

Abstract
Methylation profiling has radically transformed our understanding of tumors previously called central nervous system primitive neuroectodermal tumors (CNS-PNET). While this marks a momentous step towards defining key differences, reclassification has thrown treatment into disarray. To shed light on response to therapy and guide clinical decision-making, we report outcomes and molecular features of children with CNS-PNETs from two multi-center risk-adapted studies (SJMB03 for patients ≥ 3 years; SJYC07 for patients < 3 years) complemented by a non-protocol institutional cohort. Seventy patients who had a histological diagnosis of CNS-PNET or CNS embryonal tumor from one of the new categories that have supplanted CNS-PNET were included. This cohort was molecularly characterized by DNA methylation profiling (n = 70), whole-exome sequencing (n = 53), RNA-sequencing (n = 20) and germline sequencing (n = 28). Clinical characteristics were detailed, and treatment was divided into craniospinal irradiation (CSI)-containing (SJMB03 and SJMB03-like) and CSI-sparing therapy (SJYC07 and SJYC07-like). When the cohort was analyzed in its entirety, no differences were observed in the 5-year survival rates even when CSI-containing therapy was compared to CSI-sparing therapy. However, when analyzed by DNA methylation molecular grouping, significant survival differences were observed, and treatment particulars provided suggestions of therapeutic response. Patients with CNS neuroblastoma with FOXR2 activation (CNS-NB-FOXR2) had a 5-year event-free survival (EFS)/overall survival (OS) of 66.7% ± 19.2%/83.3% ± 15.2%, and CIC rearranged sarcoma (CNS-SARC-CIC) had a 5-year EFS/OS both of 57.1% ± 18.7% with most receiving regimens that contained radiation (focal or CSI) and multidrug chemotherapy. Patients with high-grade neuroepithelial tumor with BCOR alteration (HGNET-BCOR) had abysmal responses to upfront chemotherapy-only regimens (5-year EFS = 0%), but survival extended with salvage radiation after progression [5-year OS = 53.6% ± 20.1%]. Patients with embryonal tumor with multilayered rosettes (ETMR) or high-grade glioma/glioblastoma multiforme (HGG/GBM) did not respond favorably to any modality (5-year EFS/OS= 10.7 ± 5.8%/17.9 ± 7.2% and 10% ± 9.0%/10% ± 9.0%, respectively). As an accompaniment, we have assembled this data onto an interactive website to allow users to probe and query the cases. By reporting on a carefully matched clinical and molecular cohort, we provide needed insight for future clinical management.
Introduction
With the advent of epigenomic profiling in central nervous system (CNS) tumors, numerous entities have undergone a recharacterization and a plethora of new molecular groups established [1]. In perhaps the most transformative shift, CNS-primitive neuroectodermal tumor (CNS-PNET), which was once considered the supratentorial counterpart of medulloblastoma, was rejected as an entity due to the biologic heterogeneity that was uncovered in the landmark study by Sturm et al [16]. In its place new and more descriptive entities such as CIC rearranged sarcoma (CNS-SARC-CIC), CNS neuroblastoma with FOXR2 activation (CNS-NB-FOXR2), CNS tumor with BCOR internal tandem duplication (CNS-BCOR-ITD), embryonal tumor with multilayered rosettes (ETMR), and CNS embryonal tumor not elsewhere classified/not otherwise specified (CNS-ET--NOS/NEC) now occupy the 2021 WHO CNS tumor classification [9]. But the rarity of these tumors combined with several new molecular groupings has left the neuro-oncology community struggling with treatment decisions. Recent publications have suggested that outcomes are by-and-large predicated upon the molecular makeup of the tumor [3, 6, 16, 20], but how does a physician decide what treatment to choose for a child newly diagnosed with one of these entities? To address this question, we report the molecular profiles and clinical outcome of children with tumors previously considered as CNS-PNETs treated on two prospective multi-center studies (SJMB03, SJYC07) and an institutional cohort from St. Jude Children’s Research Hospital.
Patients and Methods
Design and Study Cohort
For this study we sought to identify and include patients with any of the following histologic diagnoses: CNS-PNET, high-grade neuroepithelial tumor (HGNET), ETMR, CNS neuroblastoma, CNS embryonal tumor, as well as any who have subsequently been identified to belong to the methylation-classes of interest (CNS-NB-FOXR2, CNS-SARC-CIC, high-grade neuroepithelial tumor with BCOR alteration [HGNET-BCOR], high-grade neuroepithelial tumor with MN1 alteration [HGNET-MN1]). The cohort was assembled from two multi-center studies (SJMB03, SJYC07) and an institutional cohort of patients treated similarly off-protocol between January 1, 2003, and February 15, 2022 (non-protocol treatment plan cohort [NPTP]). SJMB03 was a multi-institutional, phase III study (NCT00085202) for patients aged 3–21 years with newly diagnosed medulloblastoma, CNS-PNET, or Atypical Teratoid Rhabdoid Tumor (ATRT) that enrolled between June 24, 2003, and March 7, 2013. SJYC07 was a multi-institutional Phase II, risk-adapted study (NCT00602667) for children younger than 3 years with newly diagnosed malignant brain tumors that enrolled between November 9, 2007, and April 19, 2017. SJMB03 and SJYC07 were approved by the Institutional Review Board (IRB) with written informed consent obtained from patients and families; the review of non-protocol patients was approved by the IRB with waiver for consent. Only patients with tissue available for molecular testing were included.
Treatment Protocols
The study approaches for SJMB03 and SJYC07 have been previously reported [8, 14, 18]. Briefly, after initial maximal safe resection and staging, SJMB03 patients were stratified into average-risk (SJMB03-AR) or high-risk (SJMB03-HR) arms, with SJMB03-HR criteria including presence of metastasis (M+) and/or bulky residual disease > 1.5 cm2 at the primary site (R+). Risk-adapted CSI (SJMB03-AR = 23.4 Gy; SJMB03-HR = 36–39.6 Gy) and focal boost to the primary site (55.8 – 59.4 Gy) and metastatic sites (SJMB03-HR) were delivered, followed by four sequential cycles of high-dose chemotherapy (cisplatin/cyclophosphamide/vincristine) with autologous stem cell rescue. Together SJMB03-AR and SJMB03-HR were categorized as “CSI-containing” primary therapy. SJYC07 patients with localized disease were enrolled on the low-risk (SYC07-LR) or intermediate-risk (SJYC07-IR) arm based on histology and/or age at diagnosis, and M+ patients were enrolled on the high-risk (SJYC07-HR) arm. All SJYC07 patients started with four cycles of induction chemotherapy (high-dose methotrexate/cisplatin/cyclophosphamide/vincristine; vinblastine added for SJYC07-HR disease) followed by risk-adapted consolidation: SJYC07-LR patients received two cycles of carboplatin, etoposide, and cyclophosphamide; SJYC07-IR patients received focal radiation (54 Gy); and SJYC07-HR patients received two cycles of cyclophosphamide and topotecan. Post-consolidation all SJYC07 patients who continued on therapy were prescribed metronomic maintenance therapy for 6 months. (Supplemental Fig. S1) Together these three arms were catalogued as “CSI-sparing” primary therapies including “CSI-sparing with focal RT” encompassing those on SJYC07-IR who received focal RT and “CSI-sparing chemotherapy only” encompassing SJYC07-LR, SJYC07-HR, and SJYC07-IR who did not get focal RT. Patients in the NPTP cohort were risk-stratified in a similar fashion and, in most cases, treated according to the above protocols. Thus, these patients were grouped into SJMB03-like, SJYC07-like therapies, and palliative care (if curative intent therapy was not attempted).
Methylation Profiling and Genomic Sequencing
DNA and RNA were extracted from formalin-fixed paraffin-embedded or fresh-frozen tumor samples. Genome-wide DNA methylation profiling was performed on tumor samples using the Infinium Methylation EPIC BeadChip array as described previously [14, 18]. Epigenomic classification was conducted using t-distributed stochastic neighbor embedding (TSNE) on the new samples together with reference profiles obtained from published brain tumor dataset [1] and placed into broader categories under “TSNE category” or more granular categories by “TSNE closest cluster”. Tumor samples were further classified using Molecular Neuropathology (MNP) brain tumor classifier version 12.5 (www.molecularneuropathology.org). Whole-exome sequencing (WES, n = 53), whole-genome sequencing (WGS, n = 11), and RNA-sequencing (RNA-seq, n = 20) data were generated for samples with adequate tumor DNA/RNA. Germline (blood) DNA from 28 patients was available for paired sequencing. Relapse samples were also analyzed on 8 patients (methylation n = 6, WES n = 7, WGS n = 5, RNA-seq n = 6).
Survival Analyses
The date of diagnosis was defined as the date of first biopsy or resection. Overall survival (OS) was defined as the duration between the date of diagnosis and date of either death from any cause or last follow-up; event-free survival (EFS) was defined as the duration between the date of diagnosis and date of either progression, relapse, second malignancy, death from any cause, or last follow-up. Survival comparisons were performed via log-rank tests. Fisher’s exact test was used to compare categorical variables. Statistical analyses were performed using R v3.6.0 (www.R-project.org).
Data Portal
We extended the capabilities of ProteinPaint [22] to interactively visualize the cohort metadata, clinical features and oncoprints using the D3.js library for all the frontend charting and interactive features on the browser. We used R’s survival package (https://cran.r-project.org/web/packages/survival/index.html) to perform survival analysis.
Results
Demographics and Clinical Characteristics
Seventy patients were included (Fig 1a). Thirty-eight (54%) were male. The median age at diagnosis was 2.69 years (range 0.57–15.56). Fifteen patients treated on from SJMB03, 34 on SJYC07, and 21 on NPTPs. Sixty-eight (97%) patients were treated with curative intent while palliation was adopted in 2 patients. Among those managed with curative intent, 20 patients received CSI with chemotherapy, and 48 received CSI-sparing therapy as part of primary management. At diagnosis, 62 (89%) were originally called CNS-PNET or HGNET or CNS embryonal tumor, 7 (10%) high-grade glioma (HGG), and 1 (1%) medulloblastoma (MB) by histological analysis. Primary tumor location was cerebral in 57 (81%), brainstem in 5 (7%), posterior fossa in 4 (6%), pineal in 3 (4%), and spinal in 1 (1%). Eight (12%) patients had M+ disease at diagnosis. Baseline risk features and stratification are detailed in Table 1.
Fig. 1.
