

Abstract
Background
Malignant gliomas commandeer dense inflammatory infiltrates with glioma-associated macrophages and microglia (GAMM) promoting immune suppression, evasion, and tumor progression. Like all cells in the mononuclear phagocytic system, GAMM constitutively express the poliovirus receptor, CD155. Besides myeloid cells, CD155 is widely upregulated in the neoplastic compartment of malignant gliomas. Intratumor treatment with the highly attenuated rhino:poliovirus chimera, PVSRIPO, yielded long-term survival with durable radiographic responses in patients with recurrent glioblastoma (Desjardins et al. New England Journal of Medicine, 2018). This scenario raises questions about the contributions of myeloid versus neoplastic cells to polio virotherapy of malignant gliomas.
Methods
We investigated PVSRIPO immunotherapy in immunocompetent mouse brain tumor models with blinded, board-certified neuropathologist review, a range of neuropathological, immunohistochemical, and immunofluorescence analyses, and RNAseq of the tumor region.
Results
PVSRIPO treatment caused intense engagement of the GAMM infiltrate associated with substantial, but transient tumor regression. This was accompanied by marked microglia activation and proliferation in normal brain surrounding the tumor, in the ipsilateral hemisphere and extending into the contralateral hemisphere. There was no evidence for lytic infection of malignant cells. PVSRIPO-instigated microglia activation occurred against a backdrop of sustained innate antiviral inflammation, associated with induction of the Programmed Cell Death Ligand 1 (PD-L1) immune checkpoint on GAMM. Combining PVSRIPO with PD1/PD-L1 blockade led to durable remissions.
Conclusions
Our work implicates GAMM as active drivers of PVSRIPO-induced antitumor inflammation and reveals profound and widespread neuroinflammatory activation of the brain-resident myeloid compartment by PVSRIPO.
Keywords: glioma, immunotherapy, interferon, macrophages, microglia
Key Points.
Polio virotherapy causes brain-wide microglia activation independent of oncolysis
Sustained type I IFN responses elicit dramatic, but transient antitumor effects, after single-dose virotherapy
Remission of murine brain tumors after single-dose polio virotherapy requires PD-L1 blockade
Importance of the Study.
Virotherapy approaches for both childhood and adult glioma patients have been translated to the clinic. While viruses developed to target gliomas generally were selected for their direct lytic capacity, the extent to which “oncolysis” versus engagement of innate and adaptive immunity contributes to anti-glioma efficacy remains unclear. Moreover, mechanisms by which productive virotherapy is mediated in the glioma microenvironment remain unresolved. Here we demonstrate that PVSRIPO induces CNS-wide microglial inflammation independent of viral oncolysis. Sustained type I interferon responses were associated with transient antitumor effects, which coincided with PD-L1 induction on microglia and tumor-associated macrophages. Combining virotherapy with PD-L1 blockade mediated durable antitumor effects after polio virotherapy, in a manner dependent on CD8+ T cells. These data demonstrate an oncolysis-independent role for polio virotherapy in inducing antitumor immunity that is limited by immune resistance.
Malignant gliomas comprise heterogeneous groups of primary Central Nervous System (CNS) neoplasms that respond poorly to chemoradiation. They feature dense inflammatory infiltrates populated with glioma-associated macrophages and microglia (GAMM).1 Microglia—derived from the embryonic yolk sac2 and capable of self-renewal in the CNS3—and CNS-associated macrophages (CaM) are the principal innate immune cells of the CNS.4 Malignant gliomas recruit and subvert these cells for tumor-accommodating pro-growth, angiogenic and immune-skewing effects.5
Treatment of glioblastoma (GBM) with PVSRIPO achieved greater long-term survival versus criteria-matched external controls with Overall Survival at 36 months of 21% versus 4%.6 Long-term survival after PVSRIPO was associated with objective radiographic responses with a median duration of >60 months. Polioviruses (PVs) use human CD155 (hCD155) as their sole host cell receptor.7 Upon natural (oral) infection, PV targets intestinal/lymphoid CD11c+ dendritic cells (DCs)/macrophages8; lymphatic structures are the principal sites of PV replication in chimpanzees.9,10 Wild type (wt) PV is cytotoxic with rampant viral growth in DCs/macrophages in vivo8 and in vitro.11 In contrast, PVSRIPO is engineered for profound attenuation,12 evident as absent neuropathogenicity after high-dose intracerebral (IC) inoculation,6,13 and a peculiar noncytopathogenic phenotype in DCs/macrophages with marginal viral translation and propagation driving sustained innate antiviral type-I interferon (IFN) dominant inflammation.14–16
We inquired if PVSRIPO—analogous to wt PV tropism for myeloid cells—targets the GAMM infiltrate and CNS-resident myeloid cells. We used immuno-competent IC mouse tumor models for comprehensive histology, immunohistochemistry (IHC), and RNAseq studies. We report profound PVSRIPO-mediated GAMM engagement and tumor regression without signs of direct viral damage to the neoplastic compartment. Beyond GAMM, we observed microglia activation immersing the tumor periphery, the tumor-bearing hemisphere, and throughout contralateral brain. This was evident as microglia with increased cytoplasm and thickened ramified processes, highlighted on Trans-membrane protein 119 (Tmem119)17 and Ionized Ca++-binding adaptor protein 1 (Iba1)18 staining, and profuse microglia proliferation. This was associated with type-I/II IFN innate signaling gene signatures, and upregulation of PD-L1 on GAMM. Drastic, but transient, antitumor effects of the acute neuroinflammatory response to PVSRIPO resulted in durable remissions upon combination with PD1:PD-L1 immune checkpoint blockade (ICB).
Materials and Methods
Virus, Cells, and Transgenic Mice
We utilized PVSRIPO produced as a good-laboratory-practice lot suitable for research purposes. Infectious cDNA12 was digested with MluI, in vitro transcribed with T7 polymerase (Megascript, Thermo Fisher) and full-length viral RNA was transfected into HeLa cells with DMRIE-C in Opti-MEM (Thermo Fisher).19 Progeny virus recovered from transfected HeLa cells was quantified by plaque assay and amplified as described previously.19 Upon manifestation of cytopathic effects, the infected cultures were harvested, subjected to 3 freeze–thaw cycles, the lysate was spun at 14,000 g, and the resulting supernatant was filtered through 0.1 μm syringe filters (Pall Corp.), and subjected to centrifugation through a 100-kDa cutoff spin column (Millipore). We used CT2AhCD155 and B16hCD155 cells for I.C. implantation into C57Bl6 mice transgenic for human CD155 (hCD155-tg mice). Parental CT2A cells (a gift from Dr. P. Fecci, Duke Univ.) and B16.F10 (ATCC) were subjected to lentiviral transduction with hCD155 and fluorescence-activated cell sorting with α-CD155-PE (BioLegend). Cells, confirmed mycoplasma negative (Duke Cell Culture Facility), were grown in Dulbecco's Modified Eagle Medium (DMEM) (Invitrogen) with 10% fetal bovine serum (Sigma, #F0926) to 60%–70% confluency and harvested for I.C. implantation. Homozygous hCD155-tg mice (a gift from S. Koike, Tokyo Metropolitan Institute of Medical Science, Japan) are maintained as a breeding colony. For survival studies, 8–12 weeks old, ~17–25 g male and female mice were used, with roughly equal distribution in each treatment group; female mice were used for nonsurvival studies. Mice were housed in the Duke University Cancer Center Isolation facility under Biosafety Level 2 (BSL2) conditions with 12-hour light/dark cycles, relative humidity of 50 ± 20%, and temperature of 21 ± 3°C.
