

Abstract
Background
Borrelia are important disease-causing tick- and louse-borne spirochaetes than can infect a wide variety of vertebrates, including humans and reptiles. Reptile-associated (REP) Borrelia, once considered a peculiarity, are now recognised as a distinct and important evolutionary lineage, and are increasingly being discovered worldwide in association with novel hosts. Numerous novel Borrelia spp. associated with monitor lizards (Varanus spp.) have been recently identified throughout the Indo-Pacific region; however, there is a lack of genomic data on these Borrelia.
Methods
We used metagenomic techniques to sequence almost complete genomes of novel Borrelia spp. from Varanus varius and Varanus giganteus from Australia, and used long- and short-read technologies to sequence the complete genomes of two strains of a novel Borrelia sp. previously isolated from ticks infesting Varanus salvator from Indonesia. We investigated intra- and interspecies genomic diversity, including plasmid diversity and relatedness, among Varanus-associated Borrelia and other available REP Borrelia and, based on 712 whole genome orthologues, produced the most complete phylogenetic analysis, to the best of our knowledge, of REP Borrelia to date.
Results
The genomic architecture of Varanus-associated Borrelia spp. is similar to that of Borrelia spp. that cause relapsing fever (RF), and includes a highly conserved megaplasmid and numerous smaller linear and circular plasmids that lack structural consistency between species. Analysis of PF32 and PF57/62 plasmid partitioning genes indicated that REP Borrelia plasmids fall into at least six distinct plasmid families, some of which are related to previously defined Borrelia plasmid families, whereas the others appear to be unique. REP Borrelia contain immunogenic variable major proteins that are homologous to those found in Borrelia spp. that cause RF, although they are limited in copy number and variability and have low sequence identities to RF variable major proteins. Phylogenetic analyses based on single marker genes and 712 single copy orthologs also definitively demonstrated the monophyly of REP Borrelia as a unique lineage.
Conclusions
In this work we present four new genomes from three novel Borrelia, and thus double the number of REP Borrelia genomes publicly available. The genomic characterisation of these Borrelia clearly demonstrates their distinctiveness as species, and we propose the names Borrelia salvatorii, ‘Candidatus Borrelia undatumii’, and ‘Candidatus Borrelia rubricentralis’ for them.
Graphical Abstract

Supplementary Information
The online version contains supplementary material available at 10.1186/s13071-023-05937-4.
Keywords: Borrelia, Reptile-associated Borrelia, Ticks, Tick-borne disease, Varanidae, Monitor lizard
Background
Borrelia are globally distributed tick- and louse-borne spirochetes that can infect a wide variety of vertebrate hosts, including humans. Borrelia spp. are ecologically diverse and broadly cluster into four distinct evolutionary lineages that reflect their host and vector associations, ecological life cycles, physiology, and human disease manifestations [1]. These lineages include the Ixodes-transmitted Lyme borreliosis (LB) clade (Borreliella), the Argasidae-transmitted and Ixodidae-transmitted relapsing fever (RF) clades, and the reptile-associated (REP) clade, which is dominated by reptile-infecting species transmitted by Metastriata tick vectors, such as Borrelia turcica, but also includes diverse species such as ‘Candidatus (Ca.) Borrelia tachyglossi’ from Australian echidnas and ‘Ca. Borrelia mahuryensis’ found in passerine birds from French Guinea [2, 3].
The RF and LB clades include important human pathogens and have been widely studied for many years. However, REP Borrelia were discovered relatively recently and have not received similar scientific attention. Nevertheless, molecular techniques have improved our ability to detect and classify Borrelia, and putative REP Borrelia are increasingly reported from tortoises, snakes, and Varanus (monitor lizards), and even the toad Rhinella horribilis [4–11].
In particular, the number of REP Borrelia associated with Varanus spp. (varanid) hosts has grown rapidly, with novel putative species found in ticks collected from Varanus exanthematicus from Tanzania [9], Varanus varius from Australia [7], Varanus salvator from Indonesia [8], V. salvator and Varanus bengalensis from Thailand [5], and V. bengalensis from Pakistan [12]. These Borrelia have been found predominantly in association with Amblyomma spp. ticks, including Amblyomma varanense and Amblyomma gervaisi, but also with the tick Bothriocroton undatum in Australia, although it has not been established whether these ticks are true Borrelia vectors as only blood-fed specimens have been analysed. Phylogenetic analysis of 16S ribosomal RNA (rRNA) and flaB sequences indicated that varanid-associated Borrelia form a monophyletic clade within the REP Borrelia lineage that is distinct from other Borrelia clades [8].
Borrelia have unique genomes that contain a single linear chromosome and a multitude of both linear and circular plasmids, which can comprise up to 40% of their total genomic content. While the chromosomes of all Borrelia species are highly conserved, their plasmids are highly variable, structurally plastic, and contain a high proportion of repetitive genomic elements [13]. In addition, some core plasmids contain critical proteins required for transmission and host invasion, while other accessory plasmids are not critical for Borrelia survival but may confer phenotypes that influence their pathogenicity and clinical manifestations [14]. Because of their structural complexity and genetic repetitivity, long-read sequencing technologies are required to properly assemble Borrelia plasmids [15].
Genomic data are a critical resource that underpin our understanding of Borrelia evolution, population dynamics, host and vector adaptation, and pathogenesis [16]. While a wealth of genomic resources exists for many Argasidae-transmitted RF species and LB species, genomic resources are lacking for most REP species due to their ad hoc discovery and the lack of cultivated isolates. However, the genomes of three REP species, Borrelia turcica, ‘Ca. Borrelia tachyglossi’, and ‘Ca. Borrelia mahuryensis’, have been sequenced [2, 3]. The genome of B. turcica is considered complete as it was sequenced from a pure culture with Illumina and PacBio technologies, while the genomes of ‘Ca. B. tachyglossi’, and ‘Ca. B. mahuryensis’ are thus far incomplete, particularly their plasmid fractions, as they were assembled from Illumina data only directly from an engorged tick and a low passage cultured isolate, respectively [2, 3].
Analysis of these genomes indicates that REP Borrelia genomes have a RF-like architecture, including chromosomal synapomorphies, such as the presence of the GlpQ antigen, a single 23S rRNA gene, and a large linear megaplasmid [2, 3]. However, REP genomes also contain distinct genomic features that differentiate them from both the RF and LB species, such as the inclusion of putative maltose metabolism genes glvA and glvC in the rRNA operon [10], and a highly reduced set of antigenic variable membrane proteins (vmps) compared to their RF counterparts, and numerous genes that are homologous to genes found in LB species that were subsequently lost by all RF species [2, 3]. Interrogating the genomes of REP Borrelia spp. is an alternative approach to thorough ecological or epidemiological studies, which have as yet not been performed for any of these species, that can be used to gain insights into their unique biology and evolution and may assist in determining their relevance for human and animal health.
Here, we sequenced four additional REP Borrelia genomes from three novel Borrelia species from varanids from Indonesia and Australia. Indonesian Borrelia were previously isolated from the midgut and salivary glands of engorged A. varanense ticks collected from an individual V. salvator in Bogor, Indonesia [8]. Australian Borrelia were identified during this study by polymerase chain reaction (PCR) on ethanol-preserved Amblyomma and Bothriocroton ticks from V. giganteus and V. varius, respectively, and their genomes reconstructed by metagenomic sequencing.
Methods
Australian varanid tick samples
Three cohorts of Australian varanid ticks were screened for Borrelia, comprising a cohort of Bothriocroton undatum from Varanus varius from the Burragorang Valley, New South Wales [17], a cohort of Amblyomma calabyi, Amblyomma fimbriatum, and Amblyomma limbatum from Varanus giganteus from the vicinity of Mt. Conner on the Curtin Springs cattle station, Northern Territory, and a cohort of A. limbatum from Varanus acanthurus from the Fitzroy River, Kimberly, Western Australia (Fig. 1; Additional file 1: Table S1). Ticks were stored in ethanol at ambient temperature until the current study, and morphologically identified to species, sex, and life stage using standard morphological keys for Australian ticks [18, 19].
Fig. 1.