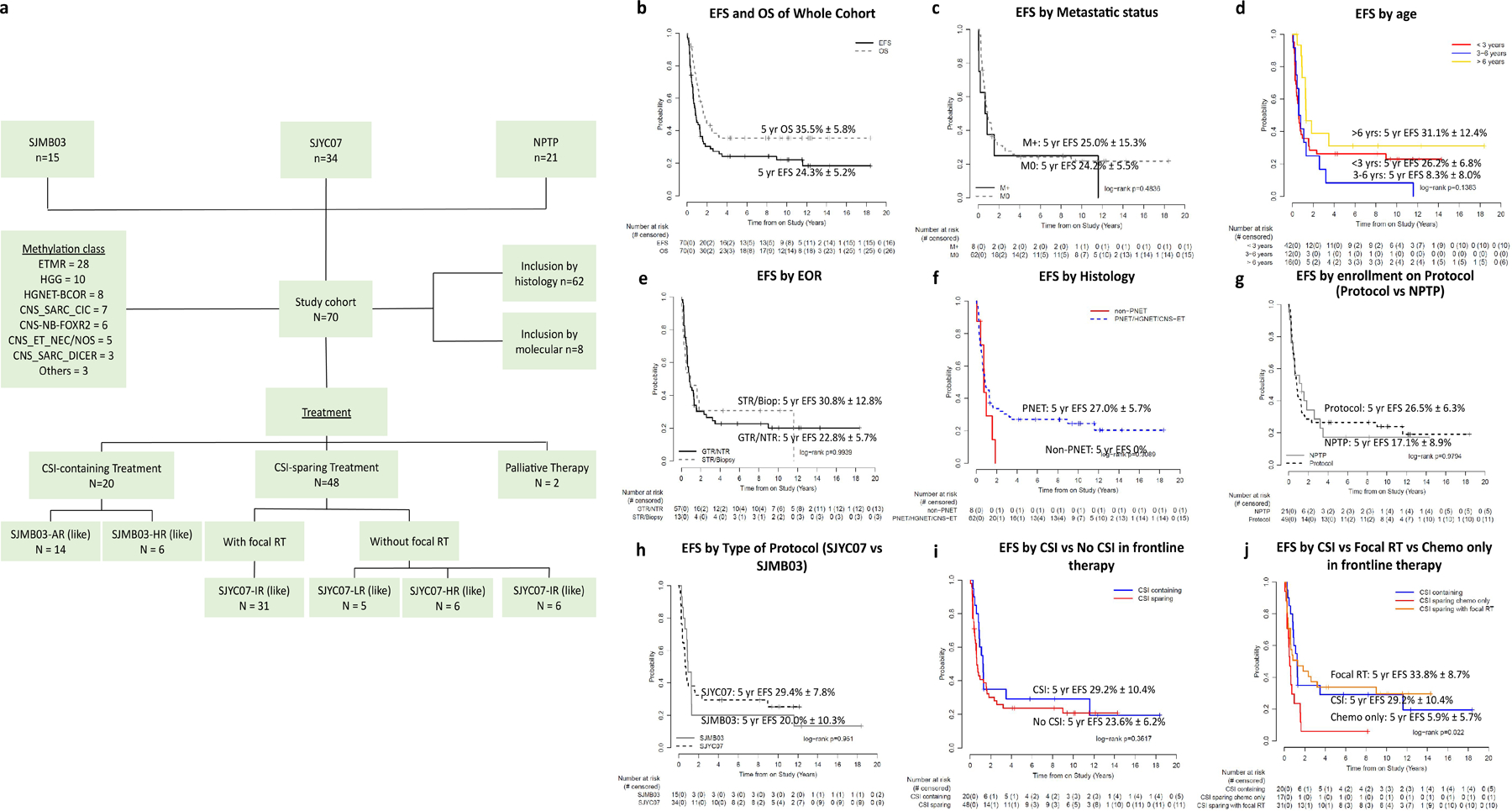
Cohort composition and key clinical outcomes. (a) The cohort was assembled from two multi-center studies (SJMB03, SJYC07) and a NPTP cohort. Patients were included based on histology and molecular grouping. All samples were molecularly characterized by DNA methylation profiling into methylation classes. Treatment was divided into CSI-containing (SJMB03 and SJMB03-like) and CSI-sparing therapy (SJYC07 and SJYC07-like) and palliative therapy. (b) EFS and OS of the whole cohort. EFS by (c) metastatic status (M0 vs M+); (d) age [< 3years (red), 3–6 years (blue) >6 years (yellow)]; (e) surgical EOR (STR/Biop vs GTR/NTR; (f) histology [PNET/HGNET/CNS-Embryonal Tumor (blue); non-PNET (red)]; (g) enrollment on protocol [Protocol, (SJMB03 or SJYC07) vs NPTP]; (h) type of protocol (SJYC07 vs SJMB03); (i) CSI (blue) vs no CSI in frontline therapy (red). (j) CSI (blue) vs Focal RT (orange) vs Chemotherapy only (red) in frontline therapy. Abbreviations: AR, average-risk; Biop, biopsy; CNS, central nervous system; CNS-ET-NEC/NOS, CNS embryonal tumor not elsewhere defined/not otherwise specified; CNS-SARC-CIC, CNS sarcoma with CIC rearrangement; CNS-SARC-DICER, CNS sarcoma with DICER mutation; CNS-NB-FOXR2, CNS neuroblastoma with FOXR2 activation; CSI, craniospinal irradiation; EFS, event-free survival; ETMR, embryonal tumor with multilayered rosettes; GBM, glioblastoma; GTR, gross-total resection; HGNET, high-grade neuroepithelial tumor; HGNET-BCOR, high-grade neuroepithelial tumor with BCOR alteration; HR, high-risk; IR, intermediate-risk; LR low-risk; M0, non-metastatic; M+, metastatic; NPTP, non-protocol treatment plan; NTR, near-total resection; PNET, primitive neuro-ectodermal tumor; RT, radiation therapy.
Table 1.
Demographic, clinical, and treatment characteristics of the cohort
| SJMB03 | NPTP (SJMB03-like) | SJYC07 | NPTP (SJYC07-Like) | Palliative | Entire Cohort | |
|---|---|---|---|---|---|---|
| n | n | n | n | n | n | |
| Age on Study | ||||||
| < 3 years | – | – | 34 | 7 | 1 | 42 |
| 3–6 years | 5 | 1 | – | 5 | 1 | 12 |
| >6 | 10 | 4 | – | 2 | 16 | |
| Gender | ||||||
| Male | 10 | 2 | 18 | 8 | 0 | 38 |
| Female | 5 | 3 | 16 | 6 | 2 | 32 |
| Histology | ||||||
| PNET/HGNET/CNS_ET | 15 | 5 | 28 | 12 | 2 | 62 |
| HGG | 0 | 0 | 5 | 2 | 0 | 7 |
| MB | 0 | 0 | 1 | 0 | 0 | 1 |
| Metastatic Status | ||||||
| Non metastatic M0 | 13 | 5 | 30 | 13 | 1 | 62 |
| Metastatic (M+) | 2 | 0 | 4 | 1 | 1 | 8 |
| Extent of Resection | ||||||
| GTR (R0) | 11 | 5 | 24 | 13 | 0 | 53 |
| NTR (R0) | 1 | 0 | 3 | 0 | 0 | 4 |
| Subtotal (R+) | 2 | 0 | 6 | 1 | 1 | 10 |
| Biopsy (R+) | 1 | 0 | 1 | 0 | 1 | 3 |
| Treatment Risk group | ||||||
| SJMB03-AR | 10 | 4 | – | – | – | 14 |
| SJMB03-HR | 5 | 1 | – | – | – | 6 |
| SJYC07-LR | – | – | 4 | 1 | – | 5 |
| SJYC07-IR | – | – | 26 | 11 | – | 37 |
| SJYC07-HR | – | – | 4 | 2 | – | 6 |
| Palliative | – | – | – | – | 2 | 2 |
| Molecular Grouping (tSNE) | ||||||
| ETMR | 5 | 0 | 16 | 5 | 2 | 28 |
| HGG/GBM | 8 | 0 | 1 | 1 | 0 | 10 |
| CNS_NB_FOXR2 | 0 | 1 | 5 | 0 | 0 | 6 |
| EFT_CIC | 0 | 2 | 3 | 2 | 0 | 7 |
| HGNET_BCOR | 1 | 0 | 3 | 4 | 0 | 8 |
| DICER1 intracranial sarcoma | 0 | 2 | 0 | 1 | 0 | 3 |
| EPN_RELA-like | 1 | 0 | 4 | 0 | 0 | 5 |
| PXA | 0 | 0 | 0 | 1 | 0 | 1 |
| EWS | 0 | 0 | 1 | 0 | 0 | 1 |
| CPC | 0 | 0 | 1 | 0 | 0 | 1 |
| Total | 15 | 5 | 34 | 14 | 2 | 70 |
Treatment Outcome and Prognostic Factors
Overall Cohort
With a median follow-up of 9.5 years (range 0.1 – 18.4), for patients who are event-free, 54 events were observed (44 deaths due to progressive disease [PD], 7 PD without death, 2 subsequent malignancies without death, 1 subsequent tumor [meningioma] without death). The 5-year EFS and OS were 24.3% ± 5.2% and 35.5% ± 5.8% respectively (Fig 1b). No significant differences were seen by presence of metastasis, age, extent of resection, histology, enrollment on protocol (protocol vs NPTP), type of protocol (SJMB03 vs SJYC07), and use of CSI (Fig 1c–i). The only significant finding was that those treated with surgery and chemotherapy without adjuvant RT (CSI or focal) had a worse EFS than those treated with RT (focal or CSI) (p = 0.022; Fig 1j).
CSI-containing therapy (SJMB03 or SJMB03-like)
CSI-containing primary therapy was given to 20 patients (SJMB03-AR/AR - like=14, SJMB03-HR/HR-like = 6). Fifteen were from SJMB03 and 5 patients were treated on NPTP. Median age was 11.20 years (range 3.17–15.56). All were histologically defined as CNS-PNET or HGNET or CNS embryonal tumor. Gross-total resection (GTR) or near-total resection (NTR) was achieved in 17 (85%) patients, subtotal resection (STR) and biopsy-only in 3 (15%). The 5-year EFS and OS for the whole cohort were 29.2 ± 10.4% and 32.5 ± 10.8%; for SJMB03-AR/AR-like were 28.6 ± 12.1% and 35.7 ± 12.8%; and for SJMB03-HR/HR-like were 33.3% ± 19.2% and 25.0% ± 20.4%. (Fig 1i and Supplemental Fig. S2–S3).
