

Abstract

In the current work, a novel vanadotungstate compound, (C6H9N2)4[V2W4O19]·2H2O (1), is isolated by a simple stepwise synthesis method and characterized by a combined experimental and computational study. Molecular docking is conducted for the first time for this kind of substituted Lindqvist polyoxometalates to elucidate for potential antidiabetic activity. Hence, the modeling results revealed a significant docking score of the reported compound to bind to the active sites of α-glucosidase with the lowest binding energy of −5.7 kcal/mol, where the standard drug acarbose (ACB) had −4.6 kcal/mol binding energy. The stability of binding was enhanced by strong H-bonding, van der Waals, and electrostatic interactions occurring in the three-dimensional (3D) supramolecular network of polyanionic vanadotungstate subunits templated with organic moieties as shown by X-ray diffraction and Hirshfeld analyses. Furthermore, density functional theory (DFT) calculations supported with photophysical measurements are also discussed to predict the most chemical and biological reactivity. In this view, the complete description of electronic and biological features of (1) is enhanced by determination of the highest occupied molecular orbital (HOMO)/least unoccupied molecular orbital (LUMO) energy, electronic density, ionization potential, electron affinity, etc. These chemical descriptors, intermolecular interactions, docking score, and binding free energy estimation are essential in understanding the reactivity of this bioactive compound offering potential inhibition of the α-glucosidase enzyme.
Short abstract
Combined experimental and computational studies of the newly synthesized Lindqvist-type vanadotungstate are reported. Interestingly, screening by molecular docking model and structure analysis well predicted a potent inhibitory activity against the α-glucosidase enzyme, thus providing evidence for the therapeutic antidiabetic potential of the reported compound against diabetes mellitus. Hopefully, Lindqvist-type polyoxometalate compounds can be promoting alternatives to standard diabetes treatments and may offer new therapeutic findings.
Introduction
Throughout the world, millions of people suffer from chronic metabolic illness, diabetes. More than 90% of the diabetes mellitus cases are type 2, which are principally due to ineffective insulin action. Up to now, the unique well-known class of antidiabetic drugs are α-glucosidase inhibitors, which are approved for the treatment of type 2 diabetes mellitus.1,2 Acarbose (ACB), miglitol, and voglibose constitute the three available α-glucosidase inhibitors, of which ACB is the widely used agent according to previous studies. Nevertheless, it is reported that ACB can be the cause of some gastrointestinal adverse effects such as flatulence, which constitute a limiting factor for prolonged therapy.1,2
For all these reasons, the discovery and development of new antidiabetic drugs are an important area of research. In recent years, polyoxometalates (POMs) have attracted significant attention due to their diverse biological activities, including potential antidiabetic properties.3,4 However, the underlying mechanisms of their biological activity are not yet fully understood.
Recent studies have shown that POMs exhibit various biological activities, including antimicrobial, antiviral, antitumor, and antioxidant properties. These activities are attributed to their unique structures, redox properties, and metal–ligand interactions. Additionally, POMs have been reported to exhibit hypoglycemic and antidiabetic activities, making them promising candidates for the development of novel antidiabetic agents.5 For instance, it has been reported that POMs can inhibit α-glucosidase, an enzyme involved in the breakdown of complex carbohydrates, resulting in reduced blood glucose levels.6 Additionally, POMs have been shown to activate the insulin signaling pathway and stimulate glucose uptake in cells.5,7
Furthermore, molecular docking studies have demonstrated that POMs can interact with key targets involved in glucose metabolism, such as glucokinase and peroxisome proliferator-activated receptor-γ (PPARγ). Glucokinase is an enzyme that regulates glucose uptake in the liver, while PPARγ is a nuclear receptor involved in glucose homeostasis and insulin sensitivity.6,8
Despite these promising findings, the potential clinical applications of POMs as antidiabetic agents have yet to be fully explored. Therefore, further research is needed to elucidate the molecular mechanisms underlying their antidiabetic activities and to optimize their therapeutic potential.6,9
On the other hand, the famous subclass of substituted Lindqvist-type HPOMs, V-substituted polyoxotungstate [VxWyO19]n−-based materials, is widely explored due to their potential physical and biological applications such as in electrical and optical areas9,10 as a potent inhibitor of HIV-1 and promising antioxidant agent due to their enhanced reducing powers.11
Nowadays, the synergistic effect of mixed metal facilitates the close surfaces to powerfully connect with other species, especially, transition metal complexes and organic cations, being potent materials with a set of scientific patent and functional claims in optical, electrical, and biological fields,12−23 namely, two wide-band gap semiconductors (H2pip)2[V2W4O19]·4H2O (Eg = 3.92 eV) and (H2pipEtOH)2[V2W4O19]·4H2O (Eg = 3.72 eV), which are previously reported and specified by their promising antibacterial and antifungal activities as well as their higher dielectric constants.24
Herein, we focus on the correlation between the experimental and theoretical studies of a novel disubstituted vanadotungstate based on 2-amino-6-methylpyridinium cations. For this reason, X-ray diffraction and spectroscopic measurements are combined with density functional theory (DFT) to enhance with Hirshfeld surface analysis and its structural, optical, and electrical properties. Additionally, to understand more the electrical behavior and the chemical reactivity of the entitled compound, the molecular electrostatic potential and the frontier molecular orbitals with their energy gaps have been also calculated. For the first time, we will also perform molecular docking studies to investigate the binding modes of Lindqvist-POMs with key targets involved in glucose metabolism, such as α-glycosidase. Molecular docking is a computational technique that predicts the binding mode and affinity of ligands with protein targets.25 Our study is a testament to the potential of these techniques in drug discovery and development, particularly in the investigation of complex molecular systems such as mixed-metal Lindqvist-type vandotungstates, which can be challenging to study experimentally.
Methods and Materials
Synthesis of (1)
For the preparation of the divanadotetratungstate salt, 0.18 g of V2O5 (1 mmol) was added with stirring to an aqueous solution (30 mL) of Na2WO4·2H2O (1.32 g, 4 mmol) previously heated. Then, 2 mL of concentrated acetic acid is added to the mixture to completely dissolve the insoluble reagents. Then, an alcoholic solution (10 mL) of 2-amino-6-methylpyridin (2 mmol) is added to the previous mixture.
The obtained mixture was kept under continuous stirring for 3 h. After about two weeks of slow evaporation at room temperature, yellowish single crystals with high quality were recovered (yield: 65% based on W). Elemental analysis confirmed its stoichiometry (Anal. Found: C, 17.84; H, 2.42; N, 6.25; V, 6.28; W, 45.47 wt %. Calc: C, 17.84; H, 2.48; N, 6.94; V, 6.31; W, 45.56 wt %). (IR, KBr, cm–1): 3352 (m), 3311 (m), 3171 (m) 3085 (s), 1635 (s), 1470 (w), 1392 (w), 1313 (w), 1305 (w), 1291 (w),1175 (w), 1039 (w), 990 (vs), 970 (vs), 939 (vs), 764 (s),579 (m), 532 (m), 432 (w) (Figure S3).
Materials and Physical Measurements
All reagents used in the synthesis of (1) were commercially purchased (Sigma Aldrich Chimie S.a.r. St. Quentin Fallavier Cedex 38297 France) and employed without further purification. The CHN analyses were recorded on a Perkin–Elmer 2400 CHN elemental analyzer, and V and W were analyzed on ICP-AES, inductively coupled plasma spectrometry. The IR spectra were obtained on a Perkin–Elmer Spectrum BXII spectrometer using a sample dispersed in a spectroscopically pure KBr pellet in the 400–4000 cm–1 region. UV—vis spectra of (1) and 2-amino-6-methylpyridin were carried out in the solid state on a Perkin Elmer Lambda 19 spectrophotometer in the 200–800 nm range. A Perkin Elmer LS55 spectrofluorometer was used to perform the photoluminescence analysis in the solid state at room temperature of the synthesized compound. The measurements of electrical conductivity were made with an HP 4192A impedance analyzer. The measurements covered a wide range of frequencies (100–107 Hz) and temperatures (315–375 K).
The single-crystal X-ray diffraction data collected with a Super Nova Atlas diffractometer (MoK = 0.71073) were used to determine the crystal structure of (1). The structure was solved and refined by using software SHELXS-9724 and SHELXL-9726 in WINGX27 based on F2 using the complete matrix least squares approach. All non-hydrogen atoms were refined isotropically and then anisotropically. All hydrogen atoms were placed geometrically and treated as riding in geometrically optimized positions. Water H atoms were refined using restraints [O–H = 0.85 (1) A, H···H = 1.44 (2) A and Uiso(H) = 1.5 Ueq(O)]. Full crystallographic data have been deposited with the Cambridge Crystallographic Data Centre (CCDC2149574).
Computational Details
The Kohn–Sham density functional theory technique for many quantum mechanics was used for optimization to examine the electronic structural features of (1). All of the investigated adducts were optimized utilizing the RB3LYP functional at the LANL2DZ basis set level of theory using GaussView 6.0.1628 and Gaussian 16.29 The strongest force and maximum displacement were compared to threshold values of 0.00045 Hartree/Bohr and 0.0018 Bohr, respectively, to achieve SCF consistency. Molecular-phase calculations reveal the lack of imaginary frequency, suggesting that the surfaces have attained their minimum potential energy. GausView was utilized to examine the geometry of the compound. To undertake NCI and molecular electrostatic potential (MEP) calculations, visual molecular dynamics (VMD),30 a molecular graphics program, and Multiwfn software31 were utilized.
Molecular Docking Procedures
The receptors used for molecular docking were downloaded from the online Protein Data Bank and prepared using Discovery Studio.32,33 The selected protein was prepared by first specifying the reactive pockets, exterminating residual water molecules and introducing explicit hydrogen atoms. The protein cavity was specified with X, Y, and Z coordinates of 23.056, 70.770, and −19.665, respectively, with the corresponding size of 30.02 Å, after which the format was converted to the pdbqt format using Autodock Vina.34,35 The selected ligand and standard α-glucosidase inhibitor agent (acarbose) were prepared by carrying out energy reduction at the DFT/RB3LYP/Lanl2Dz level of theory. The selected α-glucosidase receptor (3wyl) was then docked together with the aforementioned organic compounds. The result obtained from the docking experiment was visualized using Discovery Studio as well as PyMol.36 It was observed that the scoring function on AutoDock is primarily based on the presence of hydrogen bonds, electrostatic interactions, van der Waals interactions, and change in entropy as the ligand interacts with the protein at the estimated active site predicted. The grid parameter file (GPT) was aligned to 20 Å × 20 Å × 20 Å, 20 Å × 20 Å × 20 Å, and 20 Å × 20 Å × 20 Å within the X, Y, and Z planes.
Results and Discussion
X-ray and Hirshfeld Surface Analyses of (1)
The X-ray diffraction analysis reveals that compound (1) was crystallized in the orthorhombic system with the Fddd space group (Table 1).
Table 1. Crystallographic Data and Refinement Parameters of Compound (1).
| CCDC deposition No | CCDC2149574 |
|---|---|
| formula | (C6H9N2)4[V2W4O19]·2H2O |
| crystal color | yellow |
| FW | 1613.92 |
| crystal system | orthorhombic |
| space group | Fddd |
| a (Å) | 14.8173 (2) |
| b (Å) | 22.8032 (3) |
| c (Å) | 23.4497 (3) Å |
| V (Å3) | 7923.23 (18) |
| Z | 8 |
| Dx (Mg m–3) | 2.706 |
| crystal size (mm) | 0.19 × 0.15 × 0.11 |
| F (000) | 6000 |
| absorption coefficient (mm–1) | 12.11 |
| radiation (A °) | λ = 0.71073 |
| theta min–max (°) | 37.8° – 2.5° |
| diffractometer | superNova, atlas |
| index ranges | –25/h/25; –39/k/ 39; –39/l/40 |
| observed data [I > 2.0 sigma(I)] | 4334 |
| reflections independent, Rint | 5242 (Rint = 0.093) |
| R, wR(F), S | 0.026, 0.064, 1.22 |
| minimum and maximum resd. dens. (e/Å3) | –1.15;1.46 |
The asymmetric unit in this structure consists of one water molecule, one organic cation [C6H9N2]+, and 1/3 of the Lindqvist anion [V2W4O19]4– (Figure 1).
Figure 1.

