

Abstract
Oral-facial-digital syndromes (OFDS) are a group of clinically and genetically heterogeneous disorders characterized by defects in the development of the face and oral cavity along with digit anomalies. Pathogenic variants in over 20 genes encoding ciliary proteins have been found to cause OFDS through deleterious structural or functional impacts on primary cilia. We identified by exome sequencing bi-allelic missense variants in a novel disease-causing ciliary gene RAB34 in four individuals from three unrelated families. Affected individuals presented a novel form of OFDS (OFDS-RAB34) accompanied by cardiac, cerebral, skeletal and anorectal defects. RAB34 encodes a member of the Rab GTPase superfamily and was recently identified as a key mediator of ciliary membrane formation. Unlike many genes required for cilium assembly, RAB34 acts selectively in cell types that use the intracellular ciliogenesis pathway, in which nascent cilia begin to form in the cytoplasm. We find that the protein products of these pathogenic variants, which are clustered near the RAB34 C-terminus, exhibit a strong loss of function. Although some variants retain the ability to be recruited to the mother centriole, cells expressing mutant RAB34 exhibit a significant defect in cilium assembly. While many Rab proteins have been previously linked to ciliogenesis, our studies establish RAB34 as the first small GTPase involved in OFDS and reveal the distinct clinical manifestations caused by impairment of intracellular ciliogenesis.
Introduction
The primary cilium is a single nonmotile, microtubule-based structure that protrudes from the surface of many vertebrate cells. This sensory organelle detects both mechanical and chemical extracellular stimuli to direct a variety of downstream signaling responses (1). In particular, the primary cilium acts as a key organizing center for various signaling pathways including Hedgehog, WNT, Notch, TGFβ/BMP, growth factors and GPCRs (2). In light of this key role in numerous biological and developmental processes, it is not surprising that primary ciliary defects can lead to a broad spectrum of human diseases, collectively known as ciliopathies (3). Ciliopathies affect a range of body systems including the brain, kidneys, eyes, liver, ears, lung, heart, skeleton and limbs. Ciliopathy genetics can be complex, with clinical overlap between different syndromes and variable phenotypes and expressivity seen for mutant alleles of the same gene (3,4). Among ciliopathies, oral-facial-digital syndromes (OFDS) are a group of clinically and genetically heterogeneous disorders characterized by defects in the development of the face and oral cavity along with digit anomalies (5). Currently, 25 genes encoding ciliary proteins have been linked to OFDS (6–24). Pathogenic variants in these genes have structural or functional impacts on primary cilia at different levels: for example, in the distal basal body structures needed for cilium assembly (e.g. OFD1, C2CD3), in formation of a transition zone that enables ciliary compartmentalization (e.g. TMEM107) and in ciliary trafficking via intraflagellar transport (e.g. IFT57). In addition, many distinct subgroups of OFDS have been defined on the basis of their characteristic clinical features as well as their causative mutations (5). Despite this variation, a commonality is that most OFDS genes are needed for the assembly or function of primary cilia.
Ciliogenesis is a complex, multistep process in which the mother centriole serves as the foundation for assembly of the axonemal microtubules, the transition zone and the surrounding ciliary membrane. This assembly process is tightly regulated by extrinsic signals as well as by features of the internal cell state, such as cell cycle stage (25–27). The complexity of ciliogenesis is further illustrated by the fact that different cell types and tissues characteristically build their cilia through distinct assembly pathways (25–27). For example, epithelial cells typically use the ‘extracellular pathway’ of ciliogenesis, in which the mother centriole docks to the plasma membrane and nucleates the outgrowth of a cilium that protrudes from the cell surface (28). In contrast, mesenchymal cells typically form cilia by the ‘intracellular pathway’ of ciliogenesis, in which the nascent cilium first forms within the cytoplasm (29). In this intracellular mode of cilium assembly, ciliogenesis begins with the recruitment of pre-ciliary vesicles that are captured at the distal appendages of the mother centriole. Subsequently, these vesicles fuse to form the ciliary vesicle, which occurs in conjunction with the removal of centriolar capping proteins such as CP110. Subsequent extension of the axonemal microtubules is accompanied by growth of the ciliary vesicle and the remodeling of this membrane into two adjacent but distinct domains: the nascent ciliary membrane that faces the axoneme and the ciliary sheath that faces the cytoplasm. Lastly, fusion of the ciliary sheath with the plasma membrane exposes the mature cilium to the external environment, with cilia formed in this manner typically remaining recessed within an invagination of the plasma membrane known as the ciliary pocket (25–27,30). Formation of the ciliary membrane thus requires several vesicle trafficking, vesicle fusion and membrane remodeling processes, some of which are unique to the intracellular ciliogenesis pathway (25–27,31).
Given the complex process by which the ciliary membrane forms, it is not surprising that several Rab GTPases participate in ciliogenesis. Rab proteins represent a subfamily of the small GTPase superfamily and act as master regulators of membrane trafficking by orchestrating the biogenesis, transport, tethering and fusion of membrane-bound organelles and vesicles (32–35). Rab proteins cycle between two states: an inactive, GDP-bound state and an active, GTP-bound state that allows association with various effector proteins (35). Multiple Rab proteins are proposed to participate in ciliogenesis, including RAB8, RAB10, RAB11, RAB19, RAB23 and RAB35 (36–39). More recently, RAB34, a Rab GTPase previously implicated in lysosomal positioning, activation of macropinocytosis and phagosome fusion, was identified as a key mediator of ciliary membrane formation (36,40–44). Specifically, RAB34 is essential for the formation of the ciliary vesicle at the mother centriole. In addition, RAB34 localizes to the ciliary vesicle during its formation and is subsequently found on the ciliary sheath of the nascent intracellular cilium (40,41,45,46). Consistent with RAB34 having functional roles and localizations that are specific to the intracellular ciliogenesis pathway, RAB34 is critical for ciliogenesis in the RPE1 and NIH-3 T3 cell lines that use the intracellular pathway. However, RAB34 is dispensable for ciliogenesis in kidney cell lines that use the extracellular pathway (36,40,41,45).
Given the central role of cilia in Hedgehog signaling, Rab34 disruption causes strong defects in Hedgehog signaling in cultured cells and mutant mice (36,41,45,47). In addition, Rab34 knockout (KO) mice display ciliopathy phenotypes including polydactyly, cleft lip and palate, and variable craniofacial features in addition to lung hypoplasia, exencephaly, edema and hemorrhage. The majority of Rab34 KO mice die perinatally (45,48), revealing effects on embryonic development that are significant, but also considerably milder than in null mutants of mouse genes that are universally required for ciliogenesis, such as Ift88 or Kif3a (49,50). Collectively, these studies have revealed RAB34 as a key protein that has cell-type-specific roles in primary cilium formation whose inactivation may cause a distinct set of ciliary phenotypes at the organismal level.
In this study, we report the first description of pathogenic variants in RAB34 in humans, causing OFDS with cerebral, cardiac and anorectal defects, and shortening of long bones. In functional studies, we further show that the protein products of these pathogenic variants exhibit a strong loss of function. While some retain the ability to be recruited to the mother centriole, RAB34 mutant proteins produce a strong impairment in cilium assembly, with little to no improvement over RAB34 KO cells. Thus, our study reveals a new OFDS syndrome with characteristic clinical features that result from inactivation of a small GTPase with cell-type-specific roles in ciliary membrane formation.
