

Abstract
Purpose:
Coccidioidomycosis (CM) is a systemic fungal disease caused by the dimorphic fungi Coccidioides immitis and Coccidioides posadasii. In its endemic areas of the United States, CM is growing as a public health challenge with a marked increase in incidence in the last 15 years. Although Coccidioides infection is asymptomatic in most cases, symptomatic pulmonary disease occurs in ~40% and disseminated coccidioidomycosis (DCM) occurs in ~1% of previously healthy children and adults. DCM is markedly more common in immunocompromised people, who often experience life-threatening disease despite use of antifungal medications. Although options for antifungal therapy have improved, lifelong therapy is needed for those who develop coccidioidal meningitis. The purpose of this article was to review the state of antifungal therapy and recent studies of host–pathogen interactions in CM in light of advances in immunomodulatory therapy.
Methods:
The study included a review of PubMed and abstracts of the Coccidioidomycosis Study Group (years 2000–2019).
Findings:
Current therapy for CM relies upon azole and polyene antifungal agents. Murine models and studies of DCM in patients with monogenic primary immunodeficiency states and acquired immunodeficiency have revealed the importance of both innate and adaptive immune responses in the control of infections with Coccidioides species. In particular, defects in sensing of fungi and induction of cellular immune responses have been frequently reported. More recently, polymorphisms in key signaling pathways and in the generation of Th17 and Th1 immune responses have been linked with DCM.
Implications:
Antifungal therapy is sufficient to control disease in most cases of CM, but treatment failure occurs in cases of severe pulmonary disease and nonmeningeal disseminated disease. Lifelong therapy is recommended for meningitis in view of the very high risk of recurrence. Corticosteroid therapy is advised by some experts for severe pulmonary disease and for some neurologic complications of DCM. DCM is only rarely the result of a severe monogenic immunodeficiency. Case studies suggest that reorienting cellular immune responses or augmenting effector immune responses may help resolve DCM. Systematic investigation of immunotherapy for coccidioidomycosis is advisable and may help to address the recent marked increase in reports of the disease in endemic areas.
Keywords: Antifungal, Coccidioides, Coccidioidomycosis, Genomics, Immunology, Valley fever
INTRODUCTION
Coccidioidomycosis (CM) is a systemic disease caused by infection by the closely related dimorphic fungi Coccidioides immitis and Coccidioides posadasii, found only in the semi-arid areas of North America, Central America, and South America (Figure 1). In California, most cases occur in the central San Joaquin Valley, giving rise to the term “valley fever,” which is often used as a synonym for systemic disease due to Coccidioides infection. In the United States, most cases (95%) occur in California and Arizona.1 A marked increase in incident cases from these 2 states has occurred in the last decade, particularly in California, where the annual rate increased in 2017 to 18.8 per 100,000 population with 7466 reported cases, the highest annual number of cases ever recorded. In 2017, more than 14,000 cases were reported from all states to the Centers for Disease Control and Prevention, but this figure may represent as few as 6% of the true number of new infections. For example, in one prospective study in Arizona, 16% of individuals presenting with prolonged chest symptoms or community-acquired pneumonia between 2003 and 2004 were found to have serologic evidence of CM. Similar results were obtained in a retrospective analysis of data from 30 outpatient clinics in the city of Phoenix.2,3
Figure 1.
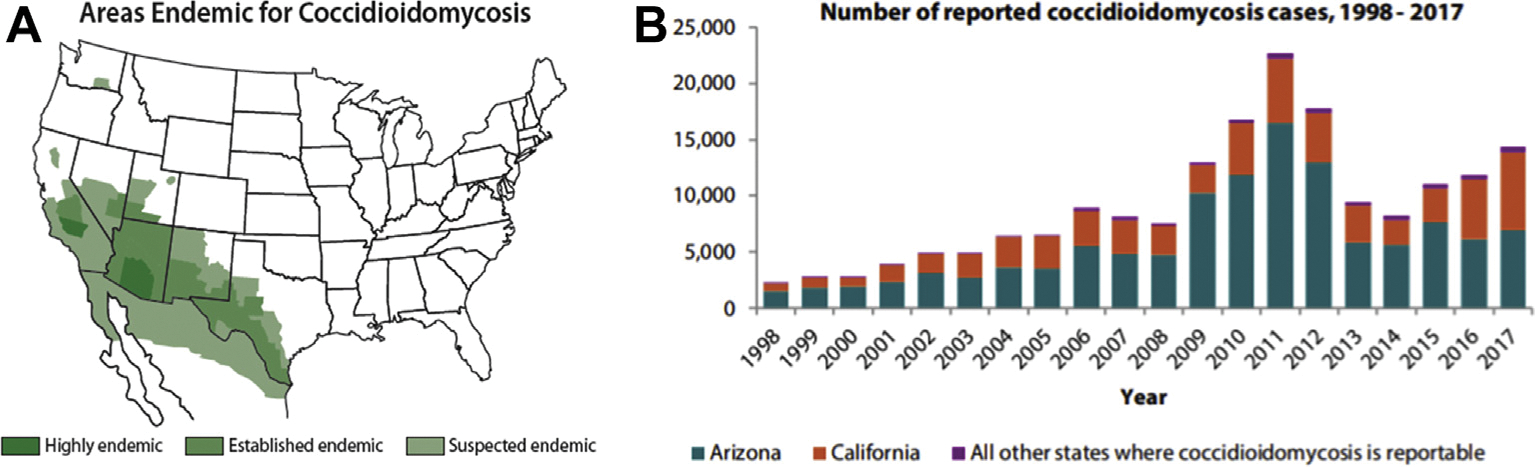
Epidemiology of Coccidioides infection. (A) Areas in which human infection and autochthonous transmission to animals have been described. Reprinted from https://www.cdc.gov/fungal/diseases/coccidioidomycosis/causes.html. (B) Cases of coccidioidomycosis reported to the Centers for Disease Control and Prevention (1998–2017). Note the predominance of case reports from California and Arizona. Reprinted from https://www.cdc.gov/fungal/pdf/valley-fever-expanding-cocci-P.pdf.
CM was first described in Argentina in 1892 followed by the recognition of 2 additional cases in 1894 in California; all 3 subjects died of similar disease that included granulomatous lesions of the skin.4 In the mid-20th century, Charles Smith and others developed serologic methods and skin test reagents used in seminal epidemiologic and clinical studies of CM. These studies clarified that respiratory acquisition of infection may be followed by severe pulmonary disease, osteomyelitis, meningitis, and other life-threatening complications, leading Smith to note “its notorious clinical and pathological mimicry of tuberculosis.”5–7 These studies and others also revealed that Filipino subjects and people of African ancestry are at greater risk of disseminated CM (DCM).6,8,9 Although this fraction remains small (~3.4% among African-American subjects in the seminal study by Smith and Beard6), DCM can have devastating consequences in previously healthy individuals. Studies in the late 20th century revealed that diabetes, HIV infection, and iatrogenic immunosuppression increase the risk of DCM.10–13
Fortunately, the antifungal medications developed in the mid-to late-20th century have reduced the morbidity and mortality of CM, including DCM. Amphotericin B (AmB) deoxycholate, a polyene antifungal agent, was developed in the 1950s and immediately shown to have utility in the treatment of coccidioidal meningitis.14 Although often poorly tolerated, intravenous AmB deoxycholate remained the mainstay of antifungal therapy for severe CM and DCM until the development and licensure of antifungal azole medications. Newer lipid formulations (eg, liposomal AmB and amphotericin lipid complex) of AmB are less toxic than the original AmB deoxycholate preparation and are often used in the initial treatment of nonmeningeal DCM in immunocompromised hosts. Ketoconazole, an imidazole, was modestly effective but has been supplanted by the more effective and less toxic triazoles (eg, fluconazole, itraconazole, posaconazole, isavuconazole).15–17 One prospective study was performed comparing the relative activity of triazoles; itraconazole appeared marginally more effective than low daily dose (400 mg/d) oral fluconazole in a multicenter comparison of the 2 drugs as treatment for progressive, nonmeningeal CM.18 Current antifungal therapy for severe pulmonary CM or DCM generally consists of oral triazole antifungal medications or intravenous lipid formulations of AmB.19,20 Unfortunately, the triazoles and polyenes are poorly fungicidal, and neither class produces a sterilizing cure of meningitis; relapse of meningitis is very common if triazole therapy is stopped,21 and lifelong, continuous antifungal therapy is recommended.19,20 These and other limitations of current antifungal therapy for CM have recently been reviewed in detail.19
Overall, CM is a disease that is unrecognized in most cases and may not require antifungal therapy if limited to minor pulmonary disease. At the opposite end of the spectrum, extrapulmonary disease occurs in ~1%, in whom antifungal therapy is not always successful. The current review analyzes recent data revealing host–pathogen interactions that may be determinants of dissemination of CM and which suggest possible therapeutic approaches. We postulate that genomic evaluation of individuals with DCM is advisable and that immunomodulatory therapy to augment or reorient immune responses is a therapeutic avenue that should be subjected to investigation.
