

Abstract
The aging process is accompanied by the onset of disease and a general decline in wellness. Insights into the aging process have revealed a number of cellular hallmarks of aging, among these epigenetic alterations, loss of proteostasis, mitochondrial dysfunction, cellular senescence, and stem cell exhaustion. Mitochondrial dysfunction increasingly appears to be a common factor connecting several of these hallmarks, driving the aging process and afflicting tissues throughout the body. Recent research has uncovered a much more complex involvement of mitochondria in the cell than has previously been appreciated and revealed novel ways in which mitochondrial defects feed into disease pathology. In this review we evaluate ways in which problems in mitochondria contribute to disease beyond the well-known mechanisms of oxidative stress and bioenergetic deficits, and we predict the direction that mitochondrial disease research will take in years to come.
Keywords: Mitochondria, atherosclerosis, neurodegeneration, stem cells, osteoporosis, sirtuins, immunity, aging
INTRODUCTION
Mitochondria deteriorate with age, losing respiratory activity, accumulating damage to their DNA (mtDNA), and producing excessive amounts of reactive oxygen species (ROS) [1]. While for decades it was believed that ROS were exclusively toxic molecules causing damage, it is now accepted that low levels of some ROS species have signaling roles [2]. Excessive and aberrant ROS generation, nonetheless, is one of the major consequences of mitochondrial dysfunction. Mitochondria are the main source of ROS in the cell, with most stemming from complexes I and III of the electron transport chain (ETC) [3, 4]. Electrons escape at these points as a side-product of oxidative respiration and in turn reduce oxygen, generating superoxide. ROS exert a plethora of detrimental effects in the cell by causing oxidative damage to nucleic acids, proteins, and lipids. The mitochondrial free radical theory of aging postulates that the damage caused by accumulating ROS produced by mitochondria is the driving force behind aging [5]. This theory is corroborated to some extent by the inverse correlation between mitochondrial ROS production and lifespan in mammals [6]. Furthermore, mitochondrially-targeted catalase has been found to have protective effects against cardiac diseases, cancer, and insulin resistance in mice [7]. ROS are not the only aspect of flawed mitochondria that contributes to degenerating health though; ATP shortage, mutations in mtDNA, mitochondrial permeability transition pore (mPTP) opening, apoptosis, Ca2+ deregulation, inflammation, and altered fusion/fission dynamics are all mitochondrial factors that, while not necessarily acting independently, become disrupted in many diseases.
Studies on caloric restriction, one of the most consistent and powerful tools to boost lifespan and healthspan across a variety of organisms, provide further support for a theory of aging centered around mitochondria through the finding that the health benefits of this method may stem from mitochondria . Genotype has been reported to be an important determinant of an individual’s response to dietary restriction, particularly when it comes to genes concerning mitochondria [8, 9]. Additionally, mitochondrial membrane potential has been found to be a predictive marker of replicative lifespan in yeast grown on media with reduced glucose [10]. Mitochondria also appear to be major signaling hubs that in part govern the molecular changes induced by dietary restriction [11].
Current evidence points to mitochondrial dysfunction as an overarching mechanism of aging and age-related disease. It is implicated in an extensive list of aging pathologies such as cancer, intestinal barrier dysfunction, depression, chronic obstructive pulmonary disease (COPD), diabetes, and others [12–16]. Here we examine the role of mitochondrial dysfunction in diseases that are common but are not typicallly associated with mitochondrial dysfunction or have more mitochondrial involvement than commonly appreciated such as vascular disease (atherosclerosis), the major neurodegenerative diseases (Alzheimer’s, Huntington’s, and Parkinson’s), and osteoporosis. We also look at diseases that we foresee may potentially become more prominent in society as we solve now-common diseases and extend our lifespan e.g. stem cell dysfunction and infection. Lastly we review evidence linking changes in the mitochondrial proteome and metabolome with the commencement of various age-related problems.
I. MITOCHONDRIAL DYSFUNCTION IN COMMON DISEASES
1.1. Atherosclerosis
Atherosclerosis is commonly thought of as a disease associated with diets high in fat and cholesterol. However, more recent findings indicate that mitochondrial dysfunction may in fact play a more integral role than previously appreciated. Leaks in the respiratory chain create ROS that set off a chain reaction that gives way to atherosclerosis by multiple pathways. ROS oxidize low density lipoprotein (LDL), which accumulates in the artery wall, leading to the formation of an atherosclerotic plaque. ROS also induce inflammation by causing damage to mtDNA. By a more complex mechanism, the oxidative modification of mitochondrial cardiolipins may contribute to progression of cardiovascular disease as well.
1.1.1. Ox-LDL
LDL functions to deliver cholesterol to cells throughout the body via the bloodstream. However, when LDL particles leave the vasculature and enter the arterial intima, they can be exposed to ROS produced by endothelial cells [17, 18]. These ROS convert LDL into its oxidized form, ox-LDL which binds to the extracellular matrix (ECM) of endothelial cells more easily than LDL [19, 20]. Adhesion of ox-LDL to the ECM triggers the recruitment of monocytes, which become macrophages to take up the accumulating lipoprotein and transform into foam cells [19]. Foam cells secrete apolipoprotein E (apoE) which aids high density lipoprotein (HDL) in the removal of fats and cholesterol from the cells in conjunction with various components of the HDL molecule [21, 22]. HDL can then transport the fat and cholesterol back to the liver for processing. However, if the LDL/HDL ratio is too high, the foam cells accumulate and die. The debris attracts more monocytes, perpetuating the cycle until a plaque forms composed of monocytes, cholesterol, and fats [17]. This progression is known as atherogenesis.
Oxidation of LDL has long been known to initiate a series of events leading to atherosclerosis since it was observed that thiol-dependent oxidation of LDL had atherogenic properties [23]. The subject of interest has since shifted to ROS and mitochondria. The ROS produced by endothelial cells are thought to have important physiological uses e.g. playing a part in sensing oxygen concentrations, and regulation of vascular tone, cell growth, and survival [18]. However, uncontrolled ROS production has damaging effects. In one study, it was found that decreased levels of the mitochondrial antioxidant enzyme superoxide dismutase 2 (SOD2) exacerbated oxidative stress and atherosclerosis in male apoE null mice fed normal chow [24]. When compared to control mice (apoE(−/−), SOD2(+/+)) fed the same diet, it was found that lesion formation was independent of cholesterol levels. Cholesterol is typically focused on as the central component in cardiovascular disease, but this finding implies that higher concentrations of ROS have the capacity to drive atherosclerotic development as well. The results were the same both in mice heterozygous for SOD2 and in an in vitro model of human vascular endothelial cells where siRNA was used to lower SOD2 levels. Another study tested the effects of deficiency in the antioxidant enzyme peroxiredoxin 1 (Prdx1) on apoE(−/−) mice fed normal chow. Again, larger lesions were observed in the Prdx1 null mice with no difference in cholesterol [25].
1.1.2. mtDNA
There are several enzymes in the cell such as superoxide dismutase, catalase, and glutathione peroxidase that offer protection against oxidative damage. Steady state concentrations of superoxide in the mitochondrial matrix are estimated to be from 10–200 pM though this is difficult to calculate due to the rapid conversion of superoxide to hydrogen peroxide by SOD [26]. Nonetheless, being the center of ROS generation, mitochondria often succumb to oxidative stress [20]. Particularly relevant to atherosclerosis is the consequent damage to mtDNA. mtDNA damage has been known to be correlated with the incidence of atherosclerotic lesions [27]. One possible and unexpected mechanism by which this may occur is through mtDNA damage-induced inflammation. While damaged mitochondria are degraded by autophagy, some mtDNA can escape. A recent study has shown that damaged mtDNA that avoids autophagy causes an inflammatory response [28]. THP-1 macrophages were treated with siRNA against DNase II, the enzyme responsible for degrading damaged mtDNA. With the resulting rise in damaged mtDNA, an increase in NOD-like receptor family pyrin domain containing 3 (NLRP3) inflammasome activation was observed. The study also showed that ROS inhibitors decrease NLRP3 activation. Collectively, these studies suggest a model in which ROS cause mtDNA damage that in turn induces inflammation, exacerbating atherosclerotic disease progression.
1.1.3. Cardiolipin Oxidation
The oxidation of mitochondrial cardiolipins (CLs) by ROS in the context of atherosclerosis is another area of research that has been gaining interest recently. CLs are phospholipids that localize to the inner mitochondrial membrane where they stabilize the association of complexes I-V of the ETC and optimize their efficiency [29, 30]. Due to their close proximity to the respiratory chain, however, CLs are vulnerable to oxidation. Based on their contributions to ETC complex function, it is not surprising that changes in CL have deleterious effects on ETC activity. When bovine heart submitochondrial particles were exposed to increased ROS, cytochrome c oxidase activity was reduced by ~40%. Cardiolipin content was also reduced by ~40% . This loss of cytochrome c activity could be completely reversed by addition of exogenous CL, but not peroxidized-CL, suggesting oxidized CLs and/or their subsequent loss from mitochondrial membranes were responsible for the observed inhibition [31].
With the potential for amplifying mitochondrial dysfunction, oxidized cardiolipins (ox-CL) could very well be an important contributor in diseases such as atherosclerosis. Indeed, not only are phospholipid oxidation products (ox-PL) present in atherosclerotic lesions, but they appear to contribute to lesion formation as well [32]. The ways in which ox-CL specifically exacerbates atherosclerosis are only recently beginning to be uncovered. It has been found that ox-CL increases mobilization of intracellular Ca2+ in human monocyte-derived macrophages (HMDM) and polymorphonuclear leukocytes (PMN) [33]. This led to the increased production of leukotriene B4 (LTB4), a pro-inflammatory agent. Also of note is the finding that treatment of human umbilical vein endothelial cells (HUVECs) with ox-CL augmented expression of the adhesion molecules intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1). This may be a significant part of plaque formation in light of the fact that adhesion molecules are involved in recruiting monocytes to the arterial intima [33]. The phospholipid-binding protein annexin A5 has been found to have anti-atherosclerotic attributes [34], and Wan et al. provide further support for its usefulness by demonstrating that annexin A5 treatment abolished the effects of ox-CL. In addition to their inflammatory influence, evidence is growing that ox-PL play a part in regulating gene function in endothelial cells and are implicated in foam cell development and instigation of apoptosis in macrophages [32].
While work on ox-CLs has opened new doors for atherosclerotic research, it is as yet unclear if mitochondria are the sole source of these compounds. Oxidized-CLs can certainly be produced in the mitochondria, and result in detrimental effects on the respiratory chain. Oxidized-CLs may also turn cells toward atherogenesis, but it has not been demonstrated that mitochondria link the two events. It would be intriguing to investigate whether ox-CL generated in the mitochondrial membrane can be released into the extracellular milieu, and if this occurs during the progression of atherosclerosis.
1.2. Neurodegenerative Disease
The brain is a center of high energy demand, consuming roughly a fifth of the body’s basal metabolic energy requirements [35]. Because glycolysis is down-regulated in neurons [36], much of the energetic needs of neurons are fulfilled by substrates supplied to them by way of astrocytes, via the so-called astrocyte-neuron lactate shuttle. In this process, astrocytic lactate produced by glycolysis is shuttled into neurons and then re-converted to pyruvate for entry into the tricarboxylic acid (TCA) cycle [37]. With such a reliance on mitochondrial respiration, as well as a high proportion of peroxidizable lipids and a dependence on astrocytes for key antioxidants, neurons are rendered particularly sensitive to mitochondrial dysfunction [38]. Mitochondrial dysfunction contributes to disease in neurons in several ways - by causing oxidative damage to cell structures through production of excessive ROS, by reducing oxidative phosphorylation (OXPHOS) capacity and the energy output of neurons, and ultimately through instigation of apoptotic pathways that in turn disrupt neuronal circuits [39, 40]. Moreover, RNA oxidation is a widespread event in neurodegenerative disease, and has been found to occur early in the progression of Alzheimer’s and Parkinson’s [41]. In addition, mutations in mtDNA have been linked to a variety of neurodegenerative diseases [42]. While mitochondria play an integral part of general neurodegeneration, here we will focus on mitochondria and their specific role in three major neurodegenerative diseases: Alzheimer’s, Huntington’s, and Parkinson’s.
1.2.1. Alzheimer’s Disease
Alzheimer’s disease (AD) is characterized by extracellular amyloid-β (Aβ) aggregates as well as intracellular neurofibrillary tangles (NFTs) consisting of hyperphosphorylated tau protein. While it was initially thought that the Aβ plaques were the cause of the disease, an idea known as the amyloid cascade hypothesis, there is now controversy as to whether they are actually causative of AD. There is a considerable amount of data that do not fit the amyloid model. For example, Aβ aggregates have been found in multiple instances to be present in the brains of cognitively intact elderly in quantities characteristic of the disease. There is also a poor correlation between plaque density and disease severity [38]. An alternate hypothesis proposes that rather than playing a causal role, Aβ represents a compensatory response to oxidative stress [43]. Indeed, higher levels of Aβ are associated with decreased levels of nucleic acid oxidation [38]. It is thought that Aβ achieves this by chelating redox-active metals such as iron and copper, and there is evidence to suggest that NFTs have a similar function.
A direct connection between Aβ and mitochondria exists as well. The amyloid precursor protein APP has both an endoplasmic reticulum (ER) and a mitochondrial targeting sequence. Upon overexpression in vitro, mimicking disease conditions, APP is directed to the mitochondria. During import, however, it gets stuck in the mitochondrial double membrane [44]. At this point APP is cleaved by Omi in the intermembrane space and mitochondrial γ-secretases at the outer mitochondrial membrane [45, 46]. Processing by γ-secretases could possibly generate Aβ, though this has yet to be demonstrated. Aβ itself can be imported into mitochondria where it complexes heme groups, which are critical for ETC function, and inhibits Aβ-binding alcohol dehydrogenase (ABAD) [47]. In AD, ABAD is upregulated in neurons, and its interaction with Aβ results in increased ROS [48, 49]. While this conflicts with the reports of lowered oxidation with elevated Aβ, it may be explained by the fact that Aβ only protects against metal-dependent oxidation [38]. Aberrant ROS generation is not the only deleterious effect of Aβ on mitochondria; incubation of mouse brain mitochondria with Aβ leads to mitochondrial swelling and apoptosis [50]. Interestingly, Aβ exerts these toxic effects despite the presence of PreP, a mitochondrial enzyme that degrades the amyloid protein [46].
While the status of Aβ as protective or harmful remains the subject of debate, it is clear that mitochondria are a central part of AD. Mitochondria are visibly affected in AD, presenting a fragmented phenotype that may be a result of increased fission, presumably for the purpose of setting aside damaged mitochondria to be degraded by autophagy [47]. Impairment of various mitochondrial enzymes, accumulation of oxidation products, and a perturbation in antioxidants in brain and fluid samples of AD patients are also evident [38, 40, 51]. Mitochondrial involvement in the disease is further implicated by the observation that in cell lines overexpressing APP, there is a marked decrease in ATP production and an increase in ROS generation [52].
