Figure S2). RNAseq was also performed so that the expression level between different actin isoforms can be compared as well. Consistent with results of realtime PCR, RNAseq identified significant compensatory increases in the expression of Acta1 and Actg2 in Acta2-null cardiac fibroblasts (Figure 5B). Interestingly, a slight but significant increase in the expression of Actb and Actg1 was also observed (Figure 5B). A similar small increase in the expression of Actc1 was identified as well, however, the difference was not significant due to its extremely low expression and high in-group variance (Figure 5B). Moreover, similar to RNAseq performed using WT fibroblasts isolated from infarct scars, the expression of cytoplasmic actin isoforms, Actb and Actg1, were several folds higher than that of Acta2 (Figure 5B). Consistently, Tcf21 lineage-traced cardiac fibroblasts sorted from the infarct scar of Tcf21MCM/+;Acta2fl/fl;R26eGFP mice at post-MI day 7 showed increased expression of Actg1 and Actg2, and a trend of increase in the expression of Acta1 compared to cells from Tcf21MCM/+;R26eGFP mice (Figure 5C). The expression of Actc1 was undetectable due to its extremely low expression. To understand the effect of Acta2 deletion on the protein level of other actin isoforms, we performed ICC using antibodies against different actin isoforms. Staining with an antibody recognizing smooth muscle actin isoforms (SMαA and SMγA) showed that in WT cardiac myofibroblasts the combined expression level of SMαA and SMγA was largely correlated with the expression level of SMαA and the combined protein level of SMαA and SMγA in Acta2-null cardiac myofibroblasts was significantly lower than that in WT cardiac myofibroblasts, suggesting that SMαA is the dominant smooth muscle actin isoform in cardiac myofibroblasts (Figures 5DE). However, some Acta2-null cardiac myofibroblasts (20.7% ± 2.8, n=3) had a total smooth muscle actin level similar to WT cardiac myofibroblasts, suggesting a strong compensatory increase in the expression of SMγA in these cells (Figure 5D). It is worth noting that due to the lack of an antibody specific to SMγA, we were unable to identify the exact percentage of cells with upregulated SMγA expression. The actual percentage of Acta2-null cardiac myofibroblasts with upregulated SMγA expression was likely much higher. Co-staining using an antibody that recognizes sarcomeric actin isoforms (SkMαA and CMαA) showed that the expression of sarcomeric actin in most Acta2-null cardiac myofibroblasts (80.1% ± 6.4, n=3) was higher than that in WT cardiac myofibroblasts (Figures 5DE). These findings are in line with the realtime PCR and RNAseq results (Figure 5BC). In addition, we noticed that the strong compensatory increase in the expression of SMγA and upregulated expression of sarcomeric actin isoforms only coexisted in a small fraction of Acta2-null cardiac myofibroblasts (4.5% ± 0.75, n=3) (Figure 5D), which suggests that the adaption of cardiac fibroblasts to the deletion of Acta2 varies among individual cells. We then tested the effect of Acta2 deletion on the expression of cytoplasmic actin isoforms in cardiac myofibroblasts. Western blot showed similar expression levels for both CyβA and CyγA between WT and Acta2-null cardiac myofibroblasts (Figure 5F). However, ICC showed that the formation of F-actin by cytoplasmic actin isoforms, especially CyγA, was enhanced in Acta2-null cardiac myofibroblasts as compared to WT cardiac myofibroblasts (Figure 5G). These results suggest the existence of compensatory effects mediated by multiple non-SMαA actin isoforms in Acta2-null cardiac myofibroblasts.
Figure 5.

The compensatory effects of non-SMαA actin isoforms in Acta2-null cardiac myofibroblasts.
