

Abstract
Aneuploidy is generally considered harmful, but in some microorganisms, it can act as an adaptive mechanism against environmental stress. Here, we use Leishmania—a protozoan parasite with remarkable genome plasticity—to study the early steps of aneuploidy evolution under high drug pressure (using antimony or miltefosine as stressors). By combining single‐cell genomics, lineage tracing with cellular barcodes, and longitudinal genome characterization, we reveal that aneuploidy changes under antimony pressure result from polyclonal selection of pre‐existing karyotypes, complemented by further and rapid de novo alterations in chromosome copy number along evolution. In the case of miltefosine, early parasite adaptation is associated with independent point mutations in a miltefosine transporter gene, while aneuploidy changes only emerge later, upon exposure to increased drug levels. Therefore, polyclonality and genome plasticity are hallmarks of parasite adaptation, but the scenario of aneuploidy dynamics depends on the nature and strength of the environmental stress as well as on the existence of other pre‐adaptive mechanisms.
Keywords: adaptation, aneuploidy, cellular barcodes, Leishmania, single‐cell genomics
Subject Categories: Chromatin, Transcription & Genomics; Evolution & Ecology; Microbiology, Virology & Host Pathogen Interaction
The dynamics of aneuploidy, a hallmark of Leishmania adaptation, depends on the nature and strength of the environmental stress as well as on the existence of other pre‐adaptive mechanisms.

Introduction
Aneuploidy, that is, a dosage imbalance between chromosomes in a cell, is commonly lethal or associated with deleterious effects, in particular in multicellular organisms (Ricke & van Deursen, 2013; Chunduri & Storchová, 2019). In some unicellular eukaryotes however, aneuploidy can be well‐tolerated or even beneficial. It can be found in pathogenic and non‐pathogenic unicellular eukaryotes, including Saccharomyces cerevisiae, Candida albicans, Cryptococcus neoformans, and Giardia intestinalis (Mulla et al, 2014; Tůmová et al, 2016; Yang et al, 2021), with specific aneuploidy changes being able to confer resistance against environmental stresses such as drug pressure (Yang et al, 2019). Additionally, aneuploidy is also a hallmark of cancer, where it is associated with the therapeutic resistance, either by promoting dosage changes of key genes or by causing delays in drug‐targeted cell cycle stages (Replogle et al, 2020; Ippolito et al, 2021).
In recent years, Leishmania spp. emerged as a unique model for studying aneuploidy and its adaptive role. These protozoan parasites cause leishmaniases and display a digenetic life cycle characterized by two main forms: the extracellular promastigote in the midgut lumen of female sand flies, and the amastigote in phagocytic cells of the vertebrate hosts (Mann et al, 2021). Leishmania spp. display several idiosyncratic genomic and molecular features compared to other eukaryotes. Their genomes lack gene‐specific RNApol II promoters and are organized in long polycistronic arrays encompassing hundreds of genes which are not functionally related (Clayton, 2019). Transcription is thus initiated at defined chromosomal locations known as strand switch regions, which flank the polycistrons. In this context, gene dosage has a nearly one‐to‐one impact on transcription (Dumetz et al, 2017) and directly affect gene expression, which otherwise is mainly controlled post‐transcriptionally.
Unlike the abovementioned organisms where euploid genomes are common, all Leishmania genomes analyzed hitherto are aneuploid, with the most basic profile being characterized by a polysomy in chromosome 31, contrasting with a disomy in the other 33–35 chromosomes (Rogers et al, 2011; Mannaert et al, 2012). Additional dosage changes affecting multiple chromosomes (up to 22 out of 36 chromosomes) are commonly observed in cultured promastigotes (Imamura et al, 2016) and are associated with a fitness gain in vitro (Bussotti et al, 2018, 2021), but they also occur—to a lower extent—in amastigotes, in vivo (Domagalska et al, 2019). Recently, a multi‐omics study demonstrated a proportional impact of polysomies on the average expression of proteins encoded by the respective polysomic chromosomes, ultimately correlated with metabolic adaptations (Cuypers et al, 2022). Moreover, aneuploidy in Leishmania is highly dynamic and changes in response to new environments, such as drug pressure, vertebrate host or vector (Dumetz et al, 2017, 2018). Interestingly, spontaneous karyotypic modifications constantly occur even among sister cells in clonal populations, a phenomenon known as mosaic aneuploidy (Sterkers et al, 2011; Negreira et al, 2021). It is postulated that mosaic aneuploidy in Leishmania leads to phenotypic heterogeneity that can serve as substrate for natural selection, facilitating adaptation to different environmental pressures (Sterkers et al, 2012), but this remains an open question. Moreover, the clonal dynamics of populations facing strong environmental stresses and its relationship with aneuploidy modifications are currently unknown.
In the present study, we aimed to address these questions using a reproducible in vitro evolutionary model to study karyotype evolution in the context of early adaptation to sudden environmental stresses, invoked here by the direct exposure to high concentrations of two drugs, trivalent antimonial (SbIII) or miltefosine (further called ‘flash selection’). By combining single‐cell genome sequencing (SCGS), clonal lineage tracing with cellular barcodes and longitudinal genomic characterization, we showed that changes in aneuploidy under SbIII pressure arise from the polyclonal selection of pre‐existing karyotypes which converge to similar aneuploidy profiles through further cumulative karyotypic alterations. In contrast, adaptation to miltefosine was initially driven by the polyclonal selection of distinct missense mutations in a miltefosine transporter gene (LdMT), with aneuploidy modifications only happening after the already‐selected populations were exposed to a 4 times higher dosage. Thus, the dynamics of aneuploidy modulations is contingent upon the specific environmental stressors, their intensity, and the presence of other pre‐adaptive mechanisms.
Results and Discussion
In the present study, we investigated the mechanisms governing the early adaptation of Leishmania promastigotes by directly exposing a L. donovani promastigote clonal strain (BPK282) to a high concentration of SbIII (382 μM) or miltefosine (25–100 μM) in vitro. For each model—which we refer as ‘flash selection’—we followed two molecular approaches: (i) bulk and SCGS, with a special focus on aneuploidy and (ii) lineage tracing with cellular barcodes (Fig EV1A and B). This allowed us to track the evolutionary dynamics of hundreds of lineages under drug pressure, revealing the bottlenecks associated with each model and the relationship between selection of lineages and the emergence of genomic adaptations.
Figure EV1. Schematic representing the cellular barcoding strategy.
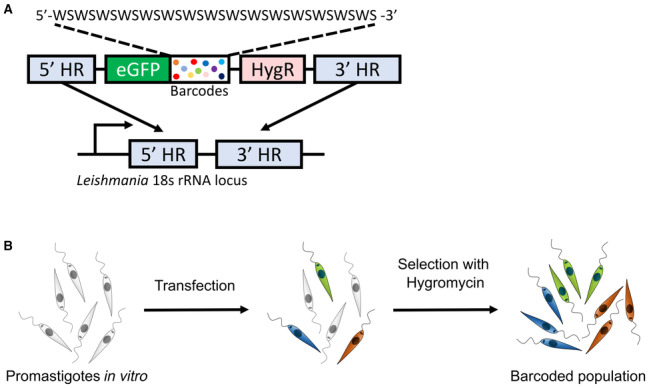
- Briefly, a double‐stranded oligonucleotide is synthesized bearing a semi‐random sequence formed by the alternation of weak (W = A or T) and strong bases (S = G or C) flanked by two fixed sequences that serve as primer binding sites for PCR amplification. This pool of DNA molecules is cloned in a vector that has homology sequences which promote the integration of the vector into the 18s rRNA locus in Leishmania genome.
- After transfection, barcoded parasites are selected with hygromycin, as the barcoding vector also has a hygromycin‐resistance gene. The eGFP gene allow to monitor by flow cytometry the potential presence of remaining non‐barcoded cells after hygromycin selection.