Importantly, oxidative damage precedes Aβ deposition and NFT formation, suggesting a possible causal role [38, 53, 54]. Strong support for involvement of oxidation in AD etiology comes from the finding that oxidation products cause an increase in production and accumulation of Aβ and hyperphosphorylation of tau [55–57]. Phosphorylated tau itself can then be oxidized, leading to fibril formation [38]. Furthermore, supplementation of various anti-oxidizing substances such as vitamin E, melatonin, copper, and omega-3 polyunsaturated fatty acid lower levels of Aβ deposition in transgenic animal models of AD [38]. Conversely, treatment of cells with ETC inhibitors results in tau phosphorylation and amyloidogenic APP processing. The use of cytoplasmic hybrids (cybrids) is another approach that has been used in AD research. The cybrid assay is one in which a cell that has been depleted of its mtDNA is fused with an enucleated cell, allowing one to separate the effects of mtDNA from nuclear genes. Cybrids transplanted with mitochondria from AD patients overproduce Aβ42 and exhibit lower complex IV (cytochrome c oxidase) activity, increased ROS, activated stress signaling and apoptotic pathways, and reduced mitochondrial membrane potential, all of which points to the mitochondria as an underlying mechanism of AD pathology [43].
Several studies reveal that a maternally-inherited factor increases incidence of AD-like traits [58]. It has been hypothesized that part of the maternal bias may be due to the unequal inheritance of COX genes; while most subunits are encoded by nuclear DNA, three are inherited from the mother’s mtDNA [43]. Other mitochondrial genes have also been linked to an increased risk of developing AD - among these a haplotype for the mitochondrial outer membrane translocation protein TOM40 [59–61]. A recent screen identified several mitochondria-related genes that are differentially expressed in astrocytes from the posterior cingulate of AD patients [62]. The results of the screen revealed an altered regulation of apoptosis, mitochondrial tRNA methylation, decreased PreP expression, and inhibition of complex I activity in this section of AD brains. Perhaps the most intriguing finding was the identification of the first known mitochondrially-encoded pseudogene, MTND1P22. Transcript levels of the pseudogene, which may be involved in transcription regulation, were found to be increased. Little is known about pseudogenes, but recent studies have suggested an increasing importance of these molecules and even hint at their involvement in disease [63, 64].
One of the earliest events in disease progression of transgenic AD mouse models is the emergence of mitochondrial dysfunction in the form of deregulation of respiratory complexes I and IV, decreased respiration, and increased oxidative stress [65, 66]. However, the consequences of dysfunction in the context of AD are still unclear. It has recently been shown that in 3-month old 3xTg mice, a transgenic model of AD overexpressing three mutations commonly associated with AD, reduced activity of complexes I and IV is concurrent with a change in cardiolipin composition of synaptic mitochondrial membranes [67]. Another study found that in the brains of 45-week-old TgCRND8 mice (a strain that overexpresses APP), there was reduced complex I activity but complex IV was unaffected, and no change in cardiolipin composition was present in this model at this age [68]. The effect on complex I was likely due to the reduced expression of one of its subunits, NDUFB8.
1.2.2. Huntington’s Disease
Huntington’s disease (HD) is caused by expansion of a trinucleotide repeat in the Huntingtin gene. Mutant huntingtin (mhtt) accumulates and forms inclusions in the striatum [42]. While this mutation is the ultimate cause of the disease, mitochondria are not idle spectators. There is a disturbance of mitochondrial energy metabolism, defects in complexes I and II, reduced aconitase activity, lowered membrane potential, abnormal depolarization, and altered structure and number of mitochondria in diseased brains [42, 51]. A bioenergetic deficit is present before the onset of symptoms, indicating mitochondrial dysfunction may be an important initiator of pathogenesis [69]. The impairment of mitochondrial bioenergetics may be due to the role of mutant huntingtin in regulation of transcription factors that are required for the transcription of mitochondrial genes. For example, it has been demonstrated that mhtt represses expression of peroxisome proliferator-activated receptor gamma coactivator-1α (PGC-1α), a transcriptional co-activator of genes involved in mitochondrial biogenesis, metabolism, and membrane structure [70, 71].
Mhtt directly interacts with mitochondria as well, activating the fission protein Drp1 and causing mitochondrial fragmentation [72]. Altered fusion/fission dynamics have been found to be a recurring theme in neurodegeneration [73]. Fragmentation can then result in reduced Ca2+-buffering capacity. Mitochondria from lymphoblasts of HD patients do in fact exhibit a lowered membrane potential and depolarize at lower levels of Ca2+ [74]. Mitochondria in a transgenic rat model of HD also show changes in Ca2+ transport and a decreased Ca2+ threshold for mPTP opening [75]. Furthermore, striatal cells expressing mhtt showed increased sensitivity to Ca2+ and hindered Ca2+ uptake capacity [76]. Considering the role of Ca2+ signaling in neuronal synaptic transmission, the changes in mitochondrial Ca2+-buffering capacity may represent another way in which altered mitochondrial dynamics impairs neuronal function. In addition to inducing fragmentation and the corresponding changes in Ca2+ handling, mhtt also impedes mitochondrial trafficking. Cytosolic mhtt aggregates physically block the transit of mitochondria along neuronal processes [77]. The accumulation of mitochondria at these roadblocks may decrease the effective size of the pool of functional mitochondria, leading to neurodegeneration.
Inhibition of succinate dehydrogenase in complex II with 3-nitropropionic acid (3-NP) has been used to model HD. 3-NP cripples energy production, leads to striatal neuronal degeneration, and invokes pathophysiological characteristics similar to those of HD [78]. The toxin’s specificity of action on striatal neurons is likely due to a combination of factors in the local cellular environment [79]. Using 3-NP to study the etiology of the disease reveals that toxin-induced mitochondrial dysfunction is followed by impaired neuronal excitability and a decrease in neurotransmitter release [80]. Neurotrophins, which aid in the survival and function of nerve cells, have been found to amplify corticostriatal synaptic transmission. However, after exposure to 3-NP, the neurotrophins BDNF, NT −4/5, and NT-3 had diminished effects on corticostriatal transmission.
1.2.3. Parkinson’s Disease
It is well established that mitochondrial dysfunction is associated with familial Parkinson’s disease (PD). Newer studies hint at the possibility that mitochondrial dysfunction might also underlie many cases of idiosyncratic PD, which account for 90% of all PD cases. PD is characterized by a loss of dopaminergic neurons and the formation of inclusions composed of α-synuclein (α-syn) called Lewy bodies. PD shares many mitochondria-related characteristics with AD including oxidative damage in affected regions, inhibited respiratory complex function, and increased incidence of mtDNA mutations [81–83].
A complex I defect is the most prominent mitochondrial feature in PD [42, 82]. This was first discovered when it was found that the compound 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) caused Parkinsonism. MPTP is capable of crossing the blood-brain barrier, and has a high affinity for a dopamine transporter, allowing it to act mostly on dopaminergic neurons [84]. At the outer mitochondrial membrane, MPTP is processed to produce MPP+ which enters into mitochondria and exerts its toxic effects by binding to and inhibiting complex I. The consequent reduction of ATP and elevation of ROS can then activate apoptotic pathways, resulting in neuronal loss [84]. The pathogenic properties of complex I inhibition are expanded further by the finding that rotenone, an inhibitor of complex I, causes aggregation of α-syn in cell culture [85]. It is thought that this is a consequence of constrained activity of the ubiquitin-protease system (UPS). Complex I dysfunction hinders proteasome activity by shrinking the energy supply and causing a rise in oxidatively damaged proteins, increasing the UPS burden [84]. As α-syn is degraded by the UPS, deterring the pathway may allow for aggregation. α-Syn is also capable of inducing dysfunction. The protein can be targeted to mitochondria under certain conditions, crippling complex I and spurring an increase in ROS production [82].
MitoPark mice illustrate the capacity of mitochondrial dysfunction to drive pathogenesis of PD. These mice were designed using Cre/Lox technology and have the mitochondrial transcription factor Tfam removed exclusively from dopaminergic neurons. Impaired mtDNA transcription in these cells leads to a loss of mtDNA-encoded respiratory complex subunits and ultimately loss of the respiratory chain. Without such a critical cellular component, the neurons die. Beginning in adulthood, the animals develop Parkinsonian symptoms and molecular markers of the disease. It should be noted that while these mice have been found to have intraneuronal inclusions, the aggregates did not contain α-syn [86]. Nonetheless, due to their ability to reproduce a large portion of the PD phenotype, MitoPark mice are widely used as a model to study PD.
A number of the proteins known to be key players in familial PD are either mitochondrial proteins or associated with mitochondria [51, 87–89]. One that has gained much interest in recent years is the mitochondrial protein PINK1. Mutations in the PINK1 gene are the main cause of one variety of familial PD [90]. Under normal conditions, PINK1 aids in regulation of mitophagy. Studies have shown that a loss of mitochondrial membrane potential causes PINK1 to accumulate at the membrane and phosphorylate Parkin, another protein that is implied in PD pathology. This then leads to mitochondrial degradation by mitophagy [90]. A PINK1 deficiency inhibits respiratory complex I, decreasing mitochondrial membrane potential and impairing transmission at neuromuscular junctions in Drosophila. This is not the only effect; dysfunction of a Na+/Ca2+ exchanger at the inner mitochondrial membrane has also been found to result from PINK1 deficiency [91]. The accumulated Ca2+ lowers the threshold for mPTP opening which can elevate ROS production through a conformational change in complex I [92]. Alternatively, complex I deficiency may result in increased ROS, which can inhibit the Na+/Ca2+ exchanger [93]. Hence, the findings of the two studies may be interrelated, but more research is needed to determine which is the primary effect of PINK1 deficiency, complex I inhibition or a Na+/Ca2+ exchanger defect. Inflammation is another feature of PD, and it has recently been discovered that PINK1 may play a pro-inflammatory role as well [90]. It therefore appears that the protein may contribute to pathology of the disease by multiple pathways.
Alzheimer’s, Huntington’s, and Parkinson’s disease all share two major things in common: mitochondrial dysfunction and the formation of soluble oligomers that often result in insoluble protein aggregates [94]. Mitochondrial dysfunction is an early event in each case, suggesting malfunctioning mitochondria may in fact be at the root of these diseases with protein oligermization or precipitation following as a consequence. It is possible that for many age-related diseases a similar pathological trajectory involving mitochondria and misfolded proteins is followed, but this has yet to be recognized. Impairment of mitochondrial processes in any cell type of any tissue could potentially create an environment in which deposition of proteins that are prone to aggregation is favored. This may occur by inhibition of the UPS, as discussed in the context of PD, or some other mechanism. In this regard, it is interesting to note that proteins that are not associated with any known deposition disease can be induced to aggregate in vitro [95]. Thus, we speculate that all tissues are likely to have their own aggregation-prone set of proteins which may vary from cell type to cell type depending on the particular proteome and cellular environment, and which may or may not be toxic. In those cases where the aggregate proteins have a toxic effect, disease would ensue. This concept is best exemplified by the MitoPark mice, which exhibit disease characteristics similar to those of PD despite the absence of α-syn from intraneuronal inclusions. This indicates there is almost certainly more than one pathway to disease, and that the main problem is not the aggregate protein itself, but rather the common factor of mitochondrial dysfunction.
1.3. Osteoporosis
The skeletal system provides the framework upon which our body’s overall structure is centered. Disruption of this framework can lead to increased risks of injury, debilitation, infection, and potentially death [96]. With age, the skeletal system naturally decays, experiencing decreased bone mineral density and bone mass, which is the cumulative effect of multiple physical, hormonal, and nutritional factors. Osteoporosis is a disease defined by the increasing loss of bone mass which leads to a porous bone structure and increased risk of fracturing [97–99]. With over 50% of people over 50 being affected, understanding the underlying causes of this disease is of great importance [100].
Normal adult bone growth is characterized by a balance between bone resorption and formation. Bone resorption is the breaking down of existing bone into base components (mainly type I collagen and inorganic salts) by osteoclast cells located on the surface of dissolving bone [101, 102]. Bone matrix formation and mineralization is undertaken by osteoblasts located on the surface of new bone, which use the broken down components made available by osteoclasts to form new bone matrices [101, 103]. These newly formed matrices are mineralized by osteoblasts that, as they get trapped within the forming matrix, then populate the matrix as osteocytes [104, 105]. Osteocytes comprise 90–95% of all bone cells, and direct the bone’s formation response to strain [106–108]. Osteoclasts and osteoblasts both come from bone marrow. With the need for nutrient metabolism required for bone remodeling, mitochondria play an important part in the creation of bone. This section will focus on the role of mitochondrial dysfunction, in particular mtDNA deletions and mitochondrial ROS, in osteoporosis.
1.3.1. mtDNA Damage
Findings from several sources, including genome-wide association studies (GWAS) in humans, studies on mice with increased mtDNA mutation rates, and from studies using cells that are defective for dysfunctional mitochondria disposal, all suggest that increasing amounts of damage to mitochondria or mtDNA are connected with osteoporosis [109–114]. In one example, a study of 2,286 US Caucasian men and women of Northern European origin from the mid-west found a significant association with single nucleotide polymorphisms in the mtDNA-encoded NADH dehydrogenase 2 and cytochrome b genes [109]. Although it is unclear precisley how these alterations translate into increased risk of osteoporosis, it is telling that both of these genes form essential parts of the mitochondrial respirtatory chain.
An early hypothesis by Varanasi [113], based on data from 15 men with symptomatic vertebral fractures, suggested that mtDNA deletions, and not oxidative stress, were the cause of osteoporosis. Varanasi suggested that the build up of lactate following mitochondrial electron transport chain dysfunction stimulated bone resorption in osteoclasts. The link between mtDNA deletions and osteoporosis has also received support by a 2004 study from Trifunovic and colleagues [112]. That study used homozygous knock-in mice that expressed a proof-reading-deficient version of PolgA, the nucleus-encoded catalytic subunit of mtDNA polymerase.. Such mice exhibited advanced aging phenotypes, including loss of bone mineral density and whole body bone mineral content, both characteristic of osteoporosis in humans [112].
Additional evidence from mice shows that osteoporosis can occur following mitochondrial dysfunction. HTRA2/OMI is an ATP-independent serine protease located in the intermembrane space of the mitochondria and is thought to function as a protein quality control protease. Its loss has been specifically shown to result in several age-related pathologies, including elevated mtDNA deletions, oesteoporosis, and neurodegeneration, among other phenotypes [110]. Mutant mitochondria in these animals are prevented from being marked for mitophagy. It is thought that as a consequence mtDNA deletions are allowed to replicate and in turn the buildup of defective mitochondria leads to impaired ATP production and an inability to meet the energy demand of normal cellular function [110]. One effect of this decrease in ATP production may be an increased rate of osteoclast resorption, leading to a decrease in bone mineral density [111].
1.3.2. ROS
In a shift from Varanasi’s hypothesis, in vitro studies and a progeric mouse model point toward mitochondrial-derived ROS as a key factor in the etiology of osteoporosis [98, 100, 115, 116]. Mice with homozygous SOD2 deficiency in connective tissue display early onset aging phenotypes, including decreased levels of bone mineral density [116]. Other studies show that the control of H2O2 is a crucial part of osteoporosis [115]. H2O2 production in cells is amplified by the adapter protein p66shc, which is released from an inhibitor complex in the inner mitochondrial membrane in response to a variety of pro-apoptotic stimuli and acts as a redox enzyme catalyzing the reduction of O2 to H2O2 through electron transfer from cytochrome c [117]. High levels of H2O2 in osteoblastic cells result in apoptosis and initiate osteoporosis by impairing osteoblast formation [98, 115]. A buildup of H2O2 conversely induces osteoclast proliferation and is required for osteoclast maturation. The attenuation of H2O2 by forkhead box O transcription factors (FoxOs) and estrogen administration promotes osteoblast formation, leading to bone growth. Increased levels of H2O2 inactivate FoxOs, and the effectiveness of these defenses decreases with age [118]. Thus, in youth, our body’s normal ROS defenses are able to maintain the balance between bone formation and resorption. However, as we age, our defenses become less effective and lead to the dominance of resorption by osteoclasts, resulting in decreased bone mineral density and osteoporosis.