Results
Aided by GeneMatcher (51), we identified four individuals from three unrelated families with suspected pathogenic variants in RAB34 (Table 1 and Fig. 1). Prenatal signs showed multiple developmental defects including short femur, polydactyly and malformations of the heart, kidney and brain, with pregnancies medically terminated in three probands (F1, F2.1, F2.2). Individual F3 was born at 39 + 5 weeks gestation with normal growth parameters after pregnancy with polyhydramnios, corpus callosum agenesis and polydactyly. He presented respiratory distress at birth requiring tracheostomy. All individuals presented typical features of ciliopathy disorders, overlapping with oral, facial and digital abnormalities (Table 1 and Supplementary Material). Oral features included bilateral cleft palate/lip (4/4), short lingual frenulum (1/4) and lobulated tongue (1/4) (Table 1 and Fig. 1). Recurrent craniofacial features included micro/retrognathia (3/4) and hypertelorism (2/4). All probands presented bilateral hand and feet polydactyly, which may be preaxial (4/4), central (2/4) or postaxial (1/4), ranging from isolated heptadactyly/ectrodactyly (2/4) to polysyndactyly (2/4), as well as deviation of the hallux (2/4) (Fig. 1). Anorectal anomalies were consistently observed (4/4), as well as cerebral malformation (2/4) including corpus callosum agenesis (2/4) and cerebellar hypoplasia (1/4) (Table 1). Variable cardiac defects (3/4) present included hypoplastic left heart, atrioventricular canal defect and ventricular septal defect (Table 1). Additional features observed included shortening of long bones (femur and humerus) (2/4) and renal anomalies (Individual F1; Table 1). The only individual alive at birth (Individual F3) required extensive care because of pneumopathy requiring mechanical ventilation and because of gastrointestinal symptoms resulting from anorectal malformations and clinical suspicion of whole colonic Hirschsprung. He presented suboptimal tolerance after surgery with bleeding episodes and abdominal distension; Individual F3 died at age 3 months because of respiratory complications.
Table 1.
Summary of clinical features
| Individual | F1 | F2.1 | F2.2 | F3 |
|---|---|---|---|---|
| Amino acid change (NM_031934.6) | p.(Arg211His) | p.(Gly202Val) | p.(Gly202Val) | p.(Glu218Lys) |
| Inheritance | Homozygous | Homozygous | Homozygous | Homozygous |
| Gender | Male | Male | Male | Male |
| Country of origin | Morocco | Czech Republic | Czech Republic | Morocco |
| Consanguineous | Yes | No | No | Yes |
| Age at last examination | 22 + 1 WG | 13 + 5 WG | 21 + 6 WG | 3 months |
| Prenatal ultrasound signs | Oral cleft, Hand-foot polydactyly, Abnormal cardiac morphology, Short fetal femur length, Renal hypertrophy | Multiple developmental defects | Multiple developmental defects | Bilateral cleft lip, Hand-foot polydactyly, Atrioventricular canal defect, Corpus callosum agenesis, Polyhydramnios |
| Neonatal issues | NA | NA | NA | Respiratory distress |
| Cranio-facial defects | Hypertelorism, Microtia | Micrognathia, Turricephaly | Hypertelorism, Synophris Micrognathia, Low-set ears | Macrocephaly, Broad forehead, Wide nasal bridge, Retrognathia |
| Oral anomalies | Bilateral oral cleft, Short lingual frenulum | Bilateral oral cleft | Bilateral oral cleft | Bilateral oral cleft, Lobulated tongue |
| Hand anomalies | Bilateral polysyndactyly | Prexial and central polydactyly (heptadactyly) | Preaxial and central polydactyly (heptadactyly) | Bilateral polysyndactyly |
| Foot anomalies | Bilateral polysyndactyly, Deviation of the hallux | Prexial and central polydactyly (heptadactyly) | Preaxial and central polydactyly (left hexadactyly, right ectrodactyly), Deviation of the right hallux | Bilateral polysyndactyly |
| Structural cerebral anomalies | Corpus callosum agenesis, Cerebellar hypoplasia | NA | NA | Corpus callosum agenesis |
| Cardiac anomalies | Ventricular septal defect, Left ventricular hypoplasia | No | Hypoplastic left heart, Persistent truncus arteriosus | Atrioventricular canal defect |
| Other clinical features | Anal atresia, Short femur | Small anus | Anal atresia, Short femur and humerus, Bilobar right lung | Anorectal anomaly, Possible Hirschsprung Dyspnea, Pneumopathy |
Overview of clinical features observed in individuals with pathogenic RAB34 variants. NA, not available.
Figure 1.

Characteristics of individuals with RAB34 variants. (A–C) Individual F1 at 13 + 5 WG presenting oral cleft (A), bilateral limb polysyndactyly (B, C) and anal atresia (C). (D–G) Individual F2.1 at 21 + 6 WG, presenting turricephaly (D), preaxial polydactyly (E), anal atresia (F) and bilateral oral cleft (G). (H–Q) Individual F2.2 at 22 + 1 WG showing preaxial and central polydactyly (H, I, K, L, N–Q) and bilateral oral cleft (J, M). X-rays confirm bilateral polydactyly and central, Y-sharped metacarpal (I, L). (R–T) Individual F3 (age 14 days) presenting bilateral oral cleft (R) and hand/feet polysyndactyly (S, T).
We identified by exome sequencing homozygous missense variants in RAB34 in all affected individuals (Table 1 and Fig. 2): RAB34: NM_031934.6: p.(Gly202Val), p.(Arg211His) and p.(Glu218Lys). All variants are absent in the gnomAD database at the homozygous state (Supplementary Material, Table S1). Sanger sequencing confirmed the recessive mode of inheritance, with each parent having a pathogenic variant in a heterozygous state. None of the probands were diagnosed with pathogenic copy number variants or with additional pathogenic or likely pathogenic variants in genes known to be mutated in ciliopathies or other developmental disorders.
Figure 2.
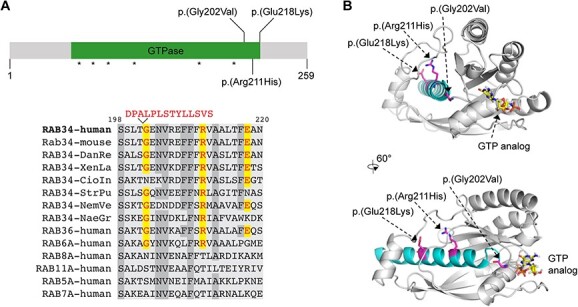
Schematic representation of RAB34 protein with identified variants and structural model. (A) Top: schematic of RAB34 protein highlighting clustered homozygous missense variants and nucleotide-contacting regions (asterisks); bottom: amino acid sequence conservation in the mutated region of RAB34 (bottom). In the sequence alignment, mutations at Gly202 (including 14aa insertion caused by altered splicing), Arg211 and Glu218 are indicated, and highly conserved residues are shaded. Abbreviations: DanRe = Danio rerio; XenLa = Xenopus laevis; CioIn = Ciona Intestinalis (sea squirt); StrPu = Strongylocentrotus purpuratus (sea urchin); NemVe = Nematostella vectensis (sea anemone); NaeGr = Naegleria gruberi. (B) Protein structure prediction for RAB34 obtained from Uniprot/AlphaFold2 Accession ID AF-Q9BZG1 (74). The C-terminal helix encoded by exon 9 is shown in cyan. Phosphate moiety of GTP analog interacting with the Switch I and Switch II regions and the phosphate binding loop is shown in orange. N-terminal (1–10 aa) and C-terminal (231–259 aa) residues with low confidence score are not included in the model.
Protein sequence analysis showed high conservation of the C-terminal region of the RAB34 protein where all three pathogenic variants are found (Fig. 2A). Two of the variants are found in the GTPase domain (p.(Gly202Val) and p.(Arg211His)) at residues that are conserved in RAB34 orthologs as well as RAB36, a close paralog of RAB34. The third variant (p.(Glu218Lys)) is at a position that exhibits slightly lower conservation (Fig. 2A). Notably, all variants are located in exon 9 (NM_031934), predicted to encode a C-terminal alpha helix (Fig. 2B). In silico mutagenesis suggested these variants may cause protein structural changes that impair protein stability, supporting a pathogenic status of these reported variants (Supplementary Material, Table S2 and Fig. S1A).