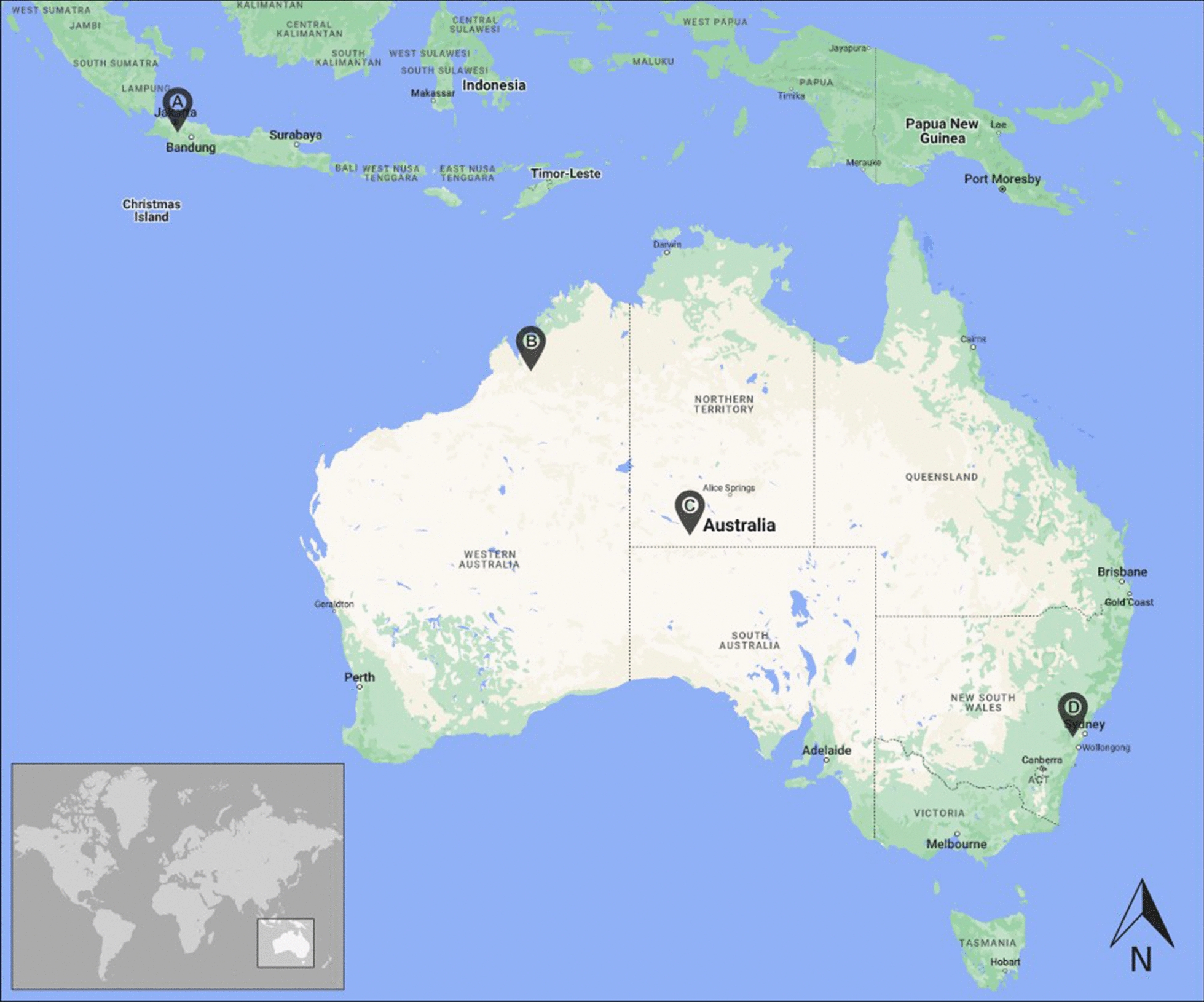
Map of the approximate collection locations of the monitor lizards from which tick cohorts were screened for Borrelia. Varanus salvator (Bogor, West Java, Indonesia) (A); Varanus acanthurus (Fitzroy River basin, Western Australia) (B); Varanus giganteus (Mt. Conner, Northern Territory) (C); Varanus varius (Burragorang Valley, New South Wales) (D)
PCR screening of Australian varanid ticks for Borrelia
Genomic DNA was extracted from individual ticks using the DNeasy Blood and Tissue Kit (QIAGEN, Germany) following the manufacturer’s protocol. DNA samples were screened for Borrelia with nested PCRs targeting the flaB and 16S rRNA genes, as previously described [20], using 0.4 M of each primer, 1.5 mM of MgCl2 and KAPA Taq DNA polymerase. PCR amplicons were electrophoresed through 1% agarose gels stained with SYBRsafe, and positive amplicons were excised from the gel, purified, and selected samples Sanger sequenced using both 5’ and 3’ PCR primers (Additional file 1: Table S1). Samples were only considered Borrelia-positive with consistent amplification of both flaB and 16S loci.
Metagenomic sequencing and assembly of Australian varanid Borrelia genomes
Individual representative Borrelia-positive tick DNA samples were prepared for shotgun metagenomic sequencing using the Illumina DNA Prep Library Kit and sequenced on a MiSeq using 300 base pair (bp) paired-end chemistry (Illumina, USA) or NextSeq using 150 bp paired-end chemistry at the Genome Discovery Unit—Australian Cancer Research Foundation Biomolecular Resource Facility, The John Curtin School of Medical Research, Australian National University. Metagenomic assembly of Borrelia spp. was performed with metaspades [21], with Borrelia contigs identified with mmseq2 [22] searching against a custom database of curated Borrelia genomes from the National Center for Biotechnology Information (NCBI) Reference Sequence Database (RefSeq). Borrelia chromosomal contigs were scaffolded to the B. turcica strain IST7 chromosome with Mauve v1.1.1 [23], and contigs were corrected and gaps closed by mapping reads back to scaffolded contigs with minimap2 [24]. Plasmid contigs were checked for completeness and circularisation by aligning plasmids to themselves and searching for terminal overlaps and terminal telomere structures.
Borrelia sp. RT1S and RT5S from V. salvator
In addition to Australian varanid Borrelia spp., we sequenced Borrelia sp. strains RT1S and RT5S that were isolated from A. varanense ticks engorged on V. salvator in Bogor, West Java, Indonesia, as reported previously [8]. Strains RT1S and RT5S were isolated from separate A. varanense salivary glands from the same host animal in BSK-H medium, and genomic DNA was extracted from low passage cultures using the Genomic-tip 100/G kit (QIAGEN, Germany).
Borrelia strains RT1S and RT5S were sequenced with Illumina and MinION technologies. DNA was prepared for Illumina sequencing using the Illumina DNA Prep Library Kit and sequenced on a MiSeq using 300 bp paired-end chemistry (Illumina, USA). MinION sequencing libraries were prepared using the Ligation Sequencing Kit [Oxford Nanopore Technologies (ONT), UK], and base calling was performed with Guppy v4.2.2. Raw Illumina and ONT sequences were quality filtered with Fastp v0.23.1 [25] (for Illumina reads), and Porechop v0.2.4 (https://github.com/rrwick/Porechop), Filtlong v0.2.1 (https://github.com/rrwick/Filtlong), and Pacasus v1.2 [26] (for ONT reads). Assembly of ONT reads was performed using Canu v2.2 [27] and Flye v2.8 [28] with five rounds of polishing with Pilon v1.24 [29]. Contigs from different assemblies were aligned together to determine the correctness and completeness of replicon contigs, and putative plasmid contigs were checked for completeness and circularisation acribed above.
Genome annotation
Plasmids in the final assemblies were named based on their topology [linear plasmid (lp), circular plasmid (cp)] and size (to the nearest kilo base); in cases where multiple plasmids shared similar topology and size, they were suffixed alphabetically (e.g. cp30, cp30-B). Genomic features were annotated using NCBI Prokaryotic Genome Annotation Pipeline v5.2 [30], and InterProScan v5.56–89.0 [31] was used to classify putative replicon partitioning proteins and vmps using the CDD, Pfam, and Superfamily databases [32–34], following the methods of Kneubehl et al. [35] and Kuleshov et al. [36]. In brief, the following gene families correlated with the subsequent Pfam and vmp gene families: cd02038 and cd02042 (PF32), PF01672 (PF49), PF02089 (PF50), PF02414 (PF57/62), PF01441 and SSF63515 [variable small proteins (vsps)] and PF00921 and SSF74748 [variable large proteins (vlps)] [36]. PF32 and PF57/62 plasmid partitioning protein genes were aligned with those from Kneubehl et al. [35] with MAFFT [37] and phylogenetic analyses were performed with IQ-TREE v2.2.0 [38] with 1000 ultrafast bootstrap approximations and near-zero branches collapsed into a polytomy. Alignment and phylogenetic analysis of vlp genes, including non-redundant vlp genes from other Borrelia spp., were performed in the same manner. All final genomes were deposited in GenBank BioProject PRJNA774887.