MATERIALS AND METHODS
The PubMed and Clinical Trials.gov databases were searched for related titles published until December 2019, using the following key words alone and in combination: Valley Fever, Coccidioides, coccidioidomycosis, and disseminated coccidioidomycosis. These were reviewed alone and in combination with at least one of the following terms: genetic, pathogenesis, meningitis, mortality, predisposition, and host. In addition, references listed in relevant articles were reviewed for other references related to the theme of host–pathogen interactions in human infections caused by Coccidioides species. Abstracts for Coccidioidimycosis Study Group were also reviewed, as available online.
OVERVIEW OF HOST–PATHOGEN INTERACTIONS IN CM
Coccidioides life cycle and biology
As noted earlier, 2 distinct species of Coccidioides are now recognized: C immitis (largely confined to California) and C posadasii (found in all other endemic areas in the Americas). Although the 2 species exhibit minor differences in their growth characteristics in vitro, their life cycle and clinical manifestations are indistinguishable.22,23 The fungus grows in filamentous form within the upper several centimeters of soil. Septation of the hyphae occurs during fungal growth, and alternate cells undergo autolysis, liberating spores (arthroconidia). Dispersal of these infectious arthroconidia occurs when the soil is disturbed by wind, excavation, recreational activities, and other phenomena24 (Figure 2). Once inhaled into the lung, the arthroconidia germinate and form spherules, which can enlarge to as much as 150 mm in diameter. Internal segmentation of these spherules produces multiple minute endospores (each 2–3 μm in diameter) within a shell composed of a variety of macromolecules, including chitin, a long-chain polysaccharide polymer. Eventually, the spherules rupture and release hundreds of endospores that may disseminate widely throughout the host, forming new spherules, with the potential to repeat the process. This form of exponential amplification is unique among endemic mycoses and is analogous to the reproduction of viruses that produce hundreds or thousands of progeny in parasitized cells in a single replicative cycle.9,25
Figure 2.
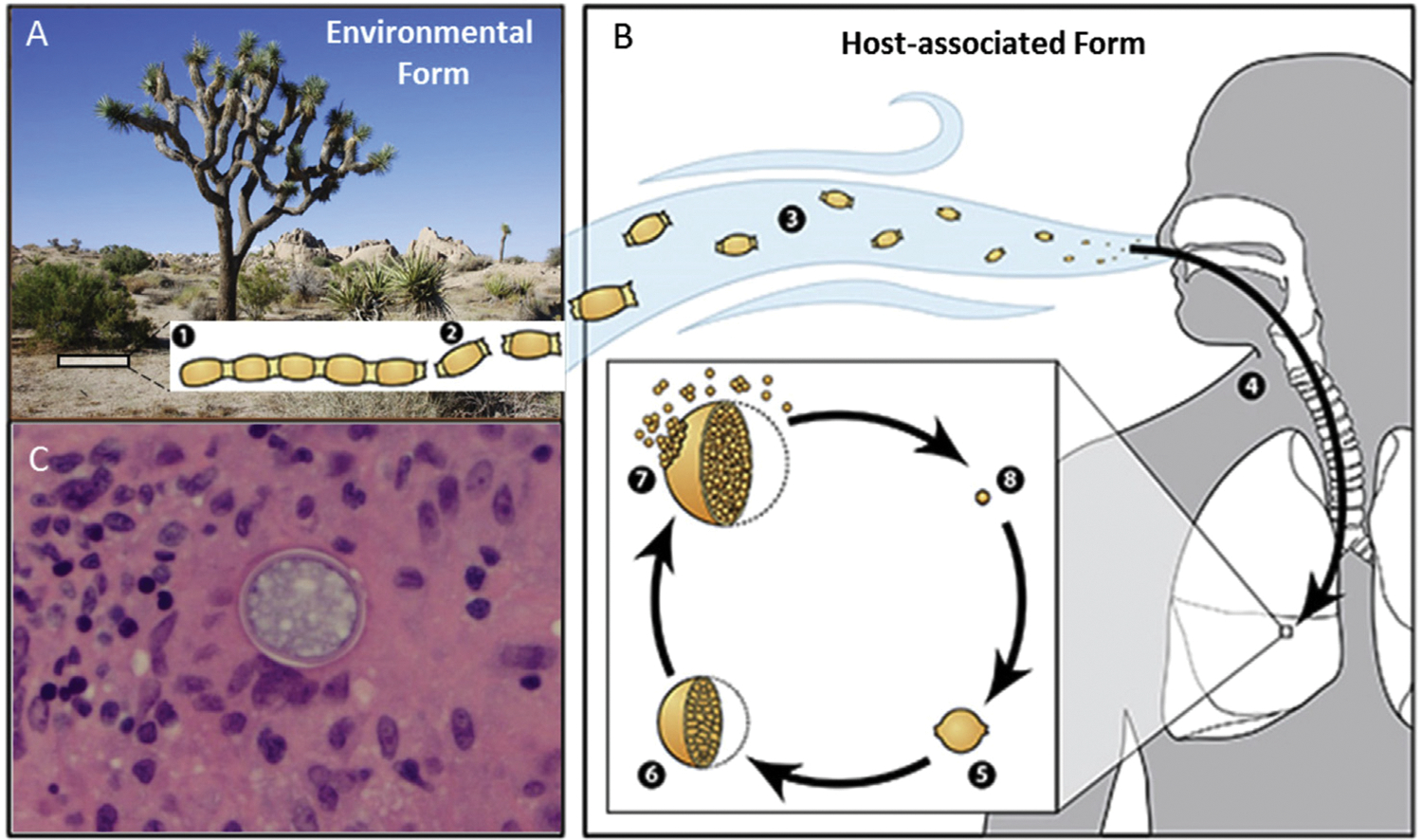
Coccidioides biology. (A) Coccidioides grows as a mold with septate hyphae near the surface of the soil (1). The hyphae fragment into arthroconidia (2) and become aerosolized when the soil is disturbed by human activity or the wind (3). (B) These arthroconidia are inhaled and settle into the lungs, where signals from the environment promote the formation of spherules (5). Spherules divide internally until they are filled with endospores (6). When spherules rupture (7), the endospores are released. Endospores are then able to disseminate and develop into new spherules and to repeat the cycle. (C) Spherule in a soft tissue scalp lesion of a boy with disseminated coccidioidomycosis. Figure modified from https://www.cdc.gov/fungal/diseases/coccidioidomycosis/causes.html#wastate.
This morphological transition of the fungus from its hyphal form to infectious endospores via spherule formation involves substantial transcriptional reprogramming. Coccidioidal arthroconidia can be induced to form spherules in vitro by an increase in environmental temperature and ambient carbon dioxide concentration. Two studies26,27 have shown that spherule formation under these conditions is associated with changes in a number of metabolic pathways, particularly those involved in the metabolism of complex carbohydrates.22,27,28 Among other findings, these in vitro studies showed that the fungal chitinase 2 and chitinase 3 genes are transcriptionally upregulated in spherules compared with mycelia, presumably due to the remodeling of the cell wall involved in spherule maturation and during the release of endospores. Interestingly, the presence of human neutrophils induces spherule differentiation during in vitro culture and also inhibits the formation of hyphae.29 Because spherules seem to promote persistence of Coccidioides, this observation suggests that specific adaptations occurred during the evolution of Coccidioides that promote survival in the face of innate and adaptive immune responses mounted by animals.22,23,27,30,31
Innate immunity
Our understanding of innate immune responses to Coccidioides is largely derived from murine models and the characterization of immune dysfunction in a handful of human cases.23,26,32–35 Immune responses to fungi are generally initiated by sensing of cell wall components, fungal DNA, and other fungal elements by pattern recognition receptors. Detection of pathogen-associated molecular patterns expressed by pathogenic fungi has been most clearly shown by examination of the phenotype of mice lacking Dectin-1. Dectin-1, a lectin expressed on neutrophils, macrophages, and dendritic cells, recognizes β-1,3-glucan, a key polysaccharide cell wall component of Coccidioides, Histoplasma, Candida, and other pathogenic fungi.36 Dectin-1 has an extracellular carbohydrate recognition domain linked via a long stalk to a transmembrane domain and a cytoplasmic tail containing a tyrosine-rich immunoreceptor tyrosine-based activation domain. Clustering of Dectin-1 molecules by contact with poly-β-1,3-glucan is followed by phosphorylation of tyrosine residues in the immunoreceptor tyrosine-based activation domain; this action triggers Syk tyrosine kinase activity that promotes downstream signaling through CARD9, leading to the elaboration of key cytokines that promote the activation and differentiation of T cells (Figure 3).
Figure 3.