As the p.(Gly202Val) variant also occurs at a splice acceptor site, we assessed the impact of this mutation on precursor mRNA processing via transcript analysis of RAB34 (Supplementary Material, Fig. S1B and C). Analysis of RAB34 transcripts in heterozygous paternal and maternal samples of Individuals F2.1 and F2.2 showed that in addition to wild-type (WT) transcript, there are also aberrant transcripts from the mutant allele not seen in controls. The most abundant aberrant transcripts resulted from in-frame retention of 42 bases from the 3′ end of intron 8 or alternatively exon 9 skipping (Supplementary Material, Fig. S1C). Exon 9 skipping would remove 36 amino acids comprising the whole C-terminal helix of the RAB34 protein, very likely leading to improper protein folding and degradation.
We next sought to assess the functional impact of OFDS-associated RAB34 variants on RAB34-dependent ciliogenesis in the RPE1 human cell line, a widely used model of the intracellular ciliogenesis pathway that requires RAB34 (36,41). Specifically, we assessed the ability of transgenes encoding RAB34 Gly202Val, Arg211His and the Gly202Val splice variant with 14 amino acids inserted to rescue the loss of ciliogenesis we previously observed in RAB34 KO RPE1 cells (41). We used lentiviral transduction to stably express these RAB34 transgenes from the PGK promoter without any epitope or fluorescent protein tags (in order to avoid any potential confounding effects of such tags on RAB34 function). As a positive control, we carried out a parallel analysis using a WT RAB34 construct; all transgenes also harbored silent mutations to confer resistance to the single-guide RNA (sgRNA) used for RAB34 KO and a fluorescent Centrin2 marker to facilitate identification of transgene-expressing cells. As expected, we observed robust ciliogenesis in WT RPE1 cells, very low ciliogenesis in RAB34 KO cells and strong rescue of ciliogenesis in RAB34 KO cells expressing a WT RAB34 transgene (Fig. 3A and B). However, all variants tested yielded significantly lower ciliation than the WT transgene (P < 0.00001 for comparison of each variant to the WT RAB34 transgene). In RAB34 KO cells expressing RAB34 Gly202Val or Arg211His, ciliogenesis was marginally increased compared with KO cells without any RAB34 (P < 0.02), whereas RAB34 Gly202Val with the 14aa insert resulted in ciliogenesis indistinguishable from the parental RAB34 KO cells (P > 0.5). Thus, these pathogenic RAB34 variants exhibit a strong loss of function, consistent with the significant clinical features observed in individuals homozygous for these variants. We also noted that expression of these variants in WT RPE1 cells did not impact ciliogenesis (data not shown), indicating that these mutants lack dominant negative activity and cause OFDS via recessive loss of function.
Figure 3.

Functional characterization of OFDS-associated RAB34 mutations. (A) Cilia (marked by ARL13B) and centrioles (marked by γ-tubulin) were stained in WT RPE1 cells and RAB34 KO RPE1 cells stably expressing the indicated RAB34 cDNAs. Scale bar: 5 μm. (B) Quantification of ciliogenesis in RPE1 cells shown in (A); bars indicate means, dots show values from > 70 cells analyzed in each of N = 5 independent experiments and error bars show standard deviation across independent experiments. * indicates P < 0.00001. (C) RAB34 was stained in RAB34 KO RPE cells transduced with the indicated RAB34 cDNAs (insets show enlargement of centriolar region). Scale bar: 5 μm (insets: 1 μm). (D) Western blot of the indicated RPE1 cell lines showing expression levels of WT RAB34 and RAB34 variants in addition to HSP90 loading control. Asterisk indicates nonspecific band detected by anti-RAB34 antibody.
To further characterize the OFDS-associated RAB34 variants, we used immunofluorescence microscopy to assess their localization when expressed in RAB34 KO RPE1 cells. As previously reported, WT RAB34 can be detected as a prominent punctum at the mother centriole during ciliogenesis (Fig. 3C). In cells expressing RAB34 Gly202Val and Arg211His, we still observed localization of RAB34 puncta to mother centrioles, even though ciliogenesis was severely compromised. In contrast, the RAB34 Gly202Val splice variant mutant was never observed at the mother centriole, consistent with its more severe ciliogenesis defect. Lastly, we evaluated the expression level of OFDS-associated RAB34 variants by western blot (Fig. 3D). Expression of WT RAB34 from the PGK promoter in RAB34 KO cells led to elevated RAB34 levels compared with endogenously expressed RAB34 in WT RPE1 cells. In contrast, all mutant forms of RAB34 showed reduced levels in whole cell lysate, ranging from a moderate reduction relative to endogenous RAB34 for the Arg211His variant to a more severe reduction for the Gly202Val-containing splice variant. These results suggest that these mutations destabilize the RAB34 protein and promote its degradation.
Discussion
Clinical delineation in a new subtype of OFDS
We report here a novel autosomal recessive ciliopathy disorder in four individuals from three unrelated families associated with pathogenic variants in the RAB34 gene. We observe the typical features characteristic of the OFDS including variable facial dysmorphism, consistent bilateral cleft lip/palate, lobulated tongue, abnormal frenulum and bilateral polydactyly or polysyndactyly of the feet and hands. Twenty-five identified OFDS genes delineate many OFDS subtypes with one or more distinctive clinical features (e.g. skeletal dysplasia, cerebral malformations, renal cysts, deafness, etc.) (6–24). OFDS-RAB34 is associated with cardiac defects, previously reported in other OFDS subtypes, and particularly in individuals with pathogenic variants in components of the CPLANE complex located at the basal body of the primary cilium (classified OFD type VI; genes INTU, WDPCP, CPLANE1) (5,10,11,52). Similarly, X-rays displayed Y-shaped metacarpal bones in OFDS-RAB34, a feature also predominantly described in OFD VI (10). Additional recurrent symptoms observed in this RAB34-cohort involve short femur and/or humerus, which is not characteristic of OFD type VI. These isolated skeletal defects have not yet been described in OFDS, although other limb anomalies are reported in several OFDS subtypes including short leg or arm in OFD types I, IV and IX (53,54), tibial dysplasia in OFD types IV and VIII (53,54) and mesomelic short stature in atypical OFDS (12,55). The anal atresia/anomaly observed in OFDS-RAB34 appears as an ultra-rare malformation in OFDS, described in five individuals presenting OFD type II, IV or VI (56–60). However, anal atresia is reported more frequently in Pallister–Hall syndrome (MIM #146510), Bardet–Biedl syndrome, McKusick–Kaufman syndrome (MIM # 236700) and Meckel syndrome (61–63). Simonini et al. (61) reported imperforate anus in 33% of fetuses with Bardet–Biedl/McKusick–Kaufman syndromes. Lastly, in our cohort, cerebral malformations were observed including agenesis of the corpus callosum (F1 and F3) and cerebellar hypoplasia (F1), which have been previously described specifically in OFD types I and VI (64). The clinical overlap with several OFD types makes OFDS-RAB34 difficult to classify and emphasizes once more the continuum of clinical spectrum in OFDS and ciliopathies. Interestingly, similar skeletal, cardiac and pulmonary defects were observed in OFD type II (Mohr syndrome), an OFDS without known molecular basis, suggesting RAB34 as a possible molecular cause of OFD type II (65). We note that most individuals of this study are fetuses with medical termination of pregnancy between 13 and 22 weeks of gestation (WG), with only one individual born and evaluated at 3 months, precluding a detailed analysis of the clinical evolution of the affected individuals, particularly at the neurodevelopmental level. However, severe respiratory distress and digestive dysfunctions were at the forefront of patient care for Individual F3. Taken together, the association of bilateral oral cleft, polydactyly/polysyndactyly, cerebral malformations, cardiac defects, anorectal anomalies and shortening of long bones represents a new subtype of OFDS caused by loss of RAB34 function. Many of these phenotypes are similar to those observed in Rab34-mutant mice, which also exhibit cleft lip/palate and preaxial polydactyly (45,48). Notably, human cases also present with heart defects, structural brain anomalies and anal atresia, which have not been reported to date in Rab34 mouse mutants. Our analysis of OFDS-RAB34 therefore expands the phenotypic spectrum associated with mutations in this gene and links RAB34-dependent cilium assembly to the development of the face, digits, long bones, heart, brain and digestive tract.
