

Abstract
Introduction:
The inositol polyphosphate-5-phosphatase D (INPP5D) gene encodes a dual-specificity phosphatase that can dephosphorylate both phospholipids and phosphoproteins. Single nucleotide polymorphisms in INPP5D impact risk for developing late onset sporadic Alzheimer’s disease (LOAD).
Methods:
To assess the consequences of inducible Inpp5d knockdown in microglia of APPKM670/671NL/PSEN1Δexon9 (PSAPP) mice, we injected 3-month-old Inpp5dfl/fl/Cx3cr1CreER/+ and PSAPP/Inpp5dfl/fl/Cx3cr1CreER/+ mice with either tamoxifen (TAM) or corn oil (CO) to induce recombination.
Results:
At age 6 months, we found that the percent area of 6E10+ deposits and plaque-associated microglia in Inpp5d knockdown mice were increased compared to controls. Spatial transcriptomics identified a plaque-specific expression profile that was extensively altered by Inpp5d knockdown.
Discussion:
These results demonstrate that conditional Inpp5d downregulation in the PSAPP mouse increases plaque burden and recruitment of microglia to plaques. Spatial transcriptomics highlighted an extended gene expression signature associated with plaques and identified CST7 (cystatin F) as a novel marker of plaques.
Keywords: Alzheimer’s disease, cystatin F, Inpp5d, microglia, oligomer, SHIP1, spatial transcriptomics
Highlights
Inpp5d knockdown increases plaque burden and plaque-associated microglia number.
Spatial transcriptomics identifies an expanded plaque-specific gene expression profile.
Plaque-induced gene expression is altered by Inpp5d knockdown in microglia.
Our plaque-associated gene signature overlaps with human Alzheimer’s disease gene networks.
1 |. BACKGROUND
Late-onset sporadic Alzheimer’s disease (LOAD) is the most common form of dementia, characterized by progressive memory decline, leading to loss of cerebrocortical function and eventual death. Genome-wide association studies (GWAS), whole genome sequencing, and gene-expression network analysis have identified hub and driver genes and their networks associated with risk of LOAD.1,2 Expression of a large proportion of these genes is specific to, or enriched in, microglia, underscoring a potential role for microglia in AD pathogenesis.3 Multiple approaches have highlighted inositol polyphosphate-5-phosphatase D (INPP5D) as an AD risk gene, and its expression in the brain is restricted to microglia.4–6 Current evidence suggests that increased INPP5D levels associated with inheritance of the common variant rs35349669 confer increased risk for LOAD.7 A second variant in INPP5D, rs61068452, was found to be protective against AD, and was significantly associated with lower cerebrospinal fluid (CSF) total tau (t-tau)/amyloid beta (Aβ)42 ratio.8
INPP5D is a dual-specificity phosphatase that can dephosphorylate both phospholipids and phosphoproteins.9,10 INPP5D encodes the 5′ phosphatase SHIP1 (Src homology 2 [SH2] domain-containing inositol polyphosphate 5′ phosphatase) protein. SHIP1 negatively regulates targets such as immune cell surface receptors by catalyzing the conversion of phosphatidylinositol (3,4,5)-trisphosphate (PI[3,4,5]P3) to phosphatidylinositol (3,4)-bisphosphate (PI[3,4]P2). This lipid dephosphorylation reaction prevents receptor activation, mitigating signal transduction through the phosphatidylinositol-3-kinase/mammalian target of rapamycin (PI3K/mTOR) pathway.10 In particular, SHIP1 inhibits phagocytic signal transduction by triggering receptor expressed on myeloid cells 2 (TREM2) and tyrosine kinase binding protein (TYROBP, also known as DAP12).11 DAP12/TYROBP was also identified as a key driver gene in AD via an unbiased, multi-scale computational network. Constitutive deletion or pharmacologic inhibition of SHIP1 increases phagocytosis of various substances, including zymosan in SHIP1 knockout (KO) macrophages and Aβ in SHIP1 inhibitor-treated microglia.12,13 Promotion of these functions in microglia has been considered a means for preventing or reducing AD pathology and improving disease outcomes, but loss of the inhibitory signal from SHIP1 could also promote inflammatory processes that are detrimental in AD.14
These studies strongly implicate a role for INPP5D expression or activity of the encoded protein, SHIP1, in AD pathophysiology. Additionally, INPP5D expression increases with progression of clinical LOAD and with aging in the 5XFAD mouse model.15 The specific role(s) that INPP5D play(s) in both early or late disease, and the mechanism(s) contributing to these altered functions, remain unknown. We therefore knocked down Inpp5d conditionally in microglia to determine early effects as amyloid pathology develops in APPKM670/671NL/PSEN1Δexon9 (PSAPP) mice at a time correlating to a preclinical level. This strategy also avoids the developmental effects that can occur in constitutive knockouts.16,17 Although the simplest rationale may support hypothesizing that reduction of INPP5D would be beneficial in AD, the complex interactions associated with inflammatory signaling and its contribution to disease progression would suggest the alternative; that is, detrimental outcomes are just as likely. Herein, we demonstrate that Inpp5d downregulation increases Aβ fibrillization, plaque burden, and number of microglia associated with Aβ plaques in the PSAPP amyloid-depositing mouse model of AD.
2 |. RESULTS
2.1 |. Inpp5d knockdown worsened amyloid plaque deposition while increasing recruitment of microglia to the plaque
We used a combination of previously established mouse lines, including the PSAPP mouse, to assess pathological consequences of Inpp5d knockdown (Figure 1A, Figure S2A in supporting information). To induce recombination, 3-month-old Inpp5dfl/fl/Cx3cr1CreER/+ mice, with and without the PSEN1 and APP transgenes, were injected for 5 consecutive days with either tamoxifen (TAM) or corn oil (CO). SHIP1 protein levels were reduced by 75% 1 month post-recombination (Figure S1 in supporting information). At 6 months of age, a reduction in SHIP1 protein levels was maintained in both TAM-treated PSAPP/Inpp5dfl/fl/Cx3cr1CreER/+ (PSAPP-TAM) and wild-type Inpp5dfl/fl/Cx3cr1CreER/+ (WT-TAM) mice compared to CO-treated mice, Inpp5dfl/fl/Cx3cr1CreER/+ (WT-CO), or PSAPP/Inpp5dfl/fl/Cx3cr1CreER/+ (PSAPP-CO; Figure 1B,C; Figure S1). Similar to findings in the 5xFAD mouse model,15 PSAPP-CO mice showed an increase in SHIP1 protein levels.
FIGURE 1. Inpp5d/ SHIP1 knockdown increased amyloid pathology and plaque-associated microglia number.
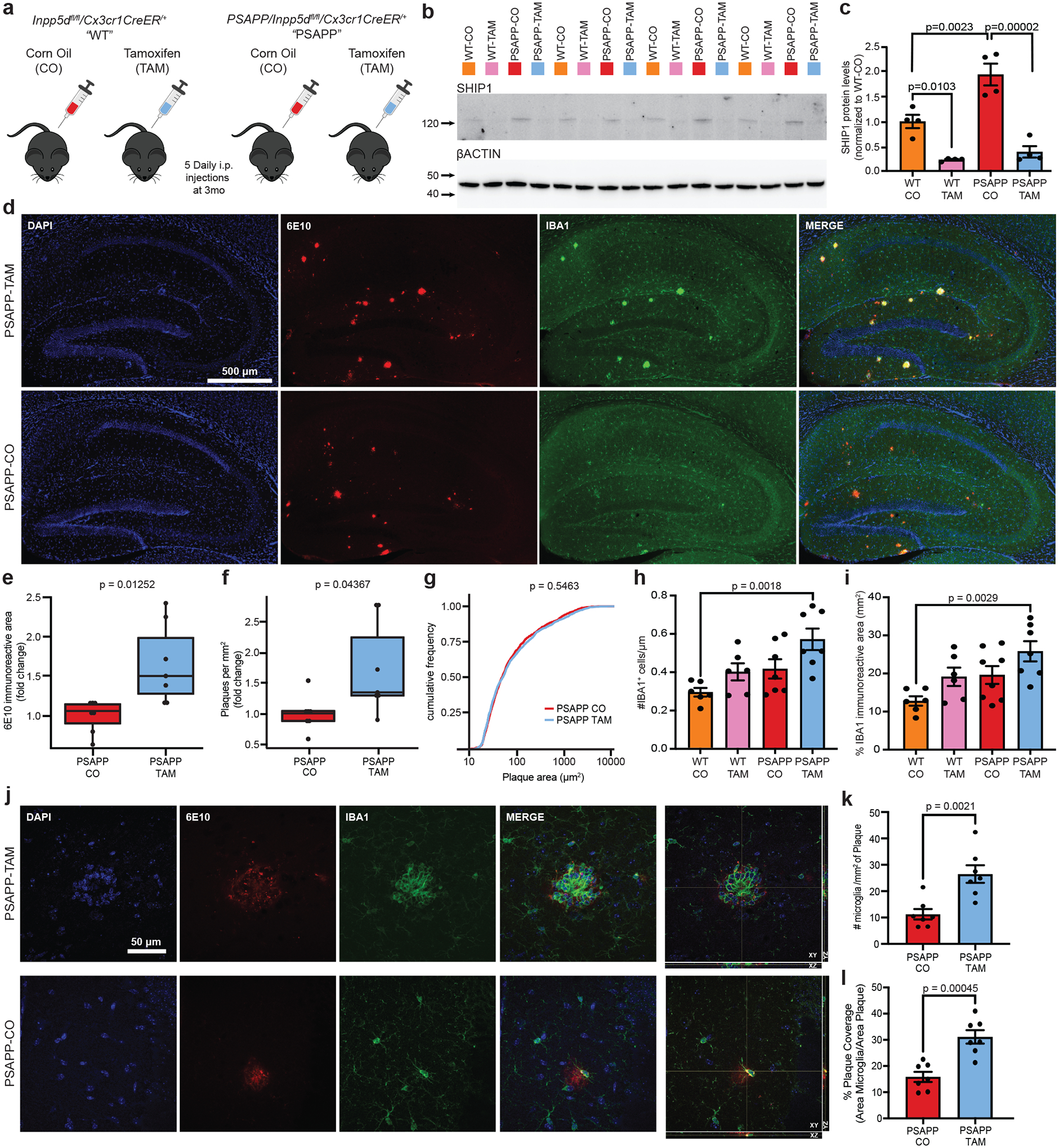
