

Abstract
To pave the road towards precision medicine in cancer, patients with similar biology ought to be grouped into same cancer subtypes. Utilizing high-dimensional multiomics datasets, integrative approaches have been developed to uncover cancer subtypes. Recently, Graph Neural Networks have been discovered to learn node embeddings utilizing node features and associations on graph-structured data. Some integrative prediction tools have been developed leveraging these advances on multiple networks with some limitations. Addressing these limitations, we developed SUPREME, a node classification framework, which integrates multiple data modalities on graph-structured data. On breast cancer subtyping, unlike existing tools, SUPREME generates patient embeddings from multiple similarity networks utilizing multiomics features and integrates them with raw features to capture complementary signals. On breast cancer subtype prediction tasks from three datasets, SUPREME outperformed other tools. SUPREME-inferred subtypes had significant survival differences, mostly having more significance than ground truth, and outperformed nine other approaches. These results suggest that with proper multiomics data utilization, SUPREME could demystify undiscovered characteristics in cancer subtypes that cause significant survival differences and could improve ground truth label, which depends mainly on one datatype. In addition, to show model-agnostic property of SUPREME, we applied it to two additional datasets and had a clear outperformance.
INTRODUCTION
Cancer is one of the deadliest diseases for which cancer-causing agents such as oncogenes, mutations, and gene regulatory associations have not been fully demystified. Cancer patients show different characteristics in terms of the progression of disease and response to treatment (1). Various biological datasets from cancer tissues have been generated to better characterize cancer biology. For instance, The Cancer Genome Atlas (TCGA) project generated over 2.5 petabytes of multiple omics (multiomics) data for thousands of patients from 33 different cancer types (data are available at https://portal.gdc.cancer.gov/). Specifically for breast cancer, the Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) has generated four types of multiomics data for thousands of breast tumor samples (2). Utilizing high-dimensional biological datasets in public databases, computational approaches have been developed to discover subtypes of various cancers (3–5). Several of the cancer subtype prediction studies rely only on one type of biological datatype (4,6,7). However, each of these datatypes captures a different part of the underlying biology, thus developing integrative computational methods has been an important research area in bioinformatics.
Breast cancer is currently the most commonly-diagnosed cancer worldwide (8). Therapeutic groups in breast cancer (i.e., estrogen receptor-positive, progesterone receptor-positive, human epidermal growth factor receptor 2 (HER2) amplified group, and triple-negative breast cancer) mainly depend on three receptors. Even though these receptors are very impactful in determining the breast cancer subtypes, they are not solely sufficient to classify a patient. Some other studies showed that genomic and clinical features such as race, age, and some mutations are also important in breast cancer subtyping (9,10).
Genomic datatypes are found informative for differentiating subgroups in breast cancer. In 2009, Parker et al. (11) found a clear difference in the expression of 50 genes for breast cancer and introduced breast cancer molecular subtypes, called PAM50 subtypes. In 2012, the TCGA group published a study analyzing breast cancer subgroups and their associations with single datatypes, obtaining subtype-specific patterns in each datatype (12) and supporting the importance of gene expression-based models such as PAM50 (11). Even though there are important signals from both clinical and genomic features to determine the subtype of a patient, relying on a single data modality is not sufficient to differentiate subtypes clearly. As we get more samples and datatypes to analyze, it is important to integrate all the available datatypes properly with advanced approaches to understand differences in the characteristics of cancer patients.
Recently several groups have developed unsupervised computational tools to integrate multiple datatypes to discover cancer subtypes. For instance, iClusterPlus (13) uses a joint latent variable model concatenating multiple datatypes with dimension reduction to cluster cancer patients. Similarity Network Fusion (SNF) (14) builds a patient similarity network based on each datatype, obtains a fused patient network by applying a nonlinear fusion step, and performs the clustering on that final network. PINSPlus (15) assumes that samples that are truly in the same subtype are clustered together despite small changes in the data. PINSPlus discovers the subtypes if the samples are highly connected for different datatypes applying data perturbation. The authors demonstrated that PINSPlus had robust results with significant survival differences across different cancer types. Those studies focus on unsupervised multiomics data integration without the utilization of found subtype labels such as PAM50 subtype labels. Furthermore, these tools utilize patient similarity networks or features, but not both simultaneously, while there are recent improvements in graph representation learning allowing the utilization of both at the same time (16–18).
Graphs (networks) are suitable data structures to store multiomics datasets, however, machine learning (ML)-based approaches are challenging on graph data. Deep learning-based architectures have been used extensively for grid-like data (e.g., image), however, these methods are not directly applicable to graph data. Graphs are unstructured as each node has a varying number of neighbors and there is no fixed ordering of nodes. To train ML models on graph data, embedding (a fixed low-dimensional vector) is used and some shallow embedding methods emerged by encoding every node into embedding, representing the position and the local relationships in the graph (19–21). However, these shallow embedding methods are not scalable for large graphs and cannot utilize the node features that we have plenty of, thus, these methods have been replaced with more advanced deep learning-based methods such as Graph Neural Networks (GNNs) (16,17). The main difference in GNN-based architectures is how the features are aggregated from the local structure. Graph Convolutional Network (GCN) is one of the most popular GNNs that uses a modified aggregation involving self edges with normalization across neighbors (18). GNNs have recently been applied to biological problems such as cancer type/subtype prediction and drug response prediction (18, 22–25).
Even though there are some studies applying convolution to graph-structured data for cancer subtyping, these models are mostly applicable to a single network or had some limitations for integrative approaches. In (26), cancer type prediction of patients from 33 cancer and non-cancer types (i.e., all normal samples from all 33 available cancer types) was performed using GCNs. The input network was based on gene coexpression or protein-protein interaction, but the convolution was done on the gene expression dataset only, thus, missing the information of multiple data modalities. Multiomics GCN (MOGONET) is a supervised multiomics integration framework using GCNs with a patient similarity network for mRNA expression, DNA methylation, and microRNA expression separately (27). MOGONET gets the label independently from three different models, then uses them to get the final prediction. However, it does not consider multiple features for networks. We call this kind of embedding datatype-specific patient embedding where the methodology generates datatype-specific networks with datatype-specific node features and considers only the prediction labels from separate GCN models. However, these embeddings could be improved by utilizing all the multiomics patient features on each local network structure, making the embedding network-specific patient embedding. Moreover, it is possible to utilize GCN not only to get the prediction label but also to obtain the embeddings and integrate them. Going further, we can also integrate the patient features (called raw features) with embeddings to capture any diluted signals from features. To utilize more from available knowledge, it is important to properly integrate multiple network representations and multiomics features simultaneously.
To address the aforementioned limitations, we developed a computational tool named SUPREME integrating multiple types of datasets using GCNs. SUPREME generated similarity networks using features from multi-modal datasets where node features include features from all data modalities, assuming that nodes with a similar local neighborhood are likely to belong to the same class. SUPREME encodes the relations on a network from each datatype and obtains network-specific node embeddings incorporating node features on each network. Then SUPREME integrates these embeddings providing extensive evaluations of all combinations of node embeddings. For each combination, SUPREME integrates the selected embeddings with raw features to utilize all the knowledge at the same time. SUPREME utilizes all available datatypes from public datasets and can interpret each datatype’s effectiveness in terms of features and networks. Being model-agnostic, SUPREME could be easily adapted to any model, any prediction task handling any number of datatypes, and could be easily modified by changing the embedding integration method, network generation strategy, and feature extraction approach.