Flash selection with SbIII leads to rapid changes in aneuploidy
The first flash selection protocol we used was previously developed in a study on resistance to trivalent antimonial (SbIII), in which resistant parasites with fully restored growth to wild‐type levels were observed after 5 weeks (and passaged every 7 days) of direct exposure to 382 μM SbIII (Dumetz et al, 2018). We reproduced the experiment with a barcoded BPK282 and determined by bulk whole genome sequencing (WGS) the genomic alterations encountered in the populations along five passages under SbIII pressure and in four replicates (further called SePOP1‐4). The search for SNPs and indels in coding regions did not reveal any consistent change associated with the SbIII pressure compared to the control populations (cPOP1‐4) which were maintained with PBS in place of SbIII (Fig EV2A). We also evaluated intrachromosomal copy number variations with a specific attention for the MRPA gene (chromosome 23) which encodes an ABC transporter involved in SbIII sequestration (Frézard et al, 2014) that is often amplified in SbIII resistant Leishmania (Dumetz et al, 2018). In 3 out of 4 replicates, the copy number of the MRPA locus remained stable at ~ 3 copies per haploid genome similarly to the initial condition as well as to cPOP1‐4, with the exception of SePOP1, which displayed an increase to almost 10 copies per haploid genome at passage 5 (Fig EV2B). When investigating aneuploidy variation, we found that BPK282 contained six chromosomes with somy higher than 2 (chr 5, 9, 16, 23, 26, and 31) at the onset of the experiment, and we found a further dosage increase affecting 5–8 chromosomes at passages 2–3 in SePOP1‐4 after SbIII exposure (Fig 1A). Interestingly, although distinct aneuploidy profiles emerged in different replicates, all four replicates consistently shared a dosage increase of chromosomes 23, 27, and 31, which could point to an adaptive advantage of the amplification of these chromosomes.
Figure EV2. Additional genomic changes associated with the SbIII‐Flash selection performed on the barcoded BPK282 population.
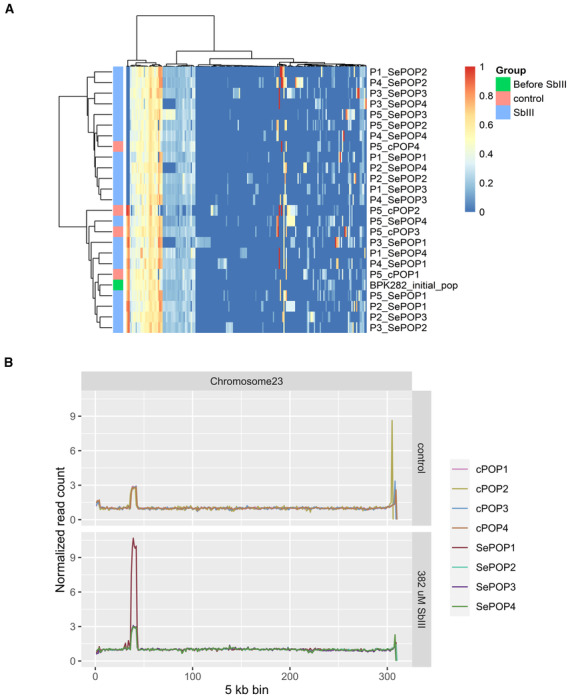
- Heatmap depicting the allele frequencies of SNPs and indels identified in protein coding regions compared to the reference genome. An additional annotation bar display to which group (control or SbIII‐exposed) a sample belongs. Samples are named as Px_cPOPy for controls, and Px_SePOPy for the SbIII‐exposed groups, with x being the number of passages and y being the replicate number. The initial population is named as ‘Before SbIII’.
- Copy number variation in the MRPA locus. The Y axis represents the median read count of 5 kb bins normalized by the median count of chromosome 23 and reflects the average copy number per haploid genome of the MRPA locus.
Figure 1. Aneuploidy changes of L. donovani BPK282 during flash selection with SbIII .
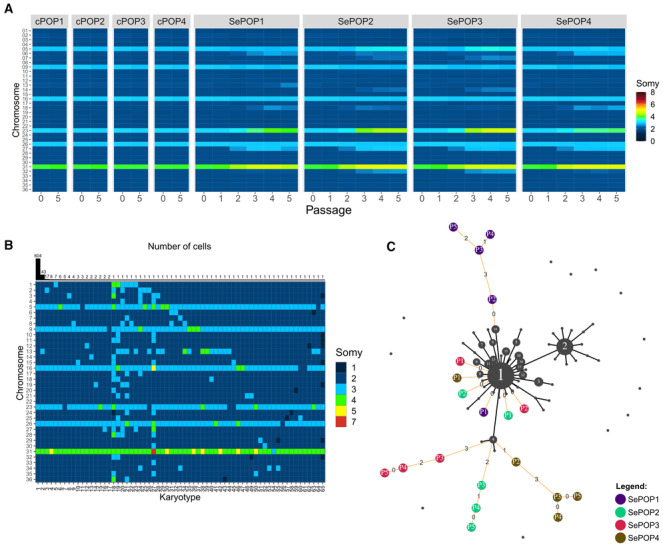
- Bulk genome sequencing: Heatmap showing the average copy number of each chromosome between passages 0 (before drug exposure) and passage 5 in cPOP1‐4 and along all 5 passages in the four replicates under SbIII flash selection (SePOP1‐4).
- Heatmap depicting all karyotypes identified in the barcoded population prior to SbIII‐exposure using SCGS. Karyotypes are ordered decreasingly based on their frequency. Bars on the top display the number of promastigotes found with each karyotype.
- Minimum spanning tree displaying the number of somy changes between the karyotypes identified in the single‐cell data (black nodes) and the rounded bulk aneuploidy of the SbIII‐selected populations (colored nodes: purple = SePOP1, green = SePOP2, red = SePOP3 and brown = SePOP4) at passages 1–5 (P1‐P5); we based this analysis on the previous observation that the rounded bulk aneuploidy profile of a given promastigote population reflects the most dominant karyotype in that population (Negreira et al, 2021). Black lines connecting two nodes indicate that these two karyotypes are different by a single somy change. Orange lines connect the bulk karyotypes of the SePOP1‐4 to the single‐cell data. Numbers in the orange lines indicate how many somy changes are between the nodes connected by them. Unconnected black nodes are single‐cell karyotypes that have 2 or more somy differences compared to any other karyotype.
Source data are available online for this figure.
The dosage increase of these chromosomes, in particular chromosome 23, is commonly reported in several antimony resistance studies, including in other L. donovani strains (Dumetz et al, 2018) as well as other Leishmania species such as L. infantum, L. guyanensis, L. braziliensis, and L. panamensis (Brotherton et al, 2013; Monte‐Neto et al, 2015; Patino et al, 2019). The line here used (BPK282) is remarkably pre‐adapted to SbIII (Dumetz et al, 2018)—like other strains of the Gangetic plain—thanks to a pre‐existing intrachromosomal amplification of MRPA genes encountered in 200 sequenced L. donovani isolates of that region (Imamura et al, 2016). This pre‐adaptation likely comes from the combination of high antimony pressure for decades, highly endemic pollution with arsenic—which can cause cross‐resistance to antimonials (Perry et al, 2011, 2013)—and anthroponotic transmission without animal reservoir. The recurrent dosage increase of chromosome 23 observed here under SbIII pressure is thus a rapid way to further amplify the MRPA gene. Noteworthy, even in parasites in which MRPA was artificially deleted, chromosome 23 still consistently displayed increase in copy number in populations selected for antimony resistance, suggesting that other genes in this chromosome beyond MRPA might be relevant to antimony tolerance (Douanne et al, 2020).
Regarding the other two chromosomes, chromosome 31 also bears a gene involved in antimony resistance, the sodium stibogluconate resistance protein gene (LdBPK_310951.1). Interestingly, the ortholog of this gene displayed an increased copy number in L. braziliensis promastigotes experimentally selected for antimony resistance in vitro compared to non‐selected lines (Patino et al, 2019). Moreover, this same study found a 50 kb intrachromosomal amplification affecting 23 genes (out of a total of 31 amplified genes) in chromosome 27 in the SbIII resistant line, with many of these genes displaying a copy number more than 10 times higher compared to the SbIII sensitive line (Patino et al, 2019). Among these genes, a WW domain/Zinc finger CCCH type‐protein gene (LdBPK_270130.1 ortholog in L. donovani) was also the gene with the most upregulated expression compared to the SbIII‐sensitive line. Importantly, CCCH type zinc finger proteins are known targets of antimony (Frézard et al, 2014), and therefore, a higher expression of this gene might mitigate its inactivation by the drug.
