

Abstract
Allergic asthma is a chronic lung disease characterized by airway hyperresponsiveness and cellular infiltration that is exacerbated by immunoglobulin E-dependent mast cell (MC) activation. Interleukin-9 (IL-9) promotes MC expansion during allergic inflammation but precisely how IL-9 expands tissue MCs and promotes MC function is unclear. In this report, using multiple models of allergic airway inflammation, we show that both mature MCs (mMCs) and MC progenitors (MCp) express IL-9R and respond to IL-9 during allergic inflammation. IL-9 acts on MCp in the bone marrow and lungs to enhance proliferative capacity. Furthermore, IL-9 in the lung stimulates the mobilization of CCR2+ mMC from the bone marrow and recruitment to the allergic lung. Mixed bone marrow chimeras demonstrate that these are intrinsic effects in the MCp and mMC populations. IL-9-producing T cells are both necessary and sufficient to increase MC numbers in the lung in the context of allergic inflammation. Importantly, T cell IL-9-mediated MC expansion is required for the development of antigen-induced and MC-dependent airway hyperreactivity. Collectively, these data demonstrate that T cell IL-9 induces lung MC expansion and migration by direct effects on the proliferation of MCp and the migration of mMC to mediate airway hyperreactivity.
INTRODUCTION
Allergic asthma is a chronic lung disease characterized by allergen-induced airway hyperresponsiveness (AHR), eosinophilic infiltration, and intermittent airway obstruction1. Airway inflammation and AHR are further exacerbated by immunoglobulin (Ig)E-dependent mast cell (MC) activation2. Upon allergen exposure, MCs degranulate to release pro-inflammatory mediators, such as histamine, proteases, leukotrienes, cytokines, and chemokines, that mediate immediate hypersensitivity and contribute to the pathological features of allergic asthma3,4.
Among the cytokines that regulate MCs, interleukin 9 (IL-9) was initially identified as an MC growth factor5,6. IL-9 signals by binding to its IL-9 receptor (IL-9R) consisting of a unique α chain (IL-9Rα) and a common γ chain. IL-9R is expressed in a variety of cells including epithelial cells, fibroblasts, granulocytes, lymphocytes, macrophages, and MCs7. The role of IL-9 in asthma pathophysiology and disease control has gained considerable attention8–10. Increasing evidence for the crucial role of IL-9 in allergic asthma is derived from clinical studies in which patients exhibited significantly elevated IL-9 and clusters of differentiation (CD4+) T helper 9 (TH9) cell numbers in the peripheral blood11–15. Consistent with these reports, elevated IL-9 expression strongly correlated with high serum IgE concentration in atopic patients, suggesting that IL-9 is associated with IgE-mediated allergic responses16. In asthma models, IL-9 is required for MC accumulation17–19, and IL-9 from T cells is required for MC expansion during allergic airway inflammation20. Yet, although MCs have the highest expression of IL-9R in the allergic lung8,9,12,21, exactly how IL-9 regulates MC expansion and function in vivo is not well described.
MCs are tissue-resident cells of hematopoietic origin that complete their differentiation in the peripheral microenvironment. Although both MCs and basophils express FcεRI and may derive from common precursors22,23, mature cells can be distinguished by their divergent expression of c-Kit and CD49b (Dx5)23,24. In mice, as MC progenitors (MCp) leave the bone marrow, they can be identified in the blood by Lineage (Lin)− c-Kithi ST2+ integrin β7 (Itgβ7)hi CD16/32 (FcγRII/III)hi with variable maturity defined by FcεRI expression25. This is consistent with multiple studies in mice utilizing expression of c-Kit, FcεRI, and Itgβ7 to distinguish MC maturation status23,26. Thus, while both MCp and mature MC (mMCs) express c-Kit and FcεRIα, there is a consensus that MC differentiation can be distinguished using flow cytometric analysis of Itgβ7, FcγRII/III, and side scatter (SSC) profile26–29.
In mouse models of allergic airway disease, MCp in the bone marrow can migrate to the allergic lung in a CCR2/CCL2, CXCR2/ CXCL3, and α4β7/VCAM-1-dependent manner30–33. Moreover, studies in mice have identified that IL-9 contributes to MC accumulation in lung tissue8,9,20,21,34. However, it is unclear if IL-9-mediated MC expansion is selective and if IL-9 only acts on tissue-resident mMCs.
Here, we define the mechanism for IL-9-mediated MC expansion in the allergic lung. We show that IL-9R is expressed at multiple stages of MC development and that proliferation and migration of MCs following allergen challenge was dependent upon IL-9 signaling. Additionally, we identify IL-9-producing CD4+ T cells as controllers of MC-dependent AHR.
RESULTS
IL-9R is constitutively expressed in multiple stages of MC development
To investigate if IL-9 affects MC at different stages of development, we first defined the expression of IL-9R on MCp and mMC within the lungs of naïve mice, identified based on published gating strategies: MCp (Lin− CD45+ c-kit+ CD49b− ST2+ Itgβ7+ CD16/32int FcεRIα+ SSClo) and mMC (Lin− CD45+ c-kit+ CD49b−/int ST2+ Itgβ7−/lo CD16/32+ FcεRIα+ SSChi) (Fig. 1A)27,28. A previous report demonstrated that in response to allergen exposure, IL-9R is upregulated in MCs compared to naïve controls8. In line with this, IL-9R was highly expressed on MCp and mMC with the greatest expression on mMC (Figs. 1B and 1C). Studies also show that inhibiting IL-9 signaling leads to significantly reduced lung MC numbers, indicating that MCs are highly responsive to IL-9 during an allergic response8,9,20. To begin to determine how IL-9 impacts MC populations in the lung, we investigated the effects of IL-9 blockade on MCp and mMC numbers in a chronic house dust mite (HDM) allergen exposure model using control or IL-9 neutralizing antibody during the last 2 weeks of HDM treatment (Fig. 1D). Blockade of IL-9 led to diminished MC accumulation in the trachea, while also significantly reducing MCp and mMC numbers in the bone marrow and lungs (Figs. 1E–G). Together, these data demonstrate that IL-9 can act on both MCp and mMC during an allergic airway inflammatory response.
Fig. 1.
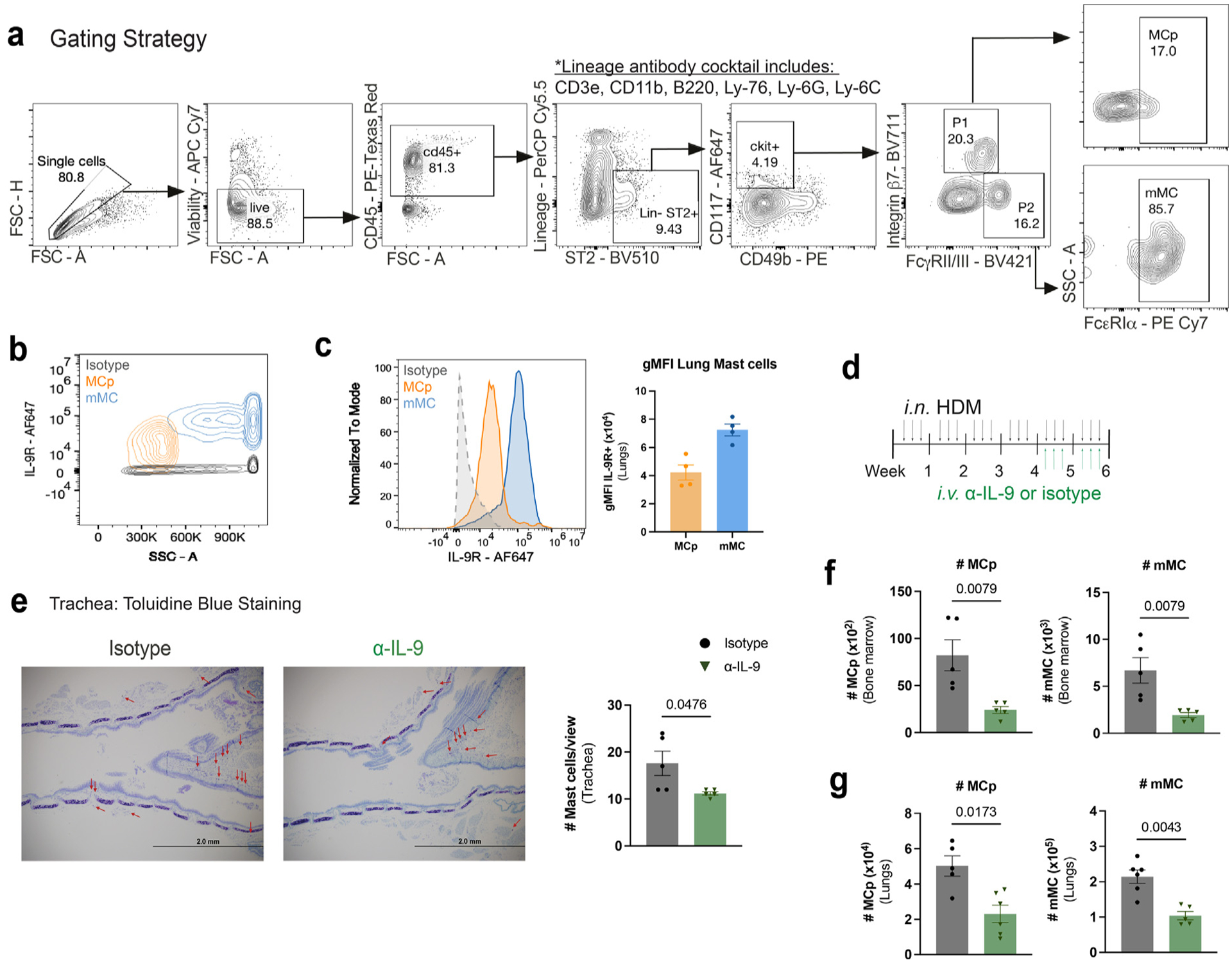
MCp and mMCs are IL-9 responders in the allergic lung. (A) Flow cytometry MC gating strategy using representative flow cytometry plots. P1 and P2 are further gated on FcεRIα+ cells. Mast cell progenitors (MCp) and mature mast cells (mMC). (B–C) Naïve WT lung MC were analyzed using flow cytometry for IL-9 receptor expression: B, representative flow cytometry contour plot of IL-9R expression on MC. (C) Histogram for IL-9R and geometric mean fluorescence intensity (gMFI) for each population was compared to isotype control (n = 3–4). (D–G) HDM-treated WT mice were treated with α-IL-9 or isotype control during the last 2 weeks of HDM treatment. (D) Schematic of experimental design. (E) MC numbers and frequencies were assessed in the trachea via toluidine blue staining; F–G: flow cytometry analysis of bone marrow (F) and lung (G) MC (n = 5–6) Data are representative of two independent experiments with similar results. Error bars indicate ± standard error of mean. Mann-Whitney U test was used for comparisons to generate p values in (C) and (E–G). APC = Allophycocyanin; CD=clusters of differentiation; FSC = forward scatter; HDM = house dust mite; IL = interleukin; MC = mast cell; MCp = MC progenitors; mMC = mature MC.
