

Abstract
Background and Objectives
The RFC1 spectrum has become considerably expanded as multisystemic features beyond the triad of cerebellar ataxia, neuropathy, and vestibular areflexia syndrome (CANVAS) have started to be unveiled, although many still require clinical replication. Here, we aimed to clinically characterize a cohort of RFC1-positive patients by addressing both classic and multisystemic features. In a second part of this study, we prospectively assessed small nerve fibers (SNF) and autonomic function in a subset of these RFC1-related patients.
Methods
We retrospectively enrolled 67 RFC1-positive patients from multiple neurologic centers in Portugal. All patients underwent full neurologic and vestibular evaluation, as well as neuroimaging and neurophysiologic studies. For SNF and autonomic testing (n = 15), we performed skin biopsies, quantitative sensory testing, sudoscan, sympathetic skin response, heart rate deep breathing, and tilt test.
Results
Multisystemic features beyond CANVAS were present in 82% of the patients, mainly chronic cough (66%) and dysautonomia (43%). Other features included motor neuron (MN) affection and motor neuropathy (18%), hyperkinetic movement disorders (16%), sleep apnea (6%), REM and non-REM sleep disorders (5%), and cranial neuropathy (5%). Ten patients reported an inverse association between cough and ataxia severity. A very severe epidermal denervation was found in skin biopsies of all patients. Autonomic dysfunction comprised cardiovascular (67%), cardiovagal (54%), and/or sudomotor (50%) systems.
Discussion
The presence of MN involvement, motor neuropathy, small fiber neuropathy, or extrapyramidal signs should not preclude RFC1 testing in cases of sensory neuronopathy. Indeed, the RFC1 spectrum can overlap not only with multiple system atrophy but also with hereditary motor and sensory neuropathy, hereditary sensory and autonomic neuropathy, and feeding dystonia phenotypes. Some clinical-paraclinical dissociations can pose diagnostic challenges, namely large and small fiber neuropathy and sudomotor dysfunction which are usually subclinical.
Introduction
Biallelic intronic AAGGG repeat expansions in the RFC1 gene were first described in 2019 by Cortese et al. as a frequent cause of late-onset ataxia, especially if the triad of cerebellar ataxia, neuropathy, and vestibular areflexia syndrome (CANVAS) was present.1 For the past 3 years, the RFC1 spectrum has become exponentially complex as multisystemic involvement beyond CANVAS, novel pathogenic RFC1 configurations, and new neuropathologic features has started to be unveiled. Although sensory ataxia and chronic cough remain the key features in RFC1-related disorder (RRD),2,3 several others require further validation to confirm a causal association with RFC14 because data in the literature mainly refer to individual reports or small-to-intermediate series retrieved from ataxia or neuropathy cohorts.5 Two studies, by Cortese et al. and Traschütz et al.,2,3 investigated the full phenotypic spectrum and natural history of RRD in 2 large cohorts of 100 and 70 patients, respectively, but only the latter focused on multisystemic presentation (beyond CANVAS).3
TAKE-HOME POINTS
→ RFC1-positive patients can present with pyramidal signs, including mild spasticity and Babinski sign, and not merely normal or brisk deep tendon reflexes.
→ Severe epidermal denervation was universal in our subset of patients, which confirms the involvement of small myelinated and unmyelinated fibers in RFC1-related disorder.
→ We found, in our cohort, an inverse association between cough severity and ataxia progression.
→ Feeding dystonia, a finding restricted to few clinical entities, is part of the RFC1 spectrum disorder.
→ Small fiber neuropathy and dysautonomia can manifest severely or, more frequently, be asymptomatic, challenging the diagnosis. They should be actively search as this may affect patients' quality of life.
This study consisted of 2 parts. Part I aimed to clinically characterize 67 Portuguese patients carrying biallelic AAGGG repeat expansions in RFC1 by addressing both classic and multisystemic features. Specifically, this part of the study sought to explore a possible inverse association between cough severity and ataxia progression, taking into account the study by Coutinho et al.6 in a series of patients with ataxia and spasmodic cough of undetermined genetic nature. Knowing that sensory and dysautonomic symptoms are frequent in RRD,2 part II aimed to prospectively assess small nerve fibers (SNF) and autonomic function in a subset of RFC1 patients.
Methods
Patient Selection and Data Collection
Patients with genetic confirmation of biallelic RFC1 (AAGGG)n expansions followed at Centro Hospitalar Universitário do Porto (CHUPorto), Portugal, and other neurologic centers across the country (6 tertiary and 11 district hospitals, in a total of 17 hospitals) were retrospectively enrolled between 2020 and 2022. Patients or medical records were systematically assessed by the local neurologist according to a standardized proform. The following data were retrieved for all patients: demographics; family history; signs and symptoms suggestive of sensory neuropathy, cerebellar dysfunction, and vestibular areflexia; presence and characteristics of cough, dysautonomia, and other neurologic features [e.g., cognitive decline and extrapyramidal and motor neuron (MN) signs]; age of symptom onset; last neurologic examination; and use of walking aids. The investigational workup was also retrospectively reviewed and included brain and cervical MRI, neurophysiologic studies [nerve conduction studies (NCS), somatosensory evoked potentials], vestibular testing [video-oculography (VOG), video head impulse test], and previous genetic analyses. Involvement of each of the 3 main systems affected in RFC1 expansions (sensory neuropathy, bilateral vestibulopathy, and cerebellar dysfunction) was classified according to clinical and paraclinical data, as previously specified by Cortese et al.2 “Full CANVAS” required clinical and/or paraclinical evidence of simultaneous involvement of the 3 systems. CANVAS phenotypes with additional features were classified as “multisystemic RFC1.”