CSI-sparing therapy [SJYC07 or SJYC07-like]
CSI-sparing therapy was given in 48 patients with 34 patients treated on SJYC07 and 14 patients on NPTPs. Median age was 2.13 years (range 0.57 – 13.83). Thirty-one patients were treated with focal RT +/− chemotherapy (SJYC07-IR/IR-like [22 on-protocol, 9 NPTP]) and 17 with chemotherapy only (5 SJYC07-LR/LR-like [4 on-protocol, 1 NPTP); 6 SJYC07-IR/IR-like who progressed or declined RT (4 on-protocol, 2 NPTP); 6 SJYC07-HR/HR-like (4 on-protocol, 2 NPTP)]. Forty tumors were histologically defined as CNS-PNET/HGNET/CNS embryonal tumor, 7 HGG, and 1 MB. GTR or NTR was achieved in 40 (83%) patients, STR or biopsy in 8 (17%). The 5-year EFS and OS were, respectively, 23.6 ± 6.2% and 38.1% ± 7.1% for the whole cohort; 33.8% ± 8.7% and 49.8% ± 9.2% for CSI sparing with focal RT; and 5.9% ± 5.7% and 17.6% ± 9.2% for CSI sparing chemotherapy only (Fig 1i–j and Supplemental Fig. S2). Five-year EFS was 0% for SJYC07-LR/LR-like; 28.2% ± 7.5% for SJYC07-IR/IR-like; and 16.7% ± 15.2% for SJYC07-HR/HR-like (Supplemental Fig. S3).
Molecular Classification, Management, and Outcome by Molecular Groups
Overall Cohort
TSNE analysis of methylation classified 28 as ETMR (40%), 10 as HGG (14%), 6 as CNS-NB-FOXR2 (9%), 7 as CNS-SARC-CIC (10%), 8 as HGNET-BCOR (11%), 3 as primary intracranial sarcoma DICER1-mutant (CNS-SARC-DICER1, 4%), 1 as EWS, 1 PXA, and 1 choroid plexus tumor (CPT). Five tumors (7%) could not be definitively classified and clustered close to but not within EPN-ZFTA [hereon referred to as CNS embryonal tumor not elsewhere classified or not otherwise specified (CNS-ET-NEC/NOS)-) (Fig. 2a). When compared to original histology non-PNET (HGG or MB) classified as HGNET-BCOR, CNS-NB-FOXR2, CNS-SARC-CIC, or ETMR. Except in a few cases, the TSNE category and TSNE closest cluster agreed with the latest MNP classification (Fig. 2b).
Fig. 2.
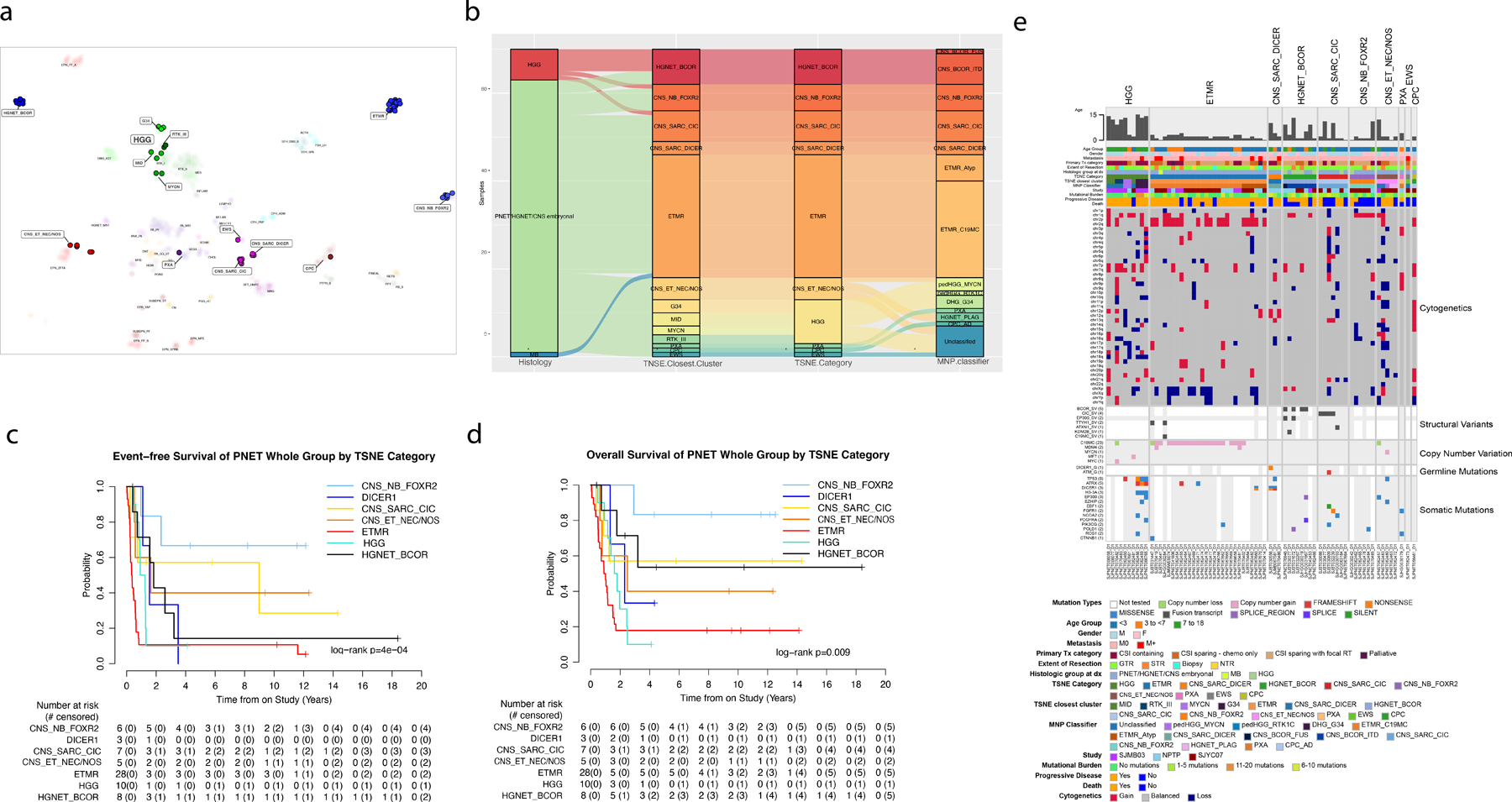
Categorization of cohort into molecular groups with outcomes and genomic alterations. (a) TSNE clustering of cohort samples against reference classes in the Capper dataset [1]. Samples from this study appear as solid-colored circles over reference samples shown in faded colors. Entities unto which our cohort samples matched or clustered near are highlighted in boxes with increased font size. (b) Alluvial plot shows the relationship between histology, TSNE clustering, TSNE category (where HGG entities are consolidated), and MNP classifier. (c-d) EFS and overall OS by TSNE category. (e) Oncoprint aligning clinical features (age, gender, metastasis, histology, primary treatment, extent of resection, type of study, progressive disease, death) with molecular findings (TSNE classification, MNP classification, mutational burden, cytogenetic whole-chromosomal arm alterations, gene fusions, focal copy number alterations, germline mutations and somatic mutations). Abbreviations: CNS, central nervous system, CNS-BCOR-FUS, CNS tumor with BCOR fusion; CNS-BCOR-ITD, CNS tumor with BCOR ITD; CNS-ET-NEC/NOS, CNS embryonal tumor not elsewhere defined/not otherwise specified; CNS-SARC-CIC, CNS sarcoma with CIC rearrangement; CNS-SARC-DICER, CNS sarcoma with DICER mutation, CNS-NB-FOXR2, CNS neuroblastoma with FOXR2 activation; CPC, choroid plexus carcinoma; CPC-AD, choroid plexus carcinoma adult subtype; CSI, craniospinal irradiation; DHG-G34, diffuse hemispheric glioma H3 G34 mutant; ETMR, embryonal tumor with multilayered rosettes; ETMR-C19MC, embryonal tumor with multilayered rosettes with C19 microRNA cluster; ETMR-Atyp, embryonal tumor with multilayered rosettes atypical; EWS, Ewing’s Sarcoma; F, female; G34, high-grade glioma subclass G34; GTR, gross-total resection; HGG, high-grade glioma; HGNET, high-grade neuroepithelial tumor; HGNET-BCOR, high-grade neuroepithelial tumor with BCOR alteration; HGNET-PLAG, high-grade neuroepithelial tumor with PLAG alteration; ITD, internal tandem duplication; M, male; M0, non-metastatic; M+, metastatic; MB, medulloblastoma; MID, high-grade glioma subclass MID; MNP, Molecular Neuropathology brain tumor classifier, MYCN, high-grade glioma subclass MYCN; NPTP, non-protocol treatment plan; NTR, near-total resection; pedHGG-MYCN, pediatric-type high grade glioma, subclass MYCN; pedHGG-RTK1C, pediatric-type high grade glioma, subclass RTK1C; PNET, primitive neuro-ectodermal tumor; PXA, pleomorphic xanthoastrocytoma; RT, radiation therapy; RTK_III, high-grade glioma subclass RTKIII; STR, subtotal resection; TSNE, t-stochastic neighbor embedding.
The survival of patients differed significantly according to molecular groups. Patients with CNS-NB-FOXR2 had the best EFS whereas patients with HGNET-BCOR, ETMR, CNS-SARC-DICER1 and HGG had inferior EFS (Fig. 2c). The same trend was shown for OS except for HGNET-BCOR where a higher proportion of patients survived after relapse (Fig. 2d). Putative driver gene alterations, chromosomal gains and loss, and general demographics are summarized by methylation group in Figure 2e.
ETMR
By methylation classification, 28 patients had ETMR with a median age of 2.36 years (0.73–4.88). Thirteen were male and 15 female. Five (18%) were metastatic at diagnosis. All were histologically defined as CNS-PNET/HGNET/CNS embryonal tumor except for one called MB. Location of the primary tumor was cerebral in 18 (64%), brainstem in 4 (14%), posterior fossa in 2 (7%), pineal in 2 (7%), and spinal in 1 (3.5%). The typical C19MC amplicon was observed in 21 (75%) samples, 17 (61%) had copy number gain of chromosome 2, and 11 (39%) had copy number loss of the X chromosome. Nineteen (68%) had tumor DNA sequencing, all displaying a low mutational burden and very few recurrent mutations. Two ATRX (D1940fs, DT836S) mutations and 2 DICER1 (E1813G, R676X) mutations (both in the same patient who did not have the C19MC amplicon) were identified. There was one CTNNB1 (T41A) mutation. Paired germline sequencing was available in 8 (28%) yet no germline mutations were detected (Fig. 3a). RNA-seq was performed in 3 (11%) samples, 2 of which showed TTYH1 fusions: one with MiR373, the other with MiR512 (Fig. 3a; Supplemental Fig. S7). The primary treatment was CSI-containing for 5 (SJMB03 AR = 3, SJMB03 HR = 2) and CSI sparing for 21 (CSI sparing with focal RT = 13, CSI sparing chemotherapy only = 8) while two were treated palliatively. The 5-year EFS and OS for the entire cohort were 10.7 ± 5.8% and 17.9 ± 7.2%, respectively. No differences among the treatment categories were observed (Fig. 2c–d; Fig. 3b–c). There were 5 long-term survivors (4 females, 1 male): 3 were nonmetastatic (all SJYC07-IR/IR-like therapy); 2 were metastatic (1 SJMB03-HR therapy, 1 SJYC07-HR therapy).