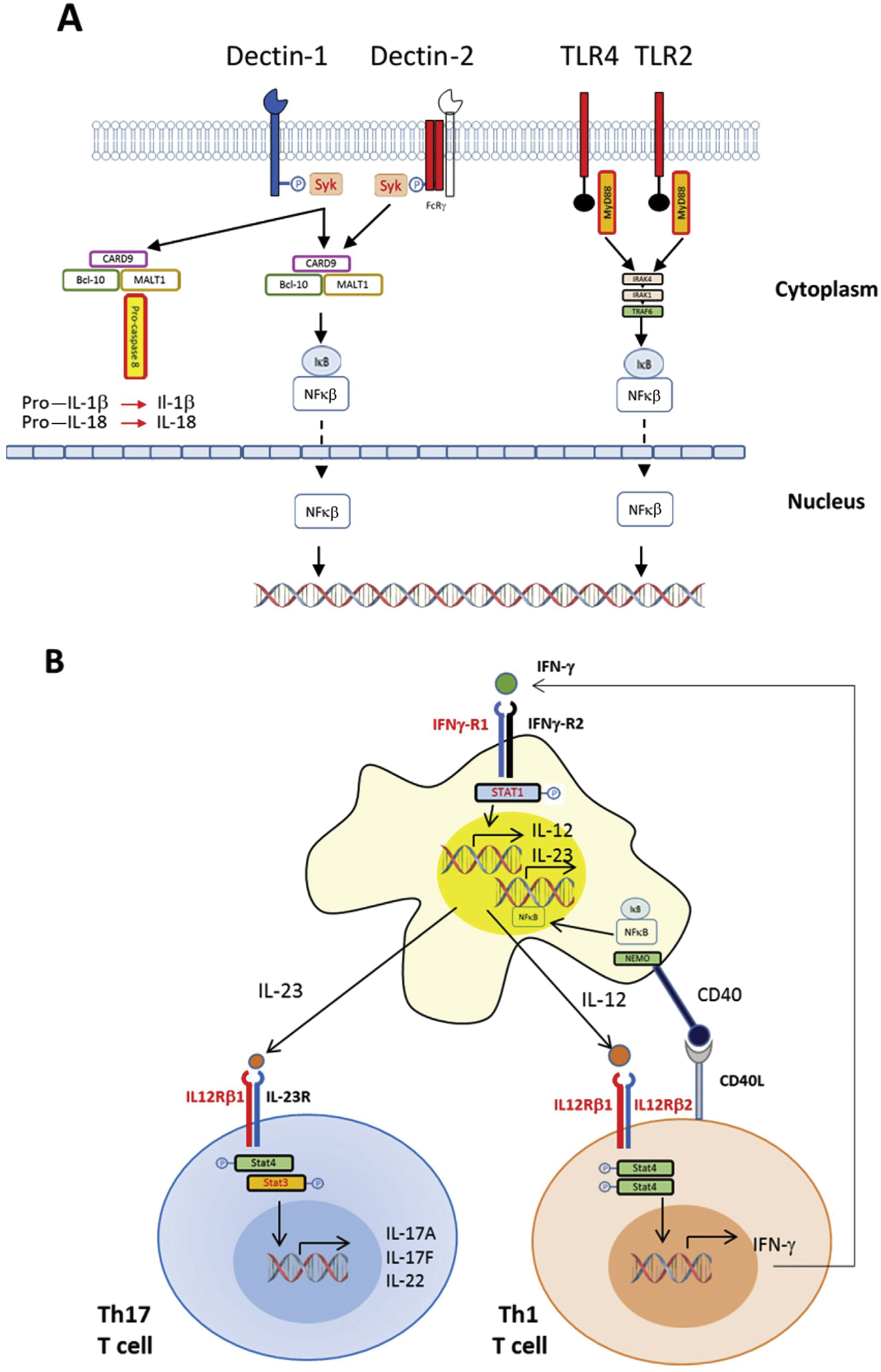
Immunobiology of coccidioidomycosis. (A) This simplified diagram outlines the recognition of fungal pathogen–associated molecular patterns by pattern recognition receptors that trigger activation of macrophages, neutrophils, and dendritic cells. Dectin-1 and Dectin-2 are examples of C-type lectin receptors involved in recognition of fungal cell wall components. Aggregation of Dectin-1 molecules in the presence of fungal polysaccharide ligands triggers phosphorylation of its cytoplasmic tail, leading to activation of Syk kinase, which induces formation of the CARD9-Bcl10-Malt1 complex, which mediates activation of nuclear factor kappa β (NF-κβ) and the production of cytokines, including interleukin (IL)-2, IL-6, IL-10, and IL-12, that activate and promote differentiation of T cells. Malt-1 can also engage and proteolytically cleave pro-caspase 8, activating the proinflammatory cytokines IL-1β and IL-18. Recognition of coccidioidal molecules by Toll-like receptor 2 (TLR2) is essential for vaccine-induced protection in murine models of coccidioidomycosis. Homodimers or heterodimers containing TLR2 are linked by an adaptor molecule to MyD88, activating IRAK4; subsequent downstream signaling leads to degradation of the IkB complex, releasing NF-κB complexes and allowing their nuclear entry and activation of cytokine transcription. Toll-like receptor 4 (TLR4) and Dectin-2 have not been confirmed to play a role in coccidioidomycosis but are important in other mycoses. (B) Differentiation of CD4 T cells and their polarization into Th1 or Th17 cells and crosstalk with macrophages and other antigen-presenting cells. As outlined in panel A, recognition of fungal cellular components leads to activation of macrophages and other phagocytes, which release IL-2, IL-6, IL-10, IL-23, and other cytokines that promote the expansion and differentiation of naive CD4 T cells. Those that become Th17 cells express IL-17A, IL-17F, and IL-22, which promote production of antimicrobial peptides in epithelial cells. CD4 T cells that differentiate along the Th1 pathway in response to IL-12 release interferon gamma (IFN-γ), which enhances phagocytosis by macrophages and other phagocytes and the release of antimicrobial molecules. Red lettering reveals receptor and signaling molecules in which deleterious mutations have been found in cases of disseminated coccidioidomycosis.33,35,91 STAT = signal transducer and activator of transcription.
Mice that lack Dectin-1 are more susceptible to experimental coccidioidal infection, as shown by greater and more rapid mortality compared with control mice.26,34,37 In addition, inbred mice differ in their susceptibility to Coccidioides infection, which maps to the differential splicing of Clec7a, the gene that encodes Dectin-1. Kirkland and Fierer38 found that the estimated lethal dose of a patient-derived Coccidioides isolate differs by up to 10,000-fold among commonly used strains of mice. C57BL/6 mice are far more sensitive to infection than DBA/2 mice and produce a truncated splice variant of the Clec7a encoding a Dectin-1 protein lacking most of the extracellular stalk. By contrast, DBA/2 mice produce a full-length Dectin-1 protein. Cytokine expression profiles also differ between the strains: C57BL/6 mice produce more interleukin (IL)-10 and IL-4 and less interferon gamma (IFN-γ) in their lungs compared with DBA/2 mice. Moreover, dendritic cells from C57BL/6 mice produce more IL-10 and IL-23 and less IL-12 than cells from DBA/2 mice in response to exposure to spherules or purified β-glucan.37 Finally, CARD9-deficient mice are highly susceptible to coccidioidal infection and cannot form protective immunity in response to inoculation with spores of a mutant C posadasii strain that lacks the ability to form spherules due to the deletion of chitinase 2 and chitinase 3.30,39 These and other animal data33 show that the innate immune responses set the stage for the successful control of Coccidioides infection. In contrast to other fungal pathogens (eg, Blastomyces dermatitidis, Candida albicans, Aspergillus species), murine models also indicate that mannose receptors, Dectin-2, Toll-like receptor 2 (TLR2), and Toll-like receptor 4 do not play a significant role in detection of coccidioidal infection.30,33,40–42
Interestingly, no cases of DCM in humans have as yet been attributed to Dectin-1 deficiency, even though polymorphisms in the Clec7a gene are known to exist and have been linked to mucocutaneous candidiasis,43 and loss-of-function in CARD9 is strongly linked to invasive infections by Candida species.44
Adaptive immunity
As noted previously, infection with Coccidioides species is often clinically inapparent.33,35 This was shown by Smith and Beard6 who found that 60% of servicemen stationed at airfields in the San Joaquin Valley developed skin test reactivity (ie, antigen-specific T-cell responses) to coccidioidal antigens without symptoms. Most of those who develop symptomatic coccidioidal disease have self-limited infection and low titers of complement-fixing antibodies to coccidioidal antigens. By contrast, the small fraction of people with extrapulmonary dissemination of coccidioidal infection typically have a high titer of complement-fixing antibodies and lack delayed-type hypersensitivity, evincing a Th2 bias in cellular immune response.7,45
This type 2 immunity represents the presence of helper T cells producing the cytokines IL-4, IL-13, and IL-5. IL-4 stimulates B cells to boost immunoglobulin E (IgE) production to high levels, which has been correlated with the severity of Coccidioides disease.46 Murine data suggest that IL-4 itself directly reduces the antifungal activity of innate immune cells as it promotes expression of arginase-1 and blocks nitric oxide production,47 which is required for fungicidal activity.48 Murine studies of cryptococcal infection indicate that IL-13 works in a similar fashion49 in promoting high expression of IL-4.