(A) Tcf21 lineage-traced cardiac fibroblasts sorted from uninjured hearts and the infarct region at 3 days and 7 days after MI were subjected to RNAseq. The normalized transcription levels (TPM) of Acta2, Acta1, Actb, Actc1, Actg1, and Actg2 are shown. n=2 for each group. *P < 0.05; ***P < 0.0001 vs uninjured. (B) The transcription levels of Acta2, Acta1, Actb, Actc1, Actg1, and Actg2 in cultured WT and Acta2-null cardiac myofibroblasts were revealed by RNAseq. n=2 for each group. **P < 0.01. (C) Tcf21 lineage-traced cardiac fibroblasts were sorted from the infarct scar of Tcf21MCM/+;R26eGFP and Tcf21MCM/+;Acta2fl/fl;R26eGFP mice at post-MI day 7. The expression level of Acta2, Acta1, Actg1, Actg2, Col1a1, Col3a1, and Postn were revealed by realtime PCR. n=4 for each group. *P < 0.05; **P < 0.01. (D-E) WT and Acta2-null cardiac myofibroblasts were subjected to ICC to identify the protein level of SMαA, the combined protein level of SkMαA and CMαA, and the combined protein level of SMαA and SMγA using specific antibodies (D). Nuclei are shown with Dapi. Higher-magnification images highlight the areas marked by solid-line boxes (color-coded). Scale bar: 100 μm. The fluorescence intensity was measured using Image J (E). n=3 for WT; n=4 for Acta2-null. **P < 0.01; ***P < 0.0001. (F-G) Western blot (F) and ICC (G) were performed to quantify SMαA, CyβA, and CyγA protein levels in WT and Acta2-null cardiac myofibroblasts using specific antibodies. Higher-magnification images highlight the areas marked by yellow solid-line boxes. n=3 for each group (F). Images represent 3 independent replications (G). Scale bar: 100 μm.
Another major function of myofibroblasts is the secretion of ECM proteins. The expression of Col1a1, Col3a1, and Postn in Tcf21 lineage-traced cardiac fibroblasts sorted from infarct scars of Tcf21MCM/+;R26eGFP and Tcf21MCM/+;Acta2fl/fl;R26eGFP mice at post-MI day 7 were measured. No difference in Col1a1 and Col3a1 expression was identified (Figure 5G), which agreed with the picrosirius red staining data (Figure 1J). However, slightly lower expression of Postn was identified in cells from Tcf21MCM/+;Acta2fl/fl;R26eGFP mice.
3.6. The compensatory increase in the expression of other muscle actin isoforms in Acta2-null cardiac myofibroblasts does not require SRF/MRTFA.
Serum response factor (SRF) and its cofactor, myocardin-related transcription factor-A (MRTFA), promote myofibroblast differentiation and stress fiber formation [39, 40]. To explore the role of SRF/MRTFA in the compensatory increase of the expression of other muscle actin isoforms in Acta2-null cardiac myofibroblasts, we first performed ATACseq, which identified SRF binding motifs in accessible chromatin regions in and around Acta2, Actg2, and Acta1 (Figures S3A–C; Table S1). Moreover, increased accessibility was identified in a region immediately downstream of Actg2 and adjacent to an SRF motif in Acta2-null cardiac myofibroblasts as compared to WT cardiac myofibroblasts (Figure S3B). These results suggest that SRF/MRTFA may regulate the expression of muscle actin isoforms and is involved in the compensatory increase in the expression of non-Acta2 actin isoforms in Acta2-null cardiac myofibroblasts. To test these possibilities, we knocked down Mrtfa in WT and Acta2-null cardiac fibroblasts using Lenti-shMrtfa and then induced myofibroblast differentiation. ICC showed that KD of Mrtfa significantly reduced the expression of SMαA and the abundance of F-actin but had no effect on the total actin level (Figures 6A–C). Realtime PCR showed that besides the reduced expression of Acta2, KD of Mrtfa also reduced the expression of Acta1 and Actg2 but did not affect the expression of cytoplasmic actin isoforms, Actb and Actg1 (Figure 6D). Interestingly, an increase in the expression of Actc1 was observed in Mrtfa KD cardiac fibroblasts likely due to a compensatory increase induced by the downregulated expression of all other muscle actin isoforms (Figure 6D). Surprisingly, despite the reduced expression of Actg2 and Acta1, Mrtfa KD did not affect the compensatory increase in the expression of these actin isoforms caused by Acta2 KO (Figure 6D). Moreover, a slight increase in the expression of Actg1 was observed in Mrtfa KD Acta2-null cardiac myofibroblasts likely due to a compensatory effect induced by the seriously disrupted expression of muscle actin isoforms by Acta2 KO and Mrtfa KD (Figure 6D). ICC performed using antibodies against specific actin isoforms largely agreed with realtime PCR results, showing that Mrtfa KD reduced the expression of muscle actin isoforms but did not affect the expression of cytoplasmic actin isoforms or the compensatory increase in the expression of sarcomeric actin isoforms (Figures 6E–G).