In this study, SUPREME was applied to three different prediction tasks from five different datasets. We applied SUPREME to predict subtypes of breast cancer patients using multiomics datasets (from TCGA and METABRIC datasets separately and together). Our results on cancer subtype prediction tasks showed that SUPREME outperformed other integrative supervised cancer (sub)type prediction tools and baseline methods. SUPREME had improved performance showing the importance of GCN-based approaches, network-specific patient embeddings, and raw feature integration. SUPREME was robust showing high and consistent prediction performance. We observed that the gene expression (EXP)-based features were the most significant features, as expected for breast cancer. Importantly, SUPREME-inferred cancer subtypes had consistently significant survival differences and were mostly more significant than the survival differences between ground truth subtypes, which were based on gene expression datatype. These results suggest that SUPREME can differentiate the characteristics of cancer subtypes properly utilizing the multiple network relations and multiple datatypes. To demonstrate the model-agnostic property of our tool, we also applied SUPREME to ACM and IMDB datasets and SUPREME outperformed other methods on both datasets.
MATERIALS AND METHODS
SUPREME is a computational tool for node classification tasks integrating multiple data modalities using GCNs. Briefly, the first step is data preparation. In the second step, SUPREME extracts features from each datatype. Using those features, SUPREME generates individual similarity networks per datatype where features from all datatypes are used as node attributes. In the third step, using the obtained networks and features, SUPREME generates the network-specific node embeddings by running GCN on each network. In the last step, SUPREME does prediction by integrating individual network-specific embeddings and raw features. In the following part, we explain each step of SUPREME in detail.
Data preparation
We applied SUPREME on three datasets for the breast cancer subtype prediction task. We collected the data and generated seven datatypes (i.e., clinical, copy number aberration, coexpression, gene expression, DNA methylation, microRNA expression, and mutation) across 1022 breast tumor samples from TCGA (12), five datatypes (i.e., clinical, copy number aberration, coexpression, gene expression, and mutation) across 1699 breast tumor samples from METABRIC (2) and three datatypes (clinical, gene expression, and mutation) across a total of 2721 breast tumor samples from the combined datasets of TCGA and METABRIC. As ground truth for the prediction task, we obtained the PAM50 subtype labels, namely Basal-like, HER2-Enriched, Luminal-A, Luminal-B, and Normal-like (11). Data preprocessing details are in Supplementary Methods 1.1.
We also collected ACM and IMDB datasets for two additional tasks: movie genre prediction from IMDB dataset (https://www.imdb.com) and paper area prediction task from ACM dataset (http://dl.acm.org). IMDB dataset has a heterogeneous network with three node types (movie, actor, and director) along with two associations: movie-actor and movie-director. The movies have three genre classes: action, comedy, and drama. ACM dataset has also three node types (paper, author, and subject) on a heterogeneous network along with two associations: paper-author and paper-subject. The papers have three classes: database, wireless communication, and data mining.
The number of features and samples for each dataset are shown in Table 1.
Table 1.
Number of features and samples for each dataset. Subtypes are abbreviated as BL: basal-like, HER2: HER2-Enriched, LA: luminal-A, LB: luminal-B, NL: normal-like
| Dataset | Number of raw features | Number of samples |
|---|---|---|
| TCGA | 3088 | 1022 samples: 172 BL (17%), 78 HER2 (8%), 538 LA (53%), 195 LB (19%), 39 NL (4%) |
| METABRIC | 1761 | 1699 samples: 199 BL (12%), 220 HER2 (13%), 679 LA (40%), 461 LB (27%), 140 NL (8%) |
| Combined (TCGA+ METABRIC) | 1229 | 2721 samples: 371 BL (14%), 298 HER2 (11%), 1217 LA (45%), 656 LB (24%), 179 NL (7%) |
| IMDB | 3066 | 4278 samples: 1135 (27%), 1584 (37%), 1559 (36%) |
| ACM | 1870 | 3025 samples: 1061 (35%), 965 (32%), 999 (33%) |
Feature extraction & network generation
Breast cancer subtyping
SUPREME incorporates seven datatypes for TCGA data, five datatypes for METABRIC data, and three datatypes for the combined data. We utilized a Random Forest-based feature selection algorithm, called Boruta (28), to extract features from high dimensional datatypes. The selected features in the data preprocessing step (i.e., multiomics features) were used to compute the similarity between patients when generating the patient similarity networks, as node features in the patient similarity networks, and to integrate as raw features before the prediction task. To compute patient similarities in datatype-specific patient similarity networks, we used Pearson correlation for gene expression, copy number aberration, DNA methylation, microRNA expression, and coexpression datatypes; the Gower metric (29) from the daisy function of cluster R package (30) for clinical features; and Jaccard distance for binary mutation features. After selecting the top edges, the edge weights were eliminated to generate an unweighted network. We used 2500 edges for the datatypes of TCGA, 4500 for METABRIC and 7000 for the combined data (having approximately 2.5 times the sample size). Details of feature extraction and network generation are in Supplementary Methods 1.2.
Movie genre prediction
We did not apply any feature selection for the IMDB dataset and used node features processed in (31). Using two associations (i.e., movie-actor and movie-director) in the data, two movie similarity networks were generated based on two meta-paths using (32): movie-director-movie with 17 446 edges and movie-actor-movie with 85 358 edges. Meta-path-based similarity networks connect nodes based on a given association. For instance, the meta-path movie-actor-movie defines similarity as the existence of at least one common actor between two movies.
Paper area prediction
For the ACM dataset, we did not apply any feature selection and used the node features processed in (31). Utilizing two associations (i.e., paper-author and paper-subject) in the data, two meta-paths were used to generate two paper similarity networks using (33): paper-author-paper with 29 281 edges and paper-subject-paper with 2 210 761 edges. The meta-path-based similarity definition is the same as in the IMDB dataset.
When there is a high number of raw features and many networks to integrate, this might affect the prediction performance, and model training could be time-consuming. Thus, we added another optional feature selection step to further reduce the number of raw features integrated with the node embeddings for the prediction task. We enabled this additional feature selection for TCGA data where we had a high number of raw features and networks and observed that it reduced running time without affecting the prediction performance. We did not apply optional feature selection for ACM and IMDB datasets since we have only two networks. Similarly, we did not apply any reduction for the number of edges for these datasets since we do not have any quantitative similarities to prioritize the edges on meta-path-based similarity networks.
Node embedding generation
After extracting features and generating networks, we obtained network-specific node embeddings, which capture the topology of the network as well as node features to be utilized in a downstream ML task.
In this study, we used the GCN model of Kipf and Welling (18) involving self edges in convolution and scaling the sum of aggregated features across the neighbors. GCN models learn the data by performing convolution on networks, considering one-hop local neighbors with equal contribution, and encoding the local topology of the network. Stacked layers involve recursive neighborhood diffusion considering more than a one-hop neighborhood.
Let’s call an undirected graph as 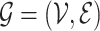 where
where 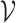 is a set of n nodes, i.e.,
is a set of n nodes, i.e., 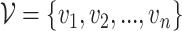 , and
, and 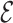 is a set of edges between nodes where
is a set of edges between nodes where 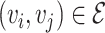 when
when 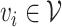 ,
, 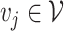 and
and 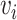 and
and 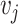 have an association based on the graph
have an association based on the graph 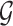 . Since the graph
. Since the graph 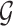 is undirected,
is undirected,  .