Single‐cell genomics reveals potential evolutionary paths that led to SbIII‐associated aneuploidy changes
To evaluate if the aneuploidy changes observed in the SbIII‐exposed populations are due to the selection of pre‐existing or de novo generated karyotypes, we submitted the same barcoded cell population to high throughput SCGS prior to flash selection. In total, 864 promastigotes were individually sequenced, with a total of 65 different karyotypes (kar1‐65) being detected (Fig 1B). These single‐cell data revealed a relatively reduced mosaicism, with almost 70% of the parasites displaying the same karyotype (kar1). None of the pre‐existing karyotypes were identical to the aneuploidy profiles observed in bulk in SePOP1‐4 (Fig 1B). However, individual somy changes consistently observed in SePOP1‐4 (tetrasomy in chromosome 23, trisomy in chromosome 27 and pentasomy in chromosome 31) were already present—in few cells—before the flash selection. For instance, kar18—one of the most aneuploid karyotypes, had a tetrasomic chromosome 23 and a trisomic chromosome 27, while kar38 and kar50 both shared amplification of chromosome 23 and 31. Other single cells showed dosage increase of one of these chromosomes only, for instance, kar15 that only showed tetrasomy of chromosome 23. However, none of the sequenced promastigotes showed amplification of chromosomes 23, 27, and 31 concomitantly, and no pre‐existing karyotype was identified with a pentasomy in chromosome 23 as observed in the SePOP3, suggesting that some of the aneuploidy changes seen in SePOP1‐4 happened either after initial exposure to SbIII or that they were present at a frequency lower than the detection limit of our SCGS data (1 in 864 or 0.116%).
To gain insights on possible evolutionary paths leading to the aneuploidy changes observed in the SePOP1‐4 (bulk data) from the initial population (single‐cell data), we built a minimum spanning tree connecting the karyotypes found in the single‐cell data (Fig 1C—black nodes) to the rounded aneuploidy profiles of SePOP1‐4 (Fig 1C—colored nodes). This approach revealed that the shortest path between pre‐existing karyotypes and the selected karyotypes in SePOP1 starts from kar16, which has the same aneuploidy profile as the one observed in this population at passage 2, characterized by a trisomic chromosome 27. For SePOP2‐4, the closest pre‐existing karyotype is kar4, which has a pentasomic chromosome 31. At passage 2, SePOP4 is almost identical to kar4, being one step away from this karyotype. This single somy difference is due to a trisomy in chromosome 27 which is not present in kar4. From passage 2–3, SePOP4 accumulated 3 extra somy changes (trisomy in chromosomes 6 and 18 and a tetrasomy in chromosome 23). For SePOP2 and SePOP3, the first aneuploidy changes emerged at passage 3 and had already 2 and 3 somy differences compared to kar4, respectively. Thus, the tree points to a process of polyclonal selection of subpopulations bearing pre‐existing chromosomal amplifications followed by cumulative karyotypic modifications in subsequent time points in SePOP1‐4.
Changes in aneuploidy in SbIII selection arise from polyclonal selection and convergent evolution
To document the cell population dynamics during adaptation to SbIII, we applied cellular barcodes to track the evolution of hundreds of clonal lineages during flash selection. In summary, we generated a barcoded promastigote population consisting of 453 different traceable lineages. The frequency of each lineage in each SePOP was monitored by amplicon sequencing. This approach revealed that the flash selection with SbIII led to a fourfold reduction in lineage diversity that stabilized between passages 3 and 4, leaving between 101 and 131 of detectable lineages (Fig EV3A).
Figure EV3. Supporting figures for main Fig 2 .
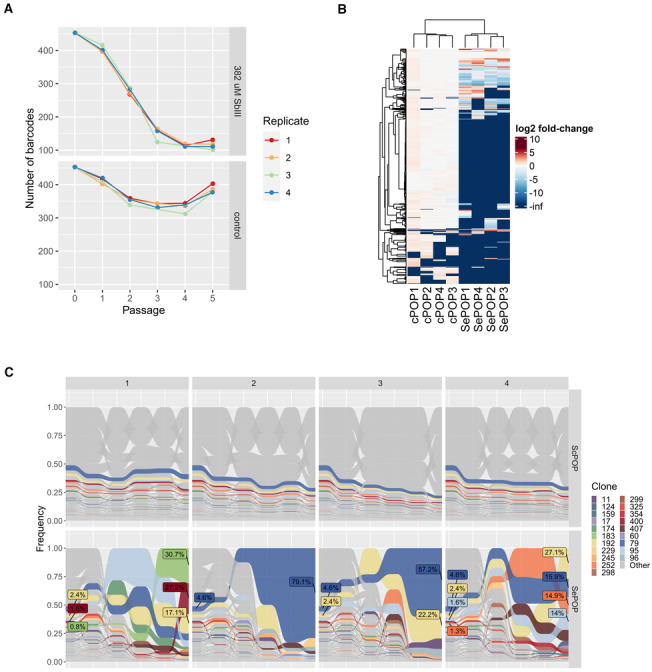
- Total number of different barcodes identified in each population at each timepoint in the SbIII‐exposed populations (top) and the controls (bottom).
- Heatmap displaying the SbIII‐associated fold‐change of each lineage (rows) in each population (columns) at passage 5.
- Frequency of each barcoded lineage along the 5 passages in the ScPOP1‐4 (top) and SePOP1‐4 (bottom) populations. This is similar to main Fig 2F but includes the ScPOP1‐4 for comparison. Only lineages that reached a frequency higher than 1% at passage 5 in at least one of the SePOP populations are colored.
To distinguish frequency changes associated with SbIII pressure from other sources of variation, such as stochastic loss due to passaging, we normalized the frequency of each lineage in each SePOP to their respective average frequency in the cPOP at each passage (Fig 2A). Here we refer to this parameter as “SbIII‐associated frequency change”. In this sense, lineages that, for example, die or are negatively affected under antimony pressure but not under standard in vitro conditions display a negative SbIII‐associated frequency change, while lineages that are eliminated over time in both cPOP and SePOP display similar values. This analysis showed that a large fraction of lineages representing a total 74.6–84.8% of the initial population was negatively affected or completely eliminated during SbIII pressure (Fig 2B). This fraction was represented by a total of 362–395 lineages, including 303 which were consistently affected negatively in all four SePOP (Fig 2C, left panel, red), suggesting that this subset of lineages had a lower tolerance to SbIII stress. Interestingly, the fraction of positively affected lineages represented 5.2–8.4% of the initial population (Fig 2B), with only 60 lineages displaying a frequency higher in the drug‐exposed group compared to the controls at passage 5. This subset became dominant to represent 77.7–97.4% of the final populations at passage 5. Moreover, most of the positively affected lineages were enriched in only one of the SePOPs (Figs 2C and EV3B). Altogether, these data suggest that (i) a subset of lineages was fitter to SbIII prior to the drug exposure and (ii) the further expansion of these surviving lineages was divergent between independent replicates.
Figure 2. Clonal dynamics of SbIII adaptation revealed with cellular barcodes.
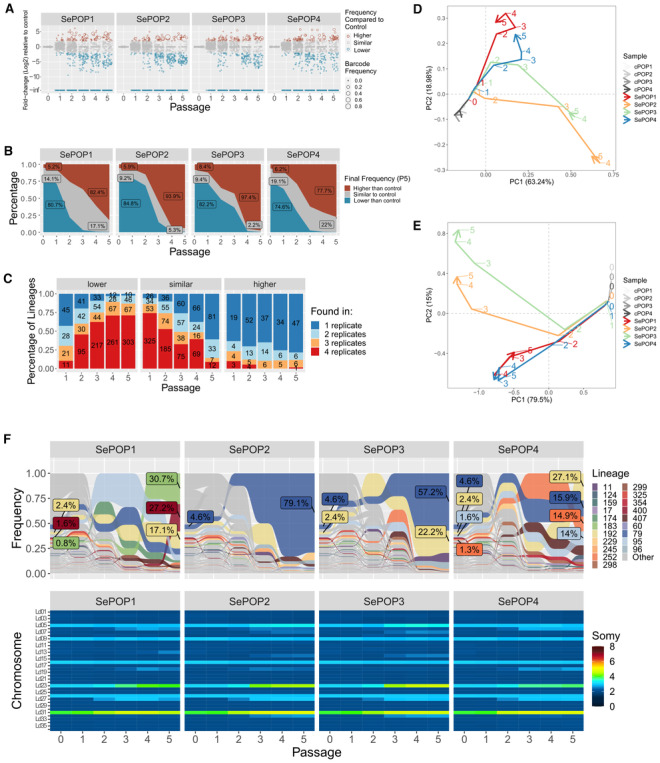
- Fold‐change of the frequency of each clone in the SbIII‐exposed group (4 replicates—SePOP1‐4) relative to their average frequency in the control group in the same time point (SbIII‐associated fold change). Each dot represents a barcoded lineage. Lineages with a log2 SbIII‐associated fold change smaller than −2 or greater than +2 were considered negatively and positively affected respectively. Lineages with fold‐change at ‐infinite are lineages that were eliminated under drug pressure.