Figure 6.

Knocking down of Mrtfa reduces the expression of muscle actin isoforms but does not affect the compensatory effects of non-SMαA actin isoforms in Acta2-null cardiac myofibroblasts.
WT and Acta2-null cardiac fibroblasts were treated with Lenti-GFP or Lenti-shMrtfa and induced for myofibroblast differentiation using TGFβ for 2 days. (A-C) ICC was performed to identify the expression of SMαA (A-B), total F-actin level (by phalloidin) (A), and total actin level (B). Scale bar: 40 μm (A), 100 μm (B). The fluorescence intensity was measured using Image J (C). n=4 for each group. Different letters indicate samples with significant differences (P < 0.05). (D) Realtime PCR was performed to identify the transcription levels of Acta2, Acta1, Actb, Actc1, Actg1, and Actg2. n=3 for each group. Different letters indicate samples with significant differences (P< 0.05). (E-G) ICC was performed to identify the expression of SMαA (F-G), SkMαA/CMαA (F), SMα/γA (F), CyβA (G), and CyγA (G). The fluorescence intensity was measured using Image J (E). Different letters indicate samples with significant differences (P < 0.05). n=4 for each group. Scale bar: 100 μm (F) and 40 μm (G).
4. DISCUSSION
Myofibroblasts with a highly developed microfilament system were first identified in skin wounds [41]. Later studies found that these cells express an elevated level of SMαA which is incorporated into stress fibers [42–44]. Ever since, SMαA stress fiber has been used as a marker for myofibroblast differentiation. Due to the specific expression of SMαA in myofibroblasts, it was speculated that SMαA stress fiber may play an important role in multiple myofibroblast activities involved in wound healing such as contraction, motility, and myofibroblast proliferation. However, in this study, using a newly generated mouse line with tamoxifen-inducible cardiac fibroblast-specific deletion of Acta2, we identified that the loss of Acta2 did not significantly affect the myofibroblast differentiation of cardiac fibroblasts. No difference in contractility, matrix stabilization ability, proliferation, or motility was observed between WT and Acta2-null cardiac myofibroblasts. In line with results obtained in vitro, mice lacking Acta2 in cardiac fibroblasts also had normal post-MI survival rate, cardiac repair, and cardiac function. The unaffected myofibroblast differentiation of Acta2-null cardiac fibroblasts was likely due to the increased expression or stress fiber formation of non-SkMαA actin isoforms.
Due to the contractile function of vascular smooth muscle cells, which also express a high level of SMαA, it is reasonably expected that SMαA is required for the contraction of myofibroblasts. In infarcted hearts, the tonic contraction of cardiac myofibroblasts is thought to negatively affect cardiomyocyte contraction [45, 46]. However, the tonic contraction of stress fibers in cardiac myofibroblasts may also stabilize the infarcted myocardium through cross-linking the surrounding matrix in cooperation with focal adhesion [15, 33, 38], which may be especially important before the scar ECM is mature enough to prevent cardiac rupture. A similar role of myofibroblast contraction may also present in other types of tissue after injury. Thus, the role of SMαA in myofibroblast contraction has received some special attention. It was found that overexpression of Acta2 increased the contractility of 3T3 fibroblasts more significantly than the overexpression of Actb, Actg1, or Actc1, even though the incorporation of all actin isoforms into stress fibers was identified [47]. Disruption of the SMαA stress fiber structure using an SMαA fusion peptide containing the N-terminal of SMαA [48] reduced adhesion and contractility of rat embryo fibroblast cell line REF-52 [16]. Besides these early studies of the function of SMαA stress fibers that were mainly performed in vitro using cell lines, the function of SMαA stress fibers in primary myofibroblasts and their role during tissue healing were recently studied by a few groups using whole-body Acta2 KO mice [18–20]. Deletion of Acta2 was reported to reduce the contractility of hepatic stellate cells-derived myofibroblasts [20]. However, another study reported that mice lacking Acta2 had normal skin wound closure and the contractility of skin fibroblasts isolated from these mice was not different from those isolated from WT mice [19], which is in line with the current study showing the lack of difference in contractility between WT and Acta2-null cardiac myofibroblasts. The unchanged myofibroblast contractility may also explain the unaffected post-MI survival rate and cardiac remodeling in cardiac fibroblast-specific Acta2 KO mice.