.
The input for a GCN model is a feature matrix 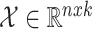 where k is the feature size, and the adjacency matrix
where k is the feature size, and the adjacency matrix 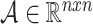 with added self edges defined as:
with added self edges defined as:
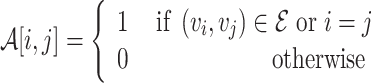 |
The iteration process is defined as:
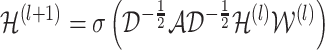 |
with 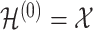 where
where
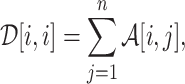 |
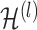 is the activation matrix in the lth layer,
is the activation matrix in the lth layer, 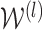 is the trainable weight matrix in the lth layer and σ is the activation function.
is the trainable weight matrix in the lth layer and σ is the activation function.
Considering breast cancer subtyping task using TCGA data, SUPREME setup for the single model generation was as follows: there were seven networks (i.e., patient similarity networks), each obtained from a different datatype. All networks had nodes as breast cancer patients and edges based on the patient similarities from the corresponding data. For instance, let us consider 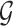 as a gene expression-derived patient similarity network. This network connects patient nodes with a high correlation between their gene expression profile. As node features,
as a gene expression-derived patient similarity network. This network connects patient nodes with a high correlation between their gene expression profile. As node features, 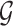 has the combined features, which were extracted from all the seven datatypes. Features of
has the combined features, which were extracted from all the seven datatypes. Features of 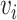 are denoted as
are denoted as 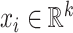 where k is the total feature size. So, the stacked feature matrix
where k is the total feature size. So, the stacked feature matrix 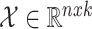 is:
is:
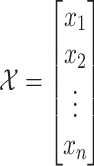 |
The local one-hop neighborhood of a node 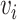 is
is 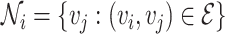 that included the set of nodes having an association with the node
that included the set of nodes having an association with the node 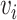 . Feature aggregation on the local neighborhood of each node was done by multiplying
. Feature aggregation on the local neighborhood of each node was done by multiplying 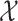 by the nxn-sized scaled adjacency matrix
by the nxn-sized scaled adjacency matrix 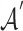 where
where
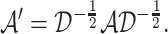 |
Using 2-layered GCN in SUPREME, we had the form of the forward model giving the output 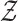 where
where
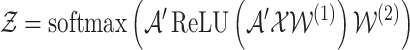 |
and 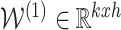 ,
, 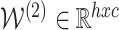 were the trainable weights for the first and second layers, respectively, where h was the hidden layer size and c was the number of classes to predict (namely, Basal-like, Luminal-A, Luminal-B, HER2-Enriched, and Normal-like, with c = 5). The loss function was calculated by cross-entropy error. Adam optimization (34) was used as the state-of-the-art for stochastic gradient descent algorithm and dropout was added for the first GCN layer. Early stopping was used with the patience of 30 forced to have at least 200 epochs.
were the trainable weights for the first and second layers, respectively, where h was the hidden layer size and c was the number of classes to predict (namely, Basal-like, Luminal-A, Luminal-B, HER2-Enriched, and Normal-like, with c = 5). The loss function was calculated by cross-entropy error. Adam optimization (34) was used as the state-of-the-art for stochastic gradient descent algorithm and dropout was added for the first GCN layer. Early stopping was used with the patience of 30 forced to have at least 200 epochs.
We split the total samples into training, validation, and test sets. This splitting was stratified, that is, keeping the same ratio of the subtype labels in the original data for each split. We kept the test set only for final evaluation of the tool. Training and validation splits are randomly selected for each run as stratified. For the breast cancer subtyping, we split 20% of the total samples as a test set. The remaining 80% of the samples were used for training (60%) and validation (20%). For IMDB and ACM datasets, we used the same data splits in (31). To tune the hyperparameters of the GCN model (i.e., hidden layer size and learning rate), for each run, SUPREME repeated an evaluation metric (i.e., macro-averaged F1 (macro F1) score) 10 times for each hyperparameter combination (Supplemental File 2) and selected the hyperparameter combination giving the best median macro F1 score on the validation data to generate the final model.
Similarly applying the methodology for other datatypes, we generated seven different GCN models on TCGA data. Repeating the same procedure for other datasets, we obtained five models on METABRIC data, three models on the combined data, and two models on ACM and IMDB data. These final models were used to extract network-specific patient embeddings to use in the downstream prediction task.
Training predictive models using node embedding combinations
For each combination of node embeddings from  datatypes, we concatenated them with the raw features and trained prediction models (having
datatypes, we concatenated them with the raw features and trained prediction models (having 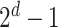 models). Specifically, we had 127, 31, seven, three, and three SUPREME models for TCGA, METABRIC, the combined data (TCGA+METABRIC), ACM, and IMDB datasets, respectively.
models). Specifically, we had 127, 31, seven, three, and three SUPREME models for TCGA, METABRIC, the combined data (TCGA+METABRIC), ACM, and IMDB datasets, respectively.
We tested SUPREME with several ML methods namely, XGBoost, Support Vector Machine (SVM), Random Forest (RF), and Multi-layer Perceptron (MLP). For all datasets, we decided to use MLP as it gave consistently high performance (Supplementary Table S1 and discussion section for the details).
We did hyperparameter tuning for the prediction task, similar to GCN hyperparameter tuning in the previous step. We used the training and validation cohort to tune the hyperparameters (e.g., hidden layer size and learning rate) of the final model, where training and validation splits were randomly selected as stratified. We repeated the SUPREME run 10 times for each hyperparameter combination and used the hyperparameter combination giving the best median macro F1 score on the validation data. Using this hyperparameter combination, the final model was built and evaluated 10 times on the test data, which was never seen during training and hyperparameter tuning. The evaluation metrics (macro F1, weighted-average F1 (weighted F1) score, and accuracy) were obtained from the median of these 10 runs.
RESULTS
We introduced a novel node classification framework, called SUPREME, that utilizes graph convolutions on multiple datatype-specific networks that are annotated with multi-modal datatypes as node features. This framework is model-agnostic and could be applied to any classification problem with properly processed datatypes and networks. In this work, SUPREME was applied specifically to the breast cancer subtype prediction problem by applying convolution on patient similarity networks constructed based on multiple biological datatypes from breast tumor samples (Figure 1). We also evaluated SUPREME on ACM and IMDB datasets demonstrating the outperformance of SUPREME in different domains.
Figure 1.

SUPREME pipeline for breast cancer subtype prediction. SUPREME extracts feature from available datatypes and generates patient similarity networks where nodes are annotated with features from all datatypes. Utilizing graph convolutions on each patient similarity network, patient embeddings are generated. To provide extensive evaluations of subtype prediction, a machine learning model is trained for each combination of patient embeddings and raw multiomics features. [uk is kth patient, i(j) is a raw feature matrix for the jth datatype where each row is 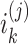 corresponding to the feature vector of the kth patient for the jth datatype. Similarly, z(j) is a node embedding matrix for the jth datatype-specific network where each row is
corresponding to the feature vector of the kth patient for the jth datatype. Similarly, z(j) is a node embedding matrix for the jth datatype-specific network where each row is 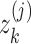 corresponding to the embedding of the kth patient.]
corresponding to the embedding of the kth patient.]