Comparative, phylogenetic, and pangenome analysis
We compared the gene content and chromosomal and plasmid synteny between varanid Borrelia chromosomes by aligning whole chromosomal sequences with Mauve v1.1.1 [23] and MUMmer [39] and aligning orthologous protein coding sequences with MCscan [40]. For pangenome analysis, protein coding sequences were identified in all genomes using Prokka v1.12 [41] and parsed into Roary v3.13 [42], which clustered gene orthologues at ≥ 60% amino acid identity and identified core and accessory genes as well as presence/absence of orthologues among the genomes. Phylogenomic analysis utilized 726 core orthologues identified by Roary v3.13 in 21 Borrelia genomes (including those from this study), with individual orthologues aligned with MAFFT, and analysis conducted in IQ-TREE v2.2.0 with proportional edge-linked partition models predicted for each orthologous gene set [43], 1000 bootstrap approximations [44], and gene and site concordance factors calculated for each node [45, 46]. 16S and flaB phylogenies were performed in the same manner with IQ-TREE v 2.2.0 with model selection and 1000 bootstrap approximations.
Results
The following results clearly demonstrate that the Borrelia spp. identified in ticks collected from V. salvator, V. varius, and V. giganteus are distinct species. Therefore, for clarity, we herein refer to these Borrelia spp. by their proposed names: Borrelia salvatorii (from V. salvator), ‘Ca. Borrelia undatumii’ (from V. varius), and ‘Ca. Borrelia rubricentralis’ (from V. giganteus).
PCR screening of Australian varanid ticks
Collectively, 230 ticks from 38 Australian lizards were screened for Borrelia by nested PCR, with 45 ticks returning positive (Additional file 1: Table S1). Overall, 5.95% (5/84) of Bothriocroton undatum ticks from V. varius in the Burragong Valley were positive for Borrelia, including male and female B. undatum from two out of 12 individual lizards. Borrelia was detected in 43.95% (40/91) of ticks from V. giganteus, including male and female A. calabyi and A. fimbriatum, and nymph A. limbatum from 10 of 14 lizards. No ticks were Borrelia-positive in the cohort of A. limbatum from V. acanthurus. Preliminary analysis of selected flaB and 16S amplicon sequences indicated that all Borrelia from Bothriocroton undatum ex V. varius and Amblyomma spp. ex V. giganteus were near-identical (flaB nucleotide similarity > 99.6%; 16S nucleotide similarity > 99.8%), with the only nucleotide difference between sequences being single nucleotide polymorphisms, all of which were non-synonymous for flaB sequences (data not shown).
Genome sequencing and assembly
Metagenomic Illumina sequencing and de novo assembly produced complete Borrelia chromosomes for both ‘Ca. Borrelia undatumii’ AG58 from Bothriocroton undatum ex V. varius and ‘Ca. Borrelia rubricentralis’ P9F1 from A. calabyi ex V. giganteus (0.912 Mb and 0.937 Mb, respectively). Each chromosomal assembly included a maximum of three contigs (minimum length 0.34 Mb), with gaps closed by mapping bridging reads across open spans < 1 kb. Putative plasmid contigs were also identified, with six putative linear plasmid contigs (6–65 kb in length) for ‘Ca. B. undatumii’, and five linear plasmid contigs (23–74 kb in length) and two complete circular plasmids (32 kb in length) for ‘Ca. B. rubricentralis’ (Table 1). For ‘Ca. B. undatumii’, plasmid contigs did not represent complete replicons, although telomere structures were identified on the right end of plasmids lp6, lp12, and lp31, confirming their linear structure. Likewise, all linear plasmids from ‘Ca. B. rubricentralis’ were incomplete, with no telomere structures identified at either end of the plasmid contigs. ‘Ca. B. rubricentralis’ plasmids cp32 and cp32-B were confirmed to be circular by overlapping terminal ends of the contigs, and were the only complete contigs identified in these assemblies. Each putative plasmid sequence in the metagenomic assemblies was represented by one continuous contig. General sequencing statistics for each assembly are presented in Additional file 1: Table S2.
Table 1.
Summary of assembled Borrelia replicons
| Replicon | Complete/partial | Accession no. | Length (base pairs) | CDS (no.) | Mean coverage (Illumina/ONT) |
|---|---|---|---|---|---|
| Borrelia salvatorii strain RT1S | |||||
| Chromosome | Complete | CP088943 | 952,250 | 864 | 1359.7/609.6 |
| lp23 | Complete | CP088944 | 23,856 | 34 | 3554.4/780.8 |
| lp25 | Complete | CP088945 | 25,673 | 32 | 5506.1/595.4 |
| cp27 | Complete | CP088946 | 27,661 | 28 | 4285.1/438.8 |
| lp27 | Complete | CP088947 | 25,416 | 32 | 5015.2/626.5 |
| lp30 | Complete | CP088948 | 30,113 | 37 | 9638.7/1231.5 |
| lp30-B | Partial | CP088949 | 30,059 | 34 | 7106.5/781.2 |
| lp51 | Complete | CP088950 | 51,384 | 61 | 2751.7/378.2 |
| lp83 | Complete | CP088951 | 82,803 | 86 | 2588.0/369.5 |
| Borrelia salvatorii strain RT5S | |||||
| Chromosome | Complete | CP088936 | 942,663 | 856 | 1464.0/388.8 |
| lp25 | Complete | CP088937 | 25,456 | 30 | 8945.1/838.0 |
| cp27 | Complete | CP088938 | 27,771 | 29 | 4716.3/318.8 |
| lp27 | Complete | CP088939 | 27,011 | 30 | 7108.5/773.9 |
| lp30 | Complete | CP088940 | 30,932 | 36 | 5415.2/573.9 |
| lp44 | Partial | CP088941 | 44,552 | 52 | 3422.8/319.1 |
| lp83 | Complete | CP088942 | 83,220 | 86 | 2669.2/399.9 |
| ‘Candidatus Borrelia undatumii’ AG58 | |||||
| Chromosome | Complete | CP086555 | 912,448 | 863 | 101.3/- |
| lp6 | Partial | CP129596 | 9,178 | 4 | 179.3/- |
| lp9 | Partial | CP129597 | 12,342 | 8 | 151.8/- |
| lp12 | Partial | CP129598 | 12,681 | 14 | 129.9/- |
| lp17 | Partial | CP129599 | 17,500 | 22 | 91.9/- |
| lp31 | Partial | CP129600 | 37,310 | 31 | 179.1/- |
| lp65 | Partial | CP129601 | 65,850 | 74 | 170.2/- |
| ‘Candidatus Borrelia rubricentralis’ P9F1 | |||||
| Chromosome | Complete | CP129407 | 936,962 | 851 | 24.4/- |
| lp23 | Partial | CP129410 | 23,742 | 25 | 21.2/- |
| lp24 | Partial | CP129411 | 24,219 | 31 | 31.3/- |
| lp25 | Partial | CP129412 | 25,027 | 26 | 25.1/- |
| cp32-B | Complete | CP129409 | 32,481 | 39 | 24.7/- |
| cp32 | Complete | CP129408 | 32,878 | 41 | 36.2/- |
| lp36 | Partial | CP129413 | 36,508 | 42 | 28.7/- |
| lp74 | Partial | CP129414 | 74,710 | 80 | 26.4/- |
lp Linear plasmid, cp circular plasmid, CDS coding sequence, ONT Oxford Nanopore Technologies
In addition to Australian varanid Borrelia, we also sequenced two Borrelia strains isolated from A. varanense ex V. salvator in Indonesia [8]. Hybrid de novo assembly of B. salvatorii strains RT1S and RT5S from MinION and Illumina sequencing produced a complete chromosome contig for each isolate (9.4–9.5 Mb), and eight and six complete plasmid contigs for strains RT1S and RT5S, respectively (Table 1), including five core plasmids that were highly conserved between strains, and four plasmids that were divergent between strains (Fig. 2).
Fig. 2.