Besides the contractile function of myofibroblasts, previous studies have also investigated the effects of SMαA stress fibers on myofibroblast motility, proliferation, and ECM production. A study inhibiting SMαA function using a neutralization antibody identified that the motility of breast tissue-derived myofibroblasts was enhanced when SMαA function was inhibited [15]. Another study found that renal fibroblasts isolated from Acta2-null mice had enhanced motility and proliferation in vitro [18]. In addition, the same study also identified an elevated level of collagen production by Acta2-null renal myofibroblasts, which exacerbated renal fibrosis. In contrast, the deletion of Acta2 reduced the collagen expression by myofibroblasts Acta2-null hepatic stellate cells [20]. Our study, however, did not identify a significant change in any of these myofibroblast functions and activities in cardiac myofibroblasts owing to Acta2 deletion. Interestingly, we found that cardiac fibroblasts from Tcf21MCM/+;Acta2fl/fl;R26eGFP mice expressed a slightly lower level of Postn compared to cells from Tcf21MCM/+;R26eGFP mice after MI. Even though the underlying mechanism remains to be studied, this slight reduction in Postn expression did not seem to alter the overall post-MI healing of the heart.
The reason for the large discrepancy among results generated by different studies of the function of SMαA may be multifaceted. First, it is well known that immortalized cell lines, which were used by many early studies, often act significantly differently from primary cells. Second, the experimental procedure of studies using SMαA fusion peptide and neutralization antibodies often depend on the treatment-induced acute effects that suddenly disrupt SMαA stress fiber function followed by functional analyses. This strategy likely did not provide other actin isoforms with enough time to develop a compensatory effect. Indeed, even though Acta2-null mice had normal skin wound contraction [19], a study applying SMαA fusion peptide to skin wounds in vivo significantly inhibited rat skin wound contraction [49]. Third, the inconsistent results of research focusing on fibroblasts or fibroblast-like cells in different organs using Acta2 KO mice likely reflect the difference in the adaptation of these cells to Acta2 deletion. Unlike Acta2-null cardiac and skin myofibroblasts that have normal contractility, a study using Acta2 KO mice reported a reduction in the contractility of myofibroblasts derived from hepatic stellate cells [20]. In the current study, significant increases in the transcription of Actg2 and Acta1, 2 other muscle actin isoforms, were identified in Acta2-null cardiac myofibroblasts as compared to WT cardiac myofibroblasts. Similar increases in the expression of other muscle actin isoforms were also identified in Acta2-null skin myofibroblasts [19]. Such increases were however not identified in myofibroblasts derived from Acta2-null hepatic stellate cells [20]. Instead, an increase in the expression of cytoplasmic actin isoforms was detected in these cells compared to their WT counterparts. It is possible that without the guidance of stress fibers formed by muscle actin isoforms the ability of non-muscle actin isoforms to form stress fibers is relatively limited, which likely contributed to the more impacted contractile function of Acta2-null hepatic stellate cell-derived myofibroblasts. Interestingly, besides the increased expression of non-SMαA muscle actin isoforms, we found that the formation of stress fibers by cytoplasmic actin isoforms was also increased in Acta2-null cardiac myofibroblasts even though the changes in the expression of these actin isoforms were rather small. Given the much lower expression level of non-Acta2 muscle actin isoforms compared to cytoplasmic actin isoforms even considering the compensatory increase in their expression, cytoplasmic actin isoforms are likely also very critical for the normal myofibroblast differentiation of Acta2-null cardiac fibroblasts. However, it should be noted that due to the unavailability of antibodies specific to several actin isoforms that work in western blot, we were not able to accurately access the levels of different actin isoforms incorporated into filamentous actin strands.