Intracranial Tumor Models
All animal procedures were performed under a Duke IACUC-approved vertebrate animal use protocol. Briefly, prior to surgery, hCD155-tg mice were shaved on the scalp and tagged on the left ear. During surgery, mice were under continuously inhaled isoflurane anesthesia and pain control (0.1 ml, 0.5% meloxicam S.C.). Mice were mounted onto stereotactic frames when there was no toe pinch response; a heating pad (37oC) was used to maintain body temperature. The scalp was sterilized (betadine + 70% ethanol), and a #10 blade scalpel incision was made along the sagittal suture. The surgical field was washed with 3% H2O2 to expose the bregma, which was set as the origin (x = 0, y = 0, and z = 0) of the coordinate system. A 50 μl microsyringe (Hamilton, #80901) prefilled with tumor cells homogenously suspended in 2.4% methylcellulose was fixed onto the frame, and a keyhole was created at (x = 2.0 mm and z = −0.5 mm) by microdrill, so the 30 Gy injecting needle could pierce through the skull freely, to a depth of 3.6 mm measuring from where the bevel tip intersects with the brain surface. Each mouse received 1 × 105 CT2AhCD155 cells (5 μl), or 1 × 103 B16hCD155 cells (5 μl), infused at 2.5 μl/min by an automatic injector. One min after completing the injection, the needle was slowly withdrawn, and the trepanation hole was sealed with bone wax (Covidien). The wound was closed with surgical glue (Vetclose, Henry Schein, Inc.) and mice were monitored during recovery until rolling over. PVSRIPO/mock treatment was performed on a day that is ~1/3 of the median survival for each tumor model (ie, day 6 for CT2AhCD155; day 5 for B16hCD155). Individual mice were divided into treatment groups by a random number generator, and intratumoral infusion of PVSRIPO (5 × 107 pfu in 5 μl serum-free DMEM)/mock [5 μl DMEM]) was given in the same manner as tumor implantation, through the keyhole created for tumor implantation and to the same depth of 3.6 mm. Following tumor implantation, mice were weighed every 2 or 3 days until reaching the humane endpoint, defined as >15% weight loss from the highest recorded weight, or until neurological symptoms (seizure, paralysis, etc.) were evident. Mice surviving long-term (3-times median survival, >60 days) were re-challenged with contralateral re-implantation with the same CT2A inoculum.
Quantification and Statistical Analysis
Survival data were analyzed by Kaplan Meier curves; when comparing 2 groups significance was determined by Log Rank test. For comparing histology measurements between treatment and control groups student t test with unequal variance was used; Cohen’s effect size of >0.8 was defined as a “large effect.” Other assay-specific statistical tests are indicated in the corresponding figure legends. P values of <.05 were considered significant. All statistical analyses were performed using Prism 9 software version 9.0; error bars represent Standard Error of the Mean. All data points reflect individual specimens, independent experimental repeats, or mice.
Results
Optimizing Mouse Intracerebral Tumor Models for Blinded Neuropathologist Review
CT2A is a methylcholanthrene-induced murine anaplastic astrocytoma20 that epitomizes key properties of human malignant glioma,21 such as intrinsic ICB resistance.22 Mouse CD155 does not function as a PV receptor; to recapitulate the clinical scenario,23 we transduced CT2A cells with hCD155. We use an established hCD155 transgenic (tg) C57Bl6 model as host for CT2AhCD155.24 hCD155-tg mice carry a transgene under control of the authentic 10 kB upstream region, exhibit hCD155 distribution similar to humans, and develop paralytic poliomyelitis upon PV infection resembling the human disease.24 PVSRIPO infection of bone-marrow-derived cells from hCD155-tg mice—generated with Granulocyte-Macrophage Colony Stimulating Factor (GM-CSF) (to mimic conventional DCs) or with FLT3 ligand (to mimic Basic leucine zipper ATF-like Transcription Factor 3 [Batf3+] DCs)—and in primary human monocyte-derived DCs, yielded similar viral translation- and host innate inflammatory patterns.16 To optimize IC tumor models for histology of all brains in all cohorts, we adjusted multiple variables, for example, needle size/use of a drill for trepanation, inoculation size and depth, for consistency and for minimizing injection tract infiltration/extra-parenchymal growth. With these measures, we achieved 100% tumor take with 93.3% of tumors contained within the injected hemisphere per gross pathological exam (Supplementary Table 2).
PVSRIPO Therapy Mediates Early Treatment Effects in CT2A Gliomas
Six days after IC CT2AhCD155 implantation in hCD155-tg mice, a single stereotactic infusion of PVSRIPO (5 × 107 pfu) or mock (DMEM) was done. This interval was chosen because CT2AhCD155 tumors reached sizeable, established striatal growths at day 6 (Figure 1). In three independent executions of this experiment, tumor-bearing brains were harvested at baseline (day 6), and from PVSRIPO/mock-treated cohorts at days 8, 10, 12, 15, or 18. Brains of all mice (n = 55) enrolled in the 3 series were collected for histology (Supplementary Table 3). Because some mice reached humane endpoints (>15% weight loss) on/before day 18, the systematic part of this study (n = 49) stopped at day 15 to avoid attrition bias. Coronal formalin-fixed, paraffin-embedded (FFPE) sections of the tumor site—using the needle track and anterior commissure as landmarks—were H&E stained for posttherapy histology in comparison with baseline and time-matched mock controls. Sample micrographs were assembled into a composite image to illustrate the treatment response to PVSRIPO over time (Figure 1). This revealed a dramatic, but transient, antitumor effect evident as a distinct histological appearance and reduced tumor size at day 8, followed by tumor relapse by day 15 (Figure 1). At baseline (day 6), the average tumor cross-sectional area was 1.2 mm2, which expanded to 6.0 mm2 by day 8 in the mock-, versus 2.5 mm2 in the PVSRIPO arm (Figure 2C). At day 15, the average cross-sectional areas of tumors were comparable in the mock and PVSRIPO arms (Figure 2C). Occasionally, the early treatment response yielded complete pathological remission (1/7 samples at day 15; Supplementary Figure 1). Divergent treatment outcomes may be due to the variable distribution of IC infusates in mice, leading to tumor satellites in some animals (Figure 1). No remissions were observed in the control cohort.
Figure 1.
