

SUMMARY
In this review, selected recent advances in the preparation and reactivity of aziridines using modern synthetic approaches are highlighted, while comparing these new strategies with more classical approaches. This critical analysis is designed to help identify current gaps in the field and is showcasing new and exciting opportunities to move the chemistry of aziridines forward in the future.
INTRODUCTION
Aziridines are three-membered, saturated nitrogen heterocycles that have been described as “epoxide’s poor relations,” since methods for their synthesis and manipulation are less developed than those for their well-studied cousins.1 Epoxide-containing natural products and metabolites are quite common in nature, while aziridine natural products are rare. Despite the challenges surrounding their synthesis and control of reactivity, advances in aziridine chemistry have seen steady growth over the last 90 years (Figure 1). In the modern era, aziridines are often found as key intermediates in the preparation of amine building blocks, chiral auxiliaries, industrially important monomers, and medicinally important compounds. Intriguingly, there has been a remarkable surge in aziridine publications within the last decade alone, which accounts for ~30% of the total number of reports.
Figure 1.
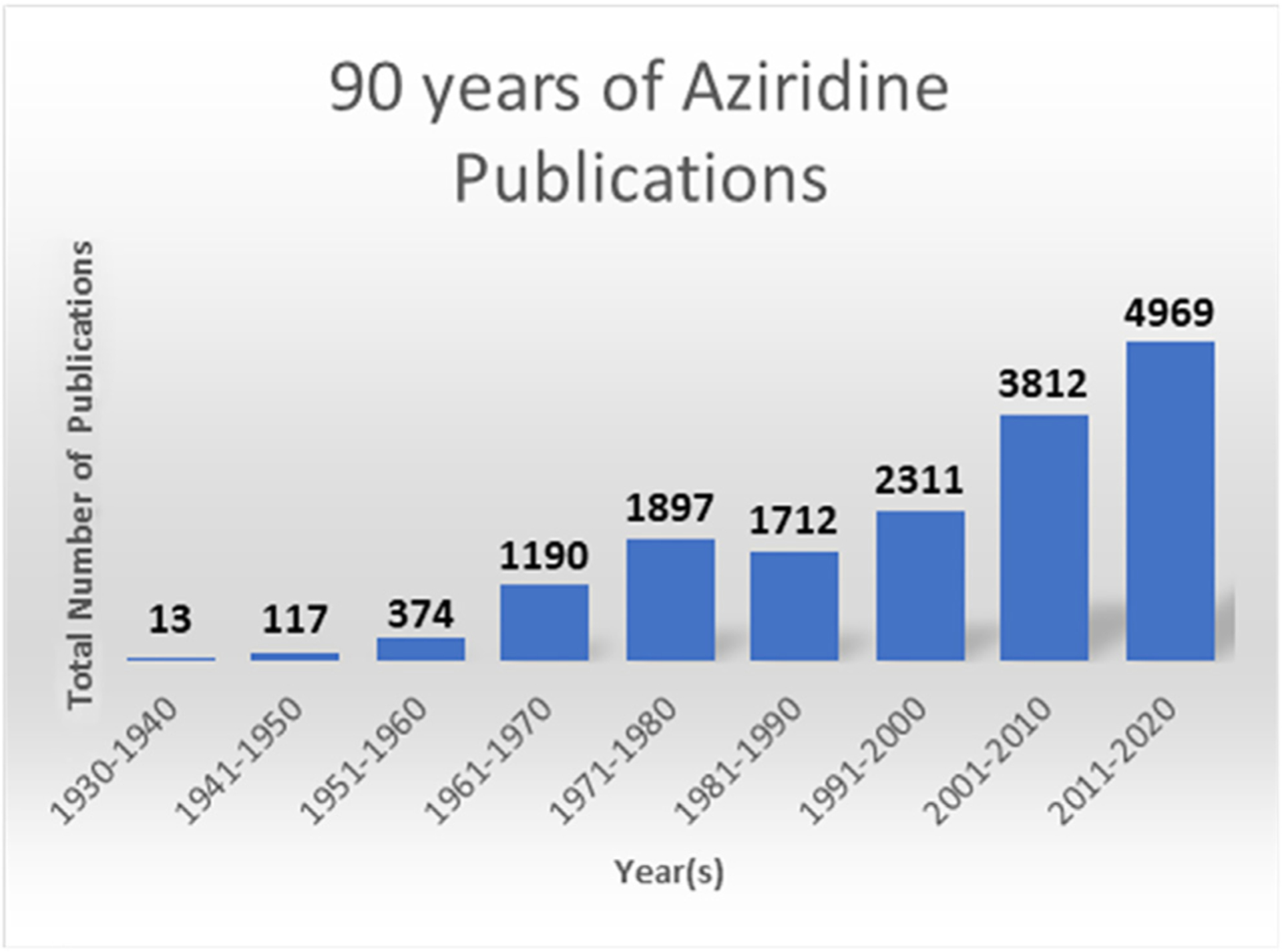
Publications from each decade with “aziridine” as the subject
An abbreviated history of aziridines
A timeline of events in the development of aziridine chemistry is highlighted against a larger world view (Figure 2). The earliest proposed assignment of an aziridine structure was in 1875 by Sabaneyev, although the compound was later revealed to be an acetamidine derivative.2 The first true synthesis of an aziridine was of the parent molecule, performed by Gabriel in 1888.2 Gabriel originally assigned the structure as vinyl amine, his initial target compound; however, subsequent work by Marckwald in 1901 allowed for the structure to be definitively assigned.2 Despite their remarkable utility, methods and transformations of aziridines have since lagged behind those of epoxides. One reason for this disparity may be that the most common method of aziridine formation in the early literature involved cyclization via nitrogen displacement of a vicinal leaving group, often using amino acid and α-amino alcohol precursors derived from epoxides. Epoxide formation had found success in the early 1900s, with reactions such as the Darzen glycidic ester condensation (generalized in 1904) and the Prilezhaev epoxidation (1909), which made a wide variety of epoxide structures readily accessible.3 The ease of synthesis of epoxides, coupled with several unique challenges in the preparation and subsequent reactivity of aziridines, likely hampered the development of robust methods for aziridine synthesis. Indeed, the first major period of significant interest in aziridines did not occur until the 1940s, stimulated by the unusual confluence of World War I (WWI) and the advent of chemotherapeutics.
Figure 2.
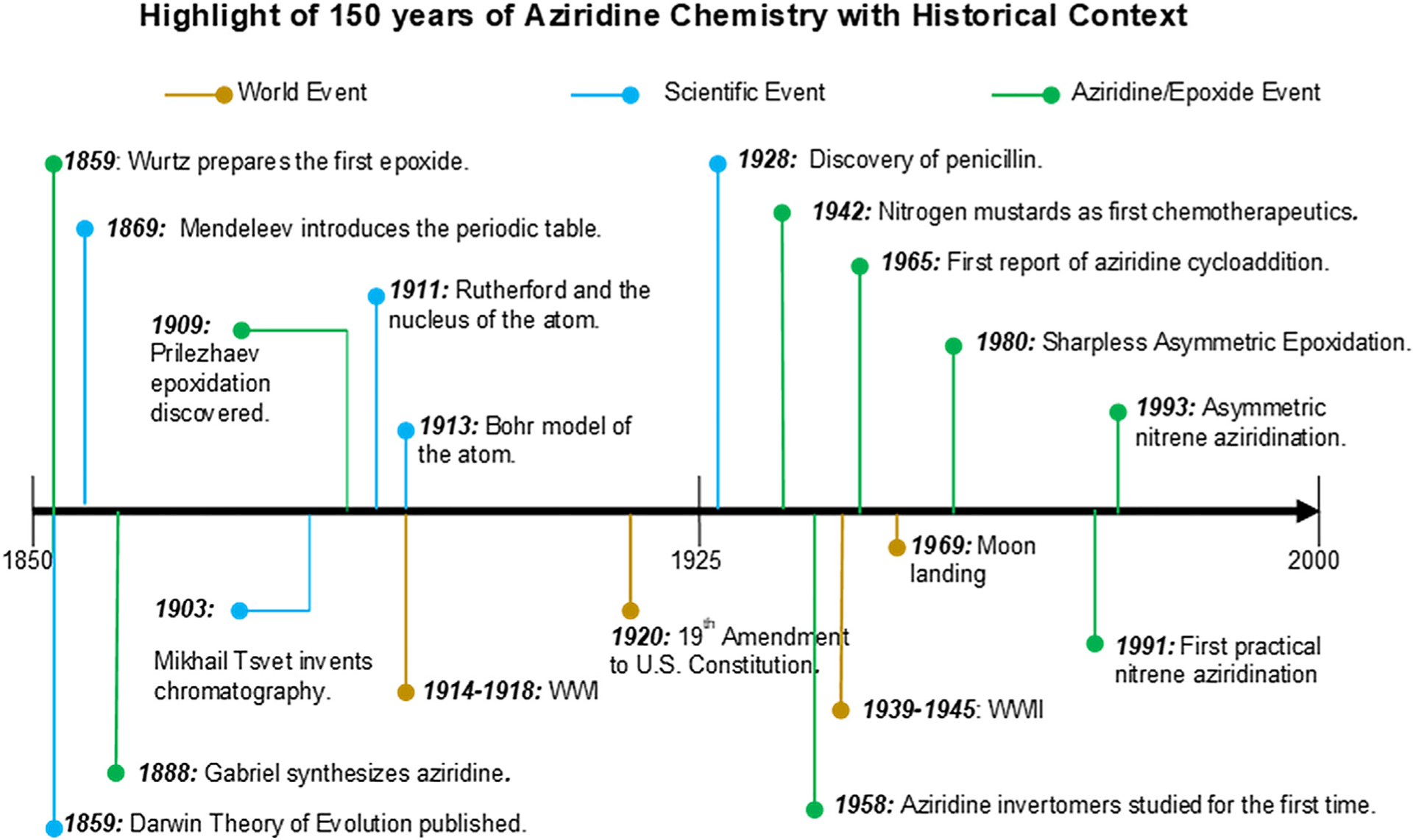
Timeline of some major advancements in aziridine chemistry and near-world events
Originally developed to remove warts, nitrogen mustards were quickly identified as potential chemical warfare agents. Their name arose from their similarities in structure and reactivity to the sulfur mustards used in WWI. The 1920s and 1930s marked the large-scale production of and investigations into the mechanism of action of these compounds. While nitrogen mustards were never used in warfare, the U.S. Department of Defense recruited Louis Goodman and Alfred Gilman to examine their potential therapeutic value.4
Noting lymphoid hypoplasia and myelosuppression in autopsies of soldiers exposed to sulfur mustard gas, the pair convinced researchers to use nitrogen mustard on a patient with advanced non-Hodgkin’s lymphoma, ultimately resulting in disease regression. This launched the field of chemotherapeutics and further research into the aziridinium species, the active intermediate; many of these results were not published until the 1950s due to the wartime secrecy of WWII. Aziridine chemistry has since continued to advance with more powerful methods for both racemic and asymmetric syntheses and creative conversions of the resulting three-membered ring into valuable chiral amines and larger heterocycles.
General properties, challenges, and classic transformations
The three-membered parent, unsubstituted aziridine ring is the smallest and simplest saturated nitrogen heterocycle (Figure 3). Aziridine, or ethyleneimine, is a water-soluble, colorless, and distillable liquid (boiling point [b.p.] 57 °C). The bond angles of aziridine are approximately 60°, which introduce considerable strain into the molecule. This strain imparts physical properties to aziridines not shared with other saturated nitrogen heterocycles. First, activated aziridine rings undergo facile ring-opening reactions, which comprise a large part of the utility of this compound class. The increased s-character of the aziridine nitrogen, which reduces its π-donor abilities, greatly reduces the basicity of the nitrogen (pKa 8 in water), compared with acyclic systems (pKa 11) and even the four-membered azetidine (pKa 11). Perhaps most interesting is the greatly increased nitrogen inversion barrier.5 This barrier in 2-methylaziridine is 17 kcal/mol, significantly higher than other secondary amines, but not high enough to resolve diastereomers at room temperature. However, introduction of an electron-withdrawing group, as shown for N-chloro-2-methylaziridine, increases this barrier to ~27 kcal/mol, making it possible to isolate diastereomers at room temperature. The enhanced electronegativity of nitrogen relative to carbon and the inherent Baeyer strain of three-membered rings facilitate ring-openings of aziridines. Examples of aziridine ring-opening are ubiquitous in the literature and yield valuable products that include 1,2 diamines, amino alcohols, chiral amines, and ring systems, such as medicinally important β-lactams.
Figure 3.
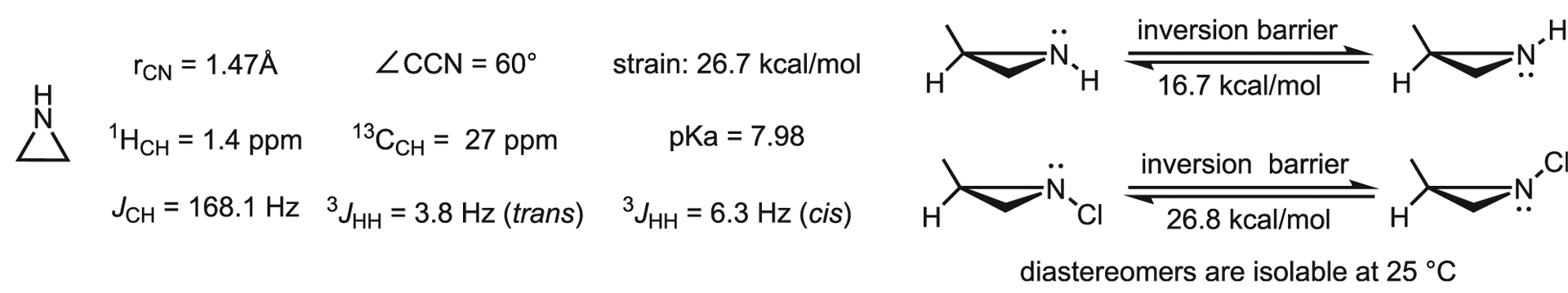
General physical properties of the parent and substituted aziridines
Challenges in aziridine chemistry
The regioselectivity and stereoselectivity of aziridine opening depend on the stability of the ring and its electronic preference, both of which are influenced by conformational preferences between invertomers. Invertomer issues can further complicate the stereoselective syntheses of aziridines and are heavily influenced by the identities of the ring substituents, most notably the N-substituent (Figure 4). The N-substituent dictates whether the aziridine is classified as “activated” or “non-activated,” biases the aziridine toward either cationic or anionic transition states that factor into solvent and nucleophile choice for ring-opening, and affects product distribution in the presence of a catalyst (Figures 4A and 4B).6
Figure 4.
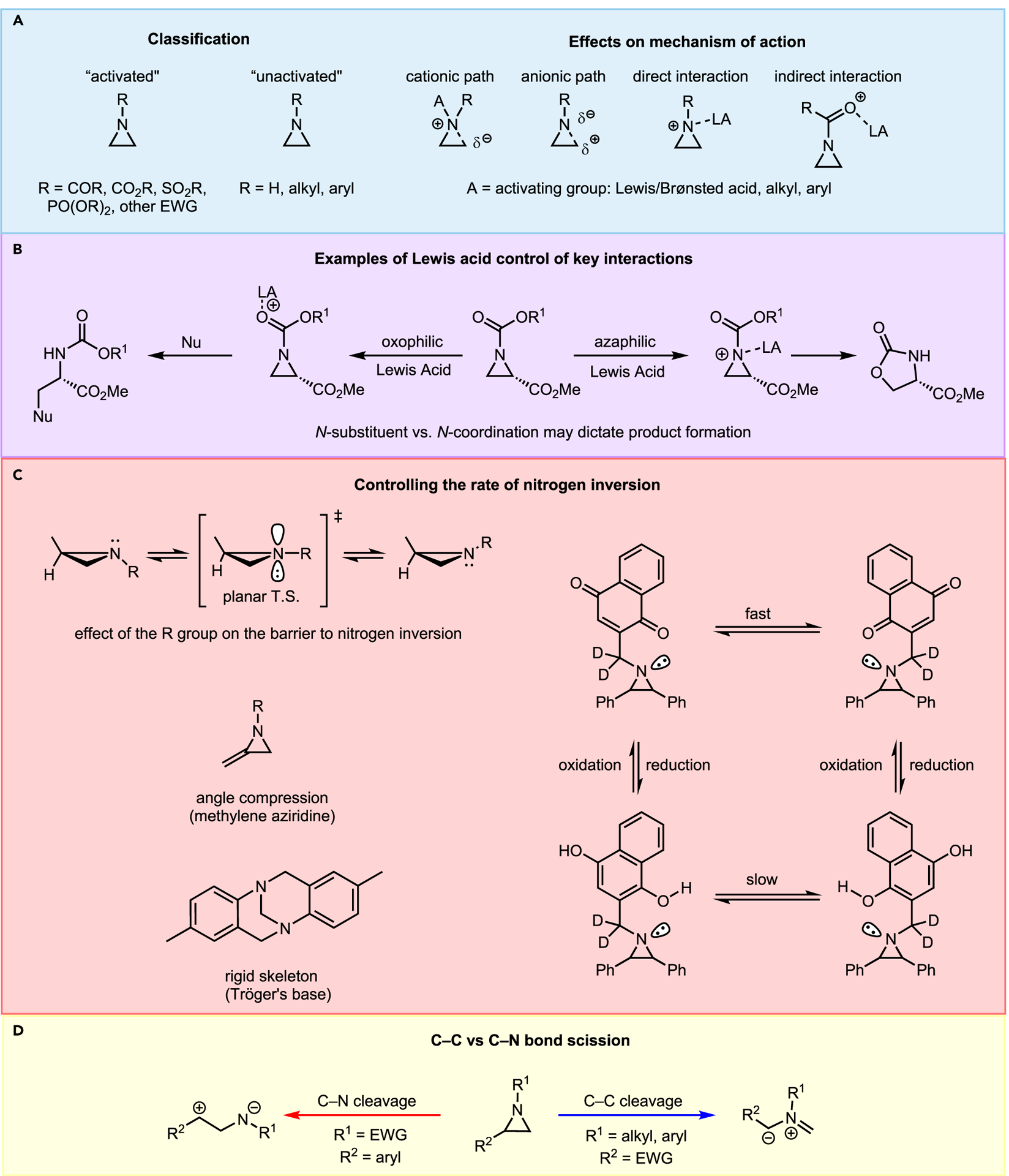
Effects and general trends of the aziridine N-substituent on the reaction outcome
The N-substituent also plays a prominent role in controlling the nitrogen inversion barrier (Figure 4C).7 High barriers to aziridine inversion can lead to interconverting enantiomers and diastereomers, which can complicate ring-opening and eventual product purification. A more subtle circumstance, which offers opportunities for reaction control, arises when the nitrogen lone pair is utilized as a directing group. For example, treatment of N-Boc-protected aziridines with organolithiums typically leads to α-lithiation products (Scheme 1A). As shown with 1.1, this is likely due to steric repulsion of the Boc group that favors 1.2 and prevents access to the aziridine lone pair, driving selectivity to the α-hydrogen in 1.3. However, the Musio lab has demonstrated that N-alkyl aziridines can direct organolithium reagents via the nitrogen lone pair, resulting in either α-lithiation of 1.6 to 1.7 or ortho-lithiation of the aromatic ring in 1.5 to give 1.4 (Scheme 1B).8 This strategy allows for selective electrophilic substitution that corresponds to the invertomer equilibrium position.
Scheme 1.
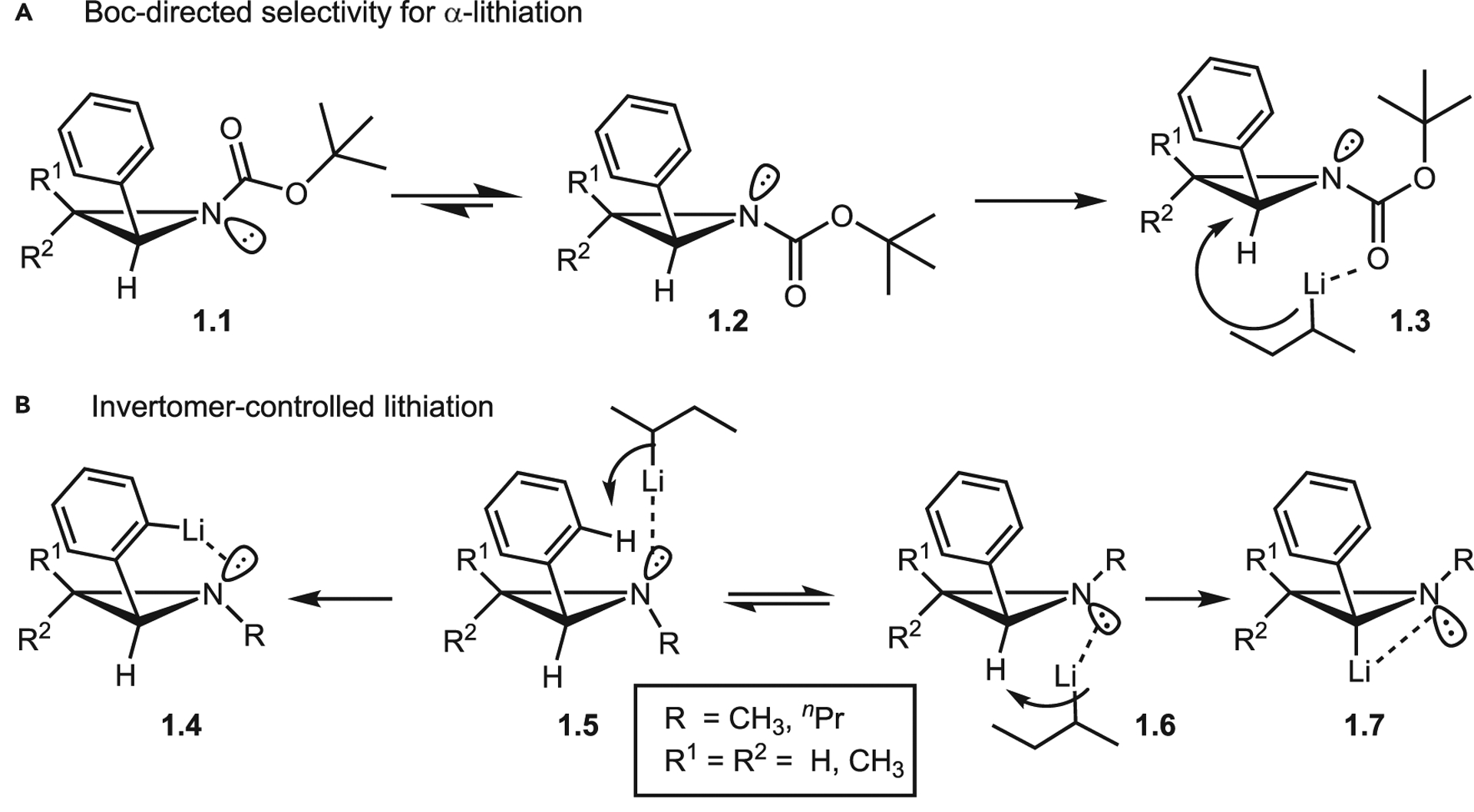
Examples of N-substituent effects on deprotonation by organolithiums
Another example of the defining role the N-substituent can play in determining reaction outcome is the regioselective opening of aziridines to 1,3-dipoles via C–N or C–C bond scission (Figure 4D). Alteration of the electronic nature of the zwitterion intermediate through judicious choice of ring substitution allows for the selective synthesis of an immense diversity of nitrogen-containing heterocycles via 1,3-dipolar cycloadditions. While the use of azomethines formed from aziridines is a very useful synthetic strategy, this topic has been covered in previous reviews, and the reader is referred to these for further information.9,10
Classic synthesis and reactivity of aziridines
In general terms, strategies for synthesizing aziridines fall into one or a combination of three categories: nitrene addition, carbene addition, or intramolecular cyclization (Figure 5A). Successful applications of these three strategies to aziridine synthesis benefit from the chemist’s skill and creativity to account for the effects described in Figure 4.
Figure 5.
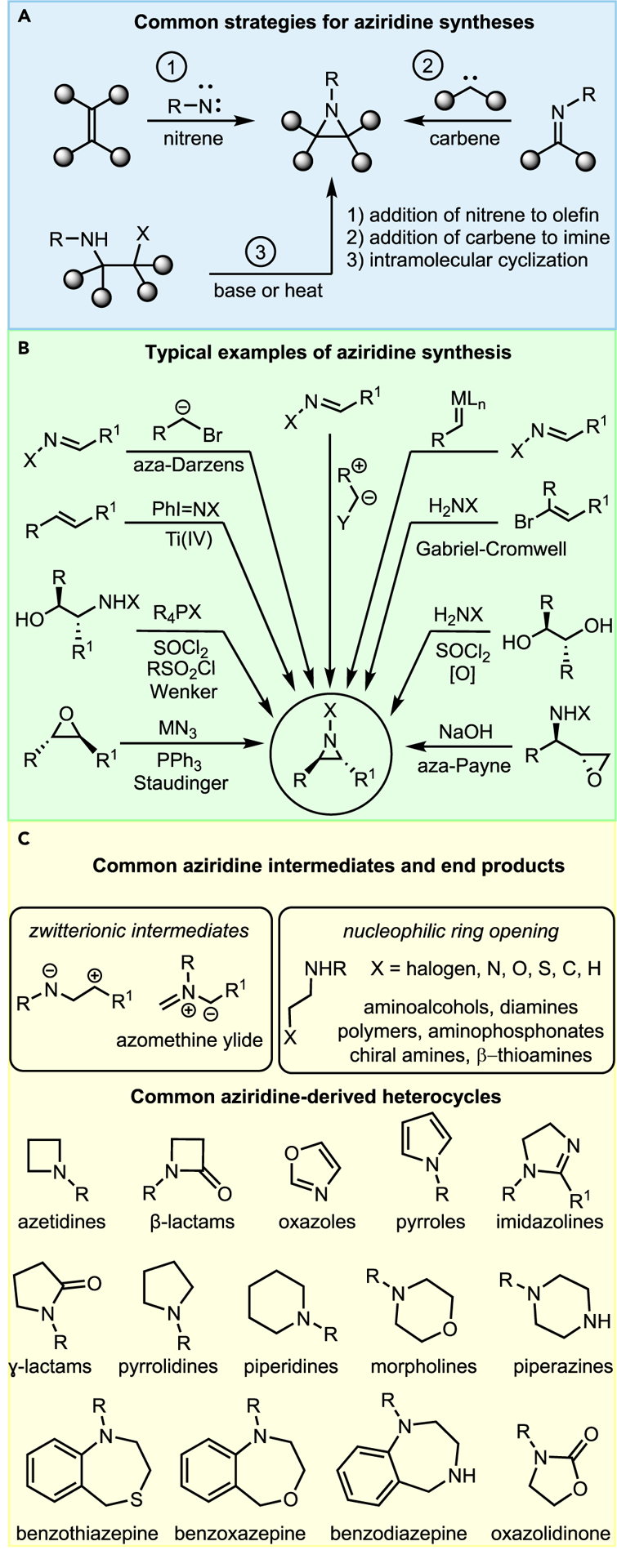
General strategies for synthesis and reactivity of aziridines
In the modern era, many reliable methods for aziridine synthesis have been reported (Figure 5B). Many have been used frequently enough to qualify as “named reactions,” and several have asymmetric counterparts. Once the aziridine ring is forged, it may react in a variety of ways, particularly nucleophilic attack on the ring to yield useful amine building blocks (Figure 5C). Alternatively, the ring may open to furnish a zwitterionic intermediate, which can be trapped by diverse dipolarophiles in a [3+2] cycloaddition reaction.
Despite the diverse motifs accessible from aziridines, there are limitations that require new strategies to manipulate these strained rings. Many oft-utilized transformations are incompatible with concerns related to green chemistry and sustainability. While the preceding section offers a short primer highlighting key challenges and known reactivity of aziridines, the remainder of this review will compare historical precedent with recent advances in aziridine chemistry. Current gaps in the field will be identified and solutions to advance aspects of green chemistry will be presented. This review is not intended to be comprehensive and instead highlights unique approaches to aziridine syntheses and reactions.
RECENT ADVANCES IN AZIRIDINE SYNTHESIS
The following sections are broadly divided into recent advances in metal-free, metal-mediated, and photo- and electrochemical methods for aziridine synthesis. Each section is written in a “stand-alone” style with a brief introduction and an overview of challenges in the area, followed by modern and selected examples of advancements.
Metal-free methods of aziridination
“Metal-free methods” of aziridination encompass numerous transformations and end products. These methods are well documented in the literature, and readers requiring more in-depth discussion are directed to several excellent reviews.9,10
Challenges and limitations of metal-free methods
Metal-free methods for aziridination were the earliest to be investigated (Figure 5B). Classical methods suffer from limitations that interfere with interests in developing more sustainable chemistry. Most strategies for metal-free aziridine synthesis utilize either an aza-Michael-initiated ring-closing (aza-MIRC) or nucleophilic attack on an imine, as exemplified by the aza-Darzens reaction (Figure 6). Challenges include the following: (1) the need exists to install a sacrificial leaving group; (2) control of facial selectivity to give enantio- and diastereoenriched aziridines can be difficult, requiring further processing and increasing waste streams; and (3) the need exists to protect sensitive functional groups.
Figure 6.