IL-9-mediated MC expansion is tissue-specific
We next sought to test whether local IL-9 production in the lungs has systemic effects on other peripheral tissues. To analyze the ability of IL-9 to mediate effects in the airways, we treated wild-type (WT) mice intranasally with recombinant IL-9 (rIL-9) for 3 consecutive days. Compared to phosphate-buffered saline (PBS)-treated mice, IL-9-treated mice exhibited MC accumulation in the trachea, but not in the skin or tongue (Fig. b and Supplementary Fig. 1A). However, bone marrow and lung MCs exhibited expansion in response to intranasal IL-9 that was not observed in the blood, mediastinal lymph node (draining lymph node), spleen or peritoneal cavity (Figs. 2B–D and Supplementary Fig. 1B). In contrast, intranasal IL-9 was insufficient to expand lung neutrophils, eosinophils, or basophils; suggesting that these cell types do not independently respond to IL-9 in this assay (Figs. 2E–G). Therefore, expansion of MC populations in the bone marrow and lungs following IL-9 administration likely reflects a cell type-specific and tissue-specific effect of IL-9 in the airways.
Fig. 2.
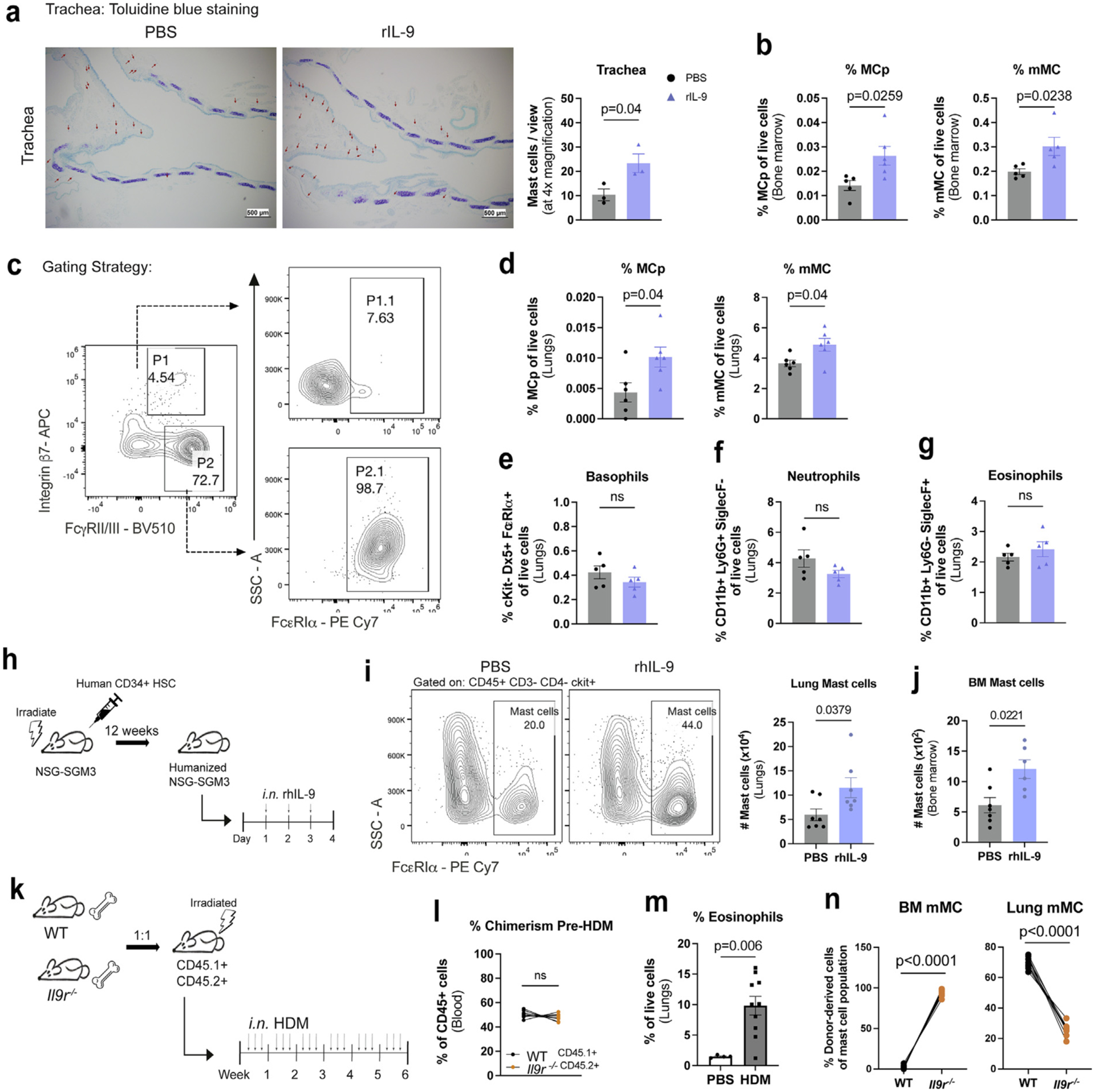
IL-9 acts intrinsically on bone marrow MC. (A–G) WT mice were treated intranasally with recombinant mouse IL-9 for 3 days: A, tracheal MC numbers were analyzed using toluidine blue staining. (B) MC frequencies in femur-derived bone marrow; C, flow cytometry gating strategy for lung MC frequencies in (D). (E–G) Flow cytometry analysis of Basophils (cKit- Dx5+ FcεRIα+), Neutrophils (CD11b+ Ly6G+ SiglecF-), and Eosinophils (CD11b+ Ly6G- SiglecF+). (n = 3–5) Data are representative of three independent experiments with similar results. (H–J) NSG mice were irradiated and reconstituted with human CD34+ cord blood cells. Following successful reconstitution of human CD45+ cells, mice were treated intranasally with recombinant human IL-9 for 3 days. (H) schematic of experimental design. (I) Flow cytometry analysis of lung MC. (J) Flow cytometry analysis of bone marrow MC. (n = 7–8). (K-N) WT and Il9r−/− bone marrow cells were transferred to lethally irradiated Boy/J x C57BL/6J F1 mice: (K) schematic of experimental design. (L) chimerism after 3 months was analyzed prior to HDM treatment using flow cytometry; M, frequency of lung eosinophils.(N) Bone marrow and lung MC were assessed for CD45.1 or CD45.2 expression by flow cytometry (PBS: n = 5; HDM: n = 10). Each data point represents an individual mouse. Data are representative of two independent experiments with similar results. Error bars indicate ± standard error of mean. Statistical significance was determined by analysis of variance, followed by Tukey’s multiple comparison test (N) or Mann-Whitney U test (A–B, D–G, I–J, and L–M). APC =; CD = clusters of differentiation; gMFI = geometric mean fluorescence intensity; HDM = house dust mite; IL = interleukin; MC = mast cell; MCp = MC progenitors; mMC = mature MC; ns = not significant; NSG = NOD-SCID, gamma chain-deficient mouse; PBS = phosphate buffered saline; PE= R-phycoerythrin; rh = recombinant human; SSC = side scatter; WT, wild type.
We then tested whether our findings are relevant for human MCs by generating humanized NSG-SGM3 mice that develop human MC and intranasally treating these mice with recombinant human IL-9 for 3 days (Fig. 2H)35. Similar to our findings in murine MC, we demonstrate that intranasal IL-9 enhances MC numbers in the lungs and bone marrow (Figs. 2I and 2J). Together, these findings support a conserved function of IL-9 in human and mouse MC expansion in the lungs and bone marrow.
IL-9 acts intrinsically on bone marrow MCs
To distinguish between direct and indirect effects of IL-9 on MCp and mMC, we utilized a mixed bone marrow chimera experiment in which C57BL/6 x Boy/J F1CD45.1+ CD45.2+ recipients were irradiated and reconstituted with bone marrow from donor WTCD45.1+ and Il9r−/− CD45.2+ (Fig. 2K). Prior to HDM treatment, chimeric mice were confirmed to express comparable frequencies of bone marrow reconstitution in the blood (Fig. 2L). After treatment with HDM, we observed greater frequencies of eosinophils, consistent with the induction of allergic airway inflammation (Fig. 2M). Il9r−/−-derived cells were enriched in the bone marrow mMC population, while lung MC were primarily WT-derived cells (Fig. 2N). The preferential increase of Il9r−/− mMC in the bone marrow could arise from altered MCp expansion and differentiation or that IL-9 signaling had an impact on mMC mobilization from the bone marrow.
IL-9 enhances MCp proliferative capacity
To directly test the impact of IL-9 signaling on MCp expansion, we analyzed mixed bone marrow chimeras to assess the contribution of WT and Il9r−/− -derived cells on MCp expansion in the bone marrow and lungs. MCp in the lungs and bone marrow were primarily WT-derived cells, indicating that MCp is largely dependent upon IL-9 signaling for expansion (Fig. 3A). From these results, we speculated that the stimulatory effects of IL-9 on MC may impact stem cell factor-dependent proliferative capacity that was observed in a previous study5. To test this, we used bone marrow-derived MC cultures grown in IL-3 and stem cell factor (SCF) and stimulated these cells with IL-9 for 2 hours. IL-9 increased bone marrow-derived MCp but not mMC proliferation (Figs. 3B and 3C). A short 3-day treatment with intranasal IL-9 was sufficient to induce lung but not bone marrow MCp proliferation (Fig. 3D).
Fig. 3.