Fig. 3.
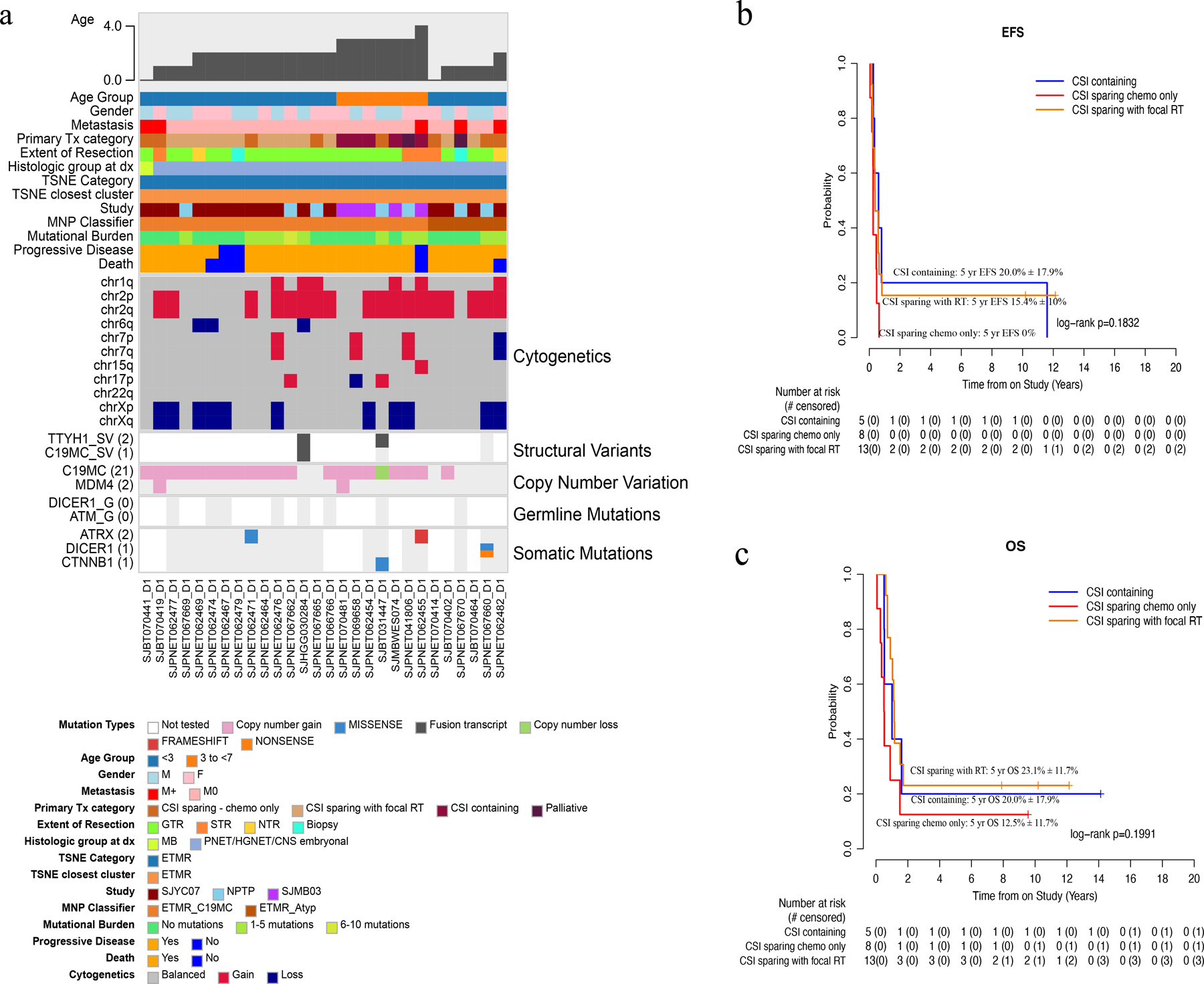
Molecular and clinical characteristics of ETMR tumors. (a) Oncoprint aligning clinical features with molecular findings. (b-c) EFS and OS by treatment category. Abbreviations: CNS, central nervous system, ETMR, embryonal tumor with multilayered rosettes; ETMR-C19MC, embryonal tumor with multilayered rosettes with C19 microRNA cluster; ETMR-Atyp, embryonal tumor with multilayered rosettes atypical; F, female; HGNET, high-grade neuroepithelial tumor; M, male; M0, non-metastatic; M+, metastatic; MB, medulloblastoma; MNP Classifier, Molecular Neuropathology brain tumor classifier; NTR, near-total resection; PNET, primitive neuro-ectodermal tumor; RT, radiation therapy; STR, subtotal resection; TSNE, t-stochastic neighbor embedding.
HGNET-BCOR
By methylation, 8 patients had HGNET-BCOR tumors. The median age was 4.55 years (1.35–13.83). Six were male and 2 were female. At diagnosis, none were metastatic. Five patients were histologically defined as HGG and 3 as CNS-PNET/HGNET/CNS embryonal tumor. Location of the primary tumor was cerebral in 4 and posterior fossa in 2. Seven (88%) had tumor sequencing, all displaying a low mutational burden and no recurrent mutations. Paired germline sequencing was available in 5 (63%) yet no germline mutations were detected. RNA-seq was performed in 6 (75%) samples: the typical BCOR-ITD was observed in 3 samples, KDM2B-NUTM1 fusion with upregulation of BCOR expression was observed in one, and BCOR-EP300 and BCOR-L3MBTL2 transcripts were concomitantly present in the other two (Fig 4a; Supplemental Fig. S7). The two patients with BCOR-EP300 were older (9.91 and 13.83 years at diagnosis) and the tumors were morphologically distinct, yet they clustered by methylation TSNE with BCOR-ITD tumors (Fig 2a). Primary treatment was CSI-containing for 1 (SJMB03-AR) and CSI-sparing for 7 (CSI-sparing with focal RT = 4, CSI-sparing chemotherapy only = 3). GTR was achieved in 6 patients, STR in 2. The 5-year EFS and OS for the entire cohort were 14.3% ± 13.2% and 53.6% ± 20.1% (Fig 2c–d), respectively. All patients, except for the 1 treated with CSI-containing and another in their first year of treatment, relapsed. Those treated with focal RT relapsed later (>1.8 years) than those receiving chemotherapy only (<1.5years) (Fig. 4b; p = 0.02). The pattern of failure at first relapse was local for 4 patients and distant in 2. Distant relapses, however, were observed with subsequent recurrences. Of the eight patients, five are alive, all of whom received RT either as part of primary management or after relapse (focal [n=3] or CSI [n=2]) (Fig. 4c–d).
Fig. 4.
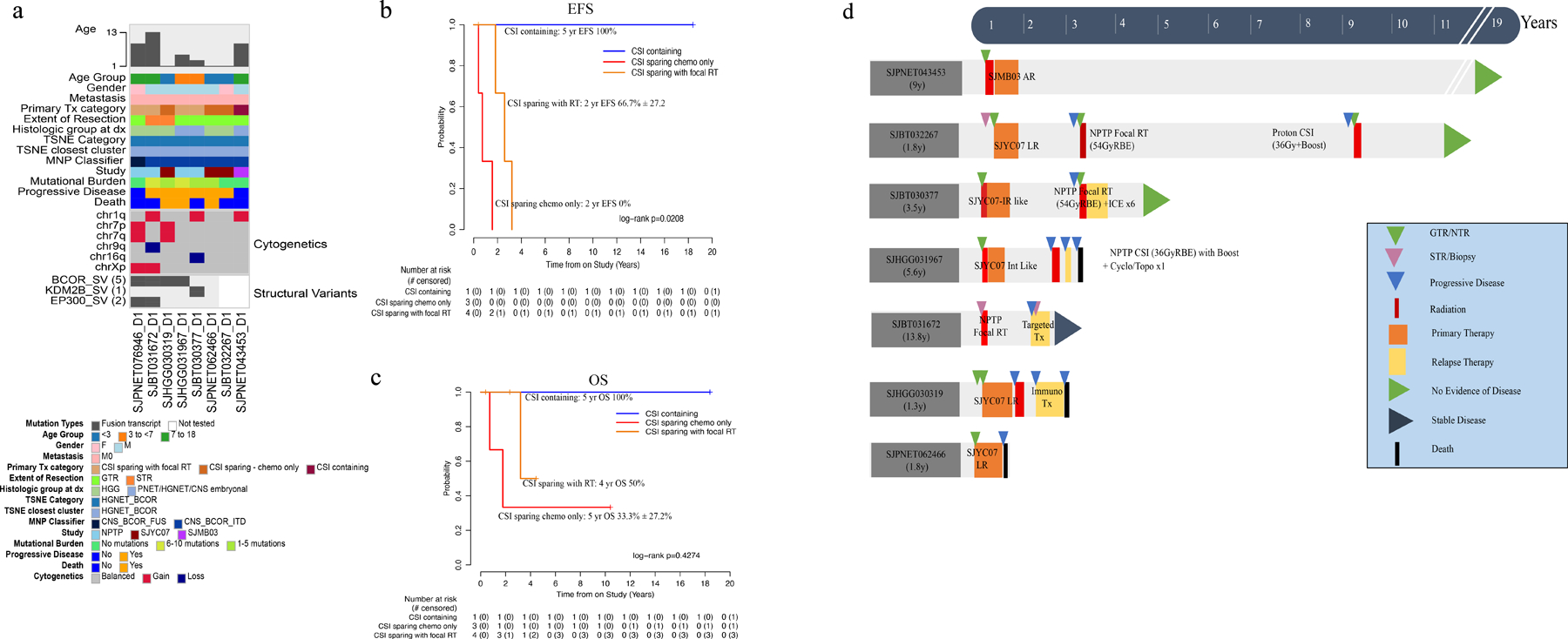
Molecular and clinical characteristics of HGNET-BCOR tumors. (a) Oncoprint aligning clinical features with molecular findings. (b-c) EFS and OS by treatment category. (d) Swimmer plot depicting all the different therapies (surgery, primary and relapse treatment) received from diagnosis through follow up by patients in this cohort. Abbreviations: AR, average risk; CNS, central nervous system, CNS-BCOR-FUS, CNS tumor with BCOR fusion; CNS-BCOR-ITD, CNS tumor with BCOR ITD; CSI, craniospinal irradiation; F, female; GTR, gross-total resection; HGG, high-grade glioma; HGNET, high-grade neuroepithelial tumor; HGNET-BCOR, high-grade neuroepithelial tumor with BCOR alteration; IR, intermediate-risk; ITD, internal tandem duplication; LR, low-risk; M, male; M0, non-metastatic; M+, metastatic; MNP classifier, Molecular Neuropathology brain tumor classifier; NPTP, non-protocol treatment plan; PNET, primitive neuro-ectodermal tumor; RT, radiation therapy; STR, subtotal resection; TSNE, t-stochastic neighbor embedding.