Polymorphisms in an intron of the IL-4 gene have been associated with high serum levels of the cytokine in those who develop paracoccidioidomycosis, another endemic mycosis found in South America.50 IL-5 promotes eosinophil development in the bone marrow, and peripheral eosinophilia has been correlated with disseminated disease, but it is not clear if there is a mechanistic relationship.51,52 Thus, there is substantial evidence that type 2 immunity attenuates antifungal immune responses. To date, only one primary immune dysregulatory disorder of type 2 immunity has been identified that promotes CM: dominant negative signal transducer and activator of transcription (STAT) 3 disease.35 However, we would expect increased susceptibility to severe CM or DCM in patients with mutations in phosphoglucomutase 3, IL-21R, and other primary atopic disorders.53,54
The main adaptive immune cells of type 1 immunity are the CD4+ T cells that produce IFN-γ (ie, Th1 cells). Vaccine models in mice have suggested that Th1 (and Th17) responses are essential for protection against DCM.31,55 Patients who have diminished type 1 immunity because of monogenic defects in either chain of the heterodimeric IL-12 receptor (IL-12Rβ1 and IL-12Rβ2) have an elevated risk of DCM35,55–57, which reflects the central role of IL-12 in the development of Th1 helper T cells. Activated Th1 cells produce IFN-γ, but individuals bearing loss-of-function defects in either of the IFN-γ receptor chains (IFN-GR1 or IFN-GR2) cannot sense this cytokine and have impaired antifungal immunity. Downstream of the IFN-γ receptor lies STAT1, and subjects with both dominant loss-of-function and dominant gain-of-function variants in STAT1 have an increased susceptibility to fungal infection.
Surprisingly, monogenic defects in the IFN-γ cytokine itself have not been identified, but individuals bearing these defects would be expected to have a profound susceptibility to fungal infections, including CM. Peripheral blood cells from patients with DCM produce less IFN-γ in response to Coccidioides antigen than cells from those with milder disease.58,59 Treatment of mice with IFN-γ was able to protect mice from systemic infection.60 A few human patients with extrapulmonary disease have been treated successfully with IFN-γ as well (as discussed later), supporting the importance of this cytokine in fighting CM. In these disorders of the IL-12/IFN-γ axis, Th1 cell development or effector function is diminished. However, broader risk factors for DCM include pregnancy, HIV infection, and the use of immunosuppressive medications, in which Th1 immunity is suppressed in favor of Th2.35
Evidence in mice and humans also supports the importance of a third program of immune responses, called Th17 immunity, for fighting fungal infections. These helper T cells produce the cytokines IL-17A, IL-17F, and IL-22 upon stimulation. Th17 cells were noted in the development of synthetic candidate vaccines to Coccidioides.31,33 The fact that depletion of Th17 cells after successful vaccination in mice removes the vaccine-induced protection supports the importance of these helper T cells in protective memory against Coccidioides.61 A major role of IL-17A is to recruit neutrophils to the site of infection, which may be critical for preventing extrapulmonary dissemination of CM. IL-17A also directs natural killer cells to produce granulocyte-macrophage colony-stimulating factor, which recruits neutrophils and enhances their fungicidal activity.62 Humans deficient in IL-17F are highly susceptible to mucocutaneous Candida, supporting the notion that this cytokine also plays a protective role in fungal immunity. IL-22 is also protective, preventing dissemination by recruitment of inflammatory innate cells and in augmenting barrier functions of infected tissues63; autoantibodies against IL-22 lead to a susceptibility to chronic infections with Candida in humans.64 IL-17A and IL-17F bind to heterodimeric receptors that comprise subunits of IL-17RA and IL-17RC. Homozygous deficiency of IL-17RA has been shown in humans to incur susceptibility to yeast infections.65,66 Notably, variants in IL-17RA are more commonly found in African-American subjects, who have a higher susceptibility to DCM.33 Together, these clinical and experimental data strongly indicate the importance of Th17 immunity in fungal infections.
The development of Th17 cells in vitro and in vivo is driven by priming naive T cells in the presence of transforming growth factor-β and an inflammatory cytokine such as IL-6. When IL-6 amounts are limiting, transforming growth factor-β instead drives the development of induced regulatory T cells. Thus, induced regulatory T cells and Th17 cells form a balance of protective and repressive immunity based on the inflammatory context at the time of priming. This balance seems to be clinically important in determining dissemination of coccidioidal infection in humans,67 although the data are limited, and it is unclear if regulatory T cells are purely suppressive of protective immunity. The complex relationship between Th17 cells and regulatory T cells offers some evidence that these cell types can, in fact, work cooperatively to promote adaptive immunity toward fungi.68 More research is needed to illuminate the immunological crosstalk in cases of infection that are controlled compared with cases of DCM.69
Evaluation of human cohorts with DCM: initial experience
Investigators at the National Institute of Allergy and Infectious Diseases (A. Hsu and S. Holland) recently reported initial results of systematic whole-exome or focused gene sequencing in 58 individuals with DCM.33 The data from this analysis were examined by filtering for rare genetic variants (defined as a frequency of ≤1% in the Exome Aggregation Consortium database70) with a high probability of having deleterious effects on protein function and likely to be associated with disseminated fungal infections. More than 100 polymorphisms were identified in genes involved in innate immune responses, including innate immune sensing and signaling pathways. These genes included elements of IL-12/IFN-γ signaling, intracytoplasmic nucleic acid sensing molecules (RIG-I), and both components of the heterodimeric IL-17 receptor (IL-17RA and IL-17RC). In vitro validation of several variants has been completed by introducing the novel permutations into cell lines. Of particular interest is the finding of polymorphisms in STAT3 and STAT4 among African-American subjects with DCM, as these participate in IL-23 signal transduction involved in differentiation of naive CD4 T cells into Th17 cells.
Similarly, we have described whole-exome sequencing analysis involving a convenience sample of ethnically and racially diverse cohort of 22 adults living in California with extrapulmonary dissemination of CM.71 Among these, 16 were men, 8 were African American, 2 were Filipino, and 2 were of Middle Eastern ancestry. In contrast to the analytic approach used by Hsu and Holland, we hypothesized that polymorphisms which predispose to dissemination of Coccidioides infection could be relatively common alleles among populations with Asian and African ancestry, yet be rare among those of European ancestry, in whom DCM is substantially less common. We identified a mean of 246,708 variants from the human reference genome per sample; of these, a mean of 6336 per sample passed quality controls, affected protein sequence or splicing, and had a frequency of ≤10% in the Exome Aggregation Consortium database (Table I). We identified 3 African-American individuals with alterations in the gene (CHIT1) encoding the human chitinase protein known to reduce or abrogate enzymatic activity.72 This finding included 1 person who was homozygous for alleles for the single amino acid changes Ala442Val and Gly354Arg, either of which abrogate chitinase activity. In addition, 2 other individuals (K09 and K11) were compound heterozygous for the Gly354Arg allele along with a deletion that activates a cryptic splice site in exon 10 leading to production of a truncated and inactive chitinase protein. Thus, 5 (27%) of 22 individuals with DCM possessed genetic variants of CHIT1 predicted to abrogate chitinase. The impact of these genetic variants on chitinase activity and the outcome of coccidioidal infection require verification, but it is conceivable that chitinase release by neutrophils and macrophages interferes with spherule formation and extensive dissemination of Coccidioides.
Table I.
Clinical manifestations of coccidioidomycosis in study participants.
| Patient Identifier | Sex | Population | Sites of Coccidioidal Disease |
|---|---|---|---|
|
| |||
| K03 | Male | AFR | OM at 1 site with epidural abscess |
| K06 | Male | AFR | Cavitary pneumonia. OM to multiple axial skeletal locations with soft tissue involvement |
| K08 | Female | AFR | Miliary pneumonia. OM to multiple axial skeletal locations. Coccidioidal meningitis |
| K09 | Male | AFR | OM at 12 axial skeletal locations |
| K11 | Male | AFR | Pulmonary disease with dissemination to 2 soft tissue sites |
| K12 | Male | AFR | OM at 3 axial skeletal locations |
| K13 | Male | AFR | Pulmonary. OM at 3 axial skeleton locations with paraspinal abscess |
| K15 | Male | AFR | Miliary pneumonia, coccidioidal meningitis, and OM with 2 axial skeletal locations |
| K14 | Male | FIL | Pulmonary disease, coccidioidal meningitis complicated by arachnoiditis. OM at 1 axial skeleton location |
| U22 | Female | FIL | OM of L4-S1 spine and right psoas muscle abscess |
| K01 | Male | LAT | Pulmonary disease with OM at 2 axial skeletal locations. Soft tissue abscess |
| K02 | Male | LAT | Miliary pneumonia. OM at 5 axial skeletal locations. Two soft tissue abscesses. Multiple cutaneous skin lesions |
| K05 | Male | LAT | Severe fibrocavitary pulmonary. OM at 2 axial skeletal locations. Prostatic abscess |
| K07 | Male | LAT | Pulmonary disease and coccidioidal meningitis. OM at 1 axial skeleton location |
| K10 | Male | LAT | Miliary pneumonia with OM at 5 different axial skeletal locations |
| K16 | Male | LAT | Pulmonary. Disseminated to skin, liver, and multiple axial skeletal foci of OM |
| K18 | Female | LAT | Coccidioidal meningitis. OM at 5 axial skeleton locations |
| K19 | Male | LAT | Pulmonary disease. Coccidioidal meningitis and OM at 2 axial skeletal locations |
| K20 | Female | LAT | OM at 12 axial skeletal locations |
| U21 | Female | LAT | Coccidioidal meningitis with hydrocephalus. Skin lesions |
| K04 | Male | ME | OM at 6 axial skeletal locations. Six soft tissue abscesses |
| K17 | Female | ME | Pulmonary disease. OM at 3 axial skeletal locations |
AFR = African American; FIL = Filipino; ME = Middle Eastern; LAT = Latino; OM = osteomyelitis.