The effect of oxidized cholesterol, or oxysterols, is another avenue by which mitochondria can influence osteoporosis [100]. Oxysterols are generated from cholesterol by P450 enzymes, the most important of which are located in mitochondria and endoplasmic reticulum (ER). Different types of oxidized cholesterol have different systemic effects and, relevant to osteoporosis, oxysterol 20(S)-hydroxycholesterol, in combination with either the 22(S) or 22(R) versions, upregulates osteoblasts and strengthens bone through increased alkaline phosphatase activity, osteocalcin gene expression, and enhanced cell mineralization [119]. Other forms of oxidized cholesterol, such as cholestan-3 β,5 α,6 β-triol, inhibit osteoblast formation [120].
II. AGE-RELATED DISEASES WITH MITOCHONDRIAL COMPONENTS THAT ARE LIKELY TO BECOME MORE PREVALENT IN SOCIETY IN THE FUTURE
2.1. Stem Cell Failure
Stem cells are generally grouped into two classes: pluripotent stem cells and tissue-specific stem cells. The more versatile pluripotent stem cells are capable of differentiating into any cell type from all three embryonic germ layers, and are found naturally in the inner cell mass of developing embryos. Tissue-specific stem cells, on the other hand, are much more limited in their differentiating capabilities and their possible fates are restricted to cell types of their tissue of origin. Almost all tissues retain cell populations with stem cell-like characteristics and these act to replenish lost cells and retain tissue function [121]. Other stem cells, called induced pluripotent stem cells (iPS cells), are artificially re-programmed somatic cells that adopt pluripotent stem cell-like characteristics. Since it is well established that tissue function declines with age, one question that comes to mind is what happens to stem cell function in the tissues of elderly individuals? Do stem cells themselves show signs of aging, does the niche in which these specialized cells reside bear the brunt of time and in turn become incapable of supporting these potentially immortal cells, or does something else occur?
Current studies indicate that aging limits the ability of stem cells to divide, self-renew, and respond to environmental signals [122]. With the stem cell differentiation pathway being determined by environmental cues, the ability of certain stem cell types to differentiate into specific tissues is affected by age. The differentiation of mesenchymal stem cells into adipogenic, chondrogenic, or osteogenic cell lines is one notable example [123]. Disruption of stem cell function underlies an increasing number of age-related pathologies, including atherosclerosis, osteoporosis, and mitochondrial respiratory chain deficiency [124–126]. Mitochondria are actively involved in the proper function of stem cells, namely by meeting varying metabolic requirements and controlling cell fate with ROS signaling [127]. Many studies now point to mtDNA mutations, altered ROS production, impaired mitochondrial metabolism, and structurally compromised mitochondria as contributing factors for some stem cell-related pathologies [128, 129].
2.1.1. Characteristics of Mitochondria in Differentiating Cells
The remarkable changes to mitochondria in stem cells can best be seen by comparing them to those in dividing somatic cells. During mitosis in somatic cells, mitochondria are pulled to the cleavage furrow via microtubules where they undergo fusion and fission to homogenize mtDNA distribution and proteome composition of the two daughter cells [130]. Cells that have defects in the mitochondrial fusion and fission proteins have a heterogeneous mitochondrial proteome, leading to reduced genetic stability, the accumulation of mtDNA mutations, and respiratory chain dysfunction [131]. Aside from the flux in organelle dynamics, there is no evidence showing a change in somatic cell mitochondria during division.
The alterations to stem cell mitochondria are in stark contrast to the relatively stable somatic cell mitochondria. Studies on mitochondrial remodeling in stem cell differentiation and reprogramming show that non-differentiating, or naive, pluripotent cells have rounded, less mature mitochondria. When pluripotent stem cells differentiate, their mitochondria become elongated and develop more mature, defined cristae [132]. As shown by studies using embryoid bodies, mitochondrial morphology appears to be a key factor in proper stem cell function. Embryoid bodies are spheres of pluripotent stem cells floating in suspension and their construction is currently the most accurate method of representing embryogenesis and studying stem cell differentiation. One study showed that the disruption of mitochondrial networks in embryoid bodies undergoing cardiac differentiation inhibited sarcomere formation [133]. The importance of morphology reflects the shift from glycolysis as the main source of energy to OXPHOS during differentiation, as an intact network is important for construction of the respiratory chain [132, 134].
It has been observed that when embryonic stem cells differentiate, mitochondrial mass and amount of mtDNA both increase [135]. Interestingly, unmodified, in vitro, human embryonic stem cells have large mtDNA deletions that are carried through to the final differentiated cells, but they do not impact the differentiation capability of the cell [136]. Human embryonic stem cells contain fewer mitochondria, produce less ATP and ROS, and have elevated ROS defenses compared to differentiating pluripotent stem cells [137]. Moreover, mitochondria in differentiated cells are tubular and located throughout the cytoplasm, while undifferentiated cell mitochondria are globular and perinuclear [138]. All these changes are important aspects of stem cell differentiation and are reversed during the reprogramming process [135]. There is no definitive answer to whether these mitochondrial changes are the same for all differentiation pathways, or if there are cell-type specific modifications.
2.1.2. Mitochondrial Role in Stem Cell Differentiation
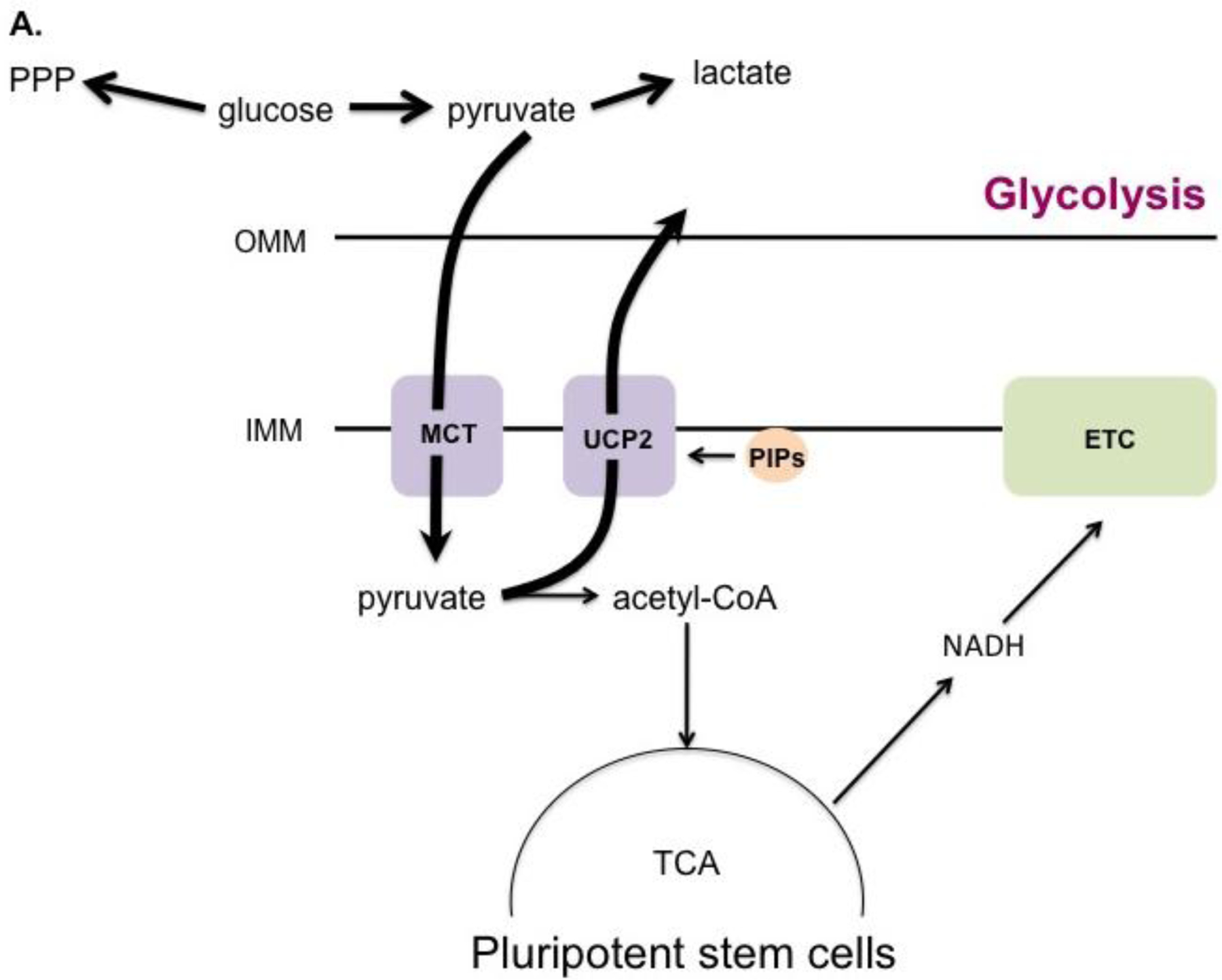
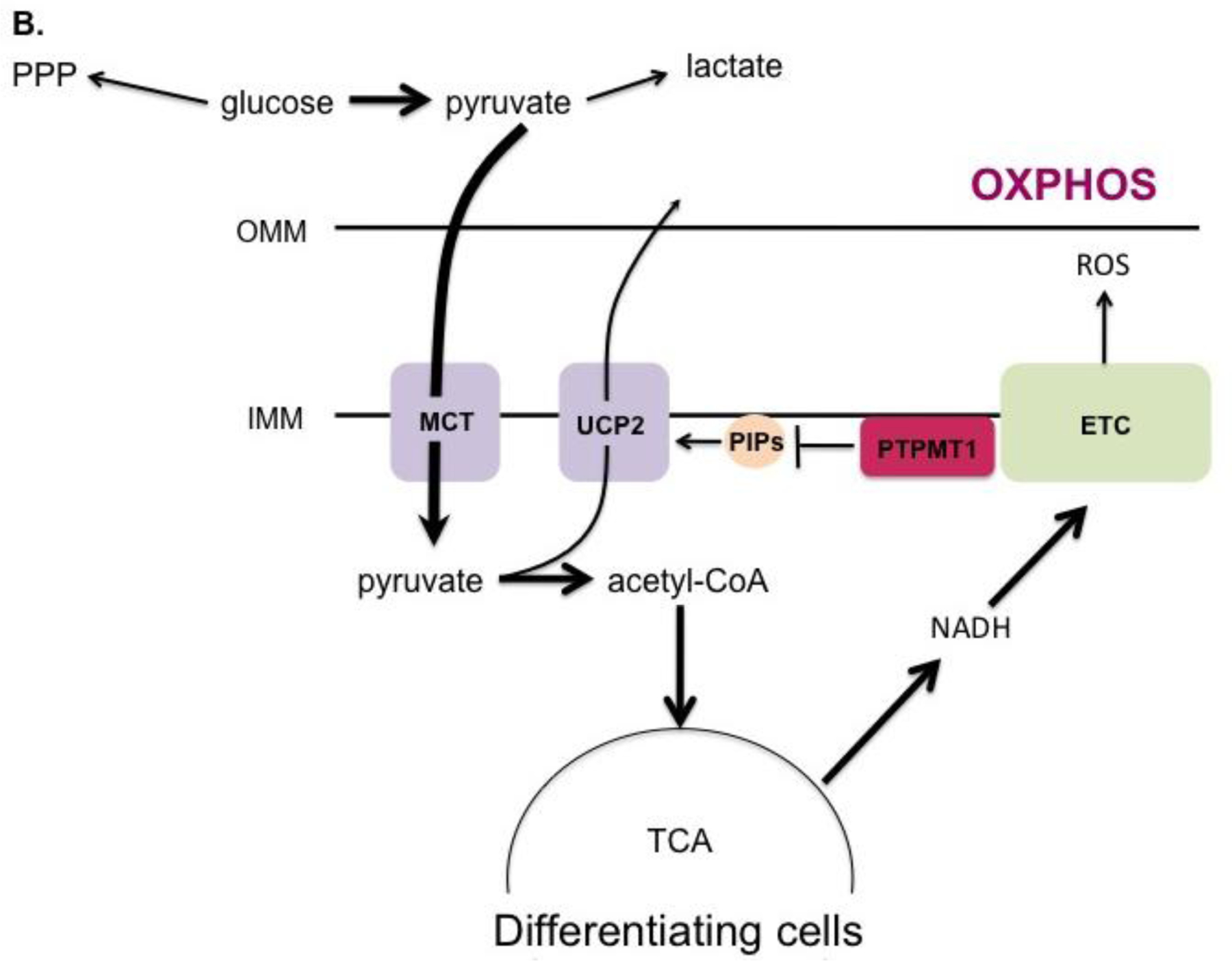
While absolute mitochondrial mass increases during differentiation as mentioned above, it has been reported that the ratio of mitochondria to total cell protein mass does not change. In fact, pluripotent and differentiated cells have similar oxygen consumption rates, suggesting their mitochondria have comparable metabolic activity [139]. How is it then that stem cells produce lower amounts of ATP compared to their differentiated counterparts? The answer lies in the metabolic programming of each cell type. Despite having fully-functioning mitochondria, pluripotent stem cells produce most of their ATP by glycolysis. This is achieved by preventing the entry of pyruvate into the TCA cycle, instead diverting glucose into other metabolic pathways, including lactate fermentation and the pentose phosphate pathway (Fig. 1A) [140]. This shunting process is mediated by uncoupling protein 2 (UCP2). UCP2 is activated by phosphatidylinositol phosphates (PIPs) [141]. During stem cell differentiation, the mitochondrial phosphatase Protein Tyrosine Phosphatase, Mitochondrial 1 (PTPMT1) dephosphorylates PIPs, inactivating UCP2 and allowing the necessary transition to mitochondrial respiration to take place (Fig. 1B)[141]. Deletion of the PTPMT1 gene, rendering the protein catalytically inactive, or removing the mitochondrial localization signal all resulted in the failure of hematopoietic stem cells to differentiate. In agreement with these findings, ectopic UCP2 expression has also been shown to impede differentiation [139]. Together these studies show that PTPMT1 is a part of the mechanism by which stem cells undergo metabolic reprogramming, serving to inactivate UCP2 and facilitate the transition to aerobic metabolism that is seen in differentiating cells.
Figure 1. ROS as determinants of stem cell differentiation.