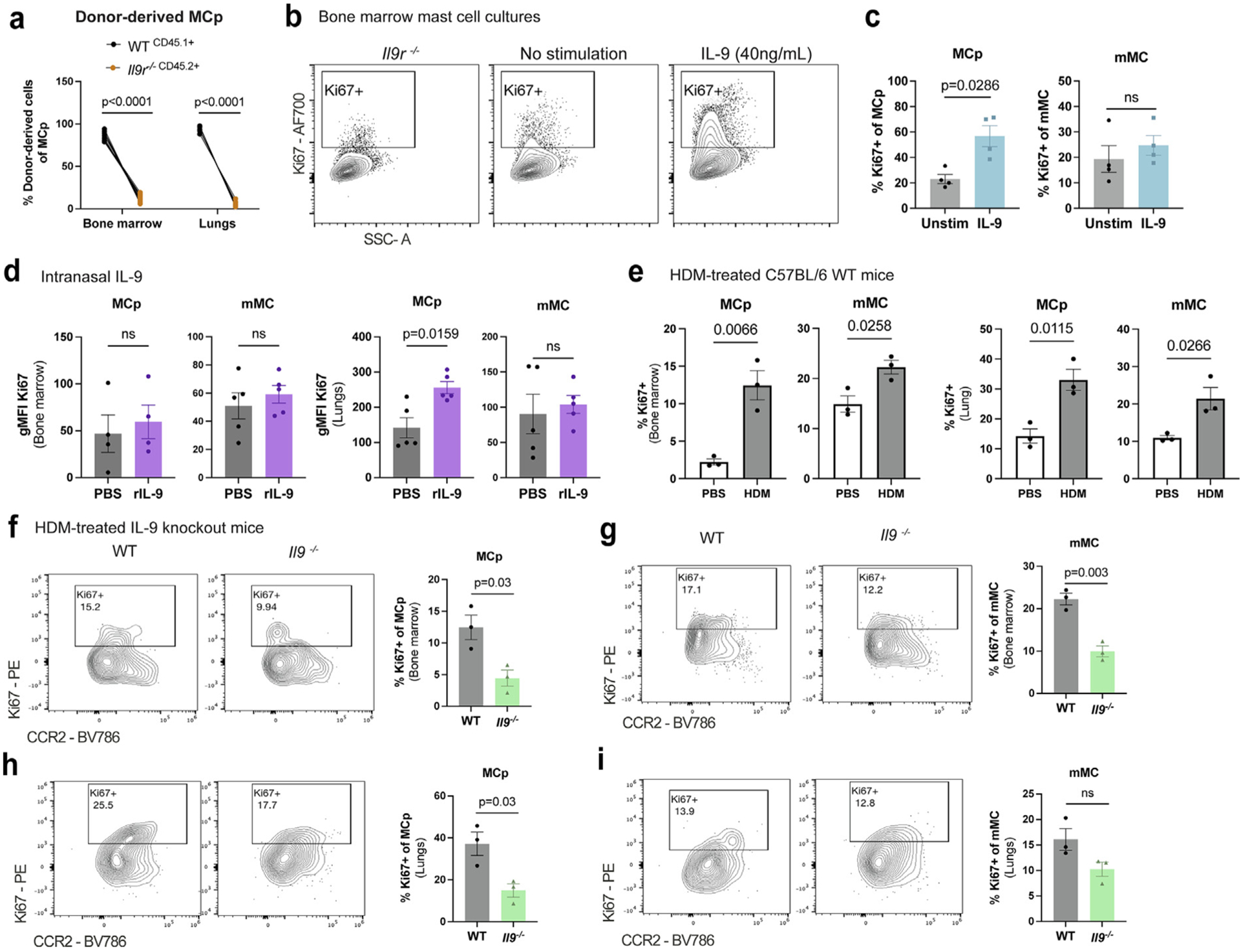
IL-9 enhances MC progenitor proliferative capacity. (A) WT (CD45.1+) and Il9r−/− (CD45.2+) bone marrow cells were transferred to lethally irradiated Boy/J x C57BL/6J F1 mice and after 3 months to allow repopulation of the immune system, mice were treated with HDM for 6 weeks. Flow cytometry analysis of CD45.1+ and CD45.2+ of lung MCp. (n = 10). (B–C) BMMC from WT mice were cultured for 2 weeks in IL-3 and SCF in RPMI. BMMC were harvested and stimulated with IL-9 (40 ng/ml) for 2 hours to assess intracellular Ki67 using flow cytometry. (B) flow cytometry plots of Ki67 staining in BMMC with WT and Il9r−/− BMMC. (C) Flow cytometry analysis of Ki67 frequencies in MCp and mMC (n = 4). (D) WT mice were intranasally treated with rIL-9 for 3 days. Flow cytometry of Ki67 gMFI was measured from bone marrow and lung MC (n = 5); E, flow cytometry analysis of Ki67 was assessed in lung MC from 6-week HDM-treated WT mice or PBS controls (n = 3). (F–I) Il9−/− and WT mice were treated intranasally with HDM 3x/week for 6 weeks. Ki67 expression was assessed via flow cytometry in (F–G) bone marrow and (H–I) lung MC (n = 3). Each data point represents an individual mouse. Data are representative of two independent experiments with similar results. Error bars indicate ± standard error of mean. Statistical significance was determined by analysis of variance, followed by Sidak’s multiple comparison test (A), Mann-Whitney U test (C–D), and Student’s t test (E–I). CD = clusters of differentiation; gMFI = geometric mean fluorescence intensity; HDM = house dust mite; IL = interleukin; MC = mast cell; MCp = MC progenitors; mMC = mature MC; ns = not significant; PBS = phosphate buffered saline; PE = R-phycoerythrin; r = recombinant; SSC = side scatter; WT, wild type.
We then compared the ability of allergen challenge to alter proliferative capacity. After a 6-week HDM chronic challenge, bone marrow and lung MCp and mMC displayed greater Ki67 expression compared to PBS-treated controls (Fig. 3E). To examine whether IL-9 contributed to allergen-dependent MCp proliferation in vivo, we utilized IL-9 deficient mice (Il9−/−) and treated the mice with HDM for 6 weeks. Il9−/− mice exhibited diminished MCp and mMC proliferation in the bone marrow (Figs. 3F and 3G). Likewise, IL-9 deficiency led to reduced lung MCp proliferation, whereas mMC displayed a trending decrease in proliferation as compared to PBS controls (Figs. 3H and 3I). This is consistent with mMC having a limited proliferative potential36. Together, these results indicate that IL-9 signaling enhances MCp proliferative capacity but that although IL-9 promotes the expansion of mMC in the lung, it does not promote the proliferation of lung mMC.
IL-9 promotes CCR2-dependent bone marrow MC recruitment to the allergic lung
Previous studies have shown that IL-9 promotes CCR2-mediated bone marrow monocyte migration to the lung and that MC can migrate in a CCR2/CCL2-dependent manner to the allergic lung in BALB/c mice8,30. Thus, we hypothesized that IL-9 may be involved in CCR2-mediated lung MC recruitment. Supporting our hypothesis, using the bone marrow chimera model in Fig. 2, we found that 10–20% of the MC in the allergic lung express CCR2 and that CCR2+ MCs were primarily WT-derived, suggesting that IL-9 impacts CCR2 migration (Fig. 4A). We then tested whether blockade of CCR2 signaling impacts MC migration in a chronic HDM allergen model (Fig. 4B). Consistent with previous reports, blockade of CCR2 led to diminished eosinophil frequencies in the lungs, demonstrating effective blockade of CCR2 (Fig. 4C)37. Notably, compared to vehicle dimethylsulfoxide (DMSO)-treated controls, blockade of CCR2 led to greater frequencies of mMC in the bone marrow while mMC numbers were profoundly diminished in the trachea and lungs, indicating that mMCs utilize CCR2 to emigrate from the bone marrow to the lungs (Figs. 4D–F). Next, we tested whether IL-9 directly impacts CCR2 expression on MC. Using bone marrow-derived MC (BMMC) cultures, stimulation with IL-9 enhanced CCR2 expression, while neutralizing IL-9 decreased CCR2 expression (Figs. 4G and 4H).
Fig. 4.
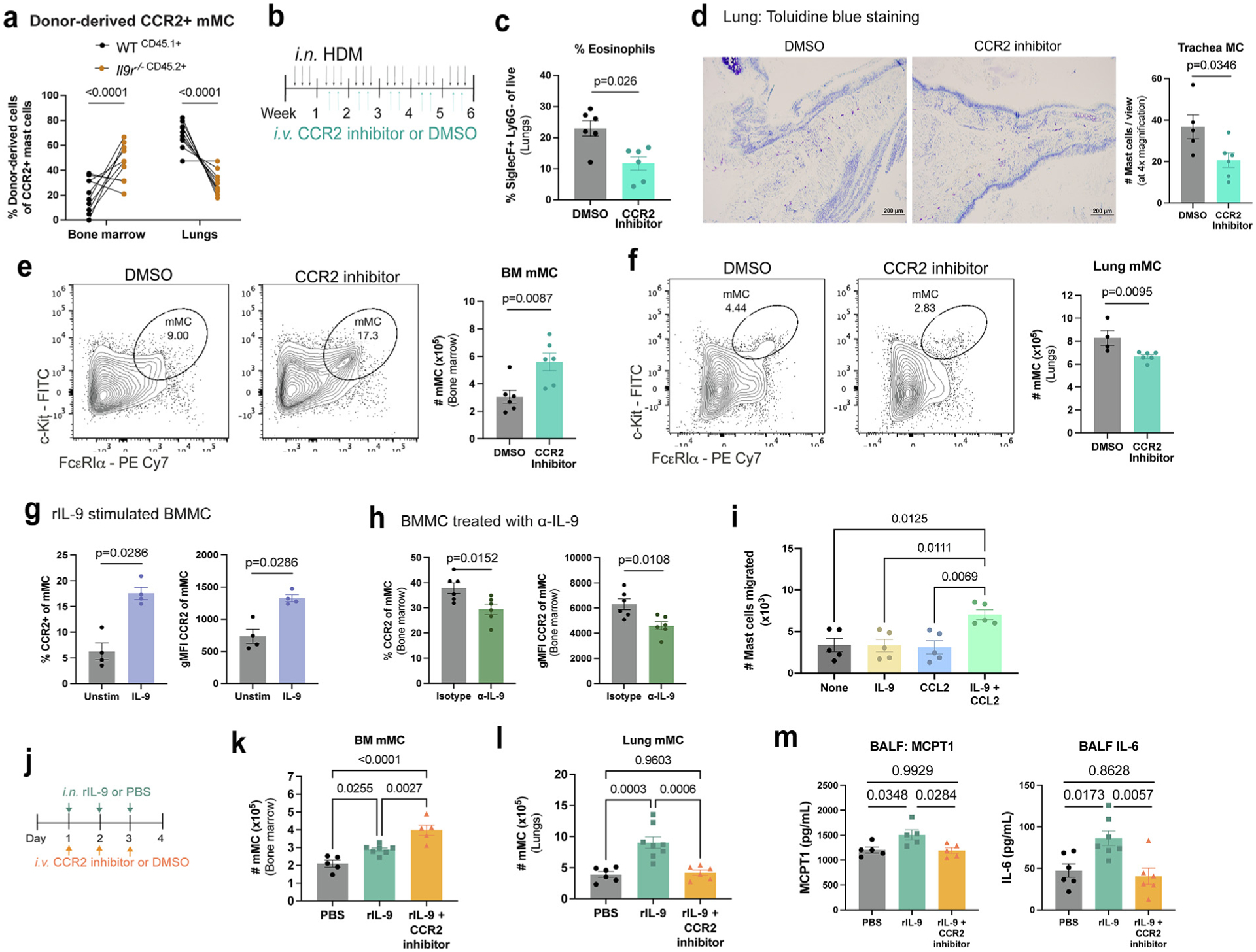
IL-9 promotes CCR2-dependent MC migration from the bone marrow to the allergic lung. (A) WT (CD45.1+) and Il9r−/− (CD45.2+) bone marrow cells were transferred to lethally irradiated BoyJ x C57BL/6J F1 mice, allowed to reconstitute immune cells, and treated with HDM for 6 weeks. Flow cytometry analysis of CCR2+ lung MC. (n = 10) Data are representative of two independent experiments with similar results. (B–F) HDM-treated WT mice were treated with CCR2 inhibitor or DMSO control as indicated in (B); C, eosinophil (CD11b+ SiglecF+ Ly6G−) frequencies were determined by flow cytometry. (D) MC in the trachea were assessed using toluidine blue staining. MC numbers were assessed in the bone marrow (E) and lungs (F) by flow cytometry (n = 6). Data are representative of two independent experiments with similar results. (G) Flow cytometry analyses of CCR2 expression in BMMC treated with IL-9 (40 ng/ml) for 2 hours (n = 4). Data are representative of two independent experiments. (H) Flow cytometry analyses of CCR2 expression in bone marrow MC from HDM-treated WT mice treated with anti-IL-9 or an isotype control (n = 6). Data are representative of three independent experiments with similar results. (I) BMMC migration assay toward cytokines and/or chemokines (n = 5). Data are representative of three independent experiments with similar results. (J–M) WT mice, treated intranasally with IL-9 for 3 days, were also intravenously treated with CCR2 inhibitor for 3 days indicated in (J). Flow cytometry analysis of bone marrow (K) and lung (L) MC. (M) MCPT1 and IL-6 protein expression was determined via enzyme-linked immunosorbent assay (n = 5–8). Data are representative of two independent experiments with similar results. Error bars indicate ± standard error of mean. Statistical significance was determined by analysis of variance, followed by Tukey’s multiple comparison test (A, I, K, and L) or Mann-Whitney U test (C, E–H, and M). CD = clusters of differentiation; DMSO = dimethyl sulfoxide; gMFI = geometric mean fluorescence intensity; HDM = house dust mite; IL = interleukin; MC = mast cell; MCp = MC progenitors; MCPT 1 = MC protease 1; mMC = mature MC; ns = not significant; PBS = Phosphate buffered saline; PE = R-phycoerythrin; r = recombinant; SSC = side scatter; WT, wild type.