RAB34-related OFD syndrome involves defective cilia formation
To functionally characterize the RAB34 protein variants associated with OFDS, we tested their ability to promote ciliogenesis in the human RPE1 cell line. RAB34 KO severely impairs ciliogenesis in RPE1 cells, and the RAB34 Gly202Val, Arg211His and Gly202Val-splice variant mutants tested here all failed to rescue ciliogenesis when expressed in RAB34 KO cells. Thus, these RAB34 variants are all strong loss-of-function mutations, with the splice variant exhibiting the greatest functional deficit, perhaps because it most severely alters RAB34 structure. Consistent with these findings, all three mutant proteins were present at significantly reduced levels compared with WT RAB34 expressed from the same promoter. Thus, the Gly202 and Arg211 residues are critical for RAB34 function, even though these amino acids are not conserved in other members of the Rab GTPase family. This finding adds to our prior evidence that non-canonical sequence features of the RAB34 GTPase domain are key regulators of its function (41). The clustering of the mutations at a C-terminal alpha helix also highlights a key functional region in RAB34 distinct from the nucleotide binding site and the Switch I and Switch II motifs that are typically critical for GTPase function. Lastly, it is interesting to note that the two point mutants are still able localize to the mother centriole, as observed for WT RAB34. Thus, recruitment of RAB34 to the mother centriole is likely not sufficient for ciliogenesis; instead, inability to form cilia was decisive in evaluating the pathogenicity of the variants.
To date, several Rab GTPases have been suggested to participate in or regulate ciliogenesis, including RAB8, RAB10, RAB11, RAB19, RAB23, RAB34 and RAB35, as well as non-canonical Rab proteins RABL2A/B, RABL3, RABL4/IFT27 and RABL5/IFT22 (37–39,66). However, analysis of mouse mutants and ciliopathy patients has not yet revealed strong ciliogenesis defects or ciliopathy phenotypes for any canonical Rab genes, apart from RAB34. RAB23 is the only Rab gene previously linked to a potential ciliopathy (Carpenter Syndrome [MIM201000]) or to ciliopathy-like phenotypes in mutant mice (67,68). However, the principle in vivo phenotype associated with Rab23 inactivation is increased Hedgehog signaling without reduced ciliation, arguing against a role in ciliogenesis (68–70). For other ciliary Rab GTPases, in some cases, mouse studies have found that even simultaneous mutation of multiple Rab genes (e.g. Rab8a and Rab8b) fails to cause ciliary defects (71). Thus, RAB34 appears to play a particularly vital role in cilium assembly, with its disruption in mouse or human producing strong ciliopathy features unique among Rab family members.
While pathogenic RAB34 variants produce significant ciliopathy features, it is noteworthy that these features likely reflect the loss of a cell-type-specific rather than universal mediator of ciliogenesis. RAB34 is perhaps the first protein shown to be required for intracellular and not extracellular ciliogenesis (40,41), and thus OFDS-RAB34 provides new insights into the cells and tissues that employ intracellular ciliogenesis. Furthermore, future identification of any genes causing a similar form of OFDS as OFDS-RAB34 may help uncover additional mediators of intracellular ciliogenesis. Finally, it is possible that further studies may reveal RAB34 variants with milder effects on function that also contribute to ciliopathies, perhaps yielding a distinct set of phenotypes that will further illuminate RAB34 function and the ciliopathy clinical spectrum.
In conclusion, we report a novel subtype of OFDS characterized by cerebral, cardiac and anorectal defects and short long bones because of recessive pathogenic variants in the RAB34 gene. Through cell-based functional studies, we establish that while some of the corresponding RAB34 mutant proteins can still localize to the mother centriole, they nonetheless exhibit a strong loss of function in cilium assembly. Given RAB34’s cell-type-specific role in assembly of primary cilia, these findings shed new light on how genes with distinct ciliary functions can cause a distinct ciliopathy syndrome.
Materials and Methods
Exome sequencing
In compliance with the local ethical guidelines and the Declaration of Helsinki, the parents provided written informed consent for their participation in the study, clinical data and specimen collection, genetic analysis and publication of relevant findings. Exome capture and sequencing were performed at Integragen SA from 1 μg of genomic DNA per individual extracted from cultured amniotic fluids (fetus) or blood samples (parents) using the Agilent_CRE kit on a HiSeq 4000 (Illumina) (Individual F1) or using SeqCap EZ MedExome Probes (Roche) (Individuals F2.1–F2.2) or xGen Exome Panel v2.0 (IDT) (Individual F3) on the NovaSeq 6000 platform (Illumina), according to manufacturer’s instructions. We generated 75-bp/100-bp paired-end reads that were aligned to the human genome reference sequence (GRCh37/hg19 assembly) using Burrows–Wheeler aligner (version 0.7.15 or 0.7.3) or Novoalign (version 3.02.13). Duplicate reads were marked using Picard MarkDuplicates (version 2.4.1/2.20.8) (http://broadinstitute.github.io/picard/), and aligned reads were then processed using GATK BaseRecalibrator and PrintReads (Genome Analysis Toolkit; version 3.8 or 3.4) to recalibrate base quality scores, according to GATK Best Practices recommendations. Quality control was performed on all BAM files by calculating depth of coverage onto the RefSeq database (release 2018-11-11) with GATK DepthOfCoverage. SNPs and indels were identified from BAM files using GATK HaplotypeCaller. All variants identified were annotated using SnpEff (version 4.3) or GEMINI framework (72). Variants were confirmed by Sanger sequencing.
Protein structural modeling
Protein structure prediction was obtained from MIZTLI software (https://miztli.biokerden.eu/) using Uniprot/AlphaFold2 Accession ID AF-Q9BZG1-F1. Structural models of RAB34 mutants and predicted effects on protein stability were generated using DynaMut2 software (73).
Transcript analysis
mRNA was isolated from blood and nasopharyngeal swab samples of both parents of Individuals F2.1 and F2.2. cDNA was prepared using SuperScript Reverse Transcriptase IV (Thermo Fisher). PCRs across RAB34 coding sequence were performed with primers located in the first and the last exon 10 (NM_031934.6). PCR was performed using Phusion polymerase (NEB) according to manufacturer’s instructions, and PCR products were Sanger sequenced.
Cell lines and cell culture
RPE1-hTERT (RPE1) and HEK293T cells were obtained from ATCC, and the clonal RAB34 CRISPR KO RPE1 cell line used was generated and characterized previously (41). Cultured cells were maintained in a humidified 37°C incubator with 5% CO2. HEK293T cells were cultured in DMEM, high glucose (Gibco) supplemented with 10% fetal bovine serum (FBS; Gemini Bio), 100 units/ml Penicillin, 100 μg/ml Streptomycin, 2 mm Glutamine and 1 mm sodium pyruvate (Gibco). RPE1 cells were cultured in DMEM/F-12 medium (Gibco) supplemented with 10% FBS, 100 units/ml of Penicillin, 100 μg/ml Streptomycin and 2 mm Glutamine. To induce ciliogenesis, RPE1 cells were serum-starved in medium containing 0.2% FBS for 48 h.