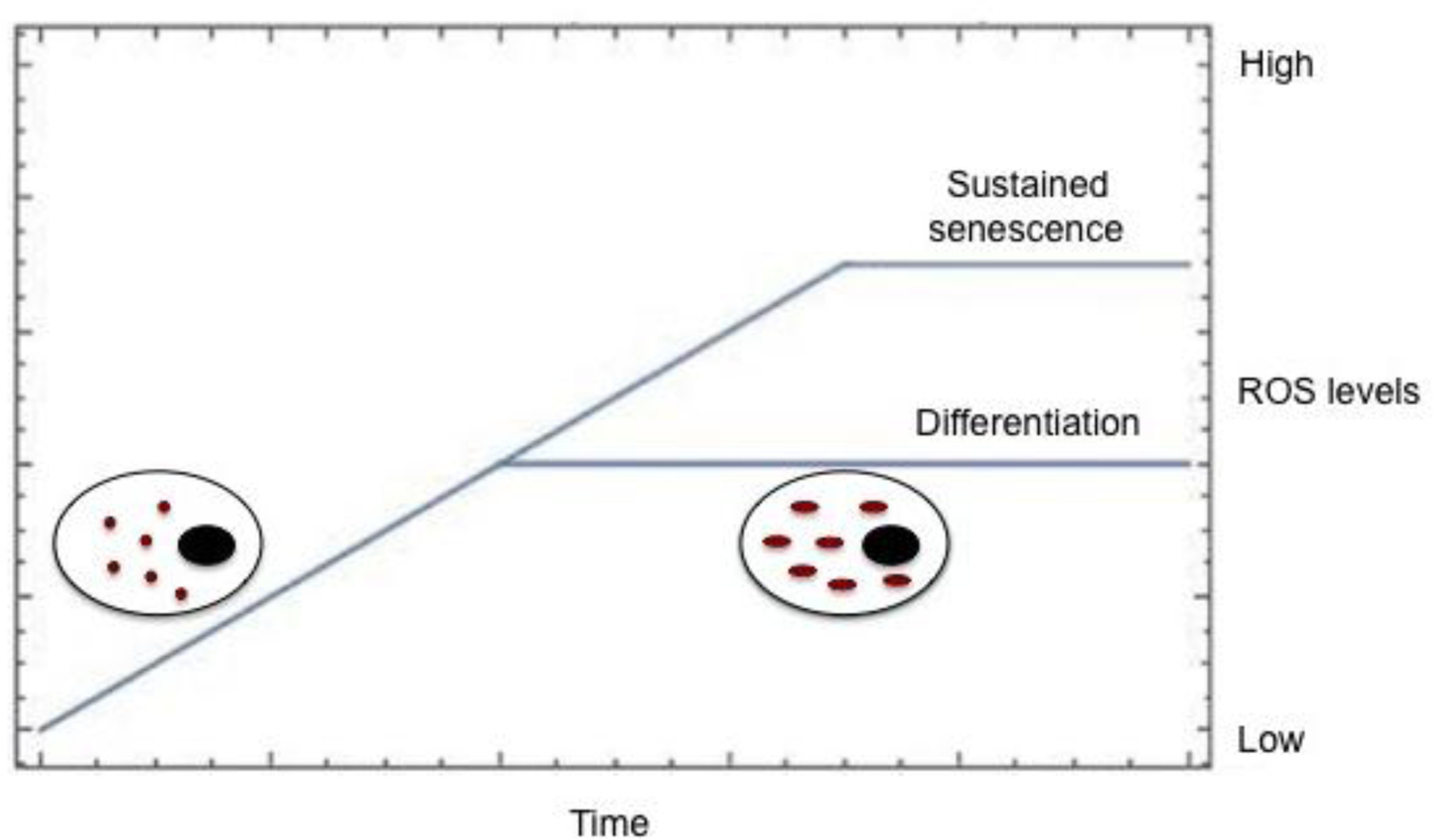
Depending on concentration, ROS can either cause a stem cell to differentiate, or guide it into a senescent state.
The importance of the metabolic state of a stem cell to its differentiation capacity is further exhibited by the finding that murine embryonic stem cells that have lower rates of metabolism, as measured by mitochondrial membrane potential and oxygen consumption, have higher differentiation capabilities in vitro and decreased tumorigenic properties in vivo [142]. This study also characterized a mechanistic target of rapamycin (mTOR)-mitochondria axis. mTOR activity correlated with metabolic activity, and when cells were treated with the mTOR inhibitor rapamycin, there was a decrease in mitochondrial oxygen consumption and an increased ability of the cells to differentiate. It has been proposed that this connection may be mediated by the interaction of mTOR with PGC-1α, a transcription factor for genes associated with mitochondrial biogenesis.
Different cell lines also have different amounts of mtDNA transcription associated with their differentiation. The timing of the increase in mtDNA transcription factors and the mitochondrial replication process is critical for the specific cell type differentiation [143]. The increase in mtDNA transcription can be timed in conjunction with changes in ROS signaling, which affects proliferation and differentiation in some stem cells, especially hematopoietic stem cells [127]. ROS signaling as a marker for differentiation was seen in Drosophila as well; lowering ROS concentrations led to delayed differentiation in fly hematopoietic stem cells, and an increase had the opposite effect [144]. Somatic mtDNA mutations can cause stem cell dysfunction and cause premature aging in mice with impaired mtDNA proofreading [124]. The effects manifest at different points in the differentiation process for different cell lineages but there is an overall inhibition of differentiation, impaired tissue replenishment, and a reduction in lifespan. It is thought that somatic mtDNA mutations are able to affect stem cells by changing ROS signaling based on the observation that a ROS scavenger was able to rescue stem cell function [124]. It is well established that mtDNA damage accumulates with age [1]. There is a link then between mitochondrial dysfunction and ailments that result from an age-dependent decline in stem cell function.
2.1.3. Mitochondria in Regulation of the Stem Cell Life Cycle
Modulating differentiation is not the only way in which mitochondria are active in stem cells; they are also important regulators of the cell cycle. p53 and p38 signaling pathways control the increase of mitochondrial mass, membrane potential, and ROS production in cells. The effect of ROS on stem cells was tested by exposing human endometrium-derived mesenchymal stem cells to non-lethal doses of hydrogen peroxide, which led to senescence via the p53/p21/pRb and p38/MK2 DNA damage response pathways. Within one hour of exposure, intracellular ROS levels increased and remained high over nine days after exposure, maintaining cells in a state of senescence [145]. Inhibition of the p38 pathway resulted in increased levels of stem-cell proliferation. Interestingly, the recovery of proliferative ability caused by p38 inhibition was not as successful when inhibitors were added later in the senescence process, suggesting a time-sensitive ability to recover stem cell functionality [145]. This study shows that endometrium-derived mesenchymal stem cells seem to rely on increased levels of ROS to maintain the DNA damage response pathways and remain in a senescent state (Fig. 2). Surprisingly, activation of the p53/MAPK pathway also led to mitochondrial dysfunction that increased ROS levels, DNA damage, and continued use of the p53/MAPK pathway, creating a feedback loop resulting in the loss of pluripotency and self-renewal capabilities of adipose-derived stem cells [122].
Figure 2. Metabolic reprogramming of differentiating stem cells.
A. In undifferentiated pluripotent stem cells, UCP2 is activated by phosphatidylinositol phosphates (PIPs) at the inner mitochondrial membrane (IMM), which prevents entry of pyruvate into the TCA cycle. Glucose is consequently shunted into alternate metabolic pathways such as the pentose phosphate pathway (PPP). B. During differentiation, the mitochondrial phosphatase PTPMT1 dephosphorylates PIPs, downregulating UCP2 and allowing pyruvate to be metabolized by the TCA cycle to fuel OXPHOS.
The growth factor TGF-β, commonly used to induce cardiomyocyte differentiation in bone marrow-derived mesenchymal stem cells, also induces senescence of these cells, partly by decreasing the expression of mitochondrial SOD2, resulting in a rise in ROS [146]. ROS levels were also found to regulate stem cell quiescence and self-renewal in hematopoietic stem cells [140]. Collectively, these results show that ROS are major signaling molecules involved in controlling multiple parts of the stem cell cycle.
The integrity of mitochondria in undifferentiated embryonic stem cells is also important in avoiding apoptosis. Disrupting the ETC or membrane permeability led to increased rates of mitochondria-mediated apoptosis [147]. Excessive mitochondrial fragmentation also results in decreased cell survival in mouse embryonic stem cells [148].
2.1.4. Mitochondria in Stem Cell-Mediated Tissue Repair
Loss of tissue-specific stem cell function, including a decreased ability to differentiate, or outright death, all lead to a decrease in the ability of a tissue to recover following homeostatic challenge [149]. Reprogramming tissue-specific stem cells or somatic cells into iPS cells has the ability to rejuvenate cells to a more youthful state, providing an avenue for stem cell-based therapies. During iPS reprogramming, cells undergo metabolic re-structuring to attain a pluripotent-like state [134, 137]. Mitochondrial morphology during the transition consists of both tubular and globular type mitochondria. While iPS cells are reliant on glycolysis, evidence suggests they have different expression profiles of genes involved in glucose metabolism and O2 consumption relative to normal pluripotent (embryonic) stem cells [138]. Nonetheless, iPS cells exhibit many of the characteristics seen in these cells, including elongated telomeres and gross gene expression profiles, but often display varying levels of rejuvenation, even between cells from the same donor [137]. Some of these differences seem to stem from the reprogramming process. One inherent issue with reprogramming has been the appearance of nonspecific point mutations in mtDNA. Oddly, no phenotype has been identified from these mutations, and they do not seem to affect the reprogrammed stem cell [134]. While it has not been fully determined whether the original age of the reprogrammed tissue-specific stem cell affects the severity of these mutations, the restructuring of mitochondria to the underdeveloped embryonic-like state described above is independent of the original cell’s age [137].
Amazingly, stem cells have their own method of restoring the respiratory function of somatic epithelial cells. Mesenchymal stem cells can target cells that have respiratory dysfunction and transfer their mitochondria through gap junctions via tunneling nanotubes made of actin, restoring mitochondrial function and decreasing ROS production. Respiratory dysfunction in the receiving cell is required for transfer to take place, both in vivo and in vitro. Miro1, an enzyme involved in mitochondrial transport along microtubules, was shown to be needed for the transfer between cells [150].
2.2. Infections
One of the concerns that arises with aging is the increased susceptibility to infections. In future research on treating immunological deficits, a mitochondria-centered approach may be in order. Beyond mediating apoptosis of infected cells, mitochondria are emerging as critical components of the innate immune response. It has been shown that the ATP needed for purinergic signaling, T-cell regulation, and initial activation of neutrophils comes from mitochondria [151, 152]. ATP production and mitochondrial Ca2+ buffering are needed for antigen presentation and processing, and ROS are a part of the signaling pathway that activates inflammatory proteins [153, 154]. With the current rise of multidrug-resistant “superbugs”, other ways of combating infections grow increasingly crucial [155]. Based on accumulating evidence, mitochondria may be viable therapeutic targets.
2.2.1. Bacterial and fungal infection
ROS form an integral component of innate immunity signaling pathways. One study found that signaling by cell surface toll-like receptors (TLRs) resulted in recruitment of mitochondria to macrophage phagosomes and stimulated mitochondrial ROS production with the aid of TLR adapter protein TRAF6 [156]. Mitochondrial ROS production was dependent on translocation of TRAF6 to mitochondria and its subsequent ubiquitination of ECSIT (evolutionarily conserved signaling intermediate in Toll pathways). ECSIT is a part of respiratory complex I assembly and typically localizes to the inner mitochondrial membrane but there is a fraction that associates with the outer membrane. TRAF6 triggered an increase in the proportion of outer membrane ECSIT. When mitochondrial ROS levels were dampened, either by mitochondrial expression of catalase or by the absence of ECSIT or TRAF6, macrophages showed an accumulation of infecting bacteria. TRAF6 and ECSIT are therefore critical to the mitochondrially-mediated immune response [156]. ROS serve other infection-fighting functions as well. Management of mitochondrial ROS concentrations by SOD2 aids in innate immunity by regulating the production of phagocytes and facilitating bacterial clearance. Importantly, a mitochondrial ROS scavenger mitoTEMPO can make up for SOD2 deficiency, opening up new possibilities for the treatment of bacterial infections [157]. In line with the idea that mitochondria may be an influential focal point of future antibacterial research endeavors, one group has found that experimentally inducing ROS production in E. coli increases the bacteria’s vulnerability to antibiotics [158]. Another recent study found that the essential oil of Monarda punctata, known for its antibacterial effects, acts at least in part by triggering an increase in bacterial ROS production, causing damage to the membrane [159]. Whether the ECSIT pathway can be exploited as a means for pharmaceutically-controlling mitochondrial ROS generation to enhance bacterial killing remains an open possibility.
Microorganisms may, however, have begun to evolve a way to not only protect themselves from their host’s immune response, but use it to their advantage. One strain of the fungus Cryptococcus gattii infects healthy individuals by subverting the host’s ROS-mediated defense. Within host macrophages, some of the infecting fungi are able to convert their own mitochondria into a tubular conformation in response to oxidative stress. These cells do not divide very frequently but are resistant to killing and appear to allow co-infecting cells that do not tubularize their mitochondria to divide rapidly [160].
2.2.2. Viral infection
Viral RNA is detected by pattern recognition receptors (PRRs) such as retinoic acid-inducible gene I (RIGI)-like receptors (RLRs) and TLRs. Once activated, these proteins interact with mitochondrial anti-viral signaling protein (MAVS), which inserts its caspase activation and recruitment domain (CARD) into the outer mitochondrial membrane. This causes a signaling cascade that activates proteins of the innate immune system [161]. During infection, there is an increased association of the endoplasmic reticulum with mitochondria, particularly elongated mitochondria. It is thought that mitochondrial elongation promotes RLR signaling by allowing interaction of MAVS with an ER protein, stimulator of interferon genes (STING). For example, a lack of mitochondrial fission proteins results in elongation and increased RLR signaling, whereas fragmentation diminishes signaling [162]. However, fission may still be important for antiviral response. A recent study established a RIP1-RIP3-DRP1 signaling axis in which the serine-threonine kinases RIP1 and RIP3 form a complex in response to infection by various RNA viruses and activate DRP1. DRP1 translocates to mitochondria and promotes activation of the NLRP3 inflammasome by inducing mitochondrial fission and ROS production [163].
Mitochondrial cell death pathways also exhibit anti-viral properties. A new model now suggests that there are two pathways by which cell-death mediators Bax and Bak can potentially act: the cell death pathway, and the viral resistance pathway. In the cell death pathway, permeabilization of the mitochondrial outer membrane by Bax and Bak causes the release of cytochrome c, leading to apoptosome and caspase activation. Alternately, it has recently been found that in caspase knockout mice, permeabilization by Bax and Bak can also instigate an antiviral response by allowing the release of mtDNA into the cytosol to activate the cGAS/STING pathway, resulting in the production of type I interferons (IFNs) [164]. This pathway is inhibited by caspases, though it is not known how, nor is it known in which cells mtDNA-mediated activation of the cGAS/STING pathway occurs in vivo.
As mediators of immunity, mitochondria are consequently targeted by several viruses: Influenza A viral protein PB1-F2 induces mitochondrial dysfunction as a mechanism of crippling the innate immune response. The protein translocates across the outer membrane, accumulates in the inner membrane space, and causes a drop in mitochondrial membrane potential. This results in mitochondrial fragmentation, inhibits NLRP3 activation, and induces apoptosis [165, 166]. Accordingly, PB1-F2 translocation correlates with subdued innate immunity [166]. The SARS virus also targets mitochondria. The virus-encoded protein ORF-9b localizes to mitochondria and triggers degradation of DRP1, MAVS, TRAF3, and TRAF6, thus evading the host immune responses [167]. There is also evidence that hepatitis C affects mitochondria by suppressing mitophagy, triggering mitochondrial fission, and preventing apoptosis [168, 169].
