

Summary
Understanding the basis for cellular growth, proliferation, and function requires determining the roles of essential genes in diverse cellular processes, including visualizing their contributions to cellular organization and morphology. Here, we combined pooled CRISPR/Cas9-based functional screening of 5,072 fitness-conferring genes in human HeLa cells with microscopy-based imaging of DNA, the DNA damage response, actin, and microtubules. Analysis of >31 million individual cells identified measurable phenotypes for >90% of gene knockouts, implicating gene targets in specific cellular processes. Clustering of phenotypic similarities based on hundreds of quantitative parameters further revealed co-functional genes across diverse cellular activities, providing predictions for gene functions and associations. By conducting pooled live-cell screening of ~450,000 cell division events for 239 genes, we additionally identified diverse genes with functional contributions to chromosome segregation. Our work establishes a resource detailing the consequences of disrupting core cellular processes that represents the functional landscape of essential human genes.
Keywords: CRISPR/Cas9, functional genomics, microscopy, high-content screening, in situ sequencing, optical pooled screening, essential genes, morphology, mitosis, cell division
Introduction
For a human cell to grow, proliferate, and function, it must carry out multiple essential processes, including transcription, mRNA splicing, translation, vesicle trafficking, proteolysis, DNA replication, and cell division. CRISPR/Cas9-based pooled genetic screens have revolutionized the ability to test the functional requirements for cell proliferation and survival by enabling the potent disruption of thousands of individual genetic elements.1 However, most current screening approaches, including those based on fluorescence-activated cell sorting (FACS),2,3 produce a single scalar measurement of barcode enrichment or depletion that summarizes the contributions of each perturbation to cellular phenotypes at the population level. Defining the specific contributions of essential genes across core cellular processes requires quantitative analysis of complex cellular phenotypes. Recent studies have combined pooled functional genetic screens with single-cell profiling of transcript abundance.4–6 However, many phenotypes corresponding to core cellular functions, particularly regarding cellular morphology or subcellular localization, instead require direct visualization using microscopy. Arrayed screens have analyzed genetic contributions to microscopy phenotypes,7 but are difficult to scale as each perturbation is tested individually. To scalably identify relationships between specific gene knockouts and their corresponding cellular phenotypes using microscopy, recent pooled approaches have utilized either in situ perturbation genotyping8,9 or the physical selection of cells for image-based phenotypes.10–14 However, these latter enrichment approaches probe pre-defined phenotype bins and thus do not enable in-depth exploration of perturbation-phenotype associations. The ability to interrogate and systematically compare a large and diverse array of cell biological phenotypes, especially morphology and spatial organization, across thousands of genomic perturbations represents an important unmet goal for functional studies. Here we use optical pooled screening8,15 to combine large-scale Cas9-based targeting of essential genes with comprehensive single-cell phenotyping and perturbation genotyping by microscopy to resolve a diversity of complex phenotypes in tens of millions of cells (Figure 1A).
Figure 1. Large-scale image-based pooled CRISPR screen identifies essential genes with roles in genome integrity.
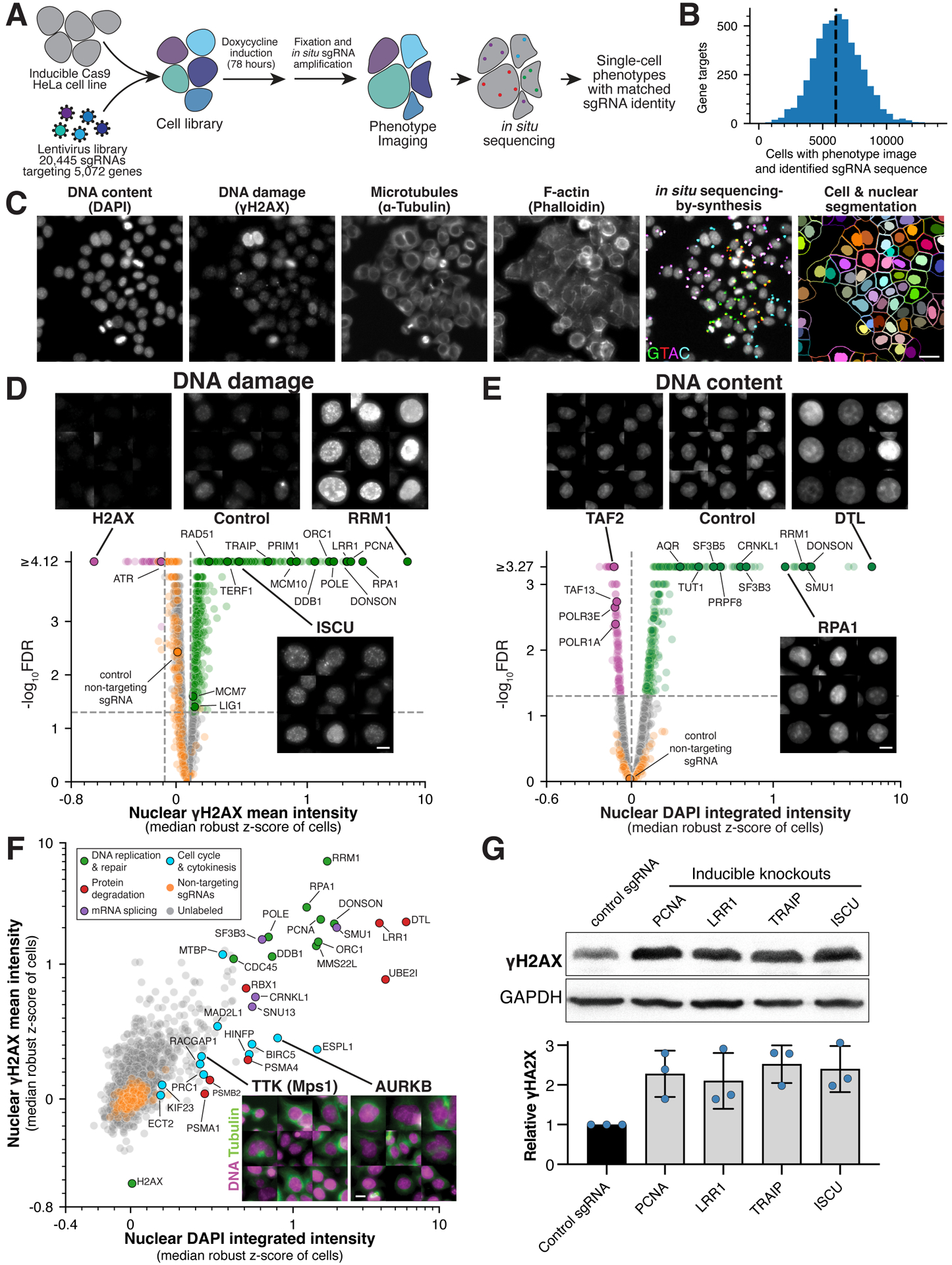
(A) Workflow for image-based pooled CRISPR screen (also see STAR Methods). (B) Histogram showing the number of cells analyzed for each gene target with image and single sgRNA sequence mapped. (C) Example image from the pooled screen showing the indicated stains together with fluorescent in situ sequencing (Laplacian-of-Gaussian filtered) and cell segmentation. Scale bar, 25 μm. (D) Volcano plot for mean nuclear γH2AX intensity across gene targets and selected images to highlight specific targets whose knockout results in increased (green) or decreased (magenta) γH2AX relative to random samples of cells expressing targeting sgRNAs (orange; FDR<0.05; STAR Methods). The median robust z-score is calculated relative to cells expressing non-targeting sgRNAs and is plotted on a symmetric log scale (linear between −1 and 1). Scale bar, 10 μm. (E) Volcano plot and selected images as in (D) for changes in integrated nuclear DAPI intensity relative to random samples of cells expressing non-targeting sgRNAs (STAR Methods). Scale bar, 10 μm. (F) Scatter plot comparing the relationship between DNA damage and content. Labeled genes are colored by functional category. Example images show tubulin (green) and DNA (magenta) to highlight multinucleated cells. Scale bar, 10 μm. (G) Western blot (top) and quantification (bottom) confirming the presence of increased DNA damage in individual knockout cell lines targeting either the indicated genes or a single copy locus control. Blue data points indicate independent replicates. For each sample, γH2AX intensity was referenced to its GAPDH loading control and normalized by negative control γH2AX relative intensity. Error bars indicate SD.