Alignment of Borrelia salvatorii strains RT1S and RT5S showing A pairwise alignment plots of plasmid sequences with colinear nucleotide regions identified by MUMmer v3.1, and B an MCscan karyotype plot showing regions of orthologous macrosynteny between protein coding sequences
Borrelia salvatorii strains RT1S and RT5S shared five core plasmids: lp83, lp30, lp27, lp25, and cp27. Pairwise alignment of these plasmids to each other showed almost identical gene synteny and nucleotide similarity (83.5–99.5%) between strains (Fig. 2; Additional file 1: Fig. S1). Plasmid cp27, the only circular plasmid identified, had the lowest overall level of average nucleotide identity (83.5%) between strains RT1S and RT5S; however, almost all of this variance occurred in one 5-kb region containing immunogenetic vlps that are known to be highly divergent between strains in other Borrelia spp. [47] (Additional file 1: Fig. S1). Both strains RT1S and RT5S contained almost identical (99.5% nucleotide similarity) copies of plasmid lp30; however, strain RT1S contained two divergent copies of lp30 (lp30 and lp30-B) that were almost completely syntenic, but shared only 63.4% nucleotide identity and 56.6% amino acid identity (Fig. 2; Additional file 1: Fig. S1). Both strains contained nearly identical lp27 plasmids, although this plasmid in strain RT1S was actually ~ 25 kb long due to the contraction of two hypothetical proteins and a noncoding region on the right side of the plasmids (Fig. 2; Additional file 1: Fig. S1). Borrelia salvatorii strain RT1S lp23 was not present in strain RT5S, although it did share orthologues with genes found on lp30 (Fig. 2; Additional file 1: Fig. S1). In addition, lp51 from strain RT1S and lp44 from strain RT5S shared orthologues at each end of the replicons, but the internal portion of each replicon was highly divergent (Fig. 2; Additional file 1: Fig. S1).
Phylogenetic analysis
Maximum likelihood phylogenetic analysis of flaB and 16S sequences strongly supports the grouping of B. salvatorii, ‘Ca. B. undatumii’, and ‘Ca. B. rubricentralis’ within a monophyletic varanid-associated clade that includes all previously described varanid genotypes from the Indo-Pacific region. (Fig. 3A–C). In agreement with previous studies, flaB and 16S phylogenies placed all REP Borrelia, including those from tortoise, snakes, and varanids into a monophyletic clade that is distinct from the hard and soft tick RF clade and the LB clade (Fig. 3A, B). Additionally, within the reptile clade there is strong support for distinct host-associated lineages related to tortoise, snake, and varanid hosts, with the exception of two recently described Borrelia spp. from Boa constrictor and Rhinella horribilis in Mexico [4, 6], which clustered within the tortoise clade (Fig. 3A, B).
Fig. 3.

Maximum likelihood phylogenies of reptile-associated (REP) Borrelia spp. based on alignment of flaB nucleotide sequences (> 301 bp) (A), 16S rRNA nucleotide gene sequences (> 1033 bp) (B), and 712 shared single copy nucleotide orthologues (C). All phylogenies were produced in IQ-TREE v2.2.0 with model selection and 1000 ultrafast bootstrap approximations. Node labels in A represent bootstrap support/gene concordance/site concordance; open circle node shapes in b and C indicate bootstrap support < 70% open circles. Borrelia sequences from this study are shown in bold. Root branches are not shown
Two distinct 16S sequences were previously identified in B. undatum from a single individual V. varius that differed by 1.5% nucleotide identity [7]. ‘Ca. B. rubricentralis’ 16S sequences grouped most closely with one of these sequences, while ‘Ca. B. undatumii’ 16S sequences from this study grouped strongly with the other one (Fig. 3B). Borrelia salvatorii 16S and flaB sequences clustered strongly with sequences from V. salvator and V. bengalensis from Thailand [5] and Pakistan [12] (Fig. 3A, B).
The results of the phylogenetic analysis based on 712 single copy orthologues also concurred with the 16S and flaB phylogenies and with previous whole genome-based Borrelia phylogenies [2, 3, 36, 48], and demonstrated overwhelmingly that the REP Borrelia clade (with the inclusion of ‘Ca. B. mahuryensis’ and ‘Ca. B. tachyglossi’) is monophyletic with respect to the RF and LB clades (Fig. 3C). Within the REP clade, all varanid-associated species were monophyletic, with B. turcica and ‘Ca. B. mahuryensis’ basally positioned to the exclusion of ‘Ca. B. tachyglossi’ (Fig. 3C). This suggests that the evolutionary pattern of distinct host-associated REP lineages observed in 16S and flaB phylogenies may hold true with the addition of more powerful genomic data. However, genomic data from additional tortoise- and snake-associated Borrelia spp. are needed to confirm this hypothesis.
Borrelia chromosomal synteny
Mauve and MCscan alignments demonstrated that varanid Borrelia spp. have highly conserved colinear chromosomes, with > 98% of all orthologous protein coding genes in synteny with other Borrelia chromosomes. Alignment of whole chromosomes, however, showed several instances of gene insertions and deletions that were synapomorphic for the REP Borrelia clade. The insertion of phosphotransferase system lactose/cellobiose transporter subunit IIA, flagellar filament outer layer protein FlaA, and Cof-type HAD-IIB family hydrolase genes (at positions 36,260, 184,184, and 200,384, respectively), and the deletion of a transfer RNA gene [(guanosine-2'-O-)-methyltransferase; at position 41,559] were synapomorphic for the REP clade with the inclusion of ‘Ca. B. mahuryensis’ and ‘Ca. B. tachyglossi’. In addition, the deletion of chromosome partition protein genes smc and parB, and a putative cytosolic protein gene (at positions 38,399, 455,067, and 133,084, respectively) were synapomorphic for REP Borrelia to the exclusion of ‘Ca. B. tachyglossi’. Finally, the only synapomorphies specific to varanid Borrelia spp. were the deletion of the DNA-3-methyladenine glycosylase gene (position 444,418) from the rRNA operon and the insertion of a small hypothetical protein (position 773,872) (Additional file 1: Fig. S2). The rRNA operon of all varanid Borrelia spp. contained three single copy rRNA genes (5S, 23S, 16S) and seven protein-coding genes, including the horizontally acquired putative maltose metabolism genes glvA and glvC, which are also found in B. turcica and ‘Ca. B. tachyglossi’ and other REP species, but are absent from all other Borrelia spp. [3, 10]. All varanid genomes also included a homologue of the RF antigenic protein glpQ, which is a major immunogenic protein expressed during RF Borrelia infection. All of the nucleotide positions given above refer to nucleotide positions along the Borrelia sp. RT1S chromosome (CP088943).
Plasmid sequence analysis
Plasmids from B. salvatorii, ‘Ca. B. undatumii’, and ‘Ca. B. rubricentralis’ had an overall low degree of structural conservation between species, but a high degree of shared orthologous gene content, with 57% of plasmid-encoded proteins shared between all three species. Some plasmids were conserved between species, such as lp30-B and lp24 from B. salvatorii and ‘Ca. B. rubricentralis’, respectively, and cp27, lp25, and lp12 from B. salvatorii, ‘Ca. B. rubricentralis’, and ‘Ca. B. undatumii’, respectively (Fig. 4). The large linear plasmids of each species (lp83, lp74, and lp65) were also highly conserved, not only between B. salvatorii, ‘Ca. B. rubricentralis’, and ‘Ca. B. undatumii’, but also with the large linear plasmids of RF species (Additional file 1: Fig. S3).
Fig. 4.

MCscan karyotype plots showing regions of orthologous macrosynteny of protein coding sequences between Borrelia salvatorii and ‘Ca. Borrelia rubricentralis’ (A), B. salvatorii and ‘Ca. Borrelia undatumii’ (B), and ‘Ca. B. rubricentralis’ and ‘Ca. Borrelia undatumii' (C) plasmids represent the complete set of plasmids present in both strains RT1S and RT5S
All REP Borrelia also contained linear or circular plasmids (or plasmid contigs) that are related to the LB cp26 plasmid, which is essential for LB Borrelia survival (Additional file 1: Fig. S4). The complete REP cp26-like plasmids analysed (B. salvatorii cp27, B. turcica lp35) contained three highly syntenic but structurally inverted homologous regions to LB cp26 plasmids, which included homologous genes including resT, which is essential for successful replication of linear replicons (Additional file 1: Fig. S4). Varanid Borrelia, however, lacked an identifiable variable tick protein/ospC homolog on cp26, which has been shown to be important for host and vertebrate infection, while B. turcica lp35 does contain a variable tick protein/ospC homolog. The gene content and ubiquitous nature of cp26-like plasmids in REP Borrelia suggest that, like in their LB homologues, they are essential for Borrelia survival.
Plasmid partitioning genes are essential for the inheritance and maintenance of plasmids, and five paralogous gene families (PF) have been identified as putative plasmid partitioning genes in Borrelia: PF32, PF49, PF50, and P57/62 [13, 49]. These genes are also used to classify plasmid compatibility for LB, and have been used for some RF groups [13, 35, 36]. We identified putative plasmid partitioning genes in our assemblies, as well as the other REP species B. turcica, ‘Ca. B. tachyglossi’, and ‘Ca. B. mahuryensis’, and compared the PF32 and PF57/62 loci to determine the relationships of REP Borrelia plasmids. Because the plasmid assemblies from ‘Ca. B. undatumii’ and ‘Ca. B. rubricentralis’ are incomplete, we did not identify any putative plasmid partitioning genes in three plasmids from ‘Ca. B. undatumii’ (lp17, lp12, and lp6). All other plasmids contained at least one PF57/62 locus, with B. salvatorii strain RT5S lp51 containing two distinct PF57/62 copies (Additional file 1: Fig. S5).