Not only are mitochondria targeted for attack by invading viruses, but they contribute to virulence. For example, mitochondria are key devices in HIV-1 infection. When an HIV-1 infected T cell makes contact with an uninfected cell, mitochondria are actively recruited by the infected cell to the site of cell-cell contact and these mitochondria buffer intracellular Ca2+, an important signal for the infection process. Perhaps most importantly, it was found that disrupting mitochondrial re-localization by inhibiting mitochondrial trafficking and Ca2+ dynamics impaired the spread of the virus at the virological synapse [170].
III. NOVEL WAYS IN WHICH MITOCHONDRIAL DISRUPTION RESULTS IN AGE-RELATED PROBLEMS
3.1. Covalent modification
Post-translational modifications represent one way in which cells respond to an ever-changing environment. The epigenetic landscape changes with age, and a disruption of epigenetic dynamics can culminate in metabolic disease [171, 172]. In mitochondria, we have only just begun to unveil the post-synthetic changes that define this organelle’s DNA, RNA and protein landscapes. Already, disruptions to these processes have been linked with disease. There is a great amount of acetylation, succinylation, and malonylation of mitochondrial proteins (Table 1). Covalent modifications such as these are regulated by the activity of sirtuins. Sirtuins have arisen as anti-aging genes, providing a link between aging and metabolism with mitochondria as a focal point [176]. There are three sirtuins that are active in mitochondria: SIRT3, SIRT4, and SIRT5. SIRT3 deacetylates many major enzymes involved in lipid metabolism and the TCA cycle, SIRT4 is a lipoamidase acting on pyruvate dehydrogenase, and SIRT5 has demalonylase, desuccinylase, and deglutarylase activity in mitochondria [174, 177–179].
Table 1.
Post-translational modifications of mitochondrial proteins.
| Type of modification | Enzymes | Targets | Reference |
|---|---|---|---|
| Phosphorylation | PKA, Src-kinases, c-Src, EGFR, ErbB2, Fgr, PDH-kinase, PINK1, Abl | TFAM, CREB, complexes I-V, aconitase hydratase, citrate synthase, glycerol-3-phosphate dehydrogenase, hexokinases 1 & 2, PDH, MPTP, Drp1, Miro, Parkin, BAD, BCL-xL | 173 |
| Deacetylation | SIRT3, SIRT4, SIRT5 | Cyclophilin D, mitoribosome, complex I, II, & V, LCAD, acetyl-CoA synthetase 2, HMGCS2, AceCS2, MCD, OTC, PDH, GDH1, CPS1, UOX, IDH2, SOD1, SOD2, aldehyde dehydrogenase 2, MPTP, OGG1, Foxo3, LBK1, Ku70 | 172,173 |
| Desuccinylation | SIRT5 | Complex II, PDH, SOD1 | 172 |
| Ubiquitination | Parkin, RNF5, Usp30, Huwe1, MARCH5, RNF185, Usp9x | Drp1, Fis1, Mfn 1&2, BNIP1, p62, SOD1, Akt 1 & 2, MCL-1, MPTP, MAVS, RIG-1, STING/MITA, TANK | 173 |
| Deglutarylation | SIRT5 | CPS1, HADHA, GOT2 | 174 |
| ADP ribosylation | SIRT4 | GDH | 172 |
| Sumoylation | MULAN, SENP2 | Drp1 | 173, 175 |
Abbreviations: PKA - protein kinase A; TFAM - transcription factor A, mitochondrial; CREB - cAMP response element-binding protein; EGFR - epidermal growth factor receptor; PDH-kinase - pyruvate dehydrogenase kinase; PINK1 - PTEN induced putative kinase 1; PDH - pyruvate dehydrogenase; BAD - BCL2-associated agonist of cell death; BCL-xL - B-cell lymphoma-extra large; LCAD - long-chain acyl CoA dehydrogenase; HMGCS2 – 3-hydroxy-3- methylglutaryl CoA synthase 2; AceCS2 - acetyl-CoA synthetase 2; MCD - malonyl CoA decarboxylase; OTC - ornithine transcarbamoylase; GDH - glutamate dehydrogenase; CPS1 - carbamoyl phosphate synthetase 1; UOX - ureate oxidase; IDH2 - isocitrate dehydrogenase 2; SOD1 - superoxide dismutase 1; SOD2 - superoxide dismutase 2; OGG1 – 8-oxoguanine-DNA glycosylase 1; Foxo3 - forkhead box O 3; LBK1 - serine/threonine kinase 1; RNF5 - ring finger protein 5; Huwe1 - HECT, UBA, and WWE domain containing 1; MCL-1 - myeloid cell leukemia 1; MARCH5 - membrane-associated ring finger (C3HC4) 5; RNF185 - ring finger protein 185; STING (also known as MITA) - stimulator of interferon genes; TANK - TRAF family member-associated NF-KB activator; HADHA - hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase/enoyl-CoA hydratase (trifunctional protein), alpha subunit; GOT2 - glutamic-oxaloacetic transaminase 2; MULAN - mitochondrial ubiquitin ligase activator of NF- KB; SENP2 - SUMO1/sentrin/SMT3 specific peptidase 2.
3.1.1. Sirtuins
Research in this area has shown that an imbalance in the regulation of post-translational modifications in mitochondria may be an important factor in cancer [180]. Lower levels of SIRT3 and SIRT4 expression occur in multiple cancers, and overexpression of SIRT3 has been found in others [181, 182]. One clue as to how differential SIRT3 expression exerts these effects may lie in its regulation of SOD2, respiratory complex I, and complex III and therefore ROS concentrations and energy homeostasis [183–185]. Furthermore, hyperacetylation in mitochondria is another characteristic of cancer as well as cardiac hypertrophy [173].
Abnormal lysine deacetylase activity in mitochondria has been linked to neurodegeneration as well. Acetylation is important for modulating mitochondrial morphology, biogenesis, trafficking, and mitophagy, all of which are affected in neurodegenerative diseases [186]. In a PINK1 knockout and α-syn overexpression double mutant mouse model of PD, there is a marked increase in deacetylation of mitochondrial matrix proteins in the brain. Since these changes in acetylation precede mitophagy and neuronal loss, the development of antibodies to detect such changes may be a useful tool for diagnosing PD early on [181]. SIRT3 has been shown to have neuroprotective properties in AD, guarding against the many deleterious effects of ROS [187].
Disruption of post-translational modification by SIRT3 appears to contribute to a variety of widespread age-related maladies. SIRT3 levels are known to decline with age [188]. A loss of SIRT3 in mice results in many markers of mitochondrial dysfunction, and these mice are more prone to developing age-related pathologies after being treated with stress factors [182]. Among these pathologies are cardiac hypertrophy, carcinogenesis, fatty liver, radiation-induced liver damage, and age-related hearing loss. It has recently been found that a decrease in SIRT3 expression also contributes to dysfunction of the central auditory system by causing an accumulation of ROS [188]. Additionally, the ability to recover from ischemia-reperfusion injury is reduced in cells depleted of SIRT3 [189]. These cells also showed low rates of oxygen consumption, and decreased complex I and SOD2 activity.
Manipulation of sirtuin expression poses an intriguing therapeutic option. Initial studies suggest that compounds that upregulate sirtuins have positive effects on metabolic, neurodegenerative, cardiovascular, and cancer conditions [172]. However, so far only a limited number of compounds that increase sirtuins have been identified and those that exist have limited effectiveness [190].
3.1.2. The TCA cycle and covalent modification
Beyond sirtuins, there are other ways in which mitochondria and epigenetic changes are interconnected. The TCA cycle intermediates α-ketoglutarate, succinate, and fumarate exert regulatory functions on demethylases [171]. α-ketoglutarate is a co-substrate of the 2-oxoglutarate Fe-dioxygenase (2-OGDO) class of enzymes that demethylate DNA and histones, while succinate and fumarate act as inhibitors. It has been shown that loss-of-function mutations, or chemical inhibition, of succinate dehydrogenase and fumarate hydratase, exacerbate cancer progression due to the resulting accumulation of succinate and fumarate, leading to alterations in histone methylation. It has also been found that there is hypermethylation of cytosine residues in AD brains [191]. Given that priming for cytosine demethylation is controlled by enzymes of the 2-OGDO family known as TETs [192], this may reflect yet another way in which mitochondrial dysfunction contributes to AD progression. Dysfunctional TCA enzymes are also implicated in cancer due to their link with TET activation. Specifically, mutations in isocitrate dehydrogenase and succinate dehydrogenase result in improper DNA methylation and can promote tumorigenesis [193]. Citrate is another important mitochondrial metabolite. Its activation of ATP-citrate lyase (ACLY) results in increased acetyl-CoA levels which, in turn, lead to histone acetylation and thereby alterations in gene expression [171]. A recent study has found that ACLY knockdown triggers cellular senescence and activation of tumor-suppressor p53 [194]. Maintenance of mitochondrial metabolites and enzymes and their role in covalent modifications may therefore be one aspect of cancer treatment in need of further investigation.
CONCLUSIONS
Mitochondria have emerged as focal constituents of different aging-related pathologies. A recurring theme in many diseases is mitochondrial dysfunction, mainly in the form of mtDNA mutations and ETC inefficiencies that give way to the loss of energy production and excessive ROS formation. The result of such shortcomings is differing pathologies, depending on the affected cell type. The exact link between mitochondrial dysfunction and aging has not been fully elucidated, but understanding this connection is certain to be an important step toward developing better methods of prevention and treatment of the multitude of ailments that loom over our ever-aging population.
Highlights
Mitochondrial dysfunction may be at the root of protein aggregation
ROS signaling and metabolic restructuring are determinants of stem cell fate
Mitochondria are central components of the innate immune response
Deregulation of covalent modifications of mitochondrial proteins has been found in multiple diseases
ACKNOWLEDGEMENTS
The authors wish to thank Dr. Megan Borror (UTHSCSA) and Erin Munkacsy (UTHSCSA) for critical comments on the manuscript. Funding support was provided by the National Institute for Aging (R21AG047561 (SLR)).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- [1].Chistiakov DA, Sobenin IA, Revin VV, Orekhov AN, Bobryshev YV, Mitochondrial aging and age-related dysfunction of mitochondria, Biomed Res Int, 2014 (2014) 238463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Figueira TR, Barros MH, Camargo AA, Castilho RF, Ferreira JC, Kowaltowski AJ, et al. Mitochondria as a source of reactive oxygen and nitrogen species: from molecular mechanisms to human health. Antioxid Redox Signal (2013); 18(16): 2029–74. [DOI] [PubMed] [Google Scholar]
- [3].Kudin AP, Bimpong-Buta NY, Vielhaber S, Elger CE, Kunz WS, Characterization of superoxide-producing sites in isolated brain mitochondria, J Biol Chem, 279 (2004) 4127–4135. [DOI] [PubMed] [Google Scholar]
- [4].Turrens JF, Mitochondrial formation of reactive oxygen species, J Physiol, 552 (2003) 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Harman D, The biologic clock: the mitochondria?, J Am Geriatr Soc, 20 (1972) 145–147. [DOI] [PubMed] [Google Scholar]
- [6].Ku HH, Brunk UT, Sohal RS, Relationship between mitochondrial superoxide and hydrogen peroxide production and longevity of mammalian species, Free Radic Biol Med, 15 (1993) 621–627. [DOI] [PubMed] [Google Scholar]
- [7].Wanagat J, Dai D, Rabinovitch P, Mitochondrial oxidative stress and mammalian healthspan, Mech Ageing Dev, 131 (2010) 527–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Schleit J, Johnson SC, Bennett CF, Simko M, Trongtham N, Castanza A, Hsieh EJ, Moller RM, Wasko BM, Delaney JR, Sutphin GL, Carr D, Murakami CJ, Tocchi A, Xian B, Chen W, Yu T, Goswami S, Higgins S, Holmberg M, Jeong KS, Kim JR, Klum S, Liao E, Lin MS, Lo W, Miller H, Olsen B, Peng ZJ, Pollard T, Pradeep P, Pruett D, Rai D, Ros V, Singh M, Spector BL, Vander Wende H, An EH, Fletcher M, Jelic M, Rabinovitch PS, MacCoss MJ, Han JD, Kennedy BK, Kaeberlein M, Molecular mechanisms underlying genotype-dependent responses to dietary restriction, Aging Cell, 12 (2013) 1050–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Weindruch RH, Cheung MK, Verity MA, Walford RL, Modification of mitochondrial respiration by aging and dietary restriction, Mech Ageing Dev, 12 (1980) 375–392. [DOI] [PubMed] [Google Scholar]
- [10].Delaney JR, Murakami C, Chou A, Carr D, Schleit J, Sutphin GL, An EH, Castanza AS, Fletcher M, Goswami S, Higgins S, Holmberg M, Hui J, Jelic M, Jeong KS, Kim JR, Klum S, Liao E, Lin MS, Lo W, Miller H, Moller R, Peng ZJ, Pollard T, Pradeep P, Pruett D, Rai D, Ros V, Schuster A, Singh M, Spector BL, Wende HV, Wang AM, Wasko BM, Olsen B, Kaeberlein M, Dietary restriction and mitochondrial function link replicative and chronological aging in Saccharomyces cerevisiae, Exp Gerontol, 48 (2013) 1006–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Long YC, Tan TM, Takao I, Tang BL, The biochemistry and cell biology of aging: metabolic regulation through mitochondrial signaling, Am J Physiol Endocrinol Metab, 306 (2014) E581–591. [DOI] [PubMed] [Google Scholar]
- [12].Ahmad T, Sundar IK, Lerner CA, Gerloff J, Tormos AM, Yao H, Rahman I, Impaired mitophagy leads to cigarette smoke stress-induced cellular senescence: Implications for chronic obstructive pulmonary disease, FASEB J, (2015). [DOI] [PMC free article] [PubMed]
- [13].Anderson G, Maes M, Oxidative/nitrosative stress and immuno-inflammatory pathways in depression: treatment implications, Curr Pharm Des, 20 (2014) 3812–3847. [DOI] [PubMed] [Google Scholar]
- [14].Boland ML, Chourasia AH, Macleod KF, Mitochondrial dysfunction in cancer, Front Oncol, 3 (2013) 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Montgomery MK, Turner N, Mitochondrial dysfunction and insulin resistance: an update, Endocr Connect, 4 (2015) R1–R15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Rera M, Clark RI, Walker DW, Intestinal barrier dysfunction links metabolic and inflammatory markers of aging to death in Drosophila, Proc Natl Acad Sci U S A, 109 (2012) 21528–21533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Insull W, The pathology of atherosclerosis: plaque development and plaque responses to medical treatment, Am J Med, 122 (2009) S3–S14. [DOI] [PubMed] [Google Scholar]
