

Abstract
Alzheimer’s disease (AD) is a major cause of dementia inducing memory loss, cognitive decline, and mortality among the ageing population. While the amyloid aggregation of peptide Aβ has long been implicated for neurodegeneration in AD, primarily through the production of toxic polymorphic aggregates and reactive oxygen species, viral infection has a less explicit role in the etiology of the brain disease. On the other hand, while the COVID-19 pandemic is known to harm human organs and function, its adverse effects on AD pathobiology and other human conditions remain unclear. Here we first identified the amyloidogenic potential of 1058HGVVFLHVTYV1068, a short fragment of the spike protein of SARS-CoV-2 coronavirus. The peptide fragment was found to be toxic and displayed a high binding propensity for the amyloidogenic segments of Aβ, thereby promoting the aggregation and toxicity of the peptide in vitro and in silico while retarding the hatching and survival of zebrafish embryos upon exposure. Our study implicated SARS-CoV-2 viral infection as a potential contributor to AD pathogenesis, a little explored area in our quest for understanding and overcoming Long Covid.
Keywords: amyloid β, SARS-CoV-2, spike protein, virus, Alzheimer’s disease
Graphical Abstract
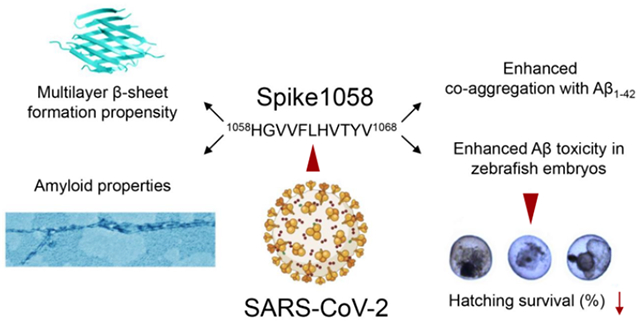
1. Introduction
Alzheimer’s disease (AD) is a debilitating human condition and a major culprit for dementia. The underling physiopathology of AD is not yet resolved, where the leading amyloid hypothesis attributes neurodegeneration to the extracellular aggregation of amyloid beta (Aβ) and intracellular formation of tau tangles, which then fosters a further decline in memory and loss of cognitive function in the host.1–2 Aside from the amyloidosis of Aβ and its downstream tauopathy, as well as the accomplice of apolipoprotein E which possesses a genetic bias, inflammation due to bacterial and viral infection and their stimulation to cerebral immunity (e.g., microglial activities) are other factors pertinent to the etiology of AD.3
The COVID-19 pandemic has caused immense damage to global human health, whose social and economic impact is not fully understood. The high rate of infection elicited by the novel SARS-CoV-2 coronavirus and its genetic variants firstly incites inflammation in the respiratory tract, causing acute as well as long-term conditions (i.e., the so-called “Long Covid”) in the bloodstream, the heart, the pancreas, the liver, the kidneys, and the brain of the infected host.4–8 Specifically, infections by influenza A, hepatitis C, HIV as well as SARS-CoV-2 viruses have been found to elevate the potential of developing brain disorders such as Parkinsonism, via processes which have not been deciphered.9–10
Mechanistically, recognition of the surface spike protein of SARS-CoV-2 by ACE-2 receptor of host cells plays a decisive role in viral infection. In vivo, the spike protein may be cleaved off by proteases to render a series of peptide fragments. The short peptide fragments differ in sequence and amphiphilicity to entail diverse capacities in influencing the aggregation kinetics of amyloid proteins such as Aβ, alpha synuclein (associated with Parkinson’s disease) and human islet amyloid polypeptide (associated with type 2 diabetes).11–17 To assess the potential effect of SAR2-CoV-2 viral infection on AD pathogenesis, in this study we examined the aggregation and toxicity of Aβ in the presence of 1058HGVVFLHVTYV1068 (or “Spike1058” in abbreviation), a 11-residue amyloidogenic fragment of SARS-CoV-2 spike protein identified by our computational analysis (Scheme). Spike1058 elevated the aggregation of Aβ in a dose-dependent manner, evidenced by a thioflavin T (ThT) fluorescence kinetic assay, transmission electron microscopy (TEM), Fourier transform infrared (FTIR), circular dichroism (CD), size exclusion-high-performance liquid chromatography (SEC-HPLC), and surface plasmon resonance (SPR) spectroscopies, and corroborated by discrete molecular dynamics (DMD) simulations (Scheme). In addition, Spike1058 was found to be toxic itself, further promoting the toxicity induced by Aβ in SH-SY5Y neuroblastoma cells while hampering the hatching and survival of zebrafish embryos. This study offered evidence for implicating the SARS-CoV-2-AD axis in connection with Long Covid.
Scheme.
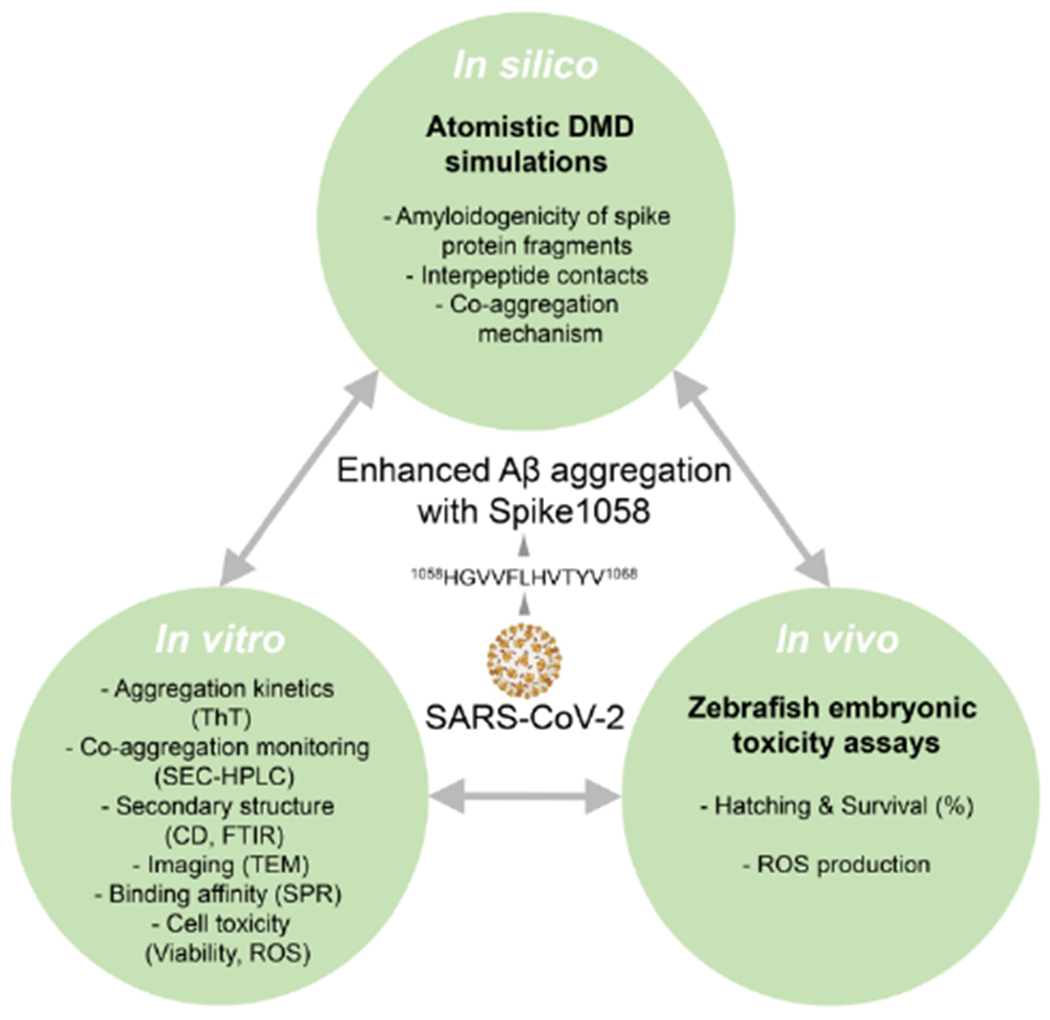
Synergistic in silico, in vitro, and in vivo methodologies for probing the effect of Spike1058 on Aβ amyloidogenesis.
2. Materials and methods
All-atom DMD simulations.
Discrete molecular dynamics (DMD) is a rapid and predictive molecular dynamics algorithm18–22 that has been widely employed to investigate protein and peptide aggregation. In all-atom DMD, all heavy atoms and polar hydrogen atoms of biomolecules are explicitly modelled, based on interaction potentials encompassing van der Waals, solvation energy, electrostatic, and hydrogen bonding interactions.23 In this study, the van der Waals parameters used in the DMD simulations were obtained from CHARMM19,24 while the Lazaridis-Karplus effective energy function, EEF1,25 was applied to calculate the corresponding solvation energy using an implicit solvent model. A reaction-like algorithm was employed to capture distance- and angle-dependent hydrogen bond interactions.23 Screened electrostatic interactions were estimated using the Debye-Hückel approximation, with a Debye length of 1 nm due to a water dielectric constant of 80 and a monovalent electrolyte concentration of approximately 0.1 M. To account for counterion condensation effects26 and maintain a net zero charge in the simulated system, counter ions (Cl− or Na+) were added as needed. The Andersen thermostat was utilized to keep the system at a constant temperature of 300 K during DMD simulations.27 A DMD simulation engine is accessible at http://www.moleculesinaction.com.
Computational peptide array method.
To examine the amyloidogenicity of the spike protein, we employed the computational peptide array method, similarly to our previous study.28 The full-length spike protein sequence obtained from the Uniprot database (P0DTC2) was divided into a series of overlapping 20-residue fragments, with each fragment being shifted by 10 residues along the sequence. For each split sequence, we performed five independent DMD simulations of two identical spike protein fragments at 300 K inside a 10 nm cubic simulation box. Periodic boundary condition (PBC) was used. The minimum initial intermolecular distance was set to be 1.5 nm. Each independent simulation started with randomized initial position, orientation, and velocity and lasted approximately 0.4 μs. All five independent simulations were used to evaluate the propensity to self-associate and form inter-peptide β-sheets.
Replica exchange DMD simulations.