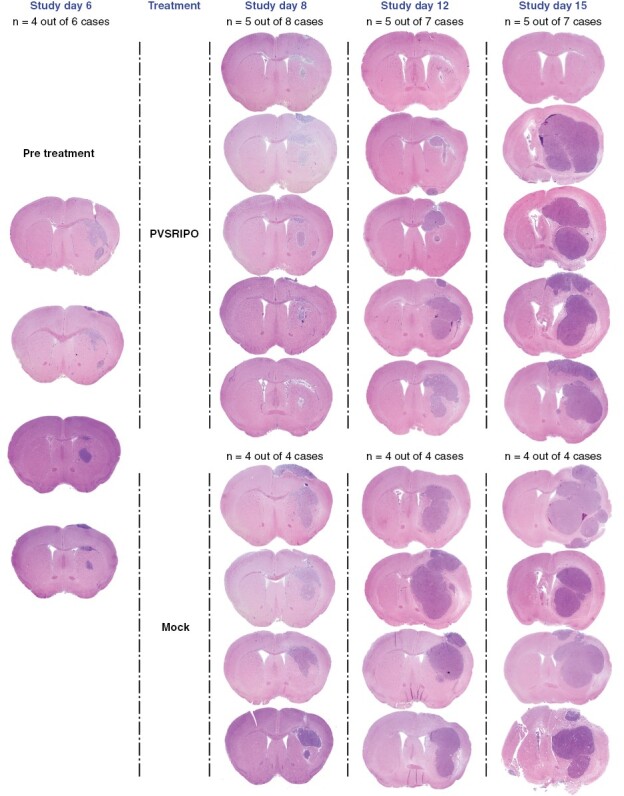
PVSRIPO therapy mediates drastic, but transient antitumor effects. CT2AhCD155 glioma cells implanted into the right hemisphere (day 0) gave rise to established gliomas on day 6 (baseline), at which time intratumor infusion of PVSRIPO (5 × 107 pfu; in 5 μl DMEM) or mock (5 μl DMEM) was instituted. Tumor-bearing brains were collected at baseline (n = 6), and post therapy (PVSRIPO/mock) at days 8 (n = 12), 10 (n = 9), 12 (n = 11), and 15 (n = 11) for histological analysis (Supplementary Table 3 for a listing of all samples). In each sample, a coronal FFPE section through the tumor site was H&E stained; tumors were evident as right hemisphere hypercellular masses. Micrographs showing the typical therapy response over time are displayed in the composite image, together with samples from baseline and matched controls.
Figure 2.
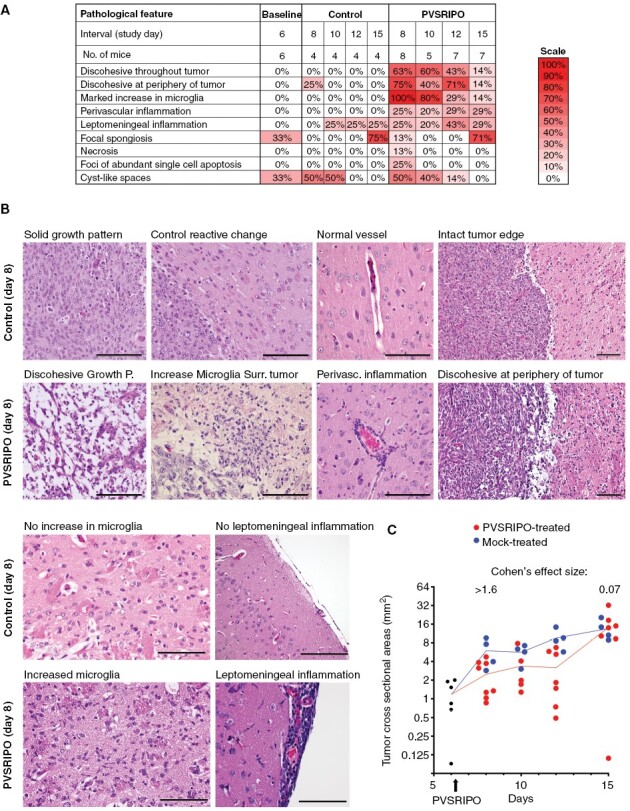
PVSRIPO treatment induces a transient histological response with sustained microglia inflammation. (A) To characterize the histologic features of PVSRIPO therapy in the CT2AhCD155 model (Figure 1), H&E-stained coronal FFPE sections at the tumor implantation site from each tumor-bearing brain, collected at the indicated intervals, were subjected to blinded review by a board-certified pathologist. Each slide was evaluated for 9 defined pathological features, and the results were summarized in color scales showing the prevalence of these features among all treated/control samples. (B) Certain histologic features were characteristic for PVSRIPO therapy, as shown in exemplary microscopic images compared to matched controls. Scale bars = 100 μm. (C) In each slide, the tumor cross-sectional area (mm2) was calculated and plotted along the time course.
PVSRIPO Induces Marked Microglia Activation
To evaluate the PVSRIPO treatment effect noted in H&E staining of whole brain sections, FFPE sections from 49 cases (Supplementary Table 3) were de-identified and submitted for blinded Board-certified neuropathologist (G.Y.L.) review (Figure 2). This analysis revealed four key features distinguishing PVSRIPO-treated brains from mock controls: (1) marked increase in microglia; (2) discohesiveness throughout tumor; (3) discohesiveness at periphery of tumor; and (4) perivascular inflammation (Figure 2A and B).
The predominant histological characteristic of the PVSRIPO response, present in all samples, was marked increase in microglia (Figure 2A). Microglia were evaluated in peritumoral brain parenchyma. Microglial activation was defined as aggregates of rod-shaped nuclei located immediately adjacent to the tumor. To meet the criteria for an increased density of activated microglia, exclusion of other key cell types was required. The cells with rod-shaped nuclei had to lack visible cytoplasm. Abundant eosinophilic cytoplasm with ramified processes would support activated astrocytes, while abundant foamy cytoplasm would raise a differential of bone-marrow-derived macrophages. Neither feature was present in regions identified as having activated microglia. In mock-treated tumors and at baseline, only a mild increase in microglia surrounding the needle track was found relative to contralateral normal brain.
PVSRIPO Causes Diffuse Activation of Microglia Throughout the CNS
Histological evidence for a microglia response to PVSRIPO requires rigorous confirmation of microglia identity, for example, with IHC markers Iba1 and Tmem119. Since Iba1 is also induced in activated CaM or infiltrating peripheral monocytes,25 we relied on Tmem119, which specifically marks microglia,17 for example, in the context of RNA virus CNS infection.25
To eliminate the tumor as a source for Iba1+ monocytic infiltration, we assessed microglia in nontumor-bearing brains treated with PVSRIPO versus mock (Figure 3). Iba1 and Tmem119 IHC demonstrated diffuse microglial activation, characterized as increased cytoplasm and thickened ramified processes, throughout the PVSRIPO-treated hemisphere (Figure 3A). In mock-treated brains, Iba1+ cells occurred at the injection site only; these cells likely represent infiltrating monocytes, as they did not stain for Tmem119 in PVSRIPO/mock-treated mice (Figure 3A). CNS-wide distribution of Iba1+/Tmem119+ cells in PVSRIPO-treated brains, excluding the injection site (Figure 3A), provides definitive evidence for diffuse microglia activation by PVSRIPO. Higher magnification images of Iba1 (Figure 3B) and Tmem119 (Figure 3C) staining in cortex (distant from the injection site) revealed Tmem119+ cells with morphologic changes characteristic of activated microglia only in PVSRIPO-treated brains (Figure 3C).