- [18].Li JM, Shah AM, Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology, Am J Physiol Regul Integr Comp Physiol, 287 (2004) R1014–1030. [DOI] [PubMed] [Google Scholar]
- [19].Kaplan M, Aviram M, Retention of oxidized LDL by extracellular matrix proteoglycans leads to its uptake by macrophages: an alternative approach to study lipoproteins cellular uptake, Arterioscler Thromb Vasc Biol, 21 (2001) 386–393. [DOI] [PubMed] [Google Scholar]
- [20].Madamanchi NR, Runge MS, Mitochondrial dysfunction in atherosclerosis, Circ Res, 100 (2007) 460–473. [DOI] [PubMed] [Google Scholar]
- [21].Mazzone T, Reardon C, Expression of heterologous human apolipoprotein E by J774 macrophages enhances cholesterol efflux to HDL3, J Lipid Res, 35 (1994) 1345–1353. [PubMed] [Google Scholar]
- [22].Toth PP, Reverse cholesterol transport: high-density lipoprotein’s magnificent mile, Curr Atheroscler Rep, 5 (2003) 386–393. [DOI] [PubMed] [Google Scholar]
- [23].Heinecke JW, Rosen H, Suzuki LA, Chait A, The role of sulfur-containing amino acids in superoxide production and modification of low density lipoprotein by arterial smooth muscle cells, J Biol Chem, 262 (1987) 10098–10103. [PubMed] [Google Scholar]
- [24].Harrison CM, Pompilius M, Pinkerton KE, Ballinger SW, Mitochondrial oxidative stress significantly influences atherogenic risk and cytokine-induced oxidant production, Environ Health Perspect, 119 (2011) 676–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Kisucka J, Chauhan AK, Patten IS, Yesilaltay A, Neumann C, Van Etten RA, Krieger M, Wagner DD, Peroxiredoxin1 prevents excessive endothelial activation and early atherosclerosis, Circ Res, 103 (2008) 598–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Murphy MP, How mitochondria produce reactive oxygen species, Biochem J, 417 (2009) 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Ballinger SW, Patterson C, Knight-Lozano CA, Burow DL, Conklin CA, Hu Z, Reuf J, Horaist C, Lebovitz R, Hunter GC, McIntyre K, Runge MS, Mitochondrial integrity and function in atherogenesis, Circulation, 106 (2002) 544–549. [DOI] [PubMed] [Google Scholar]
- [28].Ding Z, Liu S, Wang X, Dai Y, Khaidakov M, Deng X, Fan Y, Xiang D, Mehta JL, LOX-1, mtDNA damage, and NLRP3 inflammasome activation in macrophages: implications in atherogenesis, Cardiovasc Res, 103 (2014) 619–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Paradies G, Petrosillo G, Paradies V, Ruggiero FM, Role of cardiolipin peroxidation and Ca2+ in mitochondrial dysfunction and disease, Cell Calcium, 45 (2009) 643–650. [DOI] [PubMed] [Google Scholar]
- [30].Schwall CT, Greenwood VL, Alder NN, The stability and activity of respiratory Complex II is cardiolipin-dependent, Biochim Biophys Acta, 1817 (2012) 1588–1596. [DOI] [PubMed] [Google Scholar]
- [31].Paradies G, Petrosillo G, Pistolese M, Ruggiero FM, The effect of reactive oxygen species generated from the mitochondrial electron transport chain on the cytochrome c oxidase activity and on the cardiolipin content in bovine heart submitochondrial particles, FEBS Lett, 466 (2000) 323–326. [DOI] [PubMed] [Google Scholar]
- [32].Lee S, Birukov KG, Romanoski CE, Springstead JR, Lusis AJ, Berliner JA, Role of phospholipid oxidation products in atherosclerosis, Circ Res, 111 (2012) 778–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wan M, Hua X, Su J, Thiagarajan D, Frostegård AG, Haeggström JZ, Frostegård J, Oxidized but not native cardiolipin has pro-inflammatory effects, which are inhibited by Annexin A5, Atherosclerosis, 235 (2014) 592–598. [DOI] [PubMed] [Google Scholar]
- [34].Boersma HH, Kietselaer BL, Stolk LM, Bennaghmouch A, Hofstra L, Narula J, Heidendal GA, Reutelingsperger CP, Past, present, and future of annexin A5: from protein discovery to clinical applications, J Nucl Med, 46 (2005) 2035–2050. [PubMed] [Google Scholar]
- [35].Mink JW, Blumenschine RJ, Adams DB, Ratio of central nervous system to body metabolism in vertebrates: its constancy and functional basis, Am J Physiol, 241 (1981) R203–212. [DOI] [PubMed] [Google Scholar]
- [36].Herrero-Mendez A, Almeida A, Fernández E, Maestre C, Moncada S, Bolaños JP, The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1, Nat Cell Biol, 11 (2009) 747–752. [DOI] [PubMed] [Google Scholar]
- [37].Pellerin L, Magistretti PJ, Sweet sixteen for ANLS, J Cereb Blood Flow Metab, 32 (2012) 1152–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Nunomura A, Castellani RJ, Zhu X, Moreira PI, Perry G, Smith MA, Involvement of oxidative stress in Alzheimer disease, J Neuropathol Exp Neurol, 65 (2006) 631–641. [DOI] [PubMed] [Google Scholar]
- [39].McManus MJ, Murphy MP, Franklin JL, Mitochondria-derived reactive oxygen species mediate caspase-dependent and -independent neuronal deaths, Molecular and cellular neurosciences, 63c (2014) 13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Santos RX, Correia SC, Zhu X, Smith MA, Moreira PI, Castellani RJ, Nunomura A, Perry G, Mitochondrial DNA oxidative damage and repair in aging and Alzheimer’s disease, Antioxid Redox Signal, 18 (2013) 2444–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Nunomura A, Honda K, Takeda A, Hirai K, Zhu X, Smith MA, Perry G, Oxidative damage to RNA in neurodegenerative diseases, J Biomed Biotechnol, 2006 (2006) 82323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Schon EA, Manfredi G, Neuronal degeneration and mitochondrial dysfunction, J Clin Invest, 111 (2003) 303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Swerdlow RH, Burns JM, Khan SM, The Alzheimer’s disease mitochondrial cascade hypothesis, J Alzheimers Dis, 20 Suppl 2 (2010) S265–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Lin MT, Beal MF, Alzheimer’s APP mangles mitochondria, in: Nat Med, United States, 2006, pp. 1241–1243. [DOI] [PubMed] [Google Scholar]
- [45].Pavlov PF, Wiehager B, Sakai J, Frykman S, Behbahani H, Winblad B, Ankarcrona M, Mitochondrial γ-secretase participates in the metabolism of mitochondria-associated amyloid precursor protein, FASEB J, 25 (2011) 78–88. [DOI] [PubMed] [Google Scholar]
- [46].Pinho CM, Teixeira PF, Glaser E, Mitochondrial import and degradation of amyloid-β peptide, Biochim Biophys Acta, 1837 (2014) 1069–1074. [DOI] [PubMed] [Google Scholar]
- [47].Santos RX, Correia SC, Wang X, Perry G, Smith MA, Moreira PI, Zhu X, A synergistic dysfunction of mitochondrial fission/fusion dynamics and mitophagy in Alzheimer’s disease, J Alzheimers Dis, 20 Suppl 2 (2010) S401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lustbader JW, Cirilli M, Lin C, Xu HW, Takuma K, Wang N, Caspersen C, Chen X, Pollak S, Chaney M, Trinchese F, Liu S, Gunn-Moore F, Lue LF, Walker DG, Kuppusamy P, Zewier ZL, Arancio O, Stern D, Yan SS, Wu H, ABAD directly links Abeta to mitochondrial toxicity in Alzheimer’s disease, Science, 304 (2004) 448–452. [DOI] [PubMed] [Google Scholar]
- [49].Yan SD, Fu J, Soto C, Chen X, Zhu H, Al-Mohanna F, Collison K, Zhu A, Stern E, Saido T, Tohyama M, Ogawa S, Roher A, Stern D, An intracellular protein that binds amyloid-beta peptide and mediates neurotoxicity in Alzheimer’s disease, Nature, 389 (1997) 689–695. [DOI] [PubMed] [Google Scholar]
- [50].Kim HS, Lee JH, Lee JP, Kim EM, Chang KA, Park CH, Jeong SJ, Wittendorp MC, Seo JH, Choi SH, Suh YH, Amyloid beta peptide induces cytochrome C release from isolated mitochondria, Neuroreport, 13 (2002) 1989–1993. [DOI] [PubMed] [Google Scholar]
- [51].Johri A, Beal MF, Mitochondrial dysfunction in neurodegenerative diseases, J Pharmacol Exp Ther, 342 (2012) 619–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Krako N, Magnifico MC, Arese M, Meli G, Forte E, Lecci A, Manca A, Giuffrè A, Mastronicola D, Sarti P, Cattaneo A, Characterization of mitochondrial dysfunction in the 7PA2 cell model of Alzheimer’s disease, J Alzheimers Dis, 37 (2013) 747–758. [DOI] [PubMed] [Google Scholar]
- [53].Praticò D, Uryu K, Leight S, Trojanoswki JQ, Lee VM, Increased lipid peroxidation precedes amyloid plaque formation in an animal model of Alzheimer amyloidosis, J Neurosci, 21 (2001) 4183–4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Resende R, Moreira PI, Proença T, Deshpande A, Busciglio J, Pereira C, Oliveira CR, Brain oxidative stress in a triple-transgenic mouse model of Alzheimer disease, Free Radic Biol Med, 44 (2008) 2051–2057. [DOI] [PubMed] [Google Scholar]
- [55].Gómez-Ramos A, Díaz-Nido J, Smith MA, Perry G, Avila J, Effect of the lipid peroxidation product acrolein on tau phosphorylation in neural cells, J Neurosci Res, 71 (2003) 863–870. [DOI] [PubMed] [Google Scholar]
- [56].Misonou H, Morishima-Kawashima M, Ihara Y, Oxidative stress induces intracellular accumulation of amyloid beta-protein (Abeta) in human neuroblastoma cells, Biochemistry, 39 (2000) 6951–6959. [DOI] [PubMed] [Google Scholar]
- [57].Paola D, Domenicotti C, Nitti M, Vitali A, Borghi R, Cottalasso D, Zaccheo D, Odetti P, Strocchi P, Marinari UM, Tabaton M, Pronzato MA, Oxidative stress induces increase in intracellular amyloid beta-protein production and selective activation of betaI and betaII PKCs in NT2 cells, Biochem Biophys Res Commun, 268 (2000) 642–646. [DOI] [PubMed] [Google Scholar]
- [58].Edland SD, Silverman JM, Peskind ER, Tsuang D, Wijsman E, Morris JC, Increased risk of dementia in mothers of Alzheimer’s disease cases: evidence for maternal inheritance. Neurology 1996;47:254–256. [DOI] [PubMed] [Google Scholar]
- [59].Corral-Debrinski M, Horton T, Lott MT, Shoffner JM, McKee AC, Beal MF, Graham BH, Wallace DC, Marked changes in mitochondrial DNA deletion levels in Alzheimer brains, Genomics, 23 (1994) 471–476. [DOI] [PubMed] [Google Scholar]
- [60].Coskun PE, Beal MF, Wallace DC, Alzheimer’s brains harbor somatic mtDNA control-region mutations that suppress mitochondrial transcription and replication, Proc Natl Acad Sci U S A, 101 (2004) 10726–10731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Potkin SG, Guffanti G, Lakatos A, Turner JA, Kruggel F, Fallon JH, Saykin AJ, Orro A, Lupoli S, Salvi E, Weiner M, Macciardi F, A.s.D.N. Initiative, Hippocampal atrophy as a quantitative trait in a genome-wide association study identifying novel susceptibility genes for Alzheimer’s disease, PLoS One, 4 (2009) e6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Sekar S, McDonald J, Cuyugan L, Aldrich J, Kurdoglu A, Adkins J, Serrano G, Beach TG, Craig DW, Valla J, Reiman EM, Liang WS, Alzheimer’s disease is associated with altered expression of genes involved in immune response and mitochondrial processes in astrocytes, Neurobiol Aging, (2014). [DOI] [PMC free article] [PubMed]
- [63].Pink RC, Wicks K, Caley DP, Punch EK, Jacobs L, Carter DR, Pseudogenes: pseudo-functional or key regulators in health and disease?, RNA, 17 (2011) 792–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Sen K, Ghosh TC, Pseudogenes and their composers: delving in the ‘debris’ of human genome, Brief Funct Genomics, 12 (2013) 536–547. [DOI] [PubMed] [Google Scholar]
- [65].Rhein V, Song X, Wiesner A, Ittner LM, Baysang G, Meier F, Ozmen L, Bluethmann H, Dröse S, Brandt U, Savaskan E, Czech C, Götz J, Eckert A, Amyloid-beta and tau synergistically impair the oxidative phosphorylation system in triple transgenic Alzheimer’s disease mice, Proc Natl Acad Sci U S A, 106 (2009) 20057–20062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD, Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease, Proc Natl Acad Sci U S A, 106 (2009) 14670–14675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Monteiro-Cardoso VF, Oliveira MM, Melo T, Domingues MR, Moreira PI, Ferreiro E, Peixoto F, Videira RA, Cardiolipin Profile Changes are Associated to the Early Synaptic Mitochondrial Dysfunction in Alzheimer’s Disease, J Alzheimers Dis, (2014). [DOI] [PubMed]
- [68].Francis BM, Yang J, Song BJ, Gupta S, Maj M, Bazinet RP, Robinson B, Mount HT, Reduced levels of mitochondrial complex I subunit NDUFB8 and linked complex I + III oxidoreductase activity in the TgCRND8 mouse model of Alzheimer’s disease, J Alzheimers Dis, 39 (2014) 347–355. [DOI] [PubMed] [Google Scholar]
- [69].Quintanilla RA, Johnson GV, Role of mitochondrial dysfunction in the pathogenesis of Huntington’s disease, Brain Res Bull, 80 (2009) 242–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D, Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration, Cell, 127 (2006) 59–69. [DOI] [PubMed] [Google Scholar]
- [71].Lai L, Wang M, Martin OJ, Leone TC, Vega RB, Han X, Kelly DP, A role for peroxisome proliferator-activated receptor γ coactivator 1 (PGC-1) in the regulation of cardiac mitochondrial phospholipid biosynthesis, J Biol Chem, 289 (2014) 2250–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Song W, Chen J, Petrilli A, Liot G, Klinglmayr E, Zhou Y, Poquiz P, Tjong J, Pouladi MA, Hayden MR, Masliah E, Ellisman M, Rouiller I, Schwarzenbacher R, Bossy B, Perkins G, Bossy-Wetzel E, Mutant huntingtin binds the mitochondrial fission GTPase dynamin-related protein-1 and increases its enzymatic activity, Nat Med, 17 (2011) 377–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Knott AB, Bossy-Wetzel E, Impairing the mitochondrial fission and fusion balance: a new mechanism of neurodegeneration, Ann N Y Acad Sci, 1147 (2008) 283–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Panov AV, Gutekunst CA, Leavitt BR, Hayden MR, Burke JR, Strittmatter WJ, Greenamyre JT, Early mitochondrial calcium defects in Huntington’s disease are a direct effect of polyglutamines, Nat Neurosci, 5 (2002) 731–736. [DOI] [PubMed] [Google Scholar]