We applied replica exchange DMD (rxDMD) simulations to identify the binding hotspots of Aβ42 with Spike1058. The molecular system was consisted of a full-length Aβ42 monomer and a Spike1058 monomer in a 10 nm cubic simulation box with PBC. The initial Aβ42 structure was taken from the solution structure with the PDBID 1Z0Q.29 The Spike1058 monomer was initialized in a coil structure. 16 replicas were utilized with temperatures in the range of 275-395 K. The replica exchange approach allowed for enhanced exploration of different conformational states and facilitated the sampling of diverse structural ensembles. In replica exchange simulations, temperatures were exchanged every 50 ps between replicas of adjacent temperatures according to the Metropolis criterion to overcome local free-energy barriers that may limit sampling of the conformational states. Each replica simulation lasted ~0.8 μs.
Aggregation simulations.
Aggregation DMD simulations with 12 peptides were performed for each of the five targeted spike protein fragments: 116SLLIVNNATNVVIK129, 196NIDGYFKIYSK206, 608VAVLYQDVNCT618, 1173NASVVNIQKE1182, and 1058HGVVFLHVTYV1068. Each fragment was studied by running 30 independent trajectories at a constant temperature of 300 K with PBC. For each independent trajectory, the simulation was conducted with a 15-nm cubic simulation box. Counter ions were added for charge neutralization. The starting configuration of each peptide and ion was randomly assigned in terms of relative position and orientation with an intermolecular distance of at least 1.5 nm. The spike protein fragments were initialized in fully extended conformations, which rapidly relaxed into coil structures in simulations. Each trajectory lasted at least ~0.3 μs with a total accumulative simulation time of ~9 μs for each spike protein fragment. Additionally, 196NIDGYFKIYSK206 (Spike196) and 1058HGVVFLHVTYV1068 (Spike1058) were selected to continuously run longer DMD simulations so that their trajectories lasted ~0.7 μs. The last 0.450 μs of these trajectories were found to reach their steady states and thus used for analyzing the equilibrium aggregation dynamics.
The co-aggregation of Aβ12-22 or Aβ27-37 peptides with Spike1058 was also investigated. For each of the two Aβ fragments, 8 peptides were simulated with and without the presence of 8 Spike1058 peptides. For each molecular system, 30 independent trajectories were generated in a cubic simulation box of 200 nm with PBC and the corresponding starting configuration had all the peptides and ions randomly positioned with an intermolecular distance of at least 1.5 nm, and all peptides were initialized in the coil conformation. Each trajectory of the co-aggregation DMD simulations lasted at least 0.9 μs, corresponding to over 27 μs of total accumulated simulation time.
Computational analysis methods.
The secondary structures of the peptides were determined using the DSSP algorithm.30 A backbone hydrogen bond between two residues was recognized if the distance between the nitrogen (N) and oxygen (O) atoms was less than 3.5 Å and the angle between NH and O was greater than 150°. Two peptides were considered to form a β-sheet if (i) at least two consecutive residues in each chain adopted the β-strand conformation and (ii) they formed at least two backbone hydrogen bonds. The size of a β-sheet was the number of β-strands. We assessed the secondary structure content of the peptides using a time-averaging approach. Determination of the secondary structure content per residue involved calculating the average occurrence of a specific secondary structure for each residue within the peptide. To evaluate the secondary structure propensity of individual residues, we counted their frequency of adopting a particular type of secondary structure. By averaging these counts over time, we obtained the secondary structure propensity values for each residue.
Two atoms were considered in contact if their distance was less than the cutoff of 5.5 Å. Two residues were recognized to be in contact if they had at least one atomic contact between them, resulting in a residue contact. Accordingly, residue contact frequency between residues A and B can be evaluated by:
where T is the time period for the residue contact frequency evaluation; NA, NB refer to the number of heavy atoms in residues A and B, respectively; dcutoff denotes the coutoff distance of 5.5 Å, while dij(t) corresponds to the distance between atoms i and j at moment t.
We introduced a simple amyloidogenic score to quantify the propensity of a residue to contribute to amyloid formation. The score of a residue was determined by multiplying its β-sheet propensity, which represented its inherent tendency to adopt β-sheet structures in simulations, with its binding frequency to the other peptide. This multiplication captured the likelihood of the interaction between the residue and the peptide leading to the stabilization of a β-sheet structure.
Peptide aggregation was evaluated using the single-linkage clustering algorithm, with a peptide being assigned to a cluster if it was in contact with any of the component peptides. The size of a cluster was defined as the number of composite peptides in the cluster. To evaluate average cluster size of the system at a particular frame, the mass weighted cluster size was defined as:
Here, K denotes the number of clusters, N represents the total number of peptides, and ni corresponds to the number of peptides in the i-th cluster.
We identified a β-sheet cluster as a group of peptides in which every member formed a β-strand and belonged to a β-sheet. A β-sheet cluster can comprise multiple β-sheets. To determine the average size of β-sheets within a cluster, we used the following formula:
where nβ denotes the number of β-sheets in the β-sheet cluster, and ni refers to the number of β-strands in the i-th β-sheet. We estimated the number of β-sheet layers in a simulation frame by dividing the size of the largest β-sheet cluster (i.e., the number of peptides) with the average size of β-sheets within the cluster (i.e., ).
To visualize the free-energy landscape of the aggregation dynamics of an aggregating system in a two-dimensional configuration space, we used the potential of mean force (PMF) as a function of two certain parameters, x and y, throughout the entire simulation duration:
Here, kBT represents the thermal energy at 300 K and P(x,y) the probability of the system having configuration parameters x and y.
The Weighted Histogram Analysis Method (WHAM)31–34 was used to evaluate the mean values of parameters obtained from rxDMD simulations at different temperatures. Here, the last 400 ns of trajectories from all 16 replicas were analyzed with WHAM to obtain the mean value of a specific parameter as a function of temperature in the range of 275~395 K.
Thioflavin T (ThT) fluorescence kinetics assay.
Spike1058 (1058HGVVFLHVTYV1068) and Aβ42 (DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGVVIA), with purity ⩾95% for both peptides, were acquired from China Peptides Co. Ltd. Firstly, Aβ42 (or simply abbreviated as “Aβ”, unless specified otherwise) and Spike1058 were treated with hexafluoro-2-propanol (HFIP) to breakdown pre-existing small aggregates, and freeze-dried. The lyophilized powder of Aβ42 was dissolved in 0.01% NH4OH for dissolution purpose and left in the open for 20 min to evaporate NH4OH, and then stored at −20 °C for further experiments. For the ThT kinetic assay, a 100 μL aqueous solution mixed with 25 μM Aβ42 and 25 μM ThT was incubated with or without different concentrations of Spike1058 (12.5, 25 and 50 μM) in a 96-well plate with a transparent bottom (Corning Costar 3880) at 37 °C. The ThT fluorescence was measured on a SPARK microplate reader (TECAN, Australia) with excitation at 445 nm and emission at 488 nm every 30 min for up to 90 h. ThT (25 μM) was regarded as a control. The kinetic parameter values (lag time, t½, and k) were calculated from the performed ThT assays following the literature.19, 35
Circular dichroism (CD) spectroscopy.
Aβ42 was mixed with or without different concentrations of Spike1058 as for the ThT assay and incubated for 48 h at 37 °C. Far-ultraviolet spectra were recorded by Spectra Manager 2.0 in a Jasco J-815 spectropolarimeter at room temperature. Each spectrum was acquired from 260 to 190 nm with a 1 nm step size. The CD signal from water was subtracted from the data of each sample solution and averaged from three scans. The data were plotted in GraphPad Prism 7.0 and the secondary structural contents were determined by the Beta Structure Selection method.36
Fourier transform infrared (FTIR) spectroscopy.
Aβ42 mixed with or without Spike1058 was incubated for 48 h at 37 °C and lyophilized. The lyophilized powder was redissolved with 10 μL deionized water to prepare the FTIR samples. FTIR spectra were recorded by an IRTracer-100 (Shimadzu, Japan) spectrometer under liquid nitrogen cooling. All spectra were collected for 1580-1700 cm−1 wavenumbers. The secondary structures of all samples were analyzed by Origin Software (Origin Lab) using the built-in Peak Deconvolution application as in our previously study.35
Transmission electron microscopy (TEM).
Aβ42 samples from the ThT assay were dropped onto 300-mesh amorphous carbon-coated copper grids and allowed to incubate for 10 min. Excess aqueous was removed and the grids were negatively stained with 3% phosphotungstic acid (w/v, pH = 7) for 30 s. The grids were allowed to dry overnight at room temperature. TEM imaging was performed using a Jeol JEM1400 Flash TEM microscope, operated at 80 kV.
Size exclusion-high-performance liquid chromatography with UV-Vis (SEC-HPLC-UV).
An UHPLC system (Agilent, USA) comprising a sample manager set at 4 °C and a column oven set at 30 °C was coupled to a multichannel rapid scanning UV-VIS detector (Agilent, USA) for online SEC/UV instrumentation. Aβ42 (25 μM) was mixed with or without Spike1058 (25 μM) incubated for 4 h at 37 °C and separated by an AdvanceBio SEC column (4.6 × 150 mm, 1.9 μm particle size, 200 A pore size) (Agilent, USA) at 25 °C, and the mobile phase was equilibrated with phosphate-buffered saline (PBS) where the pH was adjusted to 5.6. The flow rate was 0.25 mL min−1, and the detection was performed at the wavelength of 220 nm. Calculations of the amounts of Aβ42 and Spike1058 were carried out based on intensity peak areas acquired for the samples.
Surface plasmon resonance (SPR) analysis.
The Aβ42-Spike1058 binding kinetics was probed using Biacore T200 (GE Healthcare). The running buffer composed of 25 mM HEPES (pH = 7.4) and 1% DMSO, and 0.05% surfactant P20 was prepared, vacuum filtered, and degassed immediately prior to use. Spike1058 (50 μg/mL) was immobilized on a CM5 sensor chip via immobilization buffer containing 10 mM sodium acetate (pH = 4.5) to a level of ~10,000 response units. Serial concentrations of Aβ42 (0, 156.25, 312.5, 625, 1250, 2500, 5000 and 10000 nM) were injected to the Fc1 (control)-Fc2 (active) channels at a flow rate of 30 μL/min for an association phase of 120 s, followed by 300 s of dissociation at 25 °C. 8 cycles of analytes were repeated according to analyte concentrations in ascending order. After each cycle, the sensor chip surface was regenerated completely by 10 mM glycine-HCl as injection buffer at a flow rate of 30 μL/min for 30 s to remove the analyte. The resulting data were fit to a 1:1 binding model using Biacore Evaluation Software (GE Healthcare).