ORTEP diagram of (1) (thermal displacement ellipsoids drawn at 50% probability; M = W/V).
Notably, the two vanadium cations are disordered among all the metallic sites in the whole Lindqvist anion. Each metal site is statistically occupied by 2/3 W and 1/3 V, similarly to the same materials previously described.12,16,18,23,24 The well-known polyoxidoanion [V2W4O19]4– shaped by six distorted octahedra sharing edges is isostructural with the [M6O19]2– isopolyanion.23,24 The geometric characteristics of the two MO6 octahedra of the asymmetric unit are grouped in Table S1.
Examination of the M–O distances revealed that the shortest distances are those that bind the metal ion to the terminal oxygen atoms with distances of 1.678 (4) and 1.650 (6) Å, whereas those of type M–Oc are considered the longest, ranging from 2.2943 (2) to 2.3093 (3) Å. The bonds involving the metal ion and bridging oxygen M–Ob have average distances of 1.910 (3)–1.934 (3) Å. For bond angles of O–M–O, the values range from 77.03 (11) to 179.09 (13)° (Table S1). It should be noted that these parameters are close comparable to those of the previously reported for the same Lindqvist-type clusters.23,24 The diversity of the distances highlights a distorted octahedral geometry with metal displaced from the equatorial plane of the MO6 octahedra toward the terminal oxygen atoms with distortion index (DI) values of 0.317 and 0.340, respectively, for M1O6 and M2O6. These values are in good agreement with those found for our reported compounds based on the [V2W4O19]4– polyanion, as mentioned in Table S2.
With regard to the packing diagram of the structure (Figure 2a), it exhibits that each [V2W4O19]4– cluster and two water molecules are strongly H-bonded via O1W–H1W1···O1 with O···O distances equal to 2.21(2) Å (Table S3) giving rise to [V2W4O19(H2O)2]4– inorganic clusters (Figure 2b). It should be noted that the Hirshfeld surface analysis coupled with the 2D fingerprint37,38 well highlights that this hydrogen bond interaction type attributed the highest contributions to O···H/H···O intercontacts (51.3%). Furthermore, the [V2W4O19(H2O)2]4– subunit is encapsulated by neighboring 2-amino-6-méthylpyridinium moieties (Figure 2b) involving a set of N–H···O (N···O distances ranging between 1.96 and 2.34 Å) and C–H···O (C···O distances varying from 3.368 (6) to 3.495 (7) Å) interactions to generate a 3D grid-like framework (Figure 2a).
Figure 2.