Additional Studies (Part II)
Fifteen patients with RFC1 AAGGG biallelic expansions from CHUPorto underwent prospective assessment for small fiber neuropathy (SFN) and dysautonomia. For autonomic nervous system (ANS) assessment, 13 patients underwent heart rate during deep breathing (HRDB; parasympathetic function) test and 12 sudoscan and sympathetic skin response (SSR; sympathetic cholinergic function). Tilt table test (TTT; sympathetic adrenergic function) was performed for 6 patients. For SNF assessment, 8 patients underwent quantitative sensory testing (QST) and 8 skin biopsies for intraepidermal nerve fiber density (IENFD) quantification. On the same day of skin biopsy, patients underwent clinical assessment for dysautonomia and neuropathy using the Composite Autonomic Symptom Score (COMPASS-31; Portuguese version)7 and Toronto Clinical Neuropathy Score (TCNS)8 scales, respectively. Ataxia severity was assessed through the Scale for Assessment and Rating of Ataxia (SARA).9
Details of skin biopsies and autonomic and sensory testing protocols of study part II and RFC1 genetic analysis are provided in the eMethods (links.lww.com/CPJ/A458).
Standard Protocol Approvals, Registrations, and Patient Consents
This study was approved by the Ethics Committee of CHUPorto (2020.227[179-DEFI/180-CE]) and by local institutional review boards. Written informed consent was obtained from patients participating in this study.
Data Availability
Anonymized study data can be provided by the authors on reasonable request.
Results
Part I
Clinical Features
A total of 67 patients with genetically confirmed biallelic RFC1 (AAGGG)n expansions were enrolled, 37 (55%) of whom were female. The median age at the time of the study was 71.0 years (range 30–87). All patients were Caucasian.
Positive family history was identified in 40 (60%) patients; 27 were from 9 RFC1-positive families, and the remaining 13 had relatives with suggestive but not genetically confirmed history. For most patients, the family pedigree consisted of at least 1 diseased sibling. In 4 families, hereditability was evocative of pseudodominance, as the mother and cousins were affected. The disease was sporadic in 27 patients, of whom 1 patient had a family history of amyotrophic lateral sclerosis.
Three patients had a history of cranial neuropathy (peripheral facial palsy, bilateral trigeminal neuralgia, and bilateral optic neuropathy), 4 patients had sleep apnea (SA), and 1 patient experienced 2 episodes of transient global amnesia (TGA) a few months before ataxia onset.
Symptoms at Disease Onset and During Progression
The clinical features of this cohort are shown in Figure 1. Patients had a median age of 55.5 years (range 26–73) at the time of disease onset, and gait imbalance was the most common symptom, reported by 59 (88%) patients, either isolated (n = 15) or combined with other symptoms (mostly sensory alterations; n = 24). Of the 8 patients without gait imbalance at onset, 5 presented with isolated sensory symptoms, 2 presented with isolated slurred speech and/or swallowing difficulties, and 1 was asymptomatic.
Figure 1. Clinical Features at Disease Onset and During Progression.

(A) Symptoms at onset and during progression in 67 RFC1-positive patients. Patients could have multiple combinations of symptoms. (B) Age of onset of each symptom. The vertical line in boxes represents median age, in years, and box edges represent lower and upper quartiles. Whiskers correspond to maximum and minimum ages.
During disease progression, 63 (94%) patients developed gait imbalance [median age of onset (MAO) 57.0 years, range 26–73] and 49 (73%) complained of sensory symptoms (MAO 59.0 years, range 39–73), including distal paresthesia (n = 26), distal numbness (n = 20), and neuropathic pain (n = 17). Among the 4 patients who did not report balance issues, 2 had isolated neuropathic pain [age of onset (AO) 39 and 44 years, respectively], which was severe and refractory to pharmacologic treatment in one; one had oromandibular dystonia (OMD) and complained of slurred speech, swallowing and feeding difficulties, and involuntary movements of the tongue (AO 62 years); and one remained asymptomatic (current age 62 years). Thirty-four (51%) and 27 (40%) patients reported dysarthria and dysphagia symptoms starting at 59.0 (range 26–82) and 63.0 (range 26–81) years, respectively. Dysarthria and dysphagia were less frequent at disease onset and tended to be generally mild during disease progression, except in 3 patients. Of these, one had very severe cerebellar dysarthria with unintelligible speech and pseudobulbar affect, and 2 had severe dysphagia requiring percutaneous endoscopic gastrostomy (PEG) after 19 and 25 years of disease, respectively. One patient with OMD had dystonic, rather than cerebellar, dysarthria. Oscillopsia was the least frequent CANVAS symptom, reported in 8 (12%) patients at onset and 19 (28%) patients during follow-up (MAO 65.0 years, range 52–75).
Dysautonomic symptoms were reported in 15 (22%) patients at disease onset and in 29 (43%) patients during disease progression (MAO 64.5 years, range 39–94). They comprised syncope (n = 11), constipation (n = 10), orthostatic hypotension (n = 9), urinary dysfunction (n = 8), erectile dysfunction (n = 3), cold feet (n = 2), and hypohidrosis (n = 2), and despite being mild in most patients, 3 patients with constipation developed severe paralytic ileus, with one requiring surgery.
Two-thirds of patients (n = 45) had chronic cough, with a MAO of 40 years (range 25–69; not specified in 13 cases), which was usually the initial symptom, presenting up to 3 decades before neurologic symptoms. Only in 4 patients the cough developed simultaneously or subsequently to neurologic symptoms. In 5 patients, cough had been previously attributed to asthma/bronchitis or gastroesophageal reflux. Details of cough features were only available for some patients. When specifically asked, 14/18 patients reported aggravating factors (meals, long chats, temperature changes, dust, cleaning products, touching the ear), and 7/12 reported alleviating factors (drinking water). Cough attacks were brief (lasting seconds to few minutes; n = 16/16) and with variable frequency among patients (daily n = 13, weekly n = 4, monthly n = 2), typically occurring both in diurnal and nocturnal periods (n = 10/16). Most patients classified the severity of attacks as moderate to intense (n = 16/21), and for some (n = 13/18), attacks lead to breathing difficulties, choking, vomiting, syncope, and feelings of “dry throat.” Notably, in 10/17 patients, the frequency and/or severity of cough attacks decreased with ataxia progression.