Cytotoxicity and reactive oxygen species (ROS) assays.
SH-SY5Y cell lines were purchased from IMMOCELL (Guangzhou, China) (ATCC® CRL-2266™) and cultured in DMEM supplemented with 10% fetal bovine serum and 1% penicillin and streptomycin (Gibco, USA) under 5% CO2. Cells were seeded at the density of 2×104 cells per well in 96-well plates and cultured for 24 h. Then 25 μM of Aβ42 mixed with or without different concentrations of Spike1058 (12.5, 25 and 50 μM) was added to the wells, respectively. After 24 h of treatment, cell viability was evaluated by Enhanced Cell Counting Kit-8 (Beyotime, China). The optical density (OD) at 450 nm of each well was recorded, and the well with only medium was set as the normal control. Cell viability was calculated according to:
For the reactive oxygen species (ROS) assay, SH-SY5Y cells were seeded at a density of 25 × 104 cells per well in 24-well plates and cultured for 24 h. Aβ (25 μM) with or without Spike1058 as in the cellular toxicity assay was added to the wells after 24 h incubation. To determine ROS generation, the fluoroprobe DCFH-DA (Beyotime, China) was used. The wells treated with medium were set as the normal control. Images were obtained with a fluorescence microscope (Leica, Wetzlar, Germany) and the DCF fluorescence was quantified at excitation/emission wavelengths of 495 nm/529 nm on a SPARK microplate reader (TECAN, Australia).
Toxicity assessment in zebrafish embryos.
AB wild-type adult zebrafish (Danio rerio) were sourced from China Zebrafish Resource Center and housed in a fish breeding circulating system (Hai Sheng, Shanghai, China) on a 14 h:10 h light/dark cycle at 28 ± 0.5 °C. Two pairs of male/female zebrafish were placed into the mating box with a divider before spawning. Spawning was triggered by removing the divider in the morning and the embryos were collected after 1 h. Embryos rinsed with 0.5 mg/L methylene blue solution were then transferred to Holtfreter′s medium (composed of 7 g NaCl, 0.40 g NaHCO3, 0.235 g CaCl2 and 0.1 g KCl in 2 L deionized water, pH = 7) in a Petri-dish at 28 °C. Healthy and fertilized embryos at the same developmental stages were selected for further experiments under a stereomicroscope (Olympus-SZ61, Olympus Ltd., Japan).
Microinjections were performed with a finely calibrated needle using a pneumatic microinjection system (PV830 Pneumatic Picopump, WPI) at an injection pressure of 30 psi. Healthy zebrafish embryos were positioned on a 10 mg/mL agarose gel plate and microinjected with Aβ42 (100 μM) or Spike1058 (12.5, 25, 50 μM) or their mixtures. A volume of 3.5 nL of the above solution was injected into the yolk of embryos at 1 hour-post-fertilization (hpf), and the same volume of Holtfreter′s medium was injected as the vehicle control. After the injection, the embryos were transferred to 96-well plates, with each well containing 200 μL of Holtfreter′s medium. Toxicity endpoints, including hatching interference and mortality, were assessed at 72 hpf using a bright field stereomicroscope (Olympus-SZ61, Olympus Ltd., Japan). Three replicates were carried out for each treatment, with each replicate consisting of 8 embryos.
ROS assays in zebrafish embryos.
To evaluate the generation of ROS, zebrafish embryos were collected after 12 h post-injection. The embryos were then incubated with 10 μM of 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) at 28 °C in darkness for 60 min, followed by three washes with Holtfreter’s medium. Tissue homogenates were obtained by grinding the embryos using a hybrid grinding machine (Biheng Bio-Technique Co. Ltd., Shanghai, China) and 200 μL of homogenates in each replicate were loaded into each well of a 96-well black/clear bottom plate. ROS production was determined by measuring the fluorescence intensity of DCF, which was excited at 485 nm and emitted at 530 nm, using a microplate reader (Varioskan LUX, Thermo Fisher Scientific, USA). Three replicates were carried out for each treatment, with each replicate consisting of 8 embryos. All zebrafish related experiments were conducted in accordance with the Animal Ethics Committee at Tongji University (Protocol #TJAD00723B02).
Statistical analysis.
In vitro experiments were performed in triplicate and data points were depicted as mean values of repeated measurements (n=3) ± standard deviation (SD). One-way analysis of variance (ANOVA) was conducted for ThT, cellular viability (%) and DCF fluorescence intensity values to evaluate the statistically significant differences between the values among the multiple groups, followed by Tukey’s multiple-comparison test. Statistically significant differences were considered when the p values were lower than 0.033 for data in Fig. 3 and lower than 0.05 for data in Fig. 4. For the zebrafish assays, all treatments were performed with three replicates, and the data were reported as average ± standard deviations (SD). One-way analysis of variance (ANOVA) was conducted to evaluate the statistically significant differences of hatching rates, survival rates, and ROS production among the multiple groups, followed by Tukey’s multiple-comparison post hoc test. Statistically significant differences were considered when the p values were lower than 0.05.
Fig. 3. Spike1058 accelerated Aβ fibrillization.
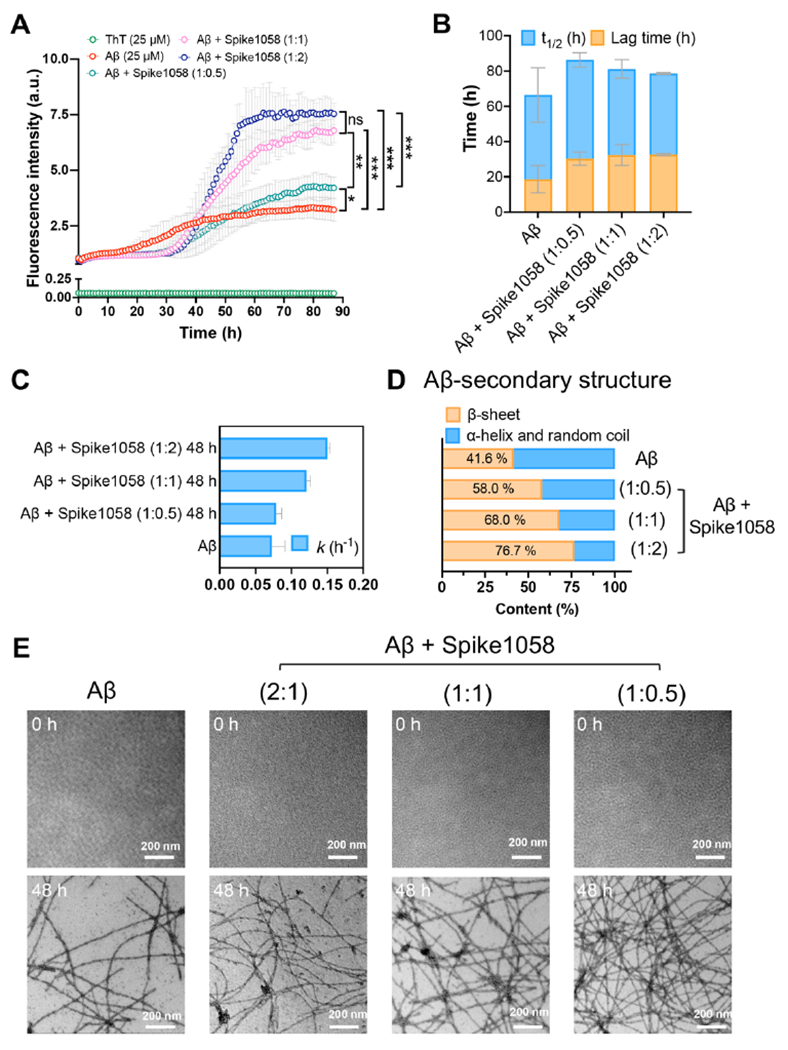
(A) A ThT fluorescence kinetic assay for determining the amyloid aggregation of Aβ (25 μM) in the presence and absence of Spike1058 (12.5, 25 and 50 μM). (B-C) ThT-derived kinetic aggregation parameters (t1/2, lag time, and apparent aggregation constant k values) over time (h) for Aβ in the presence and absence of Spike1058 (12.5, 25 and 50 μM). (D) CD-derived secondary structure profiles for non-incubated (0 h) and incubated (48 h) Aβ (25 μM) with and without Spike1058 (12.5, 25 and 50 μM). (E) TEM imaging of amyloid aggregation for non-incubated (0 h) and incubated (48 h) Aβ (25 μM) with and without Spike1058 (12.5, 25 and 50 μM). Scale bars: 200 nm. For panels A-C, data points are depicted as mean values of repeated measurements (n=3) ± standard deviation (SD). Statistical analysis was performed through Tukey’s multiple comparisons test: * p < 0.033, ** p < 0.002 and *** p < 0.001.
Fig. 4. Spike1058 accelerated Aβ-induced adverse effects in SH-SY5Y human neuroblastoma cells.
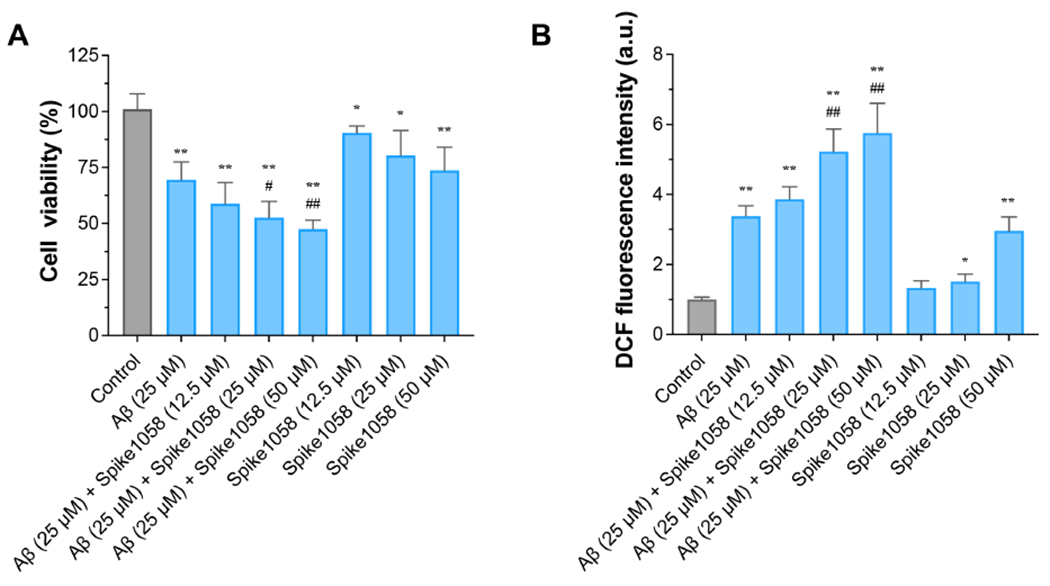
(A) SH-SY5Y cell viability (%) induced by Aβ with or without Spike1058 for 24 h. (B) ROS levels were determined induced by Aβ with or without Spike1058 for 12 h by DCFH-DA. Data were expressed as the mean ± S.D. (n=3). Significant: * p < 0.05, ** p < 0.01 vs. normal group; # p < 0.05, ## p < 0.01 vs. Aβ group.