Phylogenetic and clustering analysis of PF32 loci identified four main REP plasmid clusters, and four orphan REP replicons that did not cluster with other REP plasmids. Two orphan PF32 loci from B. turcica lp34 and ‘Ca. B. tachyglossi’ clustered within known RF plasmid families (F23 and F27, respectively, as defined by Kneubehl et al. [35]). Borrelia turcica lp32-B was closely related to the RF F26 plasmid family, while ‘Ca. Borrelia mahuryensis’ lp54 PF32 was within a polyphyletic group containing LB lp54 and cp32 plasmids as well as Borrelia turicatae BTE5EL lp32 (Fig. 5A). The remaining REP PF32 loci formed four distinct monophyletic clusters, designated REP plasmid families (RPF) 1-4, with RPF1 associated with LB cp26 plasmids, RPF2 positioned next to RF F5 plasmids, RPF3 positioned next to RF F8-9 and LB cp32 plasmids, and RPF4 representing the long linear megaplasmids and positioned sister to the RF F6 megapasmids and LB lp54 plasmids (Fig. 5A).
Fig. 5.

Maximum likelihood phylogenies of REP Borrelia plasmids based on PF32 (A) and PF57/62 (B) nucleotide sequences among a subset of Lyme borreliosis (LB) and relapsing fever (RF) sequences as used by Jones et al. [31] to define RF plasmid compatibility groups. Branches are colour coded according to bootstrap support and taxa labels are colour coded according to their phylogenetic lineage [cyan (LB), purple (RF), orange (REP)]. The outer rings in A show the plasmid families based on analysis of PF32 sequences, with purple rings indicating RF plasmid families (F4-29) as defined by Jones et al. [31] and orange rings indicating REP plasmid families (RPF1-4) as defined by our analysis. Labelled outer rings in B show plasmid families that are concordant between PF32 and PF57/72 loci, and unlabelled outer rings indicate putative REP plasmid families that were disconcordant with PF32 loci or for which no PF32 loci existed. The isolate prefixes, gene locus, and plasmid are given for each PF32 and PF57/62 locus. See Supplementary Information for details of isolate prefixes
As many plasmids lacked a PF32 loci, we also used the PF57/62 loci, which were identified on all plasmids except those that lacked any putative plasmid partitioning genes (likely due to incompleteness). Phylogenetic analysis of REP PF57/62 genes had limited congruency with the PF32 analysis; however, it demonstrated an agreement with the PF32 analysis that most REP plasmids form monophyletic lineages that are distinct from RF and LB plasmid families. There was broad congruency between the RPF1 and RPF4 families between the two phylogenies, indicating a more stable inheritance pattern for these plasmids throughout all Borrelia, including REP species (Fig. 5B). Other REP PF57/62 loci clustered into four distinct monophylies, although these clusters did not concur with the PF32 phylogeny (Fig. 5). These groups included two clades that were sister to RF F20 plasmids, one clade that was sister to LB lp17 plasmids, and one clade that was sister to a large cluster of unclassified RF plasmids (Fig. 5B). In addition, two orphan PF57/62 genes were identified from ‘Ca. B. tachyglossi’, with one from cp6 clustering with RF F27, in agreement with the PF32 analysis.
We also investigated the phylogenetic relatedness of REP vlp to the immunogenic vlp and vsp proteins of RF species which underpin their complex polyphasic antigenic switching phenotype. Immunogenic vlps loci were found on plasmids cp27, lp25, and lp12 from B. salvatorii, ‘Ca. B. rubricentralis’, and ‘Ca. B. undatumii’, respectively, although they had low (< 44%) levels of homology to RF vlp amino acid sequences, and even less to LB vlp homologs. No vsp sequences were identified. Unlike their homologues in RF species, REP vlp sequences were highly reduced in number, with a maximum of five vlps found on individual plasmids. B. salvatorii cp27 contained five vlp loci, ‘Ca. B. rubricentralis’ lp25 contained five vlp loci, and ‘Ca. B. undatumii’ contained just two vlp loci (Additional file 1: Fig. S5). ‘Ca. B. rubricentralis’ lp25, B. salvatorii cp26, and B. turcica lp35 all contained clusters of 5–11 vlps which cantered on a homologous vlp1 sequence (Additional file 1: Fig. S6). This vlp1 may represent a putative expression site for antigenic switching; however, there was limited homology between REP vlp1 and known vlp expression sites in RF and LB species, and upstream and downstream homology sequences were not identified. No ospC, vtp, or vlsE homologs were identified in varanid Borrelia. Phylogenetic analysis indicated that all varanid vlp proteins clustered together into a single monophyletic cluster in which vlps from individual species largely grouped together into distinct clades (Additional file 1: Fig. S7). This is in stark contrast to the vlp sequences of RF Borrelia which were highly variable and paraphyletic across the phylogeny (Additional file 1: Fig. S7).
Pangenome
Pangenome analysis included all available REP Borrelia genomes, including B. turcica, ‘Ca. B. mahuryensis’, and ‘Ca. B. tachyglossi’, in addition to the four genomes sequenced in this study (Fig. 6A). Pangenome analysis identified a core genome of 839 gene clusters that were present in at least six of the seven genomes, and an accessory genome of 1429 gene clusters that were present in less than six of the seven genomes. While the number of core genes stabilized quickly after the addition of just three genomes to the pangenome, the accessory genome continued to increase with each additional genome (Fig. 6B). This has been observed previously for other Borrelia groups and is likely attributable to the high degree of plasmid diversity and numerous paralogous gene families characteristic of Borrelia genomes [35]. The pangenome matrix illustrates the pattern of gene presence/absence among REP Borrelia and demonstrates that there is minimal sharing of accessory gene clusters between species, and that distinct blocks of accessory genes are differentially present between species, and even between B. salvatorii strains (Fig. 6A). These differentially present gene blocks are likely linked to diverse plasmids that contain unique gene content between species, and may represent genetic elements important for host-specific adaptations.
Fig. 6.

Pangenome matrix showing the presence/absence of 2268 orthologues among REP Borrelia species created with Roary v3.11.2 (A) and graph of the number of core genes and total genes as pangenome size increases (B)
Proposed candidate names
Due to their unique genomic properties, vertebrate and tick associations, and phylogenetic distinctness as described above, we propose the designations Borrelia salvatorii, ‘Candidatus Borrelia undatumii’, and ‘Candidatus Borrelia rubricentralis’, for these novel putative species identified in ticks from V. salvator, V. varius, and V. giganteus, respectively. The specific designations relate to (1) the putative host species of B. salvatorii, i.e. V. salvator; (2) the putative tick vector of ‘Ca. Borrelia undatumii’, i.e. Bothriocroton undatum; and (3) the ‘red centre’, which describes the arid inland desert of Australia in which V. giganteus (the putative host of ‘Ca. Borrelia rubricentralis’) occurs.
Discussion
We describe herein the genomes of three novel Borrelia species associated with Varanus hosts and provide the first genomic resources for this unique Borrelia clade. Although the first varanid Borrelia was identified over a decade ago from Amblyomma exornatum ex V. exanthematicus [9], only recently have molecular studies begun uncovering their true diversity, with at least 10 distinct genotypes now known from throughout the Indo-Pacific region. However, characterisation of these genotypes has relied largely on 16S rRNA and flaB gene sequence analysis, and whether indeed these genotypes all constitute putative species is unresolved, and thus requires the analysis of additional genomic data.
We also sequenced two strains of B. salvatorii, B. salvatorii RT1S and B. salvatorii RT5S, that were isolated from the salivary glands of separate A. varanense ticks engorged on the same V. salvator host [8]. The plasmid content of these two strains differed markedly, although both strains contained a core genome consisting of five plasmids (lp83, lp30, lp27, lp25, and cp27). Furthermore, duplicate copies of lp30 were found in strain RT1S, and although these plasmids are highly syntenic they are unusually dissimilar at both the nucleotide and amino acid level, although no pseudogenes were identified in either. This level of variation in plasmid content between strains is not unusual for Borrelia, and high levels of variation and structural plasticity between closely related strains are common for most RF and LB species. In comparison, the chromosomes of B. salvatorii, and all other REP Borrelia, were remarkably stable and syntenic with those of other Borrelia spp.