SUPREME outperformed the cancer subtype prediction tools and baseline methods
For the breast cancer subtyping task, we compared the performance of SUPREME on three different datasets with seven other cancer (sub)type prediction tools and baseline methods, namely Deep cancer subtype classification (DeepCC) (35), GCN-based classification (GCNC) (26), MOGONET (27), MLP, RF, SVM, and XGBoost. For each dataset combination, SUPREME builds a separate cancer subtype prediction model. For ML-based baseline methods (i.e., MLP, RF, SVM, and XGBoost), we integrated only the raw features from the selected combination and did the prediction with those features. MOGONET utilizes GCN on multiomics data utilizing datatype-specific embedding predictions. GCNC leverages GCN with gene expression features on protein-protein interaction (PPI)- or coexpression-based gene network, while DeepCC utilizes only gene expression datatype with pathway activity transformation through an MLP model. Therefore, we had only two classification models for GCNC: GCNCPPI with the PPI network and GCNCCOE with the coexpression network, and one model for DeepCC. To see the impact of the integration of raw features into the embeddings, we also trained models without integrating raw features with patient embeddings, called SUPREME-. We ran SUPREME, SUPREME-, and the other tools for all the combinations of available datatypes. Even though MOGONET is applicable to any number of datatypes, we could not run the tool for the models with more than five datatypes (waiting time was more than two days per combination), thus we had only 31 different models for TCGA data, while we had all models for METABRIC and the combined data.
SUPREME and SUPREME- outperformed all other multiomics integration methods for three datasets in terms of macro F1, accuracy, and weighted F1 (Figure 2, Supplementary Figures S3 and S4). SUPREME significantly outperformed MLP, which utilizes raw features only in all datasets, showing the importance of GCN utilization. We observed that SUPREME significantly outperformed SUPREME- for all three datasets.
Figure 2.

Classification results. Violin plot of macro F1 scores obtained from 127 different models including all different combinations of datatypes as compared to the cancer subtype prediction tools and baseline supervised methods on TCGA data. DeepCC and GCNC violin plots show the distribution of macro F1 scores of ten runs of a single model as they can only utilize gene expression datatype. The significance level was measured with respect to SUPREME (Wilcoxon rank-sum test p-value to compare the distribution of violin plots representing the significance < 0.001 by ***, else if < 0.01 by **, and else if < 0.05 by *). [MLP: Multi-layer Perceptron, RF: Random Forest, SUPREME-: SUPREME without raw feature integration, SVM: Support Vector Machine]
We ran the tools that utilize only gene expression datatype and evaluated their performance (Supplementary Table S2). For TCGA data, SUPREME achieved significantly higher performance than DeepCC and GCNC models, while performance on METABRIC and the combined data was comparable or superior (Figures 2 and S3).
In addition, we checked the subtype-specific F1 scores, and had consistent and higher performance across all subtypes, mostly having significant differences (Supplementary Figure S5). Specifically, on TCGA data, we had significantly better performance than all other tools for all subtypes in terms of subtype-specific F1 scores. Particularly, SUPREME had a significantly higher subtype-specific F1 score than all other tools on the Normal-like subtype for all three datasets. Considering that the Normal-like subtype had the smallest sample size in all three datasets (4% of the samples from TCGA, 8% from METABRIC, and 7% from the combined data), achieving this performance increase indicates SUPREME’s robustness even for minority classes.
SUPREME had consistently high performance even with single models
To see SUPREME’s performance with a single datatype, we investigated models generated with only one datatype, called single model. We compared SUPREME with an MLP-based model trained using a single datatype to show the impact of our GCN-based approach. To show the impact of different approaches with one datatype, we compared our single models against MOGONET and our EXP-based model with DeepCC and GCNC models. We conducted these experiments for all three datasets.
Based on the single model results, SUPREME outperformed MOGONET for all single models from all three datasets (Table 2, Supplementary Tables S3–S5). Also, SUPREME outperformed MLP (six out of seven models for TCGA data, three out of five for METABRIC data, and two out of three models for the combined data), or had comparable performance, while MLP had extremely poor performance on some datatypes, showing the importance of GCN-based approach.
Table 2.
Single model results on TCGA data. Macro F1 scores for each model with a single dataype. See Figure 1 for the abbreviations of the datatypes. [MLP: multi-layer perceptron]
| Method | CLI | CNA | COE | EXP | MET | MIR | MUT |
|---|---|---|---|---|---|---|---|
| SUPREME | 0.68 ± 0.04 | 0.80 ± 0.03 | 0.76 ± 0.04 | 0.84 ± 0.02 | 0.79 ± 0.03 | 0.73 ± 0.02 | 0.75 ± 0.03 |
| SUPREME- | 0.72 ± 0.02 | 0.77 ± 0.02 | 0.77 ± 0.04 | 0.77 ± 0.05 | 0.79 ± 0.04 | 0.70 ± 0.02 | 0.74 ± 0.04 |
| MLP | 0.46 ± 0.07 | 0.53 ± 0.04 | 0.59 ± 0.02 | 0.82 ± 0.03 | 0.69 ± 0.04 | 0.74 ± 0.04 | 0.28 ± 0.06 |
| MOGONET | 0.41 ± 0.01 | 0.52 ± 0.01 | 0.57 ± 0.01 | 0.75 ± 0.01 | 0.61 ± 0.03 | 0.71 ± 0.03 | 0.34 ± 0.01 |
There was no clear winner for the comparison of the SUPREME EXP-based model with DeepCC and GCNC models. In terms of macro F1 score, SUPREME outperformed both methods on TCGA data and GCNC (1 draw, 1 win) on the combined data. (Table 2, Supplementary Tables S3–S5). This could be because DeepCC and GCNC utilize pathway activation, PPI network, or coexpression network in addition to gene expression datatype. Nonetheless, by utilizing more datatypes SUPREME outperformed or was on par with both tools for all datasets (Figure 2, Supplementary Figures S3 and S4).
EXP-based models had the highest macro F1 score for all three datasets for all methods (Table 2, Supplementary Tabless S3, S4, and S5). The only exception is that SUPREME- MET-based model had slightly higher performance than SUPREME- EXP-based model on TCGA data. High performance of EXP-based models is not surprising as the breast cancer subtype labels are based on gene expression data. We observed that SUPREME usually outperformed SUPREME- on single models, which indicates that utilizing raw features usually improves the model performance. On the other hand, there were few cases where adding raw features dropped the performance (e.g., CLI-based models on TCGA data). By examining SUPREME- and MLP model performances, we compared the predictive power of patient embeddings with raw features. We observed that patient embedding features were more useful than raw features with few exceptions, such as microRNA expression- (MIR) and EXP-based models on TCGA data, copy number aberration (CNA)-based on METABRIC data, and CLI-based model on the combined data. Specifically on TCGA, we see that CLI-based embedding was more informative than CLI-based features. For CNA- and mutation (MUT)-based models, embeddings were more useful than raw features, but we observed that integrating raw features to embeddings further improved the performance. Similarly, although for the EXP-based model on TCGA data, embeddings were less informative than raw features, integrating them improved the performance.