A, Inpp5dfl/fl/Cx3xr1CreER+ mice were crossed with the amyloid-depositing APPKM670/671NL/PSEN1Δexon9 mouse model of Alzheimer’s disease (AD) to obtain either heterozygous PSAPP mice, that is, PSAPP/Inpp5dfl/fl/Cx3xr1CreER+ (PSAPP) or wild-type littermates Inpp5dfl/fl/Cx3xr1CreER+ (WT). At age 3 months, male and female mice of both genotypes received daily injections of tamoxifen (TAM) or corn oil (CO) for 5 days before aging to the experimental timepoint of ages 6 to 6.5 months. B, Western blot of hippocampal tissue at 6 to 6.5 months of age using anti-SHIP1 (D1163) antibody normalized to β-actin. Quantification in (C). D, Representative images from 6-month PSAPP-TAM (top) and PSAPP-CO (bottom) mice stained with 6E10 (amyloid, red) or anti-IBA1 (myeloid cells, green). Scale bar = 500 μm. E, Relative fold change of 6E10-immunoreactive area in the hippocampi of male and female PSAPP-CO and PSAPP-TAM mice (n = 7 mice per group). F, Relative fold change of plaque numbers per mm2 in the hippocampi of male and female PSAPP-CO and PSAPP-TAM mice (n = 7 mice per group). For both (F) and (G), fold changes were calculated separately for each sex. G, Cumulative frequency of plaque areas of male and female PSAPP-CO and PSAPP-TAM mice (plaques from 8 mice per group). H, Quantification of number of IBA1+ microglia and (I) area of IBA1+ area in hippocampus from WT-CO, WT-TAM, PSAPP-CO and PSAPP-TAM (n = 6 to 7 mice per group). J, Confocal Z-stack and orthogonal views of plaques from PSAPP-TAM and PSAPP-CO mice. Quantification of plaque-associated microglia shown in (K). Scale bar = 50 μm. L, Quantification of plaque coverage by microglia (% area of IBA1 staining overlapping area of 6E10+ plaques) for PSAPP-CO and PSAPP-TAM (n = 7 mice per group). Bar graphs represent the mean and error bars represent standard error of the mean. P-values were calculated using unpaired t test (B, E, F, K, L), one-way analysis of variance with Tukey’s multiple comparison (C H, I), or by a Kolmogorov–Smirnov test (G)
At 6 months of age, the percent area and number of 6E10 immunoreactive deposits were increased by >66% and 73%, respectively (Figure 1D–F; Figure S2) in the hippocampi of PSAPP-TAM compared to PSAPP-CO littermates. There was no difference in the size distribution of individual amyloid plaques in PSAPP-TAM compared to PSAPP-CO mice (Figure 1G). Staining with the Congo red derivative Methoxy-XO4 (XO4), which binds specifically to β-pleated sheet-containing structures, confirmed the increase in amyloid burden associated with the Inpp5d/SHIP1 knockdown (Figure S3A–B in supporting information). Fibrillar deposits were increased despite unchanged levels of Aβ40, Aβ42, or Aβ42/Aβ40 ratios as determined by enzyme-linked immunosorbent assays (ELISAs) of Tris-buffered saline (TBS)-, Triton-X-, and formic acid extracts of mouse brain homogenates (Figure S4 in supporting information).
As both microglia and astrocytes contribute to the immune response in AD,18,19 we next measured whether Inpp5d knockdown altered their numbers. Microglia numbers are increased in PSAPP mice with prominent peri-plaque localization.20 In the WT-CO and WT-TAM mice, the number of IBA1+ cells or IBA1 immunoreactive areas in the hippocampus was unchanged after Inpp5d depletion (Figure 1H–I, Figure S3E). Similarly, these parameters did not change when comparing PSAPP-CO to PSAPP-TAM animals (Figure 1D, H–I). There were more IBA1+ cells and IBA1 immunoreactive areas in PSAPP-TAM mice compared to WT-CO mice (Figure 1H–I), whereas the difference between PSAPP-CO and WT-CO was not significant. Most striking was a spatially constrained increase in microglia associated with Aβ plaques (Figure 1C,J–L). We did not observe a significant increase of glial fibrillary acidic protein (GFAP)+ cell numbers or immunoreactive areas in the hippocampus in association with Inpp5d knockdown (Figure S3A, C–D).
Given reports of synapse density loss early in AD21 and a decrease in synaptogenesis in some AD-associated reactive astrocytes,22 we next investigated changes in synaptic markers in our mice. Western blotting of protein extracts from the hippocampi of female WT-CO, WT-TAM, PSAPP-CO, and PSAPP-TAM mice at age 6 months using anti-PSD95 and anti-synaptophysin antibodies showed no obvious effect of any manipulation or parameter on brain levels of either synaptic marker (Figure S5 in supporting information). However, epitomic characterization23 of the Aβ conformers (i.e., A11, OC, NU4) present in the brains of the various groups (Figure S5B) revealed a ≈50% increase in A11/6E10 ratio (i.e., ratio of prefibrillar oligomer content to total Aβ content). No such elevation was detected for OC/6E10 or NU4/6E10 (Figure S5).
2.2 |. PSAPP mice expressed a unique plaque-induced gene cluster that was exacerbated by Inpp5d knockdown
We next performed spatial transcriptomics to analyze brain region–resolved differential gene expression due to the presence of the APP and PSEN1 transgenes and the knockdown of Inpp5d (Figure 2, Figures S6–11 in supporting information). We sequenced a total of 12 coronal brain sections from WT-CO, WT-TAM, PSAPP-CO, and PSAPP-TAM animals (Figure 2A) and reliably identified brain regions based on gene expression profiles (Figure 2B,C). We found that one cluster, Cluster 26, was exclusively expressed in PSAPP mice (Figure 2D). Cluster 26 showed a highly cluster-specific gene expression profile as calculated using a Wilcoxon rank sum test comparing the gene expression in Cluster 26 to all other clusters (Figure 2E) and showed that it co-localized with 6E10-positive plaques of adjacent sections (Figure 2F). Gene ontology (GO) analysis of Cluster 26 genes included “Microglia pathogen phagocytosis pathway” (example genes include: C1qa/b/c, Fcer1g, Vamp8), “Tyrobp causal network” (Tyrobp, Cd37, Cd84, Itgam, Spp1), “Lysosome” (Cd63, Hexa/b, Ly86), and “Antigen processing and presentation” (B2m, H2-D1/K1, Axl; Figure 2G, Tables S1–S5 in supporting information). Given that Aβ plaques have been associated with increased inflammation,24 we next asked whether previously reported reactive microglial or astrocyte substates had similar gene expression profiles and were present in the same spatial location as Cluster 26. Indeed, Cluster 26 had a unique gene expression profile, but we found overlapping gene sets compared to previously reported, biologically defined reactive glial substates. This included integration with differentially expressed genes (DEGs) from disease associated microglia, both early (DAM1) and late (DAM2),25 disease associated astrocytes (DAA),26 plaque-induced genes (PIGs),27 and our previously identified interferon-responsive reactive astrocytes (IRRAs;28 Figure 2H). While DAMs and IRRAs co-localized with Cluster 26, DAAs had a broad expression throughout the brain, which was not surprising given the many non-specific homeostatic astrocytic genes present within the module. We also recovered 50 of the 57 previously identified PIGs and found that the PIG module co-localized with plaques and Cluster 26 (Figure 2H).
FIGURE 2. Spatial transcriptomics identified plaque-specific gene expression profiles exacerbated in Inpp5d/SHIP1 knockdown animals.