Pangenome analysis demonstrated that the REP pangenome had 2268 gene clusters, and was thus smaller than the RF pangenome (2576–2514 gene clusters), but larger than the LB pangenome (~ 1859 gene clusters) [35, 50, 51]. However, analysis of RF genomes likely overestimates the number of gene clusters due to the large number of paralogous gene clusters and high numbers of paralogous vsp and vlp genes [35]. Overall, REP Borrelia had a relatively closed pangenome with most genes shared between most isolates, and relatively few species-specific genes, with the majority of accessory genes inherited in blocks, which likely represent coinherited sets of genes on plasmids.
Classification of LB plasmid compatibility groups has largely relied on the analysis of PF32 loci [13]. Initial attempts to apply this methodology to RF species were successful and demonstrated that there exists a mix of shared and unique plasmid types between LB and RF clades [35, 36]. The results of our phylogenetic analysis of REP plasmids based on PF32 and PF57/62 genes are similar to those of Kneubehl et al. [35], and demonstrate that REP Borrelia contains at least four distinct plasmid families based on PF32 loci (designated RPF1-4), and perhaps as many as six based on PF57/62 loci. Our analysis indicated that while most REP plasmids are distinct from those of LB and RF, there exists a shared ancestry between them, which likely represents core genomic elements that are required for survival. For example, sections of the PF32 phylogeny representing the cp26/RPF1/F28 and lp54/RPF4/F6 subtrees mirror the whole genome-based phylogeny presented in Fig. 3 indicating the shared inheritance of these genomic elements with the chromosome. In addition, a limited number of orphan REP plasmids clustered within RF plasmid families, and may be a result of more recent horizontal gene transfer.
Many REP plasmids were found to lack PF32 genes (some through incomplete assembly), and analysis showed that PF57/62 loci had limited phylogenetic congruency with PF32loci but demonstrated a similar general patterns of plasmid relatedness to the PF32 phylogeny, with most REP plasmids clustering into distinct REP-specific clades. This was also noted by Kneubehl et al. [35] in their analysis of RF plasmids, and although analysis of PF57/62 loci somewhat assists the classification of these plasmids, a more a robust systematic methodology is likely needed to resolve the relationship between REP and RF Borrelia plasmids. The incongruence between PF32 and PF57/62 phylogenies is likely rooted in the rapid primordial diversification and functional conservation of PF32 genes, which provides suitable genetic variability for phylogenetic analysis, compared to PF57/62 genes, which have much higher levels of genetic variability that may obscure a phylogenetic signal [52].
The vlp and vsp genes identified in REP Borrelia had limited homology to those of RF species, and were also severely reduced in overall number and in their amino acid diversity. In each genome, vlps were clustered on one plasmid, with the cluster beginning with vlp1 that was homologous in B. turcica, B. salvatorii, and ‘Ca. B. rubricentralis’. Conservation of vlp1 between REP species suggest that it may be an expression site for the expression of other vlps. However, there was limited homology between vlp1 and known RF and LB vlp expression sites, and upstream and downstream homology sequences were not identified. Further analysis of REP vlp is needed to resolve the antigen switching mechanisms of REP Borrelia. Phylogenetic analysis of vlp amino acid sequences (Additional file 1: Fig. S7) also demonstrated that REP Borrelia vlps cluster into one monophyletic group and that vlps from each REP species cluster in a species-specific manner.
The role of varanids as true biological reservoirs of Borrelia has not been established, as all Borrelia isolates characterised to date have been from engorged ticks, including Amblyomma varanense, Amblyomma helvolum, Amblyomma gervaisi, Amblyomma exornatum, Amblyomma calabyi, Amblyomma fimbriatum, Amblyomma limbatum, and Bothriocroton undatum. Likewise, the role of these ticks as true Borrelia vectors is unknown, and while all of these tick species are reptile specialists (and some of them Varanus specialists), they often parasitize multiple reptile species, including snakes, varanids, and other squamates [53, 54]. Nevertheless, there appears to be a high level of host adaptation among these Borrelia. For example, although several varanid- and snake-associated Borrelia genotypes have been identified in A. varanense ticks from different hosts, including V. salvator, V. bengalensis, Python bivittatus, and Python reticulatus [5, 11], varanid-associated Borrelia genotypes have never been recovered from A. varanense collected from snakes, or vice versa.
Moreover, the phylogenomic and phylogenetic analyses presented here and previously [5, 8] all demonstrate convincingly that varanid-associated Borrelia, in addition to tortoise-associated and snake-associated Borrelia, separately group into distinct monophyletic clades within the REP lineage. This pattern is indicative of a high level of host adaptation and Borrelia-host coevolution within the REP lineage, which is not observed in other Borrelia lineages, where other evolutionary forces are hypothesized to drive speciation [55–57].
The data presented here suggest that varanid-associated Borrelia likely occur throughout the entire geographic range of the Varanidae, which encompasses much of central and southern mainland Asia; the Middle East; Africa; the Malay Archipelago, including Indonesia and Papua New Guinea; and Australia [58]. Although Varanus is hyperdiverse in Australia compared to other biogeographic regions [58], molecular evidence indicates an Asian origin for the Varanidae, with dispersal to Australia ca. 39–26 million years ago via dispersal events before the collision of Australian and Asian landmasses [59]. Given this biogeographic scenario, it seems probable that ancestral Varanus-associated Borrelia, and indeed perhaps also ancestral Amblyomma spp., colonized Australia at the same time. With > 74 species of Varanus worldwide, and 30 in Australia alone, there are undoubtedly numerous additional Varanus-associated Borrelia species and genotypes awaiting discovery.
Conclusions
REP Borrelia were once considered biological peculiarities and gained little scientific attention compared to their more enigmatic disease-causing cousins. However, novel REP Borrelia are increasingly being discovered around the world in association with new hosts and putative metastriate tick vectors. We now understand that REP Borrelia represents a distinct lineage that is evolutionarily, ecologically, and genetically unique compared to LB and RF Borrelia. This work addresses important gaps in genomic knowledge of REP Borrelia and provides crucial resources that will likely underpin the investigation of important evolutionary hypotheses regarding Borrelia evolution. This work has doubled the number of publicly available REP Borrelia genomes, including the first from Varanus hosts, and two strains of the same species, B. salvatorii. Through our investigation of intra- and interspecies genomic diversity among all available REP genomes, including plasmid diversity and relatedness, we have produced, to the best of our knowledge, the most complete phylogenetic analysis to date of REP Borrelia. Collectively, these genomic resources and our comparative characterisation of these novel REP genomes are likely foundational for future investigations designed to untangle the complex evolutionary history of Borrelia, and have important implications for our understanding of Borrelia-tick coevolution, host adaptation, and the ecological drivers of speciation.
Supplementary Information
Additional file 1: Table S1. Summary of Australian varanid tick samples tested for Borrelia. Fig. S1. Pairwise alignment plots of Borrelia salvatorii strains RT1S and RT5S core plasmids (A), accessory plasmids (B), and duplicated plasmids (C). Fig. S2. Schematic alignment of the ribosomal RNA operon of relapsing fever (RF), reptile-associated (REP), and Lyme borreliosis (LB) Borrelia highlighting the deletion of the mag gene, which is synapomorphic for varanid-associated Borrelia spp. Fig. S3. MCscan karyotype orthologous protein coding genes between the conserved large linear megaplasmids of Borrelia salvatorii and ‘Candidatus (Ca.) Borrelia rubricentralis’, ‘Ca. Borrelia undatumii’, Borrelia turcica, ‘Ca. Borrelia tachyglossi’, Borrelia miyamotoi, and Borrelia hermsii. These large linear megaplasmids appear to be highly conversed among all REP and RF Borrelia. Fig. S4. MCscan plot showing orthologous protein-coding genes between Borrelia burgdorferi B31 plasmid cp26 and Borrelia turcica IST7 plasmid lp25 and Borrelia salvatorii RT1S cp27. Fig. S5. Schematic of varanid-associated Borrelia spp. and the approximate locations of putative plasmid partitioning genes, and variable large proteins. Fig. S6. Schematic gene map of vlp gene clusters in REP Borrelia replicons, including vlp1 (green), which was homologous in all genomes. Fig. S7. Phylogenetic reconstruction of REP and RF Borrelia vlp amino acid sequences. Extended caption for Fig. 5. The isolate, open reading frame number, and plasmid name is given for each sequence.
Acknowledgements
The authors would like to acknowledge Alexander Kneubehl for helpful discussions and for supplying PF32 and PF57/62 sequences for analysis, Gabriele Margos and Volker Fingerle for their helpful insights, and all those involved in the field collection, transportation, and stewardship of tick samples.