Figure 3.
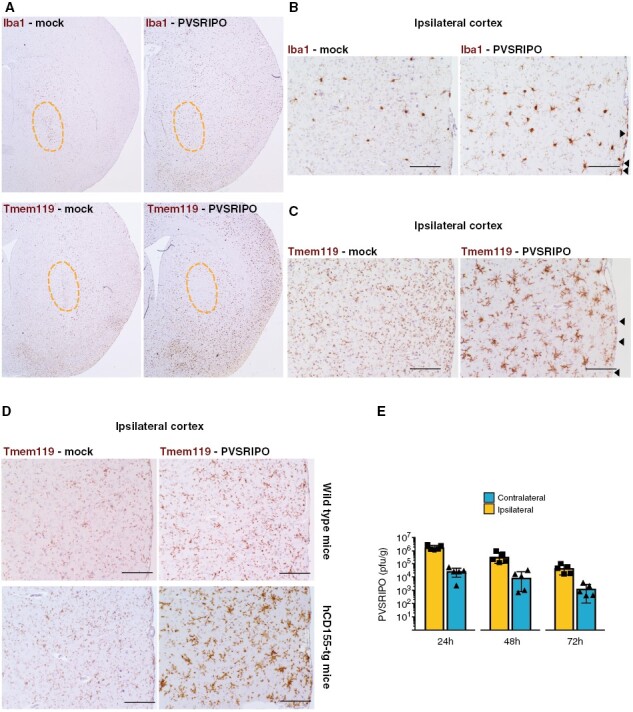
PVSRIPO induces diffuse microglia activation. (A–C) Coronal brain sections after PVSRIPO or mock injection (day 8) stained for Iba1 or Tmem119. (A) The injected hemispheres of mock-treated brains show Iba1+ (top left), Tmem119- (bottom left) cells clustering around the needle track (orange outline). In PVSRIPO-treated brains, there was diffuse hemispheric Iba1+ (top right) and Tmem119+ signal (bottom right); the needle track was devoid of Tmem119+ cells. (B, C) In ipsilateral cortex distant from the needle track, Iba1 (B) and Tmem119 (C) marked microglia with increased cytoplasm and broad, thickened ramified processes in PVSRIPO-treated brains (right panels). Note Iba1+ meningeal macrophages in PVSRIPO-treated brains (B; arrowheads) that were Tmem119- (C; arrowheads). A microglial reaction and Iba1+ meningeal macrophages were absent in mock-treated brains. (D) Non-tumor bearing wild type (top) and hCD155-tg (bottom) mice received IC inoculations of mock vs. PVSRIPO and ipsilateral cortex was analyzed by Tmem119 IHC 2 days after. (E) PVSRIPO recovery from the ipsi- and contralateral hemispheres of CT2AhCD155-harboring mice after intratumoral PVSRIPO 1-3 days post treatment.
Microglia physiologically express CD15515 and, thus, are susceptible to PVSRIPO infection. To examine infection as a factor in microglia morphological change, we compared Tmem119 staining in PVSRIPO/mock-treated (nontumor bearing) wild-type versus hCD155-tg mice. The microglia activation pattern induced by PVSRIPO (Figure 3C) was recapitulated only in hCD155-tg mice (Figure 3D), indicating that direct microglia infection is at least partially responsible for this phenotype. PVSRIPO was readily recovered from ipsi- and contralateral hemispheres at 24–72 hours after intratumoral inoculation in CT2A-bearing hCD155-tg mice (Figure 3E), possibly explaining global microglia activation engulfing virtually the entire brain.
Confirmation of PVSRIPO Antitumor Effects in an Intracerebral B16 Model
To corroborate our findings in CT2AhCD155 murine glioma, we selected the IC B16 (spontaneous mouse melanoma) model transduced with hCD155.15 PVSRIPO/mock treatment of IC B16hCD155-harboring mice was done on day 5 (vs. day 6 with CT2AhCD155) and brains were harvested on day 7 (vs. day 8) due to shorter median survival (~14.5 days vs. ~19.5 days for CT2AhCD155). Blinded neuropathologist review of mock (n = 2) versus PVSRIPO-treated (n = 3) B16hCD155-bearing brains revealed treatment effects similar to CT2AhCD155: mock-treated tumors had clearly delineated borders with minimal inflammation, whereas PVSRIPO-treated tumors were smaller, discohesive, admixed with TAMM inflammation, and prominent peritumoral and perivascular inflammation (Supplementary Figure 2A and B). The PVSRIPO antitumor effect noted in CT2AhCD155 was evident in Ki67 IHC (Supplementary Figure 2C) and CD68 IHC in the tumor bed and surrounding brain (Supplementary Figure 2D). CD68 and Iba1 mark both macrophages and microglia. CD68 and Iba1 staining patterns were similar in intracerebral B16hCD155 tumors pre/post PVSRIPO (compare Supplementary Figures 2 and 3).
PVSRIPO Engages GAMM and Causes Widespread Microglia Proliferation
Having confirmed the microglial nature of Iba1+ cells by highly ramified morphology and Tmem119 positivity (Figure 3), PVSRIPO/mock-treated CT2AhCD155 gliomas dissected on day 8 (n = 2 mock/4 PVSRIPO) were studied with multiplex IF staining of Iba1, Ki67, PD-L1 (Figure 4A and B) and confirmatory Iba1 IHC (Figure 4C and D). We focused on three regions: tumor core, tumor-adjacent brain parenchyma close to the anterior commissure (Figure 4A–D), distant contralateral cortex (Figure 4C and D). Iba1 staining delineated tissue-resident microglia with typical ramified processes that emanate from a cell body in peritumoral brain (Figure 4A–D). With PVSRIPO, the presence of activated microglia was pervasive—both in peritumoral brain and in contralateral cortex—evident in all virus-treated brains, but none of the controls (Figure 4B and D). We confirmed microglia identity by Tmem119 staining in adjacent sections (data not shown). Furthermore, in mock-treated animals, ipsilateral normal brain was devoid of Ki67 staining (Figure 4A), while in PVSRIPO-treated animals Iba1+ cells with highly ramified processes co-stained with Ki67 (Figure 4B), indicating widespread active microglia proliferation.
Figure 4.