3. Results and discussion
Computational screening of amyloidogenic spike protein fragments.
To identify potential amyloidogenic fragments of the spike protein, we employed DMD simulations using the computational peptide array method and determined an amyloidogenic score for each residue (see Methods). Additionally, we utilized available amyloid-predicting algorithms, including ANuP, PASTA, AGGRESCAN, and Waltz,37–40 to identify sequence amyloidogenicity and compared our results with those obtained by other algorithms, as shown in Supporting Information, Fig. S1. Based on experimental studies by Nyström and Per Hammarström15 as well as the consensus of different predictors, we selected five spike protein fragments: 116SLLIVNNATNVVIK129, 196NIDGYFKIYSK206, 608VAVLYQDVNCT618, 1058HGVVFLHVTYV1068 and 1173NASVVNIQKE1182 for further study. Here, fragments of residues 196-206, 608-618, and 1173-1182 have been experimentally confirmed to form fibrils,15 validating the suitability of our computational peptide array method (Fig. S1) and the amyloid-predicting algorithms.37–40
Next, we performed aggregation simulations with 12 peptides for each of the selected five fragment sequences (see Methods). To monitor the fibrillization in silico, we analyzed the mass-weighted cluster size and β-sheet content per peptide averaged over all independent simulations as a function of time, as illustrated in Fig. S2. Our results revealed that residues 116-129, 196-206, and 1058-1068 exhibited rapid increases in the cluster size and β-sheet content, maintaining β-sheet structures within the growing aggregates. This suggested a stronger fibrillization capability of these fragments compared to residues 608-618 and 1173-1182. Among the three peptides with promising fibrillization propensity in silico, we specifically focused on 196NIDGYFKIYSK206, referred to as Spike196, and 1058HGVVFLHVTYV1068, referred to as Spike1058, for further study. Both fragments adopted β-sheet conformations buried in the full-length spike protein (Fig. S3) and may only self-assemble after proteolytic cleavage. Spike196 has already been reported to form amyloid fibrils in experiments,15 while Spike1058 is a de novo spike protein fragment predicted to be amyloidogenic. Next, we continued additional DMD simulations for Spike196 and Spike1058 to reach equilibria, as depicted in Fig. S4.
By evaluating the equilibrated aggregate structures, we observed that Spike1058 had a greater β-sheet content than Spike196 (Fig. 1A&1B). This difference can be explained by a greater hydrophobicity of the Spike1058 sequence (HGVVFLHVTYV) as compared to Spike196 (NIDGYFKIYSK), containing more hydrophobic and aromatic residues as highlighted in bold. This stronger inter-peptide interactions of Spike1058 resulted in a more favorable fibril-like state and fewer intermediate states than Spike196 in the two-dimensional potential of mean force for aggregation (2D-PMF, i.e., the effective aggregation free-energy landscape; (Fig. 1C). Furthermore, we discovered that the aggregates of Spike1058 were usually comprised of multilayer β-sheets (Fig. 1D) due to extensive interpeptide and inter-sheet interactions, forming the cross-β like fibril core. In contrast, Spike196 predominantly formed single-layer sheets (Fig. 1D) and its aggregates were easy to dissociate, leading to a greater number of intermediate states (e.g., low free-energy for small β-sheet rich aggregates in Fig. 1C).
Fig. 1. Self-aggregation of Spike196 and Spike1058.
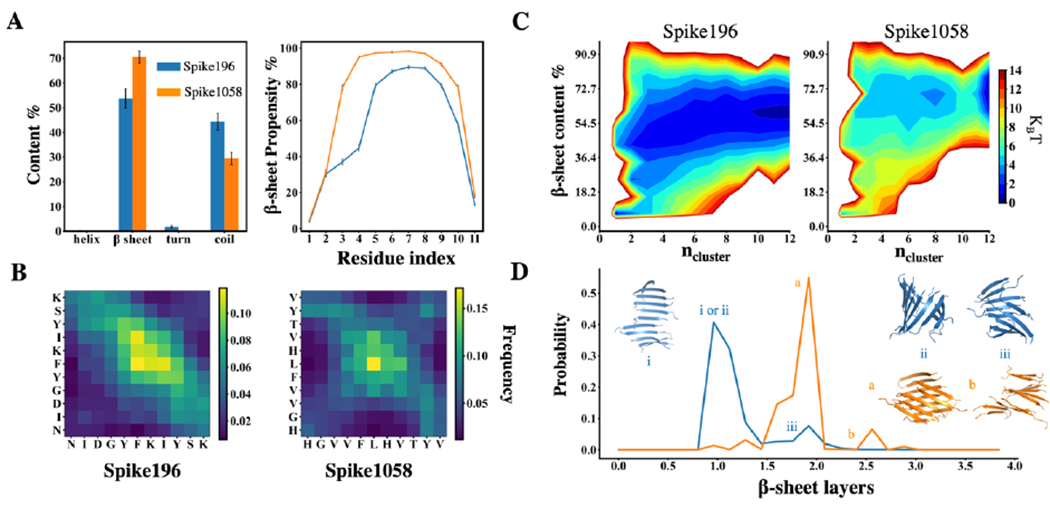
(A) Secondary structure content per peptide and residue-wise β-sheet propensity. The error bars correspond to the standard error of mean, averaged over each peptide and independent simulations. (B) Interpeptide contact maps. (C) Aggregation free-energy landscapes using 2D potential of mean force (PMF) of Spike fragments as a function of β-sheet content per peptide and the number of peptides in an aggregation cluster, ncluster. (D) Probabilities of observing different numbers of β-sheet layers for Spike196 (blue) and Spike1058 (orange). The representative snapshots correspond to the peaks labelled as i, ii, iii for Spike196 and a, b for Spike1058.
Spike1058 accelerated Aβ42 aggregation in DMD simulations.
Based on the observed greater self-aggregation propensity, we selected Spike1058 as a candidate to assess its influence on Aβ aggregation, both in silico and in vitro. To address the computational challenges of directly observing the aggregation and formation of fibril-like aggregates of full-length Aβ with and without the presence of Spike1058, we adopted a divide-and-conquer strategy. Specifically, we first identified the interaction hotspots of full-length Aβ42 with Spike1058,28, 41–42 and then used the Aβ fragments as surrogates in co-aggregation simulations to evaluate the impact of Spike1058 on Aβ aggregation.
Based on rxDMD simulations of a full-length Aβ42 monomer interacting with a Spike1058 monomer (see Methods), we computed the contact frequency map between residues of Spike1058 and Aβ42 (Fig. 2A) using WHAM analysis. Two Aβ fragments of residues 12-22 (Aβ12-22) and residues 27-37 (Aβ27-37) were observed to form prominent interactions with Spike1058. Moreover, these Spike1058-binding hotspots of Aβ overlapped with two well-known amyloidogenic regions of Aβ – i.e., the central hydrophobic cores of residues 16-22 and residues 25-35.43–44 Next, we selected two fragments, Aβ12-22 and Aβ27-37, to study the effect of Spike1058 on Aβ aggregation. We investigated the co-aggregation dynamics by performing DMD simulations of these Aβ fragments with and without an equal molar of Spike1058 peptides. Notably, the presence of Spike1058 significantly accelerated the aggregation of both Aβ fragments, evidenced by the faster increase in β-sheet content as a function of simulation time (Fig. 2B). Moreover, the β-sheet contents of the co-aggregation systems after reaching steady states (>500 ns) were greater compared to the control simulations of Aβ fragments alone, particularly for Aβ27-37. Additionally, we assessed the equilibration of the simulations by computing the time evolution of the ensemble-averaged potential energy and mass-weighted cluster size over independent simulations (Fig. S5).
Fig. 2. Co-aggregation of Spike1058 and Aβ peptides.
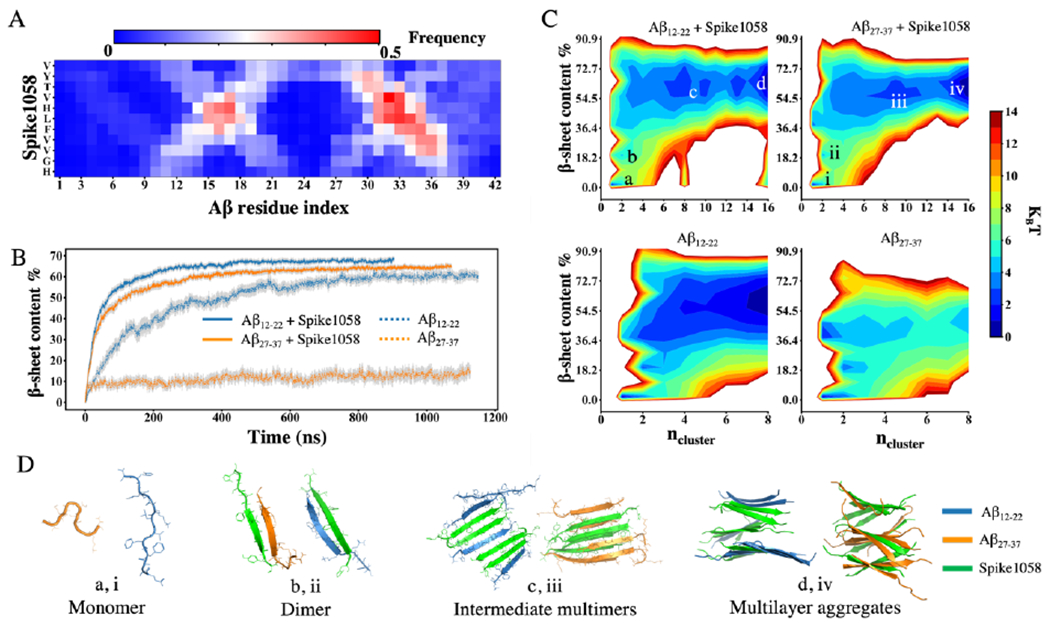
(A) Heatmap generated using WHAM on rxDMD simulations between Spike1058 and an Aβ42 monomer at 300 K. (B) Effect of equal amount Spike1058 on the β-sheet content per peptide of Aβ12-22 and Aβ27-37 over time. Error bars represent standard errors from the ensemble average over all independent trajectories. (C) Aggregation free-energy landscape using 2D PMF as a function of β-sheet content per peptide and the number of peptides in an aggregation cluster, including Aβ12-22 and Aβ27-37 with and without Spike1058. (D) Representative snapshots of conformations from the correspondingly labelled effective free-energy potential basins.