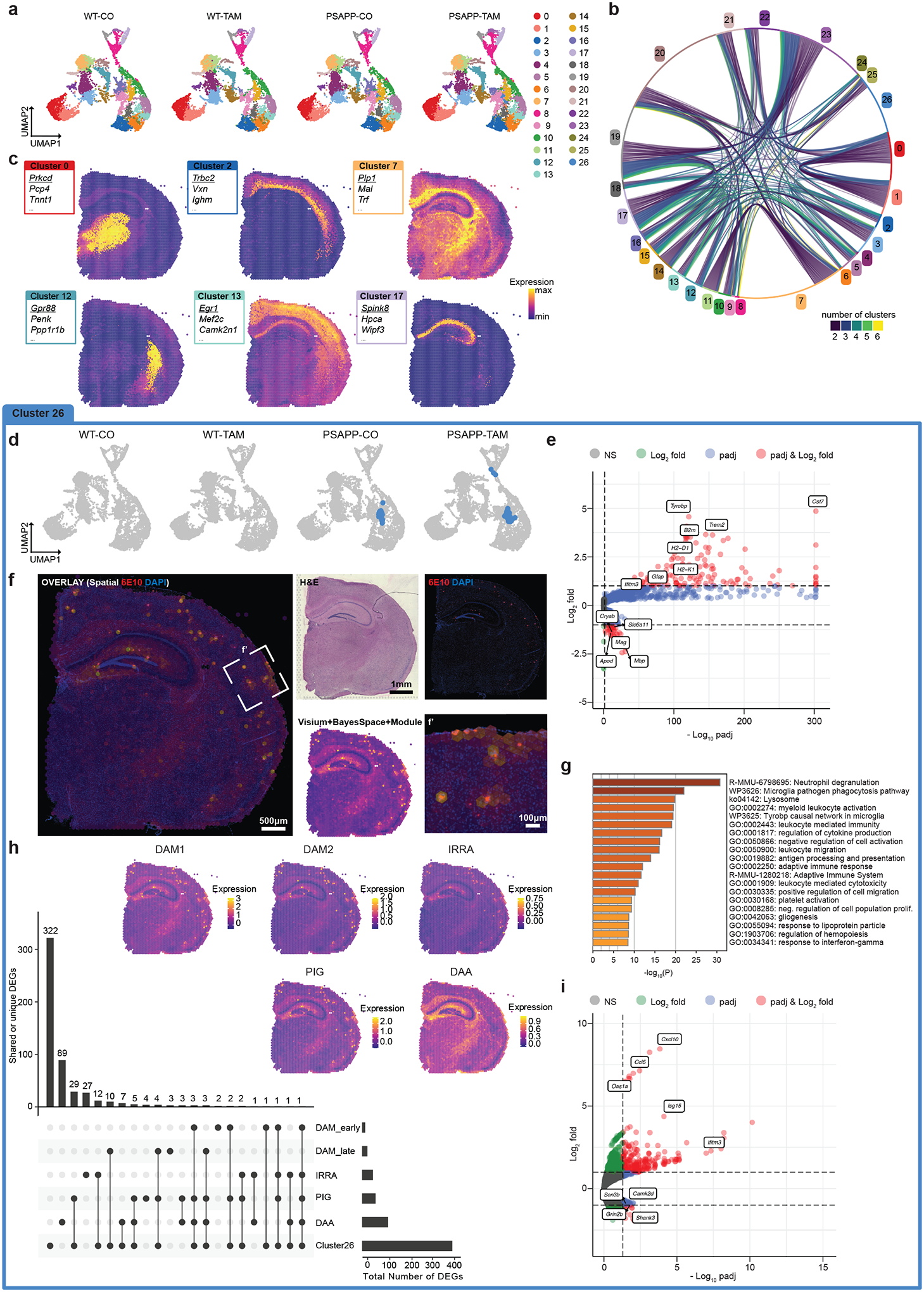
A, Uniform manifold approximation and projection (UMAP) dimensional reduction of wild-type (WT)-corn oil (CO) (8037 spots, 3 sections), WT-tamoxifen (TAM; 8882 spots, 3 sections), PSAPP/Inpp5dfl/fl/Cx3xr1CreER+ (PSAPP)-CO (7748 spots, 3 sections), and PSAPP-TAM (9366 spots, 3 sections). Each dot represents a Visium transcriptomics spot. The 27 color-coded clusters correspond to brain areas/regions (see below). B, Chord diagram of genes enriched > 1 log2 fold (l2f) in each cluster and adjusted P-value < 0.05. Each string represents a gene shared between two and six other clusters. C, Example clusters highlighted in a Visium brain section. Boxes show top three enriched genes per cluster. Underlined gene is highlighted in the SpatialFeaturePlot. D, UMAP with Cluster 26 highlighted showing that it is only present in PSAPP animals. E, Volcano plot of genes enriched in Cluster 26 as calculated with Seurat’s FindAllMarkers function. Red genes are l2f > 1 and padj < 0.05, blue l2f < 1 and padj < 0.05, green genes are l2f > 1 and padj > 0.05, gray genes are l2f < 1 and padj > 0.05. F, Module score of Cluster 26 and 6E10 (amyloid) staining of the adjacent brain section shows colocalization. f’ shows higher magnification of box in (F). G, Gene Ontology term analysis of genes enriched l2f > 1 in Cluster 26 using metascape.73 H, UpSet plot highlighting the overlapping and unique genes between previously defined reactive microglia and astrocyte subtypes: Disease-associated microglia, DAM1 (early) and DAM2 (late),25 interferon-responsive reactive astrocytes (IRRAs),28 plaque-induced genes (PIGs),27 as well as disease associated astrocytes (DAA).26 Insets are module scores of the genes in each reactive subtype highlighted on a PSAPP-TAM spatial transcriptomics brain section. For IRRAs, DAAs and Cluster 26, genes were filtered for padj < 0.05 and l2f > 0.5. For DAM1, DAM2, and PIGs, genes were selected based on gene sets provided in the original publication. I, Genes differentially expressed in Cluster 26 between APP-CO and APP-TAM as calculated with edgeR on sum of counts using muscat (red genes are l2f > 1 and padj < 0.05, blue l2f < 1 and padj < 0.05, green genes are l2f > 1 and padj > 0.05, gray genes are l2f < 1 and padj > 0.05).
We next asked whether Inpp5d knockdown altered peri-plaque gene expression patterns. We used muscat,29 a cluster-resolved, pseudobulk-based DEG analysis approach for multi-sample, multi-condition experiments. We used edgeR within muscat to calculate genes differentially expressed between PSAPP-CO and PSAPP-TAM brains. We found that the gene expression profile in Cluster 26 was extensively altered by the knockdown of Inpp5d (Figure 2I). Of the 231 genes induced in Cluster 26 upon knockdown of Inpp5d (padj < 0.05, l2f > 1), 126 were specific to PSAPP-TAM plaques. GO term analysis of these genes included an enrichment for “Defense response to virus” (Cxcl10, Ifit3, Oas1a, Ccl5, Oasl1), “Type I and II Interferon signaling” (Stat1, Ifit2, Irf5/6, Isg15), and “Chemokine signaling pathway” (Plcb2, Rac2, Ccl9/12,Txnip, H2-T23; Tables S6, S7 in supporting information). While we discovered most DEGs in Cluster 26 due to its plaque association (Figure 2F, Figures S7–S9, Tables S1–S5), muscat also uncovered DEGs outside Cluster 26 when comparing WT-TAM to PSAPP-TAM as well as PSAPP-CO to PSAPP-TAM. Given the recent evidence for Cre-mediated activation of microglia,30 we calculated genes that were changed in WT-CO versus WT-TAM mice and found that while tamoxifen injection alone drove a modest numbers of DEGs, they did not fall into categories associated with interferon signaling (Figure S10, Table S2). Last, we used NICHES, a recently developed package based on the fantom5 database,31 to interrogate which putative receptor–ligand interactions were enriched within Cluster 26 and therefore plaques. We confirmed that NICHES reliably detected receptor–ligand pairs along anatomical regions. In Cluster 26, we observed receptor–ligand pairs primarily involved in phagocytosis and lysosome activity, including (ligand – receptor): C1qb – Lrp1, C3 – Lrp1, Timp1 – Cd63, Pros1 – Axl, and Ly86 – Cd180 (Figure S11, Table S8 in supporting information).
2.3 |. Cluster 26 and Inpp5d knockdown signatures in PSAPP intersected human AD gene networks
To expand our NICHES receptor–ligand interaction spatial transcriptomic maps, we used network analysis on cluster-specific DEG lists. Leveraging high-throughput molecular profiling techniques, we generated a cohort of matched whole-genome sequencing (WGS) and RNA-seq data across four brain regions, from a set of 364 well-characterized AD and control brains in the Mount Sinai Brain Bank cohort.32 Using Bayesian probabilistic causal network (BN) analysis,33–35 we built integrative network models to organize genome-wide gene expression features into regulatory networks.32 When projecting the Cluster 26 expression signature and cluster-specific DEG list onto the network neighborhood of INPP5D in the parahippocampal gyrus region BN, we found that both were significantly enriched in the subnetwork within a path length of 6 of INPP5D (up to 16-fold enrichment, adjusted P-value = 3.1E-30; Table S9 in supporting information; Figure 3A). DAM genes like Gpnmb, Trem2, and Tyrobp were among the DEGs present in the network neighborhood of INPP5D. Figure 3A displays the expression changes in human AD brain and in Inpp5d knockdown mice within the subnetwork of up to four steps from INPP5D. In this subnetwork, half of the genes (59/119) were detected in the mouse study, among which 18 were significantly regulated by Inpp5d (19.9-fold, adjusted P-value = 3.3E-18). We therefore argue that Cluster 26 as well as Inpp5d knockdown signatures in PSAPP mice mirrored those observed in human AD gene networks. Indeed, when probing for human AD nodes from the INPP5D network, we found that these were regulated by the knockdown of Inpp5d and co-localized with the plaque-associated Cluster 26 (Figure 3B, Figure S12 in supporting information).
FIGURE 3. Top Cluster 26 differentially expressed gene Cst7 was a plaque-specific marker and was exacerbated by Inpp5d knockdown.