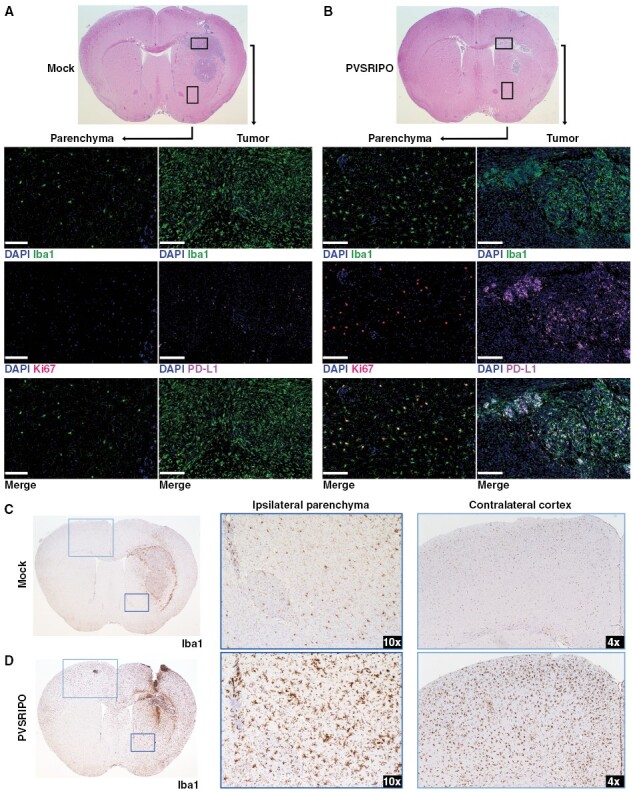
PVSRIPO therapy engaged GAMM and caused widespread activation and proliferation of tissue-resident microglia. (A, B) Representative cases from day 8 post mock (A) or PVSRIPO (B) (Figure 1) were analyzed by IF. Areas representing the malignant lesion and ipsilateral brain parenchyma are indicated by boxes. Staining with DAPI (nuclear stain), Iba1, Ki67 (proliferation marker), PD-L1 in isolation or merged is indicated. Overlapping Iba1 and PD-L1 staining is evident as white signal in the merged (B, lower right) panel. Scale bars = 100 μm. (C, D) Representative cases from day 8 post mock (C) or PVSRIPO (D) were analyzed for Iba1 IHC to document microglia morphology. Two areas studied at higher magnification in ipsilateral parenchyma (dark blue frame), or contralateral cortex (light blue frame) are indicated, highlighting widespread microglia activation with increased cytoplasm and thickened processes in the PVSRIPO-treated brains (B, D) compared to mock-treated brains (A, C).
Beyond microglia activation, PVSRIPO induced profound pro-inflammatory engagement in the tumor microenvironment (TME). In mock-treated animals, PD-L1 staining in the tumor proper was sparse and punctate (Figure 4A). In contrast, abundant PD-L1 co-staining with Iba1 in the tumor bed in PVSRIPO-treated mice indicated type-I IFN activation of GAMM (PD-L1 is an IFN-stimulated gene; PVSRIPO induces PD-L1 in human DCs in vitro16;Figure 4B). Iba1+ cell activation extending to ipsilateral brain and the contralateral hemisphere (Supplementary Figure 3B and D), and microglia proliferation in normal brain parenchyma (Supplementary Figure 3F) were also observed in all cases of IC B16hCD155 treated with PVSRIPO that were submitted for multiplex IF analysis (n = 3).
Next, we analyzed sections from PVSRIPO/mock-treated CT2AhCD155 gliomas with multiplex IF of CD45, PD-L1, and Ki67 (Figure 5). In mock-treated animals, the CD45+ compartment remained well organized, interwoven with and mostly restricted to the neoplastic lesion itself (Figure 5A), with sparse and punctate PD-L1 signal (Figure 5B). Accordingly, Ki67 staining was intense in the neoplastic lesion, with some scattered signal in the peritumoral periphery (Figure 5C). In PVSRIPO-treated animals CD45 staining was no longer confined to the malignant lesion, but extended far into brain parenchyma, forming a vast, undulating inflammatory reaction that engulfed much of the tumor-bearing hemisphere (Figure 5D), replete with PD-L1 signal (Figure 5E). Ki67+ cells were greatly diminished in the tumor itself (in accordance with reduced tumor size; Figure 2C) and were present throughout the hemisphere (Figure 5F). In PVSRIPO-treated animals, Ki67+ cells located outside the tumor region largely co-stained with Iba1 and demonstrated highly ramified processes, suggesting proliferating microglia (Figure 4B; Supplementary Figure 3F); or Ki67+ cells co-stained with CD3 in a tight perinuclear pattern, suggesting proliferating T cells (Supplementary Figure 6B). Calculation of the density of CD45+ cells in CT2AhCD155 tumor-adjacent normal brain demonstrated that PVSRIPO treatment induced a ~10-fold influx of such cells into peritumoral parenchyma; the same effect was observed in the B16hCD155 model (Supplementary Figure 4A). Automated quantification of CD45, Iba1, PD-L1, CD3, and Ki67 signal in CT2AhCD155 tumor and in ipsilateral brain of mice treated with mock/PVSRIPO are shown in Supplementary Figure 4B and C. These findings were corroborated by flow cytometry of mock vs. PVSRIPO-treated CT2A gliomas, where an overall increase in tumor-infiltrating CD45+ cells was mainly due to CD11b+ cells (ie, macrophages/microglia) that expressed elevated levels of Ki67+ and PD-L1+ (Supplementary Figure 5A–D). Consistent with histological evaluations, these proinflammatory changes exhibited by CNS/glioma myeloid compartments on study day 2 were diminished on study day 6 post PVSRIPO (Supplementary Figure 5E–H).
Figure 5.
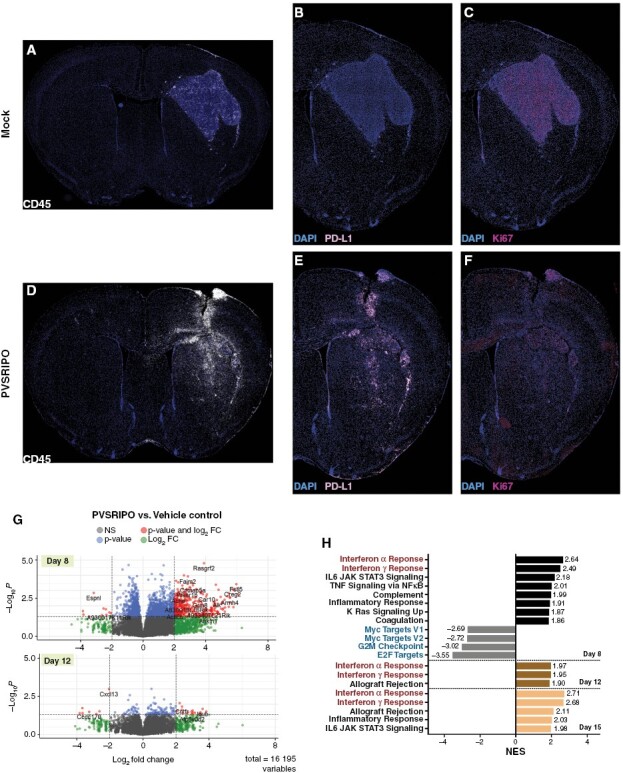
PVSRIPO immunotherapy induces type-I IFN dominant inflammatory responses in the TME. Mock (A–C) and PVSRIPO-treated (D–F) brains (day 8) were stained for CD45, Ki-67 and PD-L1 as shown. (G) Volcano plots depicting differentially expressed genes 2- and 6 days post PVSRIPO (log2 fold change cutoff = 2.0; adjusted P-value cut off = .05). (H) GSEA Hallmark genesets altered by PVSRIPO therapy over time, ranked by normalized enrichment score (NES). Only significant (false discovery rate-adjusted) genesets with NES>1.8 are shown.