To gain further insights into the effect of Spike1058 on the aggregation dynamics of the Aβ fragments from isolated monomeric states, we computed the 2D-PMF as a function of the aggregation cluster size and β-sheet content utilizing data from the entire duration of the simulations (Fig. 2C). Representative snapshots of low-energy states were also shown to provide structural visualization of the corresponding lower-energy basins in the PMF (Fig. 2D). Both Aβ fragments in the presence of Spike1058 preferred to form hetero-dimers initially. The hetero-dimers were stabilized by inter-peptide hydrogen bonds and formed β-sheets. These early heterodimers grew by continuously incorporating free monomers or undergoing merging events with other aggregates, ultimately resulting in the formation of stable multilayer β-sheets.
Importantly, the presence of Spike1058 exerted a significant acceleration on the aggregation of Aβ fragments, which can be attributed to its strong binding interactions with these fragments as well as rapid formation of β-sheet rich hetero-complexes. Compared to Aβ12-22, the self-aggregation of Aβ27-37 was significantly slower (Fig. 2B). In most of the independent simulations, Aβ27-37 tended to remain in a monomeric state in coils as well as adopting β-hairpin and turn conformations (e.g., the average secondary contents shown in Fig. S6A). Only in a few out of the 30 independent simulations, Aβ27-37 were observed to form large β-sheet rich aggregations. To validate the aggregation of Aβ27-37 in DMD simulations, we further performed aggregation simulations of Aβ27-37 in a higher concentration. Specifically, we performed additional aggregation simulations for eight Aβ27-37 peptides using a smaller cubic simulation box of 10 nm (i.e., 8× greater peptide concentration than that in Fig. 2). Indeed, with a higher peptide concentration, Aβ27-37 were able to form stable β-sheet rich aggregates (Fig. S7). We also computed the inter-peptide contact frequency maps of Spike1058 co-aggregating with the two Aβ fragments (Fig. S6B). In both cases, hydrophobic Leu-Leu pairwise contacts were found to play a crucial role in facilitating the inter-peptide interactions. Additionally, the presence of adjacent hydrophobic residues – e.g., Phe, Ile, and Val in both Aβ and Spike1058 – also contributed to their interactions. The observed cross-like patterns in the contact maps indicated formation of both parallel and anti-parallel β sheets. Hence, these multilayer β-sheet rich co-aggregates (Fig. 2D) were stabilized by strong hydrophobic interactions between Aβ and Spike1058. Taken together, our computational results suggested that Spike1058 may bind and accelerate the aggregation of Aβ42.
Spike1058 accelerated Aβ42 fibrillization in vitro.
To corroborate our in silico findings regarding the conformation of Spike1058 and its effect on Aβ aggregation, we employed several experimental techniques including transmission electron microscopy (TEM), circular dichroism (CD) spectroscopy, Fourier transform infrared (FTIR) spectroscopy, a thioflavin T (ThT) fluorescence kinetic assay, size-exclusion-high performance liquid chromatography (SEC-HPLC), and a surface plasmon resonance (SPR) kinetic binding analysis. Specifically, the ThT kinetic assay was used to monitor real-time amyloid aggregation of Aβ at 25 μM in the presence and absence of Spike1058 at several Aβ:Spike1058 molar ratios of 1:0.5, 1:1 and 1:2. The aggregation of amyloid peptides was thoroughly analyzed, which constituted an initial lag phase with slow rates of aggregation, when amyloid peptides underwent structural rearrangements and conformational changes to render small nucleation centers.45 Herein, Aβ at 25 μM exhibited a typical sigmoidal curve with a lag phase of 18.7 ± 7.8 h and t½ of 47.9 ± 15.5 h (Fig. 3A–B), consistent with our previous reports.35,46 Nevertheless, once incubated with Spike1058 at 12.5, 25 and 50 μM, the lag phase of Aβ increased significantly to 30.3 ± 3.8, 32.3 ± 5.9 and 32.7 ± 0.58 h, respectively, implying a dose-dependent prolongment of Aβ nucleation at increasing Spike1058 concentrations (Fig. 3A–B). This result was accompanied by a change of t½ to 56.0 ± 4.1, 48.9 ± 5.2 and 46.0 ± 0.5 h further supporting the interpretation. On the other hand, the apparent aggregation constant k value increased from 0.072 ± 1.8×10−2 (Aβ control) to 0.078 ± 7.9×10−3, 0.121 ± 5.6×10−3 and 0.150 ± 3.8×10−3 h−1 upon the addition of different concentrations of Spike1058 (12.5, 25 and 50 μM) (Fig. 3C), indicating a significant dose-dependent pattern of accelerating the elongation and saturation kinetics of Aβ. This was further supported by the prolonged elongation phase from 17-42 h for Aβ control to 31-62 h, 33-59 h and 37-55 h for Aβ with increased Spike1058 concentrations (12.5, 25 and 50 μM) (Fig. 3A). This result implied that Spike1058 exposed their hydrophobic segments to enhance elongation of the Aβ species through co-aggregation. On the other hand, Spike1058 over the concentration range of 12.5, 25 and 50 μM did not notably enhance the fluorescence intensity of ThT over 90 h of incubation (Fig. S8) despite their amyloid-like morphology (Fig. S9), in accordance with the inability of ThT in tracing the aggregation kinetics of other short amyloidogenic peptide fragments.47 The effect of Spike1058 on amyloid morphology was further observed by TEM (Fig. 3E), where a typical twisted morphology of Aβ fibrils was rendered upon 48 h of incubation. In comparison, a denser fibrillar network consisted of Aβ amyloid fibrils was formed upon 48 h incubation with increased stoichiometric ratios of Spike1058. Fig. S10&Table S1 were acquired with SEC-HPLC to confirm the co-aggregation of Aβ and Spike1058. Specifically, while 4 h of incubation altered little the amounts of Aβ and Spike1058 individually, the amount of Aβ was increased by 26% while that of Spike1058 was decreased by 39.54%, respectively in the mixed Aβ-Spike1058 sample (molar ratio 1:2). Moreover, no other peaks were detected. Thus, it can be inferred that Spike1058 complexed with Aβ to render the dynamic aggregates recorded in SEC-HPLC. In addition, an SPR analysis revealed strong binding between Spike1058 and Aβ (binding constant kD of 82.6 nM, comparable to the strength of antibody-antigen association) (Fig. S11), corroborating our computational and experimental findings (Figs. 2 &3).
FTIR spectroscopy revealed the effect of Spike1058 on the secondary structure of Aβ upon co-aggregation (Fig. S12). In FTIR, the amide I band (1605-1695 cm−1), which primarily (70-85%) relies on the stretching vibrations of the C=O bond, constitutes the most intense absorption band of proteins.48 In our experiment, specifically, a characteristic shift of the maxima from higher wavenumber regions of 1648-1674 cm−1 to lower ones of 1623-1641 cm−1 provided qualitative evidence regarding the conformational transition of amyloid proteins from α-helical/random coils to β-sheets indicating the eventual occurrence of highly-ordered amyloid fibrils.48 By implementing this information and utilizing a band deconvolution tool (Methods) applied in our previous studies,19, 35 we were able to quantify and define the secondary structural profile of Aβ into α-helical/random coils and β-sheets, respectively. The acquired FTIR spectra (Fig. S12A) revealed that the β-sheet structure formed by Aβ was increased by 7.4% upon 48 h of incubation (Fig. S12E). Co-incubation with Spike1058 further enhanced Aβ’s β-sheet content by 7-33% (Fig. S12B–E). Notably, Spike1058 due to its hydrophobic character exhibited a characteristic band in the 1620-1625 cm−1 region in aqueous indicating adoption of a great number of β structures via intermolecular interactions and corroborating our computational data (Fig. 1, Figs. S2&S4). However, to exclude the likelihood of Spike1058’s intervention in the β-sheet content of Aβ during their co-incubation, we employed CD to offer additional information on the types of β-sheets formed. As shown in Fig. S13A, the CD spectrum of Aβ monomers displayed a minimum at ~ 200 nm before incubation, implying a typical random coil structure. Nevertheless, upon 48 h incubation, a negative peak at ~220 nm and a positive peak around ~198 nm appeared in the CD spectrum (Fig. S13A), indicating that Aβ transformed from its initial random coils to a β-sheet-rich conformation. Upon co-incubation of Aβ with Spike1058, the β-sheet content of Aβ was gradually increased by 16-35% in a dose-dependent manner (Table 1, Fig. 3D, Fig. S13B–D), supporting the data derived from FTIR spectroscopy. As control samples, CD spectra for non- and 48 h incubated Spike1058 were acquired in the range of 12.5-50 μM (Fig. S14). Specifically, the CD spectra indicated that the viral fragment did not significantly interfere as the background band at 210-220 nm (Fig. S14) was comparable to the Aβ-containing CD spectra (Fig. S13).
Table 1.