- Fraction of lineages that by passage 5 were either positively affected (red) or negatively affected (blue) by SbIII pressure.
- Evaluation of the consistency of the SbIII‐associated fold change scores among replicates. The bars represent the proportion of clones that had a particular SbIII‐associated fold‐change effect (higher, lower or same as in control) in 1, 2, 3, or 4 replicates (dark blue, light blue, orange, and red, respectively). The numbers in the bars indicate the absolute number of barcodes.
- Trajectory principal component analysis (PCA) of the changes in clonal composition over time in each population. This was done by performing a regular PCA on each dataset and connecting the data points of each population with an arrow line indicating their trajectories from passage 0 to passage 5 The PCA was based on the frequency of each lineage identified in each sample. The numbers indicate the passages at which each sample was collected, while colors indicate the populations.
- Trajectory PCA depicting the changes in aneuploidy in all samples. Numbers indicate the passage number at which a sample was collected for WGS. Due to strong similarity between controls, they are not well visible in the PCAs as they cluster very close to each other.
- Frequency of each barcoded lineage along the five passages under SbIII pressure (top panel). Only barcodes that reached frequencies higher than 1% at passage 5 in at least one of the replicates were colored. A repetition of Fig 1A is included for comparison (bottom panel).
Source data are available online for this figure.
In an attempt to link the evolution of lineages as revealed by barcoding sequencing with the aneuploidy modifications observed in the bulk WGS of SePOP1‐4, we processed each dataset with a ‘trajectory’ principal component analysis (PCA) and compared them (Fig 2D and E). The former one revealed that lineage composition progressively diverged between replicates, with SePOP1/4 deviating further from SePOP2/3 at later passages (Fig 2D). Interestingly, this PCA based on lineage composition resembled the aneuploidy‐based PCA shown in Fig 2E, with SePOP1/4 and 2/3 clustering separately, suggesting that changes in aneuploidy coincide with changes in lineage composition. This observation is supported when comparing the absolute frequency of the lineages with the aneuploidy changes observed in bulk at each timepoint (Figs 2F and EV3C). Here we observe that the major aneuploidy alterations arise at passages 2 to 3, coinciding with the moment where the most dominant lineages in the initial population are depleted while other lineages expand. It is also noticeable that the aneuploidy changes in SePOP2/3 are almost identical, and these two populations were dominated by the same lineage (lineage 79). Conversely, SePOP1/4 also share similar aneuploidy changes, though different from replicates 2 and 3, but lineage composition seems to be less similar. In addition, the lineage tracing data also indicated that lineages that were selected under SbIII exposure were already at frequencies above the detection limit of our SCGS data (see Fig 2F, passage 0). Thus, the most likely explanation for the aneuploidy changes seen in the bulk WGS data of SePOP1‐4 is that the karyotypes selected under drug pressure indeed originated from pre‐existing karyotypes but underwent further and rapid cumulative changes in chromosome number under SbIII pressure. Moreover, the observation that the same set of three chromosomes displayed dosage increases in all SePOP despite the fact that different lineages dominated each SePOP points to a process of convergent evolution which further supports the notion of these chromosomes being under positive selection.
Adaptation to miltefosine is associated with polyclonal selection of nucleotide variants
The results described above demonstrated the importance of aneuploidy for parasite adaptation to high SbIII pressure together with the polyclonality of corresponding molecular adaptations. In the next step, we investigated whether the same mechanisms would be associated with adaptation to another anti‐Leishmania drug: miltefosine. Noteworthy, BPK282 was isolated from the main population endemic in the Gangetic plain, before miltefosine was implemented in the region (in sharp contrast to SbIII). Hence, different results were expected for the scenario of genomic adaptation and clonal dynamics. In contrast to SbIII, there was—at least before present study—no pre‐adaptation known to miltefosine in the BPK282 strain, which is considered very susceptible to the drug (Shaw et al, 2016).
In order to initiate a flash selection with miltefosine, we first determined which was the highest concentration of the drug in which viable parasites could still be recovered: four replicates were considered per condition (MIL‐exposed populations, MePOP1‐4; Fig EV4A). The cultures with 50 and 100 μM of miltefosine did not display live parasites even after 1 month post addition of the drug (no passaging). However, in the cultures with 25 μM of MIL, although no live parasites could be detected by microscopy until the 10th day, viable promastigotes started to arise afterwards, and by the 17th day post addition of MIL, MePOP1‐4 displayed a relative cell density comparable to the controls as estimated by microscopy. These populations of survivors displayed such a higher tolerance to miltefosine that an attempt to determine their IC50 against the drug was not successful as the cultures did not suffer a reduction in viability even in the highest concentration used in the test (75 μM—Fig EV4B). Their respective IC50s were determined at passage 2 by including an additional concentration of 150 μM in the test and was defined as an average of 81.78 μM in MePOP1‐4 and 21.9 μM in the control group (P < 0.001, t‐test—Fig EV4C).
Figure EV4. Supporting figures for flash selection with miltefosine.
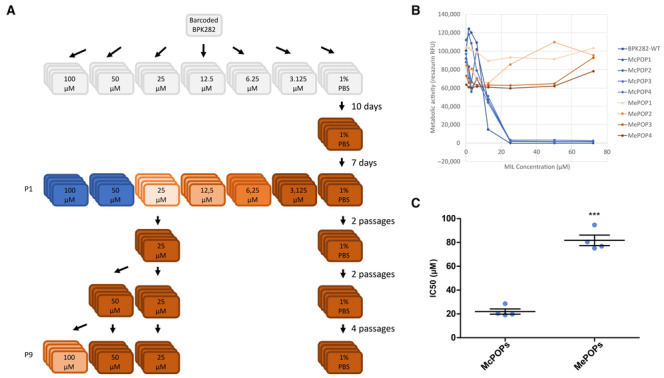
- Schematic representing the experimental design. Colors indicate the lack of viable parasites (blue) or the relative observed number of parasites at day 7 compared to the controls (darker brown = more cells).
- Dose–response to miltefosine of the metabolic activity of the populations at the first passage after exposure to the drug, estimated with the resazurin assay.
- Difference in IC50 of the same populations one passage later. ***P < 0.001 (t‐test).
When looking for genomic changes using bulk WGS, we found that the strong bottleneck associated with miltefosine exposure did not lead to any major alteration in aneuploidy, as MePOP1‐4 displayed the same profile as the initial population even after nine passages under miltefosine pressure at 25 μM (Fig 3A). However, subsequent exposure of the MePOPs to 100 μM for four passages did lead to drastic changes in aneuploidy affecting several chromosomes (Fig 3A). This demonstrated that the strong bottleneck induced by miltefosine in the first passage did not impair the potential for aneuploidy modulations in later passages and that these modifications depend on the strength of the stress caused by the drug. These observations are also in agreement with the notion of aneuploidy modulations happening de novo during adaptation to the drug as the aneuploidy profiles seen at passage 9 in the MePOPs exposed to 100 μM are also very different from the pre‐existing karyotypes identified in the single‐cell data of BPK282. In each MePOP a different profile was seen, although 3 out of 4 shared the amplification of chromosome 31, and two MePOPs displayed a dosage increase in chromosome 23. The fact that an increase in copy number of chromosome 31 was observed under strong SbIII and miltefosine pressure, as well as under pressure of other drugs (Hefnawy et al, 2022) might indicate that the dosage increase in this chromosome has also a general role against multiple types of stresses. Noteworthy, there are several ABC transporters in that chromosome (ABCC4‐7 and ABCD3) which could play a role in drug efflux (Leprohon et al, 2006). Moreover, ontology analysis of chromosome 31 in L. braziliensis demonstrated an enrichment of genes involved in iron metabolism which could play a role in general adaptation to oxidative stresses (Valdivia et al, 2015), but empirical evidence is still lacking.