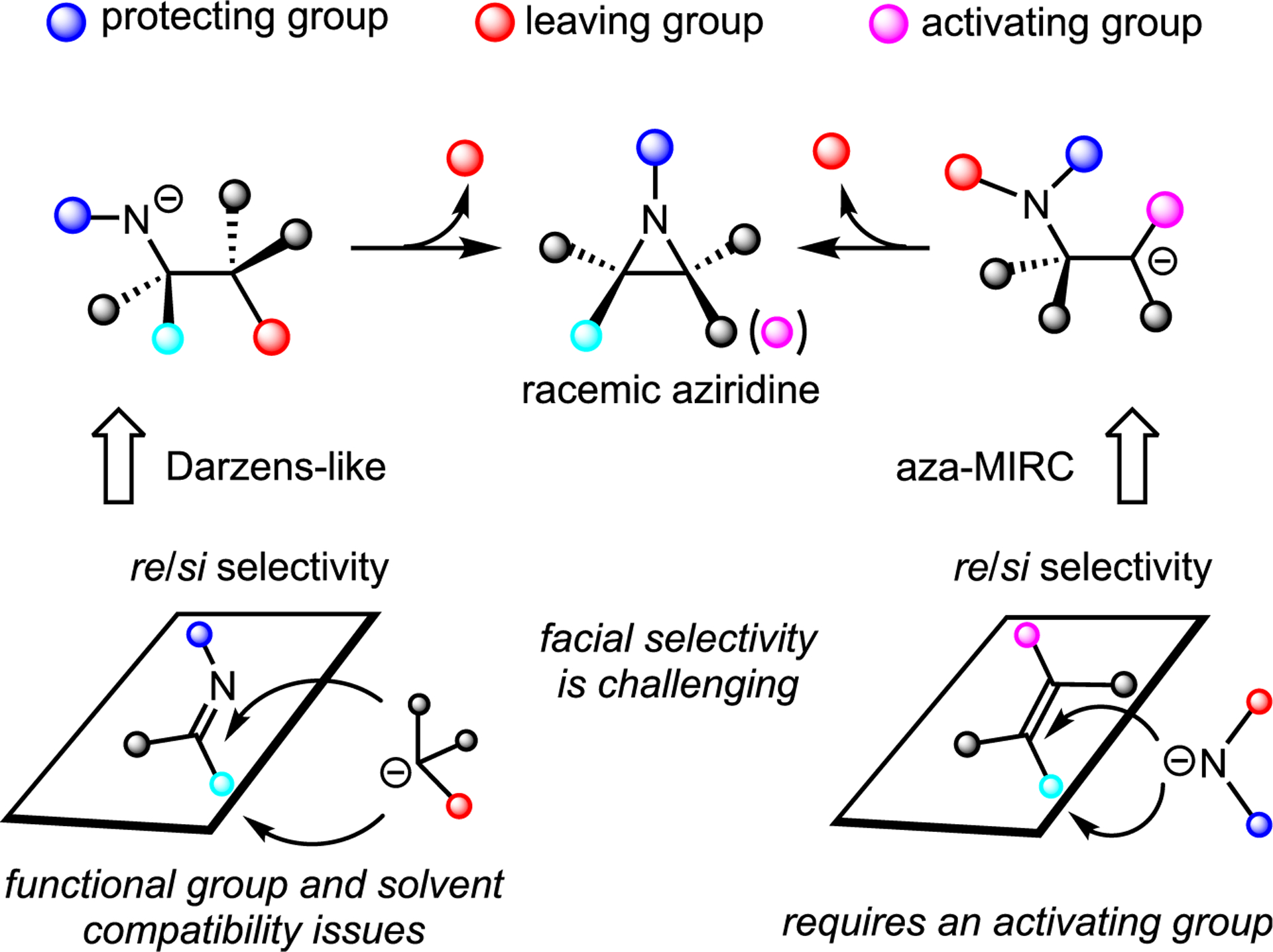
Challenges and limitations of many metal-free aziridinations
Solutions have been described for many of these challenges11,12; e.g., facial selectivity in additions to imines and olefins can be biased by utilizing chiral auxiliaries and/or organocatalysts to give high enantiomeric excess (ee) and moderate-to-excellent yields.13 Thus, advances in recent years often represent minor improvements in stereoselectivity, yield, and/or ee for specific cases. However, established methods still offer opportunities, as exemplified by the cis-selective aza-MIRC via a post-reaction isomerization (PRI) from the Albrecht lab below.14
Recent examples and lessons from “classic” aziridinations.
Aza-MIRC. The organo-catalytic aza-MIRC aziridination was first reported in 2007.15 The generally accepted mechanism (Scheme 2A) involves condensation of a chiral amine onto an unsaturated carbonyl compound to give the activated iminium 2.2, which is attacked at the β position to give 2.3. Enamine attack on the amine bearing a leaving group gives imine aziridine 2.4, which hydrolyzes to give aziridine 2.5 and re-form the catalyst 2.1. The stereochemistry is set early in the reaction cycle and predominately yields trans-substituted aziridines.
Scheme 2.
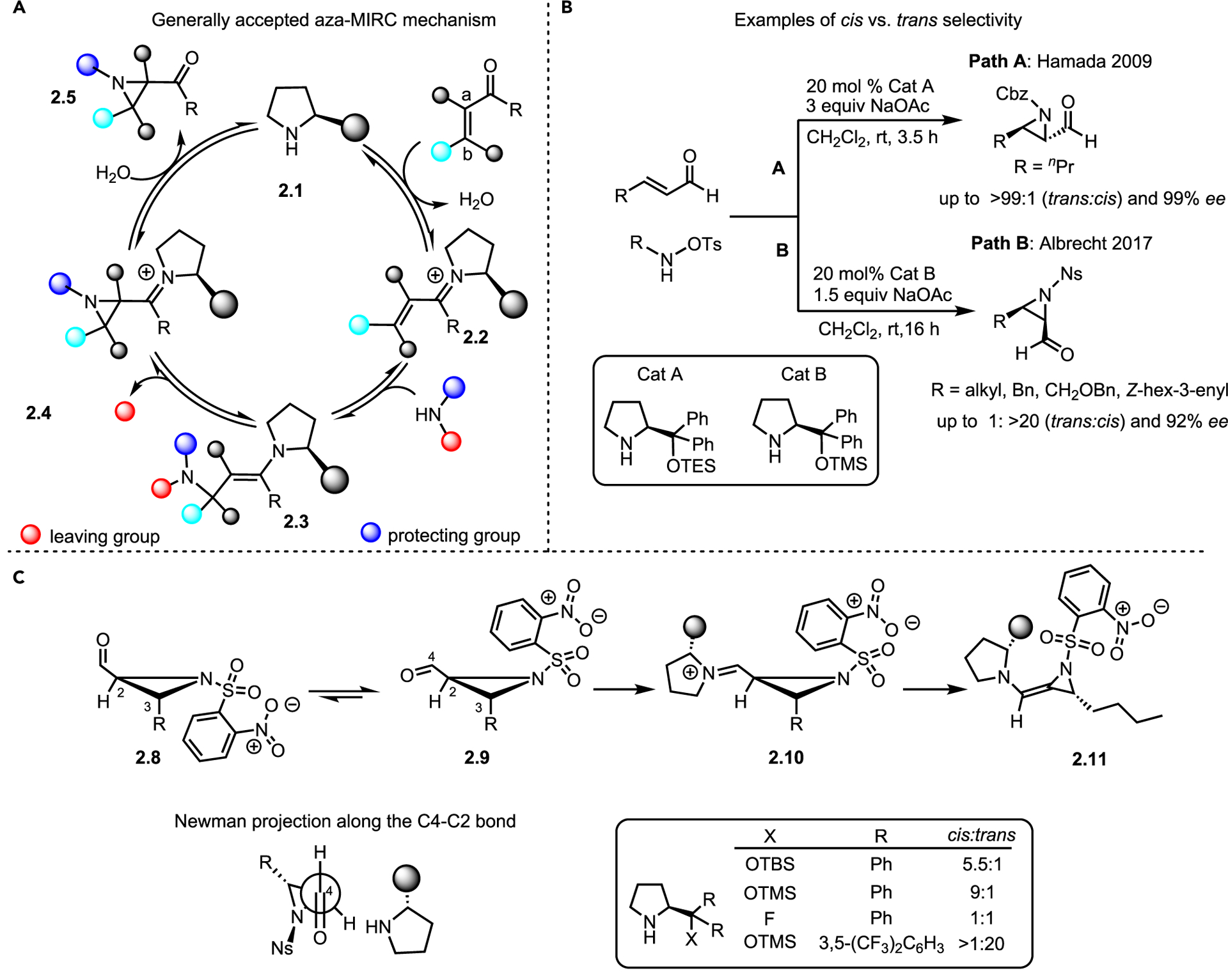
Aziridination via aza-MIRC
(A) Mechanism of aza-MIRC.
(B) Conditions to achieve stereodivergence.
(C) Conformational dynamics underlying selectivity.
Although Albrecht’s conditions (Scheme 2B, path B) are similar to typical conditions, the Hamada lab16 (Scheme 2B, path A) obtained a different isomer using slightly modified conditions. The key difference is the N-substituent; while Boc/Cbz protection yields the trans product 2.6, a nosyl (Ns) group gives near-complete isomerization to the cis form 2.7. The PRI was attributed to the greater electron-withdrawing power of the Ns group, which increases the acidity of the α proton. This was corroborated by the observed deuterium exchange at the α position during the PRI and the fact that PRI does not occur in the absence of an α proton (i.e., if H is replaced with methyl). The stereochemical outcome was attributed to “sterics” that, though true, belies the intricacies of interactions giving the observed cis aziridine (Scheme 2C).
The authors note that some degree of steric bulk is required by the C3-R group, from which it may be surmised that the equilibrium position between 2.8 and 2.9 must favor 2.9 for best results. This trans orientation between the N-substituent and the C3-R group offers a single face for chiral-pyrrolidine reattack. Note that reattack is required, as the trans form is the sole isolated product after the aza-MIRC. The most sterically accessible form in Scheme 2C (Newman projection) leads to iminium 2.10. The final few steps of the mechanism are unclear. Based on the catalyst screen (selected examples given in Scheme 2C), electronic factors appear to be at play. If Albrecht’s assertion that the final isomerization goes through 2.10 holds true, a plausible explanation would be a final inversion of the N-substituent induced by π-π interactions of the electron-poor Ns ring with the phenyl groups prior to product formation. This would obstruct the cis face relative to the C3-R group and force re-protonation on the trans side to give the observed product. This would also explain the trans-selectivity using electron-poor CF3-substituted catalysts. The poorer performance with the -OTBS (TBS, tert-butyl(dimethyl)silyl) group could be attributed to slow reattack on the enal.
The preceding work highlights that even small changes to reaction conditions can expand access to aziridines. Work in the area is likely to continue the emphasis on greener chemistry to favor sequential one-pot reactions and replace current solvents with more benign alternatives. For example, dos Santos and co-workers utilized an aza-MIRC to afford enantioenriched aldehyde aziridines, which were subjected immediately to Passerini conditions to give aziridine peptidomimetics in good yield and excellent diastereomeric ratio (dr) in an ethanol-water mixture, instead of more commonly used organic solvents (Scheme 3).17
Scheme 3.
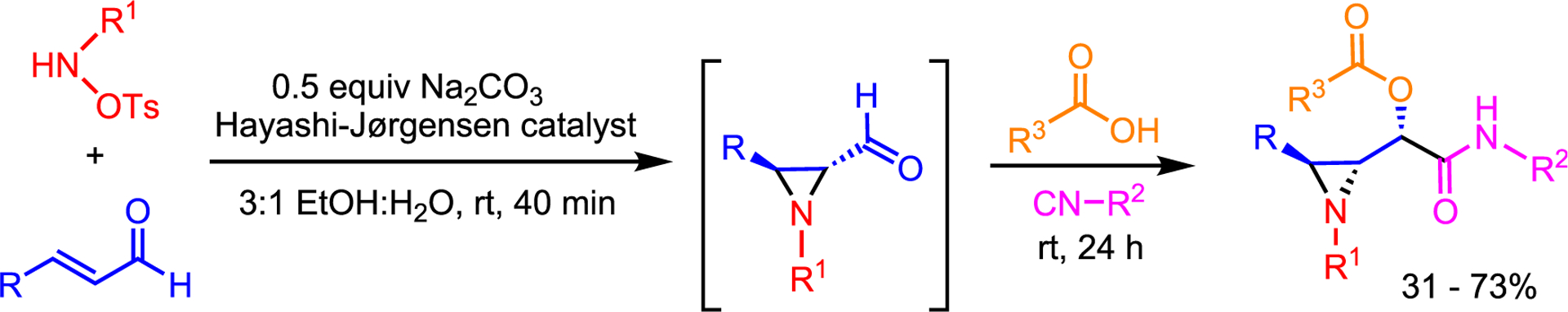
Multicomponent reactions in more environmentally friendly solvents
Multicomponent aziridination.
Multicomponent aziridinations are rare. A recent review covers advances up to 201917; thus, only a few recent examples are highlighted here to illustrate the potential of this strategy. One of the best examples of a multicomponent aziridination method is Wulff’s BOROX-controlled aza-Darzens system (Scheme 4A). Insights into the mechanism of this cis-aziridination protocol were used to synthesize a wide variety of asymmetric aziridines with excellent catalyst control. In this formal five-component reaction, the helically chiral aryl phenol ligand is treated with B(OPh)3 in the presence of amine (4.1) to yield the active boroxinate-containing (BOROX) catalyst 4.2 (Scheme 4B). Aldehyde addition generates the iminium complex 4.3 in situ. Addition of ethyl diazoacetate (EDA) leads to 4.4. The rate-determining step (RDS), SN2 attack by nitrogen, generates the aziridine 4.5, releasing the boroxinate to complete the cycle. This system is noteworthy for its practicality; the in situ generation of the imine component obviates the need for challenging purifications, while catalyst control furnishes an extensive substrate scope in high yields and good-to-excellent er (Scheme 4C).18 Recently, the Wulff lab reported trans-selective aziridinations using diazoacetamide derivatives, with the best results stemming from the use of a butyldiani-sylmethyl (BUDAM) protecting group.19
Scheme 4. Wulff’s multicomponent aziridination using BOROX-controlled aza-Darzens system.
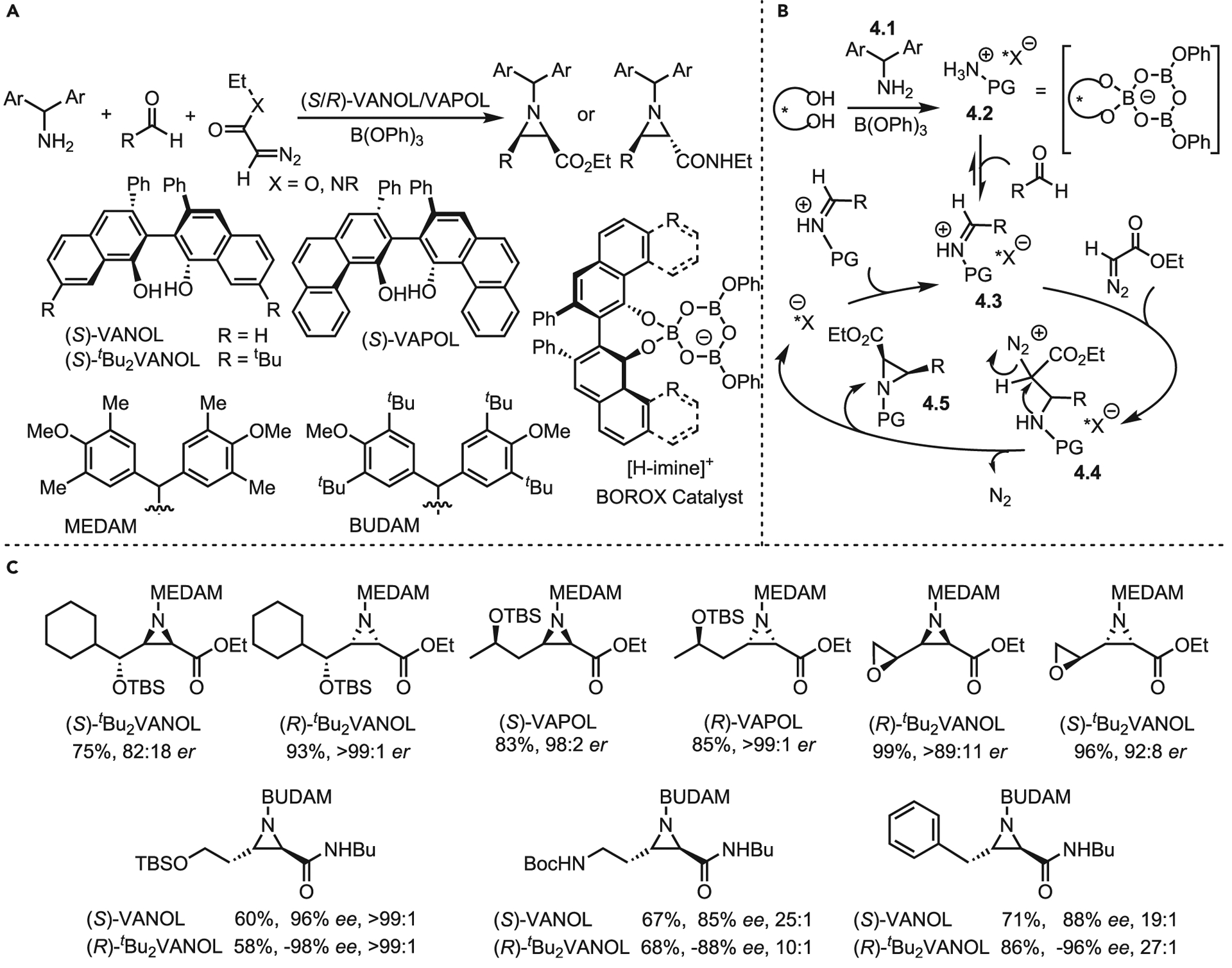
(A) General ligands and protecting groups for aziridination.
(B) Mechanism.
(C) Selected substrate scope.
The ability to selectively access cis or trans aziridines in high yield and ee is valuable, as demonstrated by Wulff’s selective preparation of all four stereoisomers of sphinganine through utilizing this method.20 Beyond these practical aspects, the nature of the catalyst control is intriguing. The iminium-boroxinate complex contains several non-covalent interactions (NCIs), including an ionic interaction between the iminium and boroxinate, π-π, and CH-π interactions between the N-protecting group and the biaryl ligand, as well as a surprisingly strong CH–O hydrogen bond in some instances. These NCIs serve to generate a tightly associated catalyst complex, while the sheer bulk of both the boroxinate and the N-protecting group control the environment around the imine in a manner reminiscent of an enzyme. Furthermore, matching the protecting group (MEDAM, tetramethyldianisylmethyl, or BUDAM) with the proper BOROX catalyst is essential to achieve maximum yield and enantiomeric and diastereomeric ratios (er/dr), although even mismatched cases generally give usable yields and stereoselectivities.
Although the BOROX catalyst system is effective, trans aziridines are only accessible through diazoacetamides, as diazoesters yield predominantly cis aziridines. This has been attributed to a flip in the ordering around the boroxinate complex, which inverts the facial selectivity and subsequent cyclization. To date, it has not been shown that the MEDAM-protecting (Scheme 4C) group can be recovered and recycled in the same manner the Wulff lab has shown with the BUDAM group. Likewise, the boroxinate catalyst is not recouped from the reaction, sacrificing otherwise excellent atom-economy. The system is also sensitive to the nature of the substrate, sometimes requiring extended incubation of the amine, aldehyde, and boroxinate prior to the introduction of the diazo species. While the scope is currently limited to disubstituted aziridines, the high yields and ee are appealing.
More recently, the Wei lab demonstrated a catalyst-free three-component coupling of EDA, alkynes, and nitrosoarenes to give di- and trisubstituted aziridines in good-to-excellent yield with high diastereoselectivity (Scheme 5).21 This is achieved via in situ generation of an N-oxide 1,3-dipole, which engages an alkyne to form a dehydroisoxazole that opens upon heating to yield a zwitterionic intermediate. Ring closure furnishes the aziridine, where the high dr is controlled by steric effects in the acyclic intermediate. While the simplicity of this system is appealing, it does require an aryl nitroso compound to stabilize the nitrogen cation through resonance and to serve as an oxygen transfer source; presumably, generation of a C=O bond is the driving force for ring-opening. The alkyne must be activated to serve as an effective dipolarophile and furnish high yields, while the scope is mostly limited to 2,3-diketo and 2,3-keto ester aziridines. Nevertheless, such aziridines are useful precursors to value-added products.
Scheme 5.
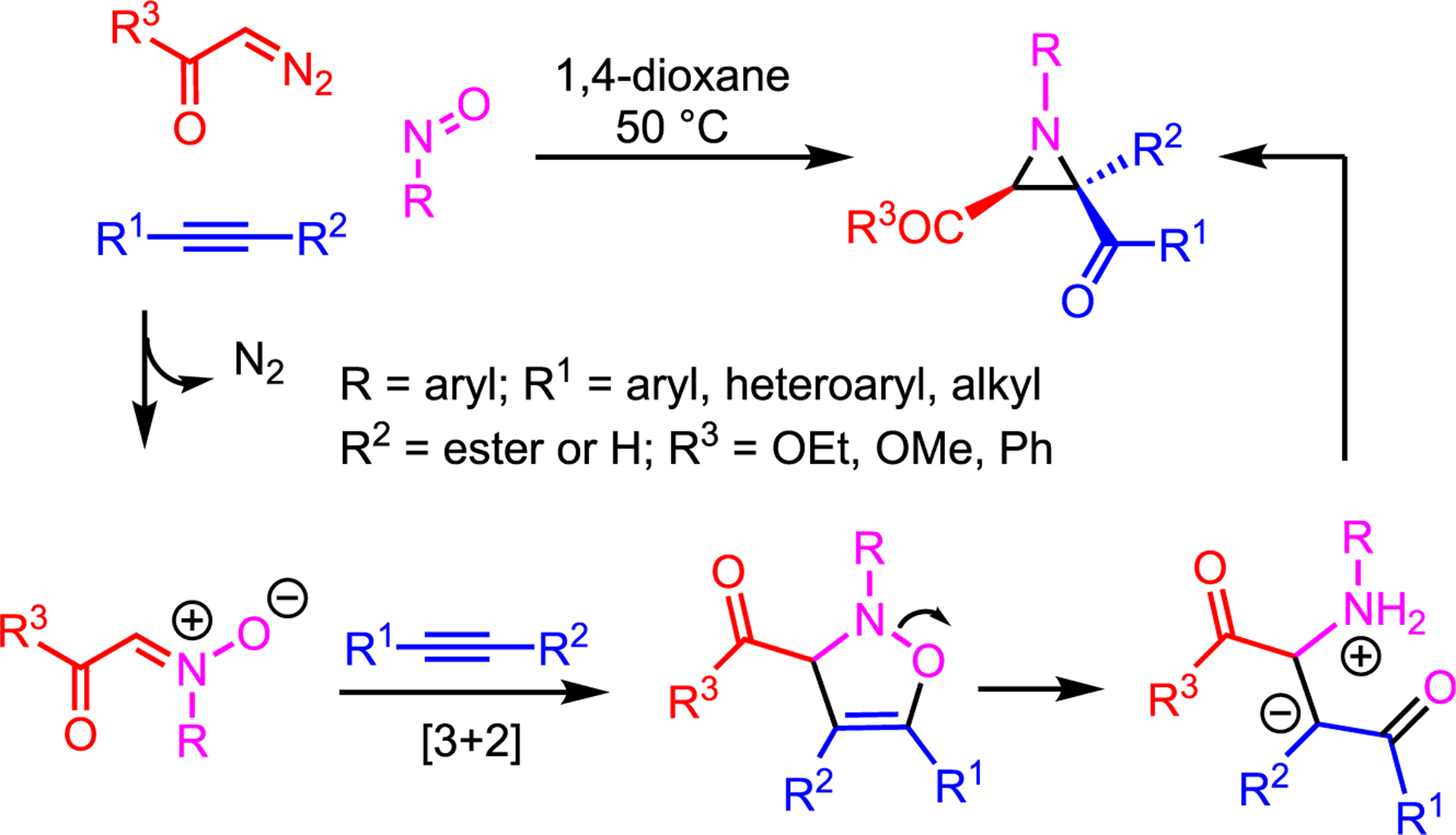
Three-component coupling to yield aziridines
Future directions and outlook for “classic aziridinations”.
Although classic, transition metal-free aziridination methods have been well studied, further investigations are warranted to explore more robust organocatalysts and reserve low-abundance precious metals for alternative uses. Combining classic approaches with photocatalytic and electrocatalytic strategies are likely to be the focus of future efforts. The continued development of one-pot cascade reactions or multicomponent reactions to yield more densely functionalized aziridines or to transform these key intermediates into useful heterocycles in short order are also attractive areas for future advances.
Recent advances in direct nitrogen addition to olefins.
Methods for aziridine formation reported in the 19th and 20th centuries mainly involve nucleophilic attack by nitrogen on activated or conjugated olefins or nucleophilic attack on imines by carbon-centered nucleophiles. However, direct addition of reactive nitrogen species to alkenes offers flexible choices of N-protecting groups to give mono-, di-, tri-, and tetrasubstituted aziridines from readily available alkenes with catalyst-controlled stereo- and chemoselectivity. Metal-free nitrene generation often requires harsh conditions incompatible with green solvents and safety considerations. Free nitrenes are also highly reactive and typically display poor chemo- and stereoselectivity. As such, productive additions of nitrenes to olefins typically employ transition metal catalysts.
A challenge in direct additions of nitrogen to olefins is the relative bond dissociation energies of N–N and N–O bonds, compared with O–O bonds. Weak O–O bonds in dioxiranes and peroxy acids have enabled olefin epoxidation for over a century, while analogous electrophilic nitrogen sources with weak N–N bonds are scarce. Oxaziridines with weak N–O bonds yield mainly epoxides or form a single C–N bond but can undergo nitrogen transfer, depending on the N-substituent.22,23 Excitingly, the past five years have seen significant advances in metal-free aziridinations of this type (Scheme 6).
Scheme 6. Overview of advances in metal-free aziridination.
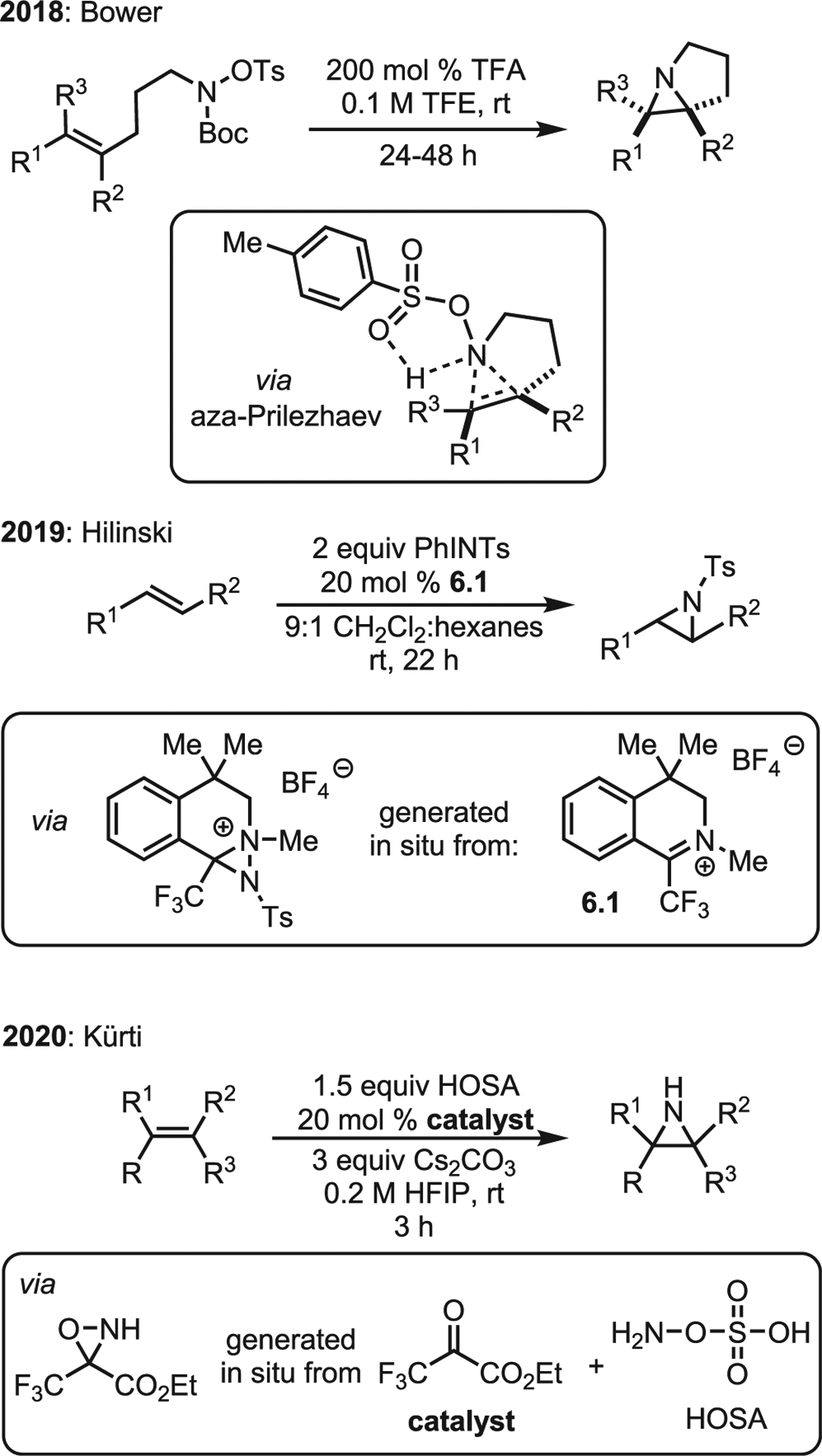
In 2018, Farndon, Young, and Bower reported an aza-Prilezhaev reaction, where in situ Boc deprotection yields an O–Ts amine, a potent aminating reagent.24 This compound is proposed to add across alkenes via a “butterfly”-like mechanism (Scheme 6, top), similar to that proposed for reactions of olefins with peroxyacids. This mechanism was supported by computational studies and the diastereospecific nature of the reaction, which suggests a concerted mechanism. Alternative stepwise mechanisms were explored computationally but had prohibitively high energy barriers. The transformation works best with neutral-to-electron-rich olefins, and electron-deficient species do not react. The authors also disclosed preliminary work on an intermolecular variant that gave stereospecific aziridination products in moderate yields.
In 2019, Johnson and Hilinski disclosed a novel organic nitrenoid-mediated aziridination (Scheme 6).25 Aziridinations employing diaziridines are known, but these utilize nucleophilic nitrogen and an α,β-unsaturated system (i.e., the aza-MIRC path). In contrast, the Hilinski system proposes an electrophilic diaziridine intermediate (Scheme 7). The iminium catalyst 7.1 is attacked by the iminoiodane, which immediately cyclizes to the active diaziridine 7.3. Conformational analysis revealed a restricted trajectory from which the olefin may attack to offer only the tosylated nitrogen as an accessible electrophilic target. Thus, issues with poor electrophilicity of diaziridines are circumvented through in situ generation of a diaziridinium ion to facilitate olefin attack to give 7.4. Presumably, the intermediate cation is persistent, as attack of a second equivalent of iminoiodane may occur (7.6) to give oxidative cleavage instead of the desired aziridine 7.5. The reaction is stereospecific to exclusively furnish trans-aziridines, albeit in moderate yield, which the authors claim as evidence of a carbocation intermediate. During optimization studies, an unexpected instability of the aziridine to the full reaction conditions was noted; removal of any single component (N-tosyliminobenzyliodinane [PhINTs], catalyst, or styrene) resulted in preservation of the aziridine. Thus, the concentration of the least soluble component, PhINTs, was limited by using a solvent system of 9:1 dichloromethane: hexane.