(a) Packing diagram of (1). (b) View of the [V2W4O19]4– subunit surrounded with organic cations and water molecules.
In this incident, as observed in Figure 3a,b, the results of HS analysis expose a moderate contribution of H···H intercontacts (21.2%), whereas other much less contributions are detected for other intercontacts such as O···O, C···C, and W···H/H···W (Figure S1).
Figure 3.

(a) Hirshfeld surfaces mapped over dnorm (b) shape index and (c) 2D fingerprint plot and relative contribution of molecular intercontacts of (1).
Additionally, π–π stacking interactions are also established between the pyridine rings of organic cations with a distance of 3.445 Å separating the centroids. These weak van der Waals-type interactions are evidenced by the existence of the red triangles on the HS plotted with shape index (Figure 3c) to hence the stability of the 3D topology.
Optimized Structure
Generally, this is the first step in the theoretical study of a molecule. This operation aims to seek an appropriate model in order to predict an approximate structure of the molecular system and by distributing to the physicochemical properties by simple correlation between the theoretical results and those given by experience. The main interatomic distances and bond angles determined by theoretical calculation of the various entities involved in the structure are reported in Table S4. The optimized structure of (1), a representative POM, was obtained using density functional theory (DFT) and is illustrated in Figure 4. It is worth noting that there is a slight discrepancy between the theoretical and experimental values, which is likely attributed to the absence of intermolecular interactions in the optimized structure, as it is considered in the gaseous phase.
Figure 4.

Optimized structure of (1).
Molecular Electrostatic Potential Surfaces
The molecular electrostatic potential (MEP) of (1) was calculated by using the RB3LYP method and LANL2DZ basis set. In fact, this map is used to identify the electron donor and acceptor sites in addition to the hydrogen bond interactions. The MEP plot of (1) is shown in Figure 5, including surface and contour maps. The brown-red negative region was specially centered on the Lindqvist anion (Figure 5), indicating a minimum electrostatic potential with a minimum value of −8.543 × 10–2 kcal/mol, which makes this region open to electrophilic attacks.
Figure 5.

(a) Molecular electrostatic potential surface and (b) contour maps of (1).
Furthermore, the blue colors in the ESP map at its center around the organic cations point to the maximum positive region with a maximum value of 8.543 × 10–2 kcal/mol, susceptible to nucleophilic activity. It should be noted that the electrophilic site is observed with a dark-blue color at the level of the hydrogen atoms for organic moieties, whereas the nucleophilic site is located at the level of oxygen atoms belonging to [V2W4O19]4– anions and water molecules. Consequently, the most reactive sites for electrophilic attack will be the oxygen atoms and the hydrogen atoms will be considered nucleophilic attack agents in the compound.
Non-covalent interaction (NCI) analysis with reduced density gradient (RDG) is a newly developed approach based on the electron density ρ(r) and its first derivative that is applied nowadays to investigate non-covalent interactions39,40 using eq 1
| 1 |
It is used to identify and visualize the non-covalent interactions (NCI) such as van der Waals interactions, hydrogen bonds, and steric repulsion through introducing the charge density as a simple color coding process. The RDG versus sign (λ2)ρ(r) (the product sign of λ2 time’s density (ρ)) plot combined with the isosurface density graph of compound (1) is given in Figure 6. It reveals the existence of a strong steric effect observed inside the Lindqvist anion [V2W4O19]4– and the six-membered rings of 2-amino-6-methylpyridinium cations when the sign (λ2)ρ is positive (red), whereas the blue regions at very low density well indicate the presence of strong H-bonding interactions (O–H···O and N–H···O) and the green spikes refer to the van der Waals interactions. Notably, it is well concurred with the results of the Hirshfeld analysis, which previously illustrates the important contribution of the O···H/H···O contacts to stabilize the structure.
Figure 6.

(a) RDG and (b) isosurface density plots along with the color-filled scale bar for compound (1).
Determination of Global Reactivity Parameters
The electronic density plots of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) are depicted in Figure 7. The frontier orbital energies are considered very important parameters to study the electronic properties and the chemical stability of materials. It is clearly observed that the EHOMO density is localized on the organic and inorganic moieties, leading to electronic transfers between them, while the ELUMO is mostly located on [V2W4O19]4–, which is attributed to that the present vacant orbitals can accept electrons. In the orbits of compound (1), the energies of the highest occupied molecular orbital (EHOMO) and the lowest unoccupied molecular orbital (ELUMO) are – 6.3132 and −2.5138 eV, respectively.
Figure 7.