Secondary structure contents of peptides determined by CD spectroscopy.
| Secondary structure contents (%) | |||||
|---|---|---|---|---|---|
|
| |||||
| 37 °C/48 h | Anti-parallel (left-twisted) | Anti-parallel (relaxed) | Anti-parallel (right-twisted) | Parallel | Total β-sheets |
| Aβ | -a | - | 6.5 | 35.1 | 41.6 |
| Aβ/Spike1058=1/0.5 | 6.7 | 14.6 | 36.7 | - | 58.0 |
| Aβ/Spike1058=1/1 | 6.1 | 21.3 | 34.9 | - | 68.0 |
| Aβ/Spike1058=1/2 | 9.1 | 39.8 | 27.8 | - | 76.7 |
a: nondetectable
To gain further insights into β-sheet orientation (parallel/anti-parallel) and twisting, we utilized the web server of a novel algorithm program, β-structure selection (BeStSel),36 which reliably distinguished parallel from anti-parallel β-sheets and quantitatively determined the twist angle distribution of β-sheets based on the definition by Ho and Curmi.49
The CD spectrum of fibrillar Aβ solution at 48 h revealed that the amyloid fibrils were highly organized consisting mainly of parallel in-register β-sheets (84% out of total β-sheet content) (Table 1), in accordance with the reports using other analytical techniques (NMR, MD, XRD).50–53 Notably, co-incubation between the viral fragment and Aβ rendered the formation of aggregates with an enhanced propensity for anti-parallel β-sheets (Table 1). This specific result is notable as anti-parallel β-sheet formation among full-length wild-type Aβ is exclusively linked with intermediate prefibrillar species exerting significant neurotoxicity, compared to parallel orientations.54–55 We also analyzed the content of parallel vs. anti-parallel β-sheet contents in the co-aggregation simulations for Spike1058 and Aβ fragments. Using the averages of simulations with the two Aβ fragments, we indeed observed an increase of anti-parallel β-sheet ratio in the co-aggregation of Spike1058 and Aβ, compared to the self-aggregation of spike1058 and the Aβ fragments alone (Fig. S15). Other than Aβ42, amyloidogenic fragments of Aβ such as 16-22 and 11-28 fragments56–58 or the Iowa mutation,59 which is implicated in cerebral amyloid angiopathy (CAA),59 have been shown to form transient fibrils with anti-parallel structure, but eventually transformed into parallel β-sheet orientations. To further assess whether the viral fragment was able to exert significant impact on amyloid protein-induced toxicity, we performed a cell viability assay with SH-SY5Y human neuroblastoma cells.
Spike1058 accelerated the toxicity of Aβ in vitro.
Neurodegenerative-related amyloid proteins such as Aβ are known to have a deleterious effect on neuronal cells, leading to a cascade of events and AD or PD-related pathologies. Although the exact role of amyloid protein-derived neurotoxicity remains unsolved, several reports correlated cell toxicity with the ability of amyloid proteins to impact cell membrane integrity through various proposed models, including the carpet, detergent, and ion-channel pore models.60–66 As shown in Fig. 4, the viability of SH-SY5Y cells was significantly reduced upon 24 h exposure to Aβ. Viable SH-SY5Y cells further exhibited a gradual decline in number at increasing molar ratios of Spike:Aβ (0.5, 1, 2:1), reaching 22% at 2:1 Spike:Aβ molar ratio compared with the Aβ control group (p < 0.01, Fig. 4A) upon 24 h incubation. However, Spike1058 control at a similar concentration to Aβ (25 μM) impacted cell viability 11% less than Aβ control (Fig. 4A). In addition, cell mortality was elevated in a dose-dependent manner when exposed to Spike1058, where the highest Spike1058 concentration (50 μM) was less deleterious than Aβ control. Furthermore, as shown in Fig. 4B, when Aβ was pre-incubated with Spike1058 (25 and 50 μM), the intracellular ROS levels were significantly increased by 55% and 70% (p < 0.01) in a dose-dependent manner compared with Aβ control. Taken together, Spike1058 could elevate the toxicity induced by Aβ in neuronal cells.
Spike1058 accelerated Aβ42 toxicity in vivo.
Microinjection of Aβ and Spike1058, either individually or in combination, was employed to investigate the impact of Spike1058 on the toxicity of Aβ in zebrafish embryos (Fig. 5A). Both the timepoint of 72 hpf for examining embryonic hatching and survival and the timepoint of 12 h peptide co-incubation for quantifying ROS production ensured the toxicity of Aβ through self-aggregation was observable, based on one of our earlier studies using this animal model.67 As illustrated in Fig. 5B–D, the pronounced hatching interference and mortality at 72 hpf were induced by Aβ at 100 μM and Spike1058 at 12.5, 25, and 50 μM, compared to the control group. In addition, Spike1058 appeared to exert a more substantial influence than Aβ at 72 hpf on the hatching and mortality rate of zebrafish embryos. Moreover, the mixtures of Aβ and Spike1058 significantly decreased the hatching rate (p < 0.01) and survival rate (p < 0.05) in comparison to the administration of Aβ or Spike1058 alone, suggesting a potential synergistic effect where Spike1058 may augment the toxicity of Aβ. Furthermore, a significant increase in the generation of biotic ROS occurred in zebrafish embryos incubated with the Aβ-Spike1058 mixtures for 12 h, surpassing the levels observed with Aβ or Spike1058 alone (Fig. 5E), corroborating the trends observed in hatching and survival rates.
Fig. 5. Spike1058 accelerated Aβ-induced adverse effects in zebrafish embryos.
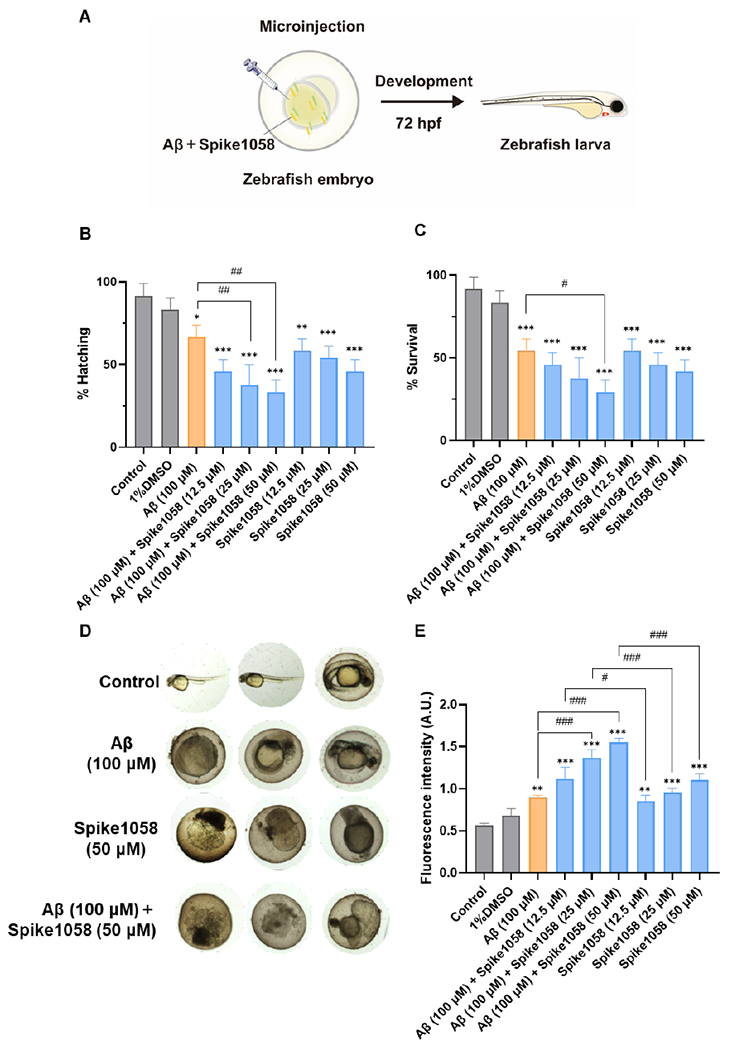
(A) Schematic of the experimental design with zebrafish embryos. (B) Hatching and (C) survival rate of zebrafish embryos induced by Aβ with or without Spike1058 at 72 hpf. (D) Representative images of healthy, dead, and abnormal embryos. (E) ROS levels of zebrafish embryos induced by Aβ with or without Spike1058 for 12 h by H2DCFDA. Data were determined by mean ± standard deviation. n = 3 (each replicate contained 8 zebrafish). Statistical analysis was performed through Tukey’s multiple-comparison post hoc test: * p < 0.05, ** p < 0.01, *** p < 0.001 vs. normal group; # p < 0.05, ## p < 0.01, ### p < 0.001 vs. Aβ group.
4. Conclusion
The COVID-19 pandemic is becoming a thing of the past after three plus years, technically, but an estimated 65 million people globally are still suffering from the aftermath of Long Covid that remains inadequately studied and poorly understood.68–69 Due to the large timescale in the development of dementia, coupled with the limited accessibility of the human brain, it is especially challenging to establish a plausible link between the pathologies of neurological disorders and viral infection.
Accordingly, in the current pilot study we first screened the spike protein of SARS-CoV-2 coronavirus computationally and identified an amyloidogenic candidate named the Spike1058 fragment (Fig. 1). We then examined Aβ amyloidosis and toxicity that were both exacerbated by the presence of Spike1058, using synergistic techniques of ThT (aggregation kinetics), TEM (aggregation morphology), FTIR (secondary structure), CD (secondary structure), cell viability and ROS production (toxicity) assays (Figs. 3–5). The molecular basis for the elevated aggregation and toxicity can be traced back to the self-aggregation potential of Spike1058 (Figs. 1&S9) as well as the strong binding affinity of Spike1058 for the amyloidogenic segments of Aβ to promote rapid dimerization of monomeric Aβ and structural transitions of the peptide to energetically more favorable toxic intermediates and (pre)fibrils that possessed anti-parallel cross-β sheets, as revealed by DMD simulations and CD, SEC-HPLC, FTIR and SPR spectroscopies (Figs. 2, 3, S6, S10–13). These in vitro and in silico findings were further corroborated by hatching, survival, and ROS assays performed with zebrafish embryos, where impaired development and increased toxicity were consistently observed with the animal model subjected to peptide co-aggregation. It should be noted that in AD animal models,70 the presence of functional proteins, cell membranes, and biological barriers could significantly slow down the self-aggregation processes of Aβ and Spike1058 and interfere with the mutual binding of the two peptide species, thereby retarding the phenomena as observed in this study. Together, our study implicated spike protein as a toxic amyloidogenic agent, whose co-aggregation with Aβ could potentially exacerbate the AD pathology (including the downstream events of Aβ amyloidosis in tauopathy, inflammation, and immune response3) and instigate Long Covid.