To further explore how IL-9 affects CCR2-dependent MC migration, we utilized an in vitro migration assay of BMMC cultures to test chemoattraction to CCL2. We found that IL-9 or CCL2 alone did not promote MC migration; however, the combination of IL-9 and CCL2 resulted in significant MC migration (Fig. 4I). IL-9-induced MC migration was CCL2-specific, since IL-9 was unable to enhance MC migration toward CXCL1, CXCL2, CXCL3, CXCL8, or CXCL12 (Supplementary Fig. 2A). Moreover, IL-4 in combination with CCL2 was unable to promote MC migration, suggesting that not all type 2 cytokines impact this response (Supplementary Fig. 2B).
We next sought to test whether IL-9 induced CCR2-dependent MC migration in vivo using intranasal IL-9 treatment and intravenous treatment with the CCR2 inhibitor (Fig. 4J). Blockade of CCR2 in rIL-9-treated mice significantly increased mMC numbers in the bone marrow and reduced mMC accumulation in the lungs (Figs. 4K and 4L). We further demonstrate that blockade of CCR2 led to diminished MC protease 1 (MCPT1) and IL-6 expression in the bronchoalveolar lavage fluid (BALF) (Fig. 4M), suggesting that the accumulation of MCs in the lung greatly contributes to the amount of MC degranulation markers: MCPT1 and IL-6 levels in the BALF. Taken together, these results indicate that bone marrow MCs respond to IL-9, enhancing CCR2 expression to facilitate migration to the allergic lung.
IL-9 has also been shown to enhance interstitial macrophage expansion and function in allergic airway inflammation in an IL-9-dependent manner8. Since monocytes and macrophages are known to migrate in a CCL2/CCR2 axis in allergic airway inflammation8,38, we wanted to confirm whether blockade of CCR2 impacted macrophage recruitment to the lungs. Indeed, blockade of CCR2 reduced macrophage frequencies in the lungs (Supplementary Fig. 3A). To test whether IL-9 responsive macrophages impact MC expansion, we depleted macrophages in WT mice using clodronate liposomes prior to intranasal IL-9 treatment (Supplementary Fig. 3B). We demonstrated that a single dose of clodronate was sufficient to reduce macrophage numbers in the lungs (Supplementary Fig. 3C). Importantly, there was a significant increase in MC numbers even when macrophages were depleted, although the absolute numbers were diminished compared to mice treated with control liposomes (Supplementary Fig. 3D). These findings, coupled with the bone marrow chimera results and in vitro assays showing that IL-9 promotes MCp proliferation and STAT5 activation (Figs. 3A–C and Supplementary Fig. 3E), suggest that IL-9 acts directly acts on MC to promote expansion in the allergic lung and that macrophages may contribute to MC expansion.
T cell-derived IL-9 promotes MC expansion in the allergic lung
CD4+ T helper 9 cells (TH9) are an important source of IL-9 in mouse models of allergic airway inflammation8,20,39,40. MC and innate lymphoid cells have been demonstrated to contribute to the pool of IL-9 in allergic airway inflammation17. Thus, we first assessed the contribution of T cell IL-9 in comparison to MC and ILCs IL-9. Using our chronic allergen model, we demonstrate that T cells contribute the majority of IL-9 in the lung after chronic allergen exposure (Supplementary Fig. 4A). Next, to define the role of TH9-derived IL-9 on MC expansion, we adoptively transferred in vitro polarized OTII-derived TH9 or TH2 cells into WT recipient mice followed by intranasal ovalbumin (OVA) challenge for 5 days to elicit allergic airway inflammation (Fig. 5A). Following allergen challenge, adoptively transferred T cells were present in the lungs but was also found in the bronchoalveolar lavage and draining lymph node (Supplementary Fig. 4B). We further demonstrate that adoptive T cell transfer successfully sensitized mice to ovalbumin by showing increased serum OVA-specific IgE in TH2 and TH9 adoptive transfer conditions. (Supplementary Fig. 4C). Adoptive T cell transfer and treatment with ovalbumin for 5 days induced allergic airway inflammation as evident by eosinophil infiltration in the lungs (Fig. 5B). As compared to TH2 cells or PBS controls, recipients of TH9 cells exhibited greater MC numbers evident within the trachea, bone marrow, and lungs (Figs. 5C–G). Furthermore, we assessed MCp proliferative capacity and showed that TH9 cell transfer led to enhanced lung MCp proliferation, in contrast to TH2 cells or PBS controls (Fig. 5H).
Fig. 5.
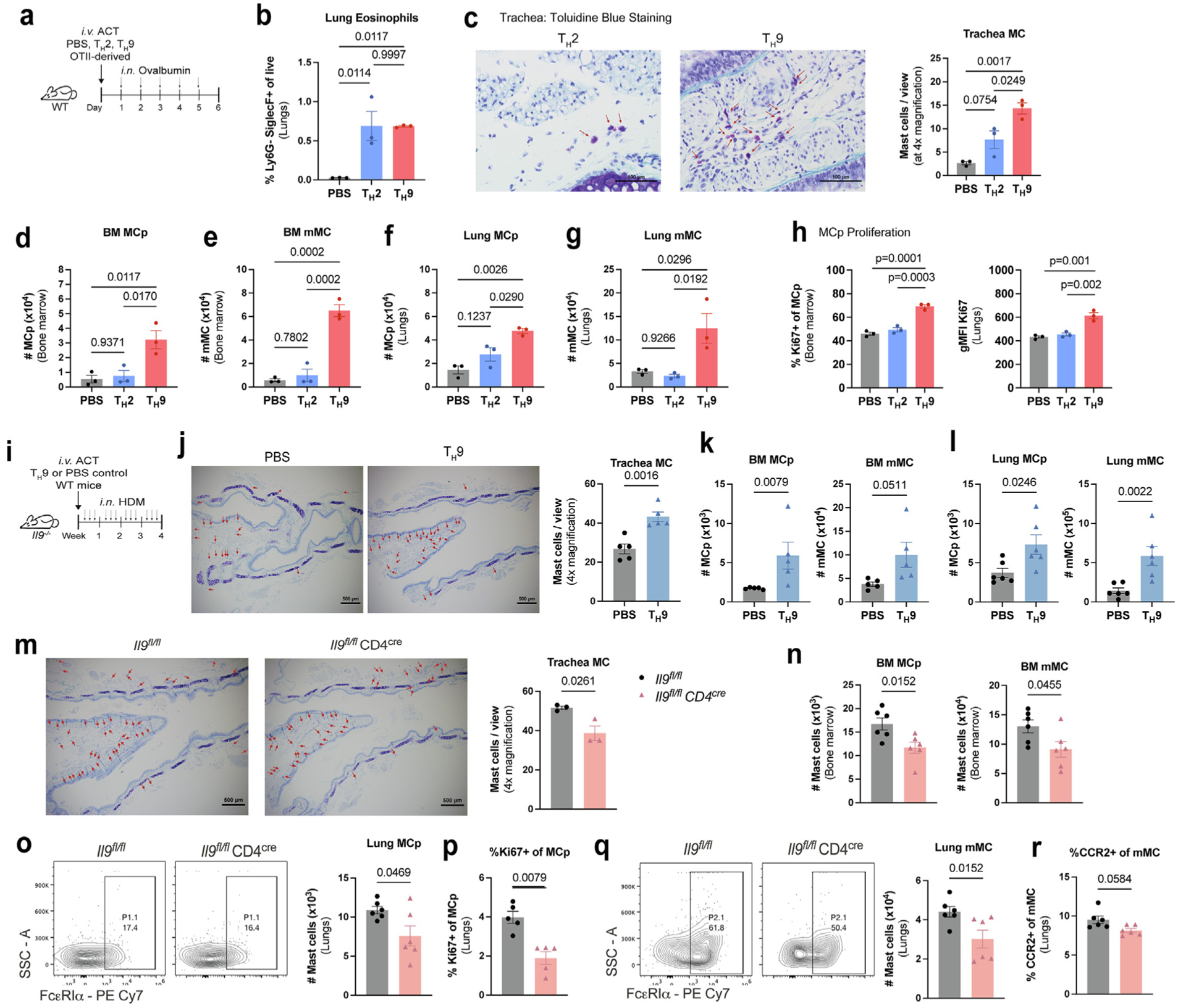
T cell IL-9 is a central source of IL-9 mediating MC expansion in the allergic lung. (A–H): A, WT mice transferred with OTII-derived TH2 or TH9 cells were treated with OVA for 5 days; B, lung eosinophil frequencies were determined by flow cytometry. MC numbers were assessed in the trachea (C) using toluidine blue staining and in the bone marrow (D–E) and lungs (F–G) using flow cytometry. (H) Flow cytometry analysis of Ki67 expression in MCp. (n = 3). Data are representative of three independent experiments with similar results. (I–L) WT-derived TH9 cells or PBS were transferred to Il9−/− recipient mice, and subsequently treated with HDM as shown in (I); (J) MC numbers were assessed in the trachea using toluidine blue staining. 4x magnification with Scale = 500 μm. (K–L) Flow cytometry analysis of bone marrow (K) and lung (L) MC numbers (n = 5–6). (M–Q) Il9fl/fl CD4cre mice and littermate controls (Il9fl/fl) were treated with HDM for 6 weeks M, toluidine blue staining of trachea. 4x magnification with Scale = 500 μm; N, flow cytometry analysis of MCs in the bone marrow; O, representative flow cytometry contour plots of lung MCp; P, Ki67 expression in lung MCp; Q, representative flow cytometry contour plots of lung mMC; R, CCR2 expression in lung mMC (n = 6). Data are representative of two independent experiments with similar results. Error bars indicate ± standard error of mean. Statistical significance was determined by analysis of variance, followed by Tukey’s multiple comparison test (B–H) or Mann-Whitney U test (J–R). ACT = adoptive cell transfer; CD = clusters of differentiation; HDM = house dust mite; IL = interleukin; MCp = mast cell progenitors; MCPT 1 = mast cell protease 1; mMC = mature mast cells; ns = not significant; OVA = ovalbumin; PBS = phosphate buffered saline; PE = R-phycoerythrin; SSC = side scatter; TH = T helper; WT, wild type.
To determine if TH9 cells are a sufficient source of IL-9 mediating MC accumulation in the allergic lung, we adoptively transferred WT-derived in vitro polarized TH9 cells into Il9−/− mice (Fig. 5I). TH9 adoptive transfer led to greater MC accumulation in the trachea compared to PBS controls (Fig. 5J), and increased numbers of MCp and mMC in the bone marrow and lungs following TH9 cell transfer and allergen challenge (Figs. 5K and 5L).
To directly test the requirement for T cell IL-9, we used a recently generated mouse strain with a conditional deletion of Il9 transgene in T cells (Il9fl/fl CD4cre)40. Following the allergen challenge, mice with T cell-specific IL-9 deficiency had diminished MC accumulation in the trachea and bone marrow (Figs. 5M and 5N). Il9fl/fl CD4cre mice exhibited reduced lung MCp numbers and decreased Ki67 expression (Figs. 5O and 5P). Moreover, we observed a decrease in lung mMC numbers and lung mMC CCR2 expression (Figs. 5Q and 5R). Thus, T cell IL-9 is a central source of IL-9 mediating MCp proliferation and CCR2-dependent mMC migration in allergic airway inflammation.