Figure 3. Flash selection with miltefosine.
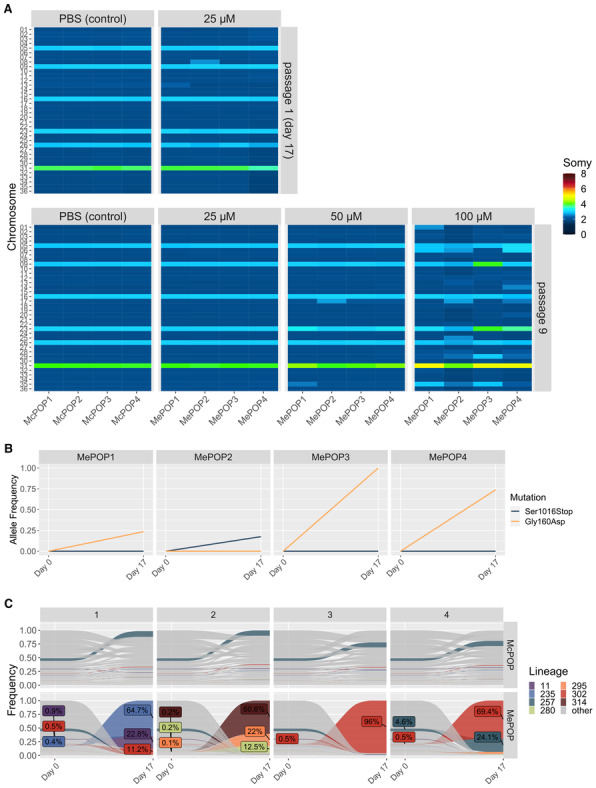
- Bulk aneuploidy profile of the populations kept under different concentrations of miltefosine (four replicates per concentration—MePOP1‐4) at passages 1 (upper panel) and passage 9 (bottom panel).
- Allele frequency of the Gly160Asp and the Ser1016Stop mutations identified in the miltefosine‐transporter gene (LdMT) in the drug‐exposed populations.
- Tracing of lineages before (day 0) and after 17 days (passage 1) under selection with 25 μM miltefosine (MePOP) or with PBS as control (McPOP). Only lineages that reached frequencies higher than 1% in at least one population in the last timepoint (day 17) are colored. Colored labels display the frequency of some lineages at day 0 and day 17.
Source data are available online for this figure.
In contrast to the SbIII model, relevant SNPs were encountered between MePOP1‐4 and the controls, already from the first passage. In particular, in the four selected populations, independent missense mutations arose in the phospholipid‐transporting ATPase1‐likeprotein gene, which is also known as the miltefosine transporter (LdMT—ID: LdBPK_131590.1). In MePOP1, 3 and 4 the same mutation promoting a substitution of a glycine by an aspartate at amino acid (Gly160Asp) was observed, while MePOP2 displayed a different mutation in the same gene leading to an insertion of a stop codon in place of a serine in the amino acid 1016 (Ser1016Stop—Fig 3B). Noteworthy, disruptive mutations in this gene are known to confer resistance to miltefosine in Leishmania (Pérez‐Victoria et al, 2006). Therefore, early adaptation to miltefosine was mainly driven by mutations in LdMT and not by aneuploidy modulations.
As the BPK282 population used in the MIL‐flash selection was the same barcoded population as in the SbIII‐flash selection, we also monitored clonal dynamics between passages 0 (before miltefosine‐exposure) and 1 (17 days under miltefosine pressure). This revealed that the bottleneck generated by miltefosine exposure was even stronger than the one associated with SbIII exposure, with only seven lineages surviving in at least one of the MePOP replicates, with one specific lineage (lineage 302) being present in 3 of the 4 replicates (MePOP1, 3 and 4) at passage 1 (Fig 3C). Interestingly, the frequency of lineage 302 in all three replicates where this lineage survived coincided with the allele frequency of the Gly160Asp mutation, suggesting this was a pre‐existing mutation present in this lineage. The different mutation seen in MePOP2 also coincides with the fact that this population was dominated by different lineages. The emergence of distinct lineages and mutants in the MePOPs thus illustrate a polyclonal origin of miltefosine resistance in this flash selection experiment.
Aneuploidy dynamics is thus clearly dependent on the nature and strength of the environmental stress. The contrast here observed between the aneuploidy dynamics under SbIII and miltefosine pressure could be explained by two main factors. (i) Aneuploidy changes are not selected under 25–50 μM miltefosine pressure, because another mechanism can promote survival of the parasites, that is, mutations in LdMT. (ii) The adaptive importance of aneuploidy may also depend on the needed gain or loss of expression for driving drug tolerance. The MRPA is responsible for sequestration of SbIII and a higher expression will increase parasite fitness under drug pressure: this can easily and rapidly be achieved by multiplying the effect of the MRPA amplification already present in BPK282, by increasing the somy of chromosome 23. In contrast, LdMT is responsible for uptake of miltefosine and in this case, a reduction in expression is driving resistance. Here chromosome 13 was already disomic and could probably not further decrease in somy and loss of function was achieved via the SNP ‘path’.
In nature, changes in environmental pressures are often abrupt rather than gradual, and therefore, demand for mechanisms which allow parasite populations to quickly adapt to the new environment. This abrupt change in environments—here represented by our flash selection models—is also a characteristic of drug treatment. In the case of antimonials, measures made in patients treated for visceral leishmaniasis estimate a peak of 10 mg/l or ~ 82 μM of Sb in the blood after only 2 h post drug administration (Chulay et al, 1988). For miltefosine, blood concentrations can be as high as 70 μg/ml, or 172 μM after 72 h (Dorlo et al, 2012). Moreover, bone marrow‐derived macrophages exposed to 10 μM of miltefosine in vitro display intracellular concentrations of the drug as high as 323 μM after 72 h (Voak et al, 2018). This illustrates that Leishmania parasites are directly exposed to sharp increases in drug concentrations—in the case of miltefosine, even higher than the concentrations used in this study—in patients upon drug administration.
In conclusion, our data support the role of mosaic aneuploidy in generating multiple pre‐adapted karyotypes that can be further modulated de novo during drug exposure. This process might depend on the nature and strength of the drug‐associated stress and might be potentiated by a stress‐induced increase in chromosome instability as seen in other organisms (Chen et al, 2012; Tan et al, 2019; Shen et al, 2020). Further research with longitudinal SCGS combined with lineage tracing is needed in order to validate these hypotheses. In addition, our studies should be complemented with in vivo models, where different environmental stresses can be encountered by the parasite and where the biology of the parasite is different (for instance low replication rate or deep‐quiescence; Kloehn et al, 2015; Jara et al, 2019). Finally, the new barcoding method developed and applied here could be used for characterization of clonal dynamics of Leishmania in experimental in vivo studies.
Materials and Methods
Abbreviations
In the present study, different types of samples are being studied and analyzed by bulk or single‐cell approaches. To avoid confusion, we list here the main terms used for sample description.
SePOP1‐4: SbIII exposed Populations; these are populations of cells that are analyzed by bulk methods (WGS, barcode amplification), with four replicates. Noteworthy, when we refer to the somy of a given chromosome in a SePOP, this represents an average value calculated on the population of sequenced cells; these values can be integers (most cells of that population show this somy) or intermediate (in that case, there are subpopulations with different somy, for instance a somy value of 2.5 may mean that ~ 50% of the population is disomic for that chromosome and ~ 50% is trisomic, or other combinations).
MePOP1‐4: idem but with miltefosine exposure.
cPOP1‐4 and McPOP1‐4: control populations not exposed to drugs and maintained in parallel to the drug‐exposed populations.