Cell line generation via lentiviral transduction
Cell lines expressing WT and mutant RAB34 cDNAs were generated by cloning sgRNA-resistant RAB34 variants into a lentiviral expression plasmid (pCW-mPgk-RAB34-hPgk-miRFP-670-Centrin2) that expresses untagged human RAB34 together with fluorescently labeled Centrin2. VSVG-pseudotyped lentiviral particles were produced by transfection of HEK293T cells with a lentiviral vector and packaging plasmids (pMD2.G, pRSV-Rev, pMDLg/RRE for sgRNAs; pCMV-ΔR-8.91 and pCMV-VSVG for protein-coding constructs). Following transfection using polyethyleneimine (Polysciences #24765-1), virus-containing supernatant was collected 48 h later, filtered through a 0.45 μm polyethersulfone filter (VWR 28145-505) and concentrated 10-fold using Lenti-X Concentrator (Takara Biosystems).
RPE1 cells were transduced by addition of viral supernatants diluted to an appropriate titer in medium containing 4 μg/ml polybrene (Sigma Aldrich H9268). Following 24-h incubation at 37°C, virus-containing medium was removed, and cells were passaged. Transduced cells were identified by miRFP-670-Centrin2 fluorescence and isolated by FACS using a FACSAria sorter (Becton Dickinson), generating polyclonal pools.
Ciliogenesis analysis by immunofluorescence microscopy
For immunofluorescence microscopy, cells were seeded on acid-washed 12-mm #1.5 coverslips (Fisher Scientific). Cells were fixed in −20°C methanol for 10 min, after which coverslips were permeabilized for 10 min in PBS containing 0.1% Triton X-100, washed with PBS and blocked for 20 min in PBS supplemented with 5% normal donkey serum (Jackson ImmunoResearch) and 3% Bovine Serum Albumin (BSA). Coverslips were then incubated with appropriate primary antibodies (see below) diluted in PBS with 3% BSA at room temperature for 1 h, washed five times, incubated with secondary antibodies (Jackson ImmunoResearch) for 30–60 min at room temperature, washed again, stained with Hoechst 33258 dye and mounted on glass slides in Fluoromount-G mounting medium (Electron Microscopy Sciences). Primary antibodies used include the following: anti-ARL13B (Proteintech 17711-1-AP), anti-RAB34 (Santa Cruz Biotechnology sc-376 710) and anti-γ-tubulin GTU-88 (Sigma T5326).
Stained cells were imaged using a Nikon Eclipse Ti-2 Widefield microscope equipped with a CMOS camera (Photometrics Prime BSI), a ×60 PlanApo oil objective (NA 1.40; Nikon Instruments) and an LED light source (Lumencor SOLA-V-NIR). Cilia were identified via ARL13B and γ-tubulin staining and manually scored only in cells displaying miRFP-670-Centrin2 that was co-expressed with various RAB34 variants.
Ciliated cells were manually scored by assessing the presence or absence of ARL13B-labeled cilia at each centrosome labeled by γ-tubulin or miRFP-670-Centrin2. Statistical analysis of differences in ciliation was calculated by t-test (two-sided, unequal variance).
SDS-PAGE and western blotting
For analysis of RAB34 levels in whole-cell lysate, RPE1 cells were lysed in RIPA buffer (50 mm Tris pH 7.4, 150 mm NaCl, 2% Igepal CA-630, 0.25% sodium deoxycholate, 0.5 mm DTT, protease inhibitors) on ice for 10 min and centrifuged at 20 000 g for 10 min. The supernatant was collected, and equal amounts of protein, as determined by Bio-Rad Protein Assay, were analyzed by SDS-PAGE. Protein samples were separated on NuPAGE 4–12% Bis-Tris gels (Invitrogen) run in MOPS buffer (50 mm Tris, 50 mm MOPS, 3.5 mm SDS, 1 mm EDTA) and transferred to a PVDF membrane (Millipore Sigma #IPVH00010). The membrane was blocked in 1:1 PBS/SeaBlock (Thermo Fisher #37527) and incubated with primary antibodies overnight at 4°C (anti-RAB34, Proteintech #27435-1-AP and anti-HSP90, Cell Signaling Technology #4877S). After washing, the membranes were incubated with Protein A-HRP for 30 min at room temperature. The blots were developed using Clarity Western ECL Substrate (Bio-Rad) and imaged on a ChemiDoc Touch imager (Bio-Rad).
Supplementary Material
Acknowledgements
We thank the patients and their families for their participation. We thank the Integragen Society, the Centre National de Gestion, the Institut de Génétique et de Biologie Moléculaire et Cellulaire, and the computer cluster of the University of Burgundy for technical support and management of the Dijon-Bourgogne computing platform. We also thank the GeneMatcher data sharing platform (https://genematcher.org/).
Conflict of Interest statement. None declared.
Contributor Information
Ange-Line Bruel, INSERM U1231 Génétique des Anomalies du Développement (GAD), University Bourgogne Franche-Comté, 21070 Dijon, France; Unité Fonctionnelle Innovation en Diagnostic Génomique des Maladies Rares, Fédération Hospitalo-Universitaire Médecine Translationnelle et Anomalies du Développement (FHU-TRANSLAD), Centre Hospitalo-Universitaire (CHU) Dijon Bourgogne, 21079 Dijon, France.
Anil Kumar Ganga, Department of Molecular, Cellular, and Developmental Biology, Yale University, New Haven, CT 06511, USA.
Lenka Nosková, Research Unit for Rare Diseases, Department of Pediatrics and Inherited Metabolic Disorders, First Faculty of Medicine, Charles University and General University Hospital in Prague, Prague 128 08, Czech Republic.
Irene Valenzuela, Department of Clinical and Molecular Genetics, Vall d'Hebron University Hospital, 08035 Barcelona, Spain; Medical Genetics Group, Vall d'Hebron Research Institute,08035 Barcelona, Spain.
Jelena Martinovic, Unit of Embryo-Fetal Pathology, AP-HP, Antoine Béclère Hospital, Paris Saclay University, 92141 Clamart, France.
Yannis Duffourd, INSERM U1231 Génétique des Anomalies du Développement (GAD), University Bourgogne Franche-Comté, 21070 Dijon, France; Unité Fonctionnelle Innovation en Diagnostic Génomique des Maladies Rares, Fédération Hospitalo-Universitaire Médecine Translationnelle et Anomalies du Développement (FHU-TRANSLAD), Centre Hospitalo-Universitaire (CHU) Dijon Bourgogne, 21079 Dijon, France.
Marie Zikánová, Research Unit for Rare Diseases, Department of Pediatrics and Inherited Metabolic Disorders, First Faculty of Medicine, Charles University and General University Hospital in Prague, Prague 128 08, Czech Republic.
Filip Majer, Research Unit for Rare Diseases, Department of Pediatrics and Inherited Metabolic Disorders, First Faculty of Medicine, Charles University and General University Hospital in Prague, Prague 128 08, Czech Republic.
Stanislav Kmoch, Research Unit for Rare Diseases, Department of Pediatrics and Inherited Metabolic Disorders, First Faculty of Medicine, Charles University and General University Hospital in Prague, Prague 128 08, Czech Republic.
Markéta Mohler, Institute of Molecular and Clinical Pathology and Medical Genetics, University Hospital Ostrava, Ostrava 708 52, Czech Republic.
Jingbo Sun, Department of Molecular, Cellular, and Developmental Biology, Yale University, New Haven, CT 06511, USA.
Lauren K Sweeney, Department of Molecular, Cellular, and Developmental Biology, Yale University, New Haven, CT 06511, USA.