SUPREME had significant survival differences between predicted subtypes consistently
To measure the ability of the supervised methods to differentiate samples based on survival, we predicted the subtype labels for each data modality combination and performed the survival analysis. In addition to the supervised methods, we also included the state-of-the-art unsupervised tools that are specifically applied to cancer subtyping (i.e., iClusterPlus (13), SNF (14), and PINSPlus (15)) and an algorithmically-relevant clustering method (i.e. affinity propagation (AP) clustering). AP clustering is relevant because it uses a message-passing strategy to find the cluster representatives and the best representative for each node. We obtained five clusters from the unsupervised methods to match the number of PAM50 subtypes and checked the survival differences for these obtained clusters. This analysis was only applied to the results on TCGA data where patient survival data were available. To check the statistical significance of survival differences between subtypes, we applied the log-rank test to compute p-values. Details of survival analysis are in Supplementary Methods 1.3.
The results showed that SUPREME’s predicted subtypes consistently had significant differences in survival rates and significantly outperformed all other nine methods in terms of the P-value (Figure 3). SUPREME had 0.0035 as the lowest P-value (when integrating CNA-, COE-, MET- and MUT-based patient embeddings) and 0.0131 as the median P-value (Supplementary Figure S6A for the Kaplan–Meier plot). Similarly for SUPREME-, we had 0.0018 as the lowest P-value (when integrating CNA- and COE-based patient embeddings), and 0.0147 as the median P-value. Interestingly, SUPREME had a more significant survival difference than the survival difference between ground truth (i.e., PAM50) labels (Supplementary Figure S6B for the Kaplan–Meier plot for PAM50 subtypes).
Figure 3.

Survival analysis results violin plot of the log-rank P-value obtained from survival analysis for the SUPREME models as compared to the cancer subtype prediction/clustering tools and baseline methods. Significance level was measured with respect to SUPREME (Wilcoxon rank-sum test P-value to compare the distribution of violin plots representing the significance <0.001 by ***, else if <0.01 by **, and else if <0.05 by *). The continuous line shows the significance level of 0.05 and the dashed line shows the ground truth’s significance level. The below figure focuses on the significant survival P-values (<0.05) [AP: affinity propagation, MLP: multi-layer perceptron, RF: random forest, SNF: similarity network fusion, SUPREME: SUPREME without raw feature integration, SVM: support vector machine].
Specifically, 106 out of 127 SUPREME models had a lower P-value than the p-value for ground truth. For 57% of those models, we had CNA-based embedding selected. It is followed by 52% from COE-, CLI- and MET-based embeddings. This might suggest that those embeddings could contribute more to differentiating survival differences between subtypes.
AP, iClusterPlus, MOGONET, and SNF methods had a wide range of P-values, while SUPREME, MLP, SVM, and XGBoost had mostly significant P-values (≤0.05) with a median lower than the significance level of the ground truth. SUPREME was better than SUPREME-, but the difference was not significant.
Using support from the predicted subtypes by each model in SUPREME, we computed an ensembled consensus subtype based on majority voting for each patient (Supplemental File 3) and checked the survival difference between these consensus subtypes. Once again, we observed a significant (P-value = 0.01) survival difference between consensus subtypes (Supplementary Figure S6C). We also observed that 882 out of 1022 patients had the same subtype prediction across all 127 models showing the robustness of SUPREME predictions.
Feature/omics importance analysis
In this section, we investigated the importance of each network-specific embedding and datatype-specific features.
Impact of network-specific patient embeddings
We investigated the contribution of each patient embedding on the model performance by comparing the models built using a patient embedding from datatype 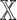 and without using that embedding. Among all
and without using that embedding. Among all 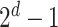 models,
models, 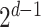 models had the patient embedding obtained from a datatype
models had the patient embedding obtained from a datatype 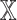 , called
, called 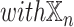 . The remaining
. The remaining 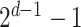 models did not have the patient embedding obtained from
models did not have the patient embedding obtained from 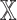 , called
, called 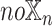 . For each datatype
. For each datatype 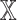 , we compared
, we compared 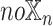 models against
models against 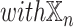 models, showing the importance of
models, showing the importance of 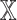 -specific patient embedding. We did this analysis on SUPREME- (i.e., without integrating the raw features) to ensure that differences were due to the patient embeddings only.
-specific patient embedding. We did this analysis on SUPREME- (i.e., without integrating the raw features) to ensure that differences were due to the patient embeddings only.
The results on TCGA data showed that the performance of models increased or stayed the same with the inclusion of patient embeddings from all datatypes except for gene expression (Figure 4). The inclusion of EXP-based embedding showed a significant decrease in the model performance. The exclusion of CLI- and CNA-based patient embeddings had a significant drop in the model performance. Those findings agree with single model results.
Figure 4.

Analysis of network-specific patient embeddings. Violin plot of macro F1 scores of SUPREME- performance for the models integrated with a specific patient embedding from each datatype (with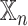 models, where
models, where 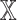 is the datatype whose embedding is included) versus excluding that embedding (no
is the datatype whose embedding is included) versus excluding that embedding (no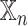 models) on TCGA data. Significance level was measured between with and no cases of the same datatype (Wilcoxon rank-sum test p-value to compare the distribution of violin plots representing the significance <0.001 by ***, else if <0.01 by **, and else if <0.05 by *). See Figure 1 for the abbreviations of datatypes. [SUPREME-: SUPREME without raw feature integration]
models) on TCGA data. Significance level was measured between with and no cases of the same datatype (Wilcoxon rank-sum test p-value to compare the distribution of violin plots representing the significance <0.001 by ***, else if <0.01 by **, and else if <0.05 by *). See Figure 1 for the abbreviations of datatypes. [SUPREME-: SUPREME without raw feature integration]
For METABRIC data, the inclusion of COE- and EXP-based embeddings increased the performance, while the other embeddings did not affect the performance much (Supplementary Figure S7A). For the combined data, MUT- and EXP-based embeddings showed higher performance when included, whereas the inclusion of CLI-based embedding did not affect the performance much (Supplementary Figure S7B).
In addition, we analyzed SUPREME results for TCGA data in terms of the best- and worst-performing models. Specifically, we had 31 top models with a macro F1 score is ≥0.88, and 30 bottom models with a macro F1 score is ≤0.83. We counted how many times each datatype occurred in the top and bottom models. CNA- and CLI-based embeddings were used for 28 and 19 out of 31 top models, respectively. The least occurred embedding was EXP-based with only six models out of 31. For the bottom models, we had 25 models from EXP-based embedding, while we had the least occurred embedding from CNA-based embedding with only five models. This analysis showed that CNA-based embedding was the most selected to have higher performance, while EXP-based embedding was rarely selected, supporting our findings in this section and in single model analysis.
Impact of features from each datatype
To see the impact of the features from each datatype, we ran SUPREME excluding the features from every single datatype separately. For each datatype 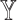 , we excluded
, we excluded 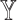 -specific node features from patient similarity networks and also did not integrate them with node embeddings during subtype prediction, called
-specific node features from patient similarity networks and also did not integrate them with node embeddings during subtype prediction, called 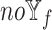 . Considering that
. Considering that 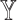 -specific patient similarity network was generated based on
-specific patient similarity network was generated based on 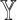 -specific features, we compared only the combinations without
-specific features, we compared only the combinations without  (
(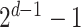 models) to ensure the differences were due to the
models) to ensure the differences were due to the 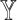 -specific features. We compared
-specific features. We compared 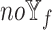 models against the corresponding SUPREME models (called
models against the corresponding SUPREME models (called 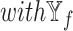 ), to show the importance of
), to show the importance of 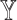 -specific features.