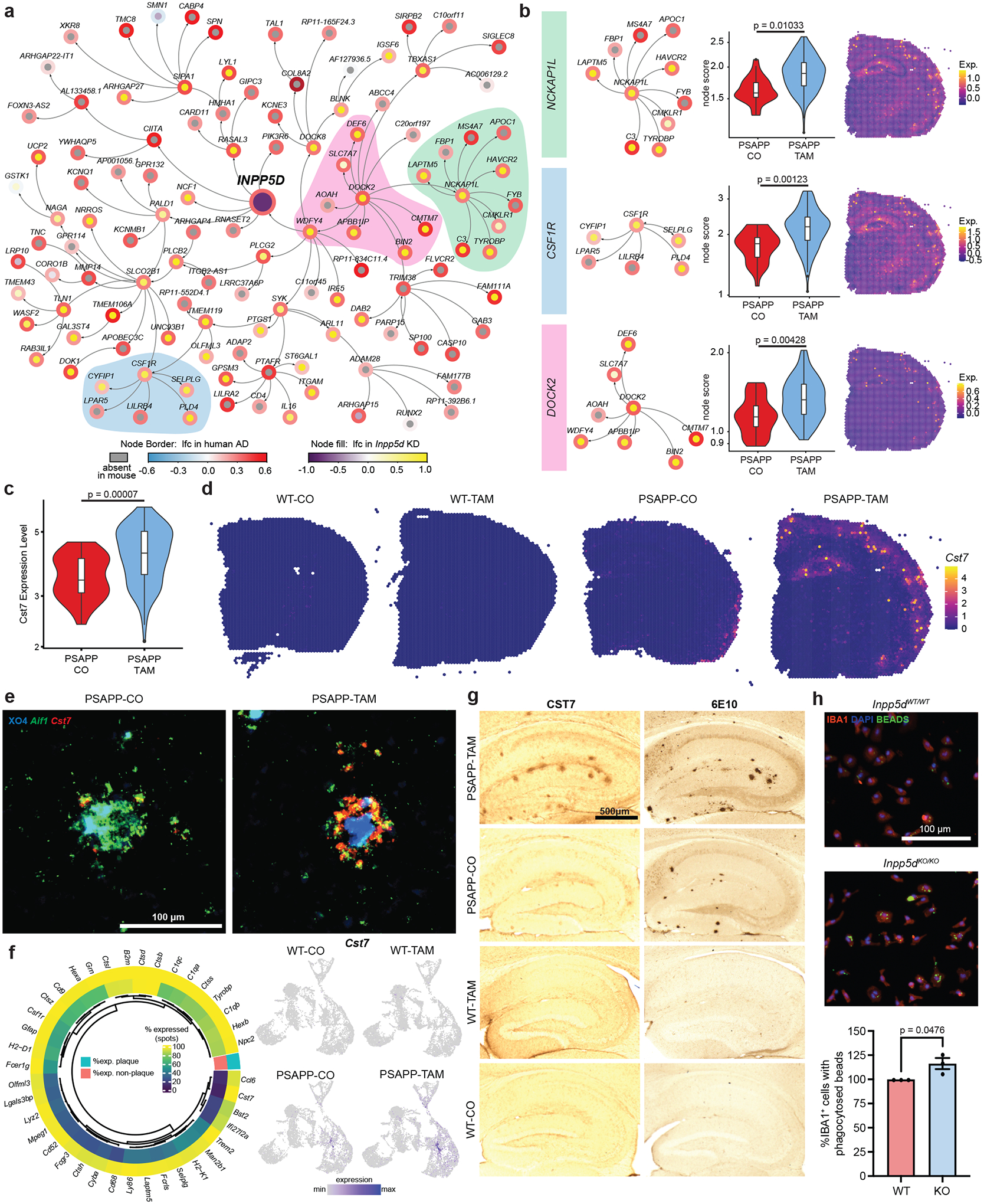
A, Network showing the gene expression change in human Alzheimer’s disease (AD) and Inpp5d knockdown mice within the neighborhood of four steps of INPP5D. Node border paint denotes the log2 fold-change (lfc) in human AD brains compared to control brains. Node fill paint denotes the log2 fold-change caused by Inpp5d in mice, with gray color denoting absence in mouse data. B, Highlight of specific nodes from network in (A). Genes associated with each node were enriched in cluster 26 from PSAPP/Inpp5dfl/fl/Cx3xr1CreER+ (PSAPP)-tamoxifen (TAM) compared to PSAPP-corn oil (CO) mice (violin plots, middle, P-values from unpaired t test) and can be spatially resolved by probing for this module in spatial transcriptomic brain sections (right). C, Violin plots of Cst7 in Cluster 26 across PSAPP-CO and PSAPP-TAM shows increased expression in PSAPP-TAM plaque spots as calculated with edgeR on sum of counts using muscat. D, Cluster 26 marker gene Cst7 highlighted in wild-type (WT)-CO, WT-TAM, PSAPP-CO, and PSAPP-TAM brain sections. E, RNAScope of Cst7 (red) and Aif1 (green) with Methoxy-XO4 (blue) staining in PSAPP-CO and PSAPP-TAM mouse cortex. Scale bar = 100 μm. F, Left: Percentage expression of the top Cluster 26 enriched genes in Cluster 26 (outer ring) versus the average percentage expression in all other clusters (inner ring). Right: FeaturePlots showing expression of Cst7 in all spots across WT-CO, WT-TAM, PSAPP-CO, and PSAPP-TAM brain sections. G, CST7 antibody staining detected with DAB and 6E10 in adjacent section from WT-CO, WT-TAM, PSAPP-CO, and PSAPP-TAM hippocampus. Scale bar = 500 μm. H, Representative images of latex bead phagocytosis assay on Inpp5d-WT mouse primary microglia and Inpp5d-knockout (KO) primary microglia with quantification of percentage IBA1+ cells with phagocytosed fluorescent bead (right). Scale bar = 100 μm. Quantification was performed from three to four wells per group; with N = 3 independent experiments. Data points represent mean normalized to Inpp5d-WT per experiment. P-values calculated with unpaired t test.
2.4 |. Novel plaque marker Cst7 was limited to plaque-associated microglia
We next probed for the gene Cst7 (cystatin F), which is specific to Cluster 26 and which has previously been identified in DAMs and PIGs (Figures 2E,H, 3C–G).[25,27] In humans, CST7 is expressed in endothelial cells in addition to microglia and macrophages.36 In our current PSAPP mice cohorts, Cst7 remained upregulated in both CO- and TAM-treated mice, suggesting that CST7 might be an excellent marker of plaques. To localize Cst7 to specific central nervous system cell types, we used RNAScope in situ hybridization (Figure 3E). Cst7 co-localized with Aif1+ microglia and with Aβ plaques in PSAPP, but not in plaque-less WT animals. This co-localization was enabled by the spatial transcriptomic analysis and would not have been detected if we had performed only bulk analysis of all microglia. Further analysis of the spatial transcriptomics data also confirmed the relative specificity of Cluster 26 genes as putative plaque markers, showing that Cst7 was expressed in 100% of plaque spots and only 8% of non-plaque spots (Figure 3F). Cst7 therefore outperformed both Trem2 as well as Tyrobp as plaque marker, both of which were highly enriched around plaques but also showed a broad expression in non-plaque tissue. We confirmed the specificity of Cst7 using immunohistochemistry and show that the CST7 protein was exclusively present around plaques in PSAPP brains (Figure 3G), leading us to propose that it is a novel marker for Aβ plaques. Previous studies associated altered Cst7 expression with microglial dysfunction leading to impairment of Aβ clearance,37 and upregulation of Cst7 in Aβ-containing microglia (but not Aβ-deficient microglia) from the 5xFAD mouse model of AD.38 These data imply that decrease of SHIP1 function after initiation of AD amyloid pathology leads to an increase in plaque burden and plaque-associated microglial gene expression.
To further assess this in light of our data suggesting an increase in plaque deposition (Figure 1E–F), we performed a phagocytosis assay in vitro to compare microglia from Inpp5d-WT to microglia from Inpp5d-KO mice. We found that phagocytosis was increased in microglia that lacked Inpp5d (Figure 3H), in line with previous reports in the literature.12 We hypothesize that under chronic stimulation from Aβ in the mouse model, microglia that lack Inpp5d undergo functional impairment in association with Aβ plaque deposition as was described previously,39 resulting in more plaques.
3 |. DISCUSSION
Here we report that conditional Inpp5d downregulation in microglia of the adult PSAPP mouse increases plaque burden and recruitment of microglia to plaques. This result was not predicted by the apparent effect of the common INPP5D polymorphisms, given that the rs35349669 single nucleotide polymorphism (SNP) confers AD risk,7 while rs61068452 is apparently protective against AD.8 Notwithstanding, INPP5D plays a role in AD pathogenesis, and remains a potential target for microglia-focused AD therapies,15 but its role in brain and disease progression requires clarification.
There are multiple reasons to have predicted that knockdown of Inpp5d might lead to an improved or delayed amyloid phenotype in mice, including perhaps decreased plaque deposition. Although SHIP1 protein has been shown to be elevated in the 5xFAD mouse,15 protein level has not been assayed in human tissue. Moreover, microglia with decreased Inpp5d or in the presence of a SHIP1 inhibitor, demonstrated increased phagocytosis.12,13 SHIP1 also blocked TREM2/TYROBP signal transduction, implying that Inpp5d knockdown should increase activity of this pathway. Finally, Inpp5d was identified as a target of miR155, and constitutive deletion of miR155, which should increase SHIP1, led to an increase in amyloid plaques.40,41 However, we observed increased plaque deposition and compaction. The conundrum of predicting the outcome in our study is similar to consideration of TREM2 action for which gain of function is predicted to be beneficial,42 yet opposite effects can be seen in a “Jekyll and Hyde” context-dependent scenario.43 Thus, SHIP1 inhibition increased phagocytosis in vitro while INPP5D expression has been reported to increase in AD patients as plaque load increases, whereas, in our study, decreased Inpp5d levels were associated with an increase in plaques. Further complicating matters, haploinsufficiency and total deficiency of TREM2 can lead to opposite effects,44 and differing outcomes have also been reported with microglial depletion.45,46 Notably, the timing of gene and/or total microglial manipulation is also critical,47 and as we noted, we deliberately used conditional knockdown early in “disease” stage to mimic what might be therapeutically viable.
Detrimental outcomes after loss of INPP5D inhibition are not entirely unexpected taken in the context of TREM2/TYROBP signaling. This pathway regulates the switch from a homeostatic to a neurodegenerative molecular signature by inducing miR155 via apolipoprotein E (APOE) signaling,48 and TYROBP has also been shown to regulate APOE signaling independent of TREM2.49 Support for beneficial outcomes associated with maintenance and/or restoration of homeostatic functions of microglia comes from the numerous studies targeting the members of this pathway, including Trem2,48,50–52 Tyrobp,53,54 and miR155.41,55 However, as we have previously seen in our studies modulating Tyrobp expression, manipulation of molecules involved in immune/microglial function in either direction may result in detrimental or exacerbated pathological outcomes.49,53,54,56 A similar interpretation could be developed from the studies of Trem2 deficiency, as differences were dependent on early versus later disease stages.50,51,57,58 It remains to be determined whether the increased plaque burden and alterations in microglia phagocytosis described herein in PSAPP mice after Inpp5d knockdown also occur in other mouse models of AD (e.g., 5xFAD), or at different stages during the progression of disease pathology.