Supplementary Material
Acknowledgements
This work was supported by the National Key Research and Development Program, Ministry of Science and Technology of China (2021YFA12009000, 2022YFC2409700), National Natural Science Foundation of China (T2250710182), National Institutes of Health (R35GM145409, P20GM121342), National Science Foundation (CBET-2030828), and Fundamental Research Funds for the Central Universities. The content is solely the responsibility of the authors and does not necessarily represent the official views of the MSTC, NSFC, NSF, and NIH.
Footnotes
Supporting Information
The Supporting Information is available free of charge. Additional details include simulations of spike protein amyloidogenicity, aggregation of five targeted spike protein fragments, co-aggregation of Spike1058 with Aβ12-22 and Aβ27-37, β-sheet propensity, and self-aggregation of Aβ27-37 (Figs. S1–S7). ThT assay, TEM imaging, SEC-HPLC, SPR, FTIR and CD spectroscopies for co-aggregation (Figs. S8–S14) and computed parallel/anti-parallel β sheet ratios for co- and self-aggregation (Fig. S15).
Conflict of Interest
The authors declare no conflict of interest.
References
- (1).Ke PC; Zhou R; Serpell LC; Riek R; Knowles TPJ; Lashuel HA; Gazit E; Hamley IW; Davis TP; Fandrich M; Otzen DE; Chapman MR; Dobson CM; Eisenberg DS; Mezzenga R Half a Century of Amyloids: Past, Present and Future. Chem Soc Rev 2020, 49, 5473–5509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Hardy JA; Higgins GA Alzheimer’s Disease: The Amyloid Cascade Hypothesis. Science 1992, 256, 184–185. [DOI] [PubMed] [Google Scholar]
- (3).Long JM; Holtzman DM Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Andrews MG; Mukhtar T; Eze UC; Simoneau CR; Ross J; Parikshak N; Wang S; Zhou L; Koontz M; Velmeshev D; Siebert CV; Gemenes KM; Tabata T; Perez Y; Wang L; Mostajo-Radji MA; de Majo M; Donohue KC; Shin D; Salma J; Pollen AA; Nowakowski TJ; Ullian E; Kumar GR; Winkler EA; Crouch EE; Ott M; Kriegstein AR Tropism of Sars-Cov-2 for Human Cortical Astrocytes. Proc Natl Acad Sci U S A 2022, 119, e2122236119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Charnley M; Islam S; Bindra GK; Engwirda J; Ratcliffe J; Zhou J; Mezzenga R; Hulett MD; Han K; Berryman JT; Reynolds NP Neurotoxic Amyloidogenic Peptides in the Proteome of Sars-Cov2: Potential Implications for Neurological Symptoms in Covid-19. Nat Commun 2022, 13, 3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Kuhn CC; Basnet N; Bodakuntla S; Alvarez-Brecht P; Nichols S; Martinez-Sanchez A; Agostini L; Soh YM; Takagi J; Biertumpfel C; Mizuno N Direct Cryo-Et Observation of Platelet Deformation Induced by Sars-Cov-2 Spike Protein. Nat Commun 2023, 14, 620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Florindo HF; Kleiner R; Vaskovich-Koubi D; Acurcio RC; Carreira B; Yeini E; Tiram G; Liubomirski Y; Satchi-Fainaro R Immune-Mediated Approaches against Covid-19. Nat Nanotechnol 2020, 15, 630–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Mavrikaki M; Lee JD; Solomon IH; Slack FJ Severe Covid-19 Is Associated with Molecular Signatures of Aging in the Human Brain. Nat Aging 2022, 2, 1130–1137. [DOI] [PubMed] [Google Scholar]
- (9).Cohen ME; Eichel R; Steiner-Birmanns B; Janah A; Ioshpa M; Bar-Shalom R; Paul JJ; Gaber H; Skrahina V; Bornstein NM; Yahalom G A Case of Probable Parkinson’s Disease after Sars-Cov-2 Infection. Lancet Neurol 2020, 19, 804–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Michiels E; Rousseau F; Schymkowitz J Mechanisms and Therapeutic Potential of Interactions between Human Amyloids and Viruses. Cell Mol Life Sci 2021, 78, 2485–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Jana AK; Lander CW; Chesney AD; Hansmann UHE Effect of an Amyloidogenic Sars-Cov-2 Protein Fragment on Alpha-Synuclein Monomers and Fibrils. J Phys Chem B 2022, 126, 3648–3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Tavassoly O; Safavi F; Tavassoly I Seeding Brain Protein Aggregation by Sars-Cov-2 as a Possible Long-Term Complication of Covid-19 Infection. ACS Chem Neurosci 2020, 11, 3704–3706. [DOI] [PubMed] [Google Scholar]
- (13).Semerdzhiev SA; Fakhree MAA; Segers-Nolten I; Blum C; Claessens M Interactions between Sars-Cov-2 N-Protein and Alpha-Synuclein Accelerate Amyloid Formation. ACS Chem Neurosci 2022, 13, 143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Castelletto V; Hamley IW Amyloid and Hydrogel Formation of a Peptide Sequence from a Coronavirus Spike Protein. ACS Nano 2022, 16, 1857–1867. [Google Scholar]
- (15).Nystrom S; Hammarstrom P Amyloidogenesis of Sars-Cov-2 Spike Protein. J Am Chem Soc 2022, 144, 8945–8950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ledford H Severe Covid Could Cause Markers of Old Age in the Brain. Nature 2022, 612, 389. [DOI] [PubMed] [Google Scholar]
- (17).Ghosh A; Pithadia AS; Bhat J; Bera S; Midya A; Fierke CA; Ramamoorthy A; Bhunia A Self-Assembly of a Nine-Residue Amyloid-Forming Peptide Fragment of Sars Corona Virus E-Protein: Mechanism of Self Aggregation and Amyloid-Inhibition of Hiapp. Biochemistry 2015, 54, 2249–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Rapaport DC The Art of Molecular Dynamics Simulation, 2nd ed.; Cambridge University Press: Cambridge, 2004. doi: 10.1017/CBO9780511816581. [DOI] [Google Scholar]
- (19).Andrikopoulos N; Song Z; Wan X; Douek AM; Javed I; Fu C; Xing Y; Xin F; Li Y; Kakinen A; Koppel K; Qiao R; Whittaker AK; Kaslin J; Davis TP; Song Y; Ding F; Ke PC Inhibition of Amyloid Aggregation and Toxicity with Janus Iron Oxide Nanoparticles. Chem Mater 2021, 33, 6484–6500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Tang H; Sun Y; Ding F Hydrophobic/Hydrophilic Ratio of Amphiphilic Helix Mimetics Determines the Effects on Islet Amyloid Polypeptide Aggregation. J Chem Inf Model 2022, 62, 1760–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ke PC; Pilkington EH; Sun Y; Javed I; Kakinen A; Peng G; Ding F; Davis TP Mitigation of Amyloidosis with Nanomaterials. Adv Mater 2020, 32, e1901690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ding F; Radic S; Chen R; Chen P; Geitner NK; Brown JM; Ke PC Direct Observation of a Single Nanoparticle-Ubiquitin Corona Formation. Nanoscale 2013, 5, 9162–9169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ding F; Tsao D; Nie H; Dokholyan NV Ab Initio Folding of Proteins with All-Atom Discrete Molecular Dynamics. Structure 2008, 16, 1010–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Brooks BR; Bruccoleri RE; Olafson BD; States DJ; Swaminathan S; Karplus M Charmm: A Program for Macromolecular Energy, Minimization, and Dynamics Calculations. Journal of Computational Chemistry 1983, 4, 187–217. [Google Scholar]
- (25).Lazaridis T; Karplus M Effective Energy Function for Proteins in Solution. Proteins: Structure, Function, and Bioinformatics 1999, 35, 133–152. [DOI] [PubMed] [Google Scholar]
- (26).Manning GS Limiting Laws and Counterion Condensation in Polyelectrolyte Solutions I. Colligative Properties. The Journal of Chemical Physics 2003, 51, 924–933. [Google Scholar]
- (27).Andersen HC Molecular Dynamics Simulations at Constant Pressure and/or Temperature. The Journal of Chemical Physics 2008, 72, 2384–2393. [Google Scholar]
- (28).Song Z; Gatch AJ; Sun Y; Ding F Differential Binding and Conformational Dynamics of Tau Microtubule-Binding Repeats with a Preformed Amyloid-Beta Fibril Seed. ACS Chem Neurosci 2023, 14, 1321–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Tomaselli S; Esposito V; Vangone P; van Nuland NA; Bonvin AM; Guerrini R; Tancredi T; Temussi PA; Picone D The Alpha-to-Beta Conformational Transition of Alzheimer’s Abeta-(1-42) Peptide in Aqueous Media Is Reversible: A Step by Step Conformational Analysis Suggests the Location of Beta Conformation Seeding. Chembiochem 2006, 7, 257–267. [DOI] [PubMed] [Google Scholar]
- (30).Kabsch W; Sander C Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 1983, 22, 2577–2637. [DOI] [PubMed] [Google Scholar]
- (31).Feig M; Karanicolas J; Brooks CL 3rd. Mmtsb Tool Set: Enhanced Sampling and Multiscale Modeling Methods for Applications in Structural Biology. J Mol Graph Model 2004, 22, 377–395. [DOI] [PubMed] [Google Scholar]
- (32).Kumar S; Rosenberg JM; Bouzida D; Swendsen RH; Kollman PA The Weighted Histogram Analysis Method for Free-Energy Calculations on Biomolecules. I. The Method. Journal of Computational Chemistry 1992, 13, 1011–1021. [Google Scholar]