Results
A large-scale, image-based pooled CRISPR screen of essential human genes
To determine the functional contributions of essential genes in cultured human cells, we first defined a set of fitness-conferring genes based on combined evidence from multiple Cas9- and transposon-based genetic screens16–24 (STAR Methods). This approach identified 5,072 genes that contribute to optimal cellular fitness, although we note that not every gene will be required in a given cell line. We selected four sgRNA sequences targeting each gene from existing sgRNA libraries,24–26 as well as 250 “non-targeting” sgRNAs, for a total library of 20,445 sgRNAs (Table S1; STAR Methods). We delivered the sgRNA library to HeLa cells containing an integrated, doxycycline-inducible Cas9 construct27 (STAR Methods). Based on trial image-based screens (Figure S1A) and an analysis of sgRNA depletion from the cell library at 3 and 5 days post-Cas9 induction (Figure S1B), we selected 78 hours post-Cas9 induction as a time point to maximize phenotype observability. This approach balances the time required for Cas9 activity and protein depletion with negative fitness effects that deplete knockout cells from the population. At this time point, we fixed the cells and amplified the sgRNA sequences in situ8,15 (Figure 1A). Following amplification, we stained and imaged cells for DNA (DAPI), the DNA damage response (γH2AX; anti-phospho-Ser139 H2AX antibody), microtubules (anti-α-tubulin antibody), and filamentous actin (phalloidin; Figure 1C). These stains were chosen to visualize diverse cell biological behaviors, including nuclear morphology, DNA damage response, cytoskeletal structures, and cell division.
Following the completion of phenotype imaging, we performed in situ sequencing-by-synthesis to identify the sgRNA present in each individual cell8,15 (Figures 1A, 1C and S1C). We extracted 1,084 phenotypic parameters from each individual cell image, including measurements of the intensity, subcellular distribution, and colocalization of stains, and cellular and nuclear size and shape (STAR Methods; Table S2). Based on their distinct morphological characteristics, we classified cells as either interphase or mitotic and conducted downstream analyses separately (STAR Methods; Figure S1D). This approach yielded microscopy images, extracted phenotypic measurements, and matched sgRNA identities for 31,884,270 individual cells with a median of 6,119 cells per gene target across each set of four sgRNAs (Figure 1B, 1E; Table S2). Image montages and phenotypic parameters for interphase and mitotic cells are available through the companion interactive web portal (https://vesuvius.wi.mit.edu/).
Interphase nuclear phenotypes identify regulators of genomic integrity
Maintaining genomic integrity is critical to ensuring proper cellular function. Cells utilize a range of DNA damage-sensors, DNA repair mechanisms, and cell cycle checkpoints to prevent, recognize, and correct genomic aberrations.28 To identify genes that are required for genome integrity, we analyzed summary phenotype scores for interphase nuclear parameters that monitor DNA damage (mean γH2AX nuclear intensity; Figure 1D) and DNA content (integrated DAPI nuclear intensity; Figure 1E; STAR Methods). Genes that displayed decreased γH2AX intensity in interphase cells relative to random samples of cells expressing targeting sgRNAs included H2AX itself and ATR, which is involved in directing the γH2AX phosphorylation event28 (Figure 1D). Reciprocally, genes whose disruption resulted in increased relative γH2AX intensity included many factors with known roles in DNA replication, DNA repair, and telomere protection28,29 (Figure 1D). Overall, we observed a clear correlation between DNA damage and DNA content (r = 0.62; Figure 1F). However, a subset of knockouts, including gene targets required for cytokinesis (e.g., ECT2, AURKB)30,31 or cell cycle control (e.g., MAD2L1, ESPL1)32displayed increases in DNA content due to increased ploidy or multinucleation, but less severe DNA damage (Figure 1F; Figure S2D–E). Amongst genes with increased γH2AX intensity, we also identified several arising from duplication events such that the corresponding sgRNAs target multiple genomic loci, and therefore likely reflect strong Cas9-associated DNA damage. This was particularly pronounced for sgRNAs whose target sites are spread across multiple chromosomes (Figure S2A). Of the 5,072 genes targeted in the screen, we observed 1,258 genes that displayed statistically increased γH2AX intensity, reflecting both direct roles for DNA replication and repair proteins, as well as indirect consequences of disrupting other cellular processes such as translation or RNA splicing (Figure S2B–C; see also Pederiva et al.33).
To validate our approach for identifying genes involved in genomic integrity, we investigated selected gene targets that displayed increased DNA damage, including the E3 ubiquitin ligase subunits LRR1 and TRAIP, and the mitochondrial iron-sulfur cluster biogenesis gene ISCU. We generated individual cell lines with inducible Cas9 and a single sgRNA targeting the corresponding gene.27 Based on Western blotting, we noted a substantial increase in γH2AX levels relative to a control sgRNA with a single genomic target site following ISCU, LRR1, and TRAIP depletion, comparable to the positive control PCNA (Figure 1G). The effect of ISCU knockout is consistent with the requirement for iron-sulfur clusters in the enzymatic activity of DNA metabolism proteins,34 and LRR1 and TRAIP have recently been reported to play roles in replisome disassembly.35 This analysis validates our image-based screening strategy to identify diverse players in genome integrity and highlights the importance of multiple genes in DNA replication and repair.
Identification of essential genes controlling cytoskeletal function
To direct cellular proliferation, organization, and mechanical force production, cells rely on dynamic cytoskeletal networks involving actin and microtubule polymers.36,37 Among the genes with strong changes in F-actin intensity in our screen, we identified established factors required for regulating actin assembly and dynamics including the actin depolymerization factor cofilin (CFL1) and F-actin capping protein CAPZB, as well as RHOA and ARHGEF7, which regulate the actin polymerization (Figure 2A). Although disrupting most established regulators altered F-actin levels as anticipated, loss of the actin-nucleating Arp2/3 complex37 resulted in a counterintuitive increase in mean actin intensity (Figure 2A). This increased actin staining was coupled with a substantial decrease in cell area (Figure S3B), suggesting that disrupting the actin cytoskeleton perturbs cellular adhesion and results in an increase in mean cytoplasmic actin intensity due to altered cell shape. Indeed, we observed similar phenotypes for adhesion components and Integrin subunits (Figures S3B–C). The gene target whose loss resulted in the largest increase of mean F-actin intensity in both interphase and mitotic cells was the E3 ubiquitin ligase KCTD10, along with its partners CUL3 and RBX1 (Figures 2A and S3A). Recent work implicated KCTD10 in restricting actin assembly during cell migration or developmentally-programmed cell fusion,38,39 but our analysis suggests a general role for this complex in regulating actin assembly. In total, we identified 460 gene knockouts with decreased F-actin intensity relative to non-targeting sgRNA controls and 899 genes with increased actin intensity (Figure 2A).
Figure 2. Identification of essential genes regulating cytoskeletal structures and cellular organization.
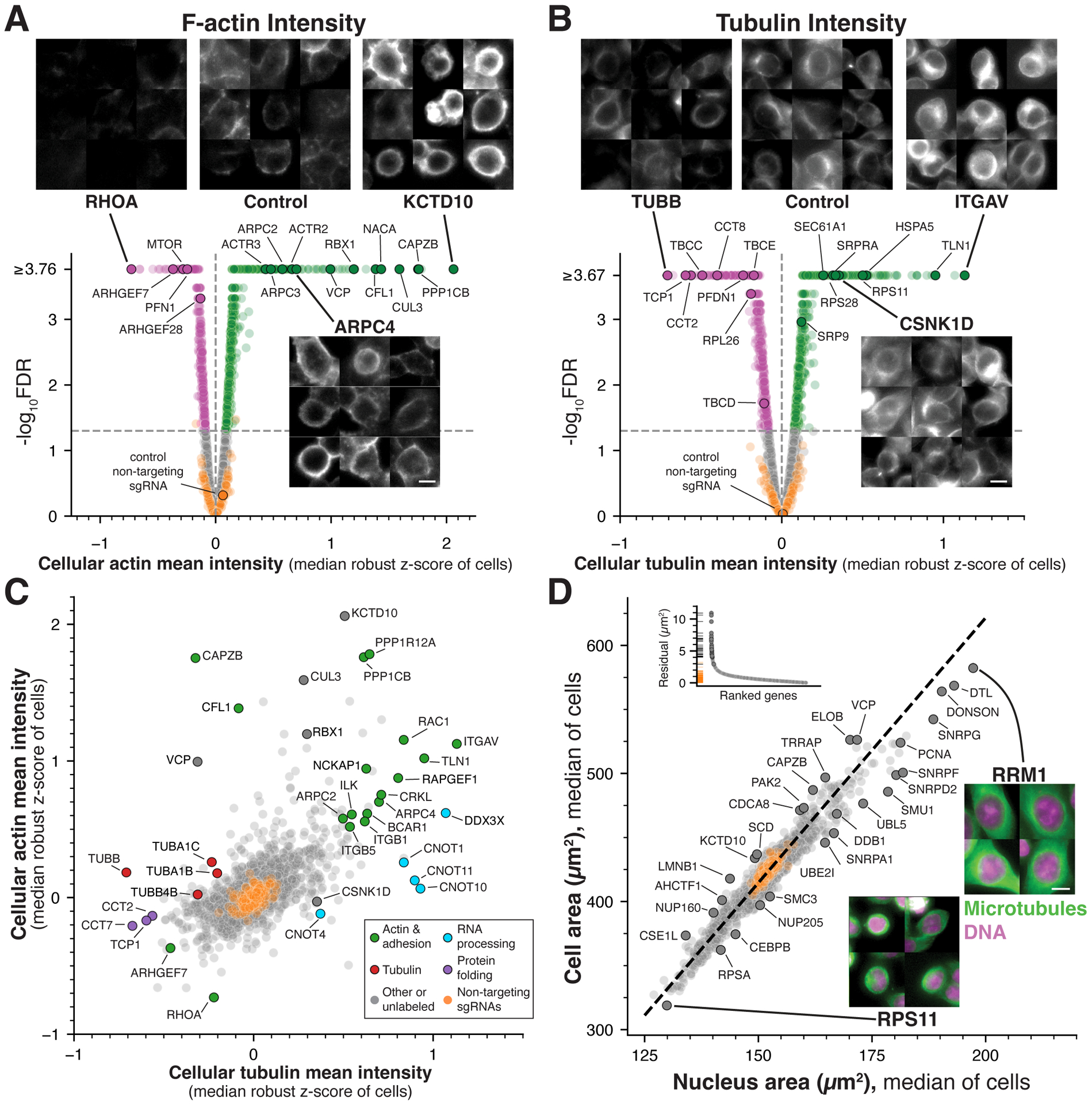
(A) Selected images and volcano plot for mean cellular F-actin (phalloidin) intensity highlighting gene targets that result in increased (green) or decreased (magenta) actin levels relative to non-targeting control cells (orange; FDR<0.05; STAR Methods). (B) Selected images and volcano plot as in (A) for mean cellular tubulin intensity. (C) Scatter plot comparing the relationship between actin and tubulin intensity highlighting targets that selectively affect one cytoskeletal element (see also Figures S3B–D). Labeled genes are colored by functional category. (D) Scatter plot comparing median cellular and nuclear area across gene targets. These morphological features are highly correlated across conditions (r = 0.96). Orthogonal regression was performed to identify targets resulting in an altered nuclear:cytoplasmic area ratio (dotted line). Labeled genes are also highlighted in the distribution of regression residuals (inset). Example images display DNA (magenta) and tubulin (green) staining for gene targets that result in altered cell and nuclear size. Scale bars, 10 μm.