Three patients complained of involuntary perioral movements. These were the presenting feature in 1 case (of OMD) and manifested at the ages of 20 and 57 years in the other 2 cases, respectively. Two patients reported symptoms of REM sleep behavior disorder (RBD).
Disability
The median disease duration (MDD) was 12 (range 0–37) years. Thirty-nine percent (n = 26) of patients required unilateral support to walk at the median age of 70 years (range 48–81) and after a median of 15.5 years (range 4–37) of disease. Six patients (9%) were wheelchair-bound at the median age of 74.5 years (range 59–84), with a MDD of 17 years (range 10–23). Three patients died at the age of 71─78 years.
Neurologic Examination
The MDD at the time of the most recent neurologic assessment was 10 (range 1–30) years. Figure 2 shows the neurologic data of RCF1-positive patients in this cohort. Gait was ataxic in 61 (91%) patients, and 57/63 (90%) had Romberg sign. Additional signs of sensory involvement were present in 65 (97%) patients, with vibration sensation loss being the most frequent, present in 95% of patients [n = 60 in the lower limbs (LL) and n = 20 in the upper limbs (UL)], followed by pinprick sensation loss in 70% (n = 45 LL, n = 22 UL, and n = 6 face), and join position loss in 63%. Concerning cerebellar signs, 33 (49%) patients had scanning dysarthria, 44 (66%) appendicular incoordination, and 54 (81%) eye movement abnormalities [nystagmus (n = 40), dysmetric saccades (n = 35), and broken pursuit (n = 28)]. HIT, performed in 58 (87%) patients, was bilaterally abnormal (i.e., presence of catch-up saccades) in 51 (88%). Motor examination revealed reduced or absent ankle reflexes in 62% of patients and mostly normal or even brisk UL and knee reflexes in 74% and 70%, respectively. Distal weakness was identified in 4 cases, of whom 2 had mild LL spasticity and 1 had gastrocnemius fasciculation and amyotrophy. Regarding movement disorders, 3 patients had orofacial dyskinesia, 3 had minor hands and feet dystonia, and 1 had OMD (feeding dystonia) associated with choreiform movements of the hands. Four patients had postural tremor of the hands, which was combined with kinetic or rest component in 2; one had chin tremor. Mild cognitive impairment was identified in 4 patients during examination (age range 65–71 years). Neurologic examination of the asymptomatic patient revealed reduced vibration sensation in LL, reduced ankle reflexes, and dysmetric saccades.
Figure 2. Neurologic Examination of RCF1-Positive Patients at Most Recent Evaluation.
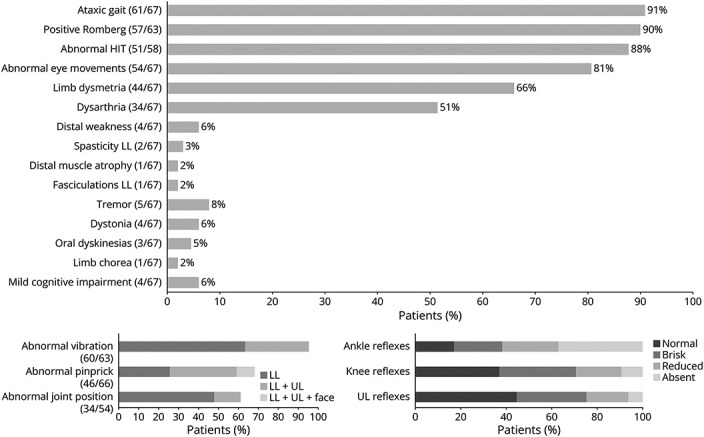
Body distribution of affected sensory modalities and reflex evaluation is detailed. HIT = head impulse test; LL = lower limbs; UL = upper limbs; ULL = upper and lower limbs.
Paraclinical Features
Patients' workup is summarized in Table 1. The median time between disease onset and NCS was 7 years (range 1–32). Sensory neuropathy was reported in all patients, with simultaneously absent or reduced sensory nerve action potentials (SNAP) in LL and UL in 83% of patients, suggesting non–length-dependent (NLD) neuropathy/ganglionopathy. In the remaining patients (17%), SNAP were absent in LL and reduced or normal in UL, indicating (LD) length-dependent neuropathy. Compound muscle action potentials (CMAP) were reduced in UL and/or LL in 8 (13%) patients, but only one of them had motor signs on examination (distal weakness). Needle electromyography (nEMG) results were not systematically assessed in this cohort, but we had (1) one patient with widespread chronic denervation (clinically presenting with distal limbs weakness and amyotrophy, gastrocnemius fasciculation, and LL spasticity) and (2) another one with isolated fasciculation potentials in distal limb muscles (clinically without motor signs); both patients did not have motor neuropathy in NCS. The other 2 patients with distal weakness (2/4) had normal NCS, but unfortunately nEMG results were not available.
Table 1.
Paraclinical Data

Cerebellar atrophy was present in brain MRI in 46 (n = 46/61, 75%) subjects. Variability was found in the presence/absence of cerebellar atrophy according to disease duration, with no significant differences in MDD between cases with and without cerebellar atrophy (8.2 ± 6.5 years vs 6.7 ± 5.3 years, t test p = 0.413). T2/FLAIR white matter hyperintensity of probable microvascular ischemic nature was observed in 41% (n = 25) of patients. Bilateral globus pallidum T2-hyperintensity was identified in 2 siblings, with concomitant dot-like areas with T2*-hyposignal in cortico-subcortical locations in one (Figure 3). Cervical MRI revealed spinal cord atrophy in 4/20 cases and T2-hyperintensity in the posterior column in one.
Figure 3. MRI Features of RFC1 Disease.