-specific features.
When we excluded features from any datatype, we observed a lower or comparable performance (Figures 5 and Supplementary Figure S8). The performance drop was significant for all the datatypes on TCGA, and gene expression and copy number aberration datatypes on METABRIC (Supplementary Figure S8A). The drop with the exclusion of the gene expression features was more drastic and it was consistent for all three datasets (Supplementary Figure S8B), supporting the importance of gene expression features for breast cancer (in agreement with findings in single model analysis).
Figure 5.

Analysis of features from each datatype. Violin plot of macro F1 scores for the models excluding the features from each datatype (no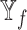 models, where
models, where 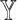 is the datatype whose features are completely excluded) versus corresponding SUPREME models (with
is the datatype whose features are completely excluded) versus corresponding SUPREME models (with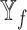 models) on TCGA data. Significance level was measured between with and no cases of the same datatype (Wilcoxon rank-sum test p-value to compare the distribution of violin plots representing the significance <0.001 by ***, else if <0.01 by **, and else if <0.05 by *). See Figure 1 for the abbreviations of datatypes.
models) on TCGA data. Significance level was measured between with and no cases of the same datatype (Wilcoxon rank-sum test p-value to compare the distribution of violin plots representing the significance <0.001 by ***, else if <0.01 by **, and else if <0.05 by *). See Figure 1 for the abbreviations of datatypes.
Ablation studies
We compared our tool with its variations when some steps were skipped to assess their importance (Table 3). A comparison of SUPREME with SUPREME- showed the importance of raw feature integration. Also, to show the importance of GCN-based approaches, we trained the same ML algorithm (MLP in our case) using only the raw features and compared it with the SUPREME, which was based on the same raw features and additional patient embeddings.
Table 3.
Ablation studies
| Comparison/Section | Measures impact of |
|---|---|
| SUPREME vs. SUPREME- | Raw feature integration |
| SUPREME vs. MLP | GCN utilization |
| Single model section | The used method with only one datatype |
| SUPREME vs. MLP in single model section | GCN utilization with only one datatype |
| This section | Node features |
To show the impact of each datatype separately, we demonstrated the performance of SUPREME models based on a single data type. We also compared SUPREME with other methods that can work with a single data modality only. To show the importance of embeddings at a single datatype level, we compared SUPREME with the MLP model trained on the features from the corresponding datatype.
In addition to these studies, here, we also checked the overall impact of node features on the prediction tasks. To do that, instead of node features, we generated one-hot encoded features and evaluated SUPREME on TCGA data. We had macro F1 score as 0.75 ± 0.01, weighted F1 score as 0.83 ± 0.01, and accuracy as 0.84 ± 0.01. These results suggest that node features were important, dropping the evaluation performance drastically. This was expected as biological features are highly effective in determining the subtypes of breast cancer.
SUPREME was model-agnostic outperforming other approaches in different domains
To show the model-agnostic feature, we evaluated SUPREME on different domains. For that purpose, we generated two meta-path-based networks from the heterogeneous network of ACM and IMDB data. Since we had only two networks for these datasets, we shared the results for individual networks and the integrated one. Based on all six evaluation metrics, SUPREME outperformed other baseline methods on both datasets (Table 4 and Supplementary Table S6). As compared to MLP, we had increased performance showing the importance of graph utilization. Single models from GCN and SUPREME were not as good as the integrated one, showing the importance of SUPREME’s integrative nature.
Table 4.
Macro F1 scores for IMDB and ACM datasets. [Macro F1: Macro-averaged F1 scores, GCNx: Result with xth network]. *Three results: First row with the first network, second row with the second network, and third row integrating the first and second networks. The first networks are based on movie-director-movie and paper-subject-paper meta-paths; and the second networks are based on movie-actor-movie and paper-author-paper meta-paths in IMDB and ACM datasets, respectively
| Method | IMDB | ACM |
|---|---|---|
| MLP | 0.53 ± 0.01 | 0.90 ± 0.01 |
| SVM | 0.55 ± 0.00 | 0.89 ± 0.00 |
| RF | 0.48 ± 0.00 | 0.89 ± 0.00 |
| GCN1 | 0.56 ± 0.00 | 0.70 ± 0.00 |
| GCN2 | 0.51 ± 0.00 | 0.91 ± 0.00 |
| 0.58 ± 0.01 | 0.91 ± 0.01 | |
| SUPREME* | 0.55 ± 0.02 | 0.92 ± 0.01 |
| 0.61 ± 0.02 | 0.94 ± 0.00 |
According to these results, the first network of IMDB data (the network based on movie-director-movie meta-path) and the second network of ACM data (the network based on paper-author-paper meta-path) were more informative. This is not surprising that movie-director-movie association was more important than movie-actor-movie on movie genre prediction task on IMDB data. This was consistent based on the GCN runs and single models of SUPREME runs. Even though there is not a big difference on individual networks of ACM data for SUPREME, we see a big difference on GCN runs, showing the importance of our methodology utilizing embedding along with node features.
DISCUSSION
In this study, we introduced SUPREME, a novel integrative approach utilizing GCNs on multiple similarity networks where nodes are attributed with multi-modal node features. We applied SUPREME to three different prediction tasks from five different datasets. We observed that SUPREME outperformed other methods on ACM and IMDB data based on six evaluation metrics (Table 4 and Supplementary Table S6). On breast cancer subtyping, we compared SUPREME with seven cancer (sub)type prediction tools and baseline methods and observed that SUPREME substantially outperformed or was on par with them based on macro F1 score, accuracy, and weighted F1 score (Figures 2, Supplementary Figures S3 and S4, and Supplementary Table S2). To demonstrate the consistency of the performance for individual SUPREME models, we shared the distribution of standard deviation of SUPREME models (Supplementary Figures S9, S10, and S11). We differentiated Normal-like subtype, which has the smallest sample size for three datasets, significantly better than all other tools on all three datasets showing SUPREME’s robustness even for minority classes (Supplementary Figure S5). We made SUPREME a publicly available tool at https://github.com/bozdaglab/SUPREME (under Creative Commons Attribution Non Commercial 4.0 International Public License) for researchers, biologists, and clinicians to utilize.
We applied survival analysis to see the power of the methods to differentiate subtypes having significant survival differences. Using TCGA data, we compared our tool with nine popular integrative cancer subtype differentiating tools and baseline methods and SUPREME had consistently significant survival differences between predicted subtypes outperforming the other tools (Figure 3).
Based on the majority of predictions, we determined ensemble subtype labels, most of which had high support from individual models (Supplemental File 3). We observed that survival difference between these ensemble subtypes was more significant than survival difference between gene expression-based ground truth (i.e., PAM50) subtypes (Supplementary Figure S6). These results suggest that some survival-related characteristics cannot be explained by gene expression data alone. SUPREME was able to extract these survival-related characteristics utilizing additional data modalities. SUPREME’s ensemble label predictions that were different from ground truth with high support could be further examined by biologists and clinicians.