SRF/MRTFA have been identified as key factors promoting myofibroblast differentiation [39, 40], Our study confirmed the important role of SRF/MRTFA in the expression of muscle actin isoforms and the formation of stress fibers in cardiac myofibroblasts. However, disruption of the SRF/MRTFA signaling failed to affect the compensatory increase in the expression of non-Acta2 actin isoforms despite the identification of SRF binding motifs in and around these genes. Such a surprising observation indicates the presence of other underlying mechanisms that remain to be identified. Our ATACseq analysis also identified the enrichment of some other motifs in actin genes such as the ones targeted by basic helix-loop-helix transcription factors or bZIP domain-containing transcription factors, whose possible contribution to the compensatory increase of actin gene expression can be studied in the future (Table S1). Other possible mechanisms also include posttranscriptional regulation that stabilizes the mRNA of actin genes.
Taken together, our results indicate that the expression of Acta2/SMαA is not required for the myofibroblast differentiation of cardiac fibroblasts and their activities in post-MI cardiac repair, which is at least partially due to the compensatory increase in the expression of other actin isoforms and the functional redundancy between SMαA and non-SMαA actin isoforms.
One limitation of this study was the suboptimal efficiency of Tcf21-MerCreMer-mediated Acta2 KO, likely due to the relatively low activity of Tcf21 promoter driving MerCreMer expression and the low chromatin accessibility around the loxP sites when Acta2 is not expressed in cardiac fibroblasts without MI. To increase the KO efficiency, we had to include another course of tamoxifen treatment starting immediately before MI. Even with prolonged tamoxifen treatment, suboptimal deletion of Acta2 in cardiac fibroblasts in the border zone was still observed in some mice. It is possible that the lower vascular permeability in the border zone compared to the scar area, owing to the lower grade of inflammation, caused a lower local concentration of active tamoxifen metabolites. Moreover, we identified GFP− cardiac fibroblasts in the scar with normal SMαA expression, which was likely also due to the presence of cardiac fibroblasts of different lineages [50, 51] and, again, the relatively low efficiency of Tcf21-MerCreMer. The possible compensation of cardiac fibroblasts without Acta2 deletion might have affected our ability to reveal some subtle effects on post-MI hearts, which can only be detected when Acta2 is deleted in all cardiac fibroblasts. A study employing a strategy inhibiting the formation of stress fibers by all actin isoforms in all cardiac fibroblasts is needed in the future to better decipher the function of actin stress fibers in myofibroblasts and post-injury cardiac repair.
Supplementary Material
6. FUNDING SOURCES
This work was supported by the Louisiana Board of Regents under grant BOR.Fu.LEQSF(2019-22)-RD-A-01 (X.F.); NIH/NIDDK under grant 1R15DK122383 (X.F.); NIH/NIGMS P20GM130555 (X.F.); and NIH/NHLBI under 1R01HL157519 (X.F.) and R01HL142217 (J.M.).
Footnotes
7. DECLARATION OF INTEREST
None.
8. REFERENCES
- [1].Sheifer SE, Gersh BJ, Yanez ND 3rd, Ades PA, Burke GL, Manolio TA, Prevalence, predisposing factors, and prognosis of clinically unrecognized myocardial infarction in the elderly, J. Am. Coll. Cardiol 35(1) (2000) 119–26. [DOI] [PubMed] [Google Scholar]
- [2].Yeh RW, Sidney S, Chandra M, Sorel M, Selby JV, Go AS, Population trends in the incidence and outcomes of acute myocardial infarction, N. Engl. J. Med 362(23) (2010) 2155–65. [DOI] [PubMed] [Google Scholar]
- [3].Yellon DM, Hausenloy DJ, Myocardial reperfusion injury, New Engl. J. Med 357(11) (2007) 1121–1135. [DOI] [PubMed] [Google Scholar]