Diagram of the HOMO and LUMO of compound (1).
These specific calculated values are used to find out the global chemical reactivity parameters of (1), such as chemical hardness (η), electrophilicity power (ω), electronegativity (χ), and potential (μ) (Table 2).
Table 2. Calculated LUMO, HOMO, Energy Values, and Global Chemical Reactivity Parameters.
| parameters | values (eV) |
|---|---|
| ELUMO (eV) | –2.5138 |
| EHOMO (eV) | –6.3132 |
| energy gap ΔE = ELUMO – EHOMO (eV) | 3.7994 |
| electron affinity: A = −ELUMO | 2.5138 |
| ionization potential: IP = −EHOMO | 6.3132 |
electronegativity: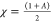
|
1.7569 |
chemical potential: 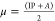
|
4.4135 |
chemical hardness: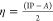
|
1.8997 |
chemical softness: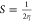
|
0.2632 |
electrophilicity index: 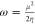
|
5.1268 |
As a result, the theoretical value of the energy gap ΔE (energy of electrons moved from the HOMO to the LUMO levels) is about 3.7994 eV, indicating a wide-band gap semiconductor behavior of the entitled compound. In addition, the electronegativity (χ) is one of the global characteristics of chemical reactivity using to forecast how and where materials will transfer electricity, energy, or heat. Moreover, the electrophilicity index value (ω = 5.1268 eV) indicates a relatively large electron flow between the donor and acceptor, establishing the energy stability when the system acquires additional electronic charge.
Density of States (DOS)
The density of states (DOS) is a concept in condensed matter physics and materials science that describes the number of electronic energy states per unit volume or per unit energy in a material. It is an important quantity for understanding the electronic properties of materials, including their conductivity, optical absorption, and thermal properties. For example, in semiconductors and insulators, the energy gap between the valence and conduction bands determines whether or not a material can conduct electricity. The DOS near the band edges can also be used to estimate the mobility of charge carriers in a material. In summary, the DOS is a fundamental quantity in condensed matter physics and materials science that provides insights into the electronic properties of materials, and it is widely used in the design and optimization of electronic and optoelectronic devices.41
Polyoxometalates (POMs) are complex molecular clusters composed of transition metal ions and oxide ions linked together by covalent bonds. The density of states (DOS) of polyoxometalates can be calculated using computational methods such as density functional theory (DFT) or ab initio methods. The DOS of POMs is typically dominated by the transition metal ions, which contribute to the electronic states near the Fermi level. The POMs have a unique electronic structure that results in a large number of electronic states, making them attractive for various applications including catalysis, sensing, and energy storage. The DOS of POMs has been studied in several recent computational studies, which have provided insights into their electronic properties. For example, it has been found that the DOS of POMs can be tuned by modifying the structure of the clusters or by introducing different types of metal ions. These studies have also demonstrated that the DOS of POMs can be used to predict their optical and redox properties. In summary, the DOS of polyoxometalates can be calculated using computational methods, and it provides insights into their electronic properties.42 Understanding the DOS of POMs is important for designing and optimizing their performance in various applications.
In this case, the density of states (DOS) versus energy plot of (1) shown in Figure 8 illustrates the presence of several energies corresponding to the isosurface distribution for the LUMO and HOMO critical number of electrons. The energy difference between the Fermi level and the previous peak can be used to calculate the activation energy of compound (1), which is estimated to be 0.5 eV, as exposed in Figure 8.
Figure 8.

Density of state (DOS) plot of the HOMO, LUMO, and energy gap of compound (1).
Molecular Docking Discussion
α-Glucosidase is a digestive enzyme that hydrolyzes the glycosidic bonds of oligosaccharides and disaccharides, thereby breaking them down into their respective monosaccharides.43 Thus, this stepwise biological reaction mechanism is essential in the absorption of carbohydrates in the small intestine of biological organisms. As such, in diabetic Homo sapiens, this process can be impaired, resulting in postprandial hyperglycemia.44 Thus, recent research focus has been on biologically generating α-glucosidase inhibitors as potential therapeutic agents for diabetes.
Furthermore, several previous studies reported that the oral administration of POMs is presumed safe with a low risk of negative health effects.4,45−50 For instance, polyoxotungstates show a potential antitumor activity and lower toxicity in vitro as well as in vivo experiments.4 Especially, vanadotungstates and polyoxovanadates have evidenced a good antitumor activity against some human cancer cells (MCF-7 human breast cells, HepG2 hepatocellular carcinoma cell, etc.) with low general cytotoxicity.45,46 In addition, they are considered inhibitors of SERCA and PMCA Ca2+-ATPase (50), as well as bioactive target agents for Alzheimer’s diabetes and infectious disease treatments.47−49
Herein, the efficacy of Lindqvist polyoxometalate as an efficient α-glucosidase inhibitor has been further examined via docking experiments to probe the binding efficiency, number of hydrogen bonds, and hydrophobic interactions formed, as this interaction would guide us in the chemistry of the proposed ligand.
The docking experiment result was compared with a standard α-glucosidase inhibitor used as a medication to treat type 2 diabetes mellitus. Acarbose (ACB) works by slowing the digestion as well as absorption of carbohydrates in the intestine of the host, thereby reducing the postprandial rise in blood glucose levels. The literature has shown that acarbose possesses a notably potential benefit; however, it is reported that ACB is one of the major causes of gastrointestinal disturbance, which limits its level of consumption.43,51 To this effect, the use of compound (1) was evaluated by docking the organic molecule against the 3wyl (α-glucosidase or diabetic receptor) biomolecule. The result obtained is compared with the standard drug (ACB) and reported in Table S5. It was observed that POM binds more effectively at the desired binding pockets identified using Molegro software, with a total binding score of −5.700 kcal/mol, whereas the standard ACB had a binding affinity of −4.600 kcal/mol. Interestingly, the number of hydrogen bond (H-bond) interactions depicted for (1) was considerably stronger than that of ACB biomolecules, respectively, as shown in Table S5 and Figure 9. As shown in Figure 9, (1) was observed to interact more closely with the 3wyl receptor than the conventional ACB drug. Nevertheless, for (1), a total of 7 H-bond intermolecular interactions were observed, while the ACB compound had a total number of 7 weak H-bonds formed as shown in Table S5 and Figure 9. The electronic property analysis from the previous section affirms that the obtained docking experimental result is in close agreement. More so, it was observed from Figure 9 that (1) formed a stronger hydrogen bond interaction inside the receptor active site, whereas acarbose or ACB had weak H-bond interactions outside the binding pocket of the protein molecule.
Figure 9.