Scheme 7.
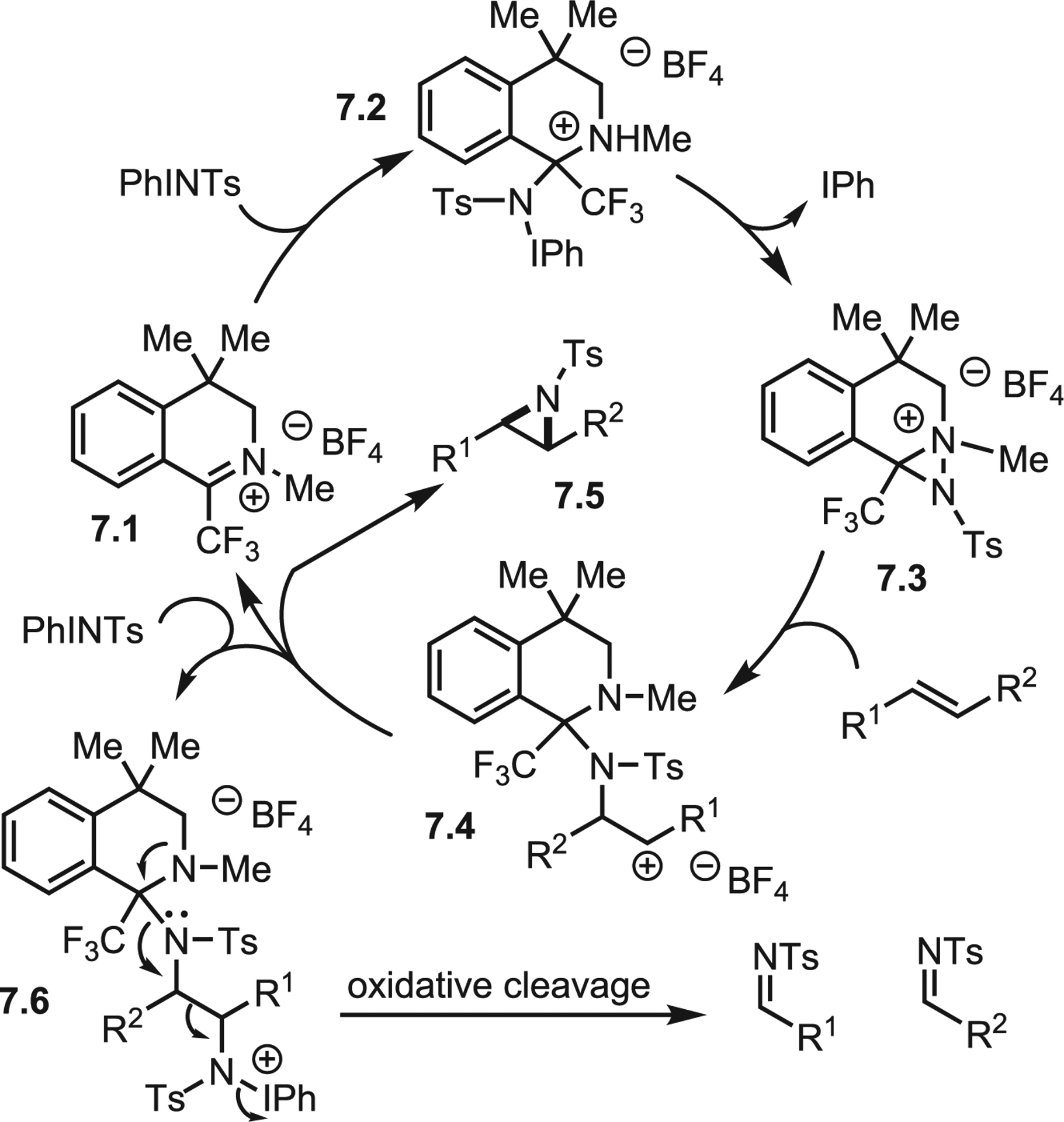
Proposed mechanism of organocatalytic nitrene transfer
Unfortunately, this method is limited to electron-rich styryl derivatives, and only monosubstituted alkenes give good yields. Nonetheless, this system is an intriguing template for using chiral counterions to induce diastereoselectivity, as well as for employing prochiral or chiral iminium species as reactive intermediates. As the diaziridine is the active species, different electrophiles, such as O-tosyl-N-Boc hydroxylamine, may also work well and tolerate the aziridine. Other achiral iminium salts may be employed with the thought that less hindered iminiums could expand the scope to encompass more substituted olefins. The iminium may be protected with a tosyl or Ns group to further activate the resultant diaziridine and allow for attack by unactivated olefins. Clearly, much about this type of reactivity remains to be addressed, and future work will expand the utility of this chemistry.
The Kürti lab recently published an exciting advance involving an organocatalytic aziridination of unactivated olefins via a transient oxaziridine.26 The initial screen was conducted with geranyl acetate 8.1 and a series of organocatalysts (Scheme 8). The most electron-poor catalyst 8a gave the best yields, with more electron-rich catalysts giving poorer results. The surprisingly low tolerance of this system to alternative conditions may explain why it has taken nearly 25 years after the disclosure of Shi’s epoxidation to develop an aza-variant. The mechanism is simple and reminiscent of the Shi epoxidation. Beginning with chiral catalyst 8d, hydroxylamine-O-sulfonic acid (HOSA) attack generates enantiomeric oxaziridines 8d′ and epi-8d′ to produce bisulfate and bicarbonate as benign by-products (Scheme 9). Extensive computations identified epi-8d′ as the slightly more favored product, leading to the proposal that the transient oxaziridine intermediate (confirmed by mass spectrometry) undergoes a concerted single-step nitrogen transfer to olefin 9.1 in order to generate aziridine 9.2. Unusually, the two epimers prefer to generate the same aziridine enantiomer (S)-9.2. The predicted selectivities in Scheme 9 assume a non-Curtin-Hammett scenario (i.e., 8d′ and epi-8d′ do not interconvert); the overall selectivity was predicted to be 84:16 er, nearly identical to the experimentally determined er of 85:15. The scope of the reaction was well explored (selected examples in Scheme 9B). Overall, the nitrogen transfer is selective for the most electron-rich olefin and favors the more sterically accessible olefin if the two are of equal electron densities (Scheme 9B, 9.4a vs. 9.4b).
Scheme 8.
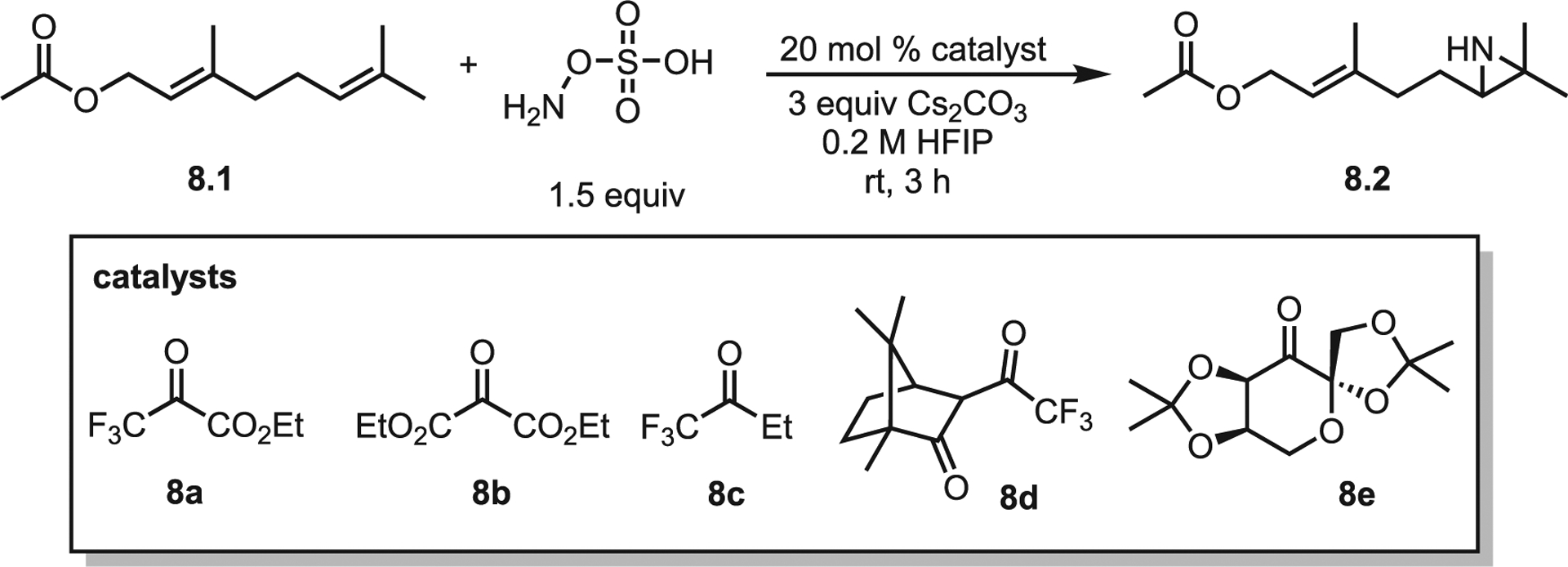
Oxaziridine-mediated aziridination of unactivated olefins
Scheme 9. Organocatalytic aziridination of unactivated olefins via a transient oxaziridine.
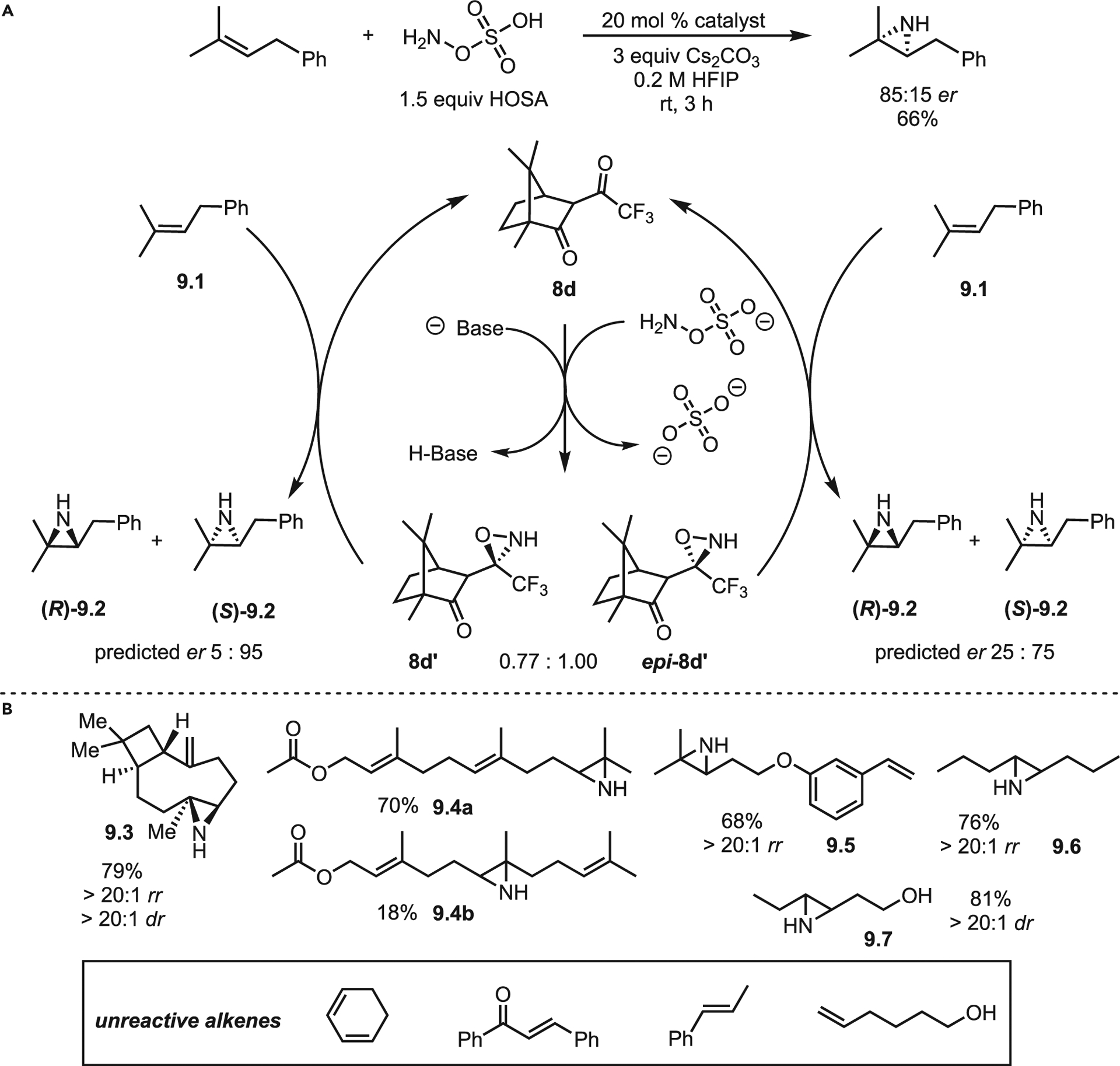
(A) Mechanism of oxaziridine-mediated aziridination.
(B) Selected scope and limitations using catalyst 8a. (dr, diastereomeric ratio and rr, regiomeric ratio).
The system is inert toward styryl derivatives and conjugated systems (Scheme 9B box and 9.5), presumably due to low nucleophilicity for productive attack on the oxaziridine. Di- and trisubstituted olefins react smoothly in good-to-excellent yields with high dr, using a chiral catalyst or substrate control; good regiomeric ratio (rr) is also observed when multiple olefins are present. Best of all, this method furnishes unprotected aziridines, removing the need for deprotection and enhancing step- and atom-economy. Limitations include unknown compatibility with more diverse heteroatom functional groups (carboxylic acids, amines, and epoxides), the lack of activity with monosubstituted olefins, and the requirement of HFIP as the solvent. Future work will shed light on all these issues as the benefits of this methodology for efficient and cheap access to NH aziridines is unparalleled.
Metal-mediated aziridination to alkenes and allenes
Methods for metal-mediated aziridination most commonly occur through the transfer of metal-supported nitrenes or nitrogen-centered radicals (NCRs). Catalytic generation of these reactive species offers advantages compared with their “free” generation, which include the following: (1) formation of reactive species under mild conditions, (2) low concentrations of active species enabling greater control of reactivity, and (3) a high degree of tunability through ligand modification. As nitrene transfer (NT) is an excellent way to generate highly valuable aziridines and amines, this outer-sphere process is under heavy investigation by the synthetic community. Numerous recent reviews discuss C–H aminations and C=C aziridinations, often concurrently, in detail. Thus, only representative aziridinations are provided to highlight recent advances in the field; interested readers are directed to detailed discussions elsewhere.27,28
A simplified general mechanism of transition-metal-mediated NT is shown in Scheme 10. A metal catalyst MLn reacts with a nitrene precursor RN=LG to give reactive intermediate metal nitrenes RNMLn, which may either react with a C=C bond to form an aziridine or with a C–H bond to generate an amine. A nitrene exists either in its singlet state, which favors concerted, asynchronous transition states, or as a triplet, favoring radical intermediates and reactivity via H-atom transfer. However, singlet and triplet states can interconvert under reaction conditions. The first successful asymmetric NT reactions were reported independently by Evans and Jacobsen in 1993, nearly 25 years after the first reported metallonitrene species by Kwart and Khan.29–31 Since these reports, numerous metals have been utilized successfully in racemic NT processes with Rh, Ru, Ir, Fe, Mn, Co, Cu, and Ag as some of the most well studied to date. However, a general asymmetric aziridination method has remained elusive.27,32
Scheme 10.
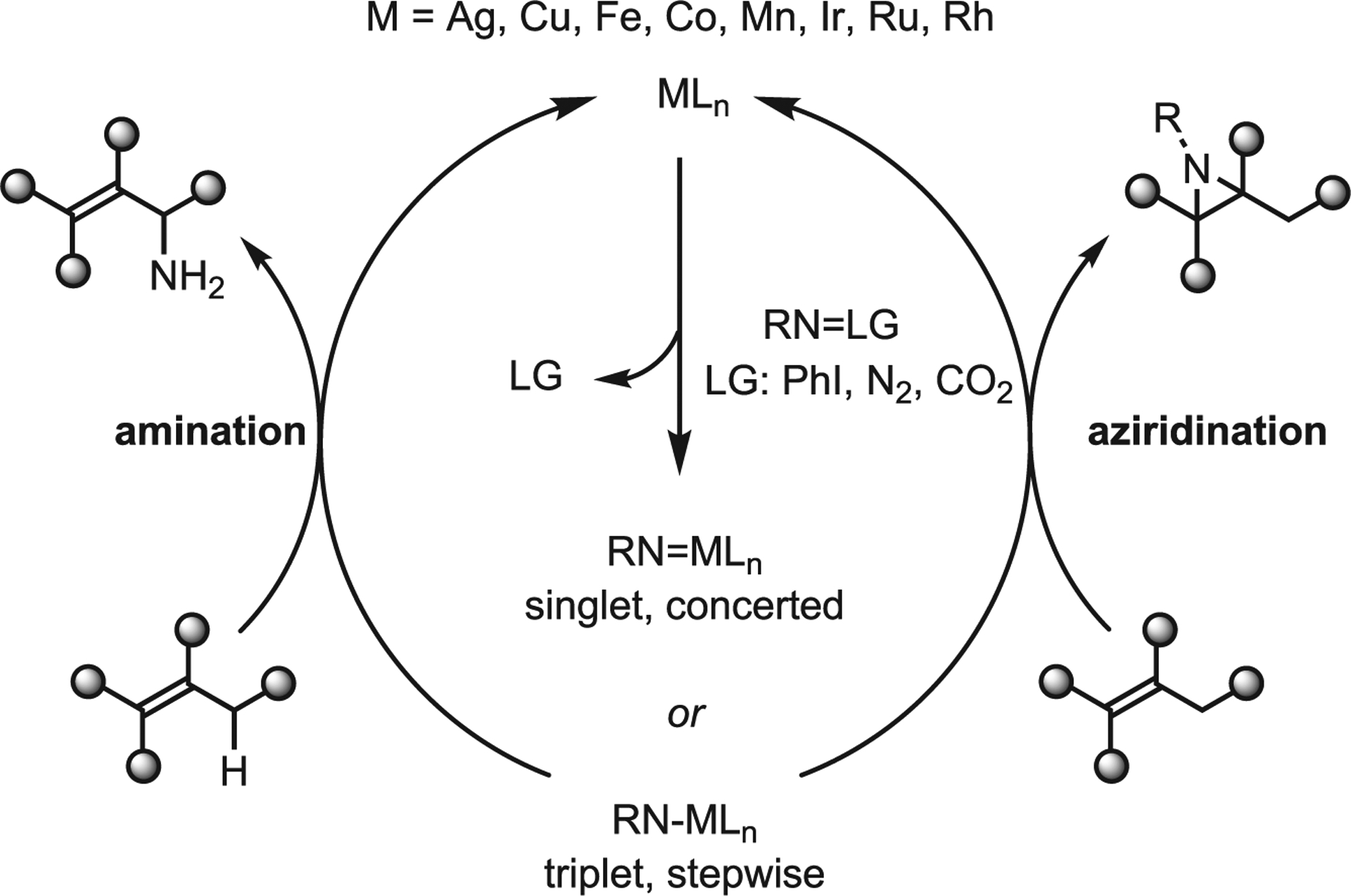
Simplified mechanism of metal-catalyzed nitrene transfer
Metal-porphyrinates are among the most frequently used catalysts for asymmetric NT. They were originally developed for C–H amination; however, inspired by early reports of stereoselective nitrene insertions, Zhang and co-workers envisioned utilizing open-shell complexes to facilitate and control reactivity through the transfer of radical character.32–34 To this end, they developed metalloradical catalysts (MRCs) based on well-defined Co(II) complexes of D2-symmetric chiral amidoporphyrins [Co(D2-Por*)]. A number of these catalysts have been reported (Scheme 11, top) and can generate aziridines in high yields and ee from a range of precursors (Scheme 11, bottom).35,36
Scheme 11. Zhang’s prior work on asymmetric aziridination using metalloradical catalysts.
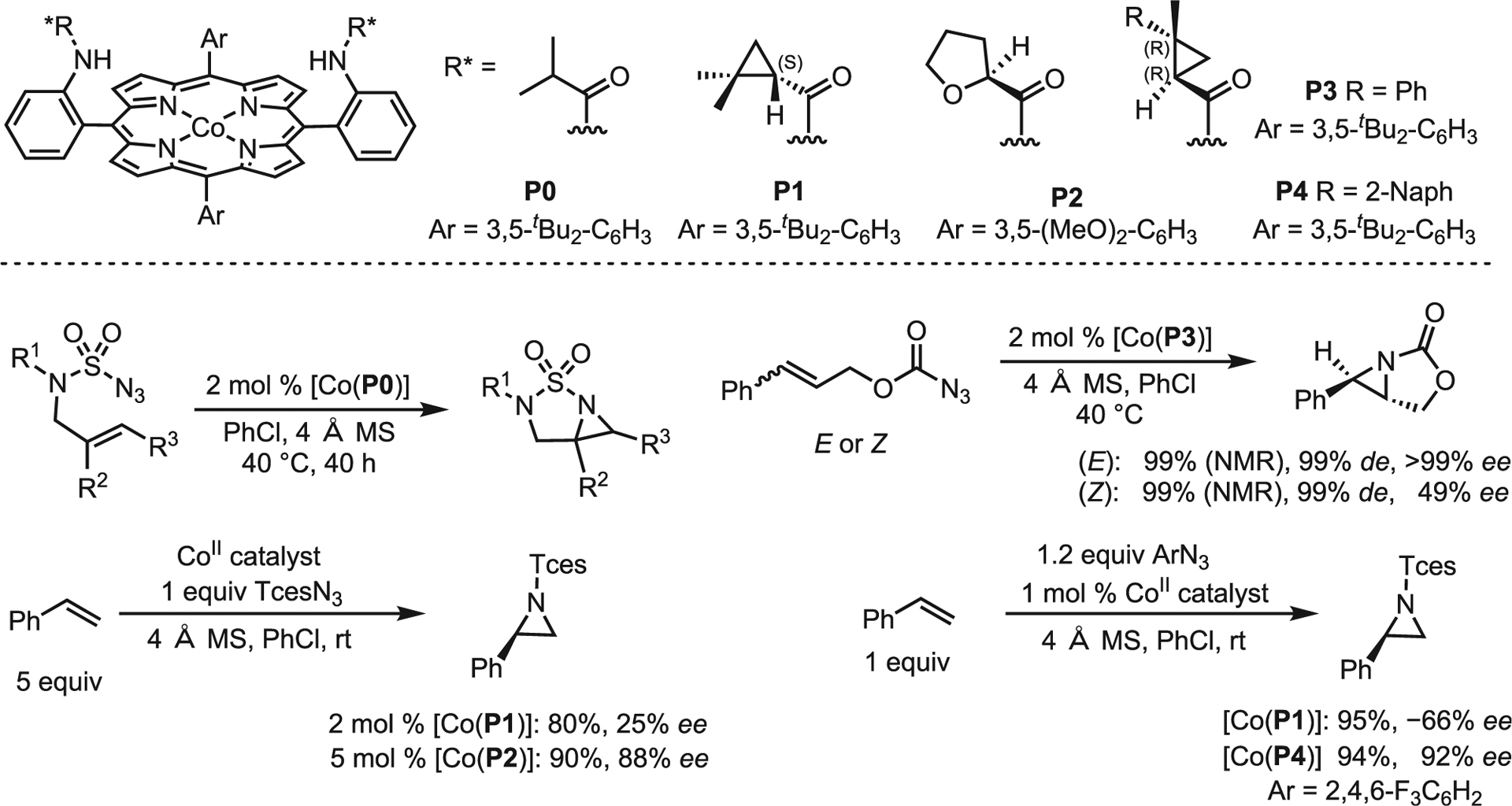
Top: cobalt porphyrin catalysts with common amide groups. Bottom: prior work by Zhang and co-workers.
An illustrative example of key attributes is given in Scheme 12, using a Troc-azide substrate. The unpaired electron (cyan orbital) rests in a well-defined d-orbital of Co(II)-porphyrin 12.1 (hence the term “metalloradical”). 12.1 induces azide decomposition to give α-Co(III)-aminyl radical 12.2. The bound species 12.2 possesses similar reactivity to a free NCR; however, the coordinating environment imposed by the metal-porphyrinate stabilizes the M–N bond to prevent homolytic release back into the medium. Combined experimental and computational experiments support the engagement of the MRC with the alkene via stepwise radial addition to give alkyl radical intermediate 12.3. In principle, the alkyl radical may undergo either 3-exo-tet cyclization to give desired aziridine 12.4a or 5-endo-trig cyclization to give oxazoline 12.4b; however, judicious choice of MRC allows for the selective production of desired aziridine. This platform is stereospecific in intermolecular cases, but it is stereoselective in intramolecular reactions. Presumably the congested chiral environment leaves insufficient room for the carbon radical intermediate 12.2 to rotate and forces rebound along the same trajectory of the initial attack in the intermolecular case. Application of the same reasoning to the intramolecular case presents only a single face for NCR attack once the side chain is in close enough proximity for subsequent cyclization. The scope is currently limited to styrene derivatives, which are well tolerated and give good yields and ee for a range of electron-donating and -withdrawing groups at all positions of the benzene ring (Scheme 12).
Scheme 12. Zhang’s more recent work using Co(II) complexes of D2-symmetric chiral amidoporphyrins.
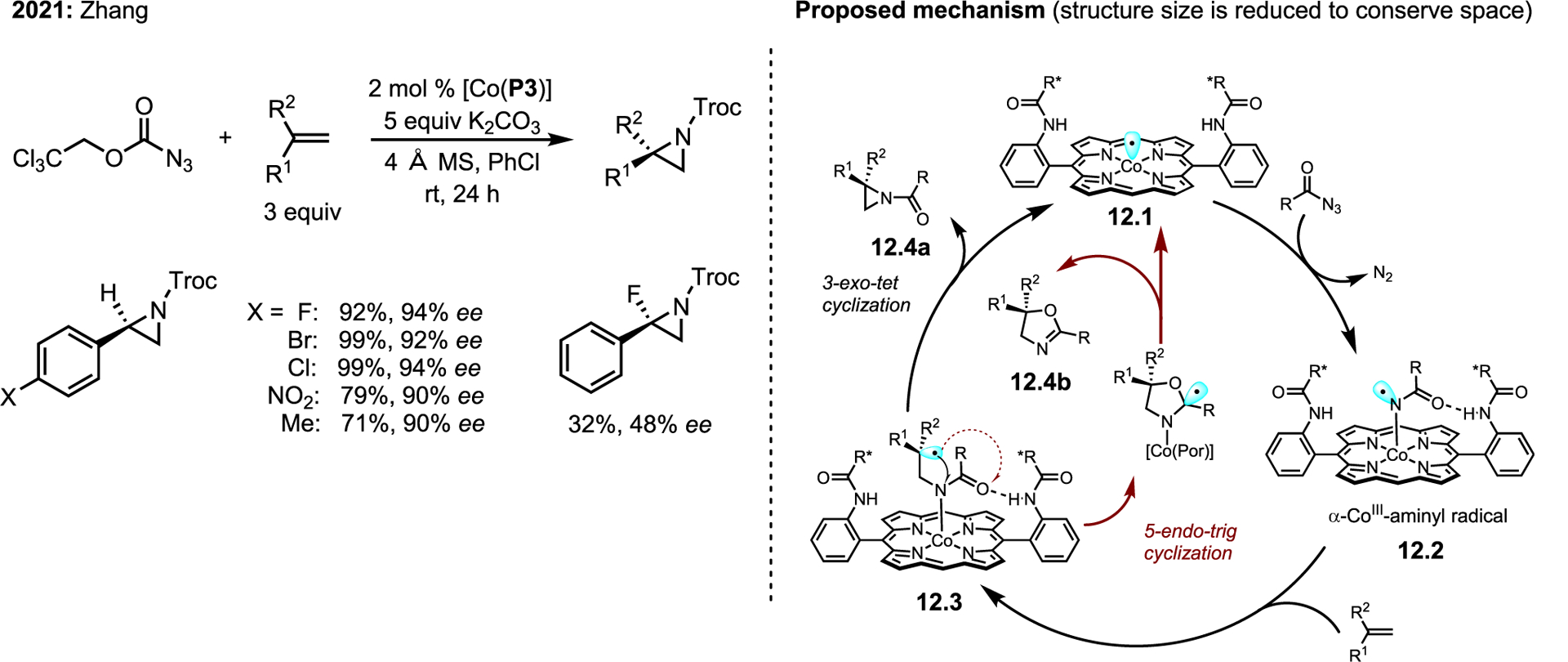
Troc azide conversion to enantioenriched azides (left). Proposed mechanism of [Co(D2-Por*)] nitrene transfer (right).
A recent addition to metal-porphyrin-catalyzed aziridinations is the use of natural porphyrins by the Arnold lab. Enzymes that utilize electrophilic nitrogen species are scarce, as sources of electrophilic nitrogen are rarely found in nature. As such, the biosyntheses of naturally occurring aziridines are believed to proceed via intramolecular cyclization. Arnold and co-workers reported that heme iron could catalyze aziridination (Figure 7A); a single mutation of a cysteine residue into a serine (C400S) of bacterial P450 promoted non-natural carbene and NT, as well as removing its native hydroxylation activity.37 The variant, dubbed P411BM3-CIS-T438S, was highly effective for C–H amination and sulfide imidation.38 This variant was chosen for continued mutation studies to optimize aziridination of 4-methoxystyrene using tosyl azide as the nitrogen source. Directed evolution resulted in several P411 variants capable of both in vitro and in vivo aziridination of electron-rich styrenes in moderate-to-excellent yields and excellent (S)-selectivity (81%–99% ee). However, particularly electron-rich styrenes proved too reactive (4-methoxystyrene, 2-methyl styrene) and hydrolyzed in the medium to give racemic hydroxylamines.38
Figure 7. Arnold’s work on heme iron-catalyzed aziridination.