Promastigote culture
A clonal promastigote population of the L. donovani BPK282/0 strain (MHOM/NP/02/BPK282/0 clone 4) was maintained at 26°C in culture with HOMEM medium (Gibco™) supplemented with 20% of heat‐inactivated fetal calf serum with regular passages being performed every 7 days with 1 in 50 dilutions.
Flash selection with SbIII
A single culture of the barcoded BPK282/0 cl4 strain at 106 promastigotes/ml was divided in 2, where one was further diluted to a final concentration of 5 × 105/ml with medium containing 764 μM of potassium antimony tartrate (final concentration 382 μM, in the text referred to as SbIII) and the other was diluted to the same parasite concentration with a medium containing 0.2% of PBS instead (final PBS concentration 0.1%) as control. These two cultures were subsequently aliquoted into 4 culture flasks each, with a final volume of 5 ml per flask. Each culture was subcultured every 7 days for a total of 5 passages by transferring 2.5 × 106 promastigotes to a new flask containing fresh medium with either 382 μM SbIII or 0.1% PBS in a final volume of 5 ml. At the end of each passage (day 7), genomic DNA was extracted from ~ 108 parasites per flask using the QIAamp DNA Mini Kit (QIAGEN) for subsequent barcode and WGS.
Flash selection with miltefosine
Flash selection with miltefosine was initiated with the same barcoded BPK282 population as in the SbIII flash selection. Flasks containing 5 × 105 promastigotes per ml (final volume of 5 ml) received miltefosine at 25, 50 and 100 μM (4 replicates per concentration) and were maintained without passaging for 1 month, with four control replicates kept in parallel. After 17 days, the cultures at 25 μM displayed a full recovery of viability and were maintained for two additional passages done every 7 days with 2.5 × 106 promastigotes being transferred to new medium at each passage. At passage 3, the 25 μM cultures were divided in 2, one maintained at 25 μM and the other being exposed to 50 μM of miltefosine. At passage 5, the 50 μM cultures were also divided into 2, this time one kept with 50 μM and another with 100 μM. Cultures were maintained for four additional passages until the experiment was stopped.
Bulk WGS
For bulk WGS, ~ 108 promastigotes were pelleted, washed 3 times with PBS and had their genomic DNA extracted with the QIAamp DNA mini kit (QIAGEN). Sequencing libraries were prepared at GenomeScan (Netherlands) and submitted to 2 × 150 pair‐end sequencing in a NovaSeq™ 6000 sequencer (Illumina) targeting a sequence depth of ~ 50× per sample. Reads were mapped to the reference LdBPK282‐V2 genome (Dumetz et al, 2017) and read count was calculated along 5 kb bins. Removal of outlier bins and correction of GC‐bias were performed as previously established (Imamura & Dujardin, 2019). Somies were estimated as the median read count per bin of chromosomes normalized by the median read count per bin of the total genome and multiplied by 2.
Barcode library construction
With cellular barcodes, individual cells are ‘tagged’ with a short, random nucleotide sequence in their genomes which is transferred to daughter cells along cell division. Thus, the progeny of each barcoded cell shares the same barcode sequence, constituting a clonal lineage (further referred simply as ‘lineage’) that can be quantified with amplicon sequencing (Fig EV1). To generate a library of semi‐random barcodes, 20 nmole of an 80‐bp single‐stranded oligonucleotide (5′‐ACTGACTGCAGTCTGAGTCTGACAGWSWSWSWSWSWSWSWSWSWSWSWSWSWSWSAGCAGAGCTACGCACTCTATGCTAG‐3′) were synthesized by Integrated DNA Technologies™. The barcodes consisted of 15 repeats of A or T (W)—G or C (S) bases and were flanked by conserved regions for barcode amplification (Bhang et al, 2015). Second strand synthesis was performed with an extension reaction using the Advantage® 2 PCR Enzyme System with 1 μM (1.2 × 1013 molecules) of the single‐stranded oligo, 2 mM dNTP mix and 500 nM of primers GD90 and GD92 in a final volume of 50 μl. The sequence information for all primers used in this study can be found in Table EV1. The PCR mix was incubated in a thermocycler with denaturation at 95°C—1 min, 4 cycles of denaturation 95°C—15 s and Annealing/Extension at 68°C—1 min, and a final extension for 1 min at 68°C. Second strand synthesis was confirmed by gel electrophoresis. This reaction also generated 15‐bp sequences at both ends of the barcode‐fragment which were necessary for the downstream cloning of the barcode library into a plasmid vector. The vector was a pLEXSY‐Hyg2.1 (Jena Bioscience EGE‐272) plasmid that was previously modified by inserting an eGFP reporter gene into the NcoI and NotI sites. This vector was digested with NotI and MluI sites and gel purified. The double‐stranded barcode library was subsequently cloned into the linearized vector using the In‐Fusion® HD Cloning Kit (Clontech® Laboratories) following instructions of the manufacturer. The estimated vector:insert ratio for the reaction was 7.25 × 1010:1.8 × 1012 (≈ 1:25) molecules, which was optimized to keep the diversity of the barcode library. The whole cloning reaction was used to transform Stellar™ chemically competent cells (Clontech® Laboratories) using the heat shock method. After transformation, bacteria were seeded in multiple LB‐agar plates (⌀ 150 mm) containing 100 μg/ml ampicillin and were grown at 37°C for 16 h. All grown colonies were pooled by adding liquid LB medium to the plates and scraping off the colonies with a cell spreader. From this collected bacterial culture plasmids were extracted using the PureLink™ HiPure Plasmid Midiprep Kit (Thermo Fisher Scientific).
Generation and characterization of the barcoded L. donovani population
The barcoded plasmid pool purified from the bacterial cells was further linearized by a SwaI digestion, whereafter the resulting DNA fragments were separated by gel electrophoresis and the required 5,821 bp band was purified using the Wizard® SV Gel and PCR Clean‐Up System (Promega). In this linear form, this vector is flanked by homology sequences that promote its genomic integration into the 18S rDNA locus through spontaneous homologous recombination. For cellular barcoding, a Leishmania donovani clonal population (BPK282/0 cl4) derived from the MHOM/NP/02/BPK282/0 strain was maintained as promastigotes at 26°C in HOMEM medium (Gibco, ThermoFisher) supplemented with 20% Foetal Bovine Serum, with regular passages done every 7 days at 1/50 dilutions. At passage 30 after cloning, 8 × 107 promastigotes were transfected with 1 μg (≈ 1.68 × 1011 molecules) of the linearized barcoding library using the Basic Parasite Nucleofector™ kit 1 (Lonza) with the U‐033 program. After 2 days post‐transfection, barcoded parasites were continuously selected with Hygromycin B at a final concentration of 25 μg/ml, and the percentage of cells bearing the barcode vector was monitored by detection of the eGFP expression with confocal microscopy and/or flow cytometry. After two passages (14 days) under hygromycin selection, 22 subclones were derived using fluorescence‐activated cell sorting (FACS) and submitted to a PCR with primers GD124 and GD125 to amplify the barcode region for a subsequent Sanger sequencing with the primer GD126 in order to determine the presence or absence of a barcode in individual promastigotes as well as to detect potential multiple insertions.
Barcodes amplification and sequencing
Barcode amplification was done using the same DNA samples used for bulk WGS. Targeted amplification of the barcodes is needed as the number of reads containing a lineage barcode (~ 50 pair end reads per sample on average in our case) in the WGS data is insufficient for the determination of the frequency of each barcoded lineage in the parasite pool. Barcodes were amplified in a two‐step PCR approach using the Kapa HiFi HotStart ReadyMix PCR kit (Kapa Biosciences). The first PCR was done with primers NtBCampF and NtBCampR at a final concentration of 10 μM each in a final volume of 100 μl per reaction containing ~ 400 ng of the template DNA. This PCR was carried out with an initial denaturation at 95°C for 3 min, followed by 18 cycles of denaturation at 98°C for 20 s, annealing at 65°C for 30 s, and extension at 72°C for 20 s, with a final extension at 72°C for 2 min. The PCRs were purified using 1.45× AMPure XP Beads (Beckman Coulter), eluted in 10 μl of water and directly transferred to a second PCR containing the primers NtIndex‐F and NtIndex‐R at 10 μM each in a final volume of 50 μl. These primers contain the adapters and index sequences needed for Illumina sequencing. The temperature cycle was 95°C for 3 min, followed by 17 cycles of 98°C for 20 s, 65°C for 30 s, and 72°C for 20 s, with a final extension at 72°C for 2 min. The final PCR product was purified once more with 1.45× AMPure XP beads and eluted in 20 μl of the Buffer EB (QIAGEN). The libraries were quantified using the KAPA Library quantification kit (Kapa Biosciences) and were pooled at equimolar ratios in a final concentration of 3 nM. The library pool was sequenced using a Illumina™ NextSeq 550 sequencer with 2 × 75 pb reads targeting on average 5 million pair‐end reads per sample.