3D plots of the docking results with the 3wyl receptor with (1) in comparison with acarbose (ACB) as well as hydrogen bond and SAS interaction for both proposed and conventional drugs.
In order to evaluate the propensity of penetration of (1) into the receptor cavity, solvent-accessible area analysis (SAS) was considered. Thus, as presented in Figure 9, it is conspicuous that (1) embedded more into the active site of the receptor protein than the ACB molecule, forming a strong intermolecular interaction at the acceptor cavity of the said biomolecule.
Optical Properties
The absorption spectra of (1) as well as the curves obtained by Gaussian-type deconvolution are given in Figure 10. It shows three absorption bands (Figure 10). The presence of these bands is proven by Gaussian deconvolution of the electronic spectrum, which highlights the presence of three bands with maxima located at 267, 320, and 380 nm.
Figure 10.

UV–visible spectra and energy gap determination of (1).
The first band around 267 nm corresponds to the π → π* and n → π* transitions, which clearly confirmed with the absorption band of pure 2-amino-6-methylpyridine, while the other two bands that appeared at 320 and 380 nm are characteristics of electronic transitions of the ligand metal charge transfer (LMCT) type within the polyanion,22−24 which result from charge transfer from oxo O2– ligands to metal ions (V+5 and W+6). Furthermore, the band gap (Eg) value was obtained according to the Tauc model.52 As depicted in Figure 10, the experimental band gap value is 3.72 eV, very close to the calculated one obtained from the electronic density plots of molecular orbitals.
The emission spectrum of (1) in the solid state upon excitation at 310 nm is recorded and given in Figure 11. Blue luminescence is detected by the presence of several emission bands located at 400, 417, and 461 nm. These emissions are referred to the π → π* and n → π* transitions observed within 2-amino-6-methylpyridinum cations in addition to ML (metal–ligand) charge transfer V/W → O, which have already been detected by UV–visible spectroscopy. These results are in agreement with the literature.22−24
Figure 11.

Solid-state emission spectrum of (1) (λexcitation = 310 nm).
Thermoelectrical Study
DSC Analysis
Differential scanning calorimetry (DSC) study of (1) was recorded by heating the sample from room temperature to 450 k at a rate of 5 k min–1. The DSC curve obtained is displayed in Figure S3. It illustrates that the melting process of (1) starts after 380 K. For this reason, we have fixed the temperature range between 315 and 375 K to undergo an electrical study of the synthesized material.
Impedance Analysis
The diagrams of Z’’ (the imaginary part of impedance) versus Z’ (the real part of impedance) of (1) at different temperatures from 315 to 375 K are shown in Figure 12.
Figure 12.

Nyquist plots of impedance data at different temperatures for the (1) simple.
The radius of the arc corresponding to the bulk resistance decreases as temperature increases, indicating an activated thermal conduction mechanism.53 In addition, the semicircles shift to a lower value of resistance, resulting in the enhancement of electrical conductivity at higher temperatures, signifying that the compound (1) follows the Cole–Cole law.54 The Nyquist plots can be ascribed to an equivalent circuit comprising a polarization resistance R and capacitor C in parallel combination,55 where the complex impedance is given by eqs 2 and 3(56)
| 2 |
| 3 |
where 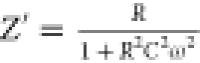 and
and 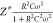
The diagrams of Z’ and Z’’ versus frequency at different temperatures are depicted in Figure 13. It is obvious that Z’ diminishes as the frequency and temperature rise, indicating an increase in the alternating current conductivity of the material. This behavior can be explained by an important contribution of the grain boundaries of the sample. With regard to the magnitude of Z’’, it is noted the appearance of the maximum peak (Z’’max), which moves to a higher frequency side with the increase in temperature, indicative of the relaxation phenomenon process.57
Figure 13.

(a) Frequency dependence of Z’ at several temperatures. (b) Plots of the imaginary part of impedance versus log(f) at various temperatures.
Conductivity Study
Figure 14 shows the temperature dependence of the conductivity in log (σT) versus 1000/T. The (σT) plot of the titled compound obeys the well-known Arrhenius law described by the expression σ.T = A exp(−Ea/KT), where A is the constant of the material, Ea is the activation energy, K is the Boltzmann constant, and T is the absolute temperature. The value of activation energy obtained from the linear fit is 0.51 eV, which is very close to the theoretical one obtained from the DOS plot.
Figure 14.

Variation of the ln(σT) versus 1000/T for (1).
Dielectric Study
The relative permittivity ε of a dielectric solid, located in an alternating electric field of angular frequency ε, is given by eq 4
| 4 |
where ε’ and ε’’ are the real and imaginary parts, respectively, which evaluate the response of these components to the external electric field. It was calculated according to eqs 5 and 6
| 5 |
| 6 |
The dependence of dielectric constants ε’ and ε’’ on frequency is shown in Figure 15. The diagrams of the real part of dielectric constant reveal that ε’ values fall sharply with increasing frequency. The high dielectric constants at low frequencies are attributed to space charge polarization.58,59 From Figure 14, it is observed that ε″ is also found to be decreasing with an increase in frequency. At high frequencies, ion vibration may be the only source of dielectric loss, which explains the low value of ε″.
Figure 15.