Núria Martínez-Gil, Department of Clinical and Molecular Genetics, Vall d'Hebron University Hospital, 08035 Barcelona, Spain; Medical Genetics Group, Vall d'Hebron Research Institute,08035 Barcelona, Spain.
Christel Thauvin-Robinet, INSERM U1231 Génétique des Anomalies du Développement (GAD), University Bourgogne Franche-Comté, 21070 Dijon, France; Unité Fonctionnelle Innovation en Diagnostic Génomique des Maladies Rares, Fédération Hospitalo-Universitaire Médecine Translationnelle et Anomalies du Développement (FHU-TRANSLAD), Centre Hospitalo-Universitaire (CHU) Dijon Bourgogne, 21079 Dijon, France; Centre de Génétique et Centre de référence maladies rares ‘Anomalies du Développement et Syndromes Malformatifs’, FHU-TRANSLAD, Hôpital d'Enfants, CHU Dijon Bourgogne, 21079 Dijon, France.
David K Breslow, Department of Molecular, Cellular, and Developmental Biology, Yale University, New Haven, CT 06511, USA.
Data Availability
The data supporting the findings of this study are present within the article or are available from the authors upon request.
Funding
We acknowledge support from the GIS-Institut des Maladies Rares (HTS); the French Foundation for Rare Diseases; the French Ministry of Health (PHRC national 2010-A01014–35 and 2013 to C.T.-R.); the Regional Council of Burgundy; the National Institutes of Health (R35GM137956 to D.K.B.); a pilot grant from the Parkinson’s Foundation (PF-RCE-1946 to D.K.B.); and the Yale Flow Cytometry Facility, which is supported in part by a National Cancer Institute Cancer Center Support Grant (P30CA016359). This work was also supported by the project National Institute for Neurological Research (Programme EXCELES, ID Project No. LX22NPO5107—funded by the European Union—Next Generation EU); the institutional program UNCE/MED/007 of Charles University in Prague; the Ministry of Health of the Czech Republic (AZV NU23-07-00281) and the National Center for Medical Genomics (LM2023067 to L.N., M.Z., F.M. and S.K.). N.M.-G. acknowledges financial support from the Spanish Ministry of Science and Innovation (MICINN) through the program Juan de la Cierva-Formación.
References
- 1. Mill, P., Christensen, S.T. and Pedersen, L.B. (2023) Primary cilia as dynamic and diverse signalling hubs in development and disease. Nat. Rev. Genet., 24, 421–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Anvarian, Z., Mykytyn, K., Mukhopadhyay, S., Pedersen, L.B. and Christensen, S.T. (2019) Cellular signalling by primary cilia in development, organ function and disease. Nat. Rev. Nephrol., 15, 199–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Valente, E.M., Rosti, R.O., Gibbs, E. and Gleeson, J.G. (2014) Primary cilia in neurodevelopmental disorders. Nat. Rev. Neurol., 10, 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Focşa, I.O., Budişteanu, M. and Bălgrădean, M. (2021) Clinical and genetic heterogeneity of primary ciliopathies (review). Int. J. Mol. Med., 48, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bruel, A.-L., Franco, B., Duffourd, Y., Thevenon, J., Jego, L., Lopez, E., Deleuze, J.-F., Doummar, D., Giles, R.H., Johnson, C.A. et al. (2017) Fifteen years of research on oral-facial-digital syndromes: from 1 to 16 causal genes. J. Med. Genet., 54, 371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Li, C., Jensen, V.L., Park, K., Kennedy, J., Garcia-Gonzalo, F.R., Romani, M., De Mori, R., Bruel, A.-L., Gaillard, D., Doray, B. et al. (2016) MKS5 and CEP290 dependent assembly pathway of the ciliary transition zone. PLoS Biol., 14, e1002416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lambacher, N.J., Bruel, A.-L., van Dam, T.J.P., Szymańska, K., Slaats, G.G., Kuhns, S., McManus, G.J., Kennedy, J.E., Gaff, K., Wu, K.M. et al. (2016) TMEM107 recruits ciliopathy proteins to subdomains of the ciliary transition zone and causes Joubert syndrome. Nat. Cell Biol., 18, 122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chevrier, V., Bruel, A.-L., Van Dam, T.J.P., Franco, B., Lo Scalzo, M., Lembo, F., Audebert, S., Baudelet, E., Isnardon, D., Bole, A. et al. (2016) OFIP/KIAA0753 forms a complex with OFD1 and FOR20 at pericentriolar satellites and centrosomes and is mutated in one individual with oral-facial-digital syndrome. Hum. Mol. Genet., 25, 497–513. [DOI] [PubMed] [Google Scholar]
- 9. Thauvin-Robinet, C., Lee, J.S., Lopez, E., Herranz-Pérez, V., Shida, T., Franco, B., Jego, L., Ye, F., Pasquier, L., Loget, P. et al. (2014) The oral-facial-digital syndrome gene C2CD3 encodes a positive regulator of centriole elongation. Nat. Genet., 46, 905–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Toriyama, M., Lee, C., Taylor, S.P., Duran, I., Cohn, D.H., Bruel, A.-L., Tabler, J.M., Drew, K., Kelly, M.R., Kim, S. et al. (2016) The ciliopathy-associated CPLANE proteins direct basal body recruitment of intraflagellar transport machinery. Nat. Genet., 48, 648–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lopez, E., Thauvin-Robinet, C., Reversade, B., Khartoufi, N.E., Devisme, L., Holder, M., Ansart-Franquet, H., Avila, M., Lacombe, D., Kleinfinger, P. et al. (2014) C5orf42 is the major gene responsible for OFD syndrome type VI. Hum. Genet., 133, 367–377. [DOI] [PubMed] [Google Scholar]
- 12. Thevenon, J., Duplomb, L., Phadke, S., Eguether, T., Saunier, A., Avila, M., Carmignac, V., Bruel, A.-L., St-Onge, J., Duffourd, Y. et al. (2016) Autosomal recessive IFT57 hypomorphic mutation cause ciliary transport defect in unclassified oral-facial-digital syndrome with short stature and brachymesophalangia. Clin. Genet., 90, 509–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Valente, E.M., Logan, C.V., Mougou-Zerelli, S., Lee, J.H., Silhavy, J.L., Brancati, F., Iannicelli, M., Travaglini, L., Romani, S., Illi, B. et al. (2010) Mutations in TMEM216 perturb ciliogenesis and cause Joubert, Meckel and related syndromes. Nat. Genet., 42, 619–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Adly, N., Alhashem, A., Ammari, A. and Alkuraya, F.S. (2014) Ciliary genes TBC1D32/C6orf170 and SCLT1 are mutated in patients with OFD type IX. Hum. Mutat., 35, 36–40. [DOI] [PubMed] [Google Scholar]