Barcode counting
Sequencing read pairs in which at least one of the reads had an average quality score lower than 20 were removed from downstream analysis. Then, read pairs were merged to form consensus sequences using the FASTP pipeline (Chen et al, 2018). The Bartender pipeline (Zhao et al, 2018) was used to generate the barcode count table, using the pattern “GACAG[29‐39]AGCAG”, and allowing for two mismatches. Downstream analyses were done in R. To distinguish barcodes originating from PCR or sequence errors from true barcodes, we submitted one sample (BPK282 population prior to drug exposure) to three independent library preps. After confirming that barcodes appearing in only one or two libraries were always detected at low frequencies, we considered as true barcodes only those found in all three libraries.
SCGS
SCGS was performed using the single‐cell CNV™ solution from 10× Genomics. Execution of the protocol and bioinformatic analysis were done as previously described (Negreira et al, 2021).
Dose–response assay against antimony and miltefosine
Dose–response assays were performed as previously described (Kulshrestha et al, 2013). In summary, logarithmic‐stage promastigotes were seeded in 96‐well plates at final concentration of 105 parasites per well in supplemented HOMEM medium per well. Parasites were exposed to different concentrations of miltefosine ranging from 3 to 75 μM (first attempt) or 150 μM (second attempt). An extra row of wells was kept with 0.2% PBS instead of antimony as a control. Parasites were exposed to the drug for 3 days at 26°C. Afterwards, each plate received 20 μg/ml of resazurin and was further incubated for 4 h at 26°C. Then, light absorption was measured at 560 nm/590 nm excitation/emission Using a Victor X2 luminometer. Assays were independently performed 3 times, with three identical plates in each execution serving as technical replicates. When possible, half maximal inhibitory concentrations (IC50) were calculated with GraphPad Prism using a sigmoidal dose–response model with variable slope.
Author contributions
Gabriel H Negreira: Conceptualization; formal analysis; investigation; visualization; methodology; writing – original draft; writing – review and editing. Robin de Groote: Investigation. Dorien Van Giel: Investigation. Pieter Monsieurs: Data curation; formal analysis; visualization; writing – review and editing. Ilse Maes: Investigation. Geraldine de Muylder: Conceptualization; methodology; writing – review and editing. Frederik Van den Broeck: Conceptualization; funding acquisition; writing – review and editing. Jean‐Claude Dujardin: Conceptualization; supervision; funding acquisition; writing – original draft; writing – review and editing. Malgorzata A Domagalska: Conceptualization; data curation; formal analysis; supervision; funding acquisition; methodology; writing – original draft; writing – review and editing.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
PDF+
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Acknowledgements
Flemish Ministry of Science and Innovation [Secondary Research Funding ITM—SOFI, Grant MADLEI]. Flemish Fund for Scientific Research [FWO, post‐doctoral grant to FVdB].
EMBO reports (2023) 24: e57413
Data availability
Raw sequencing data have been deposited in BioProject (NCBI) with accession number PRJNA970809. Custom scripts used for analysis of barcode and WGS data are available at https://github.com/gabrielnegreira/LeishBarSeqAndAneuploidy. Scripts for single‐cell data are available at https://github.com/gabrielnegreira/scgs‐somy. All data are available in the main text or the supplementary materials.
References
- Bhang HEC, Ruddy DA, Radhakrishna VK, Caushi JX, Zhao R, Hims MM, Singh AP, Kao I, Rakiec D, Shaw P et al (2015) Studying clonal dynamics in response to cancer therapy using high‐complexity barcoding. Nat Med 21: 440–448 [DOI] [PubMed] [Google Scholar]
- Brotherton M‐C, Bourassa S, Leprohon P, Légaré D, Poirier GG, Droit A, Ouellette M (2013) Proteomic and genomic analyses of antimony resistant Leishmania infantum mutant. PLoS ONE 8: e81899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussotti G, Gouzelou E, Boité MC, Kherachi I, Harrat Z, Eddaikra N, Mottram JC, Antoniou M, Christodoulou V, Bali A et al (2018) Leishmania genome dynamics during environmental adaptation reveal strain‐specific differences in gene copy variation, karyotype instability, and telomeric amplification. mBio 9: 1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussotti G, Piel L, Pescher P, Domagalska MA, Rajan KS, Cohen‐Chalamish S, Doniger T, Hiregange D‐G, Myler PJ, Unger R et al (2021) Genome instability drives epistatic adaptation in the human pathogen Leishmania . Proc Natl Acad Sci USA 118: e2113744118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Bradford WD, Seidel CW, Li R (2012) Hsp90 stress potentiates rapid cellular adaptation through induction of aneuploidy. Nature 482: 246–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Zhou Y, Chen Y, Gu J (2018) Fastp: an ultra‐fast all‐in‐one FASTQ preprocessor. Bioinformatics 34: i884–i890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chulay JD, Fleckenstein L, Smith DH (1988) Pharmacokinetics of antimony during treatment of visceral leishmaniasis with sodium stibogluconate or meglumine antimoniate. Trans R Soc Trop Med Hyg 82: 69–72 [PubMed] [Google Scholar]
- Chunduri NK, Storchová Z (2019) The diverse consequences of aneuploidy. Nat Cell Biol 21: 54–62 [DOI] [PubMed] [Google Scholar]
- Clayton C (2019) Regulation of gene expression in trypanosomatids: living with polycistronic transcription. Open Biol 9: 190072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuypers B, Meysman P, Erb I, Bittremieux W, Valkenborg D, Baggerman G, Mertens I, Sundar S, Khanal B, Notredame C et al (2022) Four layer multi‐omics reveals molecular responses to aneuploidy in Leishmania . PLoS Pathog 18: e1010848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domagalska MA, Imamura H, Sanders M, Van Den Broeck F, Bhattarai NR, Vanaerschot M, Maes I, D'Haenens E, Rai K, Rijal S et al (2019) Genomes of Leishmania parasites directly sequenced from patients with visceral leishmaniasis in the Indian subcontinent. PLoS Negl Trop Dis 13: e0007900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorlo TPC, Balasegaram M, Beijnen JH, de Vries PJ (2012) Miltefosine: a review of its pharmacology and therapeutic efficacy in the treatment of leishmaniasis. J Antimicrob Chemother 67: 2576–2597 [DOI] [PubMed] [Google Scholar]
- Douanne N, Wagner V, Roy G, Leprohon P, Ouellette M, Fernandez‐Prada C (2020) MRPA‐independent mechanisms of antimony resistance in Leishmania infantum . Int J Parasitol Drugs Drug Resist 13: 28–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumetz F, Imamura H, Sanders M, Seblova V, Myskova J, Pescher P, Vanaerschot M, Meehan CJ, Cuypers B, De Muylder G et al (2017) Modulation of aneuploidy in Leishmania donovani during adaptation to different in vitro and in vivo environments and its impact on gene expression. mBio 8: e00599–e00517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumetz F, Cuypers B, Imamura H, Zander D, D'Haenens E, Maes I, Domagalska MA, Clos J, Dujardin J‐C, De Muylder G (2018) Molecular preadaptation to antimony resistance in Leishmania donovani on the Indian subcontinent. mSphere 3: e00548‐17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frézard F, Monte‐Neto R, Reis PG (2014) Antimony transport mechanisms in resistant leishmania parasites. Biophys Rev 6: 119–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefnawy A, Negreira G, Jara M, Cotton JA, Maes I, D'Haenens E, Imamura H, Cuypers B, Monsieurs P, Mouchtoglou C et al (2022) Genomic and phenotypic characterization of experimentally selected resistant Leishmania donovani reveals a role for dynamin‐1‐like protein in the mechanism of resistance to a novel antileishmanial compound. mBio 13: e0326421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura H, Dujardin J‐C (2019) A guide to next generation sequence analysis of Leishmania genomes. In Leishmania: methods and protocols, Clos J (ed), pp 69–94. New York, NY: Humana Press; [DOI] [PubMed] [Google Scholar]