Frequency dependence of the real ε’ and imaginary ε’’ parts of the dielectric constant at various temperatures.
Conclusions
In this study, a novel V-substituted Lindqvist polyoxotungstate templated with 2-amino-6-methylpyridinium cations, (1), was characterized and qualified as a potential α-glucosidase inhibitor. X-ray diffraction combined with HS and RDG topological analyses reveals a 3D supramolecular network stabilized with a set of strong hydrogen bond interactions. The molecular electrostatic potential (MEP) map displays that negative sites in (1) are mostly located on the oxygen atoms of the Lindqvist anion. The energies of the frontier molecular orbitals are determined and used to calculate several chemical reactivity descriptors such as ionization potential, hardness, and electronic affinity. Also, the gap and the activation energy values calculated from the DOS plot are 3.7994 and 0.5 eV, respectively, very close to the experimental ones, confirming so the higher band gap semiconductor behavior of this material. For the first time, the molecular docking simulation well predicts the efficiency of V-substituted Lindqvist-type polyoxotungstate as an antidiabetic agent by comparing it with the standard ACB drug. In future, we aim to explore cytotoxicity assays and also further biomedical functions for this class of compounds in vitro as well in vivo.
Acknowledgments
The authors are extremely thankful to the Department of Chemistry, University of Carthage, Faculty of Sciences of Bizerte, for providing necessary facilities and to publish Open Access under the agreement with Ministry of Higher Education and Scientiifc Research-Tunisia.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.inorgchem.3c01651.
FT-IR and DSC analyses of (1); interatomic bond lengths and angles in (1); distortion index values for (1) and related compounds; hydrogen bond geometry; optimized geometric parameters; 2D fingerprint plot; ligand–protein complex, indicating the amino acid residues; and hydrogen bond interaction and binding affinity (PDF)
Author Contributions
All authors contributed to the study conception and design. Material preparation, spectroscopic measurements, data collection, DFT calculation, molecular docking, and analysis were performed by A.M., E.C.A., H.L., M.R., S.A., and G.E.M. The first draft of the article was written by A.M., and all authors commented on previous versions of the article. All authors read and approved the final article.
The authors declare no competing financial interest.
Supplementary Material
References
- Van de Laar F. A. Alpha-glucosidase inhibitors in the early treatment of type 2 diabetes. Vasc. Health Risk Manage. 2008, 4, 1189–1195. 10.2147/VHRM.S3119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adinortey C. A.; et al. Molecular Structure-Based Screening of the Constituents of Calotropis procera Identifies Potential Inhibitors of Diabetes Mellitus Target Alpha Glucosidase. Curr. Issues Mol. Biol. 2022, 44, 963–987. 10.3390/cimb44020064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M.; Chen X.; Chi G.; Shuai D.; Wang L.; Chen B.; Li J. Research progress on the inhibition of enzymes by polyoxometalates. Inorg. Chem. Front. 2020, 7, 4320–4332. 10.1039/D0QI00860E. [DOI] [Google Scholar]
- Bijelic A.; Aureliano M.; Rompel A. Polyoxometalates as Potential Next-Generation Metallodrugs in the Combat Against Cancer. Angew. Chem., Int. Ed. 2019, 58, 2980–2999. 10.1002/anie.201803868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aureliano M.; Gumerova N. I.; Sciortino G.; Garribba E.; McLauchlan C. C.; Rompel A.; Crans D. C. Polyoxidovanadates’ interactions with proteins: An overview. Coord. Chem. Rev. 2022, 454, 214344 10.1016/j.ccr.2021.214344. [DOI] [Google Scholar]
- Francesco A. P.Visioli, Prevention and Treatment of Atherosclerosis 2022, p 270. [DOI] [PubMed]
- De Sousa-Coelho A. L.; Aureliano Fraqueza M. G.; Gonçalves G. S. J.; Sanchez-Lombardo I.; Link W.; Ferreira B. I.; et al. Decavanadate and metformin-decavanadate effects in human melanoma cells. J. Inorg. Biochem. 2022, 235, 111915 10.1016/j.jinorgbio.2022.111915. [DOI] [PubMed] [Google Scholar]
- Benova A.; Ferencakova M.; Bardova K.; Funda J.; Prochazka J.; Spoutil F.; Cajka T.; Dzubanova M.; Balcaen T.; Kerckhofs G.; Willekens W.; van Lenthe G. H.; Alquicer G.; Pecinova A.; Mracek T.; Horakova O.; Rossmeisl M.; opecky J. K.; Tencerova M. Novel thiazolidinedione analog reduces a negative impact on bone and mesenchymal stem cell properties in obese mice compared to classical thiazolidinediones. Mol. Metab. 2022, 65, 101598 10.1016/j.molmet.2022.101598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong X.; Zhu W.; Wu Q.; Qian X.; Liu Z.; Yan W.; Gong J. Synthesis and conductivity of heptadecatungstovanadodiphosphoric heteropoly acid with Dawson structure. J. Alloys Compd. 2011, 509, 7768–7772. 10.1016/j.jallcom.2011.04.148. [DOI] [Google Scholar]
- Yu X.; Liu S.; Wang G.; Wang X.; Wang E. Studies on magnetism and conductibility of trisubstituted tungstogalliumate containing transition metal. Acta Chim. Sin. 1996, 54, 864–868. [Google Scholar]
- Hill C. L.; Weeks M. S.; Schinazi R. F. Anti-HIV-1 activity, toxicity, and stability studies of representative structural families of polyoxometalates. J. Med. Chem. 1990, 33, 2767–2772. 10.1021/jm00172a014. [DOI] [PubMed] [Google Scholar]
- Xu S.-S.; Chen W.-L.; Wang Y.-H.; Li Y.-G.; Liu Z.-J.; Shan C.-H.; Sua Z.-M.; Wang E.-B. Co-sensitization promoted light harvesting with a new mixed-addenda polyoxometalate [Cu(C12H8N2)2]2[V2W4O19].4H2O in dye-sensitized solar cells. Dalton Trans. 2015, 44, 18553–18562. 10.1039/C5DT02992A. [DOI] [PubMed] [Google Scholar]