- [75].Gellerich FN, Gizatullina Z, Nguyen HP, Trumbeckaite S, Vielhaber S, Seppet E, Zierz S, Landwehrmeyer B, Riess O, von Hörsten S, Striggow F, Impaired regulation of brain mitochondria by extramitochondrial Ca2+ in transgenic Huntington disease rats, J Biol Chem, 283 (2008) 30715–30724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Milakovic T, Quintanilla RA, Johnson GV, Mutant huntingtin expression induces mitochondrial calcium handling defects in clonal striatal cells: functional consequences, J Biol Chem, 281 (2006) 34785–34795. [DOI] [PubMed] [Google Scholar]
- [77].Chang DT, Rintoul GL, Pandipati S, Reynolds IJ, Mutant huntingtin aggregates impair mitochondrial movement and trafficking in cortical neurons, Neurobiol Dis, 22 (2006) 388–400. [DOI] [PubMed] [Google Scholar]
- [78].Beal MF, Brouillet E, Jenkins BG, Ferrante RJ, Kowall NW, Miller JM, Storey E, Srivastava R, Rosen BR, Hyman BT, Neurochemical and histologic characterization of striatal excitotoxic lesions produced by the mitochondrial toxin 3-nitropropionic acid, J Neurosci, 13 (1993) 4181–4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Brouillet E, Jacquard C, Bizat N, Blum D, 3-Nitropropionic acid: a mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington’s disease, J Neurochem, 95 (2005) 1521–1540. [DOI] [PubMed] [Google Scholar]
- [80].Mendoza E, Miranda-Barrientos JA, Vázquez-Roque RA, Morales-Herrera E, Ruelas A, De la Rosa G, Flores G, Hernández-Echeagaray E, In vivo mitochondrial inhibition alters corticostriatal synaptic function and the modulatory effects of neurotrophins, Neuroscience, 280C (2014) 156–170. [DOI] [PubMed] [Google Scholar]
- [81].Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, Jaros E, Hersheson JS, Betts J, Klopstock T, Taylor RW, Turnbull DM, High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease, Nat Genet, 38 (2006) 515–517. [DOI] [PubMed] [Google Scholar]
- [82].Perfeito R, Cunha-Oliveira T, Rego AC, Reprint of: revisiting oxidative stress and mitochondrial dysfunction in the pathogenesis of Parkinson disease-resemblance to the effect of amphetamine drugs of abuse, Free Radic Biol Med, 62 (2013) 186–201. [DOI] [PubMed] [Google Scholar]
- [83].Winklhofer KF, Haass C, Mitochondrial dysfunction in Parkinson’s disease, Biochim Biophys Acta, 1802 (2010) 29–44. [DOI] [PubMed] [Google Scholar]
- [84].Mounsey RB, Teismann P, Mitochondrial dysfunction in Parkinson’s disease: pathogenesis and neuroprotection, Parkinsons Dis, 2011 (2010) 617472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Lee HJ, Shin SY, Choi C, Lee YH, Lee SJ, Formation and removal of alpha-synuclein aggregates in cells exposed to mitochondrial inhibitors, J Biol Chem, 277 (2002) 5411–5417. [DOI] [PubMed] [Google Scholar]
- [86].Ekstrand MI, Terzioglu M, Galter D, Zhu S, Hofstetter C, Lindqvist E, Thams S, Bergstrand A, Hansson FS, Trifunovic A, Hoffer B, Cullheim S, Mohammed AH, Olson L, Larsson NG, Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons, Proc Natl Acad Sci U S A, 104 (2007) 1325–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Burchell VS, Gandhi S, Deas E, Wood NW, Abramov AY, Plun-Favreau H, Targeting mitochondrial dysfunction in neurodegenerative disease: Part I, Expert Opin Ther Targets, 14 (2010) 369–385. [DOI] [PubMed] [Google Scholar]
- [88].Burchell VS, Gandhi S, Deas E, Wood NW, Abramov AY, Plun-Favreau H, Targeting mitochondrial dysfunction in neurodegenerative disease: Part II, Expert Opin Ther Targets, 14 (2010) 497–511. [DOI] [PubMed] [Google Scholar]
- [89].Fitzgerald JC, Plun-Favreau H, Emerging pathways in genetic Parkinson’s disease: autosomal-recessive genes in Parkinson’s disease--a common pathway?, FEBS J, 275 (2008) 5758–5766. [DOI] [PubMed] [Google Scholar]
- [90].Dzamko N, Zhou J, Huang Y, Halliday GM, Parkinson’s disease-implicated kinases in the brain; insights into disease pathogenesis, Front Mol Neurosci, 7 (2014) 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Murphy AN, In a flurry of PINK, mitochondrial bioenergetics takes a leading role in Parkinson’s disease, EMBO Mol Med, 1 (2009) 81–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Batandier C, Leverve X, Fontaine E, Opening of the mitochondrial permeability transition pore induces reactive oxygen species production at the level of the respiratory chain complex I, J Biol Chem, 279 (2004) 17197–17204. [DOI] [PubMed] [Google Scholar]
- [93].Jornot L, Maechler P, Wollheim CB, Junod AF, Reactive oxygen metabolites increase mitochondrial calcium in endothelial cells: implication of the Ca2+/Na+ exchanger, J Cell Sci, 112 ( Pt 7) (1999) 1013–1022. [DOI] [PubMed] [Google Scholar]
- [94].Gispert-Sanchez S, Auburger G. The role of protein aggregates in neuronal pathology: guilty, innocent, or just trying to help? J Neural Transm Suppl 2006(70):111–7. [DOI] [PubMed] [Google Scholar]
- [95].Pawar AP, Dubay KF, Zurdo J, Chiti F, Vendruscolo M, Dobson CM, Prediction of “aggregation-prone” and “aggregation-susceptible” regions in proteins associated with neurodegenerative diseases, J Mol Biol, 350 (2005) 379–392. [DOI] [PubMed] [Google Scholar]
- [96].Berry SD, Samelson EJ, Bordes M, Broe K, Kiel DP, Survival of aged nursing home residents with hip fracture, J Gerontol A Biol Sci Med Sci 64 (2009) 771–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Marcus R, Understanding Osteoporosis, West J Med 155 (1991) 53–60. [PMC free article] [PubMed] [Google Scholar]
- [98].Yang YH, Li B, Zheng XF, Chen JW, Chen K, Jiang SD, Jiang LS, Oxidative damage to osteoblasts can be alleviated by early autophagy through the endoplasmic reticulum stress pathway--implications for the treatment of osteoporosis, Free Radic Biol Med 77 (2014) 10–20. [DOI] [PubMed] [Google Scholar]
- [99].Zarrouk A, Vejux A, Mackrill J, O’Callaghan Y, Hammami M, O’Brien N, Lizard G, Involvement of oxysterols in age-related diseases and ageing processes, Ageing Res Rev 18C (2014) 148–162. [DOI] [PubMed] [Google Scholar]
- [100].DuSell CD, Nelson ER, Wang X, Abdo J, Modder UI, Umetani M, Gesty-Palmer D, Javitt NB, Khosla S, McDonnell DP, The endogenous selective estrogen receptor modulator 27-hydroxycholesterol is a negative regulator of bone homeostasis, Endocrinology 151 (2010) 3675–3685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Cappariello A, Maurizi A, Veeriah V, Teti A, The Great Beauty of the osteoclast, Archives of Biochemistry and Biophysics 561 (2014) 13–21. [DOI] [PubMed] [Google Scholar]
- [102].Salo J, Lehenkari P, Mulari M, Metsikko K, Vaananen HK, Removal of Osteoclast Bone Resorption Products by Transcytosis, Science 276 (1997) 270–273. [DOI] [PubMed] [Google Scholar]
- [103].Hessle L, Johnson KA, Anderson HC, Narisawa S, Sali A, Goding JW, Terkeltaub R, Millan JL, Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization, Proc Natl Acad Sci U S A 99 (2002) 9445–9449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Harada S, Rodan GA, Control of osteoblast function and regulation of bone mass, Nature 423 (2003) 349. [DOI] [PubMed] [Google Scholar]
- [105].Palumbo C, A three-dimensional ultrastructural study of osteoid-osteocytes in the tibia of chick embryos, Cell Tissue Res 246 (1986) 125–131. [DOI] [PubMed] [Google Scholar]
- [106].Knothe Tate ML, Adamson JR, Tami AE, Bauer TW, The osteocyte, The International Journal of Biochemistry & Cell Biology 36 (2004) 1–8. [DOI] [PubMed] [Google Scholar]
- [107].Lanyon LE, Osteocytes, Strain Detection, Bone Modeling, and Remodeling, Calcified Tissue International 53 (1993) S102–S107. [DOI] [PubMed] [Google Scholar]
- [108].Parfitt AM, The cellular basis of bone turnover and bone loss: a rebuttal of the osteocytic resorption-bone flow theory, Clin Orthop Relat Res (1977) 236–247. [PubMed]
- [109].Guo Y, Yang TL, Liu YZ, Shen H, Lei SF, Yu N, Chen J, Xu T, Cheng Y, Tian Q, Yu P, Deng HW, Mitochondria-wide association study of common variants in osteoporosis, Ann Hum Genet 75 (2011) 569–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Kang S, Fernandes-Alnemri T, Alnemri ES, A novel role for the mitochondrial HTRA2/OMI protease in aging, Autophagy 9 (2013) 420–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Miyazaki T, Iwasawa M, Nakashima T, Mori S, Shigemoto K, Nakamura H, Katagiri H, Takayanagi H, Tanaka S, Intracellular and extracellular ATP coordinately regulate the inverse correlation between osteoclast survival and bone resorption, J Biol Chem 287 (2012) 37808–37823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, e. al, Premature ageing in mice expressing defective mitochondrial DNA polymerase, Nature 429 (2004) 417. [DOI] [PubMed] [Google Scholar]
- [113].Varanasi SS, Francis RM, Berger CE, Papiha SS, Datta HK, Mitochondrial DNA Deletion Associated Oxidative Stress and Severe Male Osteoporosis, Osteoporos Int 10 (1999) 143–149. [DOI] [PubMed] [Google Scholar]
- [114].Trifunovic A, Larsson NG, Mitochondrial dysfunction as a cause of ageing, J Intern Med 263 (2008) 167–178. [DOI] [PubMed] [Google Scholar]
- [115].Manolagas SC, From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis, Endocr Rev 31 (2010) 266–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Treiber N, Maity P, Singh K, Kohn M, Keist AF, Ferchiu F, Sante L, Frese S, Bloch W, Kreppel F, Kochanek S, Sindrilaru A, Iben S, Hogel J, Ohnmacht M, Claes LE, Ignatius A, Chung JH, Lee MJ, Kamenisch Y, Berneburg M, Nikolaus T, Braunstein K, Sperfeld AD, Ludolph AC, Briviba K, Wlascheck M, Florin L, Angel P, Scharffetter-Kochanek K, Accelerated aging phenotype in mice with conditional deficiency for mitochondrial superoxide dismutase in the connective tissue, Aging Cell 10 (2011) 239–254. [DOI] [PubMed] [Google Scholar]
- [117].Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, et al. Electron transfer between cytochrome c and p66Shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell (2005) 122(2) 221–33. [DOI] [PubMed] [Google Scholar]
- [118].Bartell SM, Kim HN, Ambrogini E, Han L, Iyer S, Serra Ucer S, Rabinovitch P, Jilka RL, Weinstein RS, Zhao H, O’Brien CA, Manolagas SC, Almeida M, FoxO proteins restrain osteoclastogenesis and bone resorption by attenuating H2O2 accumulation, Nat Commun 5 (2014) 3773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Kha HT, Basseri B, Shouhed D, Richardson J, Tetradis S, Hahn TJ, Parhami F, Oxysterols regulate differentiation of mesenchymal stem cells: pro-bone and anti-fat, J Bone Miner Res 19 (2004) 830–840. [DOI] [PubMed] [Google Scholar]
- [120].Liu H, Yuan L, Xu S, Wang K, Zhang T, Cholestane-3beta,5alpha,6beta-triol inhibits osteoblastic differentiation and promotes apoptosis of rat bone marrow stromal cells, J Cell Biochem 96 (2005) 198–208. [DOI] [PubMed] [Google Scholar]
- [121].Boyette LB, Tuan RS, Adult Stem Cells and Diseases of Aging, J Clin Med, 3 (2014) 88–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Mantovani C, Terenghi G, Magnaghi V, Senescence in adipose-derived stem cells and its implications in nerve regeneration, Neural Regen Res, 9 (2014) 10–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Pittenger MF, Mackay AM, Beck SC, Jaiswal RK, Douglas R, Mosca JD, Moorman MA, Simonetti DW, Craig S, Marshak DR, Multilineage potential of adult human mesenchymal stem cells, Science, 284 (1999) 143–147. [DOI] [PubMed] [Google Scholar]
- [124].Baines HL, Turnbull DM, Greaves LC, Human stem cell aging: do mitochondrial DNA mutations have a causal role?, Aging Cell, 13 (2014) 201–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Benisch P, Schilling T, Klein-Hitpass L, Frey SP, Seefried L, Raaijmakers N, Krug M, Regensburger M, Zeck S, Schinke T, Amling M, Ebert R, Jakob F, The transcriptional profile of mesenchymal stem cell populations in primary osteoporosis is distinct and shows overexpression of osteogenic inhibitors, PLoS One, 7 (2012) e45142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Yang H, Mohamed AS, Zhou SH, Oxidized low density lipoprotein, stem cells, and atherosclerosis, Lipids Health Dis, 11 (2012) 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Shao L, Li H, Pazhanisamy SK, Meng A, Wang Y, Zhou D, Reactive oxygen species and hematopoietic stem cell senescence, Int J Hematol, 94 (2011) 24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Chen ML, Logan TD, Hochberg ML, Shelat SG, Yu X, Wilding GE, Tan W, Kujoth GC, Prolla TA, Selak MA, Kundu M, Carroll M, Thompson JE, Erythroid dysplasia, megaloblastic anemia, and impaired lymphopoiesis arising from mitochondrial dysfunction, Blood, 114 (2009) 4045–4053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Norddahl GL, Pronk CJ, Wahlestedt M, Sten G, Nygren JM, Ugale A, Sigvardsson M, Bryder D, Accumulating mitochondrial DNA mutations drive premature hematopoietic aging phenotypes distinct from physiological stem cell aging, Cell Stem Cell, 8 (2011) 499–510. [DOI] [PubMed] [Google Scholar]
- [130].Lawrence EJ, Mandato CA, Mitochondria localize to the cleavage furrow in mammalian cytokinesis, PLoS One, 8 (2013) e72886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Mishra P, Chan DC, Mitochondrial dynamics and inheritance during cell division, development and disease, Nat Rev Mol Cell Biol, 15 (2014) 634–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Choi HW, Kim JH, Chung MK, Hong YJ, Jang HS, Seo BJ, Jung TH, Kim JS, Chung HM, Byun SJ, Han SG, Seo HG, Do JT, Mitochondrial and metabolic remodeling during reprogramming and differentiation of the reprogrammed cells, Stem Cells Dev, (2015). [DOI] [PubMed]
- [133].Chung S, Dzeja PP, Faustino RS, Perez-Terzic C, Behfar A, Terzic A, Mitochondrial oxidative metabolism is required for the cardiac differentiation of stem cells, Nat Clin Pract Cardiovasc Med, 4 Suppl 1 (2007) S60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Bukowiecki R, Adjaye J, Prigione A, Mitochondrial function in pluripotent stem cells and cellular reprogramming, Gerontology, 60 (2014) 174–182. [DOI] [PubMed] [Google Scholar]
- [135].Wanet A, Remacle N, Najar M, Sokal E, Arnould T, Najimi M, Renard P, Mitochondrial remodeling in hepatic differentiation and dedifferentiation, Int J Biochem Cell Biol, 54 (2014) 174–185. [DOI] [PubMed] [Google Scholar]