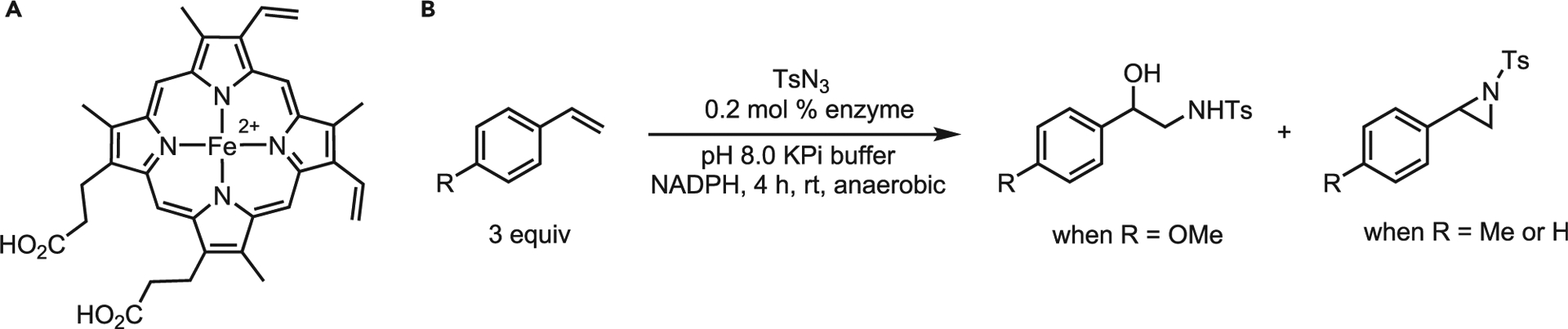
(A) Heme-iron complex scaffold.
(B) Overall aziridination under optimized conditions.
More recently, a non-heme iron enzyme capable of NT aziridination (Figure 8) was reported. Goldberg and co-workers screened seven α-ketoglutarate (αKG)-dependent iron dioxygenases for intermolecular aziridination of styrene and p-toluenesulfonyl azide.39 Only Pseudomonas savastanoi (PsEFE) effectively formed the desired aziridine. PsEFE (EFE, ethylene-forming enzyme) is unique to this family as it promotes fragmentation of αKG to ethylene in an unusually hydrophobic pocket.40 Two rounds of site-saturation mutagenesis and one round of recombination gave a variant with five mutations from the wild type to furnish the (R)-aziridine in 88% ee. While the metalloprotein catalyzes both C–H amination and aziridination, both the bacterial heme system and the current form of the metalloprotein are limited in terms of scope and yield. However, this is the first example of an enzymatic NT catalyzed by a non-heme metalloprotein. As metalloproteins comprise over 30% of all proteins, the area is open to the continued discovery and optimization of new modes of reactivity with improvements in yield and substrate scope expected soon.
Figure 8. Iron deoxygenase-catalyzed intermolecular aziridination by Goldberg.
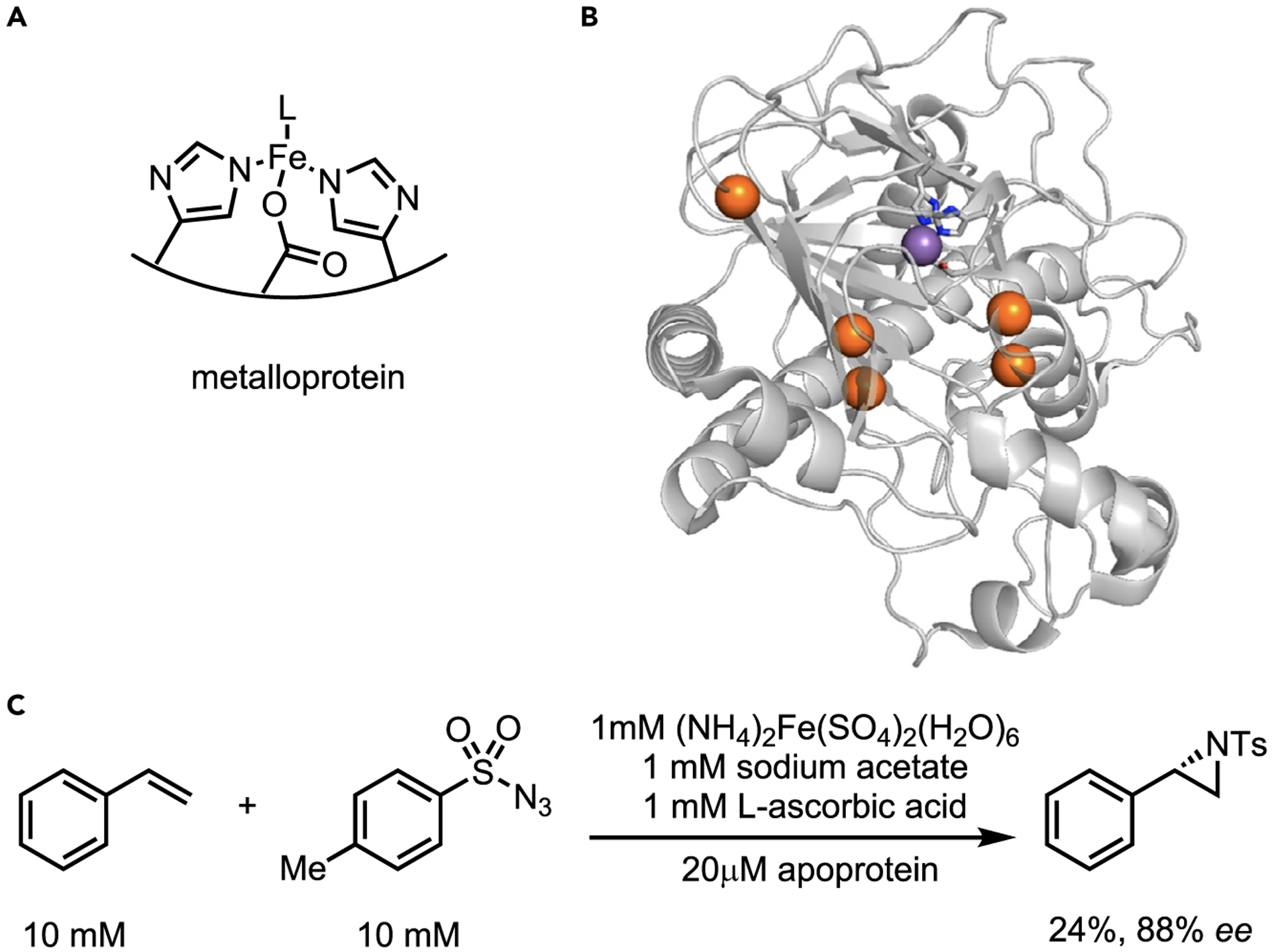
(A) Schematic of iron-bound metalloprotein.
(B) Image of most active enzyme with 5 mutations highlighted in blue.
(C) Overall transformation with optimized conditions and best reported result. Reprinted (adapted) with permission from Goldberg et al.39 Copyright 2019 American Chemical Society. PDBID: 6CBA.
Recent advances have also been made in chemocatalytic aziridinations. Chiral CF3-heterocycles are valuable scaffolds for medicinal and process chemistry.41,42 Qin published a one-step method to access chiral aziridines bearing trifluoromethyl groups via radical addition in high dr.43 Building on prior work by Buchwald and Liu,44,45 Qin and co-workers screened a Cu(I) source and commercially available ligands with a model alkenyl sulfinamide substrate and Togni’s reagent. While no consumption of starting material occurred using pyridine or phosphine ligands, L-proline furnished the desired aziridine in good yield as a single diastereomer (>25:1 dr). Initial studies were conducted in CH2Cl2; however, tBuOH was found to give the best yields. Addition of Na2CO3 increased yields by ~10% by neutralizing the 2-iodobenzoic acid by-product. Based on DFT calculations and control experiments, the authors proposed a four-stage mechanism (Scheme 13). First, the chirality of the sulfinamide induces asymmetric complex formation by orienting L-proline through a strong N–H bond. This chelates to Cu to give the Cu(I) complex 13.1. Next, the alkene π-system traps the free radical CF3•, to give 13.2. Deprotonation of the sulfanilamide N–H gives 13.3, and subsequent reductive elimination gives the aziridine in high dr. The reaction is tolerant of aryl, alkyl, heteroaryl, and H groups, which all yield single diastereomers in good-to-excellent yields (Scheme 13B). Notably, this transformation also tolerates substitution on the alkene to deliver 1,1-substituted aziridines in excellent yield and dr.
Scheme 13. Qin’s recent work on the scope and mechanism of Cu-catalyzed radical alkene aminotrifluoromethylations to furnish aziridines.
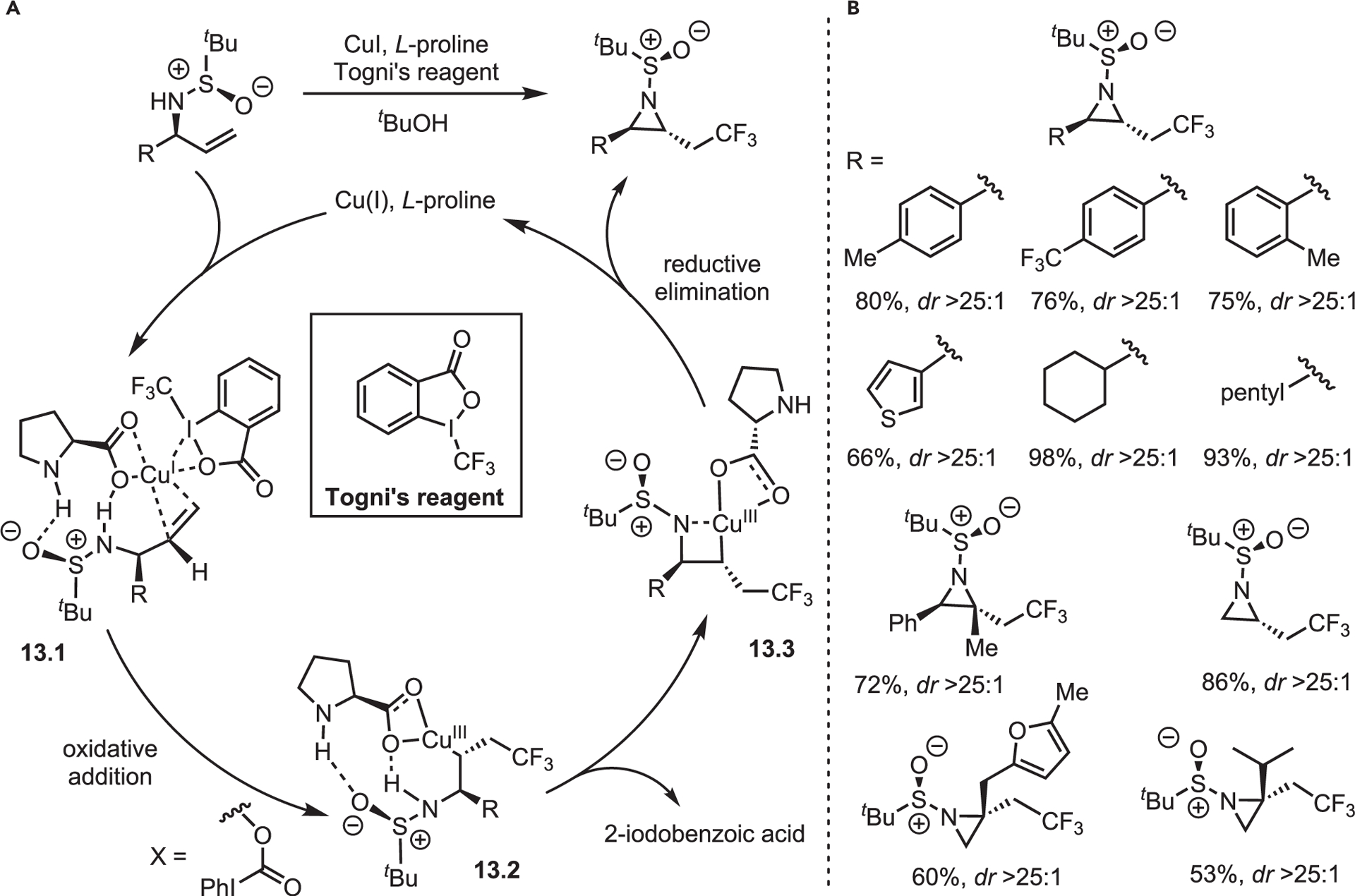
(A) Proposed mechanism of aziridination.
(B) Selected substrate scope.
Qin and co-workers’ report is an excellent demonstration of the utility of the N-substituent to dictate reaction progress and outcome. To our knowledge, the metal-mediated system with widest general scope was reported by Ess, Kürti, and Falck in 2014 (Figure 9).46 Using O-(2,4-dinitrophenyl)hydroxylamine (DPH) as an amine source, smooth conversion of a wide range of mono-, di-, tri-, and tetrasubstituted olefins to N–H and N-methyl aziridines was achieved with rhodium catalysis. Despite the general utility, the Kürti lab was unsatisfied with limitations of the method, including stoichiometric use of DPH, high NO2/C ratios of materials and by-products, and difficulty of purification. To that end, the Kürti lab recently reported the replacement of DPH with HOSA.
Figure 9. Kürti’s follow up work on olefin azirdinaition using DPH as an amine source.
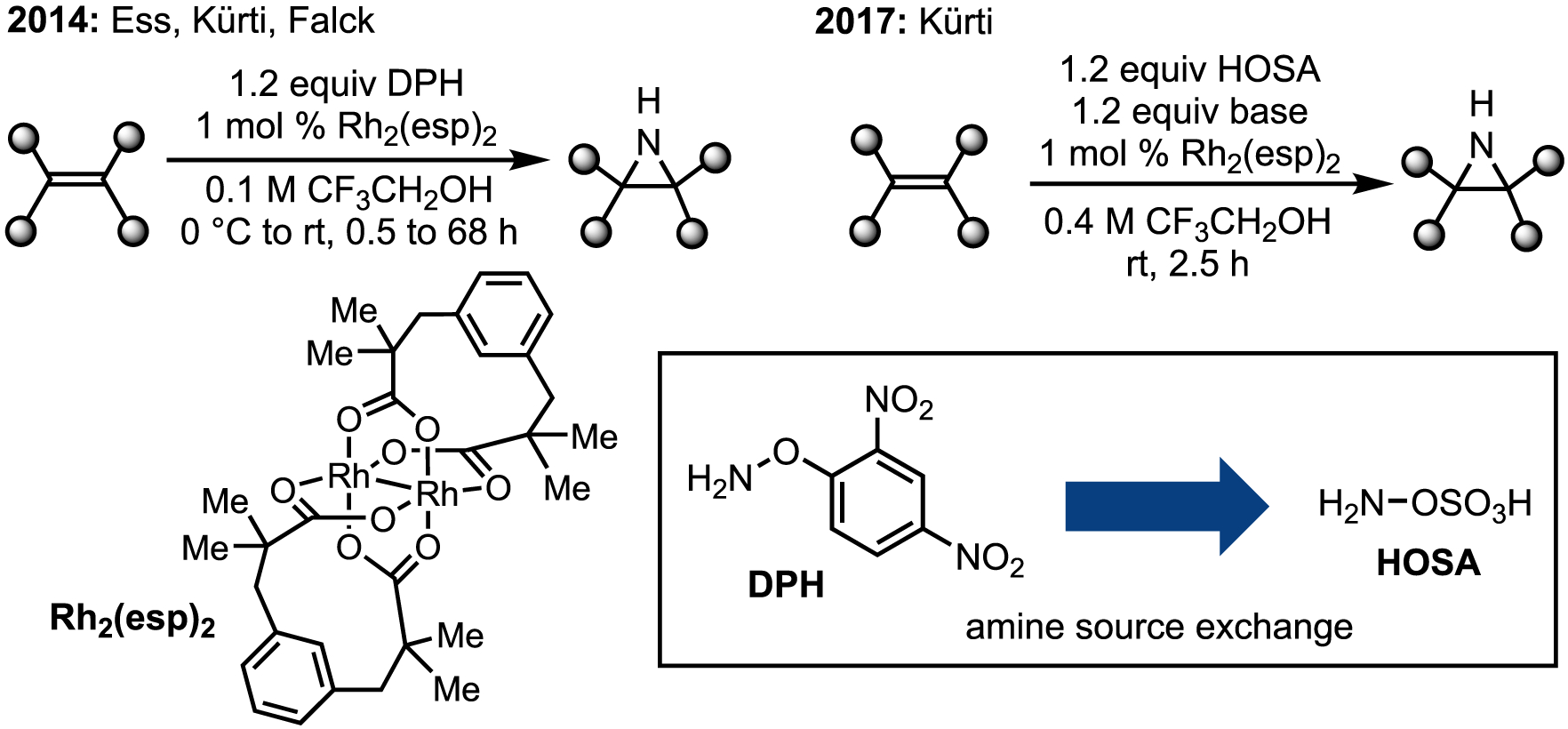
Initial conditions (left) and improved conditions (right) for Rh-catalyzed aziridination.
Under standard conditions, HOSA reacted sluggishly to give the desired aziridine in only 44% yield after 16 h. Noting the acidity of the TFE (pKa ca. 12.5) as a potential influence, they examined more basic methanol and more acidic 1,1,1,3,3,3-hexa-fluoro-2-propanol (HFIP; pKa ca. 9.3) (Scheme 14), with HFIP proving to be a superior solvent. Pyridine gave the best yields when diverse bases were examined to promote faster and more efficient aziridination. Catalyst loading and type was also investigated, with Rh2(OAc)4 giving similar yields to the Du Bois catalyst. A wide variety of substituted N–H aziridines were prepared in good-to-excellent yields; more electron-rich olefins reacted preferentially, even when potential directing groups were present (Scheme 15B). The reaction is stereospecific, yielding single enantiomers in many cases; the authors propose a mechanism similar to that reported in 2014 (Scheme 15A), where formation of a Rh-nitrenoid via amino group coordination and loss of sulfate anion occurs prior to olefin capture. Based on DFT calculations, the aziridination is proposed to proceed through a triplet transition state, leading to a singlet diradical intermediate with barrierless recombination to yield the aziridine. This improved method is more environmentally friendly and cost-effective; the general scope and straightforward operation will lead to its broad use in preparing functionally dense aziridines and amines.
Scheme 14.
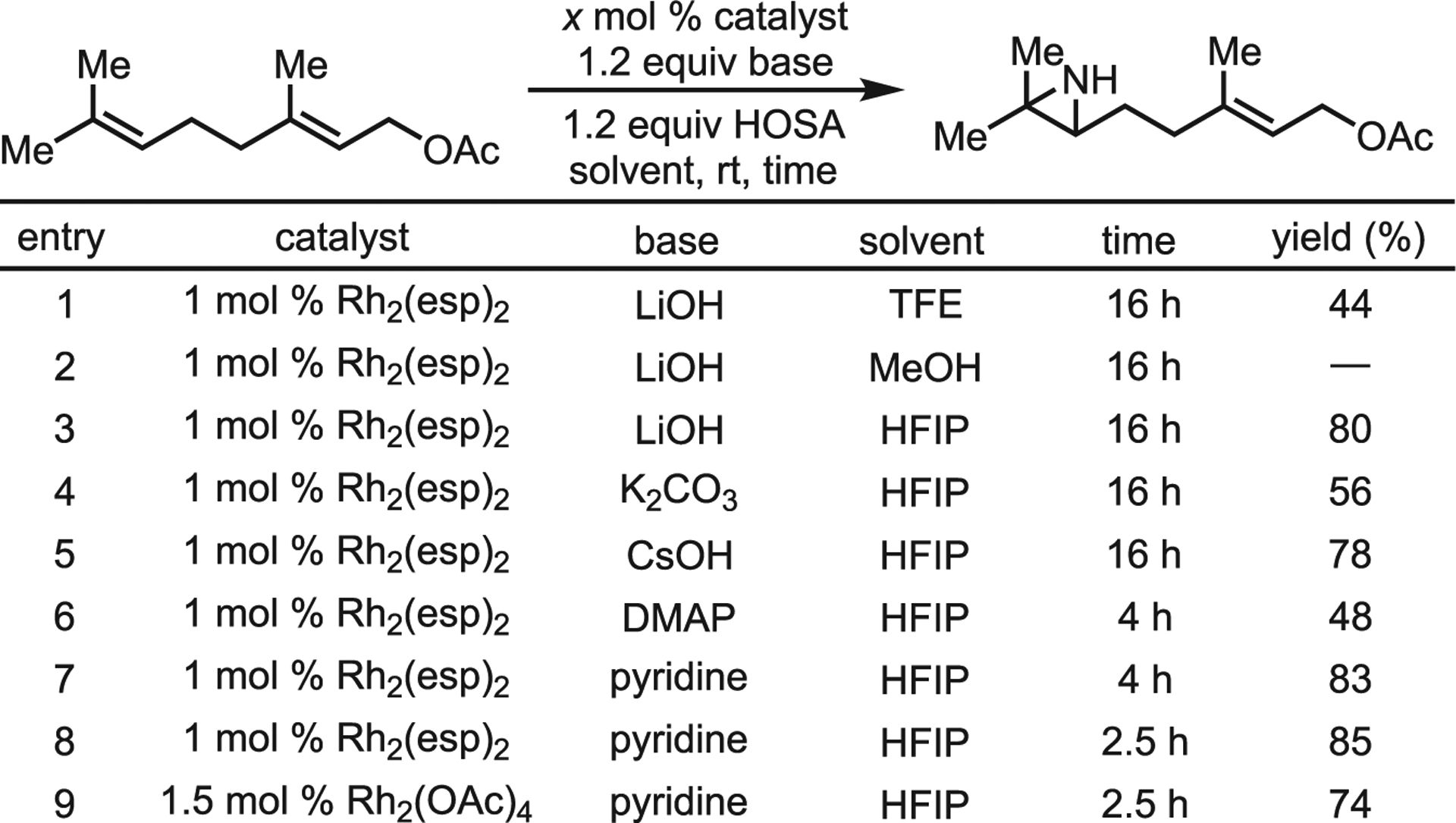
Selected optimization screen by Kürti and co-workers
Scheme 15. Kürti’s prior work on aziridination of substituted olefins using HOSA as an amine source.
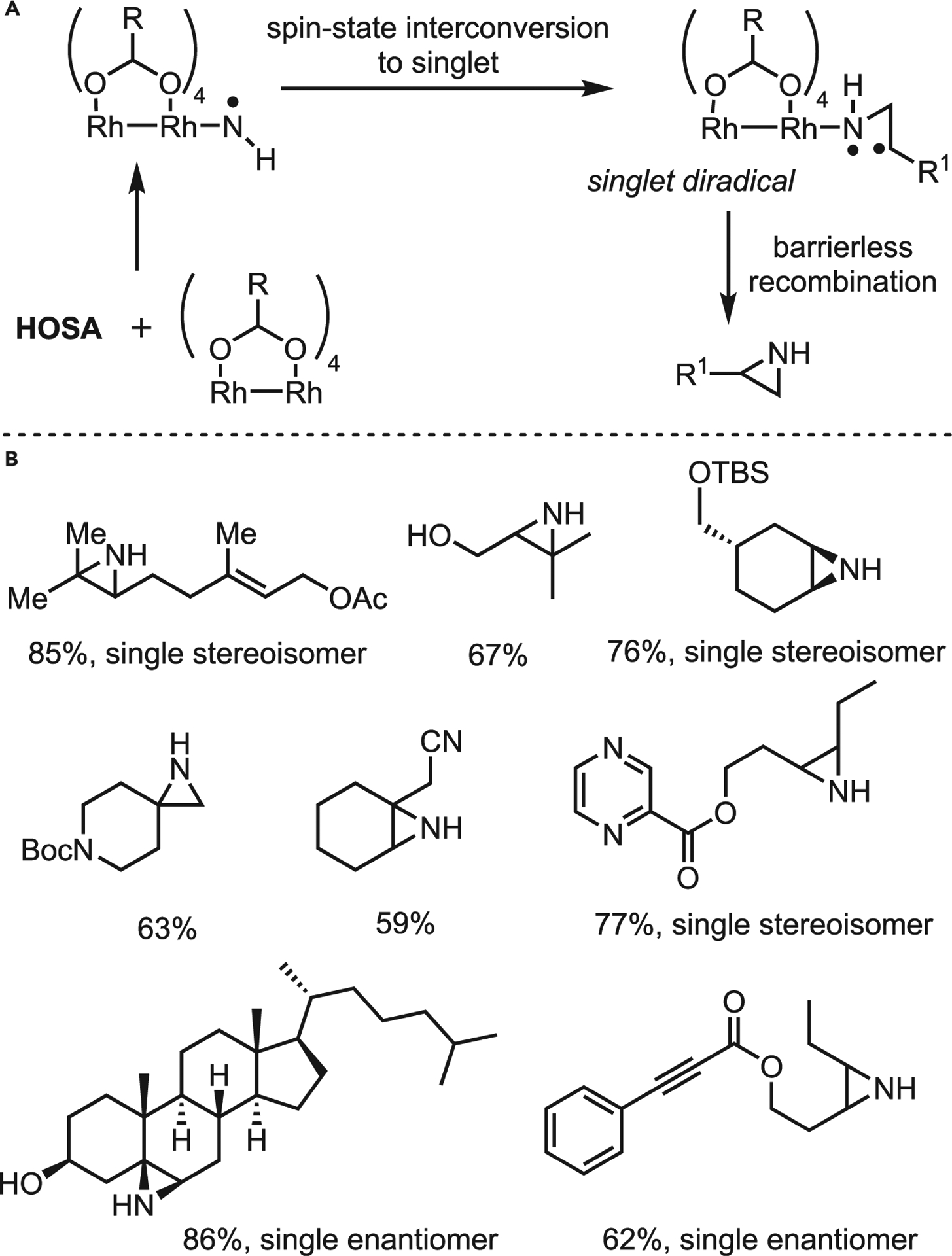
(A) Proposed mechanism.
(B) Selected scope.
The Dauban group recently reported that the C4-symmetric catalyst dirhodium(II) tetrakis[N-tetrafluorophthaloyl-(S)-tert-leucinate], (Rh2(S-tfpttl)4, promotes the asymmetric intermolecular aziridination of a variety of substituted alkenes.47 This chemistry represents a significant advance toward the challenge of developing chemo- and enantioselective catalysts for intermolecular NT. The optimal conditions (Scheme 16A) employ commercially available (Rh2(S-tfpttl)4, a phenol-based sulfamate nitrene precursor, PhI(OPiv)2, and interestingly, an equivalent of pentafluorobenzoic acid. The role of the acid additive is currently unknown. In terms of the scope (Scheme 16B), diverse monosubstituted styrenes bearing both electron-donating and -withdrawing substituents gave good yields and ee. The successful preparation of trans- and cis-methylstyrenes highlights the stereospecific nature of the chemistry. Excitingly, trisubstituted alkenes were also well tolerated in the reaction, giving the product aziridines in good yields and excellent ee. Computational investigations were carried out to provide insight into the mechanism of the reaction and suggested that the formation of the first C‒N bond is the enantiodetermining step. The triple transition state converges to a singlet product via a spin crossover that avoids the potential for olefin isomerization.
Scheme 16. Rh(II)-catalyzed asymmetric intermolecular aziridination of alkenes by Dauban.
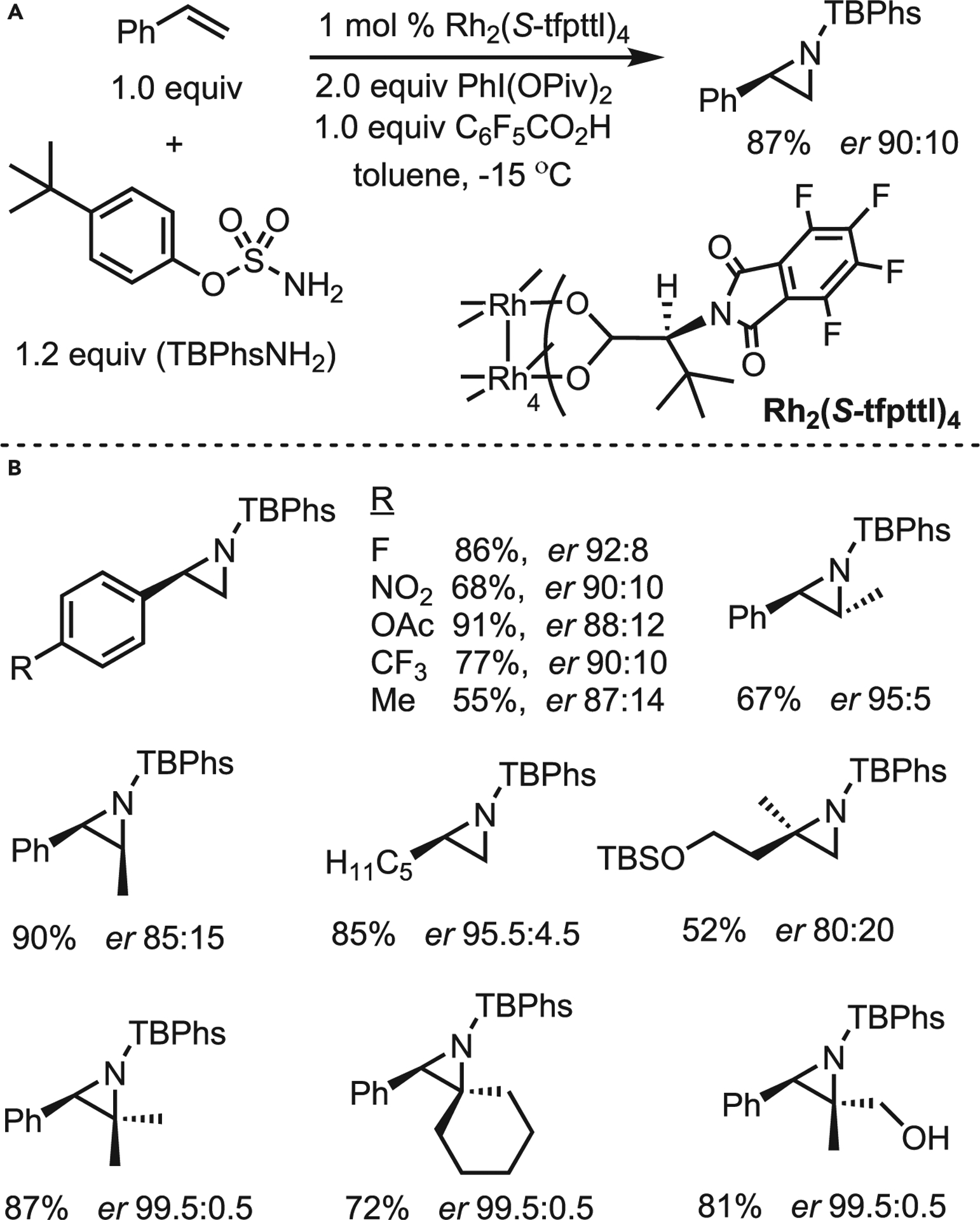
(A) Optimized conditions.