Two African-American subjects (K09 and K11) were also found to have missense variants in CLEC7A that encode Ser161Leu (heterozygous) and Ile223Ser (homozygous), respectively. Both of these changes are rare in European subjects (~0.01%) but common in African-American subjects (3%–5%). The homozygous Ile223Ser change is computationally predicted as probably damaging to protein function (Combined Annotation Dependent Depletion [CADD] score, 24.5), and the Ser161Leu variant is estimated to possibly interfere with the function of Dectin-1 (CADD score, 11.9).73
We also identified an individual with a polymorphism in the cytoplasmic tail in IL17-RA encoding a missense mutation predicted to be deleterious (Pro562Gln) (CADD score, 26.6). This variant is known to be common with an allele frequency of 7.3% across all individuals and enriched to 31% among East Asian individuals. The presence of the missense mutation was confirmed by using Sanger sequence analysis, and flow cytometric analysis revealed low expression of IL-17RA on monocytes of this individual (Figure 4). Interestingly, IL-17RA was significantly lower on the monocytes of all 5 people with DCM compared with the healthy control subjects.
Figure 4.
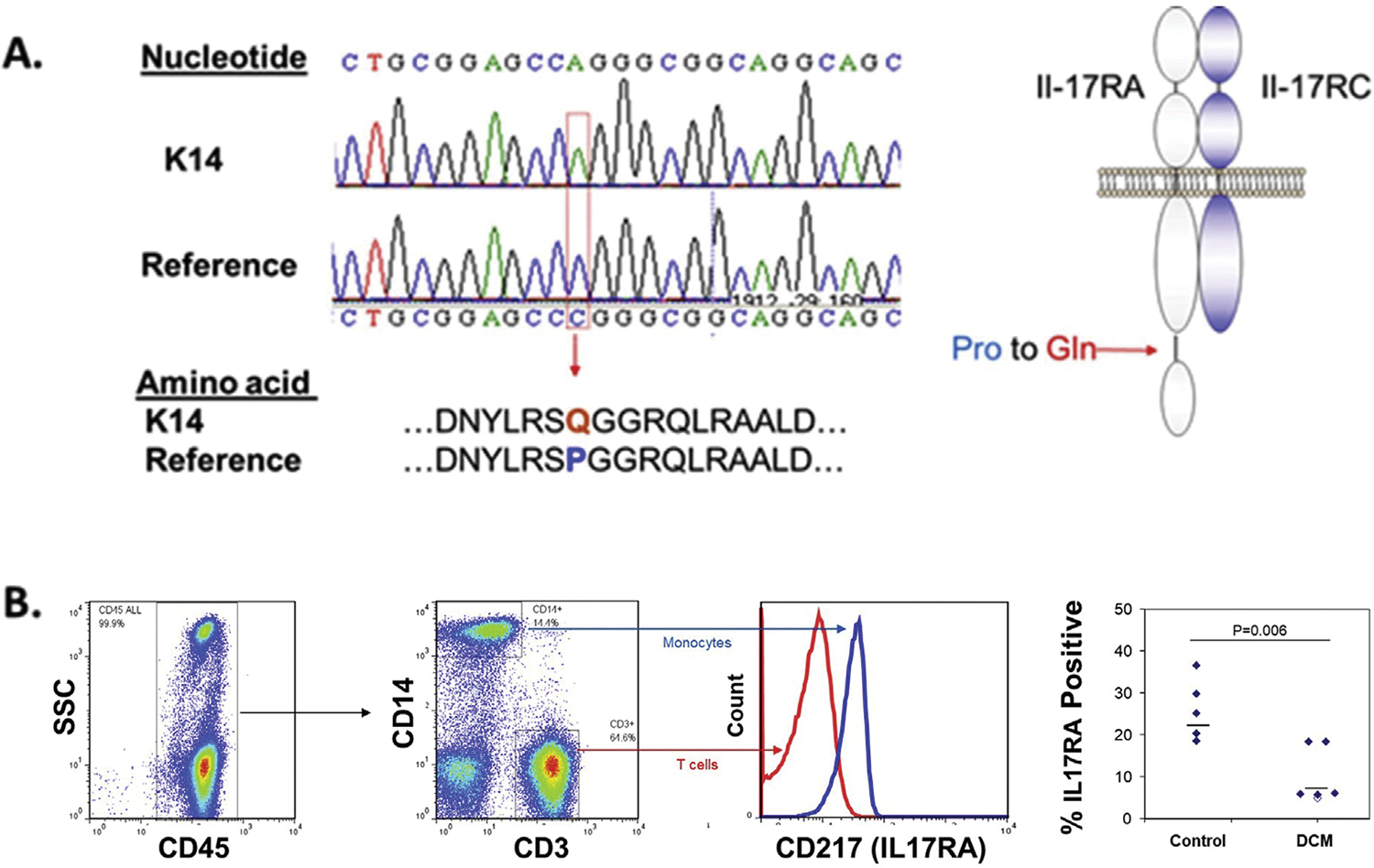
Detection and analysis of expression of an interleukin (IL)-17RA variant in coccidioidomycosis University of California Los Angeles (UCLA) Exome data. With approval from local ethics committees, DNA was extracted from peripheral blood (n = 20) and formalin-fixed tissue specimens (n = 2) of patients with extrapulmonary dissemination of coccidioidomycosis and submitted for whole-exome sequence analysis on a HiSeq 2500 instrument (Illumina, San Diego, California). The resulting data were filtered for variants in genes known or suspected to be in pathways relating to responses to fungal infections (see Supplemental Table S1). A potentially damaging homozygous variant was found in IL17-RA, an element of the heterodimeric IL-17 receptor. (A) Sanger sequencing confirmed a missense mutation in codon 562, producing a Pro562Gln change in the cytoplasmic tail of IL17-RA. (B) IL-17RA expression on monocytes of healthy control subjects and adults with disseminated coccidioidomycosis. Viably cryopreserved peripheral blood mononuclear cells from healthy young adults and 5 individuals with disseminated coccidioidomycosis (DCM) (K12,K13, K14, K17, and K18 in Table I) were thawed and analyzed by flow cytometry using fluorochrome-conjugated antibodies to CD3, CD14, CD45, and CD217 (IL-17RA). The median cell surface expression of IL-17RA (horizontal bars) was significantly reduced on monocytes from patients with DCM (Wilcoxon rank sum test) and was lowest in the cells from the individual with the Pro562Gln mutation (open symbol).
Confirmation of the significance of the chitinase, IL-17RA, and Dectin-1 polymorphisms described here has yet to be subjected to clinical study or laboratory studies of their impact on antifungal activity or signal transduction. However, these 2 studies clearly show that DCM is not usually a monogenic disorder. Instead, a number of possible mechanisms may determine the severity of pulmonary CM and the risk of extrapulmonary dissemination. Table II contains a list of established and putative risk factors for DCM.
Table II.
| Older Age | Primary Immunodeficiency |
| Male Gender | Hyper IgE syndrome (STAT3 mutations) |
| Pregnancy | IFN-γR1 polymorphisms |
| Black race and Filipino ancestry | IL12-Rβ1 or IL12-Rβ2 mutations |
| STAT1 gain of function polymorphisms | |
| Immunosuppression | Other suspected deleterious genetic lesions |
| HIV | Innate immune sensing* |
| Cancer/Chemotherapy | Signaling from innate sensing* |
| Solid Organ Transplantation | NFkB signaling* |
| Anti-inflammatory biological medications | IL-17 signaling*,** |
| Human chitinase (CHIT1)** |
Reference Hardison and Brown.36
This report.