- [4].Fu X, Khalil H, Kanisicak O, Boyer JG, Vagnozzi RJ, Maliken BD, Sargent MA, Prasad V, Valiente-Alandi I, Blaxall BC, Molkentin JD, Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart, J. Clin. Invest 128(5) (2018) 2127–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Davis J, Molkentin JD, Myofibroblasts: trust your heart and let fate decide, J. Mol. Cell. Cardiol 70 (2014) 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hinz B, Myofibroblasts, Exp. Eye Res 142 (2016) 56–70. [DOI] [PubMed] [Google Scholar]
- [7].Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, SC JL, Aronow BJ, Tallquist MD, Molkentin JD, Genetic lineage tracing defines myofibroblast origin and function in the injured heart, Nat. Commun 7 (2016) 12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Darby I, Skalli O, Gabbiani G, a-Smooth muscle actin is transiently expressed by myofibroblasts during experimental wound healing, Lab. Invest 63(1) (1990) 21–9. [PubMed] [Google Scholar]
- [9].Leslie KO, Taatjes DJ, Schwarz J, vonTurkovich M, Low RB, Cardiac myofibroblasts express alpha smooth muscle actin during right ventricular pressure overload in the rabbit, Am. J. Pathol 139(1) (1991) 207–216. [PMC free article] [PubMed] [Google Scholar]
- [10].Xiang F-L, Fang M, Yutzey KE, Loss of β-catenin in resident cardiac fibroblasts attenuates fibrosis induced by pressure overload in mice, Nat. Commun 8(1) (2017) 712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Khalil H, Kanisicak O, Vagnozzi RJ, Johansen AK, Maliken BD, Prasad V, Boyer JG, Brody MJ, Schips T, Kilian KK, Correll RN, Kawasaki K, Nagata K, Molkentin JD, Cell-specific ablation of Hsp47 defines the collagen-producing cells in the injured heart, JCI Insight 4(15) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bageghni SA, Hemmings KE, Yuldasheva NY, Maqbool A, Gamboa-Esteves FO, Humphreys NE, Jackson MS, Denton CP, Francis S, Porter KE, Ainscough JFX, Pinteaux E, Drinkhill MJ, Turner NA, Fibroblast-specific deletion of IL-1 receptor-1 reduces adverse cardiac remodeling following myocardial infarction, JCI Insight 4(17) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kaur H, Takefuji M, Ngai CY, Carvalho J, Bayer J, Wietelmann A, Poetsch A, Hoelper S, Conway SJ, Mollmann H, Looso M, Troidl C, Offermanns S, Wettschureck N, Targeted Ablation of Periostin-Expressing Activated Fibroblasts Prevents Adverse Cardiac Remodeling in Mice, Circ. Res 118(12) (2016) 1906–17. [DOI] [PubMed] [Google Scholar]
- [14].Hinz B, Celetta G, Tomasek JJ, Gabbiani G, Chaponnier C, Alpha-smooth muscle actin expression upregulates fibroblast contractile activity, Mol. Biol. Cell 12(9) (2001) 2730–2741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ronnov-Jessen L, Petersen OW, A function for filamentous alpha-smooth muscle actin: retardation of motility in fibroblasts, J. Cell Biol 134(1) (1996) 67–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Hinz B, Dugina V, Ballestrem C, Wehrle-Haller B, Chaponnier C, α-Smooth muscle actin is crucial for focal adhesion maturation in myofibroblasts, Mol. Biol. Cell 14(6) (2003) 2508–2519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zaidel-Bar R, Cohen M, Addadi L, Geiger B, Hierarchical assembly of cell–matrix adhesion complexes, Biochem. Soc. Trans 23(Pt3) (2004) 416–420. [DOI] [PubMed] [Google Scholar]
- [18].Takeji M, Moriyama T, Oseto S, Kawada N, Hori M, Imai E, Miwa T, Smooth muscle alpha-actin deficiency in myofibroblasts leads to enhanced renal tissue fibrosis, J. Biol. Chem 281(52) (2006) 40193–40200. [DOI] [PubMed] [Google Scholar]