- 15. Shamseldin, H.E., Rajab, A., Alhashem, A., Shaheen, R., Al-Shidi, T., Alamro, R., Al Harassi, S. and Alkuraya, F.S. (2013) Mutations in DDX59 implicate RNA helicase in the pathogenesis of orofaciodigital syndrome. Am. J. Hum. Genet., 93, 555–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thomas, S., Legendre, M., Saunier, S., Bessières, B., Alby, C., Bonnière, M., Toutain, A., Loeuillet, L., Szymanska, K., Jossic, F. et al. (2012) TCTN3 mutations cause Mohr-Majewski syndrome. Am. J. Hum. Genet., 91, 372–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Johnston, J.J., Lee, C., Wentzensen, I.M., Parisi, M.A., Crenshaw, M.M., Sapp, J.C., Gross, J.M., Wallingford, J.B. and Biesecker, L.G. (2017) Compound heterozygous alterations in intraflagellar transport protein CLUAP1 in a child with a novel Joubert and oral-facial-digital overlap syndrome. Cold Spring Harb. Mol. Case Stud., 3, a001321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Monroe, G.R., Kappen, I.F., Stokman, M.F., Terhal, P.A., van den Boogaard, M.-J.H., Savelberg, S.M., van der Veken, L.T., van Es, R.J., Lens, S.M., Hengeveld, R.C. et al. (2016) Compound heterozygous NEK1 variants in two siblings with oral-facial-digital syndrome type II (Mohr syndrome). Eur. J. Hum. Genet., 24, 1752–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Strong, A., Simone, L., Krentz, A., Vaccaro, C., Watson, D., Ron, H., Kalish, J.M., Pedro, H.F., Zackai, E.H. and Hakonarson, H. (2021) Expanding the genetic landscape of oral-facial-digital syndrome with two novel genes. Am. J. Med. Genet. A, 185, 2409–2416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shaheen, R., Jiang, N., Alzahrani, F., Ewida, N., Al-Sheddi, T., Alobeid, E., Musaev, D., Stanley, V., Hashem, M., Ibrahim, N. et al. (2019) Bi-allelic mutations in FAM149B1 cause abnormal primary cilium and a range of ciliopathy phenotypes in humans. Am. J. Hum. Genet., 104, 731–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Iturrate, A., Rivera-Barahona, A., Flores, C.-L., Otaify, G.A., Elhossini, R., Perez-Sanz, M.L., Nevado, J., Tenorio-Castano, J., Triviño, J.C., Garcia-Gonzalo, F.R. et al. (2022) Mutations in SCNM1 cause orofaciodigital syndrome due to minor intron splicing defects affecting primary cilia. Am. J. Hum. Genet., 109, 1828–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ferrante, M.I., Giorgio, G., Feather, S.A., Bulfone, A., Wright, V., Ghiani, M., Selicorni, A., Gammaro, L., Scolari, F., Woolf, A.S. et al. (2001) Identification of the gene for oral-facial-digital type I syndrome. Am. J. Hum. Genet., 68, 569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mascibroda, L.G., Shboul, M., Elrod, N.D., Colleaux, L., Hamamy, H., Huang, K.-L., Peart, N., Singh, M.K., Lee, H., Merriman, B. et al. (2022) INTS13 variants causing a recessive developmental ciliopathy disrupt assembly of the integrator complex. Nat. Commun., 13, 6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Shaheen, R., Szymanska, K., Basu, B., Patel, N., Ewida, N., Faqeih, E., Al Hashem, A., Derar, N., Alsharif, H., Aldahmesh, M.A. et al. (2016) Characterizing the morbid genome of ciliopathies. Genome Biol., 17, 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wang, L. and Dynlacht, B.D. (2018) The regulation of cilium assembly and disassembly in development and disease. Development, 145, dev151407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Breslow, D.K. and Holland, A.J. (2019) Mechanism and regulation of centriole and cilium biogenesis. Annu. Rev. Biochem., 88, 691–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhao, H., Khan, Z. and Westlake, C.J. (2023) Ciliogenesis membrane dynamics and organization. Semin. Cell Dev. Biol., 133, 20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sorokin, S.P. (1968) Reconstructions of centriole formation and ciliogenesis in mammalian lungs. J. Cell Sci., 3, 207–230. [DOI] [PubMed] [Google Scholar]
- 29. Sorokin, S. (1962) Centrioles and the formation of rudimentary cilia by fibroblasts and smooth muscle cells. J. Cell Biol., 15, 363–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Molla-Herman, A., Ghossoub, R., Blisnick, T., Meunier, A., Serres, C., Silbermann, F., Emmerson, C., Romeo, K., Bourdoncle, P., Schmitt, A. et al. (2010) The ciliary pocket: an endocytic membrane domain at the base of primary and motile cilia. J. Cell Sci., 123, 1785–1795. [DOI] [PubMed] [Google Scholar]
- 31. Garcia, G., Raleigh, D.R. and Reiter, J.F. (2018) How the ciliary membrane is organized inside-out to communicate outside-in. Curr. Biol., 28, R421–R434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Pfeffer, S.R. (2017) Rab GTPases: master regulators that establish the secretory and endocytic pathways. Mol. Biol. Cell, 28, 712–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Stenmark, H. (2009) Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol., 10, 513–525. [DOI] [PubMed] [Google Scholar]
- 34. Hutagalung, A.H. and Novick, P.J. (2011) Role of Rab GTPases in membrane traffic and cell physiology. Physiol. Rev., 91, 119–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Homma, Y., Hiragi, S. and Fukuda, M. (2021) Rab family of small GTPases: an updated view on their regulation and functions. FEBS J., 288, 36–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Oguchi, M.E., Okuyama, K., Homma, Y. and Fukuda, M. (2020) A comprehensive analysis of Rab GTPases reveals a role for Rab34 in serum starvation-induced primary ciliogenesis. J. Biol. Chem., 295, 12674–12685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Blacque, O.E., Scheidel, N. and Kuhns, S. (2018) Rab GTPases in cilium formation and function. Small GTPases, 9, 76–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jewett, C.E., Soh, A.W.J., Lin, C.H., Lu, Q., Lencer, E., Westlake, C.J., Pearson, C.G. and Prekeris, R. (2021) RAB19 directs cortical remodeling and membrane growth for primary ciliogenesis. Dev. Cell, 56, 325–340.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kuhns, S., Seixas, C., Pestana, S., Tavares, B., Nogueira, R., Jacinto, R., Ramalho, J.S., Simpson, J.C., Andersen, J.S., Echard, A. et al. (2019) Rab35 controls cilium length, function and membrane composition. EMBO Rep., 20, e47625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Stuck, M.W., Chong, W.M., Liao, J.-C. and Pazour, G.J. (2021) Rab34 is necessary for early stages of intracellular ciliogenesis. Curr. Biol., 31, 2887–2894.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ganga, A.K., Kennedy, M.C., Oguchi, M.E., Gray, S., Oliver, K.E., Knight, T.A., De La Cruz, E.M., Homma, Y., Fukuda, M. and Breslow, D.K. (2021) Rab34 GTPase mediates ciliary membrane formation in the intracellular ciliogenesis pathway. Curr. Biol., 31, 2895–2905.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Goldenberg, N.M., Grinstein, S. and Silverman, M. (2007) Golgi-bound Rab34 is a novel member of the secretory pathway. Mol. Biol. Cell, 18, 4762–4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kasmapour, B., Gronow, A., Bleck, C.K.E., Hong, W. and Gutierrez, M.G. (2012) Size-dependent mechanism of cargo sorting during lysosome-phagosome fusion is controlled by Rab34. Proc. Natl. Acad. Sci. U. S. A., 109, 20485–20490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang, T. and Hong, W. (2002) Interorganellar regulation of lysosome positioning by the Golgi apparatus through Rab34 interaction with Rab-interacting lysosomal protein. Mol. Biol. Cell, 13, 4317–4332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xu, S., Liu, Y., Meng, Q. and Wang, B. (2018) Rab34 small GTPase is required for hedgehog signaling and an early step of ciliary vesicle formation in mouse. J. Cell Sci., 131, jcs213710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kanie, T., Love, J.F., Fisher, S.D., Gustavsson, A.-K. and Jackson, P.K. (2023) A hierarchical pathway for assembly of the distal appendages that organize primary cilia. bioRxiv, 2023.01.06.522944. https://pubmed.ncbi.nlm.nih.gov/36711481/ https://www.biorxiv.org/content/10.1101/2023.01.06.522944v2.