To show the effect of main steps of SUPREME, we performed an ablation study. In addition, we analyzed datatype-specific embeddings and datatype-specific features. We found that gene expression features were highly important for single models and overall, as expected for breast cancer. Findings about the important embeddings of datasets were supported by SUPREME- single models, where models were fed by only one embedding. We observed that patient embeddings were mostly more informative than raw features. Integrating raw features with patient embeddings usually improved the model performance (Figures 2 and Supplementary Figure S3) except for raw features from few datatypes in single datatype-based models (Table 2, Supplementary Tables S3, S4, and S5).
To compare the performance when we do not utilize the local neighborhood, we ran SUPREME- on TCGA data with the EXP-based single model when we do not have any neighbors than the patient itself. In that model, we had a macro F1 score of 0.85 ± 0.02 for SUPREME-, which was much higher than the original EXP-based model of SUPREME-. This model was even better than the EXP-based single model of SUPREME. This might suggest that EXP-based patient features themselves could perform better than neighborhood-convolved features because the ground truth utilizes patient features themselves to decide the subtype labels. Similarly, because of that, we might see a performance improvement when we add EXP-based raw features.
SUPREME provides four options of ML algorithms to integrate embeddings and raw features, namely MLP, RF, SVM, and XGBoost. We ran SUPREME with all these choices and compared performances (Supplemental File 1, Supplementary Figures S1 and S2, and Supplementary Table S1). RF and XGBoost had a low performance for some models. Overall, SVM had a good performance on every three datasets, however, it did not converge for some models. For this study, we chose MLP due to its high and consistent prediction performance for all three datasets and its low running time.
In our experiments, we observed a high number of edges in MUT-based patient similarity networks as there were many patient pairs with the same similarity. Furthermore, the MUT-based models on TCGA data had high predictive performance, whereas these models had low predictive performance on METABRIC and the combined datasets. These discrepancies were mainly due to the sparse nature of the binary mutation features. For the special datatypes with binary-like sparse values like mutation, patient similarity networks and extracted features could be generated in a more sophisticated way such as based on the functional effect of these mutations (36–39).
SUPREME is extendable to any number of datatypes to integrate. For cases where many datasets are integrated, to avoid potential overfitting, SUPREME provides an optional feature selection step for raw features before training the final prediction model. Users could skip raw feature integration altogether when network-specific patient embeddings provide sufficient discriminatory power. Users could run SUPREME on their training/validation data by enabling/disabling these features to optimize their models. In addition, users could perform ablation studies on SUPREME to determine the most effective data modalities and their combinations. Depending on these results, for the final prediction, users could rely on the most effective model or an ensemble model utilizing the most promising features and networks.
As a future direction, SUPREME could utilize attention mechanisms (40–42), which allows getting weighted contributions from different datatypes, and also weighted neighborhoods from networks. In addition to multiomics datatypes, there are some regulatory relations such as competing endogenous RNA (ceRNA) regulation, which has been recently discovered with important insights into cancer (43). In our recent work, we inferred ceRNA interactions in breast cancer (44). To adopt this kind of regulatory relations, SUPREME could be improved to utilize patient similarity networks based on gene regulatory interactions and more complex patient relations. By improving the existing methodologies with recent advances in the literature, we can obtain more clear cancer subtype groups to pave the way for precision medicine.
DATA AVAILABILITY
TCGA data used in this article are available in The Cancer Genome Atlas project data portal at https://portal.gdc.cancer.gov/. The METABRIC data used in this article are deposited at the European Genome-Phenome Archive http://www.ebi.ac.uk/ega/, which is hosted by the European Bioinformatics Institute, under accession number EGAS00000000083 [2]. The source code for our tool is available at https://github.com/bozdaglab/SUPREME (permanent doi: 10.6084/m9.figshare.23304062).
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to thank Ethan Rebello for his contributions to testing SUPREME and working on adding GPU support to it.
Contributor Information
Ziynet Nesibe Kesimoglu, Department of Computer Science and Engineering, University of North Texas, Denton, TX, USA.
Serdar Bozdag, Department of Computer Science and Engineering, University of North Texas, Denton, TX, USA; Department of Mathematics, University of North Texas, Denton, TX, USA; BioDiscovery Institute, University of North Texas, Denton, TX, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NARGAB Online.
FUNDING
National Institute of General Medical Sciences of the National Institutes of Health [R35GM133657 to S.B.]; a summer research fellowship (to Z.N.K.) by the BioDiscovery Institute at the University of North Texas.
Conflict of interest statement. None declared.
REFERENCES
- 1. Waks A.G., Winer E.P.. Breast cancer treatment: a review. JAMA. 2019; 321:288–300. [DOI] [PubMed] [Google Scholar]
- 2. Curtis C., Shah S.P., Chin S.-F., Turashvili G., Rueda O.M., Dunning M.J., Speed D., Lynch A.G., Samarajiwa S., Yuan Y.et al.. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012; 486:346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Verhaak R.G., Hoadley K.A., Purdom E., Wang V., Qi Y., Wilkerson M.D., Miller C.R., Ding L., Golub T., Mesirov J.P.et al.. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010; 17:98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Noushmehr H., Weisenberger D.J., Diefes K., Phillips H.S., Pujara K., Berman B.P., Pan F., Pelloski C.E., Sulman E.P., Bhat K.P.et al.. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell. 2010; 17:510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Baysan M., Bozdag S., Cam M.C., Kotliarova S., Ahn S., Walling J., Killian J.K., Stevenson H., Meltzer P., Fine H.A.. G-cimp status prediction of glioblastoma samples using mRNA expression data. PloS One. 2012; 7:e47839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Vural S., Wang X., Guda C.. Classification of breast cancer patients using somatic mutation profiles and machine learning approaches. BMC Syst. Biol. 2016; 10:263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Youssef Y.M., White N.M., Grigull J., Krizova A., Samy C., Mejia-Guerrero S., Evans A., Yousef G.M.. Accurate molecular classification of kidney cancer subtypes using microRNA signature. Eur. Urol. 2011; 59:721–730. [DOI] [PubMed] [Google Scholar]
- 8. Ferlay J., Ervik M., Lam F., Colombet M., Mery L., Piñeros M., Znaor A., Soerjomataram I., Bray F.. Global cancer observatory: cancer today. Lyon: International Agency for Research on Cancer, 2018. 2020;
- 9. Anderson W.F., Chatterjee N., Ershler W.B., Brawley O.W.. Estrogen receptor breast cancer phenotypes in the Surveillance, Epidemiology, and End Results database. Breast Cancer Res. Treat. 2002; 76:27–36. [DOI] [PubMed] [Google Scholar]