We also identified multiple factors regulating interphase tubulin levels. Mean tubulin intensity was significantly decreased for genes encoding tubulin proteins, tubulin-specific chaperones, and factors required for tubulin folding and assembly (CCT chaperonins/TRiC complex and prefoldin subunits; Figure 2B). As noted above, cytoplasmic stains can display increased mean intensity under conditions where cell area is reduced due to altered substrate adhesion (Figures S3B–D). Thus, we compared actin and tubulin intensity to identify gene targets that selectively affect one stain (Figure 2C). We observed substantially increased tubulin fluorescence without corresponding increases in actin intensity for Casein kinase I delta (CSNK1D), which has been suggested to regulate microtubule-associated proteins,40 and subunits of the CCR4-NOT complex (CNOT1/4/10/11), which functions in post-transcriptional mRNA regulation.41 In summary, our analysis identifies the contributions of diverse molecular players in controlling cytoskeletal assembly and dynamics.
Analysis of morphological phenotypes reveals a tight correspondence between cellular and nuclear size
In addition to measuring stain intensities, we also analyzed morphological parameters including nuclear and cellular area. We noted substantial differences in median interphase cell area across gene targets, ranging from 319 μm2 to 583 μm2 (Figures 2D and S3E). Consistent with a role for protein production in cell growth, targeting translation-related genes resulted in substantially reduced cell area (Figure S3F). In contrast, gene targets with roles in DNA replication and repair, mRNA splicing, and proteasome function displayed increased cell area (Figure S3F), suggesting continued cell growth in the absence of division. Using individual inducible knockout cell lines, we confirmed these substantial changes in cell size for DTL and DONSON gene knockouts (Figure S3G). Nuclear size similarly varied widely across gene targets, including increased nuclear area for five genes identified in a recent photoactivation-based CRISPRi imaging screen (AURKB, CDCA8, FBXO5, TICRR, and RAD51).14 Strikingly, we observed a strong correlation between cell and nuclear area across all tested gene targets (r = 0.96; Figure 2D). Prior work has suggested that cells actively regulate their nuclear to cytoplasm ratio.42 Our analysis demonstrates that, across a wide range of cell sizes and functional perturbations, this relationship is closely maintained. However, we identified a limited number of gene targets whose depletion altered this coordinated scaling including factors with established roles in nuclear integrity and nuclear pore components (Figure 2D). Together, this analysis demonstrates that cell biological parameters from a large-scale screen can be used to provide insights into the control of cellular morphology and organization.
Phenotypic clustering of interphase cellular parameters defines co-functional genes
We next sought to take advantage of the full range of phenotypes in the rich image data from our screen to reveal gene activities required for cellular function. To represent the phenotypic landscape of essential genes, we combined summary phenotype scores from all extracted image features to create phenotypic profiles for each gene (STAR Methods). We visualized these profiles using the PHATE algorithm43 and performed Leiden clustering44 to identify genes with similar interphase phenotypes, leveraging hierarchical sub-clustering to identify relationships between individual genes in specific cases (STAR Methods; Figure 3A). Based on clear functional relationships between the genes within a given cluster, we identified clusters primarily composed of genes with roles in transcription, RNA processing, translation, protein degradation, DNA replication and damage response, cell cycle control, and other core cellular processes (Figures 3B–F and S4A–F; https://vesuvius.wi.mit.edu/). Strikingly, the clustering behaviors also allowed us to distinguish functional sub-categories with high resolution and coherence for the genes within a given cluster. For example, despite a shared role in translation, we identified separable clusters containing established 40S ribosome subunits (cluster 66), 60S ribosome subunits (cluster 23), tRNA ligases and eIF2 translation initiation subunits (cluster 14), distinct clusters for factors involved in 40S and 60S ribosome biogenesis (clusters 136 and 15, respectively), and several others which included nucleolar proteins, RNA helicases, and additional related factors (clusters 21, 112, 203, and 216; Figure 3C–D). Knockouts for the genes within each of these clusters resulted in reduced nuclear and cellular areas, but displayed differences in other phenotypic parameters, such as actin and tubulin stain intensities, enabling distinction among these functional sub-categories (Figure 3D). We also observed fine-grained functional clustering within other diverse cellular processes, including protein degradation (Figure S4A) and transcription (Figures 3E–F). Although our stains did not include membrane markers for cellular organelles, we identified multiple distinct clusters composed of vesicle trafficking components (Figure S4F). This suggests that specific cellular changes resulting indirectly from disrupting these processes can be detected by our analysis despite the limited number of stains.
Figure 3. Clustering of multi-dimensional interphase phenotypes reveals co-functional essential genes.