Increased Intra- and Extra-tumoral T Cell Density and Proliferation After PVSRIPO
Co-staining with CD3 suggested that some of the Ki67+ cells in PVSRIPO/mock-treated CT2A-bearing brains were T lymphocytes (Supplementary Figure 6). However, in contrast to mock-treated mice, where Ki67+ CD3+ cells occurred mostly within the tumor bed (Supplementary Figure 6A), such cells were disseminated throughout the peritumoral circumference of PVSRIPO-treated CT2AhCD155 (Supplementary Figure 6B) and B16hCD155-bearing mice (Supplementary Figure 7). Flow cytometry analyses of CT2A glioma-bearing hemispheres revealed T cell (Tconv, CD8+) phenotypes associated with proliferation (Ki67), cytolytic activity (GzmB), and reduced exhaustion markers (PD1, TIM3) 6 days after PVSRIPO treatment (Supplementary Figure 8).
PVSRIPO Induces Acute IFN-Dominant Inflammation
To study PVSRIPO-induced transcriptomic alterations, we selected 20 CT2AhCD155 specimens (Supplementary Table 3) collected at different intervals pre and post PVSRIPO/mock infusion. Tumor region RNA was recovered from FFPE tissue sections for bulk RNA sequencing. Differential expression analysis revealed PVSRIPO-induced up-regulation of 380 and down-regulation of 10 genes (applying a log2-fold change cutoff of 2.0 and P-value cutoff of 0.05) compared to mock controls at day 8 (Figure 5G). This ratio dropped to 18 and 7 genes, respectively, at day 12 (Figure 5G). Consistent with the antitumor and inflammatory effects observed, PVSRIPO induced a profound but transient transcriptional response at 48 hours post treatment. Gene set enrichment analysis (GSEA) interrogating Hallmark Genesets26 revealed several recurring genesets in PVSRIPO-treated brains, led by “IFNα response” and “IFNγ response.” Meanwhile, cell cycle progression genesets (“Myc targets V1/2,” “G2M checkpoint,” “E2F targets”) were downregulated (Figure 5H). These results are consistent with histological evidence for tumor regression and a broad, macrophage/microglia-centered innate antiviral inflammatory reaction.
We employed the mouse version of the Microenvironment Cell Populations (MCP)-counter algorithm27 to calculate the abundance of tissue-infiltrating immune and stromal cells over time (Supplementary Figure 10A). PVSRIPO induced infiltration of myeloid-derived innate immune cells at day 8, followed by infiltration of CD8+T cells and memory B cells on day 12. The total amount of immune/stromal infiltrates diminished over time, in step with progressive resolution of the acute inflammatory response elicited by PVSRIPO. RNAseq also confirmed the presence of viral RNA (vRNA) (Supplementary Figure 10B), consistent with virus recovery from the brains of treated mice (Figure 3E).
Combination of PVSRIPO With PD1:PD-L1 ICB Improves Long-term Antitumor Efficacy in a CD8+T Cell-Dependent Manner
Robust type-I IFN-dominant antiviral inflammation/PD-L1 induction against the backdrop of a profoundly engaged myeloid infiltrate, suggests that PVSRIPO’s antitumor effects could be buttressed with PD1:PD-L1 ICB. We assessed antitumor efficacy of PVSRIPO combined with α-PD-L1 (Figure 6A) or α-PD1 (Supplementary Figure 11). To mimic the clinical scenario with near-universal preexisting anti-PV immunity and a PV vaccine boost,6 and to evaluate if preexisting immunity precludes therapy, mice were immunized against PV 47 and 32 days prior to CT2AhCD155 implantation (Figure 6A). CT2AhCD155 tumor-bearing mice were treated with mock/PVSRIPO followed by α-PD-L1 or isotype-matched control antibodies as shown. Combining PVSRIPO with α-PD-L1 had a minor effect on median survival (24 days vs. 19 days for the mock + IgG group), but a significant effect on overall survival (OS) with 36% complete remission at day 60 (Figure 6A). PVSRIPO + α-PD-L1 therapy-induced remission was resistant to contralateral rechallenge with CT2A (Figure 6A), which was lethal in age-matched, treatment-naïve control mice. OS extension by PVSRIPO + α-PD-L1 was eliminated upon CD8+T cell depletion (Figure 6B).
Figure 6.
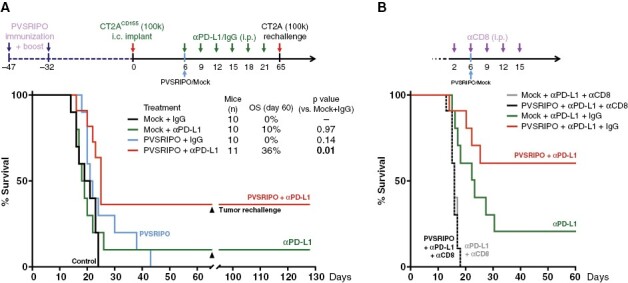
PVSRIPO combination with PD-L1 ICB yielded long-term remission in CT2AhCD155 glioma in a CD8+T cell-dependent manner. (A) hCD155-tg mice were immunized against PV, implanted with CT2AhCD155, and treated with PVSRIPO/mock combined with anti-PD-L1 or isotype control (250 μg per dose) as shown. Mice in remission were re-challenged with 105 CT2AhCD155 cells in the contralateral hemisphere (day 65) in parallel with age-matched treatment-naive mice as controls (n = 5). P-values are from Mantel–Cox log-rank test, two tailed. (B) CD8+T cell depletion eliminated PVSRIPO + anti-PD-L1 and anti-PD-L1 therapy.
Discussion
From their inception,28 viral anti-glioma strategies were predicated on a concept of viral cyto-toxicity in neoplastic cells, based on robust viral growth in in vitro tumor models. In this context, CNS resident innate immune cells often were deemed an obstacle to therapy due to their role in clearing viral agents administered intratumorally.29
PVSRIPO, by virtue of widespread upregulation of CD155 in malignancy,30 including malignant glioma,23,31 infects and damages cancerous cells via innate antiviral IFN responses32 and cytotoxic viral proteins in vitro.33,34 Yet, studies in ex vivo GBM slices, a clinically relevant model with authentic cellular heterogeneity, show that neoplastic cells are not the main target for its biological activities.15 Our studies in IC mouse tumor models confirm that PVSRIPO mainly provokes responses in the GAMM and CNS innate myeloid compartments in vivo, unleashing type-I IFN dominant engagement of the glioma immune infiltrate. This reverberates with PV’s natural cell type-specificity for migratory/lymphoid DCs and macrophages.8
PVSRIPO therapy of IC mouse tumors caused drastic antitumor effects, for example, reduced Ki67+ cells in the tumor bed, discohesive architecture, and a decline in cell cycle-related gene expression. Absent virus/vRNA accumulation in the tumor all but excludes the possibility of direct viral cell killing to account for this. This is consistent with effective PVSRIPO therapy of hCD155-tg hosts (implying PVSRIPO targeting of myeloid cells) occurring with hCD155- tumor implants.15 Indeed, CD8+T cell depletion indicates that PVSRIPO-instigated antitumor effects are immune mediated. Yet, we cannot rule out a role for viral infection and cytopathogenic damage to malignant cells in the antitumor effect, even if it is sub-lethal and nonproductive.