CIC rearranged sarcoma
By methylation, 7 patients had CNS-SARC-CIC with a median age of 1.81 years (0.65–11.88). Five were male and 2 were female. None were metastatic at diagnosis. Location of the primary tumor was cerebral in all. Seven (100%) had tumor sequencing, all displaying a low mutational burden and no recurrent mutations. Paired germline sequencing was available in 6 (86%). One ATM germline mutation was detected (Fig. 5a). All had sufficient tumor available for RNA-seq analysis and 5 fusions were detected: CIC-DUX4 in 2 patients; CIC-NUTM1, CIC-NUTM2B, ATXN1-NUTM2A were detected in 1 sample each (Fig. 5a; Supplemental Fig. S7). The primary treatment was CSI containing for 2 (SJMB03-AR = 1, SJMB03-HR = 1) and CSI-sparing with focal RT (SJYC07-IR/IR-like) for 5. GTR was achieved in 6 patients; STR in 1. Both 5-year EFS and OS for the entire cohort were 57.1% ± 18.7% (Fig. 2c–d). Three (43%) patients relapsed, all of whom had focal RT and chemotherapy (Fig. 5b–c). The pattern of failure was consistently local. The patient with an ATM germline mutation developed an in RT-field calvarial high-grade sarcoma 9 years from diagnosis. Four (57%) patients remain alive.
Fig. 5.
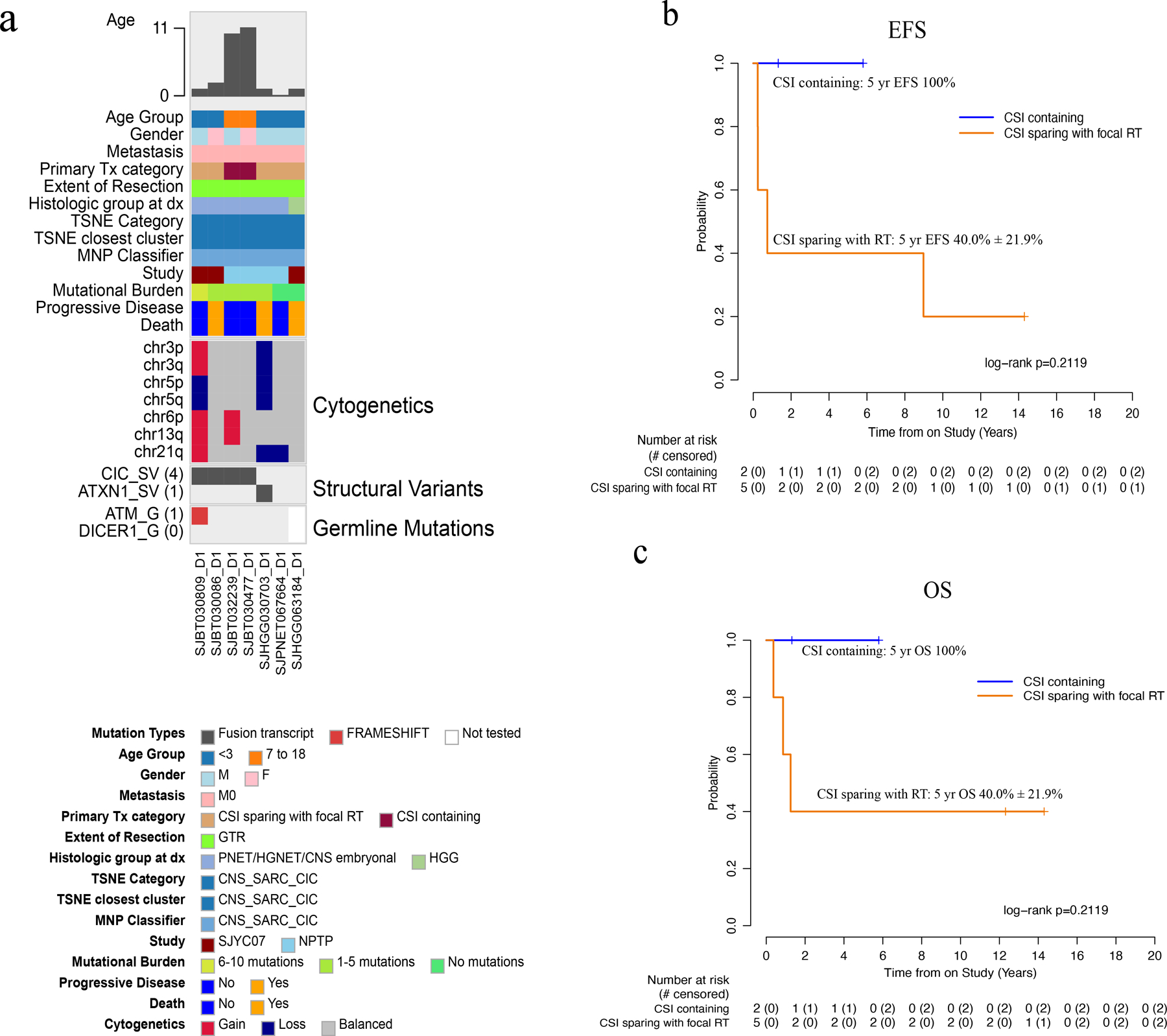
Molecular and clinical characteristics of CNS-SARC-CIC tumors. (a) Oncoprint aligning clinical features with molecular findings. (b-c) EFS and OS by treatment category. Abbreviations: CNS, central nervous system, CNS-SARC-CIC, CNS sarcoma with CIC rearrangement; CSI, craniospinal irradiation; F, female; GTR, gross-total resection; HGG, high-grade glioma; HGNET, high-grade neuroepithelial tumor; M, male; M0, non-metastatic; MNP, Molecular Neuropathology brain tumor classifier; NPTP, non-protocol treatment plan; PNET, primitive neuro-ectodermal tumor; RT, radiation therapy; TSNE, t-stochastic neighbor embedding.
CNS-NB-FOXR2
By methylation, 6 patients (3 male: 3 female) had CNS-NB-FOXR2 with an average age of 1.77 years (range 0.91–11.90). None were metastatic. At diagnosis 5 tumors were histologically defined as CNS-PNET/HGNET/CNS embryonal tumor and 1 was called HGG. Location of the primary tumor was cerebral in all patients. Five (83%) achieved GTR/NTR and one had STR. Five (83%) had copy number gain of chromosome 1q. Four (67%) had tumor sequencing, with low mutational burden and no recurrent mutations (Fig 6a). Paired germline sequencing was available in 3 (50%); no germline mutations were detected. Two had sufficient tumor available for RNA-seq analysis and high FOXR2 expression was seen in both (Fig. 6b). Primary treatment was CSI-containing for 1 (SJMB03-AR) and CSI sparing for 5 (CSI-sparing with focal RT = 4, CSI-sparing chemotherapy only = 1). The 5-year EFS and OS for the entire cohort were 66.7% ± 19.2% and 83.3% ± 15.2% (Fig. 2c–d). Two patients relapsed; one received SJYC07-LR therapy (chemotherapy only) and died; the other was treated with SJYC07-IR therapy and was salvaged with CSI (Fig. 6c–d). The pattern of failure was local in the patient treated with SJYC07-LR therapy and distant in the patient treated with SJYC07-IR therapy (with focal RT). Five (83%) of six patients are long-term survivors; three who received just focal RT and chemotherapy and two who received CSI (one as primary therapy, the other as salvage).
Fig. 6.