Notably, western blots of hippocampal extracts from female mice from each comparison group at age 6 months were probed with anti-PSD95 and anti-synaptophysin antibodies. Despite the increase in amyloid burden, the increase in plaque-associated microglia, and the appearance of DAM genes (including C1q), there was no evidence for destruction of hippocampal synapses as determined by the tissue content of these synaptic markers in mice after induction of the Inpp5d knockout. However, epitomic characterization of these same extracts revealed a ≈50% increase in A11/6E10 ratio (i.e., ratio of prefibrillar oligomer content to total Aβ content). This observation suggests that Inpp5d knockdown may alter the conformational state of Aβ in such a way as to increase plaque density while apparently reducing synaptic toxicity. Methylene blue59 and peroxisome proliferator-activated receptor gamma agonists60 have been reported to have similar conformation-altering, toxicity-modifying effects.
All the same factors altered by Trem2 manipulation are impacted by Inpp5d knockdown. Thus, it becomes very difficult to predict what will happen in vivo in the face of incipient plaque deposition, as assayed in this report, and which changes are beneficial or detrimental. Increased plaque compaction may be a protective mechanism,59,60 and there are many examples of environmental and/or microglial gene manipulation in mouse models of amyloidosis in which cognition improved in the absence of a decrease, or even in the presence of an increase, in plaques.61,62 This may have therapeutic implications, in that inhibition of SHIP1 once disease is initiated might not lead to a clinical benefit. Further, increased association of microglia with plaques reduces neuritic dystrophy.63 The existence of DEGs in regions without plaques after the knockdown of Inpp5d indicates that (at least within the time period of this study) these transcriptomic changes do not always result in plaque deposition.
We identified a plaque-specific gene expression profile, Cluster 26, which is extensively altered by the knockdown of Inpp5d, moving cells in this region toward a higher inflammatory profile. The latter results and that of our functional phagocytosis assay showing increased bead phagocytosis by constitutive Inpp5d KO primary microglia were consistent with previous results,12 and with a role for SHIP1 as a negative regulator of immune signaling.
Increased microglial phagocytosis could be predictive of a beneficial reduction in plaque burden, which is the opposite of the pathological outcomes we found in vivo. Notably, previous studies have shown that microglial function may be adversely affected by Aβ, particularly as a result of chronic exposure to pro-inflammatory cytokines, resulting in increased Aβ accumulation and potentially underpinning the contradiction between our in vitro and in vivo results.39,40 Indeed, our Inpp5d knockdown exacerbated the plaque-induced Cluster 26 profile, which overlaps with previously reported reactive glial substates, including DEGs from both early and late DAM profiles,25 and our previously identified IRRAs.28 Further studies will be required to elucidate the mechanisms underlying Inpp5d-mediated phagocytosis and plaque deposition changes in the context of AD.
There are several important caveats to consider in the interpretation of our results. SHIP1 is under investigation as a potential therapeutic target for treatment of AD, with exploration of both inhibitors and agonists. Recent work showed that a SHIP1 agonist can enhance phagocytic degradation of synaptosomes and dead neurons by microglia.64 The focus of our investigation described herein was on the inhibition of SHIP1, with the expectation that this would boost microglial functions (e.g., phagocytosis of amyloid) with the goal of reducing plaque burden: but in fact, plaque burden increased. These seemingly conflicting data emphasize the complexity in identifying an effective SHIP1-targeted therapeutic strategy that is underpinned by our results in vivo. Although our findings have multiple therapeutic implications, a caveat is our inability to associate this altered pathology with any detectable changes in brain function, which may be a result of our focus on the earliest stages of pathology and/or a dissociation between plaque load and function, which has been observed in other settings.21,60,62 Assessment of the impact of intervening with SHIP1 modulation at a range of disease stages will be critical in determining whether boosting or inhibiting SHIP1 activity is most likely to lead to a meaningful clinical benefit in patients at high risk for, or already suffering from, the clinical dementia of AD. Translating results from mouse models to human disease has been notoriously difficult, especially for AD, which has seen multiple failures of potential drugs and therapies once at the clinical trial stage. Further validation, such as with inducible CRISPR interference against INPP5D in induced pluripotent stem cell–derived microglia, could be a promising approach.65
Nonetheless, we show here that manipulating microglial SHIP1 has a profound impact on the amyloidosis and neuroinflammation of AD. This, combined with the GWAS-associated SNPs in INPP5D, highlights microglial function and/or dysfunction as a central contributor to early AD pathology. Ultimately, further research into the disease-stage–specific functional consequences of manipulating SHIP1 activity is required to determine whether SHIP1 is a viable therapeutic target to prevent or treat the clinical dementia associated with AD. Importantly, our spatial transcriptomics analysis highlights an extended DEG signature associated with plaques and identifies Cst7 as a potentially highly specific marker of plaques in the AD brain. We also took advantage of cross-species gene regulatory network analysis to highlight crossover of the INPP5D/SHIP1 pathway in human AD, as some aspects of plaque pathology can be species-specific. The network analysis and extensive spatial transcriptomics data should prove helpful for other groups seeking to localize key genes of interest in the PSAPP mouse brain for their own research.
4 |. METHODS
4.1 |. Animals
All animal procedures were conducted in accordance with the National Institutes of Health Guidelines for Animal Research and were approved by the Institutional Animal Care and Use Committee (IACUC) at the Icahn School of Medicine at Mount Sinai. Mice were housed on a 12 hour light/dark cycle and had access to food and water ad libitum. APPKM670/671NL x PSEN1Δexon9 (PSAPP), B6.129S6-Inpp5dtm1Wgk/J (Inpp5dfl/fl; stock no.: 028255), B6.129P2(Cg)-Cx3cr1tm2.1(cre/ERT2)Litt/WganJ (Cx3cr1CreER/CreER; stock no.: 021160), and B6.C-Tg(CMV-cre)1Cgn/J (CMV-Cre; stock no.: 006054) were purchased from The Jackson Laboratory. The inducible Cx3cr1CreER/CreER mice were crossed with mice carrying loxP-flanked Inpp5d to obtain Inpp5dfl/fl/Cx3cr1CreER/+ mice,66,67 in which TAM-induced Cre expression results in chronic recombination in microglia, but not in transient myeloid cells. These Inpp5dfl/fl/Cx3cr1CreER+ mice were crossed with PSAPP mice to obtain PSAPP/Inpp5dfl/fl/Cx3cr1CreER/+ and their WT, Inpp5dfl/fl/Cx3cr1CreER/+ littermates.53 Constitutive Inpp5d KO mice were generated by crossing Inpp5dfl/fl animals to a germline Cre strain, CMV-Cre, then backcrossed to remove the CMV-Cre to obtain only Inpp5dKO/WT mice. All mice were on the C57BL/6J background and experimental groups consisted of both sexes, unless otherwise noted. Spatial transcriptomic analysis was completed on brains from females.
4.2 |. Tamoxifen preparation and treatment
Tamoxifen was dissolved in CO at a concentration of 20 mg/ml by shaking overnight at 37°C. For induction of Cre recombinase, 3-month-old male and female mice received daily intraperitoneal (i.p.) injections of either TAM at 75 mg/kg body weight or CO for 5 days.
4.3 |. Tissue processing
Mice were anesthetized at 6 months of age with pentobarbital (30 mg/kg, i.p.) and transcardially perfused with 20 ml of ice-cold 0.1 M phosphate-buffered saline (PBS), pH 7.4. One hemisphere was immersion-fixed for 48 hours in 4% paraformaldehyde for immunohistochemistry analysis. The remaining hemisphere was dissected for isolation of hippocampus and cortex and snap-frozen for biochemical analysis.
4.4 |. Immunohistochemistry
Fixed hemibrains were sectioned sagittally using a vibratome (Leica) at 30 μm. Free-floating brain sections stained with 6E10 anti-Aβ antibody were pre-treated with 70% formic acid for 15 minutes. Sections were blocked for 1 hour in PBS containing 0.2% (v/v) Triton-X and 5% normal goat (or donkey) serum (NGS), then incubated overnight at 4°C in primary antibody in PBS-0.2% Triton-X with 1% NGS. The tissues were subsequently washed in PBS-0.2% Triton-X, then incubated in PBS with 1% NGS and Alexafluor-conjugated secondary antibodies for 2 hours. Sections were then counterstained as necessary and mounted onto slides. For methoxy-XO4 staining, mounted sections were dried at room temperature, then incubated sequentially (5 minutes per incubation) in PBS, 40% ethanol (EtOH), 1 μM methoxy-XO4 (Cayman Chemical) in 40% EtOH, and then in 90% EtOH before coverslipping. To perform 3,3’-diaminobenzidine (DAB) staining for CST7, sections were washed briefly in PBS, then underwent antigen retrieval using 1x Reveal Decloaker (Biocare Medical) at 80°C for 15 minutes before being allowed to cool for 5 minutes. Sections were washed again in PBS before quenching with 3% H2O2, followed by permeabilization with PBS-0.2% Triton-X. Sections were blocked with 5% NGS in PBS-0.2% Triton-X for 1 hour, then incubated overnight at 4°C in primary antibody in PBS-0.2% Triton-X with 1% NGS. The tissues were subsequently washed in PBS-0.2% Triton-X, then incubated in PBS with 1% NGS and biotinylated secondary antibody. Sections were washed in PBS, then the signal was amplified using the ABC system (VECTASTAIN Elite ABC kit, Vector Laboratories), as per the manufacturer’s instructions. The signal was then developed with a DAB Reagent (KPL), as per the manufacturer’s instructions. Sections were then mounted, dried, and dehydrated before coverslipping and imaging. Adjacent sections were used to perform 6E10 immunostaining with DAB detection, with the sections quenched as before, then treated with 70% formic acid for 15 minutes. All steps were then performed as listed previously, starting from the permeabilization step. The tissues were stained with the following primary antibodies: rabbit anti-IBA1 (1:500; Cat. # 019-19741, Wako), mouse 6E10 anti-Aβ (1:1000; Cat. #9320-500, Covance), chicken anti-GFAP (1:1000; NBP1-05198), and mouse anti-Cst7 (1:200; antibody F010; a gift from Dr. Adriano Aguzzi). Secondary antibodies included: goat anti-mouse immunoglobulin (IgG) AlexaFluor 594 or 350 (1:1000; Invitrogen), goat anti-rabbit IgG AlexaFluor 488 (1:1000; Invitrogen), goat anti-chicken 594 (1:1000; Invitrogen), and biotinylated goat anti-mouse (1:1000, Vector Laboratories). Images were acquired on a Biotek Cytation 5 Multimode Reader (Agilent) with the same gains and exposures across all animals for each stain. Automated quantification of areas and cell counts was performed using the Biotek Gen5 software (Agilent) by creating an object mask with thresholds set for intensity and object size. For total microglia counts, adjustments were performed for plaque-associated microglia based on 4’,6-diamidino-2-phenylindole (DAPI) number. Confocal images were acquired using a Confocal Zeiss LSM 510. Images were converted to Z Projects in Fiji (National Institutes of Health) based on sum intensity. Plaque-associated IBA1+ microglia were manually counted from Z Projects based on contact with a 6E10+ immunoreactive deposit.