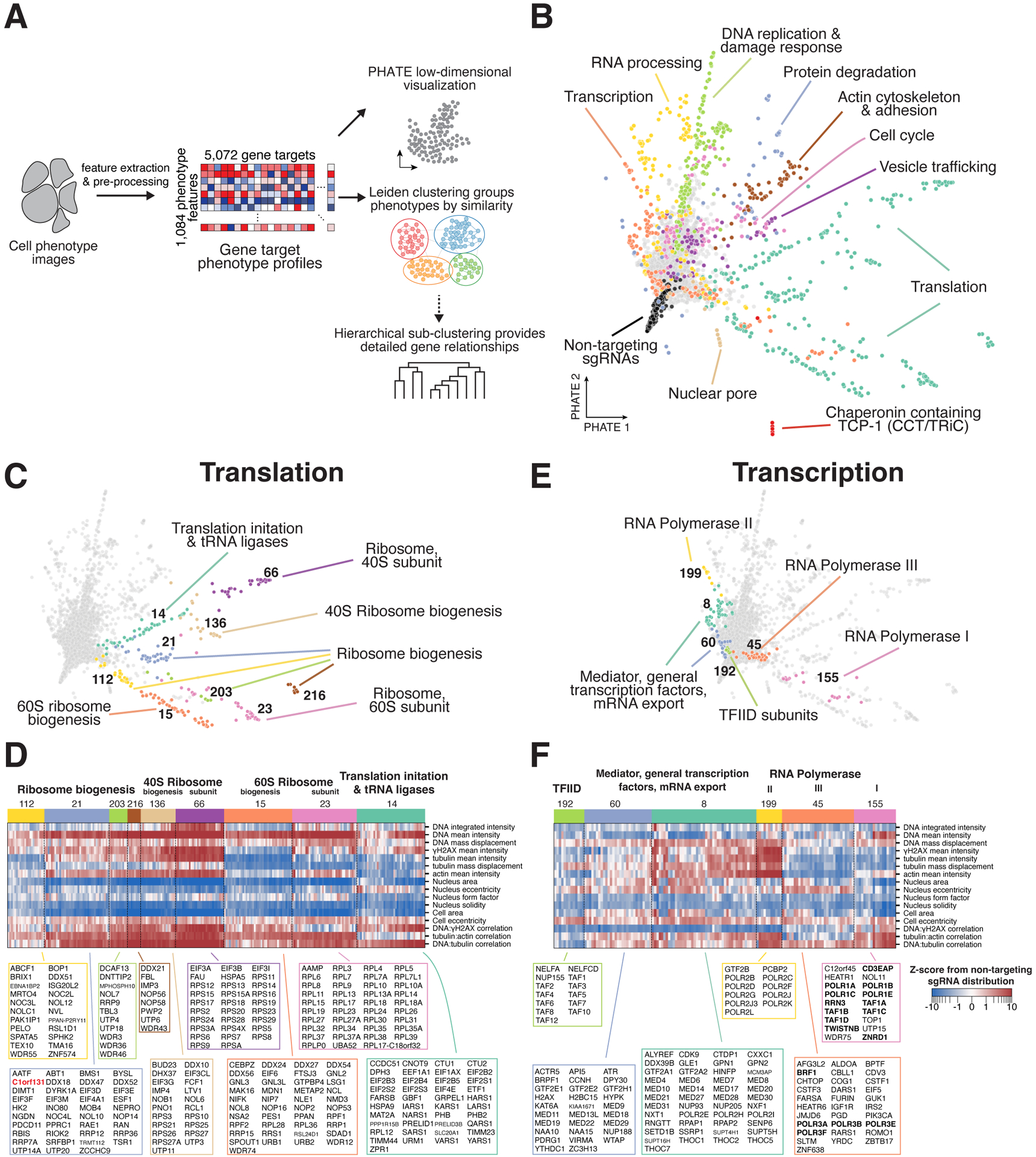
(A) Analysis workflow overview. 1,084 phenotype parameters were extracted from raw cell images and aggregated into profiles for each of the 5,072 genes. The relationships between profiles were visualized using PHATE43 and grouped by similarity using the Leiden clustering algorithm.44 In select cases, hierarchical sub-clustering was performed to identify gene-level phenotype similarities (STAR Methods). (B) Two-dimensional PHATE representation of the interphase phenotype gene profiles from the primary screen. Colors correspond to manually-annotated Leiden clusters containing the labeled functional gene categories. (C) Individual clusters related to translation from (B) identify fine-grained functional sub-categories of genes. Functional descriptions are based on manual annotations. (D) Heat map of interphase knockout phenotypes corresponding to the translation clusters in (C) for a manually-selected subset of phenotype parameters (STAR Methods). All genes from each cluster are listed. (E) Individual clusters of genes related to transcription from (B). (F) Heat map as in (D) corresponding to the clusters in (E) highlighting the phenotypic similarities that define each cluster of genes with transcriptional functions.
Based on a quantitative comparison of knockout phenotypes to non-targeting sgRNAs, 4,665 of the 5,072 tested gene targets displayed a measurable interphase phenotype (Figure S4G). Of the remaining 407 gene knockouts, only 55 genes displayed strong fitness effects at 5 days post-Cas9 induction based on sgRNA depletion from our library (Figure S4G). Thus, the 352 genes without a measurable phenotype or fitness effect are likely not required for cellular fitness in HeLa cells at the tested time point following Cas9 induction. To evaluate these behaviors, we compared our dataset with multiple orthogonal approaches to identify co-functional genes, including protein-protein interactions (BioPlex),45 co-essentiality analysis,46 curated protein complexes (CORUM),47 functional annotations (KEGG),48 and STRING associations.49 We found that the relative similarity of gene phenotype profiles measured in our screen was increased among annotated gene pairs in each of these other databases (Figure S4H). Based on our analysis, 2,058 genes (40.6%) are contained in phenotype clusters that are enriched for annotations in at least two of the tested datasets, with an additional 1,195 genes (23.6%) present in clusters that are enriched for one dataset (Figure S5A; Table S3). Although functional relationships identifiable in our approach are not limited to physically-interacting proteins, the “precision” and “recall” correspondence between our interphase phenotype clusters and curated CORUM protein complexes is similar to prior studies directly measuring protein interactions or inferring relationships using co-essentiality analysis46,50,51 (Figure S5B).
We also compared our dataset to the annotated clusters from a recently-published large-scale Perturb-seq study, which infers gene function based on transcriptional changes following Cas9 targeting.5 Both approaches identified clear clusters for core functional complexes including the TCP-1 Chaperonin complex, COP9 Signalosome, Exosome complex, NELF complex, FACT and Paf complexes, Pol I and rRNA biosynthesis factors, and others (Figures S5C–D; not shown). Similarly, both strategies identified key players in protein translation and protein degradation, although our optical screening strategy captured a larger number of these proteins and was able to achieve a more fine-grained resolution, allowing us to distinguish between ribosome core subunits and biogenesis factors, for example (Figure S5D). Notably, the Perturb-seq dataset identified clusters for mitochondrial 28S and 39S ribosomal complexes, which were likely not identified in our screen as we did not image specific markers for mitochondrial function. However, our image-based analysis captured gene contributions to core morphological processes that were not identified in the Perturb-seq results, including cytokinesis (cluster 148), nuclear transport (cluster 104), and adhesion (cluster 29).
In summary, our phenotypic clustering provides a fine-grained picture of the distinct functional contributions of specific proteins to core cellular processes. Our clustering analysis is based on functionally-defined phenotypic similarities from imaging data, and represents an orthogonal strategy for identifying co-functional genes that is not limited to physically interacting proteins or phenotypes associated with transcription effects. Thus, gene targets with measurable phenotypes present in clusters lacking prior annotations represent opportunities for identifying poorly-characterized biological pathways.
Phenotypic clustering provides insights into gene functions and pathway relationships
The coherent phenotypic clustering of known co-functional gene targets provides predictions of the cellular function for co-clustering genes. We sought to test these predictions for diverse clustering behaviors. Our interphase phenotypic clustering revealed similarities between knockouts of the key signaling proteins KRAS and BRAF with multiple mitochondrial components (cluster 149; Figure S5E). In independent experiments with stains selective for mitochondrial membrane potential, we identified a disruption of active mitochondria in KRAS and BRAF knockout cells, similar to knockouts of co-clustering mitochondrial factors (Figure S5F). This is consistent with roles for KRAS and BRAF signaling in maintaining metabolic homeostasis.52–54 Similarly, among clusters containing transcriptional regulators, we identified a cluster (121) with a distinct phenotypic profile that contains the master regulator Myc and Max transcription factors, along with multiple other transcriptional regulators, chromatin remodeling factors, and E3 ubiquitin ligase components (Figure 4A). This clustering suggests that these factors may either be specifically required for Myc expression (as is the case for ZMYND8, see Cao et al.55) or function together with Myc to promote downstream expression at its target promoters. By analyzing Myc mRNA and protein levels in individual gene knockouts from this cluster, we confirmed a role for SETD2 in regulating Myc expression (Figures 4B and S5G). Our interphase clustering analysis further implicated poorly characterized genes in specific cellular activities. For example, we nominated C1orf131 as putatively involved in ribosome biogenesis based on its membership in cluster 21 (Figure 3D), which was recently confirmed by others.56 Similarly, AKIRIN2 clustered with the 20S core particle proteasome subunits (cluster 167, Figure S4A), and was recently described as a proteasome nuclear import factor.57 In addition, HNRNPD clustered with the m6A mRNA modification writers METTL3 and METTL14 (197; Figure S5H), consistent with the emerging role of m6A in promoting HNRNPD associations with mRNA.58
Figure 4. Phenotypic clustering relationships predict gene function.