- 47. Pusapati, G.V., Kong, J.H., Patel, B.B., Krishnan, A., Sagner, A., Kinnebrew, M., Briscoe, J., Aravind, L. and Rohatgi, R. (2018) CRISPR screens uncover genes that regulate target cell sensitivity to the morphogen sonic hedgehog. Dev. Cell, 44, 113–129.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dickinson, M.E., Flenniken, A.M., Ji, X., Teboul, L., Wong, M.D., White, J.K., Meehan, T.F., Weninger, W.J., Westerberg, H., Adissu, H. et al. (2016) High-throughput discovery of novel developmental phenotypes. Nature, 537, 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Murcia, N.S., Richards, W.G., Yoder, B.K., Mucenski, M.L., Dunlap, J.R. and Woychik, R.P. (2000) The oak ridge polycystic kidney (ORPK) disease gene is required for left-right axis determination. Development, 127, 2347–2355. [DOI] [PubMed] [Google Scholar]
- 50. Marszalek, J.R., Ruiz-Lozano, P., Roberts, E., Chien, K.R. and Goldstein, L.S. (1999) Situs inversus and embryonic ciliary morphogenesis defects in mouse mutants lacking the KIF3A subunit of kinesin-II. Proc. Natl. Acad. Sci. U. S. A., 96, 5043–5048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sobreira, N., Schiettecatte, F., Valle, D. and Hamosh, A. (2015) GeneMatcher: a matching tool for connecting investigators with an interest in the same gene. Hum. Mutat., 36, 928–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yakar, O. and Tatar, A. (2022) INTU-related oral-facial-digital syndrome XVII: clinical spectrum of a rare disorder. Am. J. Med. Genet. A, 188, 590–594. [DOI] [PubMed] [Google Scholar]
- 53. Syed, S., Sawant, P.R., Spadigam, A. and Dhupar, A. (2021) Oro-facial-digital syndrome type I: a case report with novel features. Autops. Case Rep., 11, e2021315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Verma, P.K. and Bhat, N.K. (2021) Oro-facial-digital syndrome: unspecified type with the spontaneous fusion of cleft palate. Contemp. Clin. Dent., 12, 454–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Sukarova-Angelovska, E., Angelkova, N., Palcevska-Kocevska, S. and Kocova, M. (2012) The many faces of oral-facial-digital syndrome. Balk. J. Med. Genet., 15, 37–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Digilio, M.C., Giannotti, A., Pagnotta, G., Mingarelli, R. and Dallapiccola, B. (2008) Joint dislocation and cerebral anomalies are consistently associated with oral-facial-digital syndrome type IV. Clin. Genet., 48, 156–159. [DOI] [PubMed] [Google Scholar]
- 57. Mahato, P.R. and Pandey, S.B. (2012) Orofaciodigital syndrome type-VI (Varadi–Papp syndrome) with several Y-shaped metacarpals. Indian J. Hum. Genet., 18, 376–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Iaccarino, M., Lonardo, F., Giugliano, M. and Della Bruna, M.D. (1985) Prenatal diagnosis of Mohr syndrome by ultrasonography. Prenat. Diagn., 5, 415–418. [DOI] [PubMed] [Google Scholar]
- 59. Wentzensen, I.M., Johnston, J.J., Keppler-Noreuil, K., Acrich, K., David, K., Johnson, K.D., Graham, J.M., Sapp, J.C. and Biesecker, L.G. (2015) Exome sequencing identifies novel mutations in C5orf42 in patients with Joubert syndrome with oral–facial–digital anomalies. Hum. Genome Var., 2, 15045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Meinecke, P. and Hayek, H. (1990) Orofaciodigital syndrome type IV (Mohr-Majewski syndrome) with severe expression expanding the known spectrum of anomalies. J. Med. Genet., 27, 200–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Simonini, C., Floeck, A., Strizek, B., Mueller, A., Gembruch, U. and Geipel, A. (2022) Fetal ciliopathies: a retrospective observational single-center study. Arch. Gynecol. Obstet., 306, 71–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Barisic, I., Boban, L., Loane, M., Garne, E., Wellesley, D., Calzolari, E., Dolk, H., Addor, M.-C., Bergman, J.E., Braz, P. et al. (2015) Meckel–Gruber syndrome: a population-based study on prevalence, prenatal diagnosis, clinical features, and survival in Europe. Eur. J. Hum. Genet., 23, 746–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Roosing, S., Romani, M., Isrie, M., Rosti, R.O., Micalizzi, A., Musaev, D., Mazza, T., Al-gazali, L., Altunoglu, U., Boltshauser, E. et al. (2016) Mutations in CEP120 cause Joubert syndrome as well as complex ciliopathy phenotypes. J. Med. Genet., 53, 608–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Toriello, H.V., Franco, B., Bruel, A.-L. and Thauvin-Robinet, C. (2016) Oral-Facial-Digital Syndrome Type I. University of Washington, Seattle. [PubMed] [Google Scholar]
- 65. Hsieh, Y.C. and Hou, J.W. (1999) Oral-facial-digital syndrome with Y-shaped fourth metacarpals and endocardial cushion defect. Am. J. Med. Genet., 86, 278–281. [PubMed] [Google Scholar]
- 66. Kobayashi, T., Ikeda, T., Ota, R., Yasukawa, T. and Itoh, H. (2022) The atypical small GTPase RABL3 interacts with RAB11 to regulate early ciliogenesis in human cells. J. Cell Sci., 135, jcs260021. [DOI] [PubMed] [Google Scholar]
- 67. Jenkins, D., Seelow, D., Jehee, F.S., Perlyn, C.A., Alonso, L.G., Bueno, D.F., Donnai, D., Josifova, D., Mathijssen, I.M.J., Morton, J.E.V. et al. (2007) RAB23 mutations in carpenter syndrome imply an unexpected role for hedgehog signaling in cranial-suture development and obesity. Am. J. Hum. Genet., 80, 1162–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Eggenschwiler, J.T., Espinoza, E. and Anderson, K.V. (2001) Rab23 is an essential negative regulator of the mouse sonic hedgehog signalling pathway. Nature, 412, 194–198. [DOI] [PubMed] [Google Scholar]
- 69. Fuller, K., O’Connell, J.T., Gordon, J., Mauti, O. and Eggenschwiler, J. (2014) Rab23 regulates nodal signaling in vertebrate left-right patterning independently of the hedgehog pathway. Dev. Biol., 391, 182–195. [DOI] [PubMed] [Google Scholar]
- 70. Hor, C.H.H., Lo, J.C.W., Cham, A.L.S., Leong, W.Y. and Goh, E.L.K. (2021) Multifaceted functions of Rab23 on primary cilium-mediated and hedgehog signaling-mediated cerebellar granule cell proliferation. J. Neurosci., 41, 6850–6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sato, T., Iwano, T., Kunii, M., Matsuda, S., Mizuguchi, R., Jung, Y., Hagiwara, H., Yoshihara, Y., Yuzaki, M., Harada, R. et al. (2014) Rab8a and Rab8b are essential for several apical transport pathways but insufficient for ciliogenesis. J. Cell Sci., 127, 422–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Paila, U., Chapman, B.A., Kirchner, R. and Quinlan, A.R. (2013) GEMINI: integrative exploration of genetic variation and genome annotations. PLoS Comput. Biol., 9, e1003153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Rodrigues, C.H.M., Pires, D.E.V. and Ascher, D.B. (2021) DynaMut2: assessing changes in stability and flexibility upon single and multiple point missense mutations. Protein Sci., 30, 60–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Varadi, M., Anyango, S., Deshpande, M., Nair, S., Natassia, C., Yordanova, G., Yuan, D., Stroe, O., Wood, G., Laydon, A. et al. (2022) AlphaFold protein structure database: massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res., 50, D439–D444. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data supporting the findings of this study are present within the article or are available from the authors upon request.