Axial (A) and sagittal (B) T2-weighted and axial T2*–weighted (C) brain MRI of 2 RFC1-positive patients. Both have T2-hyperintensity of bilateral globus pallidum (arrow heads), more evident in the patient in Figure A; no correspondent signal abnormalities on T2* MRI or brain CT were found (not shown). The patient in Figure B also showed occipital cortico-subcortical small black dots in T2* (arrows), suggesting microhemorrhages (flow voids excluded).
Forty-eight (72%) patients underwent vestibular testing (MDD 9.5 years, range 1–30), with bilateral vestibular hypoareflexia/areflexia documented in 92% (n = 44) of patients. Patients without vestibulopathy (n = 4) were shown to be able to have long disease duration, as observed in 1 patient with negative vestibular testing at age 62 years, after 20 years of disease.
Polysomnography results were available for 2/4 patients with a history of SA, disclosing mild-to-moderate obstructive SA and periodic limb movement disorder (PLMD) of severe index (140.7/h) in one and mixed (predominantly central) SA in the other.
Part II
SFN and Dysautonomia Study
Data from SFN and ANS study are presented in Table 2. Details of patients who underwent skin biopsy and the results of their clinical evaluation scales, quantitative autonomic testing (QAT), and QST are presented in Table 3.
Table 2.
Results From Dysautonomia and Small Fiber Neuropathy Study
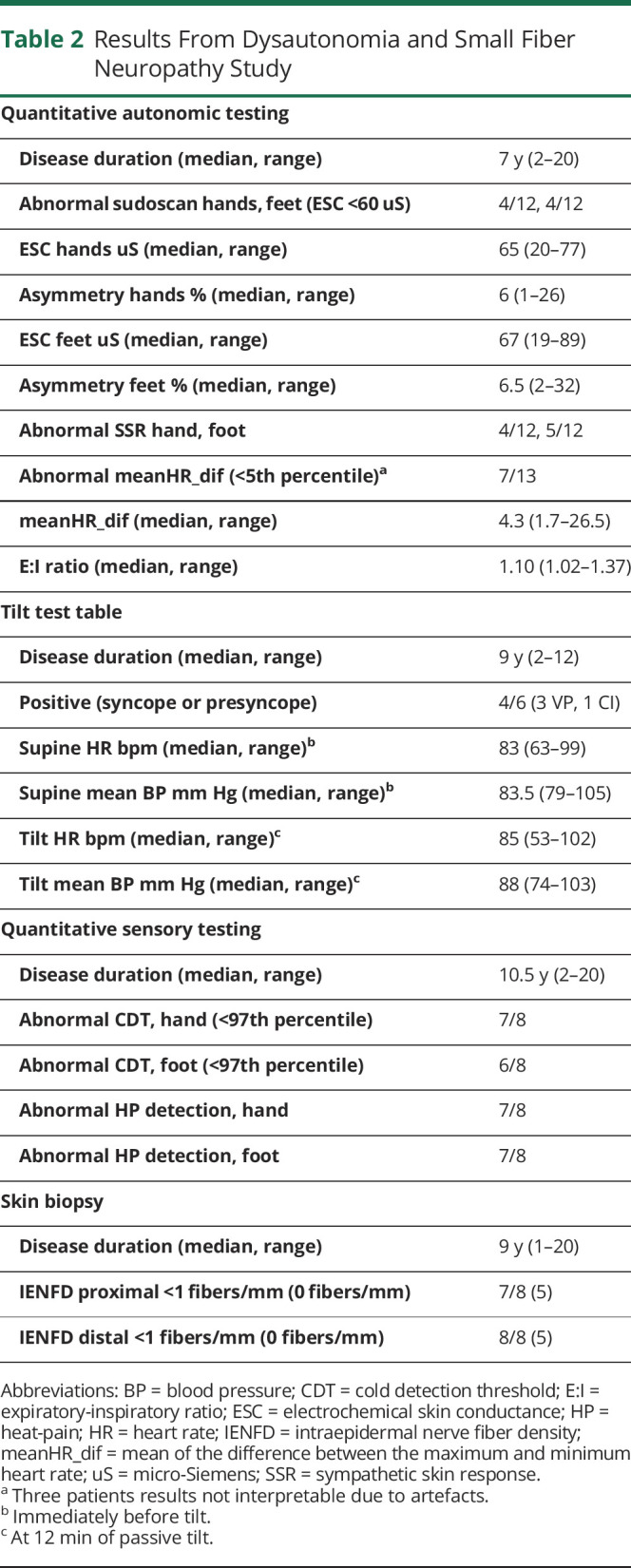
Table 3.
Clinical and Neurophysiologic Results of the 8 Patients Who Underwent Skin Biopsy

QAT
Five (n = 5/12, 42%) patients submitted to sudoscan had pathologic electrochemical skin conductance (ESC) values in the hands and/or feet; SSR was absent in the hands and/or feet of 5 (42%) patients. HRDB was significantly decreased (i.e., below the fifth percentile) in 7 (n = 7/13, 54%) cases. TTT was obtained between 0 and 8 months after QAT. Positive response was observed in 4 (n = 4/6, 67%) patients during pharmacologic tilt: 3 had vasodepressor and 1 cardioinhibitory syncope/presyncope. None had orthostatic hypotension at 12 minutes of passive tilt.
QST
All patients submitted to QST (n = 8/8) had impaired sensitivity to cold temperatures and/or to heat-pain.
Skin Biopsy
All patients (n = 8/8) presented very severe epidermal denervation, both proximally and distally, as IENFD was frequently of 0 fibers/mm (absence of epidermal fibers) or below 2 fiber/mm. The median time between disease onset and skin biopsy was 9 years (range 1–20). In addition to epidermal, also concomitant dermal denervation was observed. Of note, all these patients already presented with advanced disease at the time of biopsy, evident in abolished LL SNAP (time between NCS and skin biopsy 0–5 years).