(B) Selected scope.
The Schomaker lab has recently published several examples of Ag(I)-catalyzed NT where the diversity of coordination geometries enabled by simple sp2 N-dentate ligands leads to unexpected chemo- and site-selectivities. This flexibility was harnessed to develop a chemodivergent platform to achieve either C–H amination or aziridination by either modifying the ligand or the ligand:Ag ratio.
The Schomaker lab first reported chemodivergent aziridination in 2013 (Scheme 17A).48 Notably, allenes could be employed as substrates to furnish methyleneaziridines (MAs), a useful class of aziridines (see methyleneaziridines [MAs] and aziridinium ylides [AYs]). The utilization of Ag(I) salts was prompted by the observation that most common catalysts for NT, such as Rh2(esp)2, gave significant amounts of C–H amination over the desired aziridination in up to 17:1 excess. In 2016, the group reported catalyst-controlled intermolecular chemoselectivity, utilizing different ligands to furnish either aziridine or amine products (Scheme 17B).49 Computations suggested control was derived from the unique geometries formed in solution. Dimerization of ligand 2 leads to two non-equivalent Ag centers that facilitate a late-stage hydrogen atom transfer, followed by near-barrierless recombination to yield desired aziridines (Scheme 17B, inset image a). In 2017, the lab disclosed the first successful asymmetric NT aziridination of trisubstituted olefins, using enantioenriched bisoxazoline ligands, notably on unactivated olefins (Scheme 17C).50 The transformation tolerates a variety of functional groups, although it is currently restricted to intramolecular reactions of carbamate precursors.51,52
Scheme 17. Schomaker’s work on Ag(I)-catalyzed aziridination.
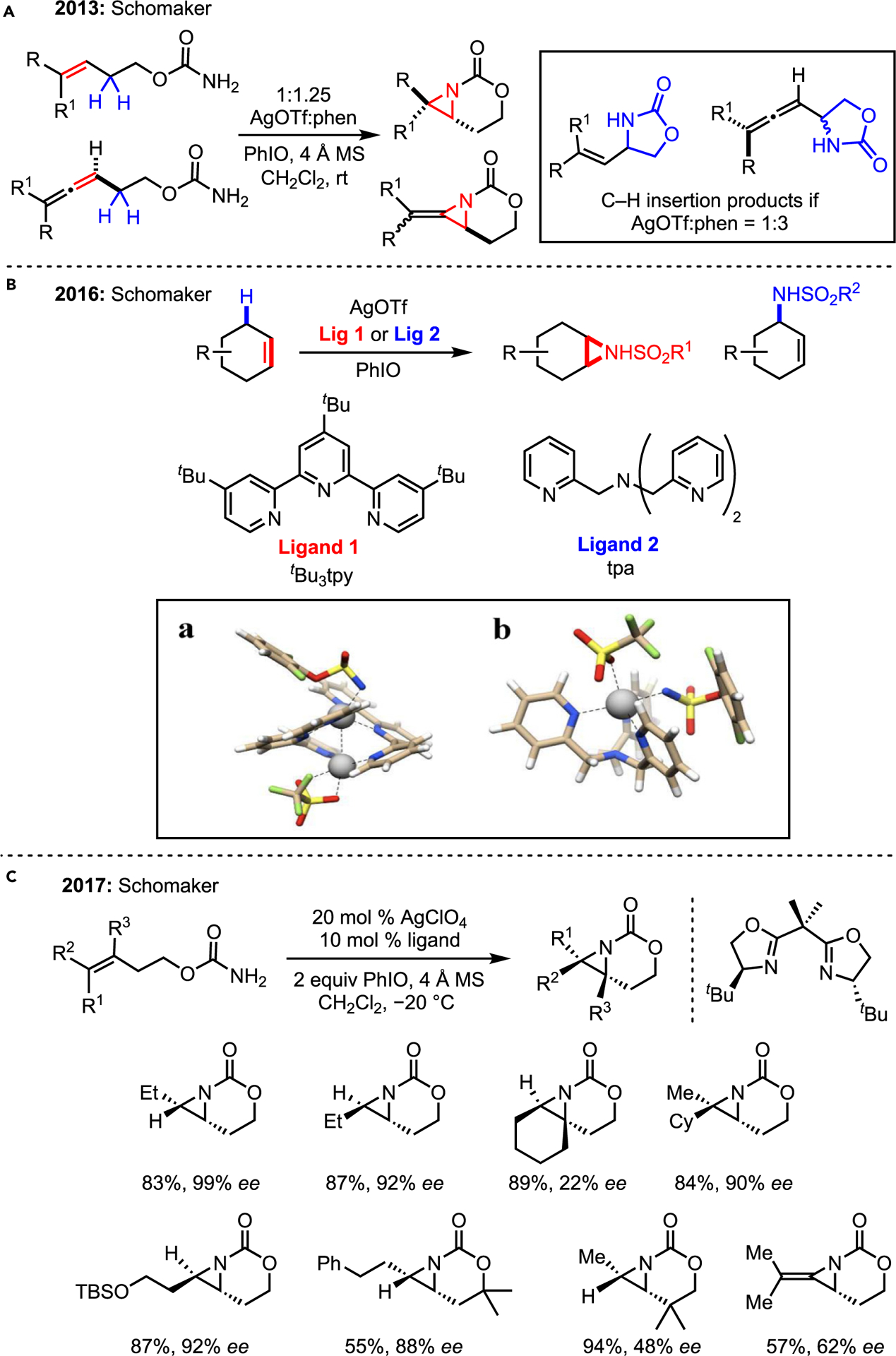
(A) Tunable Ag-catalyzed nitrene transfer.
(B) Tunable chemoselectivity via ligand.
(C) Asymmetric aziridination with chiral BOX ligands. Reprinted (adapted) with permission from Dolan et al.49 Copyright 2016 American Chemical Society.
Despite excellent progress, several challenges remain. The bulk of current methods typically begins with the aziridination of styrene to identify initial conditions, then explores scopes with substituted styrenes, and finally transitions to non-activated olefins. However, aziridinations of unusual olefins, such as those in allenes, can yield access to otherwise difficult-to-obtain products. For example, the Schomaker lab has reported chemoselective allene aziridinations to bicyclic MAs, which are readily transformed to stereochemically complex amine triads (see methyleneaziridines [MAs] and aziridinium ylides [AYs]). Allenes are easily prepared from common precursors and are becoming more mainstream as multiple groups have begun exploring their utility to access new amine chemical space.53,54
Photo and electrochemical methods for aziridination
A primary challenge in organic chemistry is the formation and control of highly reactive species in a timely, atom-economic, and sustainable fashion. To meet these demands, photochemical and electrochemical methods are re-emerging, as reported in several recent reviews.55,56 Current advances in photo- and electrochemical aziridinations rely on two complementary strategies; the former tends to focus on mild and selective methods to generate reactive nitrogen species and the latter on olefin activation and trapping with amine sources.
Photochemical aziridination
The predominant strategy in photochemical aziridination involves irradiation of a suitable precursor to generate a nitrene proximate to an unsaturated center in a manner similar to metal-mediated NT reactions. Traditionally, nitrene generation utilized harsh conditions such as high heat or UV irradiation. Recently, transition metal photocatalysts have enabled the use of visible light to indirectly form the nitrene species (Figure 10). Advantages of visible light include ease of use, less risk of photodecomposition of organic substrates and greater selectivity due to the mild formation conditions. Despite these advances, photochemical methods of aziridination are underutilized.
Figure 10.
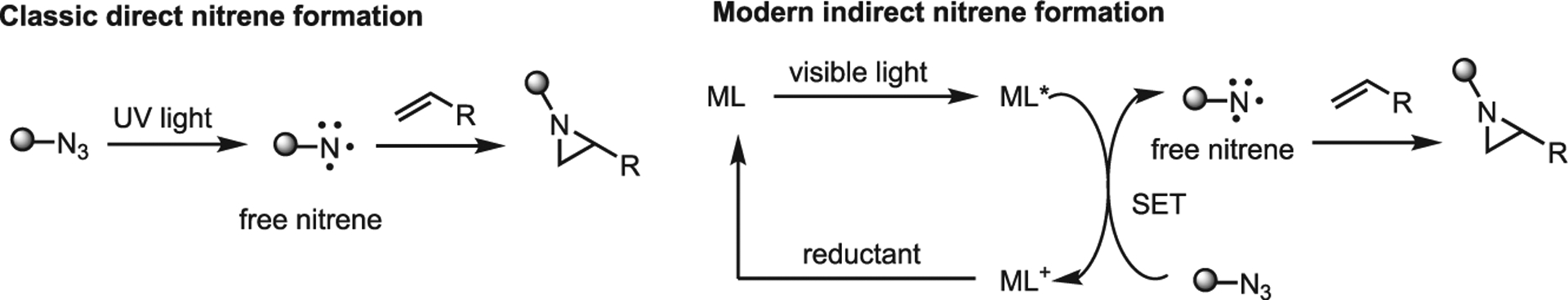
Simplified classic and modern photocatalytic methods of aziridination
The Lu lab reported visible light-induced C(sp2)–H amination and aziridination of o-allylphenyl azidoformates.57 They exposed substituted phenyl azidoformates to blue light-emitting diode (LED) irradiation in the presence of numerous transition metal photocatalysts to furnish C–H amination products. Ir[(dtbbpy)(ppy)2]PF6 was identified as the preferred catalyst; the photocatalyst enables mild generation of the reactive nitrene species that reacts as a free nitrene with a kinetic preference for olefin attack. Exploration of an extensive substrate scope revealed that allyl-substituted substrates preferentially underwent aziridination (Scheme 18). Based on prior investigation of similar transformations, the authors propose the reaction proceeds via a triplet energy transfer mechanism wherein photolysis of the azide is the RDS. The reaction is presumed to be stereospecific, although the authors only examined a single trans substrate. Regardless, this methodology produces unique [3,7,6] fused aromatic systems difficult to obtain in other ways.
Scheme 18.
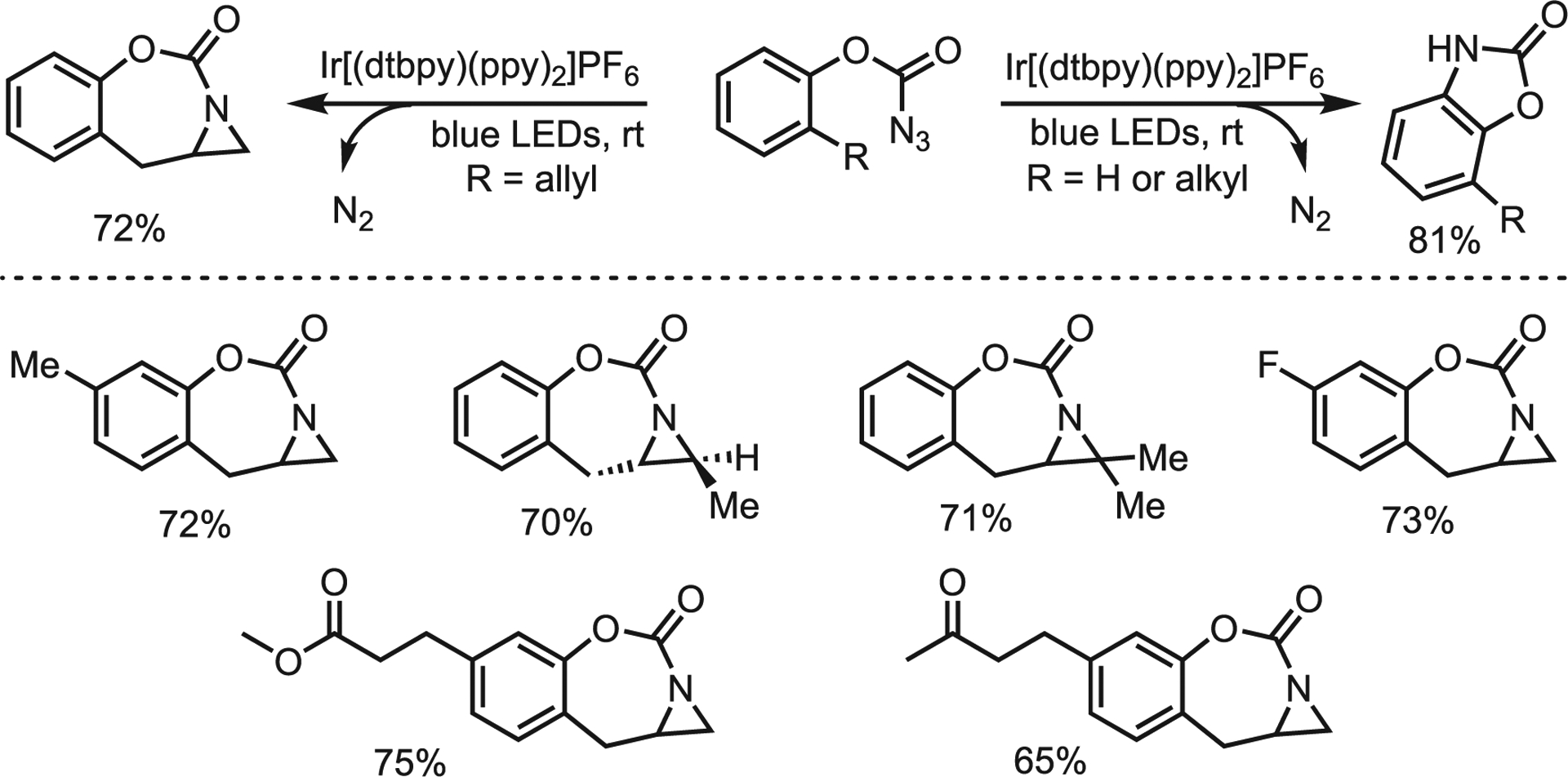
Selected scope of Ir-catalyzed aziridination of azidoformates
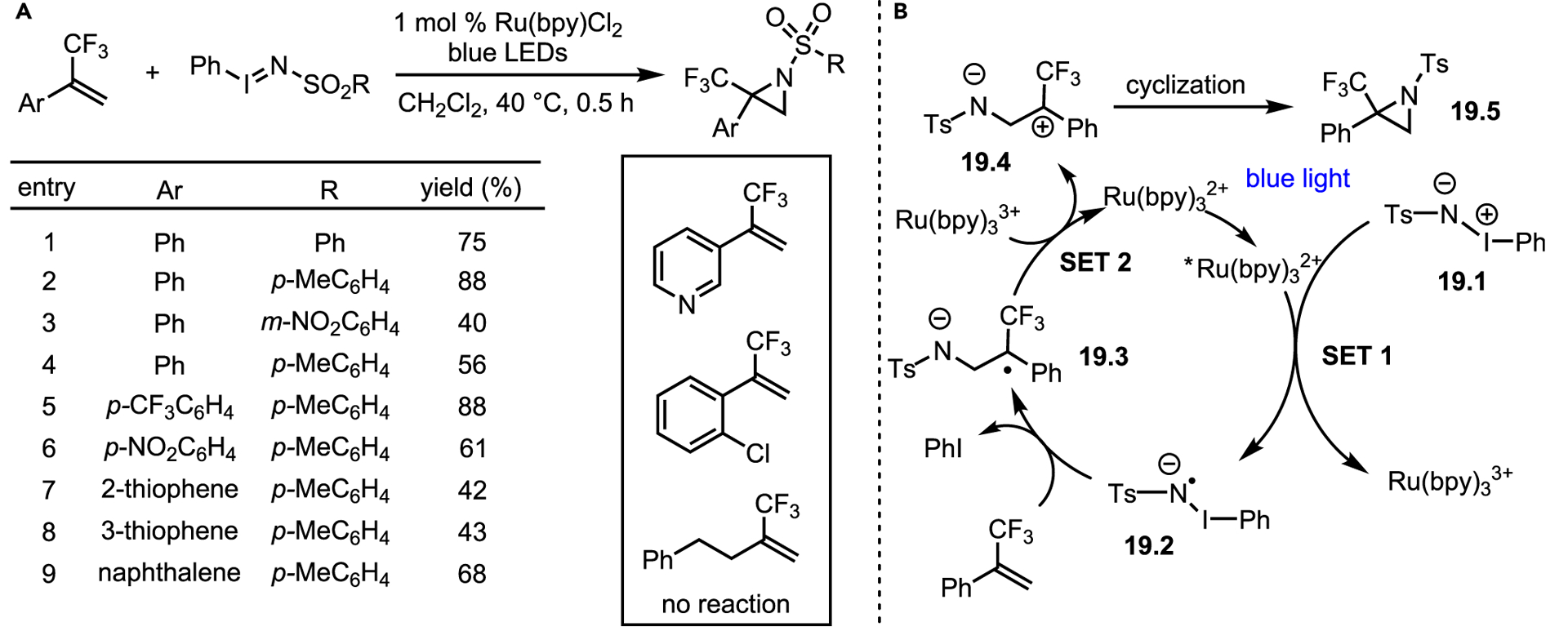
More recently, the Köenigs lab utilized photocatalysis to access valuable trifluoromethylated aziridines.58 Current approaches to these systems employ non-catalytic process, such as organolithium homologations, and/or multiple steps.59–61 Initial at tempts to generate aziridines from α-trifluoromethylated styrene with common metal-mediated and metal-free NT conditions gave no detectable product, which was attributed to the electron-withdrawing nature of the trifluoromethyl group. To combat this mismatched reactivity, Koenigs and co-workers generated a more electron-rich nitrene radical anion via photocatalyzed decomposition of N-tosyl iminoiodinane (Scheme 19A). The system was tolerant of diverse R groups attached to the sulfonyl iminoiodane substrate and of substitutions on the phenyl group of the styrene partner, including electron-withdrawing nitro groups and sensitive thiophene moieties. Unfortunately, ortho substitution was not tolerated, and isolated olefins did not undergo aziridination. Surprisingly, pyridine derivatives failed to undergo aziridination, presumably due to a single-electron transfer (SET) pathway where the pyridine may quench the radical. Control experiments showed that reductive quenchers such as DABCO, triethylamine, and the Hantzsch dihydropyridine inhibited the reaction.
Scheme 19. Photocatalyzed synthesis of trifluoromethylated aziridines by Köenigs.
(A) Selected substrate scope and limitations of methodology.
(B) Proposed mechanism.
Based on further control experiments and DFT calculations, the authors proposed that photoexcited Ru(bpy)3 undergoes a SET (SET1) with starting material 19.1 to yield intermediate 19.2 (Scheme 19B). The 19.2 decomposes to release PhI and an active nitrene radical anion, which undergoes addition to the electron-poor styrene to furnish radical anion 19.3. A second SET (SET2) oxidizes the substrate to zwitterion 19.4 and reforms the ruthenium photocatalyst. Barrierless cyclization of 19.4 gives the desired product 19.5. This methodology offers broad substrate scope with good-to-excellent yields, and more importantly, it opens the use of nitrene radical anions to further developments.
Light-induced generation of nitrene equivalents is a powerful and mild method for aziridination. However, photoexcitation chemistry is not limited to controlled decompositions. Recently Siopa, António, and Afonso demonstrated an inspired alternative in photochemical transformations of pyridinium salts to bicyclic aziridines (Scheme 20).62 Conversion of pyridinium salts to bicyclic aziridines has been known since the early 1970s63; however, Siopa and co-workers sought to scale up this method to access cyclopentenoaziridines 20.1, which reacts with secondary nucleophiles to generate bioactive aminocyclopentenes 20.2 and 20.3. A major challenge in scaling photochemical reactions is the decreasing efficiency of photon penetration with increasing reactor volume (i.e., the Beer-Lambert law of photon attenuation). To overcome this, the group crafted homemade flow reactors of different surface-to-volume ratios and tracked product formation via NMR. Optimization of starting material concentration ultimately generated the desired product 19.4 in up to 3.7 g L−1 h−1, which translates to improvement of ~28-fold, compared with previous batch reports.
Scheme 20.
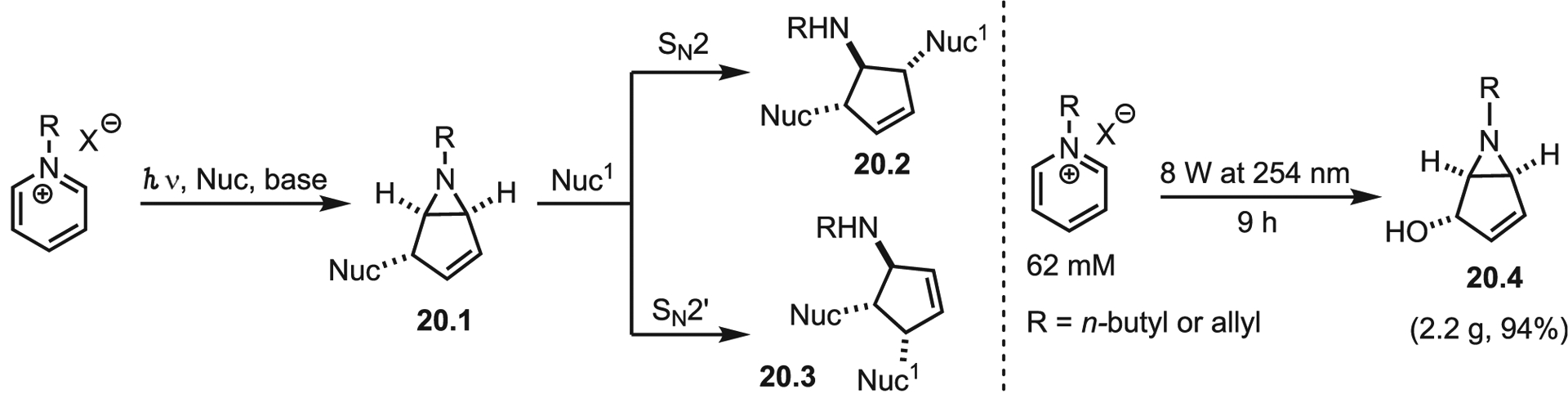
Transformation of pyridinium salts to aminocyclopentenes and results in flow
Electrochemical aziridination
The most common strategy employed for electrochemical aziridination is manipulation of the alkene partner to induce amine attack. A recent example of the complementary nature of this strategy was published by Li and co-workers who achieved the first direct aziridination of triaryl-substituted alkenes.64 Triaryl alkenes are resistant to common NT methodologies due to steric congestion of the aryl substituents hindering approach of the nitrogen source and the energetic penalty of the ring formation, which destroys conjugation in the system.
Using sulfamate 21.1, a feasibility test utilizing graphite felt electrodes was conducted in an undivided cell (Scheme 21) to give the desired aziridine in 57% yield. Switching the base to 2,6-lutidine improved yields significantly. The system was highly tolerant of aryl substitutions to furnish a range of triaryl aziridines in good-to-excellent yields. Sterics did not have much impact on yield, and alkyl alkenes also gave products in moderate-to-good yields. More electron-rich styrenes tended to give lower yields, presumably due to subsequent ring-opening of the aziridine. The proposed mechanism in Scheme 21 involves single-electron oxidation of 21.1 to intermediate 21.3. The amine partner attacks to give radical intermediate 21.4, which undergoes a second oxidation to cation 21.5. Amine cyclization gives desired product 21.2, while lutidine base removes the acidic protons generated during the reaction. The cycle is completed with a two-electron reduction to generate hydrogen. Notably, this system is limited to fluorinated sulfamates as both acetamide and tosylamine failed to give products. Regardless, the work is an excellent example of using electrochemistry to overcome limitations of other NT strategies.
Scheme 21. Li’s work on the electrochemical aziridination of triaryl-substituted alkenes.
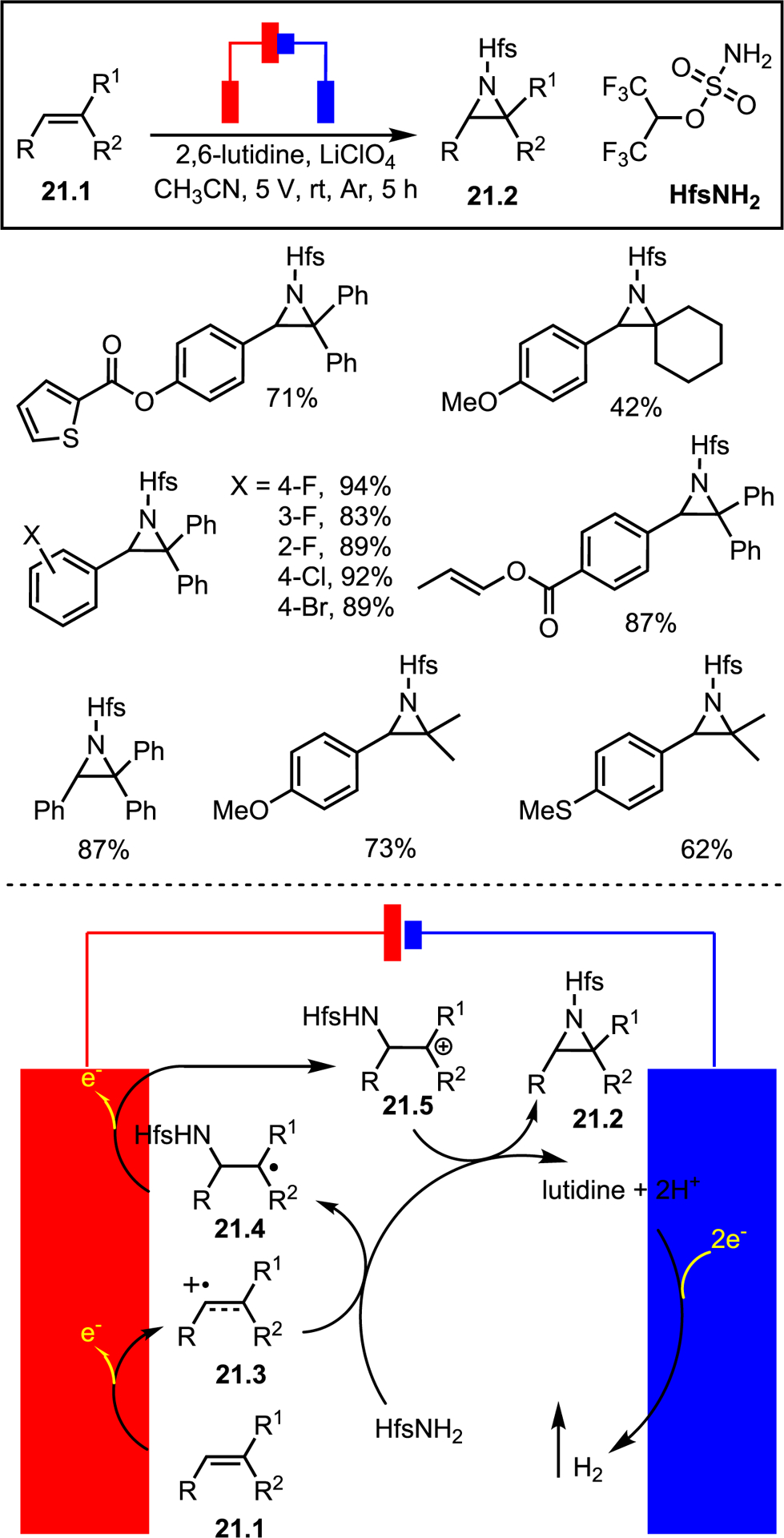
Selected scope (top); proposed mechanism (bottom).
The Wickens group recently expanded upon olefin activation to achieve aziridination.65 The nucleophilicity of over 1 million commercially available primary amines was leveraged to attack a metastable dielectrophile (Scheme 22A). Due to the difficulty of directly forming such species from olefins, they elected to explore thianthrene dicationic adducts. Electrolysis of thianthrene (TT) in the presence of olefin gave two dicationic adducts, which were characterized via X-ray crystallography (Scheme 22B). Subsequent addition of primary amine yielded desired aziridines in a two-step, one-pot reaction. In contrast to conventional dication equivalents (e.g., vicinal halides), clean aziridination was observed. The method boasts a wide substrate scope with various olefins and amines, showing good-to-excellent yields (Scheme 22C). Additionally, TT recovery stands at 98%, suggesting a highly sustainable system.
Scheme 22. Electrochemical aziridination via a dication pool strategy by Wickens.