- Imamura H, Downing T, Van den Broeck F, Sanders MJ, Rijal S, Sundar S, Mannaert A, Vanaerschot M, Berg M, De Muylder G et al (2016) Evolutionary genomics of epidemic visceral leishmaniasis in the Indian subcontinent. eLife 5: e12613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ippolito MR, Martis V, Martin S, Tijhuis AE, Hong C, Wardenaar R, Dumont M, Zerbib J, Spierings DCJ, Fachinetti D et al (2021) Gene copy‐number changes and chromosomal instability induced by aneuploidy confer resistance to chemotherapy. Dev Cell 56: 2440–2454.e6 [DOI] [PubMed] [Google Scholar]
- Jara M, Maes I, Imamura H, Domagalska MA, Dujardin JC, Arevalo J (2019) Tracking of quiescence in Leishmania by quantifying the expression of GFP in the ribosomal DNA locus. Sci Rep 9: 18951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloehn J, Saunders EC, O'Callaghan S, Dagley MJ, McConville MJ (2015) Characterization of metabolically quiescent Leishmania parasites in murine lesions using heavy water labeling. PLoS Pathog 11: e1004683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulshrestha A, Bhandari V, Mukhopadhyay R, Ramesh V, Sundar S, Maes L, Dujardin JC, Roy S, Salotra P (2013) Validation of a simple resazurin‐based promastigote assay for the routine monitoring of miltefosine susceptibility in clinical isolates of Leishmania donovani . Parasitol Res 112: 825–828 [DOI] [PubMed] [Google Scholar]
- Leprohon P, Légaré D, Girard I, Papadopoulou B, Ouellette M (2006) Modulation of Leishmania ABC protein gene expression through life stages and among drug‐resistant parasites. Eukaryot Cell 5: 1713–1725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann S, Frasca K, Scherrer S, Henao‐Martínez AF, Newman S, Ramanan P, Suarez JA (2021) A review of Leishmaniasis: current knowledge and future directions. Curr Trop Med Rep 8: 121–132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannaert A, Downing T, Imamura H, Dujardin J‐C (2012) Adaptive mechanisms in pathogens: universal aneuploidy in Leishmania . Trends Parasitol 28: 370–376 [DOI] [PubMed] [Google Scholar]
- Monte‐Neto R, Laffitte M‐CN, Leprohon P, Reis P, Frézard F, Ouellette M (2015) Intrachromosomal amplification, locus deletion and point mutation in the aquaglyceroporin AQP1 gene in antimony resistant Leishmania (Viannia) guyanensis . PLoS Negl Trop Dis 9: e0003476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulla W, Zhu J, Li R (2014) Yeast: a simple model system to study complex phenomena of aneuploidy. FEMS Microbiol Rev 38: 201–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negreira GH, Monsieurs P, Imamura H, Maes I, Kuk N, Yagoubat A, Van den Broeck F, Sterkers Y, Dujardin J‐C, Domagalska MA (2021) High throughput single‐cell genome sequencing gives insights into the generation and evolution of mosaic aneuploidy in Leishmania donovani . Nucleic Acids Res 50: 293–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patino LH, Imamura H, Cruz‐Saavedra L, Pavia P, Muskus C, Méndez C, Dujardin JC, Ramírez JD (2019) Major changes in chromosomal somy, gene expression and gene dosage driven by SbIII in Leishmania braziliensis and Leishmania panamensis . Sci Rep 9: 9485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez‐Victoria FJ, Sánchez‐Cañete MP, Seifert K, Croft SL, Sundar S, Castanys S, Gamarro F (2006) Mechanisms of experimental resistance of Leishmania to miltefosine: implications for clinical use. Drug Resist Updat 9: 26–39 [DOI] [PubMed] [Google Scholar]
- Perry MR, Wyllie S, Prajapati VK, Feldmann J, Sundar S, Boelaert M, Fairlamb AH (2011) Visceral leishmaniasis and arsenic: an ancient poison contributing to antimonial treatment failure in the Indian subcontinent? PLoS Negl Trop Dis 5: e1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry MR, Wyllie S, Raab A, Feldmann J, Fairlamb AH (2013) Chronic exposure to arsenic in drinking water can lead to resistance to antimonial drugs in a mouse model of visceral leishmaniasis. Proc Natl Acad Sci USA 110: 937–19937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Replogle JM, Zhou W, Amaro AE, McFarland JM, Villalobos‐Ortiz M, Ryan J, Letai A, Yilmaz O, Sheltzer J, Lippard SJ et al (2020) Aneuploidy increases resistance to chemotherapeutics by antagonizing cell division. Proc Natl Acad Sci USA 117: 576–30576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricke RM, van Deursen JM (2013) Aneuploidy in health, disease, and aging. J Cell Biol 201: 11–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers MB, Hilley JD, Dickens NJ, Wilkes J, Bates PA, Depledge DP, Harris D, Her Y, Herzyk P, Imamura H et al (2011) Chromosome and gene copy number variation allow major structural change between species and strains of Leishmania . Genome Res 21: 142–2142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw CD, Lonchamp J, Downing T, Imamura H, Freeman TM, Cotton JA, Sanders M, Blackburn G, Dujardin JC, Rijal S et al (2016) In vitro selection of miltefosine resistance in promastigotes of Leishmania donovani from Nepal: genomic and metabolomic characterization. Mol Microbiol 99: 148–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Wang Y‐T, Tang X‐X, Zhang K, Wang P‐M, Sui Y, Zheng D‐Q (2020) Heat shock drives genomic instability and phenotypic variations in yeast. AMB Express 10: 146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterkers Y, Lachaud L, Crobu L, Bastien P, Pagès M (2011) FISH analysis reveals aneuploidy and continual generation of chromosomal mosaicism in Leishmania major . Cell Microbiol 13: 274–283 [DOI] [PubMed] [Google Scholar]
- Sterkers Y, Lachaud L, Bourgeois N, Crobu L, Bastien P, Pagès M (2012) Novel insights into genome plasticity in eukaryotes: mosaic aneuploidy in Leishmania . Mol Microbiol 86: 15–23 [DOI] [PubMed] [Google Scholar]
- Tan Z, Chan YJA, Chua YJK, Rutledge SD, Pavelka N, Cimini D, Rancati G (2019) Environmental stresses induce karyotypic instability in colorectal cancer cells. Mol Biol Cell 30: 42–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tůmová P, Uzlíková M, Jurczyk T, Nohýnková E (2016) Constitutive aneuploidy and genomic instability in the single‐celled eukaryote Giardia intestinalis . MicrobiologyOpen 5: 560–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valdivia HO, Reis‐Cunha JL, Rodrigues‐Luiz GF, Baptista RP, Baldeviano GC, Gerbasi RV, Dobson DE, Pratlong F, Bastien P, Lescano AG et al (2015) Comparative genomic analysis of Leishmania (Viannia) peruviana and Leishmania (Viannia) braziliensis . BMC Genomics 16: 715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voak AA, Standing JF, Sepúlveda N, Harris A, Croft SL, Seifert K (2018) Pharmacodynamics and cellular accumulation of amphotericin B and miltefosine in Leishmania donovani‐infected primary macrophages. J Antimicrob Chemother 73: 1314–1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Teoh F, Tan ASM, Cao Y, Pavelka N, Berman J (2019) Aneuploidy enables cross‐adaptation to unrelated drugs. Mol Biol Evol 36: 1768–1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F, Gritsenko V, Lu H, Zhen C, Gao L, Berman J, Jiang Y (2021) Adaptation to fluconazole via aneuploidy enables cross‐adaptation to amphotericin B and Flucytosine in Cryptococcus neoformans . Microbiol Spectr 9: e00723‐21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao L, Liu Z, Levy SF, Wu S (2018) Bartender: a fast and accurate clustering algorithm to count barcode reads. Bioinformatics 34: 739–747 [DOI] [PMC free article] [PubMed] [Google Scholar]