Association With Clinical Symptoms and Scales
All patients (n = 8/8) with reduced/absent IEFND had abnormal QST results (time between skin biopsy and QST 10 months, range 0–21). Seven had reduced pinprick sense, and 5 reported sensory symptoms, although only 3 had pain complaints (Table 3). The median TCNS and SARA scores were 10 (scale maximum 19 points) and 7.5 (scale maximum 40 points), respectively. Most patients had normal sudomotor function, related or not to the secretomotor part of COMPASS-31 (Table 3). Cardiovascular adrenergic function, assessed by TTT, correlated well with the symptoms reported and/or with the orthostatic intolerance score of COMPASS-31 (Table 3). Gastrointestinal symptoms had the highest score on COMPASS-31.
RFC1 Spectrum Disorder/Multisystemic Involvement
Based on the symptoms reported and neurologic assessment, 51 (76%) patients in this cohort had “full CANVAS” phenotype, 10 (15%) had “sensory neuropathy plus cerebellar syndrome,” 4 had isolated “sensory neuropathy,” 1 had “cerebellar syndrome plus vestibular dysfunction,” and 1 had isolated OMD. In total, 8/15 patients without vestibular areflexia were not tested for HIT during neurologic assessment. After additional workup, 2 patients were reclassified as “full CANVAS” and “sensory neuropathy plus cerebellar syndrome,” respectively (Figure 4). Sensory neuropathy was subclinical in 2 cases (normal neurologic examination but sensory neuropathy on NCS), with only 1 patient with negative HIT (despite oscillopsia) presenting abnormal VOG. On the other hand, clinical assessment was more sensitive in the identification of cerebellar dysfunction than brain MRI because 12 cases with cerebellar syndrome showed no evidence of cerebellar atrophy in MRI. Asymptomatic cerebellar atrophy was found in a single patient.
Figure 4. RFC1 Spectrum Disorder.
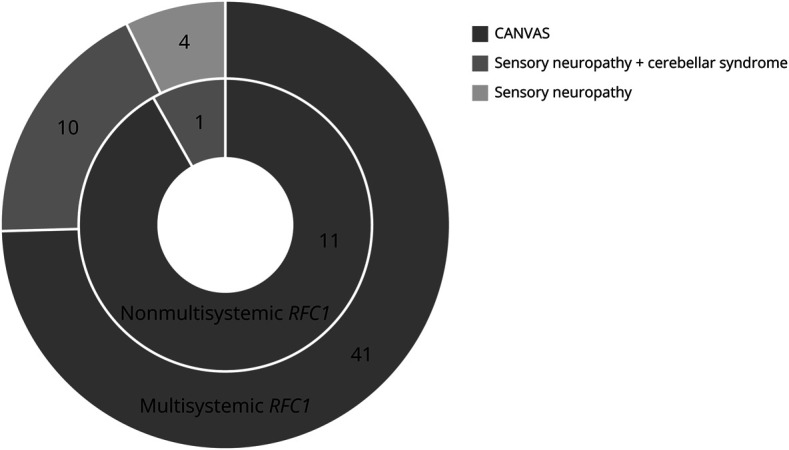
Phenotypic classification after clinical and paraclinical study, according to (1) sensory, cerebellar, and vestibular dysfunctions and (2) involvement of other systems (multisystemic RFC1, outer circle, vs nonmultisystemic RFC1, inner circle).
Overlap with other system dysfunctions (“multisystemic RFC1”) was frequent (n = 55, 82%) and included (1) chronic cough (n = 45, 66%); (2) dysautonomic symptoms (n = 29, 43%), confirmed by abnormal QAT in 11/14 (79%) patients, affecting either cardiovascular (n = 4/6, 67%), cardiovagal (n = 7/13, 54%), and/or sudomotor (n = 6/12, 50%) systems; (3) SFN (n = 8/8), as demonstrated by severely reduced IEFND and abnormal QST; (4) motor signs/abnormalities (n = 12, 18%), including fasciculation, distal weakness, atrophy and spasticity on examination, chronic denervation and fasciculation potentials on nEMG, and reduced CMAP in NCS, suggesting MN involvement and peripheral motor neuropathy; (5) hyperkinetic movement disorders (n = 11, 16%), namely minor dystonia, OMD with feeding dystonia, distal chorea, oral dyskinesias, and tremor; (6) cognitive deterioration, TGA episodes, and pseudobulbar affect (n = 6, 9%); (7) non-REM (PLMD) and REM sleep disorders (n = 3, 5%); (8) cranial neuropathy (n = 3, 5%); and (9) SA (n = 4, 6%), including of central origin (n = 1).
Discussion
As far as the authors are aware, this study investigated the RRD spectrum in a cohort of Portuguese patients and showed that the “pure CANVAS” phenotype might not be as common as previously thought because multisystemic features are frequently present. The characterization of autonomic and small fiber involvement in RRD was an additional contribution of this work and showed universal severe epidermal denervation and variable involvements of postganglionic cholinergic and adrenergic sympathetic fibers and parasympathetic fibers.