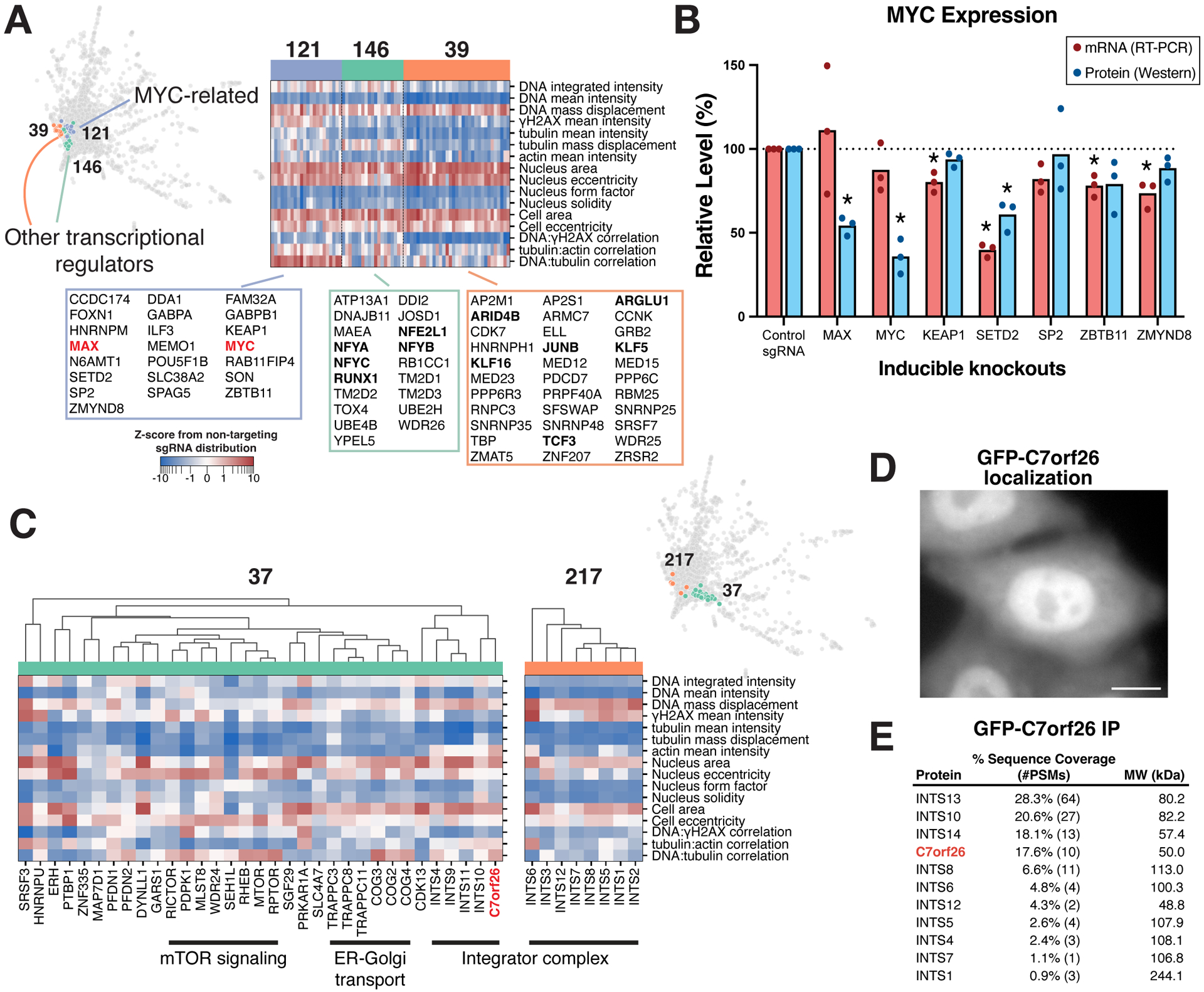
(A) Heat map of interphase phenotypes for clusters containing transcriptional regulators (STAR Methods). (B) Western blot (see Figure S5G) and mRNA quantification of MYC mRNA and protein expression following knockout of selected genes from cluster 121. *P<0.05 by two-tailed independent T-test relative to corresponding controls. (C) Phenotype heat maps of interphase clusters 37 and 217 as in (A), demonstrating the phenotypic similarity between C7orf26 knockouts with those of Integrator complex subunits. Hierarchical clustering (top) within cluster 37 using the Pearson correlation of PCA-projected phenotype profiles (STAR Methods). (D) GFP-C7orf26 localizes to the nucleus consistent with Integrator complex function. Scale bar, 10 μm. (E) Mass spectrometry from an immunoprecipitation of GFP-C7orf26 from human cells relative to controls.
We also identified the poorly characterized gene C7orf26 in a cluster containing multiple components of the mTOR signaling and ER-Golgi transport pathways, as well as subunits of the Integrator complex, an RNA endonuclease involved in RNA processing59 (cluster 37; Figure 4C). Based on hierarchical sub-clustering, we found that C7orf26 segregated closely with Integrator subunits, including INST10, and also displayed related phenotype profiles to the Integrator subunits in cluster 217 (Figure 4C). To evaluate this co-clustering relationship, we generated a cell line stably expressing GFP-C7orf26. GFP-C7orf26 localized to the nucleus (Figure 4D), consistent with Integrator complex localization.60 In affinity purifications, GFP-C7orf26 pull-downs specifically isolated multiple Integrator complex subunits, with particularly robust levels of INTS13, INTS10, and INTS14 (Figure 4E; also see ref.61,62). These proteins were recently shown to comprise a functional subunit of the Integrator complex that associates the cleavage module with target RNA.63 Our data suggests C7orf26 may interact with this sub-complex, consistent with concurrent studies.5,64 Thus, the phenotypic clustering of this dataset identifies established interacting partners and provides predictive insights to identify associations and co-functional players across key biological processes.
Analysis of mitotic phenotypes identifies requirements for proper cell division
We next analyzed the phenotypes observed in mitotic cells. In total, 2.6% of the cells visualized in our microscopy-based screen were present in the mitotic phase of the cell cycle (median of 157 mitotic cells per gene). In the presence of mitotic errors, cells activate the spindle assembly checkpoint and arrest in mitosis32 such that an increased fraction of mitotic cell images (i.e., mitotic index) can reflect a mitotic disruption. We observed an increased mitotic index for gene knockouts targeting established components of the kinetochore and mitotic spindle, but a reduced mitotic index for Spindle Assembly Checkpoint factors (Figure 5A).
Figure 5. Mitotic phenotypes uncover essential genes required for cell division.
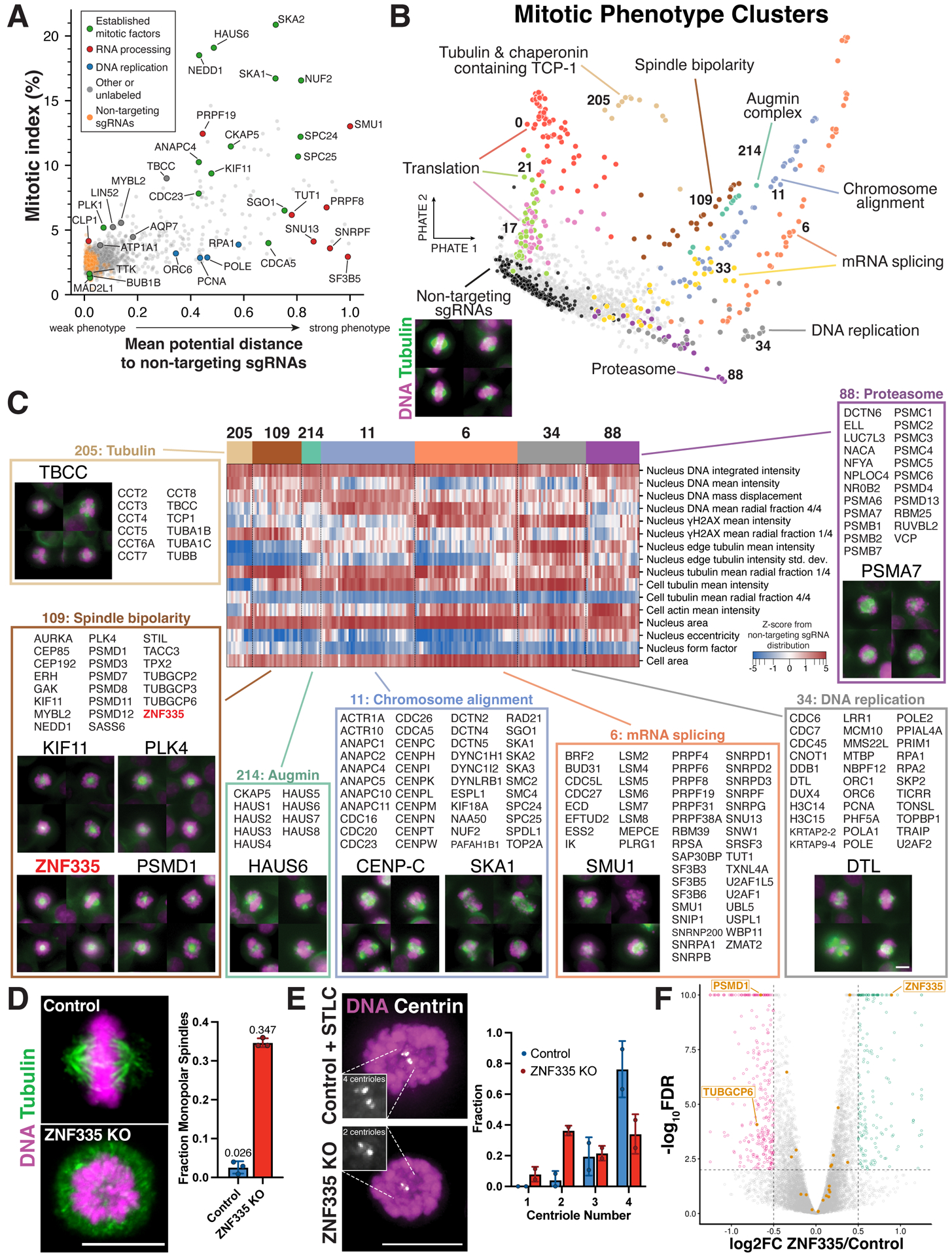
(A) Scatter plot of mitotic index for each gene target compared to a summary score of image-based mitotic phenotype strength computed by PHATE43 (STAR Methods). (B) Two-dimensional representation of the mitotic phenotype visualized using PHATE, clustered to form groups with similar phenotypes (STAR Methods). Each dot represents a single gene, colored corresponding to the indicated cluster. Functional descriptions correspond to manual annotations. (C) Selected screen images and heat map of mitotic phenotypes corresponding to the clusters in (B) for a manually-selected subset of parameters (STAR Methods). Scale bar, 10 μm. All gene targets from selected clusters are listed. (D) Left, immunofluorescence images of cell lines stably expressing a sgRNA targeting ZNF335 or control. Right, bar plot of the corresponding fraction of mitotic cells with monopolar spindles; each data point represents one experiment with >100 cells. Images are deconvolved maximum intensity projections of fixed cells stained for microtubules (anti-alpha-tubulin) and DNA (Hoechst). (E) Example images (left) of DNA (Hoechst, magenta) and Centrin (grayscale) stains of monopolar ZNF335 knockout cells along with quantification of reduced centriole numbers (right) compared to monopolar control cells generated by STLC treatment (n>88 cells per condition). Insets show magnified regions. Scale bars, 10 μm. Error bars indicate SD. (F) Volcano plot of differential gene expression in ZNF335 knockout. Yellow data points represent genes co-clustering with ZNF335 in mitotic cluster 109. 296 genes, including PSMD1 and TUBGCP6, are downregulated (magenta) and 177 genes are upregulated (green) in ZNF335 KO cells (FDR < 0.01, log2 effect size > 0.5).