- [136].Haute LV, Spits C, Geens M, Seneca S, Sermon K, Human embryonic stem cells commonly display large mitochondrial DNA deletions, Nature Biotechnology, 31 (2013) 20–23. [DOI] [PubMed] [Google Scholar]
- [137].Rohani L, Johnson AA, Arnold A, Stolzing A, The aging signature: a hallmark of induced pluripotent stem cells?, Aging Cell, 13 (2014) 2–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Varum S, Rodrigues AS, Moura MB, Momcilovic O, C.A.t. Easley, J. Ramalho-Santos, B. Van Houten, G. Schatten, Energy metabolism in human pluripotent stem cells and their differentiated counterparts, PLoS One, 6 (2011) e20914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Zhang J, Khvorostov I, Hong JS, Oktay Y, Vergnes L, Nuebel E, Wahjudi PN, Setoguchi K, Wang G, Do A, Jung HJ, McCaffery JM, Kurland IJ, Reue K, Lee WN, Koehler CM, Teitell MA, UCP2 regulates energy metabolism and differentiation potential of human pluripotent stem cells, EMBO J, 30 (2011) 4860–4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Ito K, Suda T, Metabolic requirements for the maintenance of self-renewing stem cells, Nat Rev Mol Cell Biol, 15 (2014) 243–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Yu WM, Liu X, Shen J, Jovanovic O, Pohl EE, Gerson SL, Finkel T, Broxmeyer HE, Qu CK, Metabolic regulation by the mitochondrial phosphatase PTPMT1 is required for hematopoietic stem cell differentiation, Cell Stem Cell, 12 (2013) 62–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Schieke SM, Ma M, Cao L, McCoy JP Jr., Liu C, Hensel NF, Barrett AJ, Boehm M, Finkel T, Mitochondrial metabolism modulates differentiation and teratoma formation capacity in mouse embryonic stem cells, J Biol Chem, 283 (2008) 28506–28512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].St John JC, Ramalho-Santos J, Gray HL, Petrosko P, Rawe VY, Navara CS, Simerly CR, Schatten GP, The expression of mitochondrial DNA transcription factors during early cardiomyocyte in vitro differentiation from human embryonic stem cells, Cloning Stem Cells, 7 (2005) 141–153. [DOI] [PubMed] [Google Scholar]
- [144].Owusu-Ansah E, Banerjee U, Reactive oxygen species prime Drosophila haematopoietic progenitors for differentiation, Nature, 461 (2009) 537–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Borodkina A, Shatrova A, Abushik P, Nikolsky N, Burova E, Interaction between ROS dependent DNA damage, mitochondria and p38 MAPK underlies senescence of human adult stem cells, Aging (Albany NY), 6 (2014) 481–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Wu J, Niu J, Li X, Wang X, Guo Z, Zhang F, TGF-beta1 induces senescence of bone marrow mesenchymal stem cells via increase of mitochondrial ROS production, BMC Dev Biol, 14 (2014) 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Qin H, Yu T, Qing T, Liu Y, Zhao Y, Cai J, Li J, Song Z, Qu X, Zhou P, Wu J, Ding M, Deng H, Regulation of apoptosis and differentiation by p53 in human embryonic stem cells, J Biol Chem, 282 (2007) 5842–5852. [DOI] [PubMed] [Google Scholar]
- [148].Todd LR, Damin MN, Gomathinayagam R, Horn SR, Means AR, Sankar U, Growth Factor erv1-like Modulates Drp1 to Preserve Mitochondrial Dynamics and Function in Mouse Embryonic Stem Cells, Molecular Biology of the Cell, 21 (2010) 1225–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Oh J, Lee YD, Wagers AJ, Stem cell aging: mechanisms, regulators and therapeutic opportunities, Nat Med, 20 (2014) 870–880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Las G, Shirihai OS, Miro1: new wheels for transferring mitochondria, EMBO J, 33 (2014) 939–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Bao Y, Ledderose C, Seier T, Graf AF, Brix B, Chong E, Junger WG, Mitochondria regulate neutrophil activation by generating ATP for autocrine purinergic signaling, J Biol Chem 289 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Ledderose C, Bao Y, Lidicky M, Zipperle J, Li L, Strasser K, Shapiro NI, Junger WG, Mitochondria are gate-keepers of T cell function by producing the ATP that drives purinergic signaling, J Biol Chem 289 (2014) 25936–25945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Bonifaz L, Cervantes-Silva M, Ontiveros-Dotor E, López-Villegas E, Sánchez-García F, A Role For Mitochondria In Antigen Processing And Presentation, Immunology (2014). [DOI] [PMC free article] [PubMed]
- [154].Harijith A, Ebenezer DL, Natarajan V, Reactive oxygen species at the crossroads of inflammasome and inflammation, Front Physiol 5 (2014) 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Thabit AK, Crandon JL, Nicolau DP, Antimicrobial resistance: impact on clinical and economic outcomes and the need for new antimicrobials, Expert Opin Pharmacother (2014) 1–19. [DOI] [PubMed]
- [156].West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, Ghosh S, TLR signalling augments macrophage bactericidal activity through mitochondrial ROS, Nature 472 (2011) 476–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Peterman EM, Sullivan C, Goody MF, Rodriguez-Nunez I, Yoder JA, Kim CH, Neutralization of mitochondrial superoxide by superoxide dismutase 2 promotes bacterial clearance and regulates phagocyte numbers in zebrafish, Infect Immun 83 (2015) 430–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Brynildsen MP, Winkler JA, Spina CS, MacDonald IC, Collins JJ, Potentiating antibacterial activity by predictably enhancing endogenous microbial ROS production, Nat Biotechnol 31 (2013) 160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Li H, Yang T, Li FY, Yao Y, Sun ZM, Antibacterial activity and mechanism of action of Monarda punctata essential oil and its main components against common bacterial pathogens in respiratory tract, Int J Clin Exp Pathol 7 (2014) 7389–7398. [PMC free article] [PubMed] [Google Scholar]
- [160].Voelz K, Johnston SA, Smith LM, Hall RA, Idnurm A, May RC, ‘Division of labour’ in response to host oxidative burst drives a fatal Cryptococcus gattii outbreak, Nat Commun 5 (2014) 5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Pourcelot M, Arnoult D, Mitochondrial dynamics and the innate antiviral immune response, FEBS J 281 (2014) 3791–3802. [DOI] [PubMed] [Google Scholar]
- [162].Castanier C, Garcin D, Vazquez A, Arnoult D, Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway, EMBO Rep 11 (2010) 133–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Wang X, Jiang W, Yan Y, Gong T, Han J, Tian Z, Zhou R, RNA viruses promote activation of the NLRP3 inflammasome through a RIP1-RIP3-DRP1 signaling pathway, Nat Immunol 15 (2014) 1126–1133. [DOI] [PubMed] [Google Scholar]
- [164].Rongvaux A, Jackson R, Harman CC, Li T, West AP, de Zoete MR, Wu Y, Yordy B, Lakhani SA, Kuan CY, Taniguchi T, Shadel GS, Chen ZJ, Iwasaki A, Flavell RA, Apoptotic caspases prevent the induction of type I interferons by mitochondrial DNA, Cell 159 (2014) 1563–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Chakrabarti AK, Pasricha G, An insight into the PB1F2 protein and its multifunctional role in enhancing the pathogenicity of the influenza A viruses, Virology 440 (2013) 97–104. [DOI] [PubMed] [Google Scholar]
- [166].Yoshizumi T, Ichinohe T, Sasaki O, Otera H, Kawabata S, Mihara K, Koshiba T, Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity, Nat Commun 5 (2014) 4713. [DOI] [PubMed] [Google Scholar]
- [167].Shi CS, Qi HY, Boularan C, Huang NN, Abu-Asab M, Shelhamer JH, Kehrl JH, SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome, J Immunol 193 (2014) 3080–3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Hara Y, Yanatori I, Ikeda M, Kiyokage E, Nishina S, Tomiyama Y, Toida K, Kishi F, Kato N, Imamura M, Chayama K, Hino K, Hepatitis C virus core protein suppresses mitophagy by interacting with parkin in the context of mitochondrial depolarization, Am J Pathol 184 (2014) 3026–3039. [DOI] [PubMed] [Google Scholar]
- [169].Kim SJ, Syed GH, Khan M, Chiu WW, Sohail MA, Gish RG, Siddiqui A, Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence, Proc Natl Acad Sci U S A 111 (2014) 6413–6418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Groppelli E, Starling S, Jolly C, Contact-Induced Mitochondrial Polarization Supports HIV-1 Virological Synapse Formation, J Virol 89 (2015) 14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Salminen A, Kaarniranta K, Hiltunen M, Kauppinen A, Krebs cycle dysfunction shapes epigenetic landscape of chromatin: novel insights into mitochondrial regulation of aging process, Cell Signal 26 (2014) 1598–1603. [DOI] [PubMed] [Google Scholar]
- [172].Parihar P, Solanki I, Mansuri ML, Parihar MS, Mitochondrial sirtuins: Emerging roles in metabolic regulations, energy homeostasis and diseases, Exp Gerontol 61C (2015) 130–141. [DOI] [PubMed] [Google Scholar]
- [173].Hofer A, Wenz T, Post-translational modification of mitochondria as a novel mode of regulation, Exp Gerontol 56 (2014) 202–220. [DOI] [PubMed] [Google Scholar]
- [174].Tan M, Peng C, Anderson KA, Chhoy P, Xie Z, Dai L, Park J, Chen Y, Huang H, Zhang Y, Ro J, Wagner GR, Green MF, Madsen AS, Schmiesing J, Peterson BS, Xu G, Ilkayeva OR, Muehlbauer MJ, Braulke T, Mühlhausen C, Backos DS, Olsen CA, McGuire PJ, Pletcher SD, Lombard DB, Hirschey MD, Zhao Y, Lysine glutarylation is a protein posttranslational modification regulated by SIRT5, Cell Metab 19 (2014) 605–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Fu J, Yu HM, Chiu SY, Mirando AJ, Maruyama EO, Cheng JG, Hsu W, Disruption of SUMO-specific protease 2 induces mitochondria mediated neurodegeneration, PLoS Genet 10 (2014) e1004579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Guarente L, Sirtuins in aging and disease, Cold Spring Harb Symp Quant Biol 72 (2007) 483–488. [DOI] [PubMed] [Google Scholar]
- [177].Mathias RA, Greco TM, Oberstein A, Budayeva HG, Chakrabarti R, Rowland EA, Kang Y, Shenk T, Cristea IM, Sirtuin 4 is a lipoamidase regulating pyruvate dehydrogenase complex activity, Cell 159 (2014) 1615–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Newman JC, He W, Verdin E, Mitochondrial protein acylation and intermediary metabolism: regulation by sirtuins and implications for metabolic disease, J Biol Chem 287 (2012) 42436–42443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Rardin MJ, He W, Nishida Y, Newman JC, Carrico C, Danielson SR, Guo A, Gut P, Sahu AK, Li B, Uppala R, Fitch M, Riiff T, Zhu L, Zhou J, Mulhern D, Stevens RD, Ilkayeva OR, Newgard CB, Jacobson MP, Hellerstein M, Goetzman ES, Gibson BW, Verdin E, SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks, Cell Metab 18 (2013) 920–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Kumar S, Lombard DB, Mitochondrial Sirtuins and their Relationships with Metabolic Disease and Cancer, Antioxid Redox Signal (2014). [DOI] [PMC free article] [PubMed]
- [181].Auburger G, Gispert S, Jendrach M, Mitochondrial acetylation and genetic models of Parkinson’s disease, Prog Mol Biol Transl Sci 127 (2014) 155–182. [DOI] [PubMed] [Google Scholar]
- [182].Zhu Y, Yan Y, Principe DR, Zou X, Vassilopoulos A, Gius D, SIRT3 and SIRT4 are mitochondrial tumor suppressor proteins that connect mitochondrial metabolism and carcinogenesis, Cancer Metab 2 (2014) 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, Deng CX, Finkel T, A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis, Proc Natl Acad Sci U S A 105 (2008) 14447–14452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Finley LW, Haas W, Desquiret-Dumas V, Wallace DC, Procaccio V, Gygi SP, Haigis MC, Succinate dehydrogenase is a direct target of sirtuin 3 deacetylase activity, PLoS One 6 (2011) e23295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Tao R, Vassilopoulos A, Parisiadou L, Yan Y, Gius D, Regulation of MnSOD enzymatic activity by Sirt3 connects the mitochondrial acetylome signaling networks to aging and carcinogenesis, Antioxid Redox Signal 20 (2014) 1646–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Guedes-Dias P, Oliveira JM, Lysine deacetylases and mitochondrial dynamics in neurodegeneration, Biochim Biophys Acta 1832 (2013) 1345–1359. [DOI] [PubMed] [Google Scholar]
- [187].Weir HJ, Murray TK, Kehoe PG, Love S, Verdin EM, O’Neill MJ, Lane JD, Balthasar N, CNS SIRT3 expression is altered by reactive oxygen species and in Alzheimer’s disease, PLoS One 7 (2012) e48225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Zeng L, Yang Y, Hu Y, Sun Y, Du Z, Xie Z, Zhou T, Kong W, Age-related decrease in the mitochondrial sirtuin deacetylase Sirt3 expression associated with ROS accumulation in the auditory cortex of the mimetic aging rat model, PLoS One 9 (2014) e88019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Porter GA, Urciuoli WR, Brookes PS, Nadtochiy SM, SIRT3 deficiency exacerbates ischemia-reperfusion injury: implication for aged hearts, Am J Physiol Heart Circ Physiol 306 (2014) H1602–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Westphal CH, Dipp MA, Guarente L, A therapeutic role for sirtuins in diseases of aging?, Trends Biochem Sci 32 (2007) 555–560. [DOI] [PubMed] [Google Scholar]
- [191].Coppieters N, Dieriks BV, Lill C, Faull RL, Curtis MA, Dragunow M, Global changes in DNA methylation and hydroxymethylation in Alzheimer’s disease human brain, Neurobiol Aging 35 (2014) 1334–1344. [DOI] [PubMed] [Google Scholar]
- [192].Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, Rao A, Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1, Science 324 (2009) 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Killian JK, Kim SY, Miettinen M, Smith C, Merino M, Tsokos M, Quezado M, Smith WI, Jahromi MS, Xekouki P, Szarek E, Walker RL, Lasota J, Raffeld M, Klotzle B, Wang Z, Jones L, Zhu Y, Wang Y, Waterfall JJ, O’Sullivan MJ, Bibikova M, Pacak K, Stratakis C, Janeway KA, Schiffman JD, Fan JB, Helman L, Meltzer PS, Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor, Cancer Discov 3 (2013) 648–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Lee JH, Jang H, Lee SM, Lee JE, Choi J, Kim TW, Cho EJ, Youn HD, ATP-citrate lyase regulates cellular senescence via an AMPK- and p53-dependent pathway, FEBS J (2014). [DOI] [PubMed]