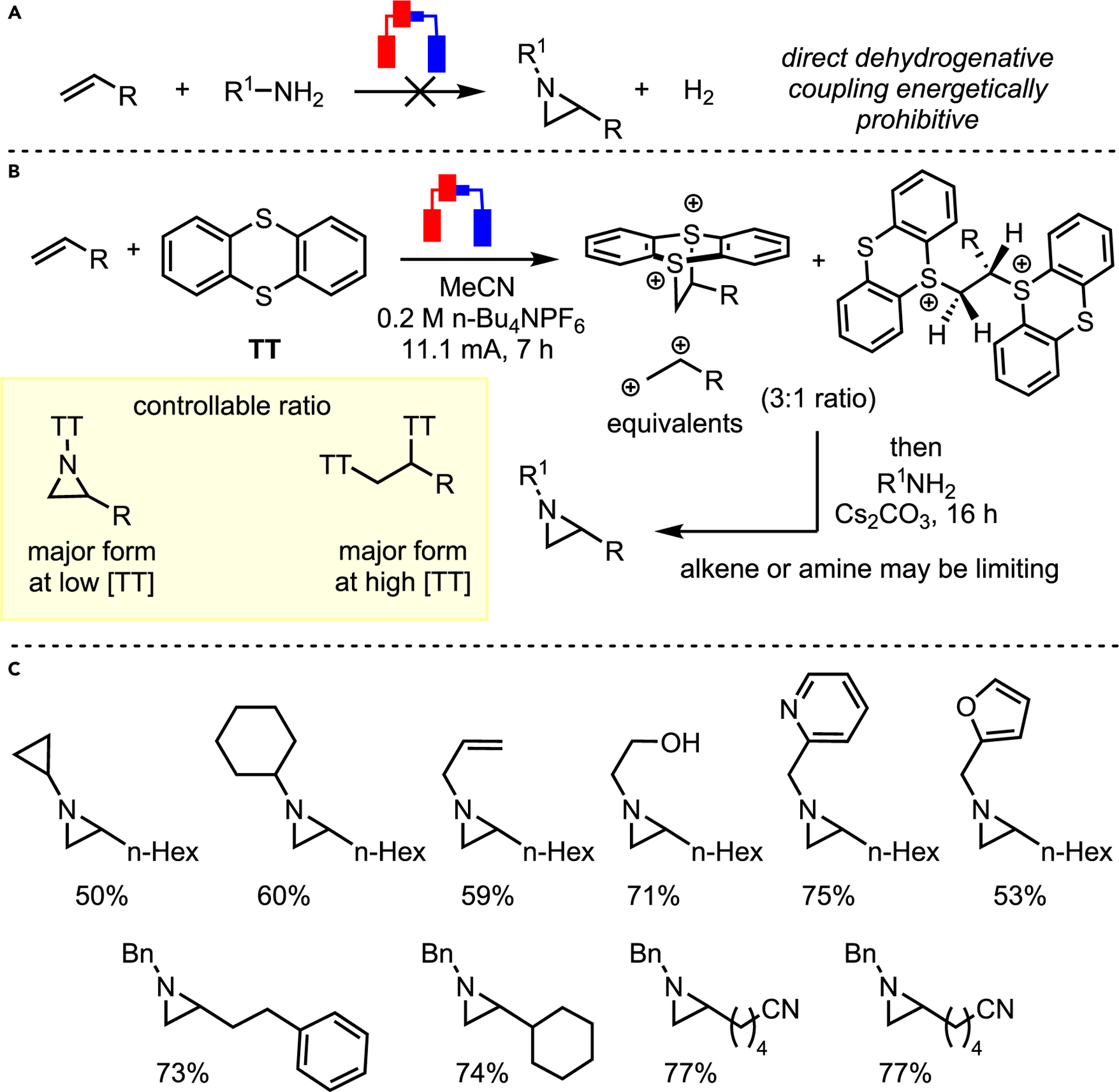
(A) Ideal electrochemical aziridination.
(B) Dication surrogate strategy for aziridination.
(C) Selected scope.
REACTIVITY OF AZIRIDINES
Regio-, chemo- and stereoselective ring-openings of aziridines with diverse nucleophiles under a variety of conditions have been well investigated. The focus in modern aziridine chemistry is on transforming these rings into increasingly complex bioactive motifs. Thus, the following sections will highlight selected aziridine motifs that have either been underutilized in the recent literature or are of utility to the wider community.
1-Azabicyclo[1.1.0]butanes
The 1-azabicyclo[1.1.0]butanes or azabicyclobutanes (ABBs) were first synthesized by Funke in 1969 and have been sporadically used to generate substituted azeti-dines.66 The unique reactivity and utility of these systems stem from the highly reactive N–C3 bond, which breaks in the presence of a suitable nucleophile to yield substituted azetidines. Azetidines have attracted recent attention for their ability to increase bioavailability and metabolic stability and are found in bioactive natural products and synthetically useful catalysts.67 ABBs are readily accessible, despite their high ring strain, via cyclopropanation of azirines or by cyclization of dibromo-propylamines. The previously limited use of these systems is likely due to the narrow scope of past methods. For example, Nagao showed that aniline adds to the C3 carbon of ABB in the presence of superstoichiometric amounts of Lewis acids; however, the same strategy failed with alkyl amines such as morpholine.68
Interest in ABBs has been reignited due to recent advances in the area to develop an “azetidinylation” of amines using Turbo-Hauser amides. The bench-stable hydrobromide salt of 2,3-dibromo-propylamine served as an ABB precursor; addition of phenyllithium generated the ABB intermediate. Treatment of the solution with Turbo-Hauser amides yielded a wide range of 3-aminoazetidine derivatives, including three late-stage pharmaceuticals, in moderate-to-good yields (Scheme 23).69 The authors noted that high yields were dependent on careful control of time, temperature, and concentration.
Scheme 23.
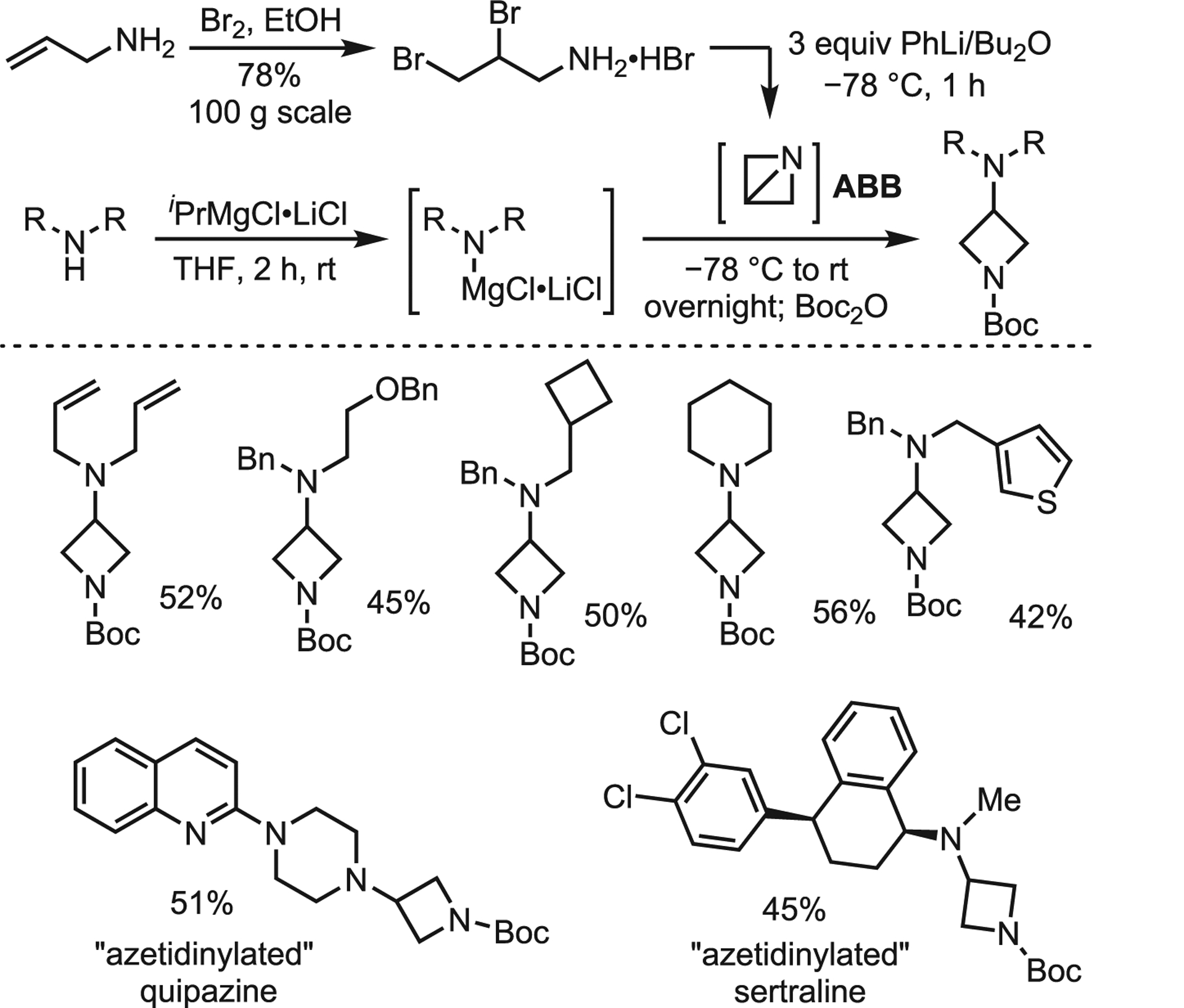
Turbo-Hauser amide addition of ABB and selected scope
Although this method is limited to secondary amines, it tolerates a range of functional groups that highlight the potential of ABBs in modern transformations. This work also inspired Gianatassio and Kadish to functionalize ABBs with carbon nucleophiles. Utilizing the same in situ ABB generation protocol as the Baran lab, carbon-centered nucleophiles (tBuLi, t-BuZnBr, and t-BuMgCl) gave little to no product. However, treatment with Gilman-type nucleophiles furnished a range of alkyl, vinyl, allyl, benzyl, and heteroaryl 3-substituted azetidines (Scheme 24).70 This method was successfully extended to the ring-opening of protected aziridines.
Scheme 24.
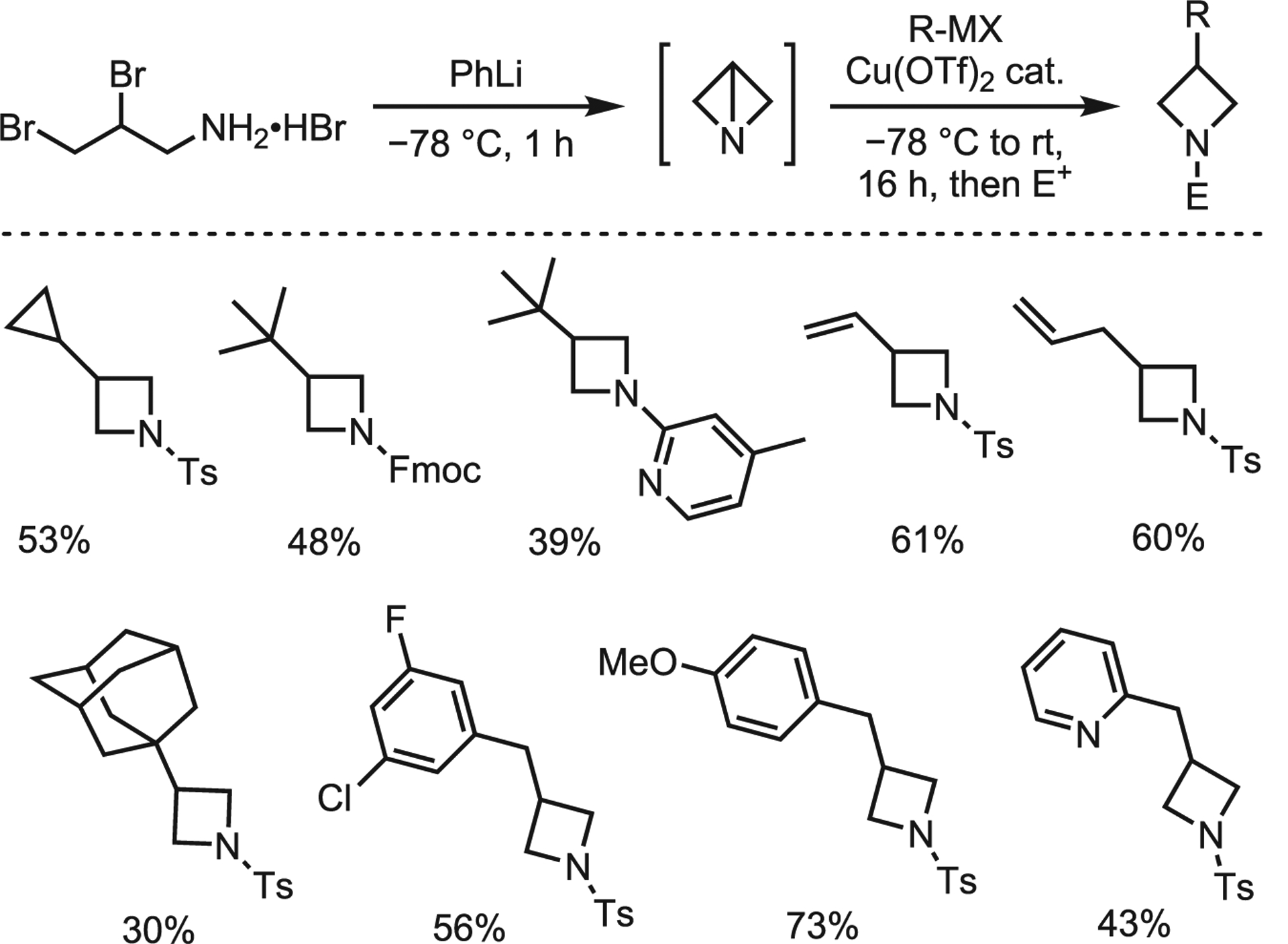
Cuprate coupling with ABBs by Gianatassio and Kadish
Aggarwal and co-workers recently reported a strain-release-driven homologation of boronic esters to give borylated azetidines.71 Driven by an interest in developing new methods for modular azetidine assembly, Aggarwal was able to lithiate ABB and trap the anionic boronic esters. A 1,2-metalate rearrangement was expected to be driven by strain-release; however, the transfer had to be electrophilically induced for good yields (Scheme 25). The initial Li-ABB was trapped as sulfoxide 25.A as a more convenient precursor than the amine salt. The robust system tolerated primary, secondary, and tertiary boronic esters to give a wide range of substituted azetidines with complete enantio- and diastereospecificity (Scheme 25). The boronic ester could also be transformed to other functional groups (Scheme 25, in box).
Scheme 25. Aggarwal’s strain-release-driven synthesis of borylated azetidines from azabicyclobutanes.
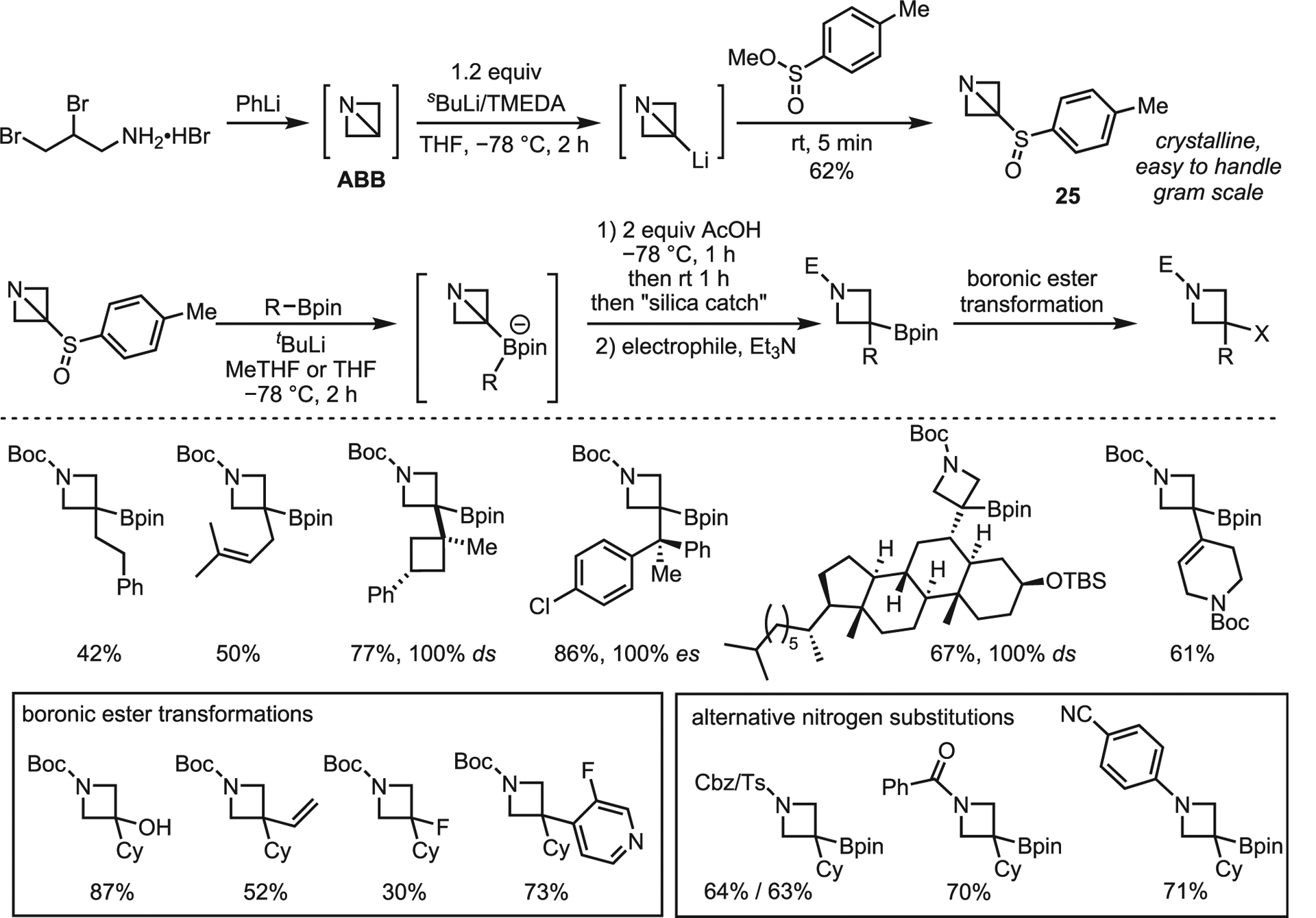
(A) Boronic ester generation from ABB precursors.
(B) Selected scope. es, enantiospecificity; ds, diastereospecificity.
As spirocycles and azetidines are both valuable motifs in medicinal chemistry, Tyler, Noble, and Aggarwal were interested in the generation of spiroazetidines using the Li-ABB system.66,72 Trapping Li-ABB with an electrophile containing a nucleophilic atom gave spirocycle formation (Scheme 26).73 Trifluoroacetic anhydride gave the best yields, high-lighting the importance of the electrophile used for subsequent N-activation. Five- and six-membered fused rings consistently performed best, while smaller four-membered and larger seven-membered ring gave modest-to-good yields.
Scheme 26.
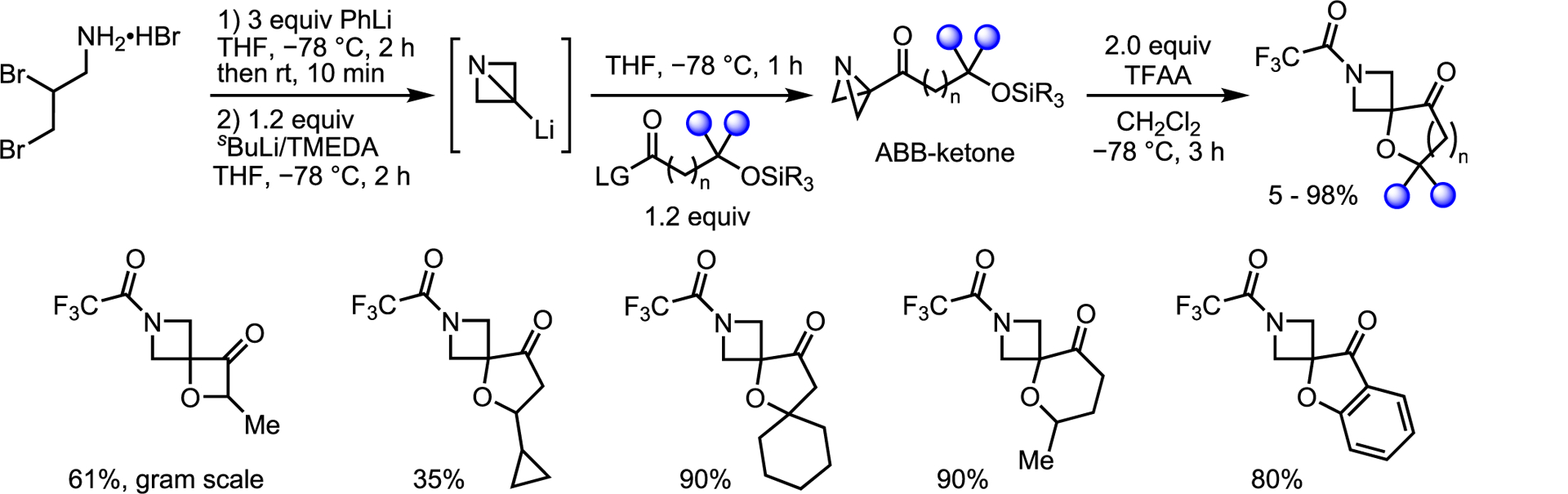
Spirocycle synthesis from ABB precursors
The recent advances in the reactivity and scope of ABBs demonstrates the synthetic relevance of these underexplored systems to modern problems, particularly in the field of medicinal chemistry. Advances in this area are expected to continue in the near future.
Methyleneaziridines (MAs) and aziridinium ylides (AYs)
Unusually strained or reactive aziridines offer opportunities to access stereochemically complex and densely functionalized amines, including N-heterocycles of varying ring sizes. Two underexplored platforms include MAs72 and the use of aziridinium ylides (AYs) as reactive intermediates. While activation of the aziridine nitrogen through quaternization to aziridinium ions has been well studied, the potential to transform AY into diverse amines by tuning reactivity has only begun to be tapped.73 MAs contain an exocyclic double bond that introduces a trigonal sp2-center into the aziridine ring. This feature enhances ring strain by 12–13 kcal mol−1 over a typical aziridine. Early methods to access MAs were generally harsh with limited substrate scope.74,75
Building on work by the Blakey and Robertson groups, the Schomaker group utilized transition metal-catalyzed aziridinations of homoallenic sulfamates and carbamates to generate bicyclic MAs (Scheme 27). The bicyclic system is rigid, limiting N-inversion and enabling stereoselective ring expansions to a host of valuable chemical entities. For example, endocyclic MAs 27.1 could be converted to S, N, O-containing cyclooctynes (SNO-OCTs) derivatives that have been utilized as tunable “orthogonal biorthogonal” labeling reagents76; such systems would be very challenging to make via alternative routes. Exocyclic MAs 27.2 further showcase the utility of these intermediates for accessing new amine chemical space. Heat or treatment with a Lewis acid opens exocyclic MAs to 2-amidoallyl cations. Recently, the Schomaker lab demonstrated a biomimetic 2-imino-Nazarov cyclization utilizing eneallene precursors to generate a corresponding 2-amidoallyl cation intermediate that furnishes [5,7]-fused sulfamate derivatives.77
Scheme 27.
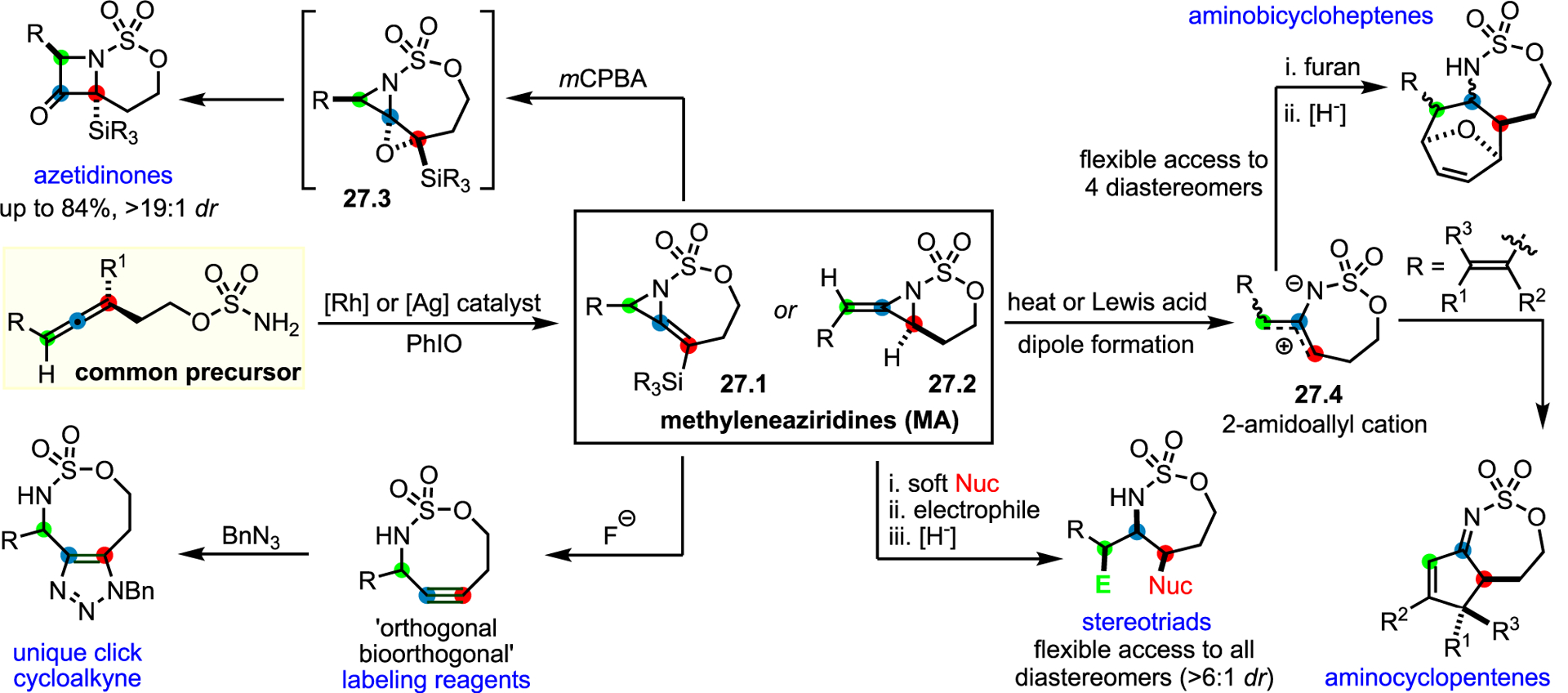
Methyleneaziridines (MAs) as intermediates to furnish diverse N-heterocycles
Finally, the innate reactivity of exocyclic MAs was exploited to construct “all-heteroatom stereotriad” motifs that contain three contiguous heteroatom-bearing stereocenters. The versatility of this platform enables the position of the nitrogen in the stereotriad, the identity of the heteroatoms (N, O, F, Cl, Br, and S), and even the relative stereochemistry to be predictably tuned. Intriguingly, the lab recently demonstrated the ability to use electrophilic fluorine to generate C–F containing amine-containing stereotriads.78
The Schomaker group also explored another mode of reactivity of bicyclic MAs, where the unexpected nucleophilicity of the nitrogen lone pair can engage a metal-supported carbene to form an intermediate AY; a detailed report on these systems has been recently published.79 While ammonium, azomethine, pyridinium, and triazolium ylides are fairly common, their AY counterparts are much rarer. AYs are conveniently generated from metal-supported carbenes, but the challenges in controlling product distribution have thus far limited investigations of these systems. Cheletropic extrusion and hydride shifts have been reported to compete with desired product formation; however, the Schomaker lab has recently made significant progress in overcoming these issues.80–83 For example, a C5H11-substituted MA engages a Rh-supported carbene to form an AY; the additional strain induced by the exocyclic olefin biases the system toward ring-opening via a concerted asynchronous [2,3]-Stevens rearrangement to yield densely functionalized fused azetidines (Scheme 28).
Scheme 28.
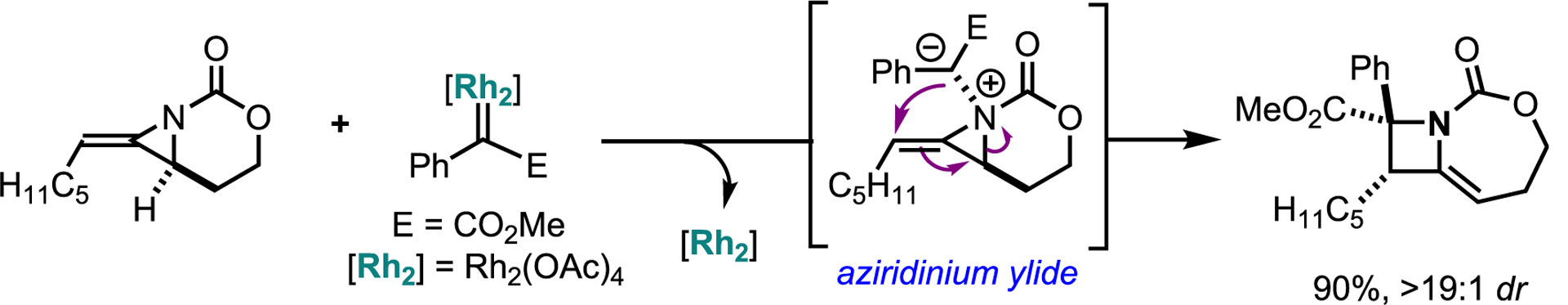
Ring expansion of methyleneaziridine via a [2,3]-Stevens rearrangement of an aziridinium ylide formed from carbene transfer Scheme
Ring expansions involving AY intermediates are not limited to MAs. The Schomaker group also showed that simple bicyclic aziridines can undergo ring expansion to access medicinally relevant dehydropiperidines (Scheme 29).84 Use of Davies’ styrenyl diazoacetate under rhodium catalysis gave products in good yields and excellent dr. The transformation tolerated a range of alkyl and aryl substituents giving generally good-to-excellent yields and dr (Scheme 28). The diversity of aziridines, carbene precursors, catalysts, and reaction conditions that can be investigated for efficient access to other novel amine scaffolds is sure to inspire other new methods based on AY intermediates.
Scheme 29.