The main histological finding after PVSRIPO was marked, diffuse microglia activation. Microglia are the bulwark of the CNS antiviral defense.35 Viral encephalitis, for example, the syndromes modeled in mice with rhabdo- (vesicular stomatitis virus),25 flavi- (eg, West Nile virus),36 or neurotropic coronaviruses,37 share common features implicated in microglia activation.35 They each infect neurons and inflict neuronal damage; they infect and damage astro- and/or oligodendrocytes; they induce peripheral monocytic infiltration in the CNS. Microglia activation occurs focally, adjacent to these events.38 PVSRIPO CNS infection lacks all these triggers. PVSRIPO is devoid of cytopathogenic potential in neurons,39 and is nonneuropathogenic in humans,6 nonhuman primates,13 and hCD155-tg mice.40 PVs do not infect astro-/oligodendrocytes. IC PVSRIPO did not induce peripheral monocyte infiltration beyond the needle track. Thus, PVSRIPO may induce microglia activation via means that are not related to CNS damage.
Unambiguously identifying microglia in the virus-infected CNS is difficult, especially differentiating microglia from Iba1+ CaM or infiltrating monocytes.35 Tmem119 staining of cells with morphological features of activated microglia defines this cell type to respond to PVSRIPO. There was profuse proliferation of such cells, evident as Ki67+ co-staining, in the brain. Microglia activation and proliferation induced by PVSRIPO was diffuse and extensive, consistent with the absence of focal viral cytopathogenicity in the CNS. Rather, this pattern of microglia activation may reflect PVSRIPO’s nonlethal phenotype in cells of the mononuclear phagocytic system yielding sustained type-I IFN dominant inflammation.14–16 Accordingly, the prevailing transcriptional signatures emerging from RNAseq analyses of the tumor region post PVSRIPO were antiviral inflammatory in nature.
The dramatic early treatment effects of PVSRIPO were transient and, in ~90% of cases, gave rise to lethal tumor progression. Our studies suggest that turning acute inflammatory antiviral reactions into durable remission may be achieved by combining PVSRIPO with immune-modulatory agents capable of blocking natural resistance points of the antiviral immune response, for example, with anti-PD-L1 or PD1 ICB.
Our investigations demonstrate that, rather than a detriment to cancer virotherapy, GAMM may be active participants in the antitumor effects elicited by virus challenge of the TME and the innate inflammatory signaling cascades this elicits. Beyond proinflammatory engagement of the tumor myeloid infiltrate, the quality, depth, and anatomical reach of PVSRIPO-induced microglia activation in peritumoral brain may be indispensable for generating functional antitumor CD8+T cell immunity in malignant gliomas, notorious immune deserts intrinsically resistant to systemic immunotherapy approaches.41 A pivotal objective of future investigations is to resolve the contributions of diverse (resident and infiltrative) myeloid subsets responding to PVSRIPO immunotherapy with endogenous type-I IFN signaling14–16 to cancer immune surveillance in the CNS.
Supplementary Material
Acknowledgments
We thank Z. Su and H. Dai for technical assistance with IHC; P. Healy for statistical assistance; S. Koike (Tokyo Metropolitan Institute of Medical Science, Japan) for providing hCD155-tg mice; S. Keir for animal training, and J. Bryant and, M. Mosaheb for assistance with animal surgeries. We thank R.E. McLendon for critical reading of the manuscript.
Contributor Information
Yuanfan Yang, Department of Pathology, Duke University School of Medicine, Durham, North Carolina, USA; Department of Neurosurgery, Duke University Medical School, Durham, NC, USA; Department of Neurosurgery, University of Alabama at Birmingham, Birmingham, Alabama, USA.
Michael C Brown, Department of Neurosurgery, Duke University Medical School, Durham, NC, USA.
Gao Zhang, Department of Pathology, Duke University School of Medicine, Durham, North Carolina, USA; Department of Neurosurgery, Duke University Medical School, Durham, NC, USA.
Kevin Stevenson, Department of Neurosurgery, Duke University Medical School, Durham, NC, USA.
Malte Mohme, Department of Neurosurgery, University Medical Center Hamburg-Eppendorf, Hamburg, Germany.
Reb Kornahrens, Department of Neurosurgery, Duke University Medical School, Durham, NC, USA.
Darell D Bigner, Department of Pathology, Duke University School of Medicine, Durham, North Carolina, USA; Department of Neurosurgery, Duke University Medical School, Durham, NC, USA.
David M Ashley, Department of Neurosurgery, Duke University Medical School, Durham, NC, USA.
Giselle Y López, Department of Pathology, Duke University School of Medicine, Durham, North Carolina, USA; Department of Neurosurgery, Duke University Medical School, Durham, NC, USA.
Matthias Gromeier, Department of Neurosurgery, Duke University Medical School, Durham, NC, USA.
Funding
This research was supported by Public Health Service Grant R01 NS108773 (M.G.); F32 CA224593 and K99 CA263021 (M.C.B.); a National Cancer Center Trainee fellowship (Y.Y.); and by the NIH National Center for Advancing Translational Sciences Award 1KL2TR002554 (G.Y.L.).
Conflict of interest statement. M.G. and D.D.B. hold equity in-, are paid consultants of- and are inventors of intellectual property licensed to Istari Oncology, Inc. M.C.B. is a paid consultant of- and an inventor of intellectual property licensed to Istari Oncology, Inc. All other authors report no competing interests.
Authorship statement. Experimental design, performance of investigations, data analyses: Y.Y., M.C.B., G.Z., K.S., M.M., R.K., M.G.; Histopathological analyses, histological scoring, immunohistochemistry analyses: Y.Y., G.Y.L.; Animal experiments: Y.Y., M.C.B., M.M., R.K.; Bioinformatics (RNAseq): Y.Y., M.C.B., G.Z., K.S.; Expert feedback, consultation: D.D.B., D.M.A.; Manuscript editing: Y.Y., G.Y.L., M.C.B., D.D.B., M.G.; Study supervision: G.Y.L., M.G.