- Hill C. L.; Delannoy L.; Duncan D. C.; Weinstock I. A.; Renneke R. F.; Reiner R. S.; Atalla R. H.; Han J. W.; Hillesheim D. A.; Cao R.; Anderson T. M.; Okun N. M.; Musaev D. G.; Geletii Y. Complex catalysts from self-repairing ensembles to highly reactive air-based oxidation systems. C. R. Chim. 2007, 10, 305–312. 10.1016/j.crci.2007.01.002. [DOI] [Google Scholar]
- Long D. L.; Tsunashima R.; Cronin L. Polyoxometalates: Building Blocks for Functional Nanoscale Systems. Angew. Chem., Int. Ed. 2010, 49, 1736–1758. 10.1002/anie.200902483. [DOI] [PubMed] [Google Scholar]
- Kaezer França M. C.; Eon J. G.; Fournier M.; Payen E.; Mentre O. (Nb2W4O19), TMA2, Na4(OH2)14(SO4): a new layered structure with Lindqvist heteropolyanions, XAS characterization of the HPAs. Solid State Sci. 2005, 7, 1533–1541. 10.1016/j.solidstatesciences.2005.07.006. [DOI] [Google Scholar]
- Son J. H.; Kwon Y. U. Crystal Engineering through Face Interactions between Tetrahedral and Octahedral Building Blocks: Crystal Structure of [ε-Al13O4(OH)24(H2O)12]2[V2W4O19]3(OH)2.27H2O. Inorg. Chem. 2004, 43, 1929–1932. 10.1021/ic035278h. [DOI] [PubMed] [Google Scholar]
- Huang W. L.; Todaro L.; Francesconi L. C.; Polenova T. 51V Magic Angle Spinning NMR Spectroscopy of Six-Coordinate Lindqvist Oxoanions: A Sensitive Probe for the Electronic Environment in Vanadium-Containing Polyoxometalates. Counterions Dictate the 51V Fine Structure Constants in Polyoxometalate. Solids J. Am. Chem. Soc. 2003, 125, 5928–5938. 10.1021/ja029246p. [DOI] [PubMed] [Google Scholar]
- Driss H.; Thouvenot R.; Debbabi M. Face-to-face interactions of metallic oxo-clusters: Synthesis and crystal structure of new isopolyvanadotungstate salts – Co(H2O)6K2V2W4O19 and [Co(H2O)6]2V2W4O19. Polyhedron 2008, 27, 2059–2064. 10.1016/j.poly.2008.03.020. [DOI] [Google Scholar]
- Wang X.; Zhou B.; Zhong C.; Ji M. Crystal structure and electrochemical behavior of a novel polyoxometalate [Ni(bpy)3]2[W4V2O19] with Lindqvist-type structure. Cryst. Res. Technol. 2006, 41, 874–879. 10.1002/crat.200510686. [DOI] [Google Scholar]
- Wang C.; Weng L.; Ren Y.; Du C.; Yue B.; Gu M.; He H. Mixed-Addenda Lindqvist-Type Polyoxoanion [V2W4O19]4- Supported Copper Complexes. Z. Anorg. Allg. Chem. 2011, 637, 472–477. 10.1002/zaac.201000332. [DOI] [Google Scholar]
- Maalaoui A.; Toumi S. A.; Rzaigui M. Bis(4-aminopyridinium) μ6-oxido-dodeca μ2-oxido-hexaoxido [rhenium VII) tetratungsten(VI)vanadium(V)]ate heptahydrate. Acta Crystallogr., Sect. E: Struct. Rep. Online 2013, 69, m661–m662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maalaoui A.; Hajsalem A.; Ramond N. R.; Akriche S. Synthesis, Characterization and Antibacterial Activity of a Novel Photoluminescent Nb-Substituted Lindqvist Polyoxotungstate Based Organic Cation. J. Clust. Sci. 2014, 25, 1525–1539. 10.1007/s10876-014-0749-6. [DOI] [Google Scholar]
- Maalaoui A.; Perez O.; Akriche S. T. Synthesis, Structural and Spectroscopic Portrayals of Tow Novel Biological and Photoluminescent Materials Based on Lindqvist and Keggin Heteropolyoxotungstates. Chem. Sci. Rev. Lett. 2014, 3, 603–607. [Google Scholar]
- Maalaoui A.; Perez O.; Rzaigui M.; Akriche S. T. Enhancement with Hirshfeld surface analysis of structural, electrical, dielectric and luminescent performance of two bioactive V-substituted polytungstates. J. Alloys Compd. 2017, 695, 1061–1072. 10.1016/j.jallcom.2016.10.231. [DOI] [Google Scholar]
- Agwupuye J. A.; Louis H.; Gber T. E.; Ahmad I.; Agwamba E. C.; Samuel A. B.; Ejiako E. J.; Patel H.; Ita I. T.; Bassey V. M. Molecular modeling and DFT Studies of Diazenylphenyl derivatives as a Potential HBV and HCV Antiviral Agents. Chem. Phys. Impact 2022, 5, 100122 10.1016/j.chphi.2022.100122. [DOI] [Google Scholar]
- Sheldrick G. M. A short history of SHELX. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Farrugia L. J.WINGX, University of Glasgow: Glasgow, 2005. [Google Scholar]
- Dennington R.; Keith T. A.; Millam J.. M.vGaussView 6.0; Semichem Inc.: Shawnee Mission, KS, 2016. [Google Scholar]
- Frisch G. E. S. M. J.; Trucks G. W.; H B Schlegel G.; Scuseria E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; Li X.; Caricato M.; Marenich A. V.; Bloino J.; Janesko B. G.; Gomperts R.; Mennucci B.; Hratchian H. P.; Ortiz J. V.; Izmaylov A. F.; Sonnenberg J. L.; Williams-Young D.; Ding F.; Lipparini F.; Egidi F.; Goings J.; Peng B.; Petrone A.; Henderson T.; Ranasinghe D.; Zakrzewski V. G.; Gao J.; Rega N.; Zheng G.; Liang W.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Throssell K.; Montgomery J. A.; Peralta J. E. Jr.; Ogliaro F.; Bearpark M. J.; Heyd J. J.; Brothers E. N.; Kudin K. N.; Staroverov V. N.; Keith T. A.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A. P.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Millam J. M.; Klene M.; Adamo C.; Cammi R.; Ochterski J. W.; Martin R. L.; Morokuma K.; Farkas O.; Foresman J. B.; Fox D. J.. Gaussian 16, (Revision B.01); Gaussian Inc: Wallingford CT, 2016. [Google Scholar]
- Humphrey W.; Dalke A.; Schulten K. VMD: visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Lu T.; Chen F. Multiwfn: a multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. 10.1002/jcc.22885. [DOI] [PubMed] [Google Scholar]
- Gao D. D.; Dou H. X.; Su H. X.; Zhang M. M.; Wang T.; Liu Q. F.; Cai H. Y.; Ding H. P.; Yang Z.; Zhu W. L.; Xu Y. C.; Wang H. Y.; Li Y. X. From hit to lead: Structure-based discovery of naphthalene-1-sulfonamide derivatives as potent and selective inhibitors of fatty acid binding protein 4. Eur. J. Med. Chem. 2018, 154, 44–59. 10.1016/j.ejmech.2018.05.007. [DOI] [PubMed] [Google Scholar]
- Dassault Systèmes . Biovia Discovery Studio 2016 Comprehensive Modeling and Simulations 2016, p 4.
- Trott O.; Olson A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardt J.; Santos-Martins D.; Tillack A. F.; Forli S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. 10.1021/acs.jcim.1c00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrodinger . PyMOL Molecular Graphics System; Schrondinger LLC, 2015https://pymol.org. [Google Scholar]