4.5 |. Western blot
Protein was extracted from cortical tissue by homogenization in RIPA buffer (Thermo Fisher Scientific) containing phosphatase/protease inhibitor cocktail (Thermo Fisher Scientific) and centrifuged at 15,000 × g for 20 minutes at 4°C before collecting the supernatant. A 50 μg aliquot of total protein was separated by electrophoresis in precast 4% to 12% Bis-Tris Gels (Bio-Rad) and transferred to nitrocellulose membranes. Membranes were blocked in 5% milk before probing with the following antibodies: rabbit anti-SHIP1 (1:1000, Cell Signaling, D1163), rabbit anti-β-actin (1:2500, Sigma, A-2066), mouse anti-PSD95 (clone 6G6-1C9, 1:2500, EMD Millipore, MAB1596), and rabbit anti-synaptophysin (1:1000, Abcam, ab16659). The secondary antibody used was peroxidase-labelled anti-rabbit IgG (1:5000, Vector Laboratories). SuperSignal West Femto ECL (Pierce, #34096) was used to reveal the immunoreactive proteins, and images were acquired using a Fujifilm ImageReader LAS-4000. Luminescent immunoreactive protein bands were quantified using Fiji software (ImageJ).
4.6 |. Spatial transcriptomics
Spatial transcriptomics analysis was performed using the 10xGenomics’ Visium platform following the manufacturer’s instructions, as we have reported.28 In short, fresh frozen brain sections from PSAPP/Inpp5dfl/fl/Cx3cr1CreER/+ and Inpp5dfl/fl/Cx3cr1CreER/+ mice either treated with CO or TAM were cryosectioned coronally to 10 μm and mounted on Visium capture areas (n = 3 for each condition, 12 sections total). After methanol fixation at −20°C for 30 minutes, sections were stained with hematoxylin and eosin, imaged on a Keyence BZ-X710 using a 20x objective, and tiled using BZ-X710 software. Sections were then enzymatically permeabilized for 12 minutes, and spatial barcodes and unique molecular identifiers were added to the captured polyA mRNA. The 12 Illumina libraries were generated using Dual Index primers and sequenced on a NovaSeq6000 using a S4 200 cycle flow cell v.1.5. The sequencing output was aligned to the mouse reference genome mm10 using SpaceRanger.
The following libraries were used to analyze and visualize the data in R (4.1.0): Seurat (4.0.6), SingleCellExperiment (1.16.0), BayesSpace (1.2.1),68 EnhancedVolcano (1.10.0), ComplexHeatmap (2.10.), viridis (0.6.2), muscat (1.5.2),29 ggplot2 (3.3.5), dplyr (1.0.7), purrr (0.3.4), scater (1.22.0), UpSetR (1.4.0), ggraph (2.0.5), igraph (1.2.11), gtools (3.9.2), cowplot (1.1.1), ggpubr (0.4.0), NICHES (0.0.2),31 CIDER (0.99.0). SCTransform was applied to each sample separately before merging. For all downstream clustering analyses, variable features from all 12 samples were used. Thirty principal components and a resolution of 0.8 were used to cluster the spots across all samples. Seurat’s FindAllMarker function was used to calculate enriched genes in each cluster and muscat was used to calculate cluster-resolved differentially expressed genes across conditions using edgeR on the sum of counts per cluster. BayesSpace68 was used to identify sub–spot level gene expression. NICHES was used to calculate ligand–receptor pair enrichment using predefined clusters and the fantom5 database.31 Seurat’s AddModuleScore was used to visualize groups of genes in Visium brain sections.
4.7 |. Network analysis
To verify if the present mouse model gene signature mirrored that in the human AD gene network, we used the BN we previously constructed from the parahippocampal gyrus region of the Mount Sinai Brain Bank AD cohort.32,69 The cluster signatures and cluster-specific DEGs from the spatial transcriptomics analysis were intersected with the network neighborhood within a path length of up to six steps from INPP5D in the human AD BN. P value significance of the intersections at different path lengths was computed by hypergeometric test in R (v4.0.5). A subnetwork up to four steps from INPP5D was visualized by Cytoscape (v3.9.0).
4.8 |. RNA in situ hybridization
RNA in situ hybridization (ISH) was performed using the RNAscope Multiplex Fluorescent kit (ACD) as before with slight modification.49 The same hemibrains collected for spatial transcriptomics were sectioned at 10 μm on a cryostat and mounted on glass slides. Slides were fixed in 4% paraformaldehyde for 15 minutes at 4°C. After fixation, sections were dehydrated and stained with methoxy-XO4 by immersion in 50% EtOH for 2 minutes, 50% 5 μM methoxy-XO4 (Cayman Chemical) in 50% EtOH for 2 minutes, then 50% EtOH for 1 minute, before proceeding through 70% EtOH and two rounds of 100% EtOH for 5 minutes each. The mounted sections were quenched with H2O2 for 10 minutes then protease-permeabilized for 20 minutes at room temperature using reagents provided in the kit (Protease IV). Brain sections were then incubated for 2 hours at 40°C with probes followed by signal amplification steps. When the ISH assay was complete, slides were incubated with fluorophore-conjugated secondary antibody for 30 minutes, then counterstained with DAPI. The following probes were used: Cst7-C2 (Cat. #498711-C2) and Aif1-C4 (Cat. #319141-C4).
4.9 |. Aβ assays
Tissue was processed via serial detergent fractionation with ultracentrifugation to produce TBS-soluble, Triton-X–soluble, and formic-acid-soluble fractions as described previously.54 In brief, tissue was homogenized in ice-cold TBS containing protease/phosphatase inhibitor cocktail, then ultracentrifuged at 100,000 × g for 1 hour at 4°C. The supernatant containing the TBS-soluble fraction was collected, and the pellet was subsequently homogenized in Triton-X solution (TBS with 1% [v/v] Triton-X-100) containing protease/phosphatase inhibitor cocktail and ultracentrifuged as before. The supernatant containing the Triton-X–soluble fraction was collected, and the pellet was homogenized in 70% formic acid before the ultracentrifugation was repeated. The resulting formic acid fraction was neutralized with 1 M Tris buffer. To quantify Aβ levels, human/rat Aβ 1-40 and 1-42 ELISA kits (Wako, Cat. #294-64701 and #290-62601) were used according to the manufacturer’s instructions. Absolute concentrations of Aβ were normalized to initial tissue weight.
For analysis of native oligomeric Aβ protein structure, 2 μl protein samples from the TBS-soluble fraction were spotted onto activated PVDF membrane (0.22 μm; Millipore). Membranes were incubated with either rabbit pAb A11 (anti-prefibrillar oligomers; 0.5 μg/ml; gift from Charles Glabe), rabbit pAb OC (anti-fibrillar oligomers and fibrils; 0.25 μg/ml; gift from Charles Glabe), or mouse mAb NU-4 (anti-oligomers; 1 μg/ml; gift from William Klein, Northwestern University). Generation, purification, and characterization of A11, OC, and NU-4 have been described previously.70,71 Membranes were incubated with appropriate anti-mouse or -rabbit horseradish peroxidase—conjugated secondary antibody (1:20,000; Vector Laboratories) and developed as described in the western blot section. Normalization to total APP/Aβ signal was achieved by detection of human APP transgene metabolites with the anti-Aβ antibody 6E10 (1:1000, Covance).
4.10 |. Purification of primary mouse microglia and phagocytosis assay
Heterozygous Inpp5dKO/WT mice were crossed to obtain Inpp5dKO/KO (KO) and Inpp5dWT /WT mice for isolation of primary microglia. Primary microglia were isolated from cerebral cortices and hippocampi from postnatal day P0-P3 mice as described previously72 with slight modification. In brief, tissue was homogenized in ice-cold Hibernate A (BrainBits), then centrifuged at 300 × g for 5 minutes at 4°C. The pellet was resuspended in high-glucose Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% heat-inactive fetal bovine serum (FBS; Gibco), 2 mM glutamine, penicillin/streptomycin (100 U/ml and 0.1 mg/ml, respectively) and seeded in poly-L-lysine precoated flasks. Cells were maintained at 37°C and 5% CO2, during which media was changed twice a week. Microglia were collected after approximately 8 to 10 days by agitating the flasks on a orbital shaker at ~1 × g (180 rpm) for 30 minutes to detach microglial cells from the astrocytic monolayer.