Similar to our analysis of interphase cells, we created summary image-based profiles of mitotic phenotypes for each gene and then clustered gene targets with similar profiles (Figures 5B–C; STAR Methods). In parallel, we conducted a manual visual analysis for each gene, which strongly corresponded to the profile-based results (Figures S6A–B). Our computational analysis identified multiple mitotic clusters with functionally-related genes, including established factors with roles in spindle assembly, DNA replication, mRNA splicing, ribosome function, and chromosome alignment (Figures 5B–C). This high-dimensional computational analysis provides a complementary, but distinct measurement of mitotic phenotypes as compared to mitotic index (Figure 5A). Visual analysis of cell image montages further allowed us to distinguish phenotypic clusters and individual genes for their specific roles during mitosis (Figure 5C). For example, we detected reduced microtubule density following depletion of the tubulin chaperone TBCC, chromosome mis-alignment following knockout of kinetochore components and splicing factors, monopolar spindles associated with the targeting of KIF11 or PLK4, and short mitotic spindles in knockouts of CKAP5 or Augmin subunits (Figure 5C).
To evaluate our analysis of mitotic phenotypes, we compared our findings to those from MitoCheck,7 a genome-wide siRNA-based arrayed microscopy screen for mitotic phenotypes in HeLa cells. Of the 293 genes identified as displaying mitotic phenotypes by MitoCheck that were also present in our screen, 79 displayed an aberrant mitotic index or measurable mitotic phenotype in our dataset, with an additional 70 genes that scored significantly in at least one interphase phenotype category that is consistent with downstream consequences of mitotic defects (Table S3). Conversely, we identified 799 genes with mitotic phenotypes in our screen that were not identified in MitoCheck. These genes are enriched for proteasome, cell cycle, and DNA replication genes among other relevant pathways (Figure S6C). The genes with mitotic phenotypes only observed in our study encode many canonical mitotic factors, such as kinetochore components, Augmin complex subunits, and centrosome proteins (Table S3). Our approach thus represents a significant improvement in sensitivity and efficiency over prior systematic approaches to identify mitotic regulators, enabled by precise Cas9-based gene perturbation and pooled phenotype acquisition.
In addition to established mitotic players, predicted mitotic roles emerged for poorly-characterized genes based on co-clustering with well-defined mitotic functions. For example, ZNF335 clustered with spindle proteins and gamma-tubulin complex subunits (cluster M109; Figure 5C). Analysis of an individual inducible knockout cell line for ZNF335 revealed a substantially increased proportion of cells with monopolar spindles (Figure 5D) and a reduction in centrioles (Figure 5E). GFP-ZNF335 localizes to the nucleus in interphase cells (Figure S6D), but did not localize to the spindle or centrioles during mitosis, and did not associate with established mitotic factors in immunoprecipitation experiments (not shown). However, RNA-sequencing analysis of gene expression following ZNF335 knockout revealed a significant decrease in the expression of the proteasome subunit PSMD1 and the gamma-tubulin ring complex subunit TUBGCP6 (Figure 5F), which both displayed similar monopolar spindle phenotypes in our screen (Figure 5C). Together, this suggests that ZNF335 promotes spindle function by regulating the expression of specific centrosome factors, and provides a potential explanation for why ZNF335 mutations are observed in human microcephaly,65 as is the case for many centrosome components.66 This highlights the value of our profile-based functional clustering approach, as this indirect role of ZNF335 in centrosome function was not identified previously based on physical interactions with centrosome components (e.g., see Gheiratmand et al.67). This analysis demonstrates the utility of pooled large-scale image-based screening to identify complex mitotic phenotypes, and also identifies dozens of genes with mitotic phenotypes that have not been implicated previously as having roles in cell division (see below).
A pooled live-cell imaging-based screen for mitotic defects
Based on the large number of genes with unexpected mitotic phenotypes and the ability of microscopy to directly visualize these phenotypes,7,68 we next performed a secondary pooled live-cell screen. We selected 228 genes with unexpected mitotic phenotypes and 11 positive controls with established roles in diverse mitotic processes. We transduced an sgRNA library containing 2 sgRNAs per gene and 50 non-targeting sgRNAs (526 total sgRNAs; see STAR Methods) into a HeLa cell line containing doxycycline-inducible Cas9 and a constitutively-expressed H2B-mCherry fusion to visualize chromatin (Figure 6A). We conducted time-lapse imaging of the pooled cell population for 24 hours with time points at 10 minute intervals, followed by fixation of the cell population and in situ readout of the sgRNA in each cell (Figure 6A; STAR Methods). After tracking cell lineages through each time course and identifying mitotic cells, we obtained time-lapse movies for 451,434 total cell division events, with a median of 1,381 division events per gene target (Figure 6B–C and S6E–F; Table S4; Movie S1; https://vesuvius.wi.mit.edu/; STAR Methods).
Figure 6. A pooled live-cell screen identifies gene targets required for mitotic progression.