Author contributions
AWG: conceptualisation, organisation, laboratory and analytical work, drafting of the manuscript. APB: analytical work and critical review of the manuscript. MM and MS: laboratory work and critical review of the manuscript. AT, KS, SS, JP, SC, RM: organisation, collection and provision of tick samples, and critical revision of the manuscript.
Funding
This work was partially funded by the Australian Commonwealth Department of Health and the Commonwealth Scientific and Industrial Research Organisation (CSIRO).
Availability of data and materials
All genomic data are available through NCBI BioProjects PRJNA783659 and PRJNA774887.
Declarations
Ethics approval and consent to participate
No specific ethics approval was required to undertake this research.
Consent for publication
No consent for publication is required.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Margos G, Gofton A, Wibberg D, Dangel A, Marosevic D, Loh S-M, et al. The genus Borrelia reloaded. PLoS ONE. 2018;13:e0208432. doi: 10.1371/journal.pone.0208432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Binetruy F, Garnier S, Boulanger N, Talagrand-Reboul E, Loire E, Faivre B, et al. A novel Borrelia species, intermediate between Lyme disease and relapsing fever groups, in Neotropical passerine-associated ticks. Sci Rep. 2020;10:10596. doi: 10.1038/s41598-020-66828-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gofton AW, Margos G, Fingerle V, Hepner S, Loh S-M, Ryan U, et al. Genome-wide analysis of Borrelia turcica and ‘Candidatus Borrelia tachyglossi’ shows relapsing fever-like genomes with unique genomic links to Lyme disease Borrelia. Infect Genet Evol J Mol Epidemiol Evol Genet Infect Dis. 2018;66:72–81. doi: 10.1016/j.meegid.2018.09.013. [DOI] [PubMed] [Google Scholar]
- 4.Colunga-Salas P, Sánchez-Montes S, Ochoa-Ochoa LM, Grostieta E, Becker I. Molecular detection of the reptile-associated Borrelia group in Amblyomma dissimile. Mexico Med Vet Entomol. 2021;35:202–206. doi: 10.1111/mve.12478. [DOI] [PubMed] [Google Scholar]
- 5.Kaenkan W, Nooma W, Chelong I-A, Baimai V, Trinachartvanit W, Ahantarig A. Reptile-associated Borrelia spp. in Amblyomma ticks. Thailand. Ticks Tick-Borne Dis. 2020;11:101315. doi: 10.1016/j.ttbdis.2019.101315. [DOI] [PubMed] [Google Scholar]
- 6.Morales-Diaz J, Colunga-Salas P, Romero-Salas D, Sánchez-Montes S, Estrada-Souza IM, Ochoa-Ochoa LM, et al. Molecular detection of reptile-associated Borrelia in Boa constrictor (Squamata: Boidae) from Veracruz. Mexico Acta Trop. 2020;205:105422. doi: 10.1016/j.actatropica.2020.105422. [DOI] [PubMed] [Google Scholar]
- 7.Panetta JL, Šíma R, Calvani NED, Hajdušek O, Chandra S, Panuccio J, et al. Reptile-associated Borrelia species in the goanna tick (Bothriocroton undatum) from Sydney, Australia. Parasit Vectors. 2017;10:616. doi: 10.1186/s13071-017-2579-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Supriyono TA, Kuwata R, Shimoda H, Hadi UK, Setiyono A, et al. Detection and isolation of tick-borne bacteria (Anaplasma spp., Rickettsia spp., and Borrelia spp.) in Amblyomma varanense ticks on lizard (Varanus salvator) Microbiol Immunol. 2019;63:328–333. doi: 10.1111/1348-0421.12721. [DOI] [PubMed] [Google Scholar]
- 9.Takano A, Fujita H, Kadosaka T, Konnai S, Tajima T, Watanabe H, et al. Characterization of reptile-associated Borrelia sp. in the vector tick, Amblyomma geoemydae, and its association with Lyme disease and relapsing fever Borrelia spp. Environ Microbiol Rep. 2011;3:632–637. doi: 10.1111/j.1758-2229.2011.00280.x. [DOI] [PubMed] [Google Scholar]
- 10.Takano A, Goka K, Une Y, Shimada Y, Fujita H, Shiino T, et al. Isolation and characterization of a novel Borrelia group of tick-borne Borreliae from imported reptiles and their associated ticks. Environ Microbiol. 2010;12:134–146. doi: 10.1111/j.1462-2920.2009.02054.x. [DOI] [PubMed] [Google Scholar]
- 11.Trinachartvanit W, Hirunkanokpun S, Sudsangiem R, Lijuan W, Boonkusol D, Baimai V, et al. Borrelia sp. phylogenetically different from Lyme disease- and relapsing fever-related Borrelia spp. in Amblyomma varanense from Python reticulatus. Parasit Vectors. 2016;9:359. doi: 10.1186/s13071-016-1629-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Khan M, Islam N, Khan A, Islam ZU, Muñoz-Leal S, Labruna MB, et al. New records of Amblyomma gervaisi from Pakistan, with detection of a reptile-associated Borrelia sp. Ticks Tick-Borne Dis. 2022;13:102047. doi: 10.1016/j.ttbdis.2022.102047. [DOI] [PubMed] [Google Scholar]
- 13.Casjens SR, Mongodin EF, Qiu W-G, Luft BJ, Schutzer SE, Gilcrease EB, et al. Genome stability of Lyme disease spirochetes: comparative genomics of Borrelia burgdorferi plasmids. PLoS ONE. 2012;7:e33280. doi: 10.1371/journal.pone.0033280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lemieux JE, Huang W, Hill N, Cerar T, Freimark L, Hernandez S, et al. Whole genome sequencing of Borrelia burgdorferi isolates reveals linked clusters of plasmid-borne accessory genome elements associated with virulence. BioRxiv. 2023 doi: 10.1101/2023.02.26.530159v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Margos G, Hepner S, Mang C, Marosevic D, Reynolds SE, Krebs S, et al. Lost in plasmids: next generation sequencing and the complex genome of the tick-borne pathogen Borrelia burgdorferi. BMC Genomics. 2017;18:422. doi: 10.1186/s12864-017-3804-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Keeffe KR, Oppler ZJ, Brisson D. Evolutionary ecology of Lyme Borrelia. Infect Genet Evol. 2020;85:104570. doi: 10.1016/j.meegid.2020.104570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pascoe JH, Flesch JS, Duncan M, Le Pla M, Mulley RC. Territoriality and seasonality in the home range of adult male free-ranging lace monitors (Varanus varius) in south-eastern Australia. Herpetol Conserv Biol. 2019;14:97–104. [Google Scholar]
- 18.Barker SC, Walker AR. Ticks of Australia. The species that infest domestic animals and humans. Zootaxa. 2014;3816:1–144. doi: 10.11646/zootaxa.3816.1.1. [DOI] [PubMed] [Google Scholar]
- 19.Roberts FHS. Australian ticks [Internet]. Melbourne, VIC, Australia: CSIRO; 1970 https://publications.csiro.au/rpr/pub?list=BRO&pid=procite:c6130083-c6c7-4f07-82a2-f94cbad708bb. Accessed 23 Mar 2021.