IMMUNE-BASED ADJUNCTIVE THERAPIES
Systemic corticosteroids
The use of high-dose systemic corticosteroid medications, as well as disease-modifying antirheumatic drugs and antineoplastic chemotherapy, represents a risk factor for exacerbation or dissemination of unrecognized CM.20,74 However, this does not mean that the systemic corticosteroids are unequivocally contraindicated by CM. Azadeh et al75 performed a retrospective study of 74 individuals treated with a short course of corticosteroid therapy for rash, wheezing, or cough associated with primary pulmonary CM. No increase in complications, clinical relapses, or extrapulmonary dissemination occurred among those treated for a median of 6 days with typical doses of prednisone or its equivalent (median total dose, 150 mg prednisone equivalents). Most of these patients, and control subjects without corticosteroid treatment, received antifungal therapy. It should also be noted that it was unclear if corticosteroid treatment was uniformly beneficial.
In contrast to this outcome, a number of reports show that systemic corticosteroids may represent an important adjunct to antifungal medications in the treatment of some cases of severe or complicated CM. Improvement has been described in severe acute pulmonary CM, including cases of adult respiratory distress syndrome.76,77 The indications for this therapy and the duration of therapy have yet to be defined; however, some experts recommend a tapering corticosteroid regimen similar to that used in cases of moderate to severe Pneumocystis jirovecii pneumonia for primary pulmonary CM with adult respiratory distress syndrome.78
Similarly, corticosteroids have been advocated as an adjunctive therapy during treatment of central nervous system complications of coccidioidal meningitis, including cranial neuropathy, arachnoiditis, vasculitic infarction, encephalitis, and the development of a syrinx, or to ameliorate symptoms.20,79,80 Intrathecal corticosteroids are also given along with intrathecal injection of AmB deoxycholate to address the headache, fever, and vomiting of this treatment.80,81
IFN-γ therapy
Systemic IFN-γ treatment has been described as an adjunctive therapy in several reports of intractable cases of DCM. Duplessis et al82 administered subcutaneous IFN-γ to 2 previously healthy African-American men who developed extensive, destructive, polyostotic coccidioidal osteomyelitis involving the axial skeleton. In both cases, DCM was clinically and radiographically refractory to prolonged therapy (5–8 months) with both azole antifungal medications (itraconazole and posaconazole) and liposomal AmB. Stimulation of peripheral blood mononuclear cells with the mitogen phytohemagglutinin along with IL-12 yielded IFN-γ release that was lower than expected. DNA sequencing of IFN-γ receptor-1, IFN-γ receptor-2, and STAT1 genes revealed no abnormalities, and the T lymphocyte subset analysis was unremarkable. After months without clear improvement with antifungal therapy alone, IFN-γ1b injections were given 3 times weekly, with marked clinical improvement. Kuberski et al83 described similar clinical improvement and eventual survival of an African-American woman after addition of IFN-γ to antifungal therapy. The underlying mechanism by which IFN-γ appeared beneficial in these cases is unclear, but enhancing the antifungal activity of macrophages might be involved. Expression of chitinase, which is upregulated by IFN-γ, may be among these effector molecules.84 This possibility is intriguing because it exhibits antifungal activity, and recombinant human chitinase has been shown to reduce mortality in experimental fungal infections in mice.85
Refocusing of cellular immunity
As noted earlier, several types of data suggest that severe CM and DCM are associated with a Th2-biased cellular immune response.
Dupilumab is a monoclonal antibody that binds to the alpha subunit of the IL-4 receptor, antagonizing IL-4 and IL-13 signaling.86 Dupilumab is approved by the US Food and Drug Administration for the treatment of patients with moderate to severe atopic dermatitis whose disease is not adequately controlled with topical prescription therapies; it is also approved as an add-on therapy for uncontrolled eosinophilic asthma or in patients with a dependence on oral glucocorticoids for asthma control.87,88 In 2 recent trials, subcutaneous dupilumab given every 2 weeks was associated with a reduction in severe exacerbations of asthma and decreased corticosteroid usage. This therapy was tolerated over periods of 24–52 weeks; transient eosinophilia was noted in both trials and hypothesized to be the result of the blockade of effects of IL-4 on eosinophil activation and egress from peripheral blood.89,90
In view of the type 2 inflammatory state that often accompanies DCM, we recently tested the idea that dupilumab might bring about improvement in cases of DCM found to be refractory to antifungal therapy alone (Tsai et al, submitted for publication). A healthy 4-year-old boy presented with fevers and a 3-week history of enlarging subcutaneous nodules on his forehead that proved to be foci of coccidioidal osteomyelitis. Bony involvement was also identified in vertebral bodies, the right tibia and fibula and the right radius. He had no history of recurrent or severe infections but was found to have a polymorphism in the IL-12Rβ1 (IL-12 receptor subunit β1) gene. Treatment with antifungal therapy (posaconazole and liposomal AmB) and IFN-γ failed to stop the progression of bony disease. Weekly administration of dupilumab was begun and was associated with resolution of the patient’s polyostotic osteomyelitis. Further experience is needed, but the current availability of immunomodulatory agents that refocus cellular immunity away from the type 2 pathway and toward IFN-γ–focused T-cell responses suggest novel therapeutic approaches to refractory DCM, including coccidioidal meningitis. Targeting IL-5, IL-13, and IgE may also be fruitful in this regard. Clinical trials might preferentially involve individuals with eosinophilia, elevated IgE levels, and other markers of Th2 activity at the time of presentation with severe CM or DCM.
CONCLUSIONS
We have far to go before we fully understand the unique pathogenicity of Coccidioides fungi, including their ability to disseminate widely, produce marked bony destruction, and to persist indefinitely. Moreover, apart from rare cases of monogenic defects in immunity, we do not yet know what factors permit dissemination of CM in the ~1% of those who become infected. The long-standing evidence that DCM occurs more frequently among otherwise healthy people of African and Filipino ancestry indicates that Coccidioides species exploit an otherwise benign difference in immune function to escape immune recognition and control. Although most cases of CM are self-limited, DCM is life-threatening and often responds poorly to antifungal therapy. Corticosteroid therapy may be beneficial in severe pulmonary and central nervous system disease to reduce inflammation as therapy is initiated, but augmenting or refocusing immune responses may be needed to induce remission or cure of coccidioidomycosis in some individuals. Systematic study of immune responses in acute CM and cases of DCM, and genetic evaluation of both those who control coccidioidal infection and those with severe disease, may help us distinguish those most likely to benefit from immunomodulatory therapy.
Supplementary Material
ACKNOWLEDGMENTS
Research mentioned in this review was supported by unrestricted grants from Chevron Foundation, USA and Nielsen Biosciences, USA. No other authors contributed to the preparation of this manuscript. No other sources of support, apart from those cited. PK conducted the literature search and produced initial manuscript drafts for all authors’ review. PK, AH, and RJ performed the studies summarized in the text, Table I, and Figure 2. MB, M-GL, and PK collaborated in the care of the patient treated with immunomodulatory therapy described briefly in text.
Footnotes
DISCLOSURES
Royce Johnson, MD, is affliated with Valley Fever Foundation of the Americas. The authors have indicated that they have no other conflicts of interest regarding the content of this article.
The sponsors had no role in the preparation of the manuscript.