- [19].Tomasek JJ, Haaksma CJ, Schwartz RJ, Howard EW, Whole animal knockout of smooth muscle alpha-actin does not alter excisional wound healing or the fibroblast-to-myofibroblast transition, Wound Repair Regen. 21(1) (2013) 166–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Rockey DC, Du Q, Shi Z, Smooth Muscle α-Actin Deficiency Leads to Decreased Liver Fibrosis via Impaired Cytoskeletal Signaling in Hepatic Stellate Cells, Am. J. Pathol 189(11) (2019) 2209–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Acharya A, Baek ST, Banfi S, Eskiocak B, Tallquist MD, Efficient inducible Cre-mediated recombination in Tcf21 cell lineages in the heart and kidney, Genesis 49(11) (2011) 870–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yamamoto M, Shook NA, Kanisicak O, Yamamoto S, Wosczyna MN, Camp JR, Goldhamer DJ, A multifunctional reporter mouse line for Cre- and FLP-dependent lineage analysis, Genesis 47(2) (2009) 107–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].van Berlo JH, Kanisicak O, Maillet M, Vagnozzi RJ, Karch J, Lin SC, Middleton RC, Marbán E, Molkentin JD, c-kit+ cells minimally contribute cardiomyocytes to the heart, Nature 509(7500) (2014) 337–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sriramula S, Haque M, Majid DSA, Francis J, Involvement of tumor necrosis factor-alpha in angiotensin II-mediated effects on salt appetite, hypertension, and cardiac hypertrophy, Hypertension (Dallas, Tex. : 1979) 51(5) (2008) 1345–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liu Y, Cseresnyés Z, Randall WR, Schneider MF, Activity-dependent nuclear translocation and intranuclear distribution of NFATc in adult skeletal muscle fibers, J. Cell Biol 155(1) (2001) 27–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, Vagnozzi RJ, Liu R, Huynh T, Lee S-J, Karch J, Molkentin JD, Fibroblast-specific TGF-β-Smad2/3 signaling underlies cardiac fibrosis, J. Clin. Investig 127(10) (2017) 3770–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Davis J, Salomonis N, Ghearing N, Lin SC, Kwong JQ, Mohan A, Swanson MS, Molkentin JD, MBNL1-mediated regulation of differentiation RNAs promotes myofibroblast transformation and the fibrotic response, Nat. Commun 6 (2015) 10084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Jiang W, Hua R, Wei M, Li C, Qiu Z, Yang X, Zhang C, An optimized method for high-titer lentivirus preparations without ultracentrifugation, Sci. Rep 5 (2015) 13875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Fu X, Zhu M, Zhang S, Foretz M, Viollet B, Du M, Obesity Impairs Skeletal Muscle Regeneration Through Inhibition of AMPK, Diabetes 65(1) (2016) 188–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Zhu MJ, Ford SP, Means WJ, Hess BW, Nathanielsz PW, Du M, Maternal nutrient restriction affects properties of skeletal muscle in offspring, The Journal of physiology 575(Pt 1) (2006) 241–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Li C, Sun J, Liu Q, Dodlapati S, Ming H, Wang L, Li Y, Li R, Jiang Z, Francis J, Fu X, The landscape of accessible chromatin in quiescent cardiac fibroblasts and cardiac fibroblasts activated after myocardial infarction, Epigenetics (2021) 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS, Model-based analysis of ChIP-Seq (MACS), Genome Biol. 9(9) (2008) R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Lee KY, Kong HJ, Larson RG, Mooney DJ, Hydrogel formation via cell crosslinking, Advanced Materials 15(21) (2003) 1828–1832. [Google Scholar]
- [34].Tunick MH, Malin EL, Smith PW, Shieh JJ, Sullivan BC, Mackey KL, Holsinger V, Proteolysis and rheology of low fat and full fat Mozzarella cheeses prepared from homogenized milk, Journal of Dairy Science 76(12) (1993) 3621–3628. [Google Scholar]
- [35].Jung JP, Lin WH, Riddle MJ, Tolar J, Ogle BM, A 3D in vitro model of the dermoepidermal junction amenable to mechanical testing, Journal of Biomedical Materials Research Part A 106(12) (2018) 3231–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Van Den Bulcke AI, Bogdanov B, De Rooze N, Schacht EH, Cornelissen M, Berghmans H, Structural and rheological properties of methacrylamide modified gelatin hydrogels, Biomacromolecules 1(1) (2000) 31–38. [DOI] [PubMed] [Google Scholar]
- [37].Grattoni CA, Al-Sharji HH, Yang C, Muggeridge AH, Zimmerman RW, Rheology and permeability of crosslinked polyacrylamide gel, J. Colloid Interface Sci 240(2) (2001) 601–607. [DOI] [PubMed] [Google Scholar]