Green fluorescent latex beads (Sigma #L1030) were used to assess phagocytosis in the primary microglia. Beads were pre-opsonized in heat inactivated FBS (Gibco) for 1 hour at 37°C before use.72 Cells were submitted to serum deprivation for 1 hour prior to addition of the beads, and then fluorescent latex beads were added to the media for an additional 1 or 4 hours. The final concentrations for beads and FBS in serum-free DMEM were 0.01% (v/v) and 0.05% (v/v), respectively. Cultures were then washed three times with PBS and fixed in 4% paraformaldehyde before cells were stained with anti-IBA1 and DAPI. Images were obtained using a Biotek Cytation 5 Multimode Reader (Agilent) to generate (at minimum) a 3 × 3 tiled montage of the well. Automated quantification of primary microglia number and dual labeling was performed using the Biotek Gen5 software (Agilent) by creating an object mask with thresholds set for intensity and object size for IBA1+ cells with a secondary mask to count the subpopulation of dual-labeled cells for their content of green fluorescent beads. Quantification was performed from three to four wells per group; data points represent experimental mean normalized to Inpp5d-WT with N = 3 independent experiments.
4.11 |. Human brain information and experiments
For human AD gene network analysis, we used the multi-Omics data we previously generated from a set of 364 well-characterized AD and control brains spanning the full spectrum of LOAD-related cognitive and neuropathological disease severities represented in the Mount Sinai Brain Bank AD cohort. Details about the cohort population information, generation of -Omics data (including WGS and RNA sequencing), and data preprocessing are described in Wang et al.69
4.12 |. Statistical analyses
All data, excluding spatial transcriptomics results, were analyzed with GraphPad Prism 9 and are presented as means ± standard error of the mean. Sample sizes and statistical tests are indicated in figure legends. Analyses used include one-way analyses of variance, and Student t test.
Supplementary Material
RESEARCH IN CONTEXT.
-
5.
Systematic review: The authors reviewed previous publications using traditional sources, including PubMed and preprint server databases. The mechanism of INPP5D/SHIP1 has been more frequently studied in the periphery. There are relatively few peer-reviewed publications regarding INPP5D/SHIP1 biology in the brain and/or within the context of Alzheimer’s disease (AD). We have cited relevant publications.
-
6.
Interpretation: Our results show that conditional Inpp5d downregulation increases plaque deposition, microglial recruitment to plaques, and plaque-induced gene expression during early stages in disease development in the APPKM670/671NL/PSEN1Δexon9 (PSAPP) mouse model of AD.
-
7.
Future directions: Our observation that specific knockdown of Inpp5d in the microglia of the PSAPP mouse model of cerebral amyloid beta amyloidosis causes complex changes in the neuropathological features associated with human AD should provide context for further investigation of its biology. These insights should help clarify role(s) for INPP5D in the brain and in AD that have diagnostic and/or therapeutic implications.
ACKNOWLEDGMENTS
The authors would like to thank Dr. Adriano Aguzzi for generously providing us with the anti-CST7 antibody F010. The authors were supported as follows: E.L.C. (P30 AG066514 to Mary Sano with Developmental Pilot Award), S.G. (U01AG046170, RF1AG058469, RF1AG059319, R01AG061894, P30 AG066514 to Mary Sano, and Cure Alzheimer’s Fund); M.E.E. (U01AG046170, RF1AG058469, RF1AG059319, R01AG061894, and Cure Alzheimer’s Fund); M.W. (U01AG046170, RF1AG057440); B.Z. (U01AG046170, RF1AG057440); C.G. (R56AG056507); S.A.L. (R01EY033353, NYU Grossman School of Medicine, the Blas Frangione Foundation, the Neurodegenerative Diseases Consortium from MD Anderson, Alzheimer’s Research UK, the Gifford Family Neuroimmune Consortium as part of the Cure Alzheimer’s Fund, the Alzheimer’s Association, the Alzheimer’s Disease Resource Center at NYU Langone Medical Center; he also acknowledges generous anonymous donors, and the financial support of Paul Slavik). P.H., R.D.K., and S.A.L. acknowledge the Genome Technology Center at NYU Langone Medical Center for sequence alignment and data pre-processing.
Footnotes
CONFLICTS OF INTEREST
SAL is a founder of AstronauTx Ltd; all other authors declare no competing interests.
SUPPORTING INFORMATION
Additional supporting information can be found online in the Supporting Information section at the end of this article.
Data availability
All sequencing data generated in association with this study are available on our searchable website portal https://liddelowlab.shinyapps.io/Hasel_Castranio_AD_Spatial/. Raw data are available in the Gene Expression Omnibus (GSE No. 203424).
REFERENCES
- 1.Zhang B, Gaiteri C, Bodea LG, et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell. 2013; 153:707–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jin SC, Benitez BA, Karch CM, et al. Coding variants in TREM2 increase risk for Alzheimer’s disease. Hum Mol Genet. 2014;23:5838–5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Efthymiou AG, Goate AM. Late onset Alzheimer’s disease genetics implicates microglial pathways in disease risk. Mol Neurodegener. 2017;12:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Y, Chen K, Sloan SA, Bennett ML, Scholze AR, O’Keeffe S, et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 2014;34:11929–11947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron. 2016;89:37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marsh SE, Walker AJ, Kamath T, et al. Dissection of artifactual and confounding glial signatures by single-cell sequencing of mouse and human brain. Nat Neurosci. 2022;25:306–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yoshino Y, Yamazaki K, Ozaki Y, et al. INPP5D mRNA expression and cognitive decline in Japanese Alzheimer’s disease subjects. J Alzheimers Dis. 2017;58:687–694. [DOI] [PubMed] [Google Scholar]
- 8.Yao X, Risacher SL, Nho K, Saykin AJ, Wang Z, Shen L. Targeted genetic analysis of cerebral blood flow imaging phenotypes implicates the INPP5D gene. Neurobiol Aging. 2019;81:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stenmark H, Aasland R, Driscoll PC. The phosphatidylinositol 3-phosphate-binding FYVE finger. FEBS Lett. 2002;513:77–84. [DOI] [PubMed] [Google Scholar]
- 10.Rohrschneider LR, Fuller JF, Wolf I, Liu Y, Lucas DM. Structure, function, and biology of SHIP proteins. Genes Dev. 2000;14:505–520. [PubMed] [Google Scholar]
- 11.Peng Q, Malhotra S, Torchia JA, Kerr WG, Coggeshall KM, Humphrey MB. TREM2- and DAP12-dependent activation of PI3K requires DAP10 and is inhibited by SHIP1. Sci Signal. 2010;3:ra38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pedicone C, Fernandes S, Dungan OM, et al. Pan-SHIP1/2 inhibitors promote microglia effector functions essential for CNS homeostasis. J Cell Sci. 2020:133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Horan KA, Watanabe K-i, Kong AM, et al. Regulation of FcγR-stimulated phagocytosis by the 72-kDa inositol polyphosphate 5-phosphatase: sHIP1, but not the 72-kDa 5-phosphatase, regulates complement receptor 3–mediated phagocytosis by differential recruitment of these 5-phosphatases to the phagocytic cup. Blood. 2007;110:4480–4491. [DOI] [PubMed] [Google Scholar]
- 14.Wendeln A-C, Degenhardt K, Kaurani L, et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature. 2018;556:332–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsai AP, Lin PB-C, Dong C, et al. INPP5D expression is associated with risk for Alzheimer’s disease and induced by plaque-associated microglia. Neurobiology of Disease. 2021;153:105303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Q, Sasaki T, Kozieradzki I, et al. SHIP is a negative regulator of growth factor receptor-mediated PKB/Akt activation and myeloid cell survival. Genes Dev. 1999;13:786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Helgason CD, Damen JE, Rosten P, et al. Targeted disruption of SHIP leads to hemopoietic perturbations, lung pathology, and a shortened life span. Genes Dev. 1998;12:1610–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fakhoury M. Microglia and astrocytes in Alzheimer’s disease: implications for therapy. Curr Neuropharmacol. 2018;16:508–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liddelow SA, Guttenplan KA, Clarke LE, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541:481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Manocha GD, Floden AM, Rausch K, Kulas JA, McGregor BA, Rojanathammanee L, et al. APP regulates microglial phenotype in a mouse model of Alzheimer’s disease. J Neurosci. 2016;36:8471–8486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Masliah E, Mallory M, Hansen L, DeTeresa R, Alford M, Terry R. Synaptic and neuritic alterations during the progression of Alzheimer’s disease. Neurosci Lett. 1994;174:67–72. [DOI] [PubMed] [Google Scholar]
- 22.Ullian EM, Christopherson KS, Barres BA. Role for glia in synaptogenesis. Glia. 2004;47:209–216. [DOI] [PubMed] [Google Scholar]
- 23.Reyes-Ruiz JM, Nakajima R, Baghallab I, et al. An “epitomic” analysis of the specificity of conformation-dependent, anti-Aß amyloid monoclonal antibodies. J Biol Chem. 2021;296:100168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li Y, Wang J, Sheng JG, et al. S100 beta increases levels of beta-amyloid precursor protein and its encoding mRNA in rat neuronal cultures. J Neurochem. 1998;71:1421–1428. [DOI] [PubMed] [Google Scholar]
- 25.Keren-Shaul H, Spinrad A, Weiner A, et al. A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 2017;169:1276–1290. [DOI] [PubMed] [Google Scholar]
- 26.Habib N, McCabe C, Medina S, et al. Disease-associated astrocytes in Alzheimer’s disease and aging. Nat Neurosci. 2020;23:701–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen WT, Lu A, Craessaerts K, Pavie B, Corthout N, et al. Spatial transcriptomics and in situ sequencing to study Alzheimer’s disease. Cell. 2020;182:976–991. [DOI] [PubMed] [Google Scholar]
- 28.Hasel P, Rose IVL, Sadick JS, Kim RD, Liddelow SA. Neuroinflammatory astrocyte subtypes in the mouse brain. Nat Neurosci. 2021;24:1475–1487. [DOI] [PubMed] [Google Scholar]