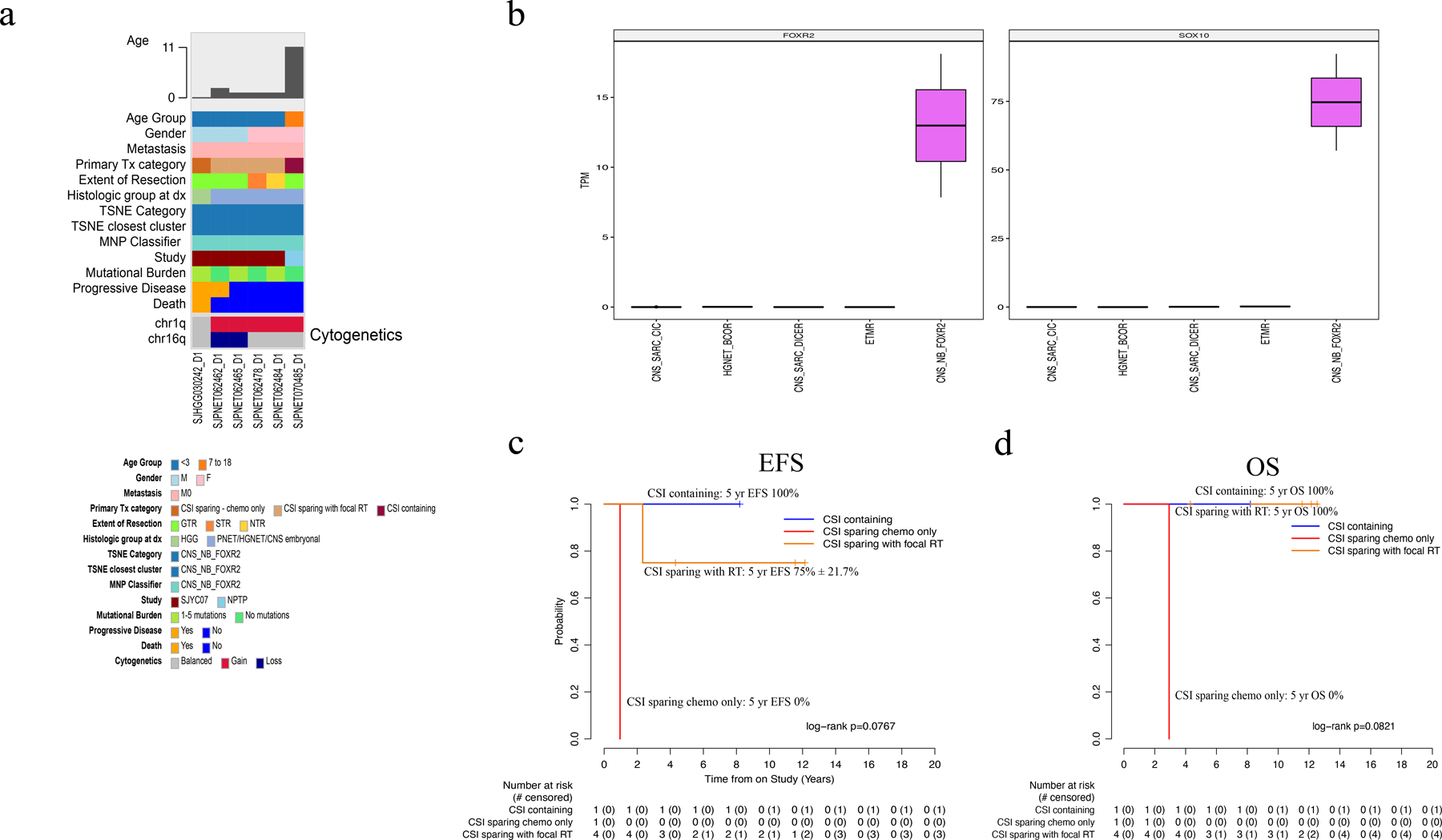
Molecular and clinical characteristics of CNS-NB-FOXR2 tumors. (a) Oncoprint aligning clinical features with molecular findings. (b) Box plots for FOXR2 and SOX10 genes show higher expression (transcript per million, TPM) in CNS-NB-FOXR2 samples than in other CNS embryonal tumors (c-d) EFS and OS by treatment category. Abbreviations: CNS, central nervous system, CNS-SARC-CIC, CNS sarcoma with CIC rearrangement; CNS-SARC-DICER, CNS sarcoma with DICER mutation, CNS-NB-FOXR2, CNS neuroblastoma with FOXR2 activation; CSI, craniospinal irradiation; ETMR, embryonal tumor with multilayered rosettes; F, female; GTR, gross-total resection; HGG, high-grade glioma; HGNET, high-grade neuroepithelial tumor; HGNET-BCOR, high-grade neuroepithelial tumor with BCOR alteration; M, male; M0, non-metastatic; MNP classifier, Molecular Neuropathology brain tumor classifier; NPTP, non-protocol treatment plan; NTR, near-total resection; PNET, primitive neuro-ectodermal tumor; RT, radiation therapy; STR, subtotal resection; TSNE, t-stochastic neighbor embedding.
Primary intracranial sarcoma, DICER1
Three tumors on TSNE formed a small cluster near CNS-SARC-CIC (Fig. 2a) and aligned with CNS-SARC-DICER1 on the MNP classifier. Median patient age was 4.71 years (range 2.69–10.52). One tumor was metastatic. Location of the primary tumor was cerebral in 2 and pineal in 1 patient. Two had sufficient tumor available for analysis; composite DICER1 mutations were found in both. All patients had germline sequencing performed and one had a pathogenic DICER1 mutation. The primary treatment was CSI-containing for 2 (SJMB03-AR) and CSI-sparing for 1 (CSI-sparing chemotherapy only) (Supplemental Fig. S4). The 5-year EFS and OS for the entire cohort were 0% and 33% ± 27%, respectively (Fig. 2c–d). Only one patient remains alive. This patient has germline DICER1 syndrome and, subsequent to the CNS tumor, has been diagnosed with a spontaneously resolved pleuropulmonary blastoma, thyroid carcinoma, and a Sertoli-Leydig carcinoma yet the original CNS-SARC-DICER1 remained in remission after CSI-containing therapy.
HGG
By methylation, 10 patients (6 male: 4 female) had HGG. More precisely these tumors separated into diffuse hemispheric glioma, H3-G34 mutant (n=3; DHG_G34) and diffuse pediatric-type high-grade glioma, H3-wildtype and IDH-wildtype (n=7; pHGG_MID, pHGG_MYCN, pHGG_RTK3). [9]. The median age was 13.03 years (range 2.14–15.56). At diagnosis all patients were histologically defined as CNS-PNET/HGNET/CNS embryonal tumor. Only one (10%) tumor was metastatic. Primary tumor was cerebral in all. Six (60%) had tumor sequencing but no germline or RNA sequencing could be performed. The most common recurrent mutations were TP53 (n=4), ATRX (n=3), and H3–3A G34R (n=3). The primary treatment was CSI-containing for 8 (SJMB03-AR = 5; SJMB03-HR = 3) and CSI sparing for 2 (CSI-sparing with focal RT = 1, CSI-sparing chemotherapy only = 1) (Supplemental Fig. S5). The 4-year EFS and OS for the entire cohort were 10.0%±9.0% and 10.0%±9.0%, respectively (Fig 2c–d). There was only one long-term survivor from this group with a diagnosis of pHGG_MID and treated with CSI-sparing therapy with focal RT (SJYC07-IR/IR-like).
CNS-ET-NEC/NOS
Five patients (3 male, 2 female) had tumors that were not easy to classify but clustered together on TSNE near EPN-ZFTA (Fig. 2a). Two of these were identified as HGNET-PLAGL1 on the MNP classifier but the others did not classify with this grouping. The median age of this cluster was 2.90 years (range 0.64–12.66). None were metastatic. Location of the primary tumor was cerebral in 4 and brainstem in 1. Five (100%) had tumor sequencing. Paired germline sequencing was available in 2 (40%) and none had sufficient tumor available for RNA sequencing. No recurrent mutations were identified. One had an EZHIP mutation in a previously described hotspot (D81A) [11]. The primary treatment was CSI-containing for 1 (SJMB03-AR = 1) and CSI-sparing for 4 patients (CSI-sparing with focal RT = 2, CSI-sparing chemotherapy only = 2) (Supplemental Fig. S6). The 5-year EFS and OS for the entire cohort were 40.0% ± 21.9% and 40.0 ± 21.9% (Fig 2c–d), respectively. There were 2 long-term survivors: 1 received SJYC07-IR/IR-like therapy and the other SJMB03 therapy.
Others
Three other tumor types observed in this collection were PXA (n=1), EWS (n=1) and CPT (n=1) by methylation. All are long-term survivors and each received slightly different therapy post-surgery with 2 receiving focal radiation (PXA and CPT) and one chemotherapy only (EWS).
Discussion
Here we describe a collection of 70 tumors, gathered over a 20-year timeframe, that fell within the now defunct histomorphologic category of CNS-PNET. When studied as a whole, survival was poor (range: 20–35%) even with therapies that are risk-stratified by clinical characteristics (age, stage, and histology). Notably, survival benefit was not apparent with the use of CSI-containing regimen for the overall cohort, despite such approach being the current standard-of-care for non-infant patients with CNS-PNET. This is distinct from the behavior of other pediatric embryonal tumors, such as medulloblastoma, ATRT, and pineoblastoma where survival improves with more expansive fields of radiation, albeit with increases in long-term neurocognitive morbidities [8, 14, 19]. Nevertheless, because recent studies have shown former CNS-PNETs to be a heterogenous collection of tumors, we suspected that analysis based on the collective grouping of these tumors was producing misleading results. In other words, we reasoned that the lumping of very different tumor types into a single group, while allowing for broad generalizations, like the above-mentioned lack of benefit from CSI, was in fact drowning out the finer details and producing questionable results. Hence, to gain better insights into the more specific entities that have supplanted CNS-PNET and document their response to conventional therapeutic options, we characterized this cohort by methylation profiling, performed next-generation sequencing on all available samples, and chronicled the treatment that was received. This said, while we believe the best way to interpret the results is by individual molecular class, we must also emphasize that due to the resulting small sample size of each group, our conclusions are limited and should be interpreted as such.
All things considered, we did find that more than half of this cohort was comprised of two highly aggressive tumors: ETMR (n=28) and HGG (n=10). The overall survival from these tumors was < 18% and, as such, contributed to the low survival of the entire group.
In ETMR we found many of the expected molecular characteristics, confirming that this group of tumors formed a prototypic cohort. Three-quarters harbored the C19MC amplicon or, when not present, rare DICER1 mutations as previously reported [6, 7]. These patients were very young and their outcomes dismal. There were a few survivors (n=5: 18%), yet, despite our efforts, we could not identify any unique or unifying characteristics among them to explain their survival. Notably, neither administration of CSI nor any form of RT was predictive of better survival, even though four of five survivors did receive some form of RT. Considering the recent Rare Brain Tumor Registry study describing benefit of GTR, location, and radiation therapy [4], it was unexpected that of the five surviving patients 2 had STR, 1 had a brainstem tumor, and 2 were metastatic at diagnosis, but our sample size was small. Overall, this data supports the dire need for novel therapeutic options for patient with ETMR who may be best served by enrolling on clinical trials that are informed by preclinical testing on ETMR model systems [6, 7].
In agreement with other studies, we observed that several HGG/GBMs can masquerade as embryonal tumors [3, 16]. In this cohort all were originally classified as CNS-PNET/HGNET/CNS embryonal tumor by histologic assessment, yet, by methylation, they classified into one of the many HGG/GBM categories. Whenever available, next generation sequencing identified mutations and aberrations in associated genes, such as H3–3A, TP53, ATRX or MYCN, that supported the methylation assignment over the original histologic grouping. Like ETMR, prognosis remained poor regardless of the type of therapy received. Only one of the ten patients survived and all patients who got CSI-containing therapy died. Nonetheless, while survival is not currently expected to improve with a more accurate diagnosis, it behooves us to correctly categorize these tumors to improve prognostication for families and to avoid more toxic therapies, like CSI or myeloablative chemotherapy, with limited impact on efficacy.
Of the remaining cases that did not classify with ETMR or HGG (n=32), we recorded many interesting observations but, most important, we found that many of these tumors are survivable. This suggests that, with better diagnostic tools, we can focus on better understanding these entities, tailor our current therapies to provide the most benefit and, hopefully, improve outcomes for the next generation with these rare diseases.