- [38].Oakes PW, Beckham Y, Strieker J, Gardel ML, Tension is required but not sufficient for focal adhesion maturation without a stress fiber template, J. Cell Biol 196(3) (2012) 363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Velasquez LS, Sutherland LB, Liu Z, Grinnell F, Kamm KE, Schneider JW, Olson EN, Small EM, Activation of MRTF-A-dependent gene expression with a small molecule promotes myofibroblast differentiation and wound healing, Proc. Natl. Acad. Sci. U. S. A 110(42) (2013) 16850–16855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Small EM, Thatcher JE, Sutherland LB, Kinoshita H, Gerard RD, Richardson JA, Dimaio JM, Sadek H, Kuwahara K, Olson EN, Myocardin-related transcription factor-a controls myofibroblast activation and fibrosis in response to myocardial infarction, Circul. Res 107(2) (2010) 294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Gabbiani G, Ryan G, Majno G, Presence of modified fibroblasts in granulation tissue and their possible role in wound contraction, Experientia 27(5) (1971) 549–550. [DOI] [PubMed] [Google Scholar]
- [42].Darby I, Skalli O, Gabbiani G, a-Smooth muscle actin is transiently expressed by myofibroblasts during experimental wound healing, Lab. Invest 63(1) (1990) 21–29. [PubMed] [Google Scholar]
- [43].Jester JV, Rodrigues MM, Herman IM, Characterization of avascular corneal wound healing fibroblasts. New insights into the myofibroblast, Am. J. Pathol 127(1) (1987) 140–148. [PMC free article] [PubMed] [Google Scholar]
- [44].Leavitt J, Gunning P, Kedes L, Jariwalla R, Smooth muscle alpha-action is a transformation-sensitive marker for mouse NIH 3T3 and Rat-2 cells, Nature 316(6031) (1985) 840–2. [DOI] [PubMed] [Google Scholar]
- [45].Weber KT, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC, Myofibroblast-mediated mechanisms of pathological remodelling of the heart, Nat. Rev. Cardiol 10(1) (2013) 15–26. [DOI] [PubMed] [Google Scholar]
- [46].Kuijpers NH, ten Eikelder HM, Bovendeerd PH, Verheule S, Arts T, Hilbers PA, Mechanoelectric feedback leads to conduction slowing and block in acutely dilated atria: a modeling study of cardiac electromechanics, American Journal of Physiology-Heart and Circulatory Physiology 292(6) (2007) H2832–H2853. [DOI] [PubMed] [Google Scholar]
- [47].Hinz B, Celetta G, Tomasek JJ, Gabbiani G, Chaponnier C, Alpha-smooth muscle actin expression upregulates fibroblast contractile activity, Mol. Biol. Cell 12(9) (2001) 2730–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chaponnier C, Goethals M, Janmey PA, Gabbiani F, Gabbiani G, Vandekerckhove J, The specific NH2-terminal sequence Ac-EEED of alpha-smooth muscle actin plays a role in polymerization in vitro and in vivo, Journal of Cell Biology 130(4) (1995) 887–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Hinz B , Gabbiani G, Chaponnier C The NH2-terminal peptide of α–smooth muscle actin inhibits force generation by the myofibroblast in vitro and in vivo, J. Cell Biol 157(4) (2002) 657–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Moore-Morris T, Guimarães-Camboa N, Banerjee I, Zambon AC, Kisseleva T, Velayoudon A, Stallcup WB, Gu Y, Dalton ND, Cedenilla M, Gomez-Amaro R, Zhou B, Brenner DA, Peterson KL, Chen J, Evans SM, Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis, J. Clin. Invest 124(7) (2014) 2921–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Ali SR, Ranjbarvaziri S, Talkhabi M, Zhao P, Subat A, Hojjat A, Kamran P, Muller AM, Volz KS, Tang Z, Red-Horse K, Ardehali R, Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation, Circ. Res 115(7) (2014) 625–35. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNAseq data of cardiac fibroblasts isolated from uninjured hearts and infarct scars were from our previous study [31] (GEO #GSE186079). All new raw sequencing data have been deposited into the public database at NCBI (GEO #GSE193163).