REFERENCES
- 1.McCotter OZ, Benedict K, Engelthaler DM, et al. Update on the epidemiology of coccidioidomycosis in the United States. Med Mycol. 2019;57(Supplement_1):S30–S40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Valdivia L, Nix D, Wright M, et al. Coccidioidomycosis as a common cause of community-acquired pneumonia. Emerg Infect Dis. 2006;12:958–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang DC, Anderson S, Wannemuehler K, et al. Testing for coccidioidomycosis among patients with community-acquired pneumonia. Emerg Infect Dis. 2008;14:1053–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deresinski S, Mirels LF. Coccidioidomycosis: what a long strange trip it’s been. Med Mycol. 2019;57(Supplement_1):S3–S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith CE. Epidemiology of acute coccidioidomycosis with erythema nodosum (“San Joaquin” or “valley fever”). Am J Public Health Nations Health. 1940;30:600–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith CE, Beard RR. Varieties of coccidioidal infection in relation to the epidemiology and control of the diseases. Am J Public Health Nations Health. 1946;36:1394–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Smith CE, Saito MT, Simons SA. Pattern of 39,500 serologic tests in coccidioidomycosis. J Am Med Assoc. 1956;160:546–552. [DOI] [PubMed] [Google Scholar]
- 8.Flynn NM, Hoeprich PD, Kawachi MM, et al. An unusual outbreak of windborne coccidioidomycosis. N Engl J Med. 1979;301:358–361. [DOI] [PubMed] [Google Scholar]
- 9.Nguyen C, Barker BM, Hoover S, et al. Recent advances in our understanding of the environmental, epidemiological, immunological, and clinical dimensions of coccidioidomycosis. Clin Microbiol Rev. 2013;26:505–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fish DG, Ampel NM, Galgiani JN, et al. Coccidioidomycosis during human immunodeficiency virus infection. A review of 77 patients. Medicine (Baltimore). 1990;69:384–391. [DOI] [PubMed] [Google Scholar]
- 11.Ampel NM. Coccidioidomycosis in persons infected with HIV type 1. Clin Infect Dis. 2005;41:1174–1178. [DOI] [PubMed] [Google Scholar]
- 12.Blair JE, Ampel NM, Hoover SE. Coccidioidomycosis in selected immunosuppressed hosts. Med Mycol. 2019;57(Supplement_1):S56–S63. [DOI] [PubMed] [Google Scholar]
- 13.Blair JE, Smilack JD, Caples SM. Coccidioidomycosis in patients with hematologic malignancies. Arch Intern Med. 2005;165:113–117. [DOI] [PubMed] [Google Scholar]
- 14.Einstein HE, Holeman CW Jr, Sandidge LL, Holden DH. Coccidioidal meningitis. The use of amphotericin B in treatment. Calif Med. 1961;94:339–343. [PMC free article] [PubMed] [Google Scholar]
- 15.Graybill JR, Stevens DA, Galgiani JN, et al. Ketoconazole treatment of coccidioidal meningitis. Ann N Y Acad Sci. 1988;544:488–49. [DOI] [PubMed] [Google Scholar]
- 16.Tucker RM, Galgiani JN, Denning DW, et al. Treatment of coccidioidal meningitis with fluconazole. Rev Infect Dis. 1990;12(Supplement_3):S380–S389. [DOI] [PubMed] [Google Scholar]
- 17.Brass C, Galgiani JN, Campbell SC, Stevens DA. Therapy of disseminated or pulmonary coccidioidomycosis with ketoconazole. Rev Infect Dis. 1980;2:656–660. [DOI] [PubMed] [Google Scholar]
- 18.Galgiani JN, Catanzaro A, Cloud GA, et al. Comparison of oral fluconazole and itraconazole for progressive, nonmeningeal coccidioidomycosis. A randomized, double-blind trial. Mycoses Study Group. Ann Intern Med. 2000;133:676–686. [DOI] [PubMed] [Google Scholar]
- 19.Thompson 3rd GR, Lewis 2nd JS, Nix DE, Patterson TF. Current concepts and future directions in the pharmacology and treatment of coccidioidomycosis. Med Mycol. 2019;57(Supplement_1):S76–S84. [DOI] [PubMed] [Google Scholar]
- 20.Galgiani JN, Ampel NM, Blair JE, et al. 2016 Infectious diseases society of America (IDSA) clinical practice guideline for the treatment of coccidioidomycosis. Clin Infect Dis. 2016;63:e112–e146. [DOI] [PubMed] [Google Scholar]
- 21.Dewsnup DH, Galgiani JN, Graybill JR, et al. Is it ever safe to stop azole therapy for Coccidioides immitis meningitis? Ann Intern Med. 1996;124:305–310. [DOI] [PubMed] [Google Scholar]
- 22.Kirkland TN, Fierer J. Coccidioides immitis and posadasii; a review of their biology, genomics, pathogenesis, and host immunity. Virulence. 2018;9:1426–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fisher MC, Koenig GL, White TJ, Taylor JW. Molecular and phenotypic description of Coccidioides posadasii sp. nov., previously recognized as the non-California population of Coccidioides immitisnon-California population of Coccidioides immitis. Mycologia. 2002;94:73–84. [PubMed] [Google Scholar]
- 24.Ramras DG, Walch HA, Murray JP, Davidson BH. An epidemic of coccidioidomycosis in the Pacific beach area of San Diego. Am Rev Respir Dis. 1970;101:975–978. [DOI] [PubMed] [Google Scholar]
- 25.Drutz DJ, Huppert M. Coccidioidomycosis: factors affecting the host-parasite interaction. J Infect Dis. 1983;147:372–390. [DOI] [PubMed] [Google Scholar]
- 26.Viriyakosol S, Fierer J, Brown GD, Kirkland TN. Innate immunity to the pathogenic fungus Coccidioides posadasii is dependent on Toll-like receptor 2 and Dectin-1Toll-like receptor 2 and Dectin-1. Infect Immun. 2005;73:1553–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Whiston E, Zhang Wise H, Sharpton TJ, Jui G, Cole GT, Taylor JW. Comparative transcriptomics of the saprobic and parasitic growth phases in Coccidioides spp. PLoS One. 2012;7, e41034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Viriyakosol S, Singhania A, Fierer J, Goldberg J, Kirkland TN, Woelk CH. Gene expression in human fungal pathogen Coccidioides immitis changes as arthroconidia differentiate into spherules and mature. BMC Microbiol. 2013;13:121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Galgiani JN, Hayden R, Payne CM. Leukocyte effects on the dimorphism of Coccidioides immitis. J Infect Dis. 1982;146:56–63. [DOI] [PubMed] [Google Scholar]
- 30.Wang H, LeBert V, Hung CY, et al. C-type lectin receptors differentially induce TH17 cells and vaccine immunity to the endemic mycosis of North America. J Immunol. 2014;192:1107–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cole GT, Hung CY, Sanderson SD, et al. Novel strategies to enhance vaccine immunity against coccidioidomycosis. Plos Pathog. 2013;9, e1003768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fierer J, Walls L, Wright F, Kirkland TN. Genes influencing resistance to Coccidioides immitis and the interleukin-10 response map to chromosomes 4 and 6 in miceinterleukin-10 response map to chromosomes 4 and 6 in mice. Infect Immun. 1999;67:2916–2919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hung CY, Hsu AP, Holland SM, Fierer J. A review of innate and adaptive immunity to coccidioidomycosis. Med Mycol. 2019;57(Supplement_1):S85–S92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Viriyakosol S, Jimenez Mdel P, Gurney MA, Ashbaugh ME, Fierer J. Dectin-1 is required for resistance to coccidioidomycosis in mice. MBio. 2013;4. e00597–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Odio CD, Marciano BE, Galgiani JN, Holland SM. Risk factors for disseminated coccidioidomycosis, United States23. Emerg Infect Dis; 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hardison SE, Brown GD. C-type lectin receptors orchestrate antifungal immunity. Nat Immunol. 2012;13:817–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.del Pilar Jimenez AM, Viriyakosol S, Walls L, et al. Susceptibility to Coccidioides species in C57BL/6 mice is associated with expression of a truncated splice variant of Dectin-1 (Clec7a)Dectin-1 (Clec7a). Genes Immun. 2008;9:338–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kirkland TN, Fierer J. Inbred mouse strains differ in resistance to lethal Coccidioides immitis infection. Infect Immun. 1983;40:912–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xue J, Chen X, Selby D, Hung CY, Yu JJ, Cole GT. A genetically engineered live attenuated vaccine of Coccidioides posadasii protects BALB/c mice against coccidioidomycosis. Infect Immun. 2009;77:3196–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Awasthi S Susceptibility of TLR4-defective C3H/HeJ mice to Coccidioides posadasii infection. Med Mycol. 2010;48:470–475. [DOI] [PubMed] [Google Scholar]
- 41.Bochud PY, Chien JW, Marr KA, et al. Toll-like receptor 4 polymorphisms and aspergillosis in stem-cell transplantation. N Engl J Med. 2008;359:1766–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Viriyakosol S, Jimenez Mdel P, Saijo S, Fierer J. Neither Dectin-2 nor the mannose receptor is required for resistance to Dectin-2 nor the mannose receptor is required for resistance to Coccidioides immitis in mice. Infect Immun. 2014;82:1147–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ferwerda G, Ferwerda B, Plantinga TS, et al. Human Dectin-1 deficiency and mucocutaneous fungal infections. N Engl J Med. 2009;361:1760–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Glocker EO, Hennigs A, Nabavi M, et al. A homozygous CARD9 mutation in a family with susceptibility to fungal infections. N Engl J Med. 2009;361:1727–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pappagianis D, Zimmer BL. Serology of coccidioidomycosis. Clin Microbiol Rev. 1990;3:247–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cox RA, Baker BS, Stevens DA. Specificity of immunoglobulin E in coccidioidomycosis and correlation with disease involvement. Infect Immun. 1982;37:609–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Munder M, Eichmann K, Moran JM, Centeno F, Soler G, Modolell M. Th1/Th2-regulated expression of arginase isoforms in murine macrophages and dendritic cells. J Immunol. 1999;163:3771–3777. [PubMed] [Google Scholar]