The most favorable outcomes were seen in the CNS-NB-FOXR2 group. Notably, this high survival was achieved in a cohort whose average age was younger than what has been previously described [20]. However, a key observation was that all five of the survivors required some form of RT (2 CSI, 3 focal RT) and, while only one received chemotherapy alone, he did not survive. Therefore, given this data and that from a recent published series by von Hoff et al. where relapse was highest in the nonirradiated patients and 89% 16/18 patients (89%) who received CSI followed by chemotherapy survived event-free [20], it is our current recommendation to treat older children (≥ 3 years) with a risk-adapted CSI-containing regimen and younger patients (< 3-years) with localized disease with a CSI-sparing regimen that consolidates with focal RT.
For HGNET-BCOR we described 8 cases that were characteristically challenging to diagnose and molecularly and clinically heterogeneous. Due to this ambiguity, these patients were either originally diagnosed histologically as HGG or CNS-PNET/HGNET/CNS embryonal tumors. As a result, the youngest patients (< 3 years) with a diagnosis of HGG received a low-risk chemotherapy-only regimen whereas the CNS-PNET/HGNET/CNS embryonal tumors and older patients got regimens containing focal RT or CSI. Although largely anecdotal, patients who received radiation appeared to have more durable disease control. Given this, we currently recommend treating HGNET-BCOR with RT sooner rather than later after a maximal safe surgical resection and avoid delaying radiation with chemotherapy. For children ≥ 3 years old, a CSI containing regimen appears warranted, and for children < 3 years focal RT is recommended but late metastatic disease should be anticipated. With regards to post-RT chemotherapy the benefit is unclear, and more information is needed. Of interest, within this group, we described a new fusion (KDM2B-NUTM1) and found two patients with EP300-BCOR fusions. While the patient with KDM2B-NUTM1 fused tumor presented similarly to those with the BCOR_ITD, the patients with EP300 fused tumors were older and their tumors were radiographically more diffuse, suggesting they may be distinct [12, 17].
We identified seven patients with methylation profiled CNS-SARC-CIC tumors. Four had fusions involving CIC and one displayed a fusion in ATXN1-NUTM2A/B. This marks the third ATXN1 fusion that, to our knowledge, has been reported in this group suggesting driver genes other than CIC are present in this entity [13]. In this category, all patients received surgery, chemotherapy, and RT. The younger patients received focal RT and the older patients received CSI. While not significantly different, the older patients exhibited a higher EFS, supporting our current recommendation to treat with CSI-containing therapy when permitted by age.
A new category that has recently been identified is CNS-SARC-DICER1 [5]. Within our cohort we identified 3 patients with this and subsequently found DICER1 mutations in 2. While little can be inferred from the treatment of such a small sample size, one of these patients was found to have a germline DICER1 mutation. This led to the early detection of multiple DICER1 syndrome related neoplasms from the subsequent surveillance. Relatedly, another patient, who developed CNS-SARC-CIC at < 2 years-old, was found to have a calvarial high-grade sarcoma on surveillance imaging at nine-years post-therapy, prompting further therapy and genetic consultation which identified an ATM germline mutation. The identification of two germline predisposition syndromes in a cohort where none were previously expected, highlights the importance of expanded testing to all children with malignancy [10, 21].
Finally, we did uncover a small number of tumors that did not classify well, highlighting that all is not understood within the new categorizations of CNS embryonal tumors. Here, we labelled these as CNS-ET-NEC/NOS tumors as recommended by the 5th edition CNS tumors volume of the WHO classification [9], since they lack sufficient data to classify them and have left these in our cohort as a reference for future exploration. Of note, the newest MNP classifier (12.5 version) categorized 2 of these as HGNET_PLAG which suggests a possible relationship with this new entity [15], but the 3 remaining samples did not yield the same result (GBM_MYCN=1, unclassifiable = 2). Furthermore, an EZHIP mutation, which has been described in posterior fossa ependymoma [2], was seen in one.
As previously mentioned, the main limitation of this study is that, despite the uniformity in treatment across two main institutional clinical trials, whether on protocol or as per, the end-results are underpowered and largely descriptive, making our recommendations more akin to suggestions than certainties. Nevertheless, it is important to emphasize that CSI-containing primary therapy was administered to this cohort in a risk-adapted fashion which, in the absence of any superior outcome from higher dosing [3], should, in our opinion, continue.
Consequently, given the rarity of these data, we have constructed a visually interactive and easy to use portal named “ Rare CNS Embryonal Tumors ” as an accompaniment to this manuscript. This portal allows a user to select their cohort of interest, query mutations, search relevant molecular findings, explore treatment, and generate survival outcomes. Links to the raw molecular data are also available to facilitate comparison of our data to additional samples.
In conclusion, we have described a very rich dataset on exceedingly rare CNS embryonal tumors that advances our understanding of these tumors and facilitates clinical decision making over an area that currently lacks direction. While we acknowledge much more needs to be learned, we hope that this manuscript assists in making more informed treatment decisions and fosters collaboration while our knowledge base of these uncommon tumors continues to grow.
Supplementary Material
Supplementary Table S1 Metadata for the study cohort
Supplementary Table S2 Filtered list of variants called from whole-exome sequencing
Supplementary Fig. S1 Schematic summary of treatment approach from (a) SJMB03 and (b) SJYC07
{kind=link}
Supplementary Fig. S2 Overall survival analysis for whole cohort, by gender, metastatic status, age, extent of resection, histology, enrollment on protocol, protocol, receipt of CSI, receipt of CSI or focal RT. Abbreviations: Biop, biopsy; CNS, central nervous system; CNS-ET, CNS-embryonal tumor; CSI, craniospinal irradiation; EFS, event-free survival; EOR, extent of resection; GTR, gross-total resection; HGNET, high-grade neuroepithelial tumor; M0, non-metastatic; M+, metastatic; NPTP, non-protocol treatment plan; NTR, near-total resection; OS, overall survival; PNET, primitive neuro-ectodermal tumor; RT, radiation therapy.
{kind=link}
Supplementary Fig. S3 Event-free survival and overall survival by primary treatment risk. Abbreviations: AR, average risk, EFS, event-free survival; HR, high-risk; IR, intermediate-risk; LR, low-risk; OS, overall survival; PNET, primitive neuro-ectodermal tumor.
{kind=link}
Supplementary Fig. S4 Molecular and clinical characteristics of CNS-SARC-DICER tumors. (a) Oncoprint aligning clinical features with molecular findings. (b-c) EFS and OS by treatment category. Abbreviations: CNS, central nervous system, CNS-SARC-DICER, CNS sarcoma with DICER mutation; CSI, craniospinal irradiation; F, female; GTR, gross-total resection; HGNET, high-grade neuroepithelial tumor; M, male; M0, non-metastatic; M+, metastatic; MNP classifier, Molecular Neuropathology brain tumor classifier; NPTP, non-protocol treatment plan; PNET, primitive neuro-ectodermal tumor; TSNE, t-stochastic neighbor embedding.
{kind=link}
Supplementary Fig. S5 Molecular and clinical characteristics of HGG tumors. (a) Oncoprint aligning clinical features with molecular findings. (b-c) EFS and OS by treatment category. Abbreviations: CNS, central nervous system; CSI, craniospinal irradiation; DHG-G34, diffuse hemispheric glioma H3 G34 mutant; F, female; G34, high-grade glioma subclass G34; GTR, gross-total resection; HGG, high-grade glioma; HGNET, high-grade neuroepithelial tumor; M, male; M0, non-metastatic; M+, metastatic; MID, high-grade glioma subclass MID; MNP classifier, Molecular Neuropathology brain tumor classifier; MYCN, high-grade glioma subclass MYCN; NPTP, non-protocol treatment plan; NTR, near-total resection; pedHGG-MYCN, pediatric-type high grade glioma, subclass MYCN; pedHGG-RTK1C, pediatric-type high grade glioma, subclass RTK1C; PNET, primitive neuro-ectodermal tumor; RT, radiation therapy; RTK_III, high-grade glioma subclass RTKIII; STR, subtotal resection; TSNE, t-stochastic neighbor embedding.
{kind=link}
Supplementary Fig. S6 Molecular and clinical characteristics of CNS-ET-NEC/NOS tumors. (a) Oncoprint aligning clinical features with molecular findings. (b-c) EFS and OS by treatment category. Abbreviations: CNS, central nervous system; CSI, craniospinal irradiation; F, female; GTR, gross-total resection; HGNET, high-grade neuroepithelial tumor; HGNET-PLAG, high-grade neuroepithelial tumor with PLAG alteration; M, male; M0, non-metastatic; MNP classifier, Molecular Neuropathology brain tumor classifier; pedHGG-MYCN, pediatric-type high grade glioma, subclass MYCN; PNET, primitive neuro-ectodermal tumor; RT, radiation therapy; STR, subtotal resection; TSNE, t-stochastic neighbor embedding.
{kind=link}
Supplementary Fig. S7 Fusion plots for (a) ETMR: TTYH1 is fused with MIR512 (SJBT031447) and MIR373 (SJHGG030284) on chromosome 19; (b) HGNET-BCOR: BCOR-ITD is shown in 3 cases (SJBT032267, SJBT032239, SJHGG030319); KDM2B-NUTM1 is shown in 1 (SJBT30377); and BCOR-EP300 and BCOR-L3MBTL2 are shown in 2 (SJBT031672, SJBT076946). (c) CNS-SARC-CIC: CIC is involved in 3 cases (SJBT030809, SJBT030477, SJBT030086, SJBT032239) and ATXN1 in one (SJBT030703).
{kind=link}
Acknowledgements
The authors acknowledge patients and families, nursing, and research staff from all participating institutions. In particular, we thank the St. Jude Biorepository for archiving and preservation of patient samples, the Cancer Biomarkers Lab for the processing of samples, the St. Jude Hartwell Center for next-generation sequencing, the St Jude Center for Applied Bioinformatics for assistance with bioinformatic analysis. Funding was provided by American Lebanese Syrian Associated Charities and National Cancer Institute Cancer Center Grant (P30CA021765), St. Jude Comprehensive Cancer Center Developmental Funds, and The Press On Fund.