A comprehensive clinical assessment of these patients was conducted, which proved to be in line with previous studies reporting that sensory neuropathy is a unanimous component of RFC1-positive cases, emerging early in the course of the disease.2,10 Although the typical “spatial pattern of progression” from neuropathy to full CANVAS could still be observed in this cohort, the time between the onset of imbalance/sensory symptoms and cerebellar symptoms (mainly dysarthria) was found to be shorter (<2 years for dysarthria) than that reported in the literature (6–8 years).2,3 The authors hypothesize that, rather than representing a true phenotypic variation, this is probably the result of a very indolent course of sensory ataxia, which makes it difficult for patients to precisely identify the AO of the neuropathy. Moreover, 2 formerly described clinical-paraclinical dissociations were also observed in this cohort: (1) the severe ganglionopathy in NCS was usually subclinical as most patients had mild and predominantly distal (sometimes asymmetrical) sensory symptoms and LD neuropathy on neurologic examination and (2) cerebellar dysfunction was more often clinically identified than through MRI.2,9,10 In fact, cerebellar atrophy could be identified in MRI shortly after disease onset or be absent for several years, corroborating the findings of Traschütz et al. that cerebellar atrophy has a variable “temporal pattern of progression.”3
The involvement of other systems beyond CANVAS was frequent in this cohort (82%) and mainly associated with chronic cough (66%) and dysautonomia (43%; the 5 core features of RRD—sensory neuropathy, vestibulopathy, cerebellar syndrome, chronic cough, and dysautonomia).3
Additional features were identified, which, although less common, should be highlighted because they can have diagnostic and prognostic implications. Motor signs suggestive of lower motor neuron (LMN) were present in 4 patients, having normal motor NCS: 3 in the form of distal weakness, of whom one had amyotrophy and calf fasciculations on visual inspection, and chronic denervation on nEMG, and one in the form of fasciculation potentials on nEMG. Upper motor neuron (UMN) involvement (besides brisk reflexes) was found in 2 cases with spasticity. Motor neuron involvement was initially described in association with elevated creatine kinase in non-Caucasian populations positive for the RFC1 ACAGG motif11,12 and was also observed in Caucasian individuals carrying biallelic AAGGG expansions.13-15 UMN signs (mainly spasticity, excluding brisk reflexes) seem to be rare in these patients and were reported in 6% (n = 4/70), 8% (n = 3/38), and 6% (n = 1/16) in previous cohorts,3,12,14 while LMN signs are common and were identified in 26% (n = 14/54, including distal atrophy and weakness), 18% (n = 7/38, including fasciculation, wasting, weakness, and neurogenic pattern on nEMG), and 31% (n = 5/16, including muscular atrophy and fasciculation) of patients in the same cohorts.3,12,14 Of note, for these cohorts, nEMG/NCS results were not presented for all patients, and therefore, motor neuropathy or anterior horn cell disease causing LMN signs is difficult to assess. Nonetheless, spinal cord pathology of 2 CANVAS patients (only in 1 RFC1 genetic analysis was performed) was able to show loss of MNs in the lumbar anterior horns.16,17 While intact corticospinal tracts were found in 2 neuropathologic studies14,17 and one using in vivo MRI,18 2 reports on the spinal cord pathology of patients with CANVAS have described “synaptic dysfunction between the first and second motor neurons” (Huin et al.)14 and “slight pallor of the corticospinal tracts” (Szmulewicz et al.),16 respectively, therefore suggesting UMN dysfunction, but requiring further investigation. Motor neuropathy with preserved muscle strength has been reported at variable rates in the literature (0%–40%) and was observed in 11% of patients in this study.3,10 While this is usually a strictly and mild neurophysiologic finding, some studies reported its coexistence with distal weakness and pes cavus, approaching the hereditary motor and sensory neuropathy (HMSN) phenotype.19,20
Hyperkinetic movement disorders were present in 16% of patients, similar to a previous study (11%) that did not specify patients' age of onset.3 Remarkably, in this cohort, oral dyskinesia preceded ataxia onset by 3 decades in one case, and OMD with feeding dystonia was the presenting feature in another, indicating an overlap with neuroacanthocytosis and other feeding dystonia phenotypes (full description in accepted article). Although tremors were probably of neuropathic nature, one patient presented with chin tremor and another with rest hand tremor in the absence of additional parkinsonian features. In agreement with a previous study, 2 siblings had globus pallidus T2-hyperintensity, reinforcing basal ganglia involvement in RFC1 expansions.3
Sleep disturbances affected 10% of patients in this cohort and comprised SA (including of central type), RBD, and PLMD. The first 2 have been previously described in RRD,3,21,22 particularly because they can mimic multiple system atrophy (MSA) in the presence of ataxia or parkinsonism.3 To the best of the authors' knowledge, PLMD has never been reported in RFC1-positive patients to date.
Severe presentations of usually mild RRD manifestations were found in this study, including 3 cases of severe paralytic ileus, 2 cases of severe dysphagia requiring PEG, and 1 case of refractory neuropathic pain, which presented with dysautonomia in the absence of ataxia, alluding to the hereditary sensory and autonomic neuropathy (HSAN) phenotype. One patient developed pseudobulbar palsy (featuring severe dysarthria, dysphagia, and pseudobulbar affect). Although this feature has not been formally associated with the RRD, brainstem MNs were implicated in bulbar weakness in 1 neuropathologic study of a RFC1-positive patient.17
Other features found in this study have not been previously reported in RFC1-positive patients, and thus, a causal association is difficult to establish at this point. TGA was reported in 1 patient, and cranial neuropathy was reported in 3 patients, including bilateral optic neuropathy, peripheral facial palsy, and bilateral trigeminal neuralgia. Previous neuropathologic and imaging studies have shown involvement of the trigeminal nerve and ganglion in CANVAS patients,23,24 as well as atrophy of geniculate ganglion of the facial nerve,23 which is responsible for taste sensation, therefore not explaining peripheral facial palsy. We would be extremely cautious at this point to establish an association between the presence of bilateral optic neuropathy and RFC1 positivity.