- 10. Dietze E.C., Sistrunk C., Miranda-Carboni G., O’regan R., Seewaldt V.L.. Triple-negative breast cancer in African-American women: disparities versus biology. Nat. Rev. Cancer. 2015; 15:248–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Parker J.S., Mullins M., Cheang M.C., Leung S., Voduc D., Vickery T., Davies S., Fauron C., He X., Hu Z.et al.. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009; 27:1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Koboldt D., Fulton R., McLellan M., Schmidt H., Kalicki-Veizer J., McMichael J., Fulton L., Dooling D., Ding L., Mardis E.et al.. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shen R., Olshen A.B., Ladanyi M.. Integrative clustering of multiple genomic data types using a joint latent variable model with application to breast and lung cancer subtype analysis. Bioinformatics. 2009; 25:2906–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang B., Mezlini A.M., Demir F., Fiume M., Tu Z., Brudno M., Haibe-Kains B., Goldenberg A.. Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods. 2014; 11:333–337. [DOI] [PubMed] [Google Scholar]
- 15. Nguyen H., Shrestha S., Draghici S., Nguyen T.. PINSPlus: a tool for tumor subtype discovery in integrated genomic data. Bioinformatics. 2019; 35:2843–2846. [DOI] [PubMed] [Google Scholar]
- 16. Gori M., Monfardini G., Scarselli F.. A new model for learning in graph domains. Proceedings. 2005 IEEE International Joint Conference on Neural Networks. 2005; 2:729–734. [Google Scholar]
- 17. Scarselli F., Gori M., Tsoi A.C., Hagenbuchner M., Monfardini G.. The graph neural network model. IEEE Trans. Neur. Networ. 2008; 20:61–80. [DOI] [PubMed] [Google Scholar]
- 18. Kipf T.N., Welling M.. Semi-supervised classification with graph convolutional networks. 2016; arXiv doi:22 February 2017, preprint: not peer reviewed 10.48550/arXiv.1609.02907. [DOI]
- 19. Hoff P.D., Raftery A.E., Handcock M.S.. Latent space approaches to social network analysis. J. Am. Stat. Assoc. 2002; 97:1090–1098. [Google Scholar]
- 20. Perozzi B., Al-Rfou R., Skiena S.. Deepwalk: online learning of social representations. Proceedings of the 20th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. 2014; 701–710. [Google Scholar]
- 21. Grover A., Leskovec J.. node2vec: scalable feature learning for networks. Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining. 2016; 855–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hamilton W., Ying Z., Leskovec J.. Inductive representation learning on large graphs. Adv. Neural Inf. Process. Syst. 2017; 30:https://proceedings.neurips.cc/paper_files/paper/2017/hash/5dd9db5e033da9c6fb5ba83c7a7ebea9-Abstract.html. [Google Scholar]
- 23. Rhee S., Seo S., Kim S.. Hybrid approach of relation network and localized graph convolutional filtering for breast cancer subtype classification. 2017; bioRxiv doi:15 June 2018, preprint: not peer reviewed 10.48550/arXiv.1711.05859. [DOI]
- 24. Mohamed S.K., Nováček V., Nounu A.. Discovering protein drug targets using knowledge graph embeddings. Bioinformatics. 2020; 36:603–610. [DOI] [PubMed] [Google Scholar]
- 25. Zitnik M., Agrawal M., Leskovec J.. Modeling polypharmacy side effects with graph convolutional networks. Bioinformatics. 2018; 34:i457–i466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ramirez R., Chiu Y.-C., Hererra A., Mostavi M., Ramirez J., Chen Y., Huang Y., Jin Y.-F.. Classification of cancer types using graph convolutional neural networks. Front. Phys. 2020; 8:203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wang T., Shao W., Huang Z., Tang H., Zhang J., Ding Z., Huang K.. MOGONET integrates multi-omics data using graph convolutional networks allowing patient classification and biomarker identification. Nat. Commun. 2021; 12:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kursa M.B., Rudnicki W.R.. Feature selection with the Boruta package. J. Stat. Softw. 2010; 36:1–13. [Google Scholar]
- 29. Gower J.C. A general coefficient of similarity and some of its properties. Biometrics. 1971; 27:857–871. [Google Scholar]
- 30. Maechler M., Rousseeuw P., Struyf A., Hubert M., Hornik K.. cluster: cluster analysis basics and extensions, R package version 2.1.3. 2022; https://cran.r-project.org/web/packages/cluster/citation.html.
- 31. Kesimoglu Z.N., Bozdag S.. GRAF: graph attention-aware fusion networks. 2023; bioRxiv doi:29 March 2023, preprint: not peer reviewedhttps://arxiv.org/pdf/2303.16781.pdf.
- 32. Fey M., Lenssen J.E.. Fast graph representation learning with PyTorch Geometric. 2019; bioRxiv doi:25 April 2019, preprint: not peer reviewed 10.48550/arXiv.1903.02428. [DOI]
- 33. Wang M., Zheng D., Ye Z., Gan Q., Li M., Song X., Zhou J., Ma C., Yu L., Gai Y.et al.. Deep graph library: a graph-centric, highly-performant package for graph neural networks. 2019; bioRxiv doi:25 August 2020, preprint: not peer reviewed 10.48550/arXiv.1909.01315. [DOI]
- 34. Kingma D.P., Ba J.. Adam: a method for stochastic optimization. 2014; bioRxiv doi:30 January 2017, preprint: not peer reviewed 10.48550/arXiv.1412.6980. [DOI]
- 35. Gao F., Wang W., Tan M., Zhu L., Zhang Y., Fessler E., Vermeulen L., Wang X.. DeepCC: a novel deep learning-based framework for cancer molecular subtype classification. Oncogenesis. 2019; 8:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Khurana E., Fu Y., Chen J., Gerstein M.. Interpretation of genomic variants using a unified biological network approach. PLoS Comput. Biol. 2013; 9:e1002886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kircher M., Witten D.M., Jain P., O’roak B.J., Cooper G.M., Shendure J.. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014; 46:310–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Leiserson M.D., Vandin F., Wu H.-T., Dobson J.R., Eldridge J.V., Thomas J.L., Papoutsaki A., Kim Y., Niu B., McLellan M.et al.. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat. Genet. 2015; 47:106–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tokheim C.J., Papadopoulos N., Kinzler K.W., Vogelstein B., Karchin R.. Evaluating the evaluation of cancer driver genes. Proc. Natl. Acad. Sci. U.S.A. 2016; 113:14330–14335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vaswani A., Shazeer N., Parmar N., Uszkoreit J., Jones L., Gomez A.N., Kaiser Ł., Polosukhin I.. Attention is all you need. Advances in Neural Information Processing Systems. 2017; 30:1–11. [Google Scholar]
- 41. Veličković P., Cucurull G., Casanova A., Romero A., Lio P., Bengio Y.. Graph attention networks. 2017; bioRxiv doi:4 February 2018, preprint: not peer reviewed 10.48550/arXiv.1710.10903. [DOI]
- 42. Brody S., Alon U., Yahav E.. How attentive are graph attention networks. 2021; bioRxiv doi:31 January 2022, preprint: not peer reviewed 10.48550/arXiv.2105.14491. [DOI]
- 43. Salmena L., Poliseno L., Tay Y., Kats L., Pandolfi P.P.. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language. Cell. 2011; 146:353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kesimoglu Z.N., Bozdag S.. Crinet: a computational tool to infer genome-wide competing endogenous RNA (ceRNA) interactions. Plos One. 2021; 16:e0251399. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
TCGA data used in this article are available in The Cancer Genome Atlas project data portal at https://portal.gdc.cancer.gov/. The METABRIC data used in this article are deposited at the European Genome-Phenome Archive http://www.ebi.ac.uk/ega/, which is hosted by the European Bioinformatics Institute, under accession number EGAS00000000083 [2]. The source code for our tool is available at https://github.com/bozdaglab/SUPREME (permanent doi: 10.6084/m9.figshare.23304062).