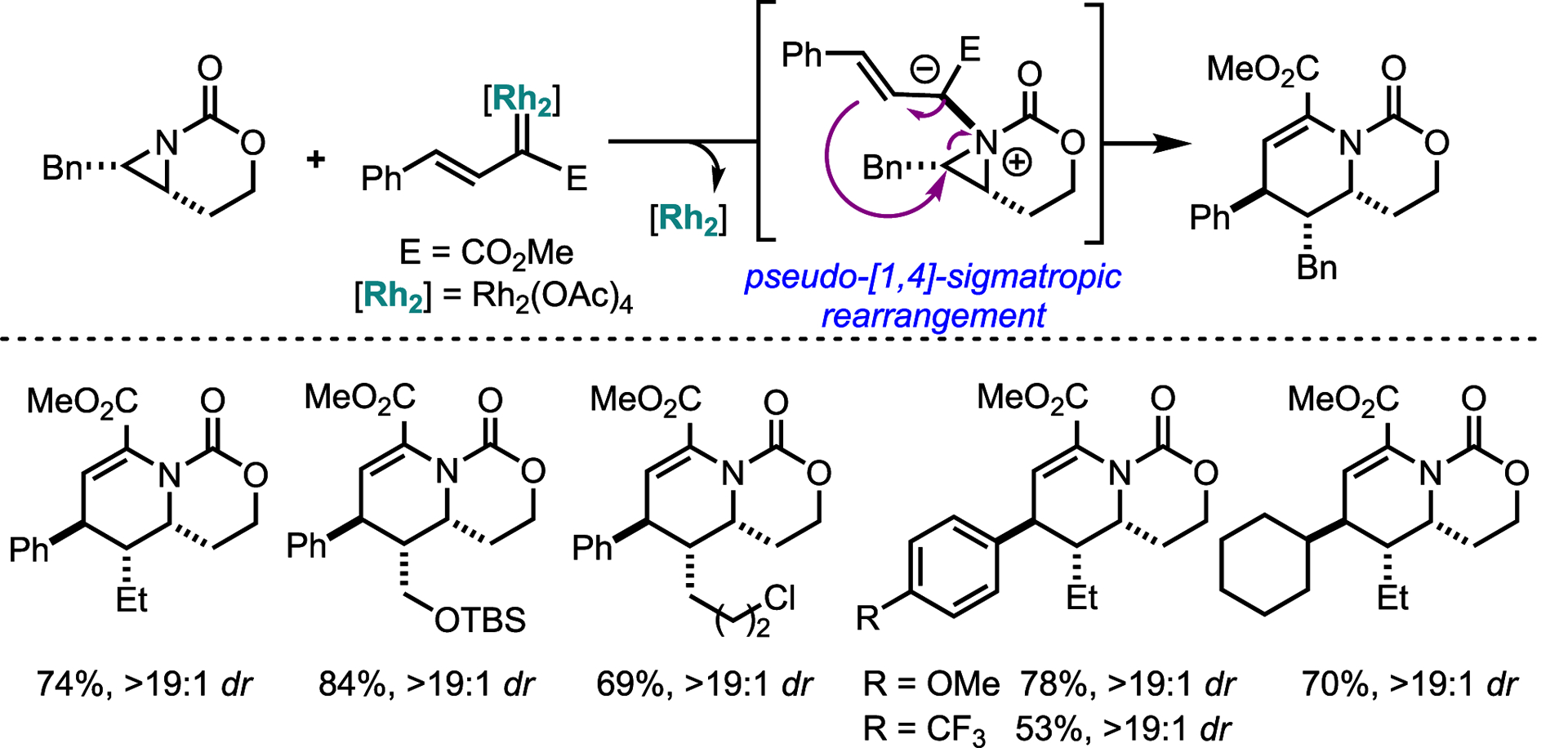
Dehydropiperidines from the carbene transfer between unbiased bicyclic aziridines and Rh-supported vinyl carbenes
APPLICATIONS OF AZIRIDINES
The preceding sections focused on general aspects of aziridine chemistry and recent advances in making and manipulating the ring. However, what is seldom discussed outside introductory paragraphs are current and future outlooks for applications of aziridines. The following sections discuss aziridines found in nature and trends in their application as medicinally relevant core structures.
Aziridines in nature
Aziridine-containing natural products are rare. Indeed, a recent study found that while epoxides are found in ~3.6% of known natural products, aziridines account for less than 0.6%.85 To our knowledge, no new aziridine-containing natural product has been discovered in the last 5 years. However, interest in aziridine natural products has been maintained over the past 60 years as the aziridine moiety has been shown to be essential to the biological function of every aziridine-containing natural product discovered thus far, including many that possess valuable antitumor, antimicrobial, or antibiotic activities.86 Nature, it seems, values quality over quantity when it comes to aziridines (Figure 11).
Figure 11.
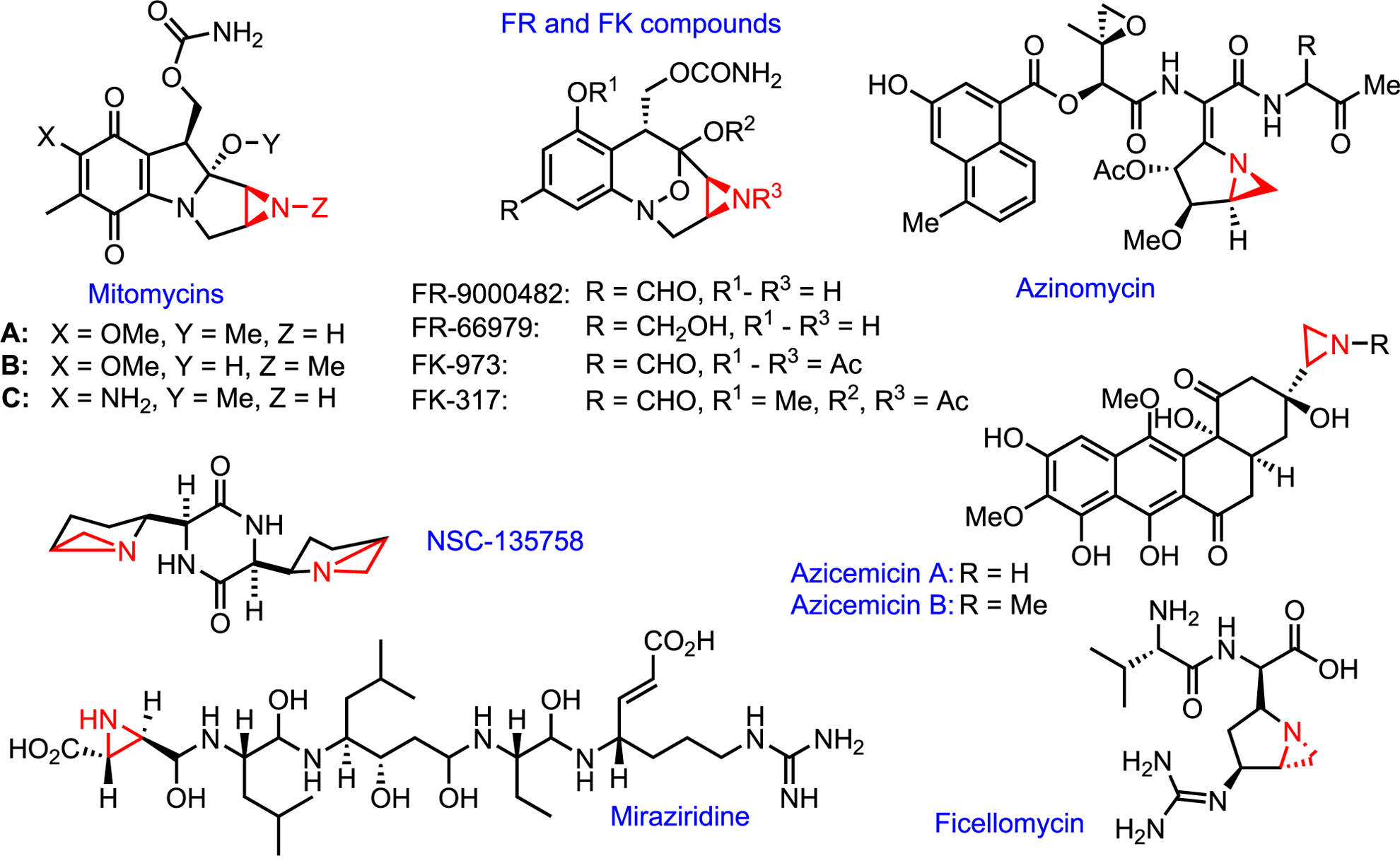
Aziridine-containing natural products
Aziridines are powerful alkylating agents that possess an inherent, though often non-specific, cytotoxicity. Nature uses aziridine’s tendency toward electrophilic ring-opening in two distinct ways. The first is straightforward utilization of the aziridine as an electrophilic warhead. The pyrrolebenzimidazole (PBI) class of natural products illustrates this activity (Scheme 30). PBIs are known DNA alkylating agents that contain an aziridine ring attached directly to a quinone. Reduction of the quinone under cellular conditions activates the aziridine via protonation, which invites attack by the phosphate backbone of DNA phosphate, leading to single-stranded DNA cleavage. Structure activity relationship (SAR) studies were carried out by Skibo and co-workers to demonstrate the necessity of the aziridine ring.87 Despite its apparently non-selective mechanism of action, the high potency against melanoma cell lines and lack of activity against leukemia are noteworthy.
Scheme 30.
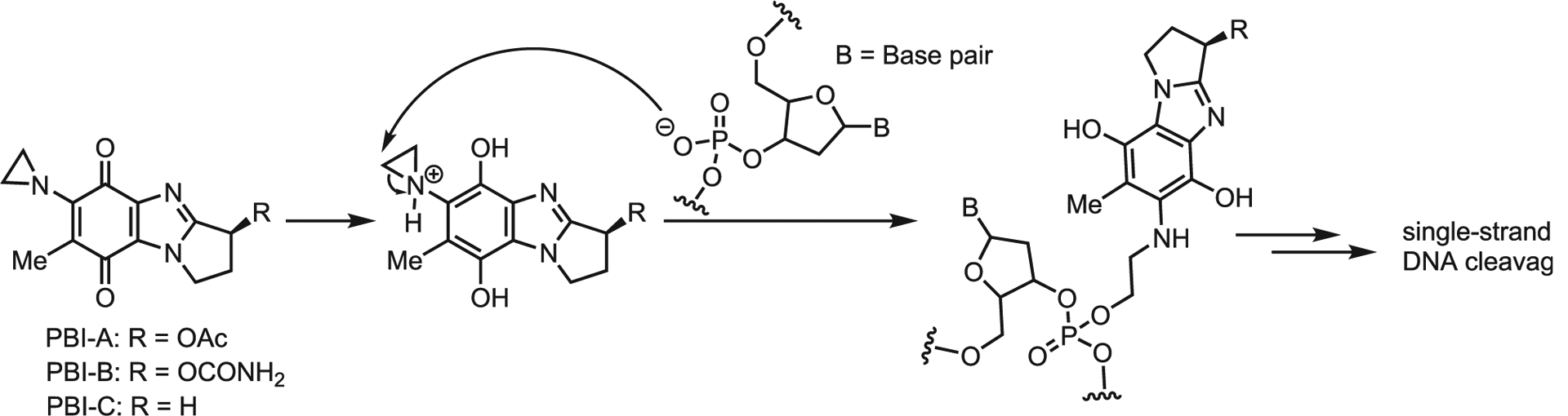
PBI proposed mechanism of action
Utilization of aziridines as “warheads” is the most common application in medicinal chemistry.88 A compelling example of the power of this strategy is the case of nitrogen mustards. Nitrogen mustards undergo intramolecular attack to generate a cationic aziridine intermediate with greatly enhanced electrophilicity (Scheme 31A). These systems are believed to operate by covalently cross-linking DNA strands to prevent cell duplication. Nitrogen mustards were the first effective anticancer agents synthesized and have enjoyed wide interest over the past 70 years, yielding several drugs and prodrugs (Scheme 31B). Major efforts in prodrug formulation have resulted in antibody-directed enzyme prodrug therapy (ADEPT), gene-directed pro-drug therapy (GDEPT), nitrogen mustards that target the central nervous system, and many other applications.
Scheme 31. Examples of aziridines used in medicinal chemistry.
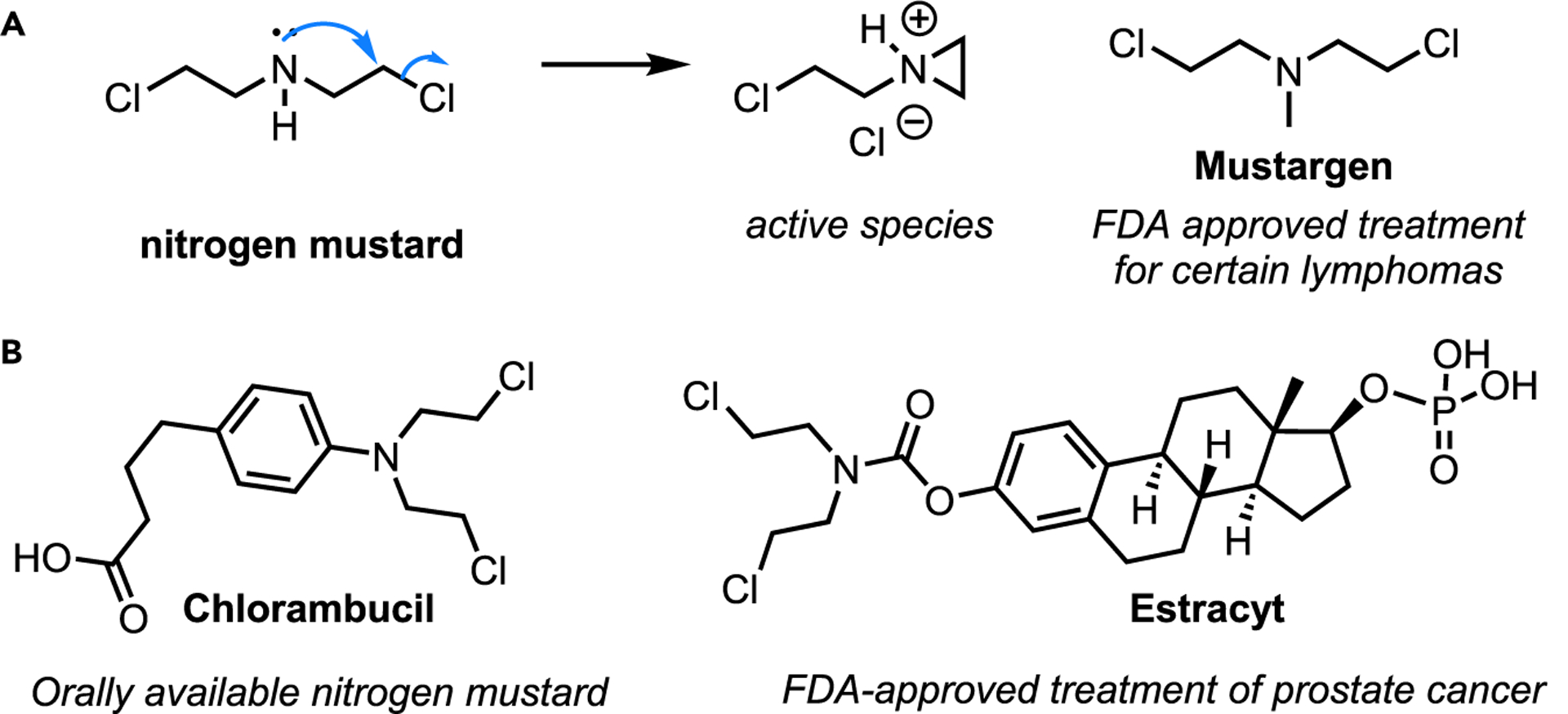
(A) Nitrogen mustard mechanism of action.
(B) Commercially available drugs.
A second way nature utilizes the aziridine ring is as a “spring release” mechanism where the aziridine is not directly attacked, but it opens as result of a secondary reaction to release molecular strain. The mitomycins, which are among the most well-studied aziridine natural products, are an excellent example of this mode of action (Scheme 32). First isolated from soil extracts of Streptomyces verticillatus, mitomycins display antitumor and antibiotic activity and are a rare example of an aziridine natural product possessing both high potency and high selectivity. Mitomycins are also noteworthy as one of the few naturally occurring antibiotics to present the same mode of action both in vitro and in vivo. Similar to the structurally related PBIs, the proposed mechanism of action of mitomycins begins with bioreductive activation by in situ conversion to the hydroxyquinone 31.2. Elimination to form an indoloaziridine intermediate 31.3 leads to 31.4, which opens at the aziridine ring to furnish the potent acceptor 31.5. The purine nitrogen of DNA is then alkylated prior to a second elimination that yields the DNA crosslinked intermediate 31.8. Final reoxidation to the quinone 31.9 is ultimately responsible for arrest of cell duplication (Scheme 32). Despite their similar activation steps, PBIs and mitomycin react quite differently; mitomycin derivatives display selective reactivity with nitrogen base pairs, but PBI systems alkylate phosphate backbones.
Scheme 32.
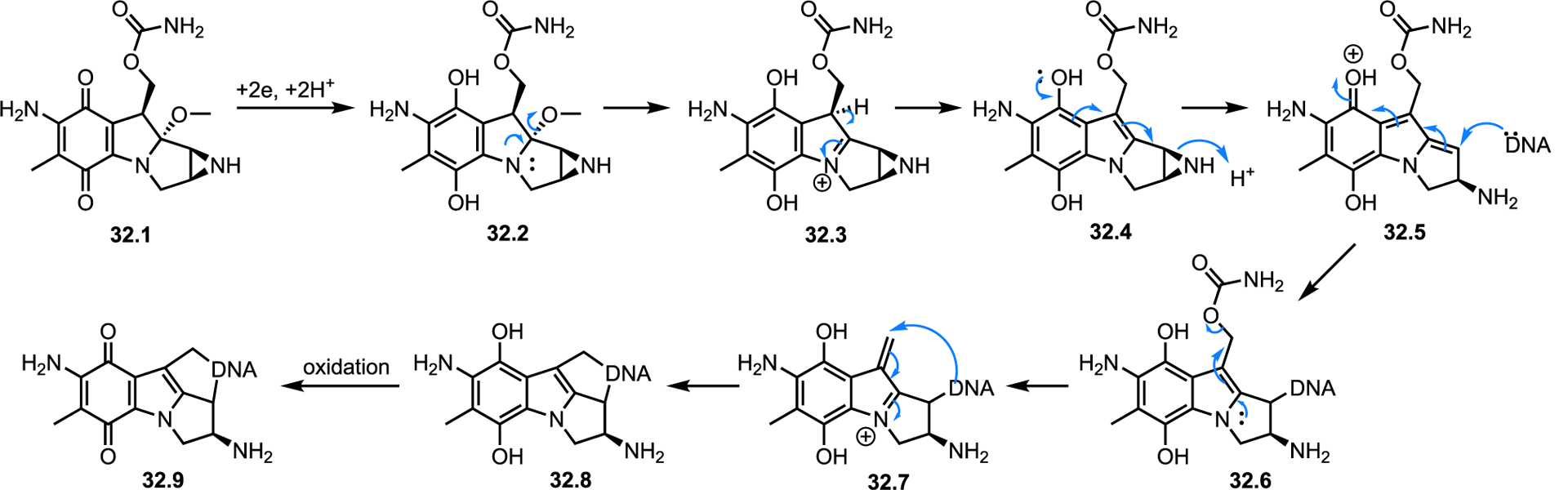
Proposed mechanism of DNA alkylation by mitomycin
When thinking of natural products, most envision structurally complex heterocycles with valuable biological activities. However, “simpler” aziridine alkaloids are commonly found in surprising places. Ethyleneimine has been detected in various foodstuffs including bakers’ yeast and the volatile flavoring constituents of cooked chicken, beef, and pork.89 More complex aziridines have been found in various plant sources. For example, aziridine-2-carboxylic acid has been isolated from mushrooms, and related compounds have found use as protease inhibitors.90 Other natural sources of aziridines in foodstuffs include onion bulbs, flue-cured tobacco, and bog rhubarb; selected structures are given in Figure 12.91,92
Figure 12.
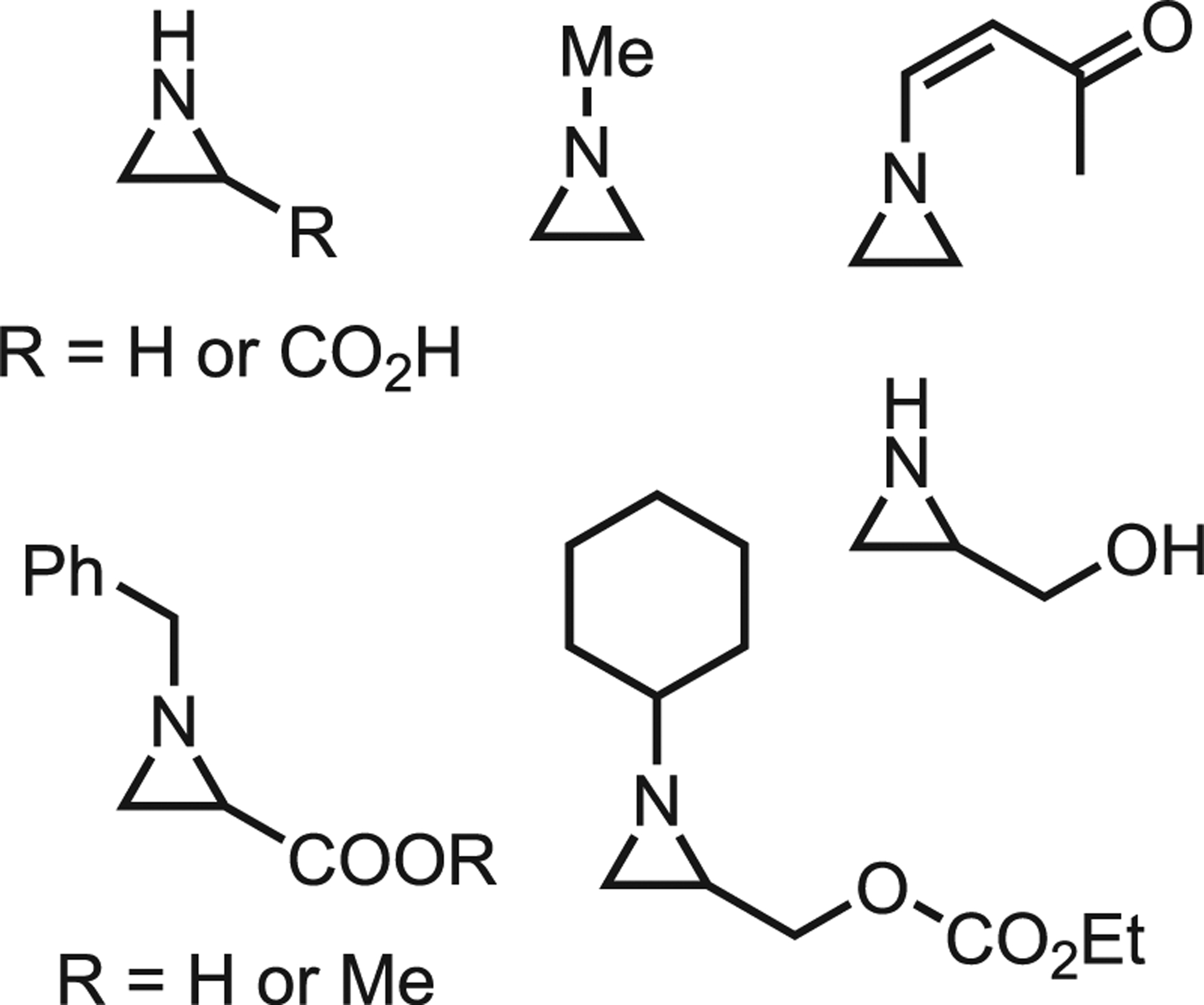
Naturally occurring aziridines
Despite the synthesis of a majority of known aziridine natural products and numerous SAR studies performed, interest in aziridines as intermediates has steadily increased (Figure 1).93 This is due to the importance of aziridines in medicinal chemistry and the increasing reliance on aziridine-based strategies to target structurally complex end products.
Medicinal relevance of aziridines
In 1900, infectious disease was the leading cause of death in the world.94 Unfortunately, infectious disease has re-emerged as an extant threat due to increasing resistance to current therapies and is now the second leading cause of death in the world, accounting for 1/5 of all deaths, and the leading cause of death in children under 5 years.95 Cancer, an umbrella term for at least 277 unique pathologies, is another leading cause of death worldwide, accounting for an estimated 9.6 million deaths in 2018.96
Aziridines possess potent antibiotic and anticancer properties. The re-emergence of bacterial threats and the persistence of cancer have led to increased incorporation of aziridine analogs in SAR campaigns. For example, Znati and co-workers treated (−)-deltoin, a natural hepatoprotective isolated from F. lueta, with various azidobenzenes to yield racemic N-aryl aziridinated analogs with good activity against multiple cancer cell lines (Scheme 33).97
Scheme 33.
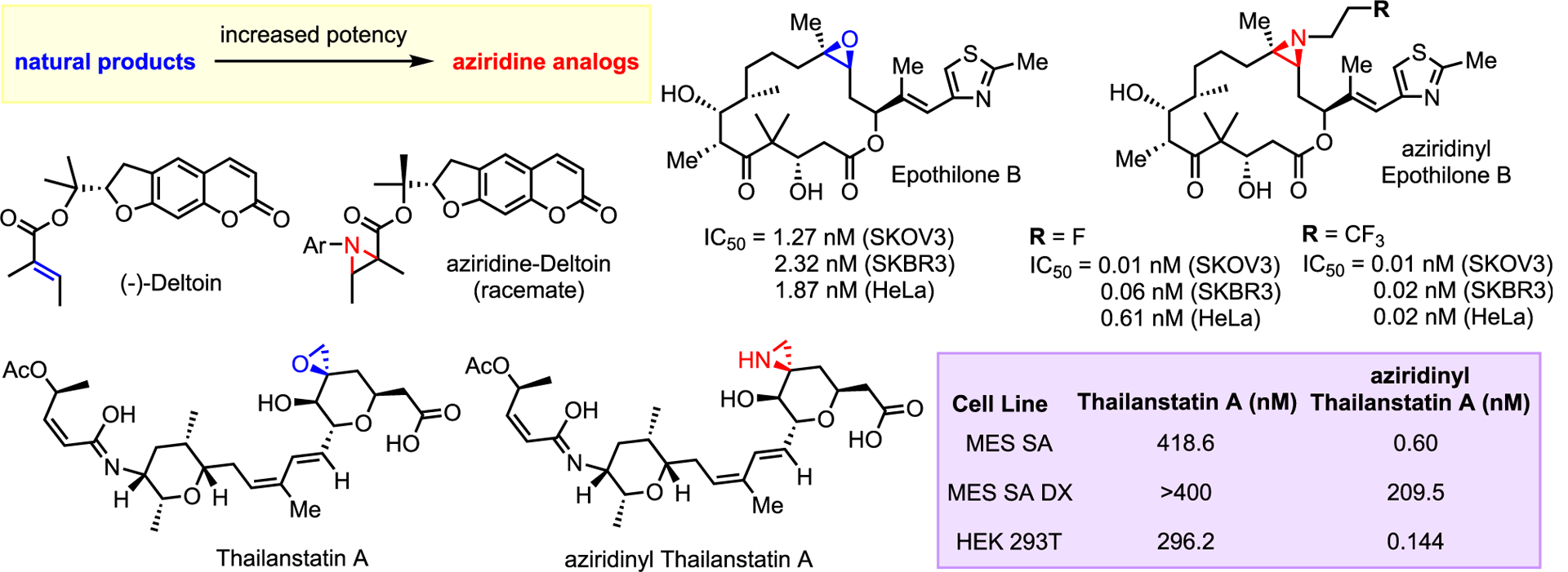
Aziridine analogs of bioactive molecules and natural products
The Nicolaou lab recently published an SAR study on epothilones, a class of well-studied macrocyclic structures that demonstrate potent cytotoxicity.98 One member, ixabe-plione, is used clinically for the treatment of breast cancer. Nicolaou and co-workers generated over 50 stereodefined analogs of epothilone B, focusing largely on aziridinyl analogs. In general, the aziridine moiety was found to be favorable for bioactivity, and in vivo assays showed that a significant portion of the generated compounds were highly potent with nanomolar IC50 values against SKBR3, SKOV3, HeLa, MDR MES SA DXE, and MES SA DX (human uterine sarcoma cell lines with marked multidrug resistance [MDR]) cancer cell lines. The most potent compounds exhibited an impressive 9,280-fold greater activity than the original epothilone B (Scheme 33). As the aziridine ring is not involved in the mechanism of cytotoxicity in this case, the activity enhancement may be due to modulation of favorable ADME (absorption, distribution, metabolism, and excretion) properties via modification of the N-substituent.
Increasing potency by exchanging an epoxide with an aziridine was also seen by Ghosh and co-workers in spliceostatins.99 Replacement of the [3,6] fused epoxide with an aziridine in thailastatin A (Scheme 33) resulted in an increase in cytotoxicity to IC50 of 0.6 nM against MES SA cell line. Unfortunately, the authors did not explore the effect of N-substitution but did note that the R configuration of the three-membered ring was essential to maintain potency.
Unique mechanisms of action are also a standout attribute of some aziridine systems. Mitomycin C was shown to enhance the efficacy of programmed death ligand 1 (PD-L1). PD-L1 immune checkpoint inhibitors are a promising class of therapeutics for treating cancer. Luo recently determined that mitomycin C works synergistically with PD-L1 inhibitors via upregulating the extracellular signal-regulated kinase (ERK) pathway, resulting in increased PD-L1 expression.100 This combination treatment was found to be more effective than individual treatments and retarded tumor growth and prolonged overall survival in a mouse model. Additionally, in April 2020, the Food and Drug Administration (FDA) approved the first therapy for treatment of low-grade upper tract urothelial cancer: a mitomycin gel, sold as Jelmyto. These recent applications of aziridine natural products are significant; the formerly unknown mechanisms of action for mitomycin C suggest other aziridines potentially possess hitherto unknown activity and effects.
Viral infections are also a major source of concern for public health and safety.101 In contrast to antibiotic strategies, antiviral strategies have largely focused on targeted countermeasures against specific viruses. Ethyleneimine and N-acetyl ethylenimine have been used for decades to inactivate viruses from several different families and for vaccine production without impairing the enzymatic or serological properties of most proteins. However, no aziridine-containing antiviral agents have been reported within the last 5 years to our knowledge.
A promising antiviral strategy is to target key proteases necessary in virus replication. At the time of writing, Pfizer announced promising drug candidate Paxlovid for combating SARS-CoV-2, which operates via the inhibition of a cystine protease. Recently, aziridine-containing compounds have been identified as selective cystine protease inhibitors,102 suggesting a promising area of expansion.103 However, the area is poorly explored, and more potent analogs and novel mechanisms of actions are needed to address the lack of broad-spectrum antivirals and combat the emerging “antibiotic resistance crisis.” Aziridines hold great potential to aid in addressing these issues. The noted increase in potency of natural product activities by exchanging epoxide for aziridine raises tantalizing questions. What about the other 4% of epoxide-containing natural products? Could potent and selective drug candidates be just an aziridination away? With the recent reported successes and increasing need in these areas, these questions certainly warrant further investigation.