- 29.Crowell HL, Soneson C, Germain P-L, et al. muscat detects subpopulation-specific state transitions from multi-sample multi-condition single-cell transcriptomics data. Nature Communications. 2020;11:6077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sahasrabuddhe V, Ghosh HS. Cx3Cr1-Cre induction leads to microglial activation and IFN-1 signaling caused by DNA damage in early postnatal brain. Cell Rep. 2022;38:110252. [DOI] [PubMed] [Google Scholar]
- 31.Raredon MSB, Yang J, Kothapalli N, Kaminski N, Niklason LE, Kluger Y, Comprehensive visualization of cell-cell interactions in single-cell and spatial transcriptomics with NICHES. bioRxiv. 2022:2022.01.23.477401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang M, Li A, Sekiya M, Beckmann ND, Quan X, Schrode N, et al. Transformative network modeling of multi-omics data reveals detailed circuits, key regulators, and potential therapeutics for Alzheimer’s disease. Neuron. 2021;109:257–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu J, Wiener MC, Zhang C, Fridman A, Minch E, Lum PY, et al. Increasing the power to detect causal associations by combining genotypic and expression data in segregating populations. PLoS Comput Biol. 2007;3:e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhu J, Zhang B, Smith EN, Drees B, Brem RB, Kruglyak L, et al. Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nat Genet. 2008;40:854–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang IM, Zhang B, Yang X, Zhu J, Stepaniants S, Zhang C, et al. Systems analysis of eleven rodent disease models reveals an inflammatome signature and key drivers. Mol Syst Biol. 2012;8:594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nuvolone M, Schmid N, Miele G, Sorce S, Moos R, Schori C, et al. Cystatin F is a biomarker of prion pathogenesis in mice. PLoS One. 2017;12:e0171923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ofengeim D, Mazzitelli S, Ito Y, et al. RIPK1 mediates a disease-associated microglial response in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2017;114:E8788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grubman A, Choo XY, Chew G, et al. Transcriptional signature in microglia associated with Aβ plaque phagocytosis. Nat Commun. 2021;12:3015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krabbe G, Halle A, Matyash V, et al. Functional impairment of microglia coincides with Beta-amyloid deposition in mice with Alzheimer-like pathology. PLoS One. 2013;8:e60921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hickman SE, Allison EK. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci. 2008;28:8354–8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Readhead B, Haure-Mirande JV, Mastroeni D, et al. miR155 regulation of behavior, neuropathology, and cortical transcriptomics in Alzheimer’s disease. Acta Neuropathol. 2020;140:295–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morenas-Rodríguez E, Li Y, Nuscher B, et al. Soluble TREM2 in CSF and its association with other biomarkers and cognition in autosomal-dominant Alzheimer’s disease: a longitudinal observational study. The Lancet Neurology. 2022;21:329–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rueda-Carrasco J, Hong S. The Jekyll and Hyde of TREM2. Trends Neurosci. 2020;43:739–740. [DOI] [PubMed] [Google Scholar]
- 44.Sayed FA, Telpoukhovskaia M, Kodama L, et al. Differential effects of partial and complete loss of TREM2 on microglial injury response and tauopathy. Proc Natl Acad Sci U S A. 2018;115:10172–10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Du Y, Brennan FH, Popovich PG, Zhou M. Microglia maintain the normal structure and function of the hippocampal astrocyte network. Glia. 2022;70:1359–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tarale P, Alam MM. Colony-stimulating factor 1 receptor signaling in the central nervous system and the potential of its pharmacological inhibitors to halt the progression of neurological disorders. Inflammopharmacology. 2022;30:821–842. [DOI] [PubMed] [Google Scholar]
- 47.Schoch KM, Ezerskiy LA, Morhaus MM, et al. Acute Trem2 reduction triggers increased microglial phagocytosis, slowing amyloid deposition in mice. Proc Natl Acad Sci U S A. 2021;118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krasemann S, Madore C, Cialic R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. 2017;47:566–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Audrain M, Haure-Mirande JV, Mleczko J, et al. Reactive or transgenic increase in microglial TYROBP reveals a TREM2-independent TYROBP-APOE link in wild-type and Alzheimer’s-related mice. Alzheimers Dement. 2021;17:149–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ulrich JD, Finn MB, Wang Y, et al. Altered microglial response to Aβ plaques in APPPS1-21 mice heterozygous for TREM2. Mol Neurodegener. 2014;9:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Parhizkar S, Arzberger T, Brendel M, et al. Loss of TREM2 function increases amyloid seeding but reduces plaque-associated ApoE. Nat Neurosci. 2019;22:191–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jay TR, Miller CM, Cheng PJ, et al. TREM2 deficiency eliminates TREM2+ inflammatory macrophages and ameliorates pathology in Alzheimer’s disease mouse models. J Exp Med. 2015;212:287–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haure-Mirande JV, Audrain M, Fanutza T, et al. Deficiency of TYROBP, an adapter protein for TREM2 and CR3 receptors, is neuroprotective in a mouse model of early Alzheimer’s pathology. Acta Neuropathol. 2017;134:769–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Haure-Mirande JV, Wang M, Audrain M, et al. Integrative approach to sporadic Alzheimer’s disease: deficiency of TYROBP in cerebral Abeta amyloidosis mouse normalizes clinical phenotype and complement subnetwork molecular pathology without reducing Abeta burden. Mol Psychiatry. 2019;24:431–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Butovsky O, Jedrychowski MP, Cialic R, et al. Targeting miR-155 restores abnormal microglia and attenuates disease in SOD1 mice. Ann Neurol. 2015;77:75–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Audrain M, Haure-Mirande JV, Wang M, et al. Integrative approach to sporadic Alzheimer’s disease: deficiency of TYROBP in a tauopathy mouse model reduces C1q and normalizes clinical phenotype while increasing spread and state of phosphorylation of tau. Mol Psychiatry. 2019;24:1383–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meilandt WJ, Ngu H, Gogineni A, et al. Trem2 deletion reduces late-stage amyloid plaque accumulation, elevates the Aβ42:Aβ40 ratio, and exacerbates axonal dystrophy and dendritic spine loss in the PS2APP Alzheimer’s mouse model. J Neurosci. 2020;40:1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jay TR, Hirsch AM, Broihier ML, et al. Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer’s disease. J Neurosci. 2017;37:637–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Necula M, Breydo L, Milton S, et al. Methylene blue inhibits amyloid Abeta oligomerization by promoting fibrillization. Biochemistry. 2007;46:8850–8860. [DOI] [PubMed] [Google Scholar]
- 60.Blume T, Deussing M, Biechele G, et al. Chronic PPARγ stimulation shifts amyloidosis to higher fibrillarity but improves cognition. Front Aging Neurosci. 2022;14:854031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jankowsky JL, Melnikova T, Fadale DJ, et al. Environmental enrichment mitigates cognitive deficits in a mouse model of Alzheimer’s disease. J Neurosci. 2005;25:5217–5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Petrov AM, Lam M, Mast N, et al. CYP46A1 Activation by Efavirenz leads to behavioral improvement without significant changes in amyloid plaque load in the brain of 5XFAD mice. Neurotherapeutics. 2019;16:710–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yuan P, Condello C, Keene CD, et al. TREM2 Haplodeficiency in mice and humans impairs the microglia barrier function leading to decreased amyloid compaction and severe axonal dystrophy. Neuron. 2016;90:724–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pedicone C, Fernandes S, Matera A, et al. Discovery of a novel SHIP1 agonist that promotes degradation of lipid-laden phagocytic cargo by microglia. iScience. 2022;25:104170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dräger NM, Sattler SM, Huang CT-L, et al. A CRISPRi/a platform in human iPSC-derived microglia uncovers regulators of disease states. Nature Neurosci. 2022;25:1149–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ayata P, Badimon A, Strasburger HJ, et al. Epigenetic regulation of brain region-specific microglia clearance activity. Nat Neurosci. 2018;21:1049–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wang JW, Howson JM, Ghansah T, et al. Influence of SHIP on the NK repertoire and allogeneic bone marrow transplantation. Science. 2002;295:2094–2097. [DOI] [PubMed] [Google Scholar]
- 68.Zhao E, Stone MR, Ren X, et al. Spatial transcriptomics at subspot resolution with BayesSpace. Nat Biotechnol. 2021;39:1375–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang M, Beckmann ND, Roussos P, et al. The Mount Sinai cohort of large-scale genomic, transcriptomic and proteomic data in Alzheimer’s disease. Sci Data. 2018;5:180185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kayed R, Head E, Thompson JL, et al. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. [DOI] [PubMed] [Google Scholar]
- 71.Lambert MP, Velasco PT, Chang L, et al. Monoclonal antibodies that target pathological assemblies of Abeta. J Neurochem. 2007;100:23–35. [DOI] [PubMed] [Google Scholar]
- 72.El Gaamouch F, Audrain M, Lin WJ, et al. VGF-derived peptide TLQP-21 modulates microglial function through C3aR1 signaling pathways and reduces neuropathology in 5xFAD mice. Mol Neurodegener. 2020;15:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou Y, Zhou B, Pache L, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Commun. 2019;10:1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequencing data generated in association with this study are available on our searchable website portal https://liddelowlab.shinyapps.io/Hasel_Castranio_AD_Spatial/. Raw data are available in the Gene Expression Omnibus (GSE No. 203424).