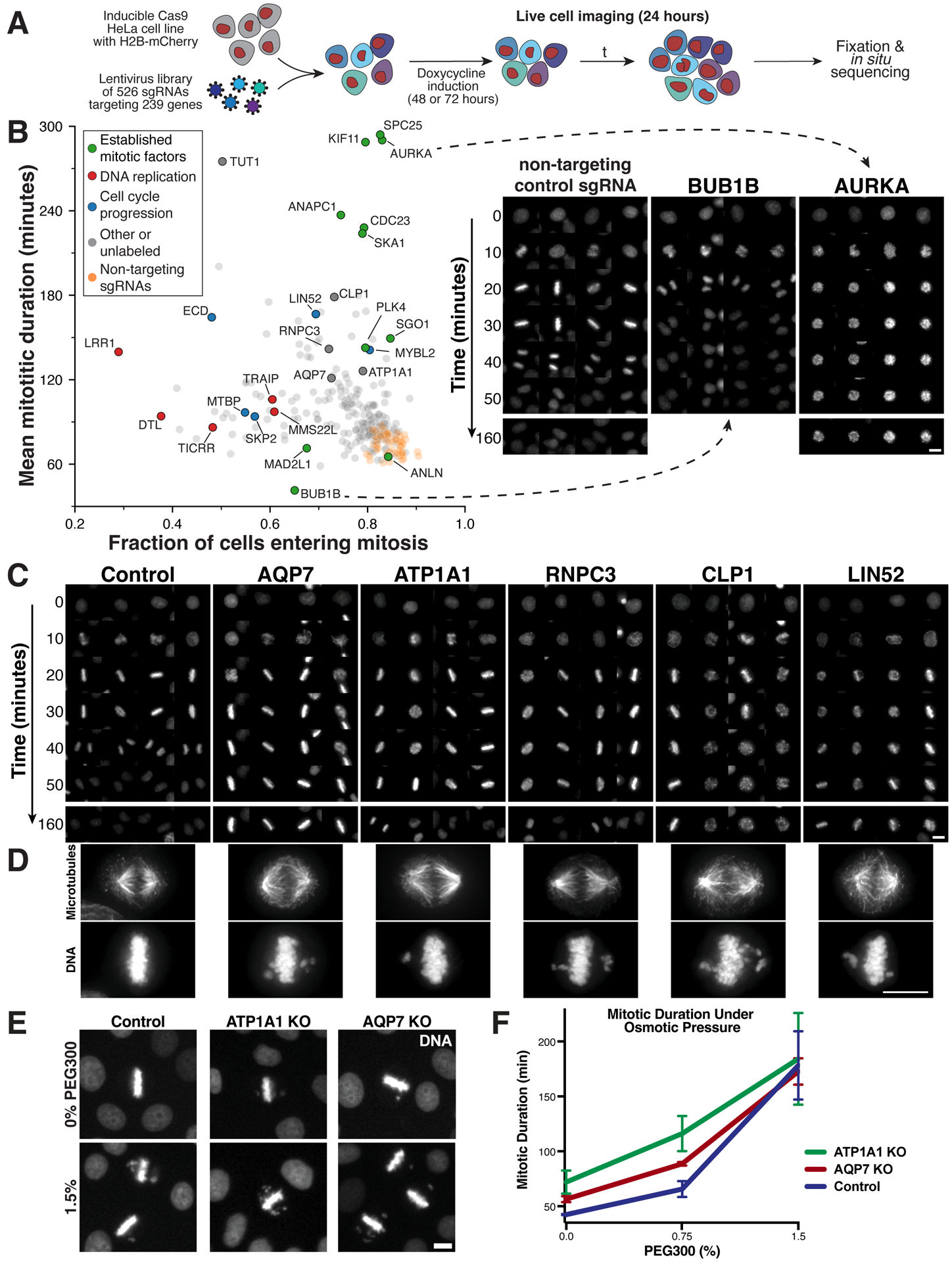
(A) Experimental workflow for the live-cell, image-based pooled CRISPR screen using a cell line expressing an H2B-mCherry fusion (STAR Methods). (B) Left, scatter plot comparing the fraction of cells that enter mitosis within the 24 hour time course and the mitotic duration of observed cell division events. Plotted values represent the mean of sgRNAs targeting the same gene. Right, example images of H2B-mCherry fluorescence at the indicated time points after mitotic entry for knockouts of established cell division components. (C) Example time course montages as in (B) demonstrating mitotic delay and mitotic defects for selected target genes. (D) Immunofluorescence images showing individual cell lines stably expressing a single sgRNA targeting the indicated genes (see also Figure S7A). Images are deconvolved maximum intensity projections of fixed cells stained for microtubules (anti-alpha-tubulin) and DNA (Hoechst). Scale bars, 10 μm. (E) Example images and (F) mitotic duration from time-lapse imaging of control, AQP7, and ATP1A1 inducible knockout cells incubated with varying PEG300 concentrations to induce hyperosmotic stress. n>50 cells per datapoint. Error bars indicate SD.
Using this automated live-cell analysis, we calculated the mean duration of cell division events for each gene target, as well as the fraction of cells that enter mitosis during the time course (Figure 6B). 197 of the 239 tested gene knockouts, including 10 of 11 established cell division factors, displayed altered mitotic duration or entry relative to non-targeting controls (Figures 6B and S6G), further supporting our ability to identify diverse mitotic players in the fixed cell analysis. We were also able to distinguish gene targets with established or predicted roles in DNA replication or repair, based on their increased mitotic duration but reduced fraction of cells entering mitosis (Figure 6B), indicative of defective mitotic entry. From visual inspection of the time-lapse montages (Figure 6C; Table S4), we selected 28 genes with chromosome alignment defects. Based on fixed and live-cell analysis of individual knockout cell lines (Figures 6D, S7A and Movie S2), each of the 28 selected gene targets displayed clear defects in chromosome alignment and segregation or spindle assembly (Figure S7A).
Unexpectedly, amongst the gene targets whose knockouts resulted in misaligned chromosomes, we identified two membrane-bound transporters - the plasma membrane-localized aquaporin AQP7 and the sodium/potassium-transporting ATPase ATP1A1 (Figure 6D). Individual AQP7 and ATP1A1 knockout cell lines displayed a reproducible delay in chromosome alignment and an extended mitotic duration (Figures 6E–F), but we did not observe defects in bipolar spindle assembly (Figure 6D) or kinetochore assembly (Figures S7B–C). As both membrane transporters are involved in maintaining a proper intracellular osmotic environment, we tested the effect of treating cells with 300 Da polyethylene glycol (PEG 300), which creates hyperosmotic stress.69 PEG300 treatment of control cells resulted in qualitatively similar mitotic phenotypes to AQP7 and ATP1A1 knockouts, including chromosome misalignment and a mitotic delay (Figures 6E–F). By titrating PEG300, we also observed an additive mitotic defect from combining hyperosmotic stress with ATP1A1 and AQP7 knockouts (Figures 6E–F). Thus, we propose that AQP7 and ATP1A1 are required to create an internal osmotic cellular environment that promotes proper chromosome segregation, revealing an unanticipated role for osmolarity and its regulation in mitotic fidelity. Our pooled live-cell screen confirms the observed mitotic defects from the primary screen and reveals the roles of diverse gene targets in mitotic progression and fidelity.
LIN52, CLP1, and RNPC3 are required for the correct expression of kinetochore assembly factors
To define the basis for the observed mitotic phenotypes, we next tested the function of the kinetochore, the key player in mediating interactions between centromere DNA and microtubule polymers during cell division.70 Of the 28 gene knockouts that we selected based on their chromosome alignment defects, 25 displayed no difference or only modest changes in the recruitment of the outer kinetochore protein Ndc80 (Figure S7B), suggesting that kinetochore assembly is largely intact. In contrast, CLP1, RNPC3, and LIN52 inducible knockouts displayed a substantial reduction in both Ndc80 localization and total Ndc80 protein levels (Figures 7A and S7D). Similarly, of the tested gene knockouts, only LIN52 resulted in a substantial reduction in the kinetochore localization or protein levels for the centromere-specific histone CENP-A (Figures 7B and S7C–D). Based on these changes in kinetochore assembly, we chose to focus on LIN52, CLP1, and RNCP3.
Figure 7. Lin52, Clp1 and RNPC3 functions to promote proper kinetochore assembly and chromosome segregation.
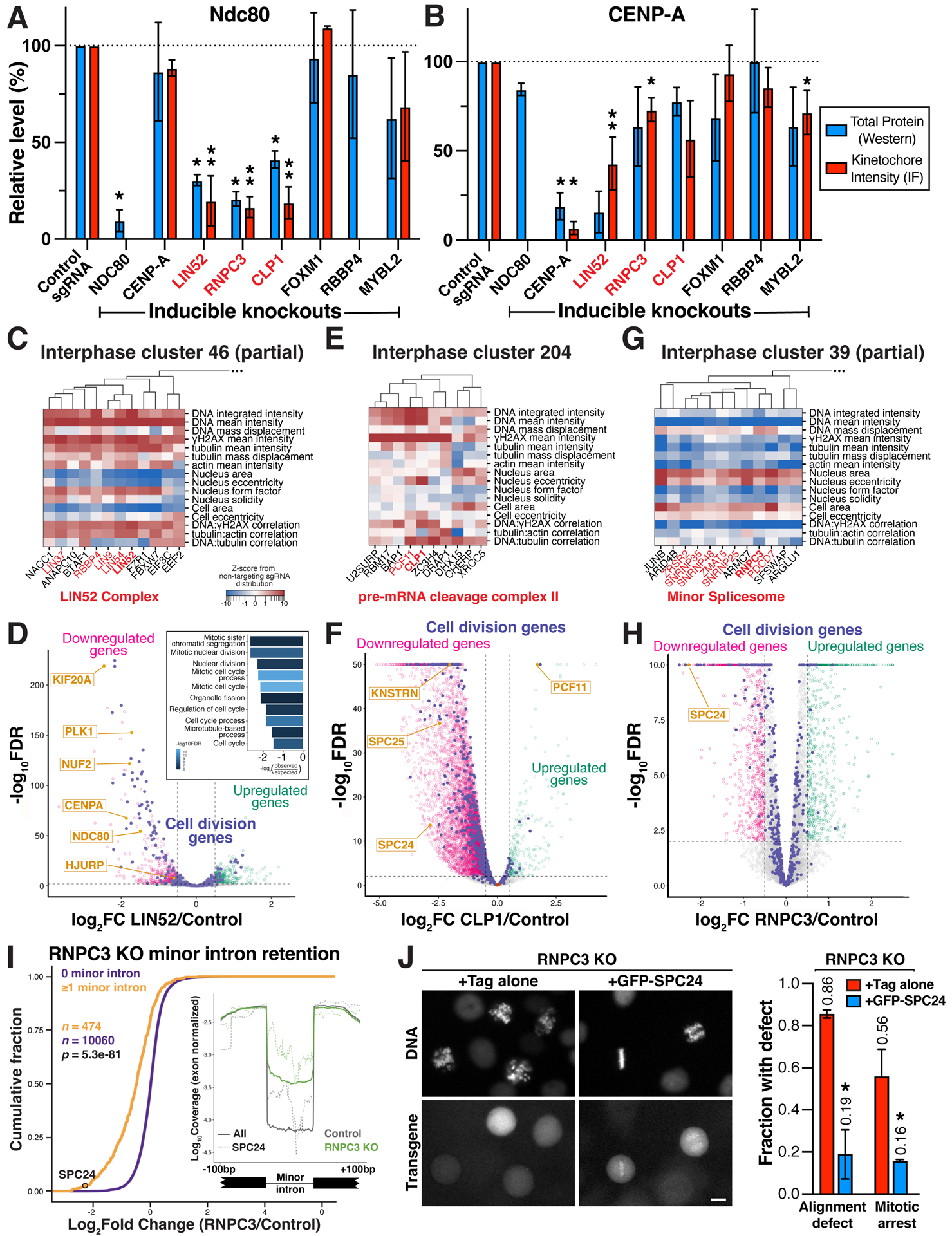
(A) Bar plot showing total protein levels (blue) and kinetochore-localized intensity (red) of the outer kinetochore microtubule-binding protein NDC80 in the indicated inducible knockout cell lines relative to a control sgRNA. N=2 biological replicates for total protein levels, which were normalized to GAPDH. N=2–10 biological replicates for kinetochore measurements, each replicate represents the median kinetochore signal from >10 cells. Both measurements were further normalized relative to controls from the same experiment. *P<0.05, **P<0.01 by two-tailed independent T-test relative to corresponding control samples. ND, no data. Error bars indicate SD. (B) Bar plot as in (A) showing total protein level (blue) and kinetochore-localized intensity for the inner kinetochore centromere-specific histone CENP-A in the indicated inducible knockout cell lines relative to a control sgRNA. Experimental design and statistical tests as in (A). (C) Phenotype heat map and hierarchical clustering for a subset of primary screen interphase cluster 46 genes (STAR Methods). (D) Volcano plot of LIN52 knockout differential expression based on RNA-seq. Genes involved in cell division processes are indicated in purple. Significance threshold FDR < 0.01, log2 effect size > 0.5 for up- (green) and down-regulated genes (magenta). Inset, GO term analysis of LIN52 downregulated genes shows a significant enrichment of mitotic genes. (E) Heat map of primary screen interphase cluster 204 phenotypes as in (C), demonstrating an association of knockout phenotypes for the pre-mRNA cleavage complex II factors CLP1 and PCF11, and the transcriptional termination factor ZC3H4. (F) Volcano plot of differential expression as in (D) following CLP1 knockout, identifying a global decrease in mRNA abundance in these cells including for SPC24 and SPC25, identified by normalizing to library spike-in control RNA (brown). (G) Heat map of a subset of primary screen interphase cluster 39 phenotypes as in (C), demonstrating tight clustering of minor spliceosome components including RNPC3. (H) Volcano plot of differential gene expression as in (D) after RNPC3 knockout, with the SPC24 outer kinetochore component significantly downregulated. (I) Cumulative distributions of mRNA fold change from RNPC3 knockout cells for transcripts containing at least 1 minor intron (orange) are significantly downregulated compared to transcripts with no minor introns (purple). Statistical significance between cumulative distributions was assessed using the Mann-Whitney U test. Inset, minor introns are retained in RNPC3 knockout cells (green), including the minor introns in SPC24 (dotted lines). (J) Left, representative images of H2B-mCherry (DNA) and transgene localization for live RNPC3 knockout cells expressing Tag only or GFP-SPC24. Right, bar plot showing fraction of mitotic RNPC3 knockout cells displaying chromosome alignment defects (n>100 cells) or arrest in mitosis (>2 hours in mitosis; n>45 cells) when expressing Tag only or GFP-SPC24. Error bars indicate SD.
LIN52 is a component of the DREAM complex, comprised of E2F family transcription factors, LIN9/37/52/54, MYBL1/2, RBL1/2, RBBP4, and TFDP1/2, which acts together with FOXM1 as a transcriptional regulator for cell cycle genes.71,72 GFP-LIN52 localizes to the nucleus (Figure S7E) and associates with LIN9/37/54, RBBP4, and RBBP7, but not other established DREAM complex proteins in immunoprecipitations (Figure S7E). Correspondingly, in our fixed-cell screen, LIN52 displayed interphase phenotypes similar to LIN9/37/54, RBBP4 and RBBP7 (cluster 46; Figure 7C), but did not co-cluster with the other DREAM-related genes present in the screen. Consistent with the phenotypic co-clustering and physical interactions, we observed chromosome misalignment, a mitotic delay, and substantial changes to kinetochore assembly in knockouts of LIN52, LIN9, and LIN54 (Figures S7F–G). RNA-seq analysis of LIN52 knockout cells revealed a pervasive decrease in the expression of diverse cell division genes (Figure 7D) including Ndc80 complex subunits, CENP-A, and the CENPA deposition machinery, providing an explanation for the broad defects in kinetochore assembly. In contrast, we did not detect altered kinetochore protein levels or chromosome misalignment for FOXM1 knockouts and only observed a modest change in CENP-A and Ndc80 localization in MYBL2 knockouts (Figures 7A–B and S7B–D), suggesting a potent role for a LIN52 sub-complex in the expression of these cell division components.
CLP1 is a component of the pre-mRNA cleavage complex II.73 In our interphase phenotypic clustering analysis, CLP1 closely associated with its interacting partner PCF1173 and the transcription termination factor ZC3H474,75 (Figure 7E). We performed RNA-sequencing analysis of CLP1 knockout cells, which revealed wide-spread defects in transcription termination (Figure S7H) and a global decrease in mRNA expression (Figure 7F). Amongst the genes that evaded downregulation, PCF11 displayed significantly increased relative gene expression, consistent with autoregulation of the pre-mRNA cleavage complex.76 Reciprocally, we observed strong downregulation of selected cell division components, including the Ndc80 complex subunits Spc24 and Spc25, explaining the selective loss of the Ndc80 complex (Figures 7A–B) and the chromosome mis-alignment defects in CLP1 knockouts (Figures 6C–D). This finding provides insights into the specific importance of 3’ end RNA processing in mitotic function.
Finally, we analyzed RNCP3. In our fixed-cell screen, RNPC3 displayed an interphase phenotype closely related with multiple components of the minor spliceosome machinery (Figure 7G), consistent with prior work.77 Based on RNA-seq analysis of RNPC3 knockouts arrested in mitosis, we found pervasive defects in the splicing of minor introns for diverse genes, coupled with a substantial down-regulation of these minor intron-containing mRNAs (Figures 7H–I). Amongst these genes, we identified the Ndc80 complex subunit SPC24 as being specifically mis-spliced and downregulated (Figures 7H–I and S7I; see also de Wolf et al.78). Strikingly, we were able to rescue the chromosome alignment defects and mitotic arrest in RNPC3 knockout cells through the exogenous expression of a spliced SPC24 cDNA (Figure 7J). Thus, the role of RNPC3 in the minor spliceosome and the selective requirement for minor intron splicing in the production of the SPC24 mRNA explains the observed outer kinetochore assembly defects and chromosome mis-segregation phenotype in RNPC3 knockouts (Figures 6C–D and 7A). Our work specifically implicates RNPC3-mediated splicing in maintaining the fidelity of chromosome segregation during cell division.
Together, these analyses provide molecular explanations for the diverse cell division phenotypes for the LIN52, CLP1, and RNPC3 knockouts observed in our large-scale optical pooled screens, with selective defects in the production of critical mitotic players resulting in chromosome mis-segregation and kinetochore dysfunction. These analyses highlight the ability of our image-based phenotypic clustering to identify functional relationships and define roles for diverse factors in complex cellular phenotypes.
Discussion
A central goal of our study was to define the phenotypic landscape for essential human gene function, including identifying unrecognized functions of well-characterized genes and assigning functional roles for poorly characterized genes. Our pooled microscopy-based analysis of tens of millions of individual knockout cells for thousands of fitness-conferring human genes identified specific contributions to core biological processes based on the resulting cellular phenotypes. Using combinations of statistically-significant changes from hundreds of quantitative parameters that are directly comparable across a large cell population, we generated phenotypic “fingerprints” for each gene target. By comparing these phenotype profiles, we defined co-functional gene relationships with sufficient resolution to distinguish related roles in specific cellular processes. Notably, the fine-grained precision and richness of the cell biological phenotype profiles we obtained using only four cellular stains were sufficient to identify functional relationships between genes across a wide range of biological pathways without analyzing specific cellular markers corresponding to each pathway or function. In addition to identifying established relationships, this work provides multiple predictions for the contributions of incompletely characterized genes to fundamental cellular processes. For example, together with concurrent studies from others,