- (33).Hamilton GL; Saikia N; Basak S; Welcome FS; Wu F; Kubiak J; Zhang C; Hao Y; Seidel CAM; Ding F; Sanabria H; Bowen ME Fuzzy Supertertiary Interactions within Psd-95 Enable Ligand Binding. Elife 2022, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Saikia N; Yanez-Orozco IS; Qiu R; Hao P; Milikisiyants S; Ou E; Hamilton GL; Weninger KR; Smirnova TI; Sanabria H; Ding F Integrative Structural Dynamics Probing of the Conformational Heterogeneity in Synaptosomal-Associated Protein 25. Cell Rep Phys Sci 2021, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Andrikopoulos N; Li Y; Nandakumar A; Quinn JF; Davis TP; Ding F; Saikia N; Ke PC Zinc-Epigallocatechin-3-Gallate Network-Coated Nanocomposites against the Pathogenesis of Amyloid-Beta. ACS Appl Mater Interfaces 2023, 15, 7777–7792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Micsonai A; Wien F; Kernya L; Lee YH; Goto Y; Refregiers M; Kardos J Accurate Secondary Structure Prediction and Fold Recognition for Circular Dichroism Spectroscopy. Proc Natl Acad Sci U S A 2015, 112, E3095–3103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Prabakaran R; Rawat P; Kumar S; Michael Gromiha M Anupp: A Versatile Tool to Predict Aggregation Nucleating Regions in Peptides and Proteins. J Mol Biol 2021, 433, 166707. [DOI] [PubMed] [Google Scholar]
- (38).Walsh I; Seno F; Tosatto SC; Trovato A Pasta 2.0: An Improved Server for Protein Aggregation Prediction. Nucleic Acids Res 2014, 42, W301–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Conchillo-Sole O; de Groot NS; Aviles FX; Vendrell J; Daura X; Ventura S Aggrescan: A Server for the Prediction and Evaluation of “Hot Spots” of Aggregation in Polypeptides. BMC Bioinformatics 2007, 8, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Oliveberg M Waltz, an Exciting New Move in Amyloid Prediction. Nat Methods 2010, 7, 187–188. [DOI] [PubMed] [Google Scholar]
- (41).Ge X; Yang Y; Sun Y; Cao W; Ding F Islet Amyloid Polypeptide Promotes Amyloid-Beta Aggregation by Binding-Induced Helix-Unfolding of the Amyloidogenic Core. ACS Chem Neurosci 2018, 9, 967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Sun Y; Ding F Thermo- and Ph-Responsive Fibrillization of Squid Suckerin A1h1 Peptide. Nanoscale 2020, 12, 6307–6317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Naldi M; Fiori J; Pistolozzi M; Drake AF; Bertucci C; Wu R; Mlynarczyk K; Filipek S; De Simone A; Andrisano V Amyloid Beta-Peptide 25-35 Self-Assembly and Its Inhibition: A Model Undecapeptide System to Gain Atomistic and Secondary Structure Details of the Alzheimer’s Disease Process and Treatment. ACS Chem Neurosci 2012, 3, 952–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Clementi ME; Marini S; Coletta M; Orsini F; Giardina B; Misiti F Abeta(31-35) and Abeta(25-35) Fragments of Amyloid Beta-Protein Induce Cellular Death through Apoptotic Signals: Role of the Redox State of Methionine-35. FEBS Lett 2005, 579, 2913–2918. [DOI] [PubMed] [Google Scholar]
- (45).Knowles TP; Vendruscolo M; Dobson CM The Amyloid State and Its Association with Protein Misfolding Diseases. Nat Rev Mol Cell Biol 2014, 15, 384–396. [DOI] [PubMed] [Google Scholar]
- (46).Tang H; Li Y; Kakinen A; Andrikopoulos N; Sun Y; Kwak E; Davis TP; Ding F; Ke PC Graphene Quantum Dots Obstruct the Membrane Axis of Alzheimer’s Amyloid Beta. Phys Chem Chem Phys 2021, 24, 86–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Bunce SJ; Wang Y; Stewart KL; Ashcroft AE; Radford SE; Hall CK; Wilson AJ Molecular Insights into the Surface-Catalyzed Secondary Nucleation of Amyloid-Beta(40) (Abeta(40)) by the Peptide Fragment Abeta(16-22). Sci Adv 2019, 5, eaav8216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Sarroukh R; Goormaghtigh E; Ruysschaert JM; Raussens V Atr-Ftir: A “Rejuvenated” Tool to Investigate Amyloid Proteins. Biochim Biophys Acta 2013, 1828, 2328–2338. [DOI] [PubMed] [Google Scholar]
- (49).Ho BK; Curmi PM Twist and Shear in Beta-Sheets and Beta-Ribbons. J Mol Biol 2002, 317, 291–308. [DOI] [PubMed] [Google Scholar]
- (50).Antzutkin ON; Balbach JJ; Leapman RD; Rizzo NW; Reed J; Tycko R Multiple Quantum Solid-State Nmr Indicates a Parallel, Not Antiparallel, Organization of β-Sheets in Alzheimer’s β-Amyloid Fibrils. Proceedings of the National Academy of Sciences 2000, 97, 13045–13050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Toyama BH; Weissman JS Amyloid Structure: Conformational Diversity and Consequences. Annu Rev Biochem 2011, 80, 557–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Chen GF; Xu TH; Yan Y; Zhou YR; Jiang Y; Melcher K; Xu HE Amyloid Beta: Structure, Biology and Structure-Based Therapeutic Development. Acta Pharmacol Sin 2017, 38, 1205–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Fitzpatrick AW; Debelouchina GT; Bayro MJ; Clare DK; Caporini MA; Bajaj VS; Jaroniec CP; Wang L; Ladizhansky V; Muller SA; MacPhee CE; Waudby CA; Mott HR; De Simone A; Knowles TP; Saibil HR; Vendruscolo M; Orlova EV; Griffin RG; Dobson CM Atomic Structure and Hierarchical Assembly of a Cross-Beta Amyloid Fibril. Proc Natl Acad Sci U S A 2013, 110, 5468–5473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Sandberg A; Luheshi LM; Sollvander S; Pereira de Barros T; Macao B; Knowles TP; Biverstal H; Lendel C; Ekholm-Petterson F; Dubnovitsky A; Lannfelt L; Dobson CM; Hard T Stabilization of Neurotoxic Alzheimer Amyloid-Beta Oligomers by Protein Engineering. Proc Natl Acad Sci U S A 2010, 107, 15595–15600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Cerf E; Sarroukh R; Tamamizu-Kato S; Breydo L; Derclaye S; Dufrene YF; Narayanaswami V; Goormaghtigh E; Ruysschaert JM; Raussens V Antiparallel Beta-Sheet: A Signature Structure of the Oligomeric Amyloid Beta-Peptide. Biochem J 2009, 421, 415–423. [DOI] [PubMed] [Google Scholar]
- (56).Klimov DK; Thirumalai D Dissecting the Assembly of Abeta16-22 Amyloid Peptides into Antiparallel Beta Sheets. Structure 2003, 11, 295–307. [DOI] [PubMed] [Google Scholar]
- (57).Balbach JJ; Ishii Y; Antzutkin ON; Leapman RD; Rizzo NW; Dyda F; Reed J; Tycko R Amyloid Fibril Formation by a Beta 16-22, a Seven-Residue Fragment of the Alzheimer’s Beta-Amyloid Peptide, and Structural Characterization by Solid State Nmr. Biochemistry 2000, 39, 13748–13759. [DOI] [PubMed] [Google Scholar]
- (58).Banerjee S; Baghel D; Hasan Ul Iqbal M; Ghosh A Nanoscale Infrared Spectroscopy Identifies Parallel to Antiparallel Beta-Sheet Transformation of Abeta Fibrils. J Phys Chem Lett 2022, 13, 10522–10526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Qiang W; Yau WM; Luo Y; Mattson MP; Tycko R Antiparallel Beta-Sheet Architecture in Iowa-Mutant Beta-Amyloid Fibrils. Proc Natl Acad Sci U S A 2012, 109, 4443–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Li Y; Tang H; Andrikopoulos N; Javed I; Cecchetto L; Nandakumar A; Kakinen A; Davis TP; Ding F; Ke PC The Membrane Axis of Alzheimer’s Nanomedicine. Adv Nanobiomed Res 2021, 1, 2000040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Sciacca MFM; Tempra C; Scollo F; Milardi D; La Rosa C Amyloid Growth and Membrane Damage: Current Themes and Emerging Perspectives from Theory and Experiments on Abeta and Hiapp. Biochim Biophys Acta Biomembr 2018, 1860, 1625–1638. [DOI] [PubMed] [Google Scholar]
- (62).Korshavn KJ; Satriano C; Lin Y; Zhang R; Dulchavsky M; Bhunia A; Ivanova MI; Lee YH; La Rosa C; Lim MH; Ramamoorthy A Reduced Lipid Bilayer Thickness Regulates the Aggregation and Cytotoxicity of Amyloid-Beta. J Biol Chem 2017, 292, 4638–4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Viles JH Imaging Amyloid-Beta Membrane Interactions: Ion-Channel Pores and Lipid-Bilayer Permeability in Alzheimer’s Disease. Angew Chem Int Ed Engl 2023, 62, e202215785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Sciacca MF; Lolicato F; Tempra C; Scollo F; Sahoo BR; Watson MD; Garcia-Vinuales S; Milardi D; Raudino A; Lee JC; Ramamoorthy A; La Rosa C Lipid-Chaperone Hypothesis: A Common Molecular Mechanism of Membrane Disruption by Intrinsically Disordered Proteins. ACS Chem Neurosci 2020, 11, 4336–4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Terakawa MS; Lin Y; Kinoshita M; Kanemura S; Itoh D; Sugiki T; Okumura M; Ramamoorthy A; Lee YH Impact of Membrane Curvature on Amyloid Aggregation. Biochim Biophys Acta Biomembr 2018, 1860, 1741–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Kotler SA; Walsh P; Brender JR; Ramamoorthy A Differences between Amyloid-Beta Aggregation in Solution and on the Membrane: Insights into Elucidation of the Mechanistic Details of Alzheimer’s Disease. Chem Soc Rev 2014, 43, 6692–6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Javed I; Peng G; Xing Y; Yu T; Zhao M; Kakinen A; Faridi A; Parish CL; Ding F; Davis TP; Ke PC; Lin S Inhibition of Amyloid Beta Toxicity in Zebrafish with a Chaperone-Gold Nanoparticle Dual Strategy. Nature Communications 2019, 10, 3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Davis HE; McCorkell L; Vogel JM; Topol EJ Long Covid: Major Findings, Mechanisms and Recommendations. Nat Rev Microbiol 2023, 21, 133–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Marshall M Long Covid: Answers Emerge on How Many People Get Better. Nature 2023. [DOI] [PubMed] [Google Scholar]
- (70).Kakinen A; Javed I; Davis TP; Ke PC In Vitro and in Vivo Models for Anti-Amyloidosis Nanomedicines. Nanoscale Horiz 2021, 6, 95–119. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.