Abnormal sudomotor function was tested through 2 different methods, sudoscan and SSR, and found to be present in 50% of patients in this study. This confirms that postganglionic cholinergic sympathetic fibers are damaged in RRD, as recently proposed by Schmitt et al.15 Similarly, this study found a discrepancy between symptoms reported and neurophysiologic abnormalities, evidenced by the low secretomotor subscore of COMPASS-31, and only 2 patients spontaneously reporting abnormal sweeting, adding another clinical-paraclinical dissociation to the RFC1 spectrum. Before the discovery of RFC1 pathogenic expansion, reduced sweat gland innervation had been documented in skin biopsies of 2 CANVAS patients.25 Sympathetic adrenergic function was tested in RFC1-positive patients through TTT for the first time. Cardiovascular dysfunction was present in the form of vasodepressor/neurogenic (pre)syncope in 67% of cases in this study; one patient with cardioinhibitory syncope underwent pacemaker implantation. Although abnormal results were only observed after pharmacologic challenge, almost all patients reported previous syncope or postural hypotension symptoms, arguing against false-positive results. Cardiac parasympathetic dysfunction (abnormal HRDB) was present in 54% of patients. Combined (sympathetic and parasympathetic) cardiac neuropathy (CAN) could only be assessed in 4/6 patients (as 2/6 patients who underwent TTT had HRDB not interpretable or not performed) and was identified in one. In the series by Schmitt et al., CAN was found in 50% of patients (n = 8/16).15 Overall, QAT can serve the following 2 main purposes: (1) documenting sudomotor dysfunction to exclude MSA, a common differential diagnosis in RRD (dysautonomia in MSA is due to central autonomic failure), and (2) educating patients with cardiac neuropathy on lifestyle measures for syncope prevention [some of whom may even require more invasive treatments (e.g., pacemaker), as one of the patients in this cohort].
All patients prospectively assessed through skin biopsy, and QST showed severe NLD loss of IEFN and abnormal cold and/or heat-pain thresholds. These results replicate the findings of Magy et al.26 and strongly suggest the involvement of A-delta and C fibers, initially hypothesized to be preserved in RRD.2 This study also confirms that QST can be a useful tool for the assessment of SNF in RFC1-positive patients. In agreement with others,2 neuropathic pain was common in this cohort (25%), but except for 1 case, it was usually mild, and no patient showed ulcerations or lesions typical of pain insensitivity, implying a clinical-paraclinical dissociation. Notwithstanding, as stated by Magy et al., SFN is probably clinically important and may affect patients' quality of life.26
The cough mechanism in RRD remains to be elucidated. Sensory neuropathy of the vagal nerve at different levels—including the nucleus solitarius and the small A-delta and C fibers of upper airways and esophagus—has been hypothesized as a possible cause.3,26 Importantly, 10/17 patients in this cohort reported a reduction in the frequency of cough episodes after ataxia onset, in agreement with the study by Coutinho et al.6 According to these authors, cerebellum-medulla networks involved in cough generation may be responsible for the “disproportionate central facilitation of cough reflex” in the beginning of the neurodegenerative process. Posteriorly, as cerebellar atrophy evolves, this central facilitation slowly attenuates, similar to what has been reported “in animals after cerebellar lesions.”6
This study has limitations that should be acknowledged. Some findings, such as cognitive impairment and sleep disturbances, are relatively frequent in the general population, and as such, their direct causality to the RFC1 expansion cannot be stablished. Others, as peripheral facial palsy, optic neuropathy and cortical microhemorrhages were never reported and could be simply coincidental. Regarding the autonomic testing, the small size of the sample included in the prospective analysis hampered more in-depth clinical correlations. Sudoscan is a substandard test to evaluated sudomotor function, and the pharmacologic intervention during the TTT lowers its specificity. Some of the autonomic symptoms could also be secondary to the patient's global condition, specifically deconditioning due to the neuropathy and cerebellar ataxia. nEMG information was not systematically collected, which comprised topographic localization of the LMN signs. Neuroradiologic assessment was performed by multiple neuroradiologists across the country, and thus, inconsistencies in conclusions regarding cerebellar atrophy cannot be ruled out. Vestibular evaluation was also not uniform, having been performed by different ENT doctors. Not all physicians inquired about chronic cough features, and patients with cognitive deterioration were not further characterized with neuropsychometry.
In conclusion, the presence of MN affection, motor neuropathy, or extrapyramidal signs should not preclude RFC1 testing in cases of sensory neuronopathy. Indeed, the RFC1 spectrum can overlap not only with MSA but also with HMSN, HSAN, and feeding dystonia phenotypes. This study also showed that SFN is a hallmark of RRD. Accordingly, neuropathic pain and cardiac and gastrointestinal dysautonomia, traditionally regarded as mild, can predominate in the course of the disease and be a source of major comorbidity in RFC1-positive patients. Study findings of an inverse association between cough and ataxia severity in some patients mainly have pathophysiologic implications and require replication. In the future, tests such as nEMG, QAT, QST, I-metaiodobenzylguanidine (MIBG) scintigraphy, skin biopsy for the assessment of intraepidermal but also dermal and glandular layers, neuropsychometry, and polysomnography should be systematically applied in large cohorts of RFC1-positive patients.
Appendix 1. Authors
Appendix 2. Coinvestigators

Study Funding
The authors report no targeted funding.
Disclosure
The authors report no relevant disclosures. Full disclosure form information provided by the authors is available with the full text of this article at Neurology.org/cp.