MC-mediated airway hyperresponsiveness is dependent upon T cell IL-9
MCs are important effector cells mediating airway hyperresponsiveness in response to allergen41,42. Having demonstrated that IL-9 promotes mMC recruitment to the lungs, we next wanted to investigate whether mMCs are required for antigen-induced airway reactivity using adoptive transfer of WT MC, or PBS as a control, into MC-deficient mice. To test this, we sorted mMCs from HDM chronically treated mice and transferred cells intranasally to MC-deficient KitWSH/WSH recipient mice. Mice were then treated with HDM for 2 weeks to allow for expansion in the allergic lung (Fig. 6A). Transfer of mMC into the airways of KitWSH/WSH recipients allowed for successful reconstitution of MC in the lung tissue (Supplementary Fig. 5A). To directly test antigen-induced airway hyperresponsiveness and avoid any confounding effects of IL-9 on IgE production, HDM-challenged mice were passively sensitized to 2,4-dinitrophenol (DNP) and challenged with DNP-BSA intratracheally (Fig. 6A). We found that the increased airway response to antigen challenge required mMC transfer, and there was minimal to no response in mice receiving PBS (Fig. 6B). We measured known human MC degranulation markers taken one hour after antigen challenge35,43. Transfer of mMC led to greater MC degranulation marker expression of MCPT1, IL-6, and CCL2 as compared to PBS transfer controls (Supplementary Fig. 5B), supporting the use of these cytokines as indicators of MC activation. These results demonstrate that mMCs respond to antigen challenge and are sufficient to induce antigen-induced airway reactivity.
Fig. 6.
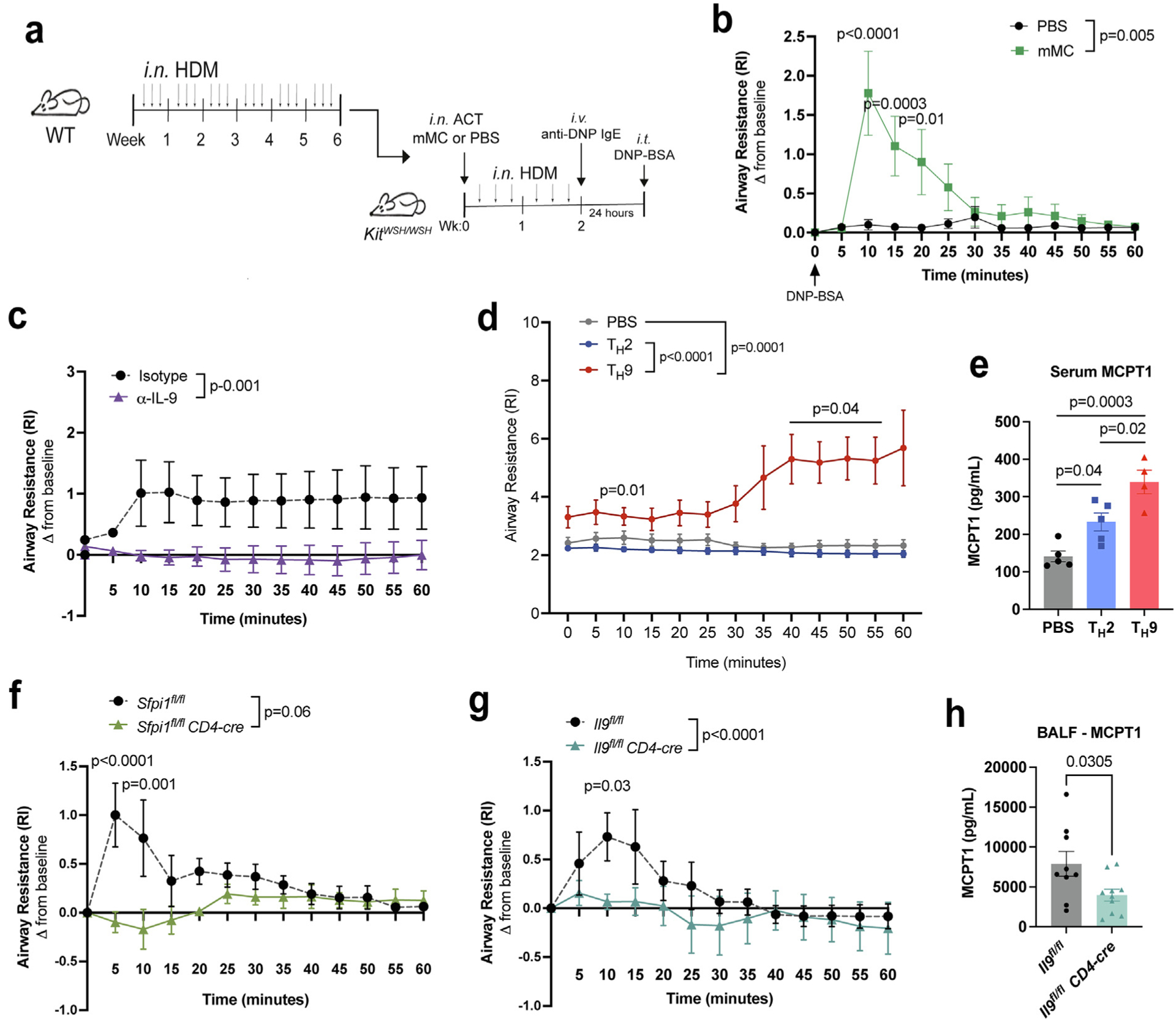
T cell IL-9 promotes antigen-induced mMC-mediated airway reactivity. (A–B) KitWSH/WSH mice were adoptively transferred with HDM-sensitized mMC from WT mice and subsequently treated with HDM for 2 weeks. Following passive sensitization to 2,4-Dinitrophenyl (DNP) and challenge intratracheally with DNP-BSA, airway reactivity was measured for one hour. Experimental design indicated in (A) and results in (B) (n = 5). Data are representative of two independent experiments with similar results. (C) HDM-treated WT mice were intravenously treated with α-IL-9 or isotype control for the last 2 weeks of treatment. Airway resistance was monitored in (C) (n = 4–5). Data are representative of two independent experiments with similar results. (D–E) WT mice transferred with OTII-derived TH2 or TH9 cells were treated with OVA for 6 days. Airway resistance was monitored for 60 minutes in (D) (PBS: n = 7, TH2: n = 10, and TH9: n = 10). Data are pooled from three independent experiments; E, serum MCPT1 expression was assessed via enzyme-linked immunosorbent assay (n = 5); F, airway resistance for HDM-treated Sfpi1fl/flCD4cre and Sfpi1fl/fl mice (n = 5). Data are representative of two independent experiments with similar results. (G–H): G, airway resistance for HDM-treated Il9fl/flCD4cre and Il9fl/fl mice; H, MCPT1 protein expression was determined via enzyme-linked immunosorbent assay (n = 5). Data are representative of two independent experiments with similar results. Error bars indicate ± standard error of mean. Statistical significance was determined by analysis of variance, followed by Tukey’s multiple comparison test (E), Sidak’s multiple comparison test (B, D, F, and G), Student’s t test in (H), and Mann-Whitney U test of area under the curve (B–D, F, and G). ACT = adoptive cell transfer; BALF = bronchoalveolar lavage fluid; CD = clusters of differentiation; HDM = house dust mite; Ig = immunoglobulin; IL = interleukin; MCp = mast cell progenitors; MCPT 1 = mast cell protease 1; mMC = mature mast cells; ns = not significant; OVA = ovalbumin; PBS = phosphate buffered saline; SSC = side scatter; TH = T helper; WT, wild type.
To determine whether T cell IL-9 contributed to antigen-specific airway hyperresponsiveness, we performed airway testing using a similar passive sensitization and challenge model described in Fig. 6A. Treatment with an α-IL-9 blocking antibody, during the last 2 weeks of allergen treatments, protects against antigen-induced airway hyperresponsiveness compared to isotype controls (Fig. 6C). Moreover, adoptive transfer of TH9 cells and OVA allergen treatment led to a consistent increase in airway resistance in response to intratracheal antigen challenge (Fig. 6D). Importantly, MC degranulation was evident following TH9 cell transfer with high expression of serum MC protease 1 (MCPT1) (Fig. 6E).
Using mouse models of TH9 deficiency (Sfpi1fl/fl CD4cre and Il9fl/fl CD4cre), we further examined the contribution of T cell-derived IL-9 on antigen-induced airway hyperresponsiveness44. Despite the decrease in airway resistance in Sfpi1fl/fl CD4cre mice upon antigen challenge, the overall response to antigen was insufficient to induce a robust protective response (Fig. 6F). Therefore, we utilized mice deficient in T cell-specific IL-9 (Il9fl/flCD4cre) and demonstrate that Il9fl/fl CD4cre mice are protected against antigen-induced airway resistance (Fig. 6G). Moreover, deficiency in T cell IL-9 led to decreased Mcpt1 transcripts in the lungs and MCPT1 levels in the bronchoalveolar lavage fluid, indicating diminished MC degranulation (Fig. 6H and Supplementary Fig. 5C). We further tested expression of additional cytokines expressed in MC including Il2, transforming growth factor-beta (Tgfb1), Vegf, and Fgf29,45. We observed that T cell-specific IL-9 deficiency did not alter Il2 or expression of profibrotic mediators, Tgfb1, Vegf, and Fgf2 transcripts (Supplementary Fig. 5C). These data suggest that our antigen challenge may be too short of a duration to observe MC induction of these cytokines. Collectively, these findings demonstrate that T cell IL-9 expansion of lung mMC regulates antigen-induced allergic airway responses.
Discussion
This study defines a mechanism for IL-9-mediated MC accumulation in the allergic lung that includes a combination of IL-9-dependent MCp proliferation in the lungs and CCR2-dependent recruitment of MCs to the lungs4,20. Our mixed bone marrow chimera model revealed that IL-9 responsive MCp were preferentially expanded in the bone marrow and the lungs, suggesting that MCp responds to IL-9 signaling. Our data also indicate that IL-9 enhances mMC CCR2 expression and contributes to MC migration from the bone marrow to the allergic lung. We further demonstrate that MCs are required to generate antigen-induced airway reactivity and that these responses are dependent upon IL-9. We also show that T cell IL-9 is an important source of IL-9 for MC expansion in the lung and allergen-induced airway reactivity.
mMCs are a highly IL-9 responsive cell type in allergic asthma9,20. However, knowledge about the impact of IL-9 signaling on MCp and how MCs are sustained within the allergic lung is limited. This study demonstrated that IL-9R is constitutively expressed in multiple stages of MC development albeit at varying intensities depending on the maturation stage. We show that mMCs express the greatest amount of IL-9R and that mMC can respond to IL-9 in the bone marrow and lungs. This is consistent with studies demonstrating that IL-9 signaling enhances MC growth in the bone marrow and that MCs are key IL-9 responders in the lungs5,9. In comparison, MCp expresses an intermediate level of IL-9R compared to mMC, thereby suggesting that IL-9 signaling can be regulated by controlling its IL-9R expression during the multiple stages of MC development. Thus, IL-9R expression is linked to the MC differentiation stage with distinct functional outcomes.