- McKinnon J. J.; Spackman M. A.; Mitchell A. S. Novel tools for visualizing and exploring intermolecular interactions in molecular crystals. Acta Crystallogr., Sect. B: Struct. Sci. 2004, 60, 627–668. 10.1107/S0108768104020300. [DOI] [PubMed] [Google Scholar]
- Wolff S. K.; Grimwood D. J.; McKinnon J. J.; Turner M. J.; Jayatilaka D.; Spackman M. A.. Crystal Explorer; University of Western Australia: Perth, Australia, 2013. [Google Scholar]
- Contreras-García J.; Boto R. A.; Izquierdo-Ruiz F.; Reva I.; Woller T.; Alonso M. A benchmark for the non-covalent interaction (NCI) index or··· is it really all in the geometry?. Theor. Chem. Acc. 2016, 135, 1–14. 10.1007/s00214-016-1977-7. [DOI] [Google Scholar]
- Saleh G.; Carlo G.; Lo Presti L.; Contreras-García J. Revealing Non-Covalent Interactions in Molecular Crystals through Their Experimental Electron Densities. Chem. – Eur. J. 2012, 18, 15523–15536. 10.1002/chem.201201290. [DOI] [PubMed] [Google Scholar]
- Chakroun R.; Jamoussi B.; Al-Mur B.; Timoumi A.; Essalah K. Impedance Spectroscopy and Dielectric Relaxation of Imidazole Substituted Palladium (II) Phthalocyanine (ImPdPc) for Organic Solar Cells. ACS Omega 2021, 6, 10655–10667. 10.1021/acsomega.1c00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillén-López A.; Espinosa-Torres N.; Cuentas-Gallegos A. K.; Robles M.; Muniz J. Understanding bond formation and its impact on the capacitive properties of SiW12 polyoxometalates adsorbed on functionalized Carbon Nanotubes. Carbon 2018, 130, 623–635. 10.1016/j.carbon.2018.01.043. [DOI] [Google Scholar]
- Hanefeld M.Treatment: Alpha Glucosidase Inhibitors, 2nd ed.; Elsevier Inc., 2018. [Google Scholar]
- Alpha-glucosidase Inhibitors. In Meyler’s Side Effects of Drugs, 16th ed.; Aronson J. K., Ed.; Elsevier: Oxford, 2016; pp 167–173. [Google Scholar]
- Liu J.; Jie-Fei W.; Qing H.; Ping S.; Lu-Lu L.; Li-Juan C.; Jun W. Z.; Carsten S.; Yu-Fei S. Multicomponent Self-Assembly of a Giant Heterometallic Polyoxotungstate Supercluster with Antitumor Activity. Angew. Chem. 2021, 133, 11253–11257. 10.1002/ange.202017318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijelic A.; Manuel A.; Annette R. The antibacterial activity of polyoxometalates: structures, antibiotic effects and future perspectives. Chem. Commun. 2018, 54, 1153–1169. 10.1039/C7CC07549A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinčić M.; Čolović M. B.; Sarić Matutinović M.; Ćetković M.; Kravić Stevović T.; Mougharbel A. S.; Todorović J.; Ignjatović S.; Radosavljević B.; Milisavljević M.; Kortz U.; Krstić D. Z. In vivo toxicity evaluation of two polyoxotungstates with potential antidiabetic activity using Wistar rats as a model system. RSC Adv. 2020, 10, 2846–2855. 10.1039/C9RA09790B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X.; Wei S.; Zhao C.; Li X.; Jin J.; Shi X.; Su Z.; Li J.; Wang J. Promising application of polyoxometalates in the treatment of cancer, infectious diseases and Alzheimer’s disease. J. Biol. Inorg. Chem. 2022, 27, 405–419. 10.1007/s00775-022-01942-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu X.; Xu K.; Zhao C.; Song X.; Li J.; Li L.; Nie W.; Bao H.; Wang J.; Niu F.; Li J. Genotoxicity and acute and subchronic toxicity studies of a bioactive polyoxometalate in Wistar rats. BMC Pharmacol. Toxicol. 2017, 18, 26. 10.1186/s40360-017-0133-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aureliano M.; Fraqueza G.; Berrocal M.; Cordoba-Granados J. J.; Gumerova N. I.; Rompel A.; Gutierrez-Merino C.; Mata A. M. Inhibition of SERCA and PMCA Ca2+-ATPase Activities by Polyoxotungstates. J. Inorg. Biochem. 2022, 236, 111952 10.1016/j.jinorgbio.2022.111952. [DOI] [PubMed] [Google Scholar]
- Bouali N.; Hammouda M. B.; Ahmad I.; Ghannay S.; Thouri A.; Dbeibia A.; Patel H.; Hamadou W. S.; Hosni K.; Snoussi M.; Adnan M.; Hassan M. I.; Noumi E.; Aouadi K.; Kadri A. Multifunctional Derivatives of Spiropyrrolidine Tethered Indeno-Quinoxaline Heterocyclic Hybrids as Potent Antimicrobial, Antioxidant and Antidiabetic Agents: Design, Synthesis, In Vitro and In Silico Approaches. Molecules. 2022, 27, 7248. 10.3390/molecules27217248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauc J. Optical properties and electronic structure of amorphous Ge and Si. Mater. Res. Bull. 1968, 3, 37–46. 10.1016/0025-5408(68)90023-8. [DOI] [Google Scholar]
- Parathan D. K.; Samantry B. K.; Chauthaey R. N.; Thakur A. K. Complex impedance studies on a layered perovskite ceramic oxide—NaNdTiO4. Mater. Sci. Eng. B 2005, 116, 7–13. 10.1016/j.mseb.2004.08.009. [DOI] [Google Scholar]
- Nadeem M.; Akhtar M. J.; Khan A. Y. Effects of low frequency near metal-insulator transition temperatures on polycrystalline La0.65Ca0.35Mn1–yFeyO3 (where y=0.05–0.10) ceramic oxides. Solid State Commun. 2005, 134, 431–436. 10.1016/j.ssc.2005.01.020. [DOI] [Google Scholar]
- Sinclair D. C.; West A. R. Impedance and modulus spectroscopy of semiconducting BaTiO3 showing positive temperature coefficient of resistance. J. Appl. Phys. 1989, 66, 3850–3856. 10.1063/1.344049. [DOI] [Google Scholar]
- Mohamed C. B.; Karoui K.; Jomni F.; Guidara K.; Rhaiem A. B. Electrical properties and conduction mechanism of [C2H5NH3]2CuCl4 compound. J. Mol. Struct. 2015, 1082, 38. 10.1016/j.molstruc.2014.11.006. [DOI] [Google Scholar]
- Cole K. S.; Cole R. H. Dispersion and absorption in dielectrics I. Alternating current characteristics. J. Chem. Phys. 1941, 9, 341–351. 10.1063/1.1750906. [DOI] [Google Scholar]
- K Singh N.; Kumar P.; Kumar H.; Rai R. Structural and dielectric properties of Dy2 (Ba0. 5R0. 5)2O7 (R= W, Mo) ceramics. Adv. Mater. Lett. 2010, 1, 79–82. 10.5185/amlett.2010.3102. [DOI] [Google Scholar]
- Mazurin O. V. Relaxation phenomena in glass. J. Non-Cryst. Solids 1977, 25, 129–169. 10.1016/0022-3093(77)90092-8. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.