References
- 1.Cortese A, Simone R, Sullivan R, et al. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat Genet. 2019;51(4):649-658. doi: 10.1038/s41588-019-0372-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cortese A, Tozza S, Yau WY, et al. Cerebellar ataxia, neuropathy, vestibular areflexia syndrome due to RFC1 repeat expansion. Brain. 2020;143:480-490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Traschütz A, Cortese A, Reich S, et al. Natural history, phenotypic spectrum, and discriminative features of multisystemic RFC1 disease. Neurology. 2021;96(9):e1369-e1382. doi: 10.1212/WNL.0000000000011528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sullivan R, Kaiyrzhanov R, Houlden H. Cerebellar ataxia, neuropathy, vestibular areflexia syndrome: genetic and clinical insights. Curr Opin Neurol. 2021;34(4):556-564. doi: 10.1097/WCO.0000000000000961 [DOI] [PubMed] [Google Scholar]
- 5.Davies K, Szmulewicz DJ, Corben LA, Delatycki M, Lockhart PJ. RFC1 -related disease. Neurol Genet. 2022;8(5):e200016. doi: 10.1212/nxg.0000000000200016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Coutinho P, Cruz VT, Tuna A, Silva SE, Guimaraes J. Cerebellar ataxia with spasmodic cough: a new form of dominant ataxia. Arch Neurol. 2006;63:553-555. [DOI] [PubMed] [Google Scholar]
- 7.Vieira B, Costa A, Videira G, Sá MJ, Abreu P. Prevalence of autonomic dysfunction in patients with multiple sclerosis. Acta Med Port. 2015;28(1):51-55. doi: 10.20344/amp.5562 [DOI] [PubMed] [Google Scholar]
- 8.Bril V, Tomioka S, Buchanan RA, Perkins BA. Reliability and validity of the modified Toronto Clinical Neuropathy Score in diabetic sensorimotor polyneuropathy. Diabetic Med. 2009;26(3):240-246. doi: 10.1111/j.1464-5491.2009.02667.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schmitz-Hübsch T, du Montcel ST, Baliko L, et al. Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology. 2006;66(11):1717-1720. doi: 10.1212/01.wnl.0000219042.60538.92 [DOI] [PubMed] [Google Scholar]
- 10.Currò R, Salvalaggio A, Tozza S, et al. RFC1 expansions are a common cause of idiopathic sensory neuropathy. Brain. 2021;144(5):1542-1550. doi: 10.1093/brain/awab072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scriba CK, Beecroft SJ, Clayton JS, et al. A novel RFC1 repeat motif (ACAGG) in two Asia-Pacific CANVAS families. Brain. 2020;143(10):2904-2910. doi: 10.1093/brain/awaa263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miyatake S, Yoshida K, Koshimizu E, et al. Repeat conformation heterogeneity in cerebellar ataxia, neuropathy, vestibular areflexia syndrome. Brain. 2022;145(3):1139-1150. doi: 10.1093/brain/awab363 [DOI] [PubMed] [Google Scholar]
- 13.Kermorvant H, Debs R, Maisonobe T, Huin V, Stojkovic T, Lenglet T. Cramp-fasciculation syndrome phenotype of cerebellar ataxia with neuropathy and vestibular areflexia syndrome (CANVAS) due to RFC1 repeat expansion. Clin Neurophysiol. 2022;134:34-36. doi: 10.1016/j.clinph.2021.11.005 [DOI] [PubMed] [Google Scholar]
- 14.Huin V, Coarelli G, Guemy C, et al. Motor neuron pathology in CANVAS due to RFC1 expansions. Brain. 2022;145(6):2121-2132. doi: 10.1093/brain/awab449 [DOI] [PubMed] [Google Scholar]
- 15.Schmitt GDS, Lima FD, Matos PCAAP, et al. Dysautonomia in RFC1-related disorder: clinical and neurophysiological evaluation. Clin Neurophysiol. 2022;142:68-74. doi: 10.1016/j.clinph.2022.07.501 [DOI] [PubMed] [Google Scholar]
- 16.Szmulewicz DJ, McLean CA, Rodriguez ML, et al. Dorsal root ganglionopathy is responsible for the sensory impairment in CANVAS. Neurology. 2014;82(16):1410-1415. doi: 10.1212/WNL.0000000000000352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reyes-Leiva D, Aldecoa I, Gelpi E, Rojas-García R. Motor neuron involvement expands the neuropathological phenotype of late-onset ataxia in RFC1 mutation (CANVAS). Brain Pathol. 2022;32(4):e13051. doi: 10.1111/bpa.13051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rezende TJR, Schmitt GS, de Lima FD, et al. RFC1-related disorder: in vivo evaluation of spinal cord damage. Mov Disord. 2022;37(10):2122-2128. doi: 10.1002/mds.29169 [DOI] [PubMed] [Google Scholar]
- 19.Tagliapietra M, Cardellini D, Ferrarini M, et al. RFC1 AAGGG repeat expansion masquerading as chronic idiopathic axonal polyneuropathy. J Neurol. 2021;268(11):4280-4290. doi: 10.1007/s00415-021-10552-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Beijer D, Dohrn MF, de Winter J, et al. RFC1 repeat expansions: a recurrent cause of sensory and autonomic neuropathy with cough and ataxia. Eur J Neurol. 2022;29(7):2156-2161. doi: 10.1111/ene.15310 [DOI] [PubMed] [Google Scholar]
- 21.Beecroft SJ, Cortese A, Sullivan R, et al. A Maori - specific RFC1 pathogenic repeat configuration in CANVAS, likely due to a founder allele. Brain. 2020;143(9):2673-2680. doi: 10.1093/brain/awaa203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsuchiya M, Nan H, Koh K, et al. RFC1 repeat expansion in Japanese patients with late-onset cerebellar ataxia. J Hum Genet. 2020;65(12):1143-1147. doi: 10.1038/s10038-020-0807-x [DOI] [PubMed] [Google Scholar]
- 23.Szmulewicz DJ, Waterston JA, Macdougall HG, et al. Cerebellar ataxia, neuropathy, vestibular areflexia syndrome (CANVAS): a review of the clinical features and video-oculographic diagnosis. Ann N Y Acad Sci. 2011;1233(1):139-147. doi: 10.1111/j.1749-6632.2011.06158.x [DOI] [PubMed] [Google Scholar]
- 24.Matos PCAAP, Rezende TJR, Schmitt GS, et al. Brain structural signature of RFC1-related disorder. Mov Disord. 2021;36(11):2634-2641. doi: 10.1002/mds.28711 [DOI] [PubMed] [Google Scholar]
- 25.Umeh CC, Polydefkis M, Chaudhry V, Zee DS. Sweat gland denervation in cerebellar ataxia with neuropathy and vestibular areflexia syndrome (CANVAS). Mov Disord Clin Pract. 2017;4(1):46-48. doi: 10.1002/mdc3.12355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Magy L, Chazelas P, Richard L, et al. Early diagnosis in cerebellar ataxia, neuropathy, vestibular areflexia syndrome (CANVAS) by focusing on major clinical clues: beyond ataxia and vestibular impairment. Biomedicines. 2022;10(8):2046. doi: 10.3390/biomedicines10082046 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Anonymized study data can be provided by the authors on reasonable request.