MCs can migrate toward the allergic lung environment in a variety of ways: α4β7 expressed on human MCp binds VCAM-1 expressed on epithelial cells, CCR3-expressing MCp migrates toward CCL5 and CCL11, and the CCR2/CCL2 axis4,30,46. However, the role of IL-9 on MC migration in allergic airway inflammation is not well defined. Our data demonstrate that IL-9 upregulates CCR2 expression on MC to promote MC migration from the bone marrow to the allergic lung. While in vitro-cultured bone marrow MCs are a valuable tool to study mucosal MC, these culture conditions have several caveats in their interpretation, particularly, on how the culture conditions differ from the MC phenotypes found in the allergic microenvironment. Still, we observed parallel effects on MC directly analyzed ex vivo from allergen-challenged mice. Despite reports that MCp migrates from the bone marrow, we were unable to identify CCR2-dependent MCp migration in mice treated with a CCR2 inhibitor (data not shown). Thus, if the IL-9/CCR2 axis does promote MCp migration to the allergic lung, an alternative pathway may function in its absence. These data indicate that the deficit in MC expansion observed in the CCR2 inhibitor-treated mice does not result from insufficient proliferation of lung-resident MC, and likely reflects a primary contribution of migratory MC. Further investigation on IL-9-dependent MC migration in vivo will define MC migration and localization within the lungs that can impact allergic responses.
IL-9 is a pleiotropic cytokine, and its intrinsic and extrinsic effects on MC responses remain to be defined. Our data support an intrinsic effect of IL-9 during multiple stages of MC development. Our mixed bone marrow chimera model revealed that IL-9R-expressing WT-derived mMC were preferentially recruited to the allergic lung, while IL-9R-deficient mMC primarily remained in the bone marrow, suggesting an effect of IL-9 on MC migration that allows a selective advantage to emigrate from the bone marrow to the lungs. We have also found that less than 10% of the bone marrow MCp were Il9r−/−-derived, indicating that IL-9 signaling on MCp is essential for MCp expansion. However, these results do not exclude the possibility that IL-9 acts on other cells in the allergic lung to contribute to MC expansion. One possibility is that IL-9 acts on CD11c+ macrophages or dendritic cells to promote an inflammatory response that can then promote MC responses. In support of this, Fu et al. demonstrate that macrophages express IL-9R at a comparable level to MC and that IL-9 expands the CD11c+ interstitial macrophages in the allergic lung8. Moreover, Dahlin et al show that CD11c+ cell depletion severely inhibited allergen-induced MC recruitment to the lungs47. These cells could be sources of, or inducers of, CCR2 ligands required for mMC recruitment. Notably, macrophage depletion diminished but did not eliminate IL-9-induced MC expansion in the allergic lung, suggesting that macrophages can contribute to MC expansion but are not required. While it is not clear how macrophages contribute to these responses, further experiments will define these circuits in the lung.
We utilize a novel method of testing MC function in vivo using antigen-induced airway testing. Our model allows us to directly test MC activation in vivo by passively sensitizing mice to a heterologous antigen and challenging intratracheally with antigen. We demonstrate that these responses are MC-dependent, as we do not observe antigen-induced airway reactivity in MC-deficient mouse models (KitWSH/WSH). Because intravenous MC adoptive transfer is insufficient to reconstitute MC in the lung48, we utilized intranasal transfer of HDM-sensitized MC followed by treatment with HDM for 2 weeks. We demonstrate that mMC intranasal transfer can reconstitute MC populations in the respiratory tract. We further demonstrate that intranasal transfer of MC was sufficient to mediate antigen-induced airway reactivity. Our findings support that IL-9 mediates MC expansion in the lower airways: trachea and allergic lung, whether IL-9 promotes MC expansion in the upper airways requires further investigation.
The source of IL-9 in these studies was also determined. Consistent with previous results in OVA-Alum allergen models, compared to TH2 cells, we demonstrate that TH9 cells have an independent role in promoting airway cellular infiltration of MC and MC-mediated airway hyperreactivity in a chronic 6-week HDM allergen model20,49,50. While TH2 cells elicited increased eosinophil infiltration compared to PBS controls, TH2 cell transfer was insufficient to significantly increase MC numbers in the trachea and lungs to similar levels as TH9 cells, and ultimately, unable to promote antigen-induced airway hyperresponsiveness. Despite the reduced MC expansion by neutralizing TH2 cytokines, IL-4 and IL-13 play important roles in maintaining IgE antibody production, and thus may indirectly contribute to mast survival51. These assays define an independent function for the IL-9/MC circuit that is distinct from the ability of TH2-dependent inflammation to increase methacholine-induced airway hyperreactivity52. Previous studies have utilized mice with a T cell-specific PU.1 deletion (Sfpi1fl/fl CD4cre) as a model for TH9 deficiency; however, these mice are not without limitations. Although PU.1-deficiency in T cells impairs T cell IL-9 production, Sfpi1 transcripts are also expressed in TH1, TH2, and TH17 cells, suggesting that it may also regulate these T cell lineages20,44. In this report, we utilize a newly generated T cell-specific IL-9 deficient mouse strain (Il9fl/fl CD4cre) that does not impact ILC2 or MC IL-9 production but eliminates T cell IL-940. Given that T cells, ILC2, and MC can contribute to IL-9 levels in the cytokine milieu53,54, that Il9fl/fl CD4cre mice still exhibited reduced MC expansion, suggests that ILC2 and MC IL-9 are insufficient and that T cells are a critical source of IL-9 promoting MC expansion in allergic airway inflammation.
The clinical implications of using anti-IL-9 antibody treatments have been demonstrated in mouse models and human clinical trials55–57. Thus far, anti-IL-9 treatment efficacy is variable; however, for individuals that benefit from this treatment, blocking IL-9 can improve asthma exacerbation rates in subjects with mild asthma55. We observed a similar greater effect of IL-9 blockade on intermittent exposures in mouse models40. Here, we further demonstrated that intravenous administration of anti-IL-9 during the last 2 weeks of allergen challenge, protected against allergen-mediated airway hyperreactivity. Data presented here and in other preclinical studies provide evidence that an IL-9/ MC pathway regulates airway inflammation and airway reactivity58. Therapies such as c-Kit kinase inhibition (Imatinib) have also been shown to reduce allergic airway hyperresponsiveness by decreasing MC numbers and tryptase expression59. Importantly, MCs offer protective roles in other diseases60. Therefore, complete ablation of MC using Imatinib raises important concerns in the design of therapeutics that have the potential to modulate MC biology. Thus, a greater understanding of which patients will benefit from targeting the IL-9/MC pathway will provide more personalized asthma therapies.
MATERIALS AND METHODS
Mice
All mice were on C57BL/6 background. WT mice (C57BL/6, Strain #002014), OTII mice (Strain #004194), and KitWSH/WSH (C57BL/6, Strain #005051) were purchased from the Jackson Laboratory. Il9r−/− mice (C57BL/6) were a gift from Dr. Jean-Christophe Renauld61. Il9−/− mice (C57BL/6) were provided by Drs. Andrew McKenzie62, Alexander Kirsch, and Sophie Paczesny. Il9fl/fl mice were generated by Jackson Laboratories using Cas9-mediated targeting as characterized in Ulrich & Kharwadkar et al40. Experiments were performed using 6–16-week-old female and male mice with no observed differences between sexes. All mice were maintained in SPF animal facilities (ambient temperature 70–72° F, humidity 50%, light/dark cycle 12/12 hour). All experiments were performed with the approval of the Indiana University Institutional Animal Care and Use Committee.
Humanized mouse model
Triple transgenic [NOD.Cg-Prkdc scid Il2rg tm1wjl Tg (CMV-IL 3, CSF2,KITLG)1Eav./MloySzJ] (NSG-SGM3) mice were purchased from the Jackson Laboratory (Strain #013062) and bred in house by the Indiana University Melvin and Bren Simon Comprehensive Cancer Center In Vivo Therapeutics Core. Mice were maintained under pathogen-free conditions in ventilated cages and were given ad libitum irradiated Teklad Uniprim Medicated Diet (Harlan Laboratories, Indianapolis, IN, TD 06596) and autoclaved, acidified water. Before transplant, cryopreserved human cord blood CD34+ cells (STEMCELL Technologies, Vancouver, Canada) were thawed and cultured for 4 hours at (5 × 105) cells/ml in StemSpan serum-free media (STEMCELL Technologies, Vancouver, Canada) supplemented with stem cell factor (SCF) at 100 ng/ml (PeproTech, Cranbury, NJ). Cells were then counted in try-pan blue and were 95 to 98% viable. Adult female NSG-SGM3 mice (6 to 8 weeks old) were sublethally irradiated (100 cGy) and injected intravenously 4 hours later with 2 × 104 human CD34+ cells.
Induction of allergic airway inflammation Acute adoptive cell transfer model
Naïve CD4+ T cells isolated from OTII mice, mice that express ovalbumin (OVA)-specific T cell receptors, were cultured ex vivo to polarize toward a TH2 (IL-4) or TH9 (IL-4 + TGFβ) phenotype. We assessed cytokine production by stimulating cells with PMA (50ng/mL) and ionomycin (1mg/mL). After 3 hours, monensin (2μM) was added to stimulated cells for another 3 hours. Cells were fixed, permeabilized, and stained for intracellular cytokines: IL-4, IL-9, and IL-13. 5×105 in vitro polarized TH2 or TH9 cells were adoptively transferred intravenously to recipient mice. Twenty-four hours after transfer, mice were intranasally treated with 100 μg OVA (Sigma, St. Louis, MO, Catalog #A5503) in 25 μl for 5 days.
Chronic HDM challenge model
House dust mite (HDM; Greer Laboratories, Lenoir, NC, Catalog #XPB91D3A2.5) was diluted with PBS. Mice were challenged intranasally with 25 μg HDM three times a week for 6 weeks. When specified, mice were treated intravenously with CCR2 inhibitor (2 mg/kg, R&D systems, Minneapolis, MN, Catalog #RS 504393) or DMSO twice a week during the last 4 weeks of HDM treatment. For some experiments, mice were treated intravenously with antibodies every other day for the last 2 weeks of HDM treatment: α-IL-9 (150μg/mouse, BioXcell, Lebanon, NH, Catalog #9C1), α-IL-13 (25 μg/ mouse, R&D, Catalog #MAB413–500), and α-IL-4 (50 μg/mouse, BioXcell, Catalog #BP0045); or its isotype control antibody: IgG2A isotype (BioXcell, Catalog #BE0085), IgG2A (R&D, Catalog #MAB006), and IgG1 isotype (BioXcell, Catalog #BP0088), respectively.
Macrophage depletion
Mice were intravenously injected with 200 μL of Clodronate liposomes (Liposoma; The Netherlands, SKU #C-010) 48 hours prior to intranasal IL-9 treatment.
Tissue harvest and processing
Mice were euthanized and lungs were lavaged with cold PBS. BAL fluid was collected, and cells were centrifuged at 1500 g for 5 minutes at 4°C for further surface staining. Lung tissue was digested in collagenase A (1 mg/ml, Roche, Indianapolis, IN, Catalog #10103586001) in DMEM (Dulbecco’s Modified Eagle Medium) (5 ml/lung, Gibco, Detroit, MI, Catalog #11965084) for 45 minutes at 37°C in rotation. After digestion, the lungs were filtered through a sieve (Bellco Glass, Vineland, NJ, SKU #1985–85000) to obtain single-cell suspensions. Cells were pelleted and red blood cells were lysed using ACK (Ammonium-Chloride-Potassium) Lysis for 5 minutes. Blood was collected for FACS analysis, and the remaining blood samples were used for plasma. Femur-derived bone marrow was flushed with cold PBS, pelleted, filtered, and proceeded to ACK lysis. Single-cell suspensions were used for flow cytometry and RNA isolation using TRIzol reagent (Invitrogen, Waltham, MA, Catalog #15596026) or the RNeasy Plus Micro Kit (QIAGEN, Germantown, MD, Catalog #74004).