References
- 1.Capper D, Jones DT, Sill M, Hovestadt V, Schrimpf D, Sturm D, Koelsche C, Sahm F, Chavez L, Reuss DE (2018) DNA methylation-based classification of central nervous system tumours. Nature 555: 469–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hübner J-M, Müller T, Papageorgiou DN, Mauermann M, Krijgsveld J, Russell RB, Ellison DW, Pfister SM, Pajtler KW, Kool M (2019) EZHIP/CXorf67 mimics K27M mutated oncohistones and functions as an intrinsic inhibitor of PRC2 function in aggressive posterior fossa ependymoma. Neuro-oncology 21: 878–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hwang EI, Kool M, Burger PC, Capper D, Chavez L, Brabetz S, Williams-Hughes C, Billups C, Heier L, Jaju A (2018) Extensive molecular and clinical heterogeneity in patients with histologically diagnosed CNS-PNET treated as a single entity: a report from the children’s oncology group randomized ACNS0332 trial. Journal of Clinical Oncology 36: 3388–3395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khan S, Solano-Paez P, Suwal T, Lu M, Al-Karmi S, Ho B, Mumal I, Shago M, Hoffman LM, Dodgshun A (2021) Clinical phenotypes and prognostic features of embryonal tumours with multi-layered rosettes: a Rare Brain Tumor Registry study. The Lancet Child & Adolescent Health 5: 800–813 [DOI] [PubMed] [Google Scholar]
- 5.Koelsche C, Mynarek M, Schrimpf D, Bertero L, Serrano J, Sahm F, Reuss DE, Hou Y, Baumhoer D, Vokuhl C (2018) Primary intracranial spindle cell sarcoma with rhabdomyosarcoma-like features share a highly distinct methylation profile and DICER1 mutations. Acta Neuropathologica 136: 327–337 [DOI] [PubMed] [Google Scholar]
- 6.Lambo S, Gröbner SN, Rausch T, Waszak SM, Schmidt C, Gorthi A, Romero JC, Mauermann M, Brabetz S, Krausert S (2019) The molecular landscape of ETMR at diagnosis and relapse. Nature 576: 274–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lambo S, von Hoff K, Korshunov A, Pfister SM, Kool M (2020) ETMR: a tumor entity in its infancy. Acta Neuropathologica 140: 249–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu AP, Gudenas B, Lin T, Orr BA, Klimo P, Kumar R, Bouffet E, Gururangan S, Crawford JR, Kellie SJ (2020) Risk-adapted therapy and biological heterogeneity in pineoblastoma: integrated clinico-pathological analysis from the prospective, multi-center SJMB03 and SJYC07 trials. Acta Neuropathologica 139: 259–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, Hawkins C, Ng H, Pfister SM, Reifenberger G (2021) The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro-oncology 23: 1231–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Newman S, Nakitandwe J, Kesserwan CA, Azzato EM, Wheeler DA, Rusch M, Shurtleff S, Hedges DJ, Hamilton KV, Foy SG (2021) Genomes for Kids: The Scope of Pathogenic Mutations in Pediatric Cancer Revealed by Comprehensive DNA and RNA Sequencing. Cancer Discovery 11: 3008–3027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pajtler KW, Wen J, Sill M, Lin T, Orisme W, Tang B, Hübner J-M, Ramaswamy V, Jia S, Dalton JD (2018) Molecular heterogeneity and CXorf67 alterations in posterior fossa group A (PFA) ependymomas. Acta Neuropathologica 136: 211–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pisapia DJ, Ohara K, Bareja R, Wilkes DC, Hissong E, Croyle JA, Kim J-H, Saab J, MacDonald TY, Beg S (2020) Fusions involving BCOR and CREBBP are rare events in infiltrating glioma. Acta Neuropathologica Communications 8: 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pratt D, Kumar-Sinha C, Cieślik M, Mehra R, Xiao H, Shao L, Franson A, Cantor E, Chinnaiyan AM, Mody R (2021) A novel ATXN1-DUX4 fusion expands the spectrum of ‘CIC-rearranged sarcoma’of the CNS to include non-CIC alterations. Acta Neuropathologica 141: 619–622 [DOI] [PubMed] [Google Scholar]
- 14.Robinson GW, Rudneva VA, Buchhalter I, Billups CA, Waszak SM, Smith KS, Bowers DC, Bendel A, Fisher PG, Partap S (2018) Risk-adapted therapy for young children with medulloblastoma (SJYC07): therapeutic and molecular outcomes from a multicentre, phase 2 trial. The Lancet Oncology 19: 768–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sievers P, Henneken SC, Blume C, Sill M, Schrimpf D, Stichel D, Okonechnikov K, Reuss DE, Benzel J, Maaß KK (2021) Recurrent fusions in PLAGL1 define a distinct subset of pediatric-type supratentorial neuroepithelial tumors. Acta Neuropathologica 142: 827–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sturm D, Orr BA, Toprak UH, Hovestadt V, Jones DT, Capper D, Sill M, Buchhalter I, Northcott PA, Leis I (2016) New brain tumor entities emerge from molecular classification of CNS-PNETs. Cell 164: 1060–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Torre M, Meredith DM, Dubuc A, Solomon DA, Perry A, Vasudevaraja V, Serrano J, Snuderl M, Ligon KL, Alexandrescu S (2019) Recurrent EP300-BCOR fusions in pediatric gliomas with distinct clinicopathologic features. Journal of Neuropathology & Experimental Neurology 78: 305–314 [DOI] [PubMed] [Google Scholar]
- 18.Upadhyaya SA, Robinson GW, Onar-Thomas A, Orr BA, Billups CA, Bowers DC, Bendel AE, Hassall T, Crawford JR, Partap S (2019) Molecular grouping and outcomes of young children with newly diagnosed ependymoma treated on the multi-institutional SJYC07 trial. Neuro-oncology 21: 1319–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Upadhyaya SA, Robinson GW, Onar-Thomas A, Orr BA, Johann P, Wu G, Billups CA, Tatevossian RG, Dhanda SK, Srinivasan A (2021) Relevance of Molecular Groups in Children with Newly Diagnosed Atypical Teratoid Rhabdoid Tumor: Results from Prospective St. Jude Multi-institutional TrialsMolecular Grouping and Outcomes for Pediatric ATRT. Clinical Cancer Research 27: 2879–2889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Von Hoff K, Haberler C, Schmitt-Hoffner F, Schepke E, De Rojas T, Jacobs S, Zapotocky M, Sumerauer D, Perek-Polnik M, Dufour C (2021) Therapeutic implications of improved molecular diagnostics for rare CNS embryonal tumor entities: Results of an international, retrospective study. Neuro-oncology 23: 1597–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, Hedges D, Ma X, Zhou X, Yergeau DA (2015) Germline mutations in predisposition genes in pediatric cancer. New England Journal of Medicine 373: 2336–2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou X, Edmonson MN, Wilkinson MR, Patel A, Wu G, Liu Y, Li Y, Zhang Z, Rusch MC, Parker M (2016) Exploring genomic alteration in pediatric cancer using ProteinPaint. Nature Genetics 48: 4–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1 Metadata for the study cohort
Supplementary Table S2 Filtered list of variants called from whole-exome sequencing
Supplementary Fig. S1 Schematic summary of treatment approach from (a) SJMB03 and (b) SJYC07
Supplementary Fig. S2 Overall survival analysis for whole cohort, by gender, metastatic status, age, extent of resection, histology, enrollment on protocol, protocol, receipt of CSI, receipt of CSI or focal RT. Abbreviations: Biop, biopsy; CNS, central nervous system; CNS-ET, CNS-embryonal tumor; CSI, craniospinal irradiation; EFS, event-free survival; EOR, extent of resection; GTR, gross-total resection; HGNET, high-grade neuroepithelial tumor; M0, non-metastatic; M+, metastatic; NPTP, non-protocol treatment plan; NTR, near-total resection; OS, overall survival; PNET, primitive neuro-ectodermal tumor; RT, radiation therapy.
Supplementary Fig. S3 Event-free survival and overall survival by primary treatment risk. Abbreviations: AR, average risk, EFS, event-free survival; HR, high-risk; IR, intermediate-risk; LR, low-risk; OS, overall survival; PNET, primitive neuro-ectodermal tumor.
Supplementary Fig. S4 Molecular and clinical characteristics of CNS-SARC-DICER tumors. (a) Oncoprint aligning clinical features with molecular findings. (b-c) EFS and OS by treatment category. Abbreviations: CNS, central nervous system, CNS-SARC-DICER, CNS sarcoma with DICER mutation; CSI, craniospinal irradiation; F, female; GTR, gross-total resection; HGNET, high-grade neuroepithelial tumor; M, male; M0, non-metastatic; M+, metastatic; MNP classifier, Molecular Neuropathology brain tumor classifier; NPTP, non-protocol treatment plan; PNET, primitive neuro-ectodermal tumor; TSNE, t-stochastic neighbor embedding.
Supplementary Fig. S5 Molecular and clinical characteristics of HGG tumors. (a) Oncoprint aligning clinical features with molecular findings. (b-c) EFS and OS by treatment category. Abbreviations: CNS, central nervous system; CSI, craniospinal irradiation; DHG-G34, diffuse hemispheric glioma H3 G34 mutant; F, female; G34, high-grade glioma subclass G34; GTR, gross-total resection; HGG, high-grade glioma; HGNET, high-grade neuroepithelial tumor; M, male; M0, non-metastatic; M+, metastatic; MID, high-grade glioma subclass MID; MNP classifier, Molecular Neuropathology brain tumor classifier; MYCN, high-grade glioma subclass MYCN; NPTP, non-protocol treatment plan; NTR, near-total resection; pedHGG-MYCN, pediatric-type high grade glioma, subclass MYCN; pedHGG-RTK1C, pediatric-type high grade glioma, subclass RTK1C; PNET, primitive neuro-ectodermal tumor; RT, radiation therapy; RTK_III, high-grade glioma subclass RTKIII; STR, subtotal resection; TSNE, t-stochastic neighbor embedding.
Supplementary Fig. S6 Molecular and clinical characteristics of CNS-ET-NEC/NOS tumors. (a) Oncoprint aligning clinical features with molecular findings. (b-c) EFS and OS by treatment category. Abbreviations: CNS, central nervous system; CSI, craniospinal irradiation; F, female; GTR, gross-total resection; HGNET, high-grade neuroepithelial tumor; HGNET-PLAG, high-grade neuroepithelial tumor with PLAG alteration; M, male; M0, non-metastatic; MNP classifier, Molecular Neuropathology brain tumor classifier; pedHGG-MYCN, pediatric-type high grade glioma, subclass MYCN; PNET, primitive neuro-ectodermal tumor; RT, radiation therapy; STR, subtotal resection; TSNE, t-stochastic neighbor embedding.
Supplementary Fig. S7 Fusion plots for (a) ETMR: TTYH1 is fused with MIR512 (SJBT031447) and MIR373 (SJHGG030284) on chromosome 19; (b) HGNET-BCOR: BCOR-ITD is shown in 3 cases (SJBT032267, SJBT032239, SJHGG030319); KDM2B-NUTM1 is shown in 1 (SJBT30377); and BCOR-EP300 and BCOR-L3MBTL2 are shown in 2 (SJBT031672, SJBT076946). (c) CNS-SARC-CIC: CIC is involved in 3 cases (SJBT030809, SJBT030477, SJBT030086, SJBT032239) and ATXN1 in one (SJBT030703).