References
- 1. Morantz RA, Wood GW, Foster M, Clark M, Gollahon K.. Macrophages in experimental and human brain tumors. Part 2: studies of the macrophage content of human brain tumors. J Neurosurg. 1979;50(3):305–311. [DOI] [PubMed] [Google Scholar]
- 2. Ginhoux F, Greter M, Leboeuf M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Huang Y, Xu Z, Xiong S, et al. Repopulated microglia are solely derived from the proliferation of residual microglia after acute depletion. Nat Neurosci. 2018;21(4):530–540. [DOI] [PubMed] [Google Scholar]
- 4. Ransohoff RM, Brown MA.. Innate immunity in the central nervous system. J Clin Invest. 2012;122(4):1164–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Poon CC, Sarkar S, Yong VW, Kelly JJP.. Glioblastoma-associated microglia and macrophages: targets for therapies to improve prognosis. Brain. 2017;140(6):1548–1560. [DOI] [PubMed] [Google Scholar]
- 6. Desjardins A, Gromeier M, Herndon JE, et al. Recurrent glioblastoma treated with recombinant poliovirus. N Engl J Med. 2018;379(2):150–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mendelsohn CL, Wimmer E, Racaniello VR.. Cellular receptor for poliovirus: molecular cloning, nucleotide sequence, and expression of a new member of the immunoglobulin superfamily. Cell. 1989;56(5):855–865. [DOI] [PubMed] [Google Scholar]
- 8. Shen L, Chen CY, Huang D, et al. Pathogenic Events in a nonhuman primate model of oral poliovirus infection leading to paralytic poliomyelitis. J Virol. 2017;91(14):e02310–e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bodian D. Emerging concept of poliomyelitis infection. Science. 1955;122(3159):105–108. [DOI] [PubMed] [Google Scholar]
- 10. Sabin AB. Pathogenesis of poliomyelitis; reappraisal in the light of new data. Science. 1956;123(3209):1151–1157. [DOI] [PubMed] [Google Scholar]
- 11. Wahid R, Cannon MJ, Chow M.. Dendritic cells and macrophages are productively infected by poliovirus. J Virol. 2005;79(1):401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gromeier M, Alexander L, Wimmer E.. Internal ribosomal entry site substitution eliminates neurovirulence in intergeneric poliovirus recombinants. Proc Natl Acad Sci U S A. 1996;93(6):2370–2375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dobrikova EY, Goetz C, Walters RW, et al. Attenuation of neurovirulence, biodistribution, and shedding of a poliovirus:rhinovirus chimera after intrathalamic inoculation in Macaca fascicularis. J Virol. 2012;86(5):2750–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Brown MC, Holl EK, Boczkowski D, et al. Cancer immunotherapy with recombinant poliovirus induces IFN-dominant activation of dendritic cells and tumor antigen-specific CTLs. Sci Transl Med. 2017;9(408):eaan4220-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Brown MC, Mosaheb MM, Mohme M, et al. Viral infection of cells within the tumor microenvironment mediates antitumor immunotherapy via selective TBK1-IRF3 signaling. Nat Commun. 2021;12(1):1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mosaheb MM, Dobrikova EY, Brown MC, et al. Genetically stable poliovirus vectors activate dendritic cells and prime antitumor CD8 T cell immunity. Nat Commun. 2020;11(1):524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bennett ML, Bennett FC, Liddelow SA, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A. 2016;113(12):E1738–E1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ahmed Z, Shaw G, Sharma VP, et al. Actin-binding proteins coronin-1a and IBA-1 are effective microglial markers for immunohistochemistry. J Histochem Cytochem. 2007;55(7):687–700. [DOI] [PubMed] [Google Scholar]
- 19. Dobrikova EY, Grisham RN, Kaiser C, Lin J, Gromeier M.. Competitive translation efficiency at the picornavirus type 1 internal ribosome entry site facilitated by viral cis and trans factors. J Virol. 2006;80(7):3310–3321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Seyfried TN, el-Abbadi M, Roy ML.. Ganglioside distribution in murine neural tumors. Mol Chem Neuropathol. 1992;17(2):147–167. [DOI] [PubMed] [Google Scholar]
- 21. Martinez-Murillo R, Martinez A.. Standardization of an orthotopic mouse brain tumor model following transplantation of CT-2A astrocytoma cells. Histol Histopathol. 2007;22(12):1309–1326. [DOI] [PubMed] [Google Scholar]
- 22. Oh T, Fakurnejad S, Sayegh ET, et al. Immunocompetent murine models for the study of glioblastoma immunotherapy. J Transl Med. 2014;12:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chandramohan V, Bryant JD, Piao H, et al. Validation of an immunohistochemistry assay for detection of cd155, the poliovirus receptor, in malignant gliomas. Arch Pathol Lab Med. 2017;141(12):1697–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Koike S, Taya C, Kurata T, et al. Transgenic mice susceptible to poliovirus. Proc Natl Acad Sci U S A. 1991;88(3):951–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chhatbar C, Detje CN, Grabski E, et al. Type I interferon receptor signaling of neurons and astrocytes regulates microglia activation during viral encephalitis. Cell Rep. 2018;25(1):118–129.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liberzon A, Birger C, Thorvaldsdottir H, et al. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1(6):417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Becht E, Giraldo NA, Lacroix L, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 2016;17(1):218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM.. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science. 1991;252(5007):854–856. [DOI] [PubMed] [Google Scholar]
- 29. Fulci G, Dmitrieva N, Gianni D, et al. Depletion of peripheral macrophages and brain microglia increases brain tumor titers of oncolytic viruses. Cancer Res. 2007;67(19):9398–9406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gromeier M, Nair SK.. Recombinant poliovirus for cancer immunotherapy. Annu Rev Med. 2018;69:289–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Merrill MK, Bernhardt G, Sampson JH, et al. Poliovirus receptor CD155-targeted oncolysis of glioma. Neuro Oncol. 2004;6(3):208–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Walton RW, Brown MC, Sacco MT, Gromeier M.. Engineered oncolytic poliovirus PVSRIPO subverts MDA5-dependent innate immune responses in cancer cells. J Virol. 2018;92(19):e00879–e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Brown MC, Bryant JD, Dobrikova EY, et al. Induction of viral, 7-methyl-guanosine cap-independent translation and oncolysis by mitogen-activated protein kinase-interacting kinase-mediated effects on the serine/arginine-rich protein kinase. J Virol. 2014;88(22):13135–13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Brown MC, Dobrikov MI, Gromeier M.. Mitogen-activated protein kinase-interacting kinase regulates mTOR/AKT signaling and controls the serine/arginine-rich protein kinase-responsive type 1 internal ribosome entry site-mediated translation and viral oncolysis. J Virol. 2014;88(22):13149–13160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chhatbar C, Prinz M.. The roles of microglia in viral encephalitis: from sensome to therapeutic targeting. Cell Mol Immunol. 2021;18(2):250–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sabouri AH, Marcondes MC, Flynn C, et al. TLR signaling controls lethal encephalitis in WNV-infected brain. Brain Res. 2014;1574:84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hohsfield LA, Tsourmas KI, Ghorbanian Y, et al. MAC2 is a long-lasting marker of peripheral cell infiltrates into the mouse CNS after bone marrow transplantation and coronavirus infection. Glia. 2022;70(5):875–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Moseman EA, Blanchard AC, Nayak D, McGavern DB.. T cell engagement of cross-presenting microglia protects the brain from a nasal virus infection. Sci Immunol. 2020;5(48):eabb1817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Merrill MK, Dobrikova EY, Gromeier M.. Cell-type-specific repression of internal ribosome entry site activity by double-stranded RNA-binding protein 76. J Virol. 2006;80(7):3147–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gromeier M, Bossert B, Arita M, Nomoto A, Wimmer E.. Dual stem loops within the poliovirus internal ribosomal entry site control neurovirulence. J Virol. 1999;73(2):958–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Grabowski MM, Sankey EW, Ryan KJ, et al. Immune suppression in gliomas. J Neurooncol. 2021;151(1):3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.