Flow cytometry
Single-cell suspensions were stained with a fixable viability dye (eBioscience, San Diego, CA, Catalog #65-0865-18) and antibodies for surface markers for 30 minutes at 4°C. After fixation with IC Fixation (eBioscience, Cat #00-8222-49) for 10 minutes dark at room temperature, cells were permeabilized with permeabilization buffer (eBioscience, Catalog #00-8333-56) for 30 minutes at 4°C and stained for cytokines for 1 hour at 4°C. For transcription factor staining, after surface staining, cells were fixed with Fixation & Permeabilization Buffer (eBioscience, Catalog # 00-5521-00) for 2 hours or overnight at 4°C, and then permeabilized with permeabilization buffer. See Table 1 for information about flow reagents.
Table 1.
Reagents for flow cytometry, cell culture, and ELISA.
| Flow cytometry antibodies | |||||
|---|---|---|---|---|---|
| Antigen/Name | Clone | Fluorochrome | Company Dilution in FACS buffer | Catalog number | |
| Fixable viability dye | eFluor 780 | eBioscience | 1:1500 | 65–0865-14 | |
| Mouse lineage antibody cocktail: | PerCP-Cy5.5 | BD Biosciences | 1:25 | 561317 | |
| CD3e | 145–2C11 | ||||
| CD11b | M1/70 | ||||
| CD45R/B220 | RA3–6B2 | ||||
| Ly-76 | TER-119 | ||||
| Ly6G and Ly6C | RB6–8C5 | ||||
| Mouse CD45 | 30-F11 | PE/Dazzle 594 | Biolegend | 1:250 | 103145 |
| Mouse CD45.1 | A20 | BV421, BV650 | Biolegend | 1:200 | 110735 |
| Mouse CD45.2 | 104 | BV605, APC | Biolegend | 1:200 | 109841 |
| Mouse CD49b | HMa2 | PE | BioLegend | 1:250 | 103506 |
| Mouse CD49b | Dx5 | BV421 | BD Biosciences | 1:200 | 563063 |
| Mouse c-Kit | 2B8 | FITC, APC | BioLegend | 1:100 | 105805, 105811 |
| Mouse ST2/IL-33R | U29–93 | BV510 | BD Biosciences | 1:100 | 745080 |
| Mouse FceRI | MAR-1 | PE-Cy7 | BioLegend | 1:200 | 134317 |
| Mouse FcyRII/III | 93 | BV510 | BioLegend | 1:200 | 101333 |
| Mouse Integrin β7 | FIB504 | APC, BV711 | BioLegend | 1:200 | 321207, 321239 |
| Mouse CCR2 | 475301 | BV786 | BD Biosciences | 1:200 | 747966 |
| Mouse Ki67 | SolA15 | PE-eFluor610, AF700 | Invitrogen | 1:200 | 61–5698-82, 56–5698-82 |
| Mouse CD3 | 145–2C11 | PerCP-Cy5.5 | BD Biosciences | 1:200 | 551163 |
| Mouse IL-4 | 11B11 | AF647 | BioLegend | 1:200 | 504110 |
| Mouse IL-9 | RM9A4 | PE | BioLegend | 1:200 | 514104 |
| Mouse IL-13 | eBio13A | AF488 | eBioscience | 1:200 | 53–7133-82 |
| Mouse Ly6G | RB6–8C5 | FITC | BD Biosciences | 1:200 | 553126 |
| Mouse SiglecF | E50–2440 | PE | BD Biosciences | 1:200 | 552126 |
| Mouse CD64 | X54–5/7.1 | FITC | BioLegend | 1:200 | 139316 |
| Mouse Mertk | 2B10C42 | PE | BioLegend | 1:200 | 151506 |
| Mouse CD11b | M1/70 | PerCP-Cy5.5 | eBioscience | 1:200 | 45–0112-82 |
| Mouse CD11c | N418 | PE-Cy7 | eBioscience | 1:200 | 25–0114-82 |
| Reagents for T helper cell differentiation | |||||
| Name (Clone) | Company | Catalog Number | |||
| anti-mouse CD3 (145–2C11) | BioXCell | BP-0001–1 | |||
| anti-mouse CD28(37.51) | BioXCell | BE0015–1 | |||
| anti-mouse IFNy (XMG1.2) | BioXCell | BE0055 | |||
| mouse IL-4 (11B11) | BioXCell | BE0045 | |||
| mouse IL-2 (JES6–5H4) | BioXCell | BE0042 | |||
| Human TGF-β1 | Miltenyi Biotec | 130–095-067 | |||
| Reagents for mast cell cultures | |||||
| mouse IL-3 | Biolegend | 575504 | |||
| mouse SCF | Biolegend | 579706 | |||
| ELISA kits | |||||
| Name | Company | Catalog Number | |||
| LEGEND MAX Mouse OVA-specific IgE ELISA | BioLegend | 439807 | |||
| Mouse MCPT1 ELISA | Invitrogen | 88–7503-88 | |||
| Mouse IL-9 ELISA MAX Deluxe | BioLegend | 442704 | |||
CD = clusters of differentiation; IL = interleukin.
Mixed bone marrow chimera
F1/CD45.1+CD45.2+ mice were irradiated at 1000 rds. One day after the irradiation, 8 million donor bone marrow cells [4 million from Boy/J mice (CD45.1+) and 4 million from Il9r−/− mice (CD45.2+)] were intravenously injected into the recipient mice. After donor cell injection, mice were rested for 12 weeks, and bone marrow reconstitution was assessed via blood. Expression of CD45.1+ and CD45.2+ cells was assessed via flow cytometry.
Bone marrow MC cultures and cell migration assay
Bone marrow MC were cultured in IL-3 (Biolegend, Cat#575504, 20 ng/ml) and stem cell factor (Biolegend, Cat#573902, 60 ng/ ml) in complete MC Roswell Park Memorial Institute (RPMI) 1640 Medium containing: FBS, L-Glutamine, sodium pyruvate, Pen/Strep, and HEPES (4-(2-hydroxyethyl)-1-piperazineethanesul fonic acid). Cells were allowed to culture for 2–5 weeks with non-adherent MC transferred into a new culture dish every few days with the addition of fresh media. Cells (5 × 105 in 100 μl serum-free RPMI containing 1% BSA) were added to the upper chamber of the transwell insert (Millipore Sigma, Carlsbad, CA, Catalog #CLS3421). The lower chamber contained 400 μl complete MC media with chemoattractants: IL-4 (40 ng/ml, Biolegend, San Diego, CA, Catalog #:574302), IL-9 (40 ng/ml, Biolegend, Catalog #:556004), CCL2 (100 ng/ml, R&D, Catalog#479-JE/CF), CXCL1 (100 ng/ml, Peprotech, Cranbury, NJ, Catalog #250-11), CXCL2 (100 ng/ml, R&D, Catalog #452-M2), CXCL8 (100 ng/ml, R&D, Catalog #208–1L), CXCL12 (100 ng/ml, R&D, Catalog #MAB310). The plates were incubated at 37°C for 3 hours, and migrated cells were counted and stained for flow cytometry.
MC adoptive transfer
WT mice were treated with HDM three times a week for 6 weeks. Lungs were processed into single-cell suspension as described above. mMC were sorted using the gating strategy described in Fig. 1. 103 mMC were transferred intranasally to KitWSH/WSH mice and were subsequently intranasally challenged with 25 μl of HDM (1 μg/μl) for 2 weeks. After the last HDM dose, mice were passively sensitized to α-DNP-IgE (3 μg/mouse) and challenged with DNP-BSA as described above.
Airway testing
Following the last allergen dose, mice were passively sensitized intravenously with 3 μg of anti-DNP IgE (Sigma-Aldrich, Burlington, MA, Catalog #D8406) and challenged intratracheally the day after with 20 μl of DNP-BSA (25 mg/ml, Life Tech Corporation, Waltham, MA, Catalog #A23018). Readings were obtained at baseline and after exposure to the allergen challenge. Data were collected for 5 minutes after 3 minutes of inhalation, and average values were expressed as airway resistance (RI). The airway response was measured for up to 60 minutes. Airway resistance was measured using a ventilator (Elan Series Mouse RC Site; Buxco Electronics, Wilmington, NC) and BioSystem XA software (Buxco Electronics, Wilmington, NC).
ELISA
MC protease-1 (MCPT1) (Invitrogen, Waltham, MA), CCL2 (Invitrogen), IL-6 (Biolegend, San Diego, CA), and Ovalbumin-specific IgE (Biolegend, San Diego, CA) enzyme-linked immunosorbent assay (ELISA) were performed according to the manufacturer’s instruction. Briefly, a 96 well-plate was coated with coating antibody overnight at 4°C. After washing three times, 300 μl ELISA blocking buffer was added to the plate and incubated at room temperature for 2 hours. After washing three times, 100 μl standards and samples were added to the plate at different dilutions (neat or 1:2) and incubated at room temperature for 2 hours. After washing three times, 100 μl diluted detection antibody was added to the plate and incubated at room temperature for 1 hour. After washing the plate three times, 100 μl of diluted Avidin-HRP solution was added to the plate and incubated at room temperature for 30 minutes in the dark. After washing three times, 100 μl substrate was added to the plate. Plates were read at absorbance 450 nm and 570 nm.
Histology
Trachea and lung samples were preserved in 10% buffered formalin for 24 hours at room temperature and then transferred into 70% ethanol. Samples were then embedded in paraffin, sectioned, and stained with toluidine blue or chloroacetate esterase. MC numbers were quantified at 4x and 40x magnification.
Statistics and data analysis
All statistics were done using Prism software version 7 (Graph-Pad, San Diego, CA, USA). Flow cytometry data were collected using a Nxt Attune flow cytometer (Life Technologies, Waltham, MA, USA) and were analyzed using FlowJo version 10 (Tree Star FlowJo, BD Bioscience, San Diego, CA, USA).
Supplementary Material
ACKNOWLEDGMENTS
We thank the members of Indiana University Melvin and Bren Simon Cancer Center Flow Cytometry Resource Facility and Drew M. Brown in the IU Histology core facility.
FUNDING
This work was supported by Public Health Service grants from the National Institutes of Health (NIH) (R01 AI057459 and AI129241 to M.H.K.). B.Z. was supported by Public Health Service grants from the NIH (R01 AI085046). A.P. was supported by NIH grant T32 AI060519. M.C. was supported by NIH grant T32 HL091816. A.C. was supported by the National Institutes of Health grant T32 DK007910. B.J.U. was supported by NIH grants T32 AI060519 and F30 HL147515. A.S.N. was supported by NIH grant T32 HL007910. The Indiana University Melvin and Bren Simon Comprehensive Cancer Center Flow Cytometry Resource Facility were supported, in part, by NIH, National Cancer Institute (NCI) grant P30 CA082709 and National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) grant U54 DK106846. The Flow Cytometry Resource Facility was supported, in part, by NIH instrumentation grant 1S10D012270.
Footnotes
DECLARATIONS OF COMPETING INTEREST
The authors declare that they have no competing interests.
APPENDIX A. SUPPLEMENTARY DATA
Supplementary data to this article can be found online at https://doi.org/10.1016/j.mucimm.2023.05.002.