- 20.Loh S-M, Gofton AW, Lo N, Gillett A, Ryan UM, Irwin PJ, et al. Novel Borrelia species detected in echidna ticks, Bothriocroton concolor, in Australia. Parasit Vectors. 2016;9:339. doi: 10.1186/s13071-016-1627-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nurk S, Meleshko D, Korobeynikov A, Pevzner PA. metaSPAdes: a new versatile metagenomic assembler. Genome Res. 2017;27:824–834. doi: 10.1101/gr.213959.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steinegger M, Söding J. MMseqs2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat Biotechnol. 2017;35:1026–1028. doi: 10.1038/nbt.3988. [DOI] [PubMed] [Google Scholar]
- 23.Darling ACE, Mau B, Blattner FR, Perna NT. Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004;14:1394–1403. doi: 10.1101/gr.2289704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 2018;34:3094–3100. doi: 10.1093/bioinformatics/bty191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen S, Zhou Y, Chen Y, Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 2018;34:i884–i890. doi: 10.1093/bioinformatics/bty560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Warris S, Schijlen E, van de Geest H, Vegesna R, Hesselink T, te Lintel HB, et al. Correcting palindromes in long reads after whole-genome amplification. BMC Genomics. 2018;19:798. doi: 10.1186/s12864-018-5164-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27:722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kolmogorov M, Yuan J, Lin Y, Pevzner PA. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol. 2019;37:540–546. doi: 10.1038/s41587-019-0072-8. [DOI] [PubMed] [Google Scholar]
- 29.Walker BJ, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, et al. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE. 2014;9:e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L, et al. NCBI Prokaryotic Genome Annotation Pipeline. Nucleic Acids Res. 2016;44:6614–6624. doi: 10.1093/nar/gkw569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones P, Binns D, Chang H-Y, Fraser M, Li W, McAnulla C, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019;47:D427–D432. doi: 10.1093/nar/gky995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lu S, Wang J, Chitsaz F, Derbyshire MK, Geer RC, Gonzales NR, et al. CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 2020;48:D265–D268. doi: 10.1093/nar/gkz991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pandurangan AP, Stahlhacke J, Oates ME, Smithers B, Gough J. The SUPERFAMILY 2.0 database: a significant proteome update and a new webserver. Nucleic Acids Res. 2019;47:D490–D494. doi: 10.1093/nar/gky1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kneubehl AR, Krishnavajhala A, Leal SM, Replogle AJ, Kingry LC, Bermúdez SE, et al. Comparative genomics of the Western Hemisphere soft tick-borne relapsing fever Borreliae highlights extensive plasmid diversity. BMC Genomics. 2022;23:410. doi: 10.1186/s12864-022-08523-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kuleshov KV, Margos G, Fingerle V, Koetsveld J, Goptar IA, Markelov ML, et al. Whole genome sequencing of Borrelia miyamotoi isolate Izh-4: reference for a complex bacterial genome. BMC Genomics. 2020;21:16. doi: 10.1186/s12864-019-6388-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30:3059–3066. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Minh BQ, Schmidt HA, Chernomor O, Schrempf D, Woodhams MD, von Haeseler A, et al. IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol Biol Evol. 2020;37:1530–1534. doi: 10.1093/molbev/msaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marçais G, Delcher AL, Phillippy AM, Coston R, Salzberg SL, Zimin A. MUMmer4: a fast and versatile genome alignment system. PLOS Comput Biol. 2018;14:e1005944. doi: 10.1371/journal.pcbi.1005944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y, Tang H, DeBarry JD, Tan X, Li J, Wang X, et al. MCScanX: a toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012;40:e49. doi: 10.1093/nar/gkr1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seemann T. Prokka: rapid prokaryotic genome annotation. Bioinformatics. 2014;30:2068–2069. doi: 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 42.Page AJ, Cummins CA, Hunt M, Wong VK, Reuter S, Holden MTG, et al. Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics. 2015;31:3691–3693. doi: 10.1093/bioinformatics/btv421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chernomor O, von Haeseler A, Minh BQ. Terrace aware data structure for phylogenomic inference from supermatrices. Syst Biol. 2016;65:997–1008. doi: 10.1093/sysbio/syw037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS. UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol. 2018;35:518–522. doi: 10.1093/molbev/msx281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Minh BQ, Hahn MW, Lanfear R. New methods to calculate concordance factors for phylogenomic datasets. Mol Biol Evol. 2020;37:2727–2733. doi: 10.1093/molbev/msaa106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mo YK, Lanfear R, Hahn MW, Minh BQ. Updated site concordance factors minimize effects of homoplasy and taxon sampling. Bioinformatics. 2023;39:btac741. [DOI] [PMC free article] [PubMed]
- 47.Barbour AG. Multiple and diverse vsp and vlp sequences in Borrelia miyamotoi, a hard tick-borne zoonotic pathogen. PLoS ONE. 2016;11:e0146283. doi: 10.1371/journal.pone.0146283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Margos G, Fingerle V, Cutler S, Gofton A, Stevenson B, Estrada-Peña A. Controversies in bacterial taxonomy: the example of the genus Borrelia. Ticks Tick-Borne Dis. 2020;11:101335. doi: 10.1016/j.ttbdis.2019.101335. [DOI] [PubMed] [Google Scholar]
- 49.Chaconas G, Norris SJ. Peaceful coexistence amongst Borrelia plasmids: getting by with a little help from their friends? Plasmid. 2013;70:161–167. doi: 10.1016/j.plasmid.2013.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Elbir H, Abi-Rached L, Pontarotti P, Yoosuf N, Drancourt M. African relapsing fever Borreliae genomospecies revealed by comparative genomics. Front Public Health. 2014;2:43. doi: 10.3389/fpubh.2014.00043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mongodin EF, Casjens SR, Bruno JF, Xu Y, Drabek EF, Riley DR, et al. Inter- and intra-specific pan-genomes of Borrelia burgdorferi sensu lato: genome stability and adaptive radiation. BMC Genomics. 2013;14:693. doi: 10.1186/1471-2164-14-693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Casjens SR, Di L, Akther S, Mongodin EF, Luft BJ, Schutzer SE, et al. Primordial origin and diversification of plasmids in Lyme disease agent bacteria. BMC Genomics. 2018;19:218. doi: 10.1186/s12864-018-4597-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Barker SC, Barker D. Ticks of Australasia: 125 species of ticks in and around Australia. Zootaxa. 2023;5253:1–670. doi: 10.11646/zootaxa.5253.1.1. [DOI] [PubMed] [Google Scholar]
- 54.Voltzit OV, Keirans JE. A review of Asian Amblyomma species (Acari, Ixodida, Ixodidae) Acarina. 2002;10:95–136. [Google Scholar]
- 55.Estrada-Peña A, Álvarez-Jarreta J, Cabezas-Cruz A. Reservoir and vector evolutionary pressures shaped the adaptation of Borrelia. Infect Genet Evol. 2018;66:308–318. doi: 10.1016/j.meegid.2018.03.023. [DOI] [PubMed] [Google Scholar]
- 56.Estrada-Peña A, Sprong H, Cabezas-Cruz A, de la Fuente J, Ramo A, Coipan EC. Nested coevolutionary networks shape the ecological relationships of ticks, hosts, and the Lyme disease bacteria of the Borrelia burgdorferi (s.l.) complex. Parasit Vectors. 2016;9:517. doi: 10.1186/s13071-016-1803-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nakao R, Kasama K, Boldbaatar B, Ogura Y, Kawabata H, Toyoda A, et al. The evolution of hard tick-borne relapsing fever Borreliae is correlated with vector species rather than geographical distance. BMC Ecol Evol. 2021;21:105. doi: 10.1186/s12862-021-01838-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pavón-Vázquez CJ, Brennan IG, Skeels A, Keogh JS. Competition and geography underlie speciation and morphological evolution in Indo-Australasian monitor lizards. Evolution. 2022;76:476–495. doi: 10.1111/evo.14403. [DOI] [PubMed] [Google Scholar]
- 59.Vidal N, Marin J, Sassi J, Battistuzzi FU, Donnellan S, Fitch AJ, et al. Molecular evidence for an Asian origin of monitor lizards followed by Tertiary dispersals to Africa and Australasia. Biol Lett. 2012;8:853–855. doi: 10.1098/rsbl.2012.0460. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. Summary of Australian varanid tick samples tested for Borrelia. Fig. S1. Pairwise alignment plots of Borrelia salvatorii strains RT1S and RT5S core plasmids (A), accessory plasmids (B), and duplicated plasmids (C). Fig. S2. Schematic alignment of the ribosomal RNA operon of relapsing fever (RF), reptile-associated (REP), and Lyme borreliosis (LB) Borrelia highlighting the deletion of the mag gene, which is synapomorphic for varanid-associated Borrelia spp. Fig. S3. MCscan karyotype orthologous protein coding genes between the conserved large linear megaplasmids of Borrelia salvatorii and ‘Candidatus (Ca.) Borrelia rubricentralis’, ‘Ca. Borrelia undatumii’, Borrelia turcica, ‘Ca. Borrelia tachyglossi’, Borrelia miyamotoi, and Borrelia hermsii. These large linear megaplasmids appear to be highly conversed among all REP and RF Borrelia. Fig. S4. MCscan plot showing orthologous protein-coding genes between Borrelia burgdorferi B31 plasmid cp26 and Borrelia turcica IST7 plasmid lp25 and Borrelia salvatorii RT1S cp27. Fig. S5. Schematic of varanid-associated Borrelia spp. and the approximate locations of putative plasmid partitioning genes, and variable large proteins. Fig. S6. Schematic gene map of vlp gene clusters in REP Borrelia replicons, including vlp1 (green), which was homologous in all genomes. Fig. S7. Phylogenetic reconstruction of REP and RF Borrelia vlp amino acid sequences. Extended caption for Fig. 5. The isolate, open reading frame number, and plasmid name is given for each sequence.
Data Availability Statement
All genomic data are available through NCBI BioProjects PRJNA783659 and PRJNA774887.