- 48.Hung CY, Castro-Lopez N, Cole GT. Card9- and MyD88-mediated gamma interferon and nitric oxide production is essential for resistance to subcutaneous Coccidioides posadasii infection. Infect Immun. 2016;84:1166–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Muller U, Stenzel W, Kohler G, et al. IL-13 induces disease-promoting type 2 cytokines, alternatively activated macrophages and allergic inflammation during pulmonary infection of mice with disease-promoting type 2 cytokines, alternatively activated macrophages and allergic inflammation during pulmonary infection of mice with Cryptococcus neoformans. J Immunol. 2007;179:5367–5377. [DOI] [PubMed] [Google Scholar]
- 50.Mendonca MS, Peracolli TS, Silva-Vergara ML, et al. High interleukin-4 expression and interleukin-4 gene polymorphisms are associated with susceptibility to human paracoccidioidomycosis. Mem Inst Oswaldo Cruz. 2015;110:781–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Harley WB, Blaser MJ. Disseminated coccidioidomycosis associated with extreme eosinophilia. Clin Infect Dis. 1994;18:627–629. [DOI] [PubMed] [Google Scholar]
- 52.Lombard CM, Tazelaar HD, Krasne DL. Pulmonary eosinophilia in coccidioidal infections. Chest. 1987;91:734–736. [DOI] [PubMed] [Google Scholar]
- 53.Lyons JJ, Milner JD. Primary atopic disorders. J Exp Med. 2018;215:1009–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wuthrich M, Gern B, Hung CY, et al. Vaccine-induced protection against 3 systemic mycoses endemic to North America requires Th17 cells in mice. J Clin Invest. 2016;126:795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vinh DC, Schwartz B, Hsu AP, et al. Interleukin-12 receptor beta1 deficiency predisposing to disseminated coccidioidomycosis. Clin Infect Dis. 2011;52:e99–e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vinh DC, Masannat F, Dzioba RB, Galgiani JN, Holland SM. Refractory disseminated coccidioidomycosis and mycobacteriosis in interferon-gamma receptor 1 deficiency. Clin Infect Dis. 2009;49:e62–e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sampaio EP, Hsu AP, Pechacek J, et al. Signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations and disseminated coccidioidomycosis and histoplasmosis. J Allergy Clin Immunol. 2013;131:1624–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Corry DB, Ampel NM, Christian L, Locksley RM, Galgiani JN. Cytokine production by peripheral blood mononuclear cells in human coccidioidomycosis. J Infect Dis. 1996;174:440–443. [DOI] [PubMed] [Google Scholar]
- 59.Ampel NM, Nelson DK, Chavez S, et al. Preliminary evaluation of whole-blood gamma interferon release for clinical assessment of cellular immunity in patients with active coccidioidomycosis. Clin Diagn Lab Immunol. 2005;12:700–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Magee DM, Cox RA. Roles of gamma interferon and interleukin-4 in genetically determined resistance to Coccidioides immitis. Infect Immun. 1995;63:3514–3519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hung C, Zhang H, Campuzano A, Ostroff GR, Yu J. An multivalent vaccine elicits protective Th17 response via activation of C-type lectin receptor- and Card9-mediated signal against pulmonary 9-mediated signal against pulmonary Coccidioides posadasii infection. J Immunol. 2018;200(1 Supplement):125.4. [Google Scholar]
- 62.Cypowyj S, Picard C, Marodi L, Casanova JL, Puel A. Immunity to infection in IL-17-deficient mice and humans. Eur J Immunol. 2012;42: 2246–2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.De Luca A, Zelante T, D’Angelo C, et al. IL-22 defines a novel immune pathway of antifungal resistance. Mucosal Immunol. 2010;3:361–373. [DOI] [PubMed] [Google Scholar]
- 64.Puel A, Doffinger R, Natividad A, et al. Autoantibodies against IL-17A, IL-17F, and IL-22 in patients with chronic mucocutaneous candidiasis and autoimmune polyendocrine syndrome type I. J Exp Med. 2010;207:291–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Puel A, Cypowyj S, Bustamante J, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity. Science. 2011;332:65–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ling Y, Cypowyj S, Aytekin C, et al. Inherited IL-17RC deficiency in patients with chronic mucocutaneous candidiasis. J Exp Med. 2015;212:619–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Davini D, Naeem F, Phong A, et al. Elevated regulatory T cells at diagnosis of Coccidioides infection associates with chronicity in pediatric patients. J Allergy Clin Immunol. 2018;142:1971–1974.e7. [DOI] [PubMed] [Google Scholar]
- 68.Whibley N, Gaffen SL. Brothers in arms: Th17 and Treg responses in Candida albicans immunity. Plos Pathog. 2014;10, e1004456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ampel NM, Robey I, Nguyen CT, et al. Ex vivo cytokine release, determined by a multiplex cytokine assay, in response to coccidioidal antigen stimulation of whole blood among subjects with recently diagnosed primary pulmonary coccidioidomycosis. mSphere. 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Krogstad P, Yourshaw M, Heidari A,Aguirre D, Grewal R, Johnson R. Whole exome sequencing identification of human genetic polymorphisms associated with extrapulmonary dissemination of coccidioidomycosis. Open Forum Infect Dis. 2015;2(suppl_1):1347. 10.1093/ofid/ofv131.138. [DOI] [Google Scholar]
- 72.Lee P, Waalen J, Crain K, Smargon A, Beutler E. Human chitotriosidase polymorphisms G354R and A442V associated with reduced enzyme activity. Blood Cells Mol Dis. 2007;39: 353–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47:D886–D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mahaffey KW, Hippenmeyer CL, Mandel R, Ampel NM. Unrecognized coccidioidomycosis complicating Pneumocystis carinii pneumonia in patients infected with the human immunodeficiency virus and treated with corticosteroids. A report of two cases. Arch Intern Med. 1993;153:1496–1498. [PubMed] [Google Scholar]
- 75.Azadeh N, Chang YH, Kusne S, et al. The impact of early and brief corticosteroids on the clinical course of primary pulmonary coccidioidomycosis. J Infect. 2013;67:148–155. [DOI] [PubMed] [Google Scholar]
- 76.Shibli M, Ghassibi J, Hajal R, O’Sullivan M. Adjunctive corticosteroids therapy in acute respiratory distress syndrome owing to disseminated coccidioidomycosis. Crit Care Med. 2002;30:1896–1898. [DOI] [PubMed] [Google Scholar]
- 77.Chang MR, Chopra N, Beenhouwer D, Goetz MB, Hoo GWS. Corticosteroids in the management of severe coccidioidomycosis. Am J Med. 2019;132:110–113. [DOI] [PubMed] [Google Scholar]
- 78.Thompson 3rd GR. Pulmonary coccidioidomycosis. Semin Respir Crit Care Med. 2011;32:754–763. [DOI] [PubMed] [Google Scholar]
- 79.D’Assumpcao C, Heidari A, Sabetian K, Johnson RH. Crescendo transient ischemic attacks due to basilar coccidioidal meningitis with coccidioma. J Investig Med High Impact Case Rep. 2018;6, 2324709618813178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Johnson R, Ho J, Fowler P, Heidari A. Coccidioidal meningitis: a review on diagnosis, treatment, and management of complications. Curr Neurol Neurosci Rep. 2018;18:19. [DOI] [PubMed] [Google Scholar]
- 81.Sievers ML, Fisher JR. Coccidioidal meningitis and intrathecal corticosteroids. Ann Intern Med. 1981;95:242–243. [DOI] [PubMed] [Google Scholar]
- 82.Duplessis CA, Tilley D, Bavaro M, Hale B, Holland SM. Two cases illustrating successful adjunctive interferon-gamma immunotherapy in refractory disseminated coccidioidomycosis. J Infect. 2011;63:223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kuberski TT, Servi RJ, Rubin PJ. Successful treatment of a critically ill patient with disseminated coccidioidomycosis, using adjunctive interferon-gamma. Clin Infect Dis. 2004;38:910–912. [DOI] [PubMed] [Google Scholar]
- 84.Malaguarnera L, Musumeci M, Di Rosa M, Scuto A, Musumeci S. Interferon-gamma, tumor necrosis factor-alpha, and lipopolysaccharide promote chitotriosidase gene expression in human macrophages. J Clin Lab Anal. 2005;19:128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.van Eijk M, van Roomen CP, Renkema GH, et al. Characterization of human phagocyte-derived chitotriosidase, a component of innate immunity. Int Immunol. 2005;17:1505–1512. [DOI] [PubMed] [Google Scholar]
- 86.Shirley M Dupilumab: first global approval. Drugs. 2017;77:1115–1121. [DOI] [PubMed] [Google Scholar]
- 87.Corren J New targeted therapies for uncontrolled asthma. J Allergy Clin Immunol Pract. 2019;7:1394–1403. [DOI] [PubMed] [Google Scholar]
- 88.Corren J, Castro M, Chanez P, et al. Dupilumab improves symptoms, quality of life, and productivity in uncontrolled persistent asthma. Ann Allergy Asthma Immunol. 2019;122:41–49.e2. [DOI] [PubMed] [Google Scholar]
- 89.Rabe KF, Nair P, Brusselle G, et al. Efficacy and safety of dupilumab in glucocorticoid-dependent severe asthma. N Engl J Med. 2018;378:2475–2485. [DOI] [PubMed] [Google Scholar]
- 90.Castro M, Corren J, Pavord ID, et al. Dupilumab efficacy and safety in moderate-to-severe uncontrolled asthma. N Engl J Med. 2018;378:2486–2496. [DOI] [PubMed] [Google Scholar]
- 91.Lee PP, Lau YL. Cellular and molecular defects underlying invasive fungal infections-revelations from endemic mycoses. Front Immunol. 2017;8:735. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.