References
- 1.Bochner BS, Undem BJ & Lichtenstein LM Immunological aspects of allergic asthma. Annu. Rev. Immunol 12, 295–335 (1994). [DOI] [PubMed] [Google Scholar]
- 2.Bradding P & Arthur G Mast cells in asthma–state of the art. Clin. Exp. Allergy 46, 194–263 (2016). [DOI] [PubMed] [Google Scholar]
- 3.da Silva EZ, Jamur MC & Oliver C Mast cell function: a new vision of an old cell. J. Histochem. Cytochem 62, 698–738 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Méndez-Enríquez E & Hallgren J Mast cells and their progenitors in allergic asthma. Front. Immunol 10, 821 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Matsuzawa S et al. IL-9 enhances the growth of human mast cell progenitors under stimulation with stem cell factor. J. Immunol 170, 3461–3467 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Renauld JC, Kermouni A, Vink A, Louahed J & Van Snick J Interleukin-9 and its receptor: involvement in mast cell differentiation and T cell oncogenesis. J. Leukoc. Biol 57, 353–360 (1995). [DOI] [PubMed] [Google Scholar]
- 7.Goswami R & Kaplan MH A brief history of IL-9. J. Immunol 186, 3283–3288 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fu Y et al. An IL-9–pulmonary macrophage axis defines the allergic lung inflammatory environment. Sci. Immunol 7, eabi9768 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kearley J et al. IL-9 governs allergen-induced mast cell numbers in the lung and chronic remodeling of the airways. Am. J. Respir. Crit. Care Med 183, 865–875 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaplan MH, Hufford MM & Olson MR The development and in vivo function of T helper 9 cells. Nat. Rev. Immunol 15, 295–307 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tsicopoulos A et al. Involvement of IL-9 in the bronchial phenotype of patients with nasal polyposis. J. Allergy Clin. Immunol 113, 462–469 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Nouri-Aria KT, Pilette C, Jacobson MR, Watanabe H & Durham SR IL-9 and c-Kit+ mast cells in allergic rhinitis during seasonal allergen exposure: effect of immunotherapy. J. Allergy Clin. Immunol 116, 73–79 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Erpenbeck VJ et al. Segmental allergen challenge in patients with atopic asthma leads to increased IL-9 expression in bronchoalveolar lavage fluid lymphocytes. J. Allergy Clin. Immunol 111, 1319–1327 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Ying S et al. Thymic stromal lymphopoietin expression is increased in asthmatic airways and correlates with expression of Th2-attracting chemokines and disease severity. J. Immunol 174, 8183–8190 (2005). [DOI] [PubMed] [Google Scholar]
- 15.Seumois G et al. Single-cell transcriptomic analysis of allergen-specific T cells in allergy and asthma. Sci. Immunol 5, eaba6087 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones CP, Gregory LG, Causton B, Campbell GA & Lloyd CM Activin A and TGF-beta promote T(H)9 cell-mediated pulmonary allergic pathology. J. Allergy Clin. Immunol 129, 1000–1010.e3 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y et al. The absence of IL-9 reduces allergic airway inflammation by reducing ILC2, Th2 and mast cells in murine model of asthma. BMC Pulm. Med 22, 180 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Temann UA, Geba GP, Rankin JA & Flavell RA Expression of interleukin 9 in the lungs of transgenic mice causes airway inflammation, mast cell hyperplasia, and bronchial hyperresponsiveness. J. Exp. Med 188, 1307–1320 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen CY et al. Induction of interleukin-9-producing mucosal mast cells promotes susceptibility to IgE-mediated experimental food allergy. Immunity 43, 788–802 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sehra S et al. TH9 cells are required for tissue mast cell accumulation during allergic inflammation. J. Allergy Clin. Immunol 136, 433–440.e1 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Knoops L, Louahed J, Van Snick J & Renauld JC IL-9 promotes but is not necessary for systemic anaphylaxis. J. Immunol 175, 335–341 (2005). [DOI] [PubMed] [Google Scholar]
- 22.Arinobu Y et al. Developmental checkpoints of the basophil/mast cell lineages in adult murine hematopoiesis. Proc. Natl Acad. Sci. U. S. A 102, 18105–18110 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu AY et al. Mast cells recruited to mesenteric lymph nodes during helminth infection remain hypogranular and produce IL-4 and IL-6. J. Immunol 190, 1758–1766 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schmetzer O, Valentin P, Church MK, Maurer M & Siebenhaar F Murine and human mast cell progenitors. Eur. J. Pharmacol 778, 2–10 (2016). [DOI] [PubMed] [Google Scholar]
- 25.Dahlin JS, Heyman B & Hallgren J Committed mast cell progenitors in mouse blood differ in maturity between Th1 and Th2 strains. Allergy 68, 1333–1337 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bankova LG, Dwyer DF, Liu AY, Austen KF & Gurish MF Maturation of mast cell progenitors to mucosal mast cells during allergic pulmonary inflammation in mice. Mucosal Immunol. 8, 596–606 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dahlin JS, Ding Z & Hallgren J Distinguishing mast cell progenitors from mature mast cells in mice. Stem Cells Dev. 24, 1703–1711 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen CC, Grimbaldeston MA, Tsai M, Weissman IL & Galli SJ Identification of mast cell progenitors in adult mice. Proc. Natl Acad. Sci. U. S. A 102, 11408–11413 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salomonsson M, Dahlin JS, Ungerstedt J & Hallgren J Localization-specific expression of CCR1 and CCR5 by mast cell progenitors. Front. Immunol 11, 321 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Collington SJ et al. The role of the CCL2/CCR2 axis in mouse mast cell migration in vitro and in vivo. J. Immunol 184, 6114–6123 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hallgren J & Gurish MF Mast cell progenitor trafficking and maturation. Adv. Exp. Med. Biol 716, 14–28 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jones TG et al. Antigen-induced increases in pulmonary mast cell progenitor numbers depend on IL-9 and CD1d-restricted NKT cells. J. Immunol 183, 5251–5260 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Banafea GH, Bakhashab S, Alshaibi HF, Natesan Pushparaj P & Rasool M The role of human mast cells in allergy and asthma. Bioengineered 13, 7049–7064 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Forbes EE et al. IL-9- and mast cell-mediated intestinal permeability predisposes to oral antigen hypersensitivity. J. Exp. Med 205, 897–913 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alakhras NS et al. Peanut allergen inhibition prevents anaphylaxis in a humanized mouse model. Sci. Transl. Med 15, eadd6373 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kambe N, Kambe M, Kochan JP & Schwartz LB Human skin–derived mast cells can proliferate while retaining their characteristic functional and protease phenotypes. Blood 97, 2045–2052 (2001). [DOI] [PubMed] [Google Scholar]
- 37.Bolus WR, Gutierrez DA, Kennedy AJ, Anderson-Baucum EK & Hasty AH CCR2 deficiency leads to increased eosinophils, alternative macrophage activation, and type 2 cytokine expression in adipose tissue. J. Leukoc. Biol 98, 467–477 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee YG et al. Recruited alveolar macrophages, in response to airway epithelial–derived monocyte chemoattractant protein 1/CCL2, regulate airway inflammation and remodeling in allergic asthma. Am. J. Respir. Cell Mol. Biol 52, 772–784 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Licona-Limón P, Kim LK, Palm NW & Flavell RA TH2, allergy and group 2 innate lymphoid cells. Nat. Immunol 14, 536–542 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Ulrich BJ et al. Allergic airway recall responses require IL-9 from resident memory CD4+ T cells. Sci. Immunol 7, eabg9296 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Williams CM & Galli SJ Mast cells can amplify airway reactivity and features of chronic inflammation in an asthma model in mice. J. Exp. Med 192, 455–462 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Carroll NG, Mutavdzic S & James AL Distribution and degranulation of airway mast cells in normal and asthmatic subjects. Eur. Respir. J 19, 879–885 (2002). [DOI] [PubMed] [Google Scholar]
- 43.Korosec P et al. Basophils, high-affinity IgE receptors, and CCL2 in human anaphylaxis. J. Allergy Clin. Immunol 140, 750–758.e15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chang HC et al. The transcription factor PU.1 is required for the development of IL-9-producing T cells and allergic inflammation. Nat. Immunol 11, 527–534 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moretti S et al. A mast cell-ILC2-Th9 pathway promotes lung inflammation in cystic fibrosis. Nat. Commun 8, 14017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.de Paulis A et al. Expression of the chemokine receptor CCR3 on human mast cells. Int. Arch. Allergy Immunol 124, 146–150 (2001). [DOI] [PubMed] [Google Scholar]
- 47.Dahlin JS, Feinstein R, Cui Y, Heyman B & Hallgren J CD11c+ cells are required for antigen-induced increase of mast cells in the lung. J. Immunol 189, 3869–3877 (2012). [DOI] [PubMed] [Google Scholar]
- 48.Ebmeyer J et al. Reconstitution of the mast cell population in W/Wv mice. Otol. Neurotol 31, 42–47 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sabbaghi F et al. In activated murine mast cells, NFATc2 is critical for the production of autocrine IL-3, thereby promoting the expression of IL-9. J. Immunol 206, 67–76 (2021). [DOI] [PubMed] [Google Scholar]
- 50.Fish SC, Donaldson DD, Goldman SJ, Williams CM & Kasaian MT IgE generation and mast cell effector function in mice deficient in IL-4 and IL-13. J. Immunol 174, 7716–7724 (2005). [DOI] [PubMed] [Google Scholar]
- 51.Fallon PG et al. IL-4 induces characteristic Th2 responses even in the combined absence of IL-5, IL-9, and IL-13. Immunity 17, 7–17 (2002). [DOI] [PubMed] [Google Scholar]
- 52.Grünig G et al. Requirement for IL-13 independently of IL-4 in experimental asthma. Science 282, 2261–2263 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wan J et al. HMGB1-induced ILC2s activate dendritic cells by producing IL-9 in asthmatic mouse model. Cell. Immunol 352:104085. [DOI] [PubMed] [Google Scholar]
- 54.Tomar S et al. IL-4–BATF signaling directly modulates IL-9 producing mucosal mast cell (MMC9) function in experimental food allergy. J. Allergy Clin. Immunol 147, 280–295 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Parker JM et al. Safety profile and clinical activity of multiple subcutaneous doses of MEDI-528, a humanized anti-interleukin-9 monoclonal antibody, in two randomized phase 2a studies in subjects with asthma. BMC Pulm. Med 11, 14 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oh CK et al. A randomized, controlled trial to evaluate the effect of an anti-interleukin-9 monoclonal antibody in adults with uncontrolled asthma. Respir. Res 14, 93 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.White B, Leon F, White W & Robbie G Two first-in-human, open-label, phase I dose-escalation safety trials of MEDI-528, a monoclonal antibody against interleukin-9, in healthy adult volunteers. Clin. Ther 31, 728–740 (2009). [DOI] [PubMed] [Google Scholar]
- 58.Oh CK, Raible D, Geba GP & Molfino NA Biology of the interleukin-9 pathway and its therapeutic potential for the treatment of asthma. Inflam. Allergy Drug Targets 10, 180–186 (2011). [DOI] [PubMed] [Google Scholar]
- 59.Berlin AA & Lukacs NW Treatment of cockroach allergen asthma model with imatinib attenuates airway responses. Am. J. Respir. Crit. Care Med 171, 35–39 (2005). [DOI] [PubMed] [Google Scholar]
- 60.Cahill KN et al. KIT inhibition by imatinib in patients with severe refractory asthma. N. Engl. J. Med 376, 1911–1920 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Steenwinckel V et al. IL-13 mediates in vivo IL-9 activities on lung epithelial cells but not on hematopoietic cells. J. Immunol 178, 3244–3251 (2007). [DOI] [PubMed] [Google Scholar]
- 62.Townsend JM et al. IL-9-deficient mice establish fundamental roles for IL-9 in pulmonary mastocytosis and goblet cell hyperplasia but not T cell development. Immunity 13, 573–583 (2000). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.