CONCLUDING REMARKS
The examples highlighted in this review show the synthetic utility and medicinal relevance of aziridines. Aziridines offer unparalleled opportunities for new methodology development and access to unexplored chemical space. The examples presented give an overview of the strategies published in recent years and highlight their significant merits and disadvantages. As the number of nitrogen atoms in agrochemicals and pharmaceuticals increases, wider applications of these compounds will go hand in hand with further development of more robust and milder synthetic methods. Recent trends indicate a growing awareness that although aziridines may not be the final frontier, they are the next frontier; aziridine scaffolds will become more prevalent as the scientific community attempts to attain sustainable development during their endeavors to identify new materials, medicines, and agrochemicals.
THE BIGGER PICTURE.
Aziridines are found in natural products, active pharmaceutical ingredients (APIs), and polymers. While well-established methods are reliable, they are generally at odds with modern considerations for sustainability and safety and often involve forcing or environmentally unfriendly conditions. Progress toward the development of milder and greener transformations requires an understanding of the challenges presented by the aziridine and a thorough knowledge of past and present developments to formulate new strategies that embody the ideals of sustainability, moving forward.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- 1.Sweeney JB (2002). Aziridines: epoxides’ ugly cousins? Chem. Soc. Rev 31, 247–258. 10.1039/B006015L. [DOI] [PubMed] [Google Scholar]
- 2.Dermer OC, and Ham GE (1969). Formation of the aziridine ring. In Ethylenimine and Other Aziridines, Chemistry and Applications (Academic Press; ), pp. 1–86. [Google Scholar]
- 3.Sienel G, Rieth R, and Rowbottom KT (2000). Epoxides. In Ullmann’s Encyclopedia of Industrial Chemistry (Wiley-VCH; ), pp. 139–154. [Google Scholar]
- 4.Goodman LS, Wintrobe MM, Dameshek W, Goodman MJ, Gilman A, and McLennan MT (1984). Nitrogen mustard therapy. Use of methyl-bis(beta-chloroethyl) amine hydrochloride and Tris(beta-chloroethyl)amine hydrochloride for Hodgkin’s disease, lymphosarcoma, leukemia and certain allied and miscellaneous disorders. JAMA 251, 2255–2261. 10.1001/jama.1984.03340410063036. [DOI] [PubMed] [Google Scholar]
- 5.Tanner D (1994). Chiral aziridines—their synthesis and use in stereoselective transformations. Angew. Chem. Int. Ed. Engl. 33, 599–619. 10.1002/anie.199405991. [DOI] [Google Scholar]
- 6.Ferraris D, Drury WJ, Cox C, and Lectka T (1998). “Orthogonal” Lewis acids: catalyzed ring opening and rearrangement of acylaziridines. J. Org. Chem 63, 4568–4569. 10.1021/jo980558d. [DOI] [Google Scholar]
- 7.Degennaro L, Mansueto R, Carenza E, Rizzi R, Florio S, Pratt LM, and Luisi R (2011). Nitrogen dynamics and reactivity of chiral aziridines: generation of configurationally stable aziridinyllithium compounds. Chemistry 17, 4992–5003. 10.1002/chem.201003424. [DOI] [PubMed] [Google Scholar]
- 8.Luisi R, Capriati V, Florio S, Di Cunto P, and Musio B (2005). Synthesis and lithiation of oxazolinylaziridines: the N-substituent effect. Tetrahedron 61, 3251–3260. 10.1016/j.tet.2005.01.045. [DOI] [Google Scholar]
- 9.Cardoso AL, and Pinho e Melo TMVD (2012). Aziridines in formal [3+2] cycloadditions: synthesis of five-membered heterocycles. Eur. J. Org. Chem 33. n/a–n/a. 10.1002/ejoc.201200406. [DOI] [Google Scholar]
- 10.Tang S, Zhang X, Sun J, Niu D, and Chruma JJ (2018). 2-Azaallyl anions, 2-Azaallyl cations, 2-Azaallyl radicals, and azomethine ylides. Chem. Rev 118, 10393–10457. 10.1021/acs.chemrev.8b00349. [DOI] [PubMed] [Google Scholar]
- 11.Singh GS (2019). Chapter four - advances in synthesis and chemistry of aziridines. In Advances in Heterocyclic Chemistry, E.F.V. Scriven and C.A. Ramsden, eds. (Academic Press; ), pp. 245–335. [Google Scholar]
- 12.Pellissier H (2014). Recent developments in asymmetric aziridination. Adv. Synth. Catal 356, 1899–1935. 10.1002/adsc.201400312. [DOI] [Google Scholar]
- 13.Roma E, Tosi E, Miceli M, and Gasperi T (2018). Asymmetric organocatalytic aziridination: recent advances. Asian J. Org. Chem 7, 2357–2367. 10.1002/ajoc.201800431. [DOI] [Google Scholar]
- 14.Frankowski S, Bojanowski J, Saktura M, Romaniszyn M, Drelich P, and Albrecht Ł (2017). Organocatalytic synthesis of cis-2,3-aziridine aldehydes by a postreaction isomerization. Org. Lett 19, 5000–5003. 10.1021/acs.orglett.7b02634. [DOI] [PubMed] [Google Scholar]
- 15.Vesely J, Ibrahem I, Zhao GL, Rios R, and Córdova A (2007). Organocatalytic enantioselective aziridination of α,β-unsaturated aldehydes. Angew. Chem. Int. Ed. Engl 46, 778–781. 10.1002/anie.200603810. [DOI] [PubMed] [Google Scholar]
- 16.Arai H, Sugaya N, Sasaki N, Makino K, Lectard S, and Hamada Y (2009). Enantioselective aziridination reaction of α,β-unsaturated aldehydes using an organocatalyst and tert-butyl N-arenesulfonyloxycarbamates. Tetrahedron Lett. 50, 3329–3332. 10.1016/j.tetlet.2009.02.095. [DOI] [Google Scholar]
- 17.da Silva AR, dos Santos DA, Paixão MW, and Corrêa AG (2019). Stereoselective multicomponent reactions in the synthesis or transformations of epoxides and aziridines. Molecules 24, 630. 10.3390/molecules24030630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mukherjee M, Zhou Y, Dai Y, Gupta AK, Pulgam VR, Staples RJ, and Wulff WD (2017). Catalyst-controlled multicomponent aziridination of chiral aldehydes. Chemistry 23, 2552–2556. 10.1002/chem.201605955. [DOI] [PubMed] [Google Scholar]
- 19.Zhou Y, Gupta AK, Mukherjee M, Zheng L, and Wulff WD (2017). Multicomponent catalytic asymmetric synthesis of transaziridines. J. Org. Chem 82, 13121–13140. 10.1021/acs.joc.7b02184. [DOI] [PubMed] [Google Scholar]
- 20.Zhou Y, Mukherjee M, Gupta AK, and Wulff WD (2017). Multicomponent cis- and trans-aziridinatons in the syntheses of all four stereoisomers of sphinganine. Org. Lett 19, 2230–2233. 10.1021/acs.orglett.7b00697. [DOI] [PubMed] [Google Scholar]
- 21.Li X, Feng T, Li D, Chang H, Gao W, and Wei W (2020). Straightforward approach toward multifunctionalized aziridines via catalyst-free three-component reactions of α-diazoesters, nitrosoarenes, and alkynes. J. Org. Chem 85, 9538–9547. 10.1021/acs.joc.0c00368. [DOI] [PubMed] [Google Scholar]
- 22.Ravindra S, Irfana Jesin CP, Shabashini A, and Nandi GC (2021). Recent advances in the preparations and synthetic applications of oxaziridines and diaziridines. Adv. Synth. Catal 363, 1756–1781. 10.1002/adsc.202001372. [DOI] [Google Scholar]
- 23.Williamson KS, Michaelis DJ, and Yoon TP (2014). Advances in the chemistry of oxaziridines. Chem. Rev 114, 8016–8036. 10.1021/cr400611n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Farndon JJ, Young TA, and Bower JF (2018). Stereospecific alkene aziridination using a bifunctional amino-reagent: an aza-Prilezhaev reaction. J. Am. Chem. Soc 140, 17846–17850. 10.1021/jacs.8b10485. [DOI] [PubMed] [Google Scholar]
- 25.Johnson SL, and Hilinski MK (2019). Organocatalytic olefin aziridination via iminium-catalyzed nitrene transfer: scope, limitations, and mechanistic insight. J. Org. Chem 84, 8589–8595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng Q-Q, Zhou Z, Jiang H, Siitonen JH, Ess DH, Zhang X, and Kürti L (2020). Organocatalytic nitrogen transfer to unactivated olefins via transient oxaziridines. Nat. Catal 3, 386–392. 10.1038/s41929-020-0430-4. [DOI] [Google Scholar]
- 27.Ju M, and Schomaker JM (2021). Nitrene transfer catalysts for enantioselective C–N bond formation. Nat. Rev. Chem 5, 580–594. 10.1038/s41570-021-00291-4. [DOI] [PubMed] [Google Scholar]
- 28.Kuijpers PF, van der Vlugt JI, Schneider S, and de Bruin B (2017). Nitrene radical intermediates in catalytic synthesis. Chemistry 23, 13819–13829. 10.1002/chem.201702537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Evans DA, Faul MM, Bilodeau MT, Anderson BA, and Barnes DM (1993). Bis(oxazoline)-copper complexes as chiral catalysts for the enantioselective aziridination of olefins. J. Am. Chem. Soc 115, 5328–5329. 10.1021/ja00065a068. [DOI] [Google Scholar]
- 30.Li Z, Conser KR, and Jacobsen EN (1993). Asymmetric alkene aziridination with readily available chiral diimine-based catalysts. J. Am. Chem. Soc 115, 5326–5327. 10.1021/ja00065a067. [DOI] [Google Scholar]
- 31.Kwart H, and Khan AA (1967). Copper-catalyzed decomposition of benzenesulfonyl azide in cyclohexene solution. J. Am. Chem. Soc 89, 1951–1953. 10.1021/ja00984a035. [DOI] [Google Scholar]
- 32.Park Y, Kim Y, and Chang S (2017). Transition metal-catalyzed C–H amination: scope, mechanism, and applications. Chem. Rev 117, 9247–9301. 10.1021/acs.chemrev.6b00644. [DOI] [PubMed] [Google Scholar]
- 33.Lai T-S, Che C-M, Kwong H-L, and Peng S-M (1997). Catalytic and asymmetric aziridination of alkenes catalysed by a chiral manganese porphyrin complex. Chem. Commun 2373–2374. 24. 10.1039/A706395D. [DOI] [Google Scholar]
- 34.Liang JL, Yuan SX, Huang JS, Yu WY, and Che CM (2002). Highly diastereo- and enantioselective intramolecular amidation of saturated C–H bonds catalyzed by ruthenium porphyrins. Angew. Chem. Int. Ed. Engl 41, 3465–3468. . [DOI] [PubMed] [Google Scholar]
- 35.Jiang H, Lang K, Lu H, Wojtas L, and Zhang XP (2016). Intramolecular radical aziridination of allylic sulfamoyl azides by cobalt(II)-based metalloradical catalysis: effective construction of strained heterobicyclic structures. Angew. Chem. Int. Ed. Engl 55, 11604–11608. 10.1002/anie.201605238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Riart-Ferrer X, Sang P, Tao J-R, Xu H, Jin LM, Lu H-J, Cui X, Wojtas L, and Zhang XP (2021). Metalloradical activation of carbonyl azides for enantioselective radical aziridination. Chem 7, 1120–1134. 10.1016/j.chempr.2021.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McIntosh JA, Coelho PS, Farwell CC, Wang ZJ, Lewis JC, Brown TR, and Arnold FH (2013). Enantioselective intramolecular C–H amination catalyzed by engineered cytochrome P450 enzymes in vitro and in vivo. Angew. Chem. Int. Ed. Engl 52, 9309–9312. 10.1002/anie.201304401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Farwell CC, Zhang RK, McIntosh JA, Hyster TK, and Arnold FH (2015). Enantioselective enzyme-catalyzed aziridination enabled by active-site evolution of a cytochrome P450. ACS Cent. Sci 1, 89–93. 10.1021/acscentsci.5b00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldberg NW, Knight AM, Zhang RK, and Arnold FH (2019). Nitrene transfer catalyzed by a non-heme iron enzyme and enhanced by non-native small-molecule ligands. J. Am. Chem. Soc 141, 19585–19588. 10.1021/jacs.9b11608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Z, Smart TJ, Choi H, Hardy F, Lohans CT, Abboud MI, Richardson MSW, Paton RS, McDonough MA, and Schofield CJ (2017). Structural and stereoelectronic insights into oxygenase-catalyzed formation of ethylene from 2-oxoglutarate. Proc. Natl. Acad. Sci. USA 114, 4667–4672. 10.1073/pnas.1617760114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nie J, Guo HC, Cahard D, and Ma JA (2011). Asymmetric construction of stereogenic carbon centers featuring a trifluoromethyl group from prochiral trifluoromethylated substrates. Chem. Rev 111, 455–529. 10.1021/cr100166a. [DOI] [PubMed] [Google Scholar]
- 42.Wang J, Sánchez-Roselló M, Aceña JL, del Pozo C, Sorochinsky AE, Fustero S, Soloshonok VA, and Liu H (2014). Fluorine in pharmaceutical industry: fluorine-containing drugs introduced to the market in the last decade (2001–2011). Chem. Rev 114, 2432–2506. 10.1021/cr4002879. [DOI] [PubMed] [Google Scholar]
- 43.Qin S, Yao L, Luo Y, Liu T, Xu J, Sun Y, Wang N, Yan J, Tang B, Yang G, and Yang C (2020). Synthesis of aziridines with multiple chiral substitutions by copper-catalyzed diastereoselective radical aminotrifluoromethylation of alkenes. Org. Chem. Front 7, 3132–3136. 10.1039/D0QO00603C. [DOI] [Google Scholar]
- 44.Zhu R, and Buchwald SL (2013). Enantioselective functionalization of radical intermediates in redox catalysis: copper-catalyzed asymmetric oxytrifluoromethylation of alkenes. Angew. Chem. Int. Ed. Engl 52, 12655–12658. 10.1002/anie.201307790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin JS, Dong XY, Li TT, Jiang NC, Tan B, and Liu XY (2016). A dual-catalytic strategy to direct asymmetric radical aminotrifluoromethylation of alkenes. J. Am. Chem. Soc 138, 9357–9360. 10.1021/jacs.6b04077. [DOI] [PubMed] [Google Scholar]
- 46.Jat JL, Paudyal MP, Gao H, Xu QL, Yousufuddin M, Devarajan D, Ess DH, Kürti L, and Falck JR (2014). Direct stereospecific synthesis of unprotected N–H and N–Me aziridines from olefins. Science 343, 61–65. 10.1126/science.1245727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boquet V, Nasrallah A, Dana AL, Brunard E, Di Chenna PH, Duran FJ, Retailleau P, Darses B, Sircoglou M, and Dauban P (2022). Rhodium(II)-catalyzed enantioselective intermolecular aziridination of alkenes. J. Am. Chem. Soc 144, 17156–17164. 10.1021/jacs.2c07337. [DOI] [PubMed] [Google Scholar]
- 48.Rigoli JW, Weatherly CD, Alderson JM, Vo BT, and Schomaker JM (2013). Tunable, chemoselective amination via silver catalysis. J. Am. Chem. Soc 135, 17238–17241. 10.1021/ja406654y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dolan NS, Scamp RJ, Yang T, Berry JF, and Schomaker JM (2016). Catalyst-controlled and tunable, chemoselective silver-catalyzed intermolecular nitrene transfer: experimental and computational studies. J. Am. Chem. Soc 138, 14658–14667. 10.1021/jacs.6b07981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ju M, Weatherly CD, Guzei IA, and Schomaker JM (2017). Chemo- and enantioselective intramolecular silver-catalyzed aziridinations. Angew. Chem. Int. Ed. Engl 56, 9944–9948. 10.1002/anie.201704786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Llaveria J, Beltrán Á , Sameera WMC, Locati A, Díaz-Requejo MM, Matheu MI, Castillón S, Maseras F, and Pérez PJ (2014). Chemo-, Regio-, and stereoselective silver-catalyzed aziridination of dienes: scope, mechanistic studies, and ring-opening reactions. J. Am. Chem. Soc 136, 5342–5350. 10.1021/ja412547r. [DOI] [PubMed] [Google Scholar]
- 52.Rodríguez MR, Besora M, Molina F, Maseras F, Díaz-Requejo MM, and Pérez PJ (2020). Intermolecular allene functionalization by silver-nitrene catalysis. J. Am. Chem. Soc 142, 13062–13071. 10.1021/jacs.0c04395. [DOI] [PubMed] [Google Scholar]
- 53.Liu L, Ward RM, and Schomaker JM (2019). Mechanistic aspects and synthetic applications of radical additions to allenes. Chem. Rev 119, 12422–12490. 10.1021/acs.chemrev.9b00312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu L, and Schomaker JM (2018). Chapter 6 - allene aziridination as a tool for the synthesis of complex amines. In Advances in Transition-Metal Mediated Heterocyclic Synthesis, D. Solé and I. Fernández, eds. (Academic Press; ), pp. 231–283. [Google Scholar]
- 55.Yan M, Kawamata Y, and Baran PS (2017). Synthetic organic electrochemical methods since 2000: on the verge of a renaissance. Chem. Rev 117, 13230–13319. 10.1021/acs.chemrev.7b00397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Twilton J, Le C, Zhang P, Shaw MH, Evans RW, and MacMillan DWC (2017). The merger of transition metal and photocatalysis. Nat. Rev. Chem 1. 10.1038/s41570-017-0052. [DOI] [Google Scholar]
- 57.Zhang Y, Dong X, Wu Y, Li G, and Lu H (2018). Visible-light-induced intramolecular C(sp2)–H amination and aziridination of azidoformates via a triplet nitrene pathway. Org. Lett 20, 4838–4842. 10.1021/acs.orglett.8b01980. [DOI] [PubMed] [Google Scholar]
- 58.Guo Y, Pei C, Jana S, and Koenigs RM (2021). Synthesis of trifluoromethylated aziridines via photocatalytic amination reaction. ACS Catal. 11, 337–342. 10.1021/acscatal.0c04564. [DOI] [Google Scholar]
- 59.Ielo L, Touqeer S, Roller A, Langer T, Holzer W, and Pace V (2019). Telescoped, divergent, chemoselective C1 and C1-C1 homologation of imine surrogates: access to quaternary chloro- and halomethyltrifluoromethyl aziridines. Angew. Chem. Int. Ed. Engl 58, 2479–2484. 10.1002/anie.201812525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mészáros Á , Székely A, Stirling A, and Novák Z (2018). Design of trifluoroalkenyl iodonium salts for a hypervalency-aided alkenylation–cyclization strategy: metal-free construction of aziridine rings. Angew. Chem. Int. Ed. Engl 57, 6643–6647. 10.1002/anie.201802347. [DOI] [PubMed] [Google Scholar]
- 61.Wang F, Zhu N, Chen P, Ye J, and Liu G (2015). Copper-catalyzed trifluoromethylazidation of alkynes: efficient access to CF3-substituted azirines and aziridines. Angew. Chem. Int. Ed. Engl 54, 9356–9360. 10.1002/anie.201503412. [DOI] [PubMed] [Google Scholar]
- 62.Siopa F, António JPM, and Afonso CAM (2018). Flow-assisted synthesis of bicyclic aziridines via photochemical transformation of pyridinium salts. Org. Process Res. Dev 22, 551–556. 10.1021/acs.oprd.8b00036. [DOI] [Google Scholar]
- 63.Kaplan L, Pavlik JW, and Wilzbach KE (1972). Photohydration of pyridinium ions. J. Am. Chem. Soc 94, 3283–3284. 10.1021/ja00764a089. [DOI] [Google Scholar]
- 64.Li J, Huang W, Chen J, He L, Cheng X, and Li G (2018). Electrochemical aziridination by alkene activation using a sulfamate as the nitrogen source. Angew. Chem. Int. Ed. Engl 57, 5695–5698. 10.1002/anie.201801106. [DOI] [PubMed] [Google Scholar]
- 65.Holst DE, Wang DJ, Kim MJ, Guzei IA, and Wickens ZK (2021). Aziridine synthesis by coupling amines and alkenes via an electrogenerated dication. Nature 596, 74–79. 10.1038/s41586-021-03717-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Funke W (1969). Synthesis and properties of 1-azabicyclo[1.1.0]butanes. Angew. Chem. Int. Ed. Engl 8, 70–71. 10.1002/anie.196900701. [DOI] [Google Scholar]
- 67.Antermite D, Degennaro L, and Luisi R (2016). Recent advances in the chemistry of metallated azetidines. Org. Biomol. Chem 15, 34–50. 10.1039/C6OB01665K. [DOI] [PubMed] [Google Scholar]
- 68.Hayashi K, Sato C, Hiki S, Kumagai T, Tamai S, Abe T, and Nagao Y (1999). Novel efficient synthesis of 1-azabicyclo[1.1.0] butane and its application to the synthesis of 1-(1,3-thiazolin-2-yl)azetidine-3-thiol useful for the pendant moiety of an oral 1b-methylcarbapenem antibiotic L-084. Tetrahedron Lett. 40, 3761–3764. 10.1016/S0040-4039(99)00603-6. [DOI] [Google Scholar]
- 69.Gianatassio R, Lopchuk JM, Wang J, Pan CM, Malins LR, Prieto L, Brandt TA, Collins MR, Gallego GM, Sach NW, et al. (2016). Organic chemistry. Strain-release amination. Science 351, 241–246. 10.1126/science.aad6252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gianatassio R, and Kadish D (2019). Direct alkylation of 1-azabicyclo[1.1.0]butanes. Org. Lett 21, 2060–2063. 10.1021/acs.orglett.9b00321. [DOI] [PubMed] [Google Scholar]
- 71.Fawcett A, Murtaza A, Gregson CHU, and Aggarwal VK (2019). Strain-release-driven homologation of boronic esters: application to the modular synthesis of azetidines. J. Am. Chem. Soc 141, 4573–4578. 10.1021/jacs.9b01513. [DOI] [PubMed] [Google Scholar]
- 72.Carreira EM, and Fessard TC (2014). Four-membered ring-containing spirocycles: synthetic strategies and opportunities. Chem. Rev 114, 8257–8322. 10.1021/cr500127b. [DOI] [PubMed] [Google Scholar]
- 73.Tyler JL, Noble A, and Aggarwal VK (2021). Strain-release driven spirocyclization of azabicyclo[1.1.0]butyl ketones. Angew. Chem. Int. Ed. Engl 60, 11824–11829. 10.1002/anie.202102754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pan B, Li F, and Zhao Y (2020). Synthesis and applications of methyleneaziridines. RSC Adv. 10, 39304–39322. 10.1039/D0RA07663E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ranjith J, and Ha HJ (2021). Synthetic applications of aziridinium ions. Molecules 26, 1774. 10.3390/molecules26061774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hu Y, Roberts JM, Kilgore HR, Mat Lani AS, Raines RT, and Schomaker JM (2020). Triple, mutually orthogonal bioorthogonal pairs through the design of electronically activated sulfamate-containing cycloalkynes. J. Am. Chem. Soc 142, 18826–18835. 10.1021/jacs.0c06725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Corbin JR, Ketelboeter DR, Fernández I, and Schomaker JM (2020). Biomimetic 2-imino-Nazarov cyclizations via eneallene aziridination. J. Am. Chem. Soc 142, 5568–5573. 10.1021/jacs.0c02441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu L, Gerstner NC, Oxtoby LJ, Guzei IA, and Schomaker JM (2017). Fluorinated amine stereotriads via allene amination. Org. Lett 19, 3239–3242. 10.1021/acs.orglett.7b01342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dequina HJ, and Schomaker JM (2020). Aziridinium ylides: underused intermediates for complex amine synthesis. Trends Chem. 2, 874–887. 10.1016/j.trechm.2020.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hata Y, and Watanabe M (1972). Fragmentation reaction of aziridinium ylids. Tetrahedron Lett. 13, 3827–3830. 10.1016/S0040-4039(01)94172-3. [DOI] [Google Scholar]
- 81.Rowlands GJ, and Kentish Barnes W (2004). Studies on the [2,3]-Stevens rearrangement of aziridinium ions. Tetrahedron Lett. 45, 5347–5350. 10.1016/j.tetlet.2004.05.087. [DOI] [Google Scholar]
- 82.Schmid SC, Guzei IA, and Schomaker JM (2017). A stereoselective [3+1] ring expansion for the synthesis of highly substituted methylene azetidines. Angew. Chem. Int. Ed. Engl 56, 12229–12233. 10.1002/anie.201705202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Schmid SC, Guzei IA, Fernández I, and Schomaker JM (2018). Ring expansion of bicyclic methyleneaziridines via concerted, near-barrierless [2,3]- Stevens rearrangements of aziridinium ylides. ACS Catal. 8, 7907–7914. 10.1021/acscatal.8b02206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Eshon J, Nicastri KA, Schmid SC, Raskopf WT, Guzei IA, Fernández I, and Schomaker JM (2020). Intermolecular [3+3] ring expansion of aziridines to dehydropiperidines through the intermediacy of aziridinium ylides. Nat. Commun 11, 1273. 10.1038/s41467-020-15134-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ertl P, and Schuhmann T (2019). A systematic cheminformatics analysis of functional groups occurring in natural products. J. Nat. Prod 82, 1258–1263. 10.1021/acs.jnatprod.8b01022. [DOI] [PubMed] [Google Scholar]
- 86.Lowden PAS (2006). Aziridine natural products – discovery, biological activity and biosynthesis. In Aziridines and Epoxides in Organic Synthesis, A.K. Yudin, ed. (Wiley-VCH; ), pp. 399–442. [Google Scholar]
- 87.Skibo EB, Islam I, Heileman MJ, and Schulz WG (1994). Structure-activity studies of benzimidazole-based DNA-cleaving agents. Comparison of benzimidazole, pyrrolobenzimidazole, and tetrahydropyridobenzimidazole analogs.J. Med. Chem 37, 78–92. 10.1021/jm00027a010. [DOI] [PubMed] [Google Scholar]
- 88.Singh GS (2016). Synthetic aziridines in medicinal chemistry: A mini-review. Mini Rev. Med. Chem 16, 892–904. 10.2174/1389557515666150709122244. [DOI] [PubMed] [Google Scholar]
- 89.Wettasinghe M, Vasanthan T, Temelli F, and Swallow K (2001). Volatile flavour composition of cooked by-product blends of chicken, beef and pork: a quantitative GC–MS investigation. Food Res. Int 34, 149–158. 10.1016/S0963-9969(00)00146-0. [DOI] [Google Scholar]
- 90.Schirmeister T, and Peric M (2000). Aziridinyl peptides as inhibitors of cysteine proteases: effect of a free carboxylic acid function on inhibition. Bioorg. Med. Chem 8, 1281–1291. 10.1016/s0968-0896(00)00058-4. [DOI] [PubMed] [Google Scholar]
- 91.Vikram A, Hamzehzarghani H, and Kushalappa AC (2005). Volatile metabolites from the headspace of onion bulbs inoculated with postharvest pathogens as a tool for disease discrimination. Can. J. Plant Pathol 27, 194–203. 10.1080/07060660509507216. [DOI] [Google Scholar]
- 92.Ismail FMD, Levitsky DO, and Dembitsky VM (2009). Aziridine alkaloids as potential therapeutic agents. Eur. J. Med. Chem 44, 3373–3387. 10.1016/j.ejmech.2009.05.013. [DOI] [PubMed] [Google Scholar]
- 93.Botuha C, Chemla F, Ferreira F, and Pérez-Luna A (2011). Aziridines in natural product synthesis. In Heterocycles in Natural Product Synthesis, K.C. Majumdar and S.K. Chattopadhyay, eds. (Wiley-VCH; ), pp. 1–39. [Google Scholar]
- 94.Yoneyama H, and Katsumata R (2006). Antibiotic resistance in bacteria and its future for novel antibiotic development. Biosci. Biotechnol. Biochem 70, 1060–1075. 10.1271/bbb.70.1060. [DOI] [PubMed] [Google Scholar]
- 95.Kraus CN (2008). Low hanging fruit in infectious disease drug development. Curr. Opin. Microbiol 11, 434–438. 10.1016/j.mib.2008.09.009. [DOI] [PubMed] [Google Scholar]
- 96.Hassanpour SH, and Dehghani M (2017). Review of cancer from perspective of molecular. J. Cancer Res. Pract 4, 127–129. 10.1016/j.jcrpr.2017.07.001. [DOI] [Google Scholar]
- 97.Znati M, Debbabi M, Romdhane A, Ben Jannet H, and Bouajila J (2018). Synthesis of new anticancer and anti-inflammatory isoxazolines and aziridines from the natural (−)-deltoin. J. Pharm. Pharmacol 70, 1700–1712. 10.1111/jphp.13013. [DOI] [PubMed] [Google Scholar]
- 98.Nicolaou KC, Shelke YG, Dherange BD, Kempema A, Lin B, Gu C, Sandoval J, Hammond M, Aujay M, and Gavrilyuk J (2020). Design, synthesis, and biological investigation of epothilone B analogues featuring lactone, lactam, and carbocyclic macrocycles, epoxide, aziridine, and 1,1-difluorocyclopropane and other fluorine residues. J. Org. Chem 85, 2865–2917. 10.1021/acs.joc.0c00123. [DOI] [PubMed] [Google Scholar]
- 99.Ghosh AK, Mishevich JL, and Jurica MS (2021). Spliceostatins and derivatives: chemical syntheses and biological properties of potent splicing inhibitors. J. Nat. Prod 84, 1681–1706. 10.1021/acs.jnatprod.1c00100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Luo M, Wang F, Zhang H, To KKW, Wu S, Chen Z, Liang S, and Fu L (2020). Mitomycin C enhanced the efficacy of PD-L1 blockade in non-small cell lung cancer. Signal Transduct. Target. Ther 5, 141. 10.1038/s41392-020-0200-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Iversen PL (2018). The threat from viruses. Molecular Basis of Resilience, 45–76. [Google Scholar]
- 102.Fey P, Chartomatsidou R, Kiefer W, Mottram JC, Kersten C, and Schirmeister T (2018). New aziridine-based inhibitors of cathepsin L-like cysteine proteases with selectivity for the Leishmania cysteine protease LmCPB2.8. Eur. J. Med. Chem 156, 587–597. 10.1016/j.ejmech.2018.07.012. [DOI] [PubMed] [Google Scholar]
- 103.Chang KO, Kim Y, Lovell S, Rathnayake AD, and Groutas WC (2019). Antiviral drug discovery: norovirus proteases and development of inhibitors. Viruses 11, 197. 10.3390/v11020197. [DOI] [PMC free article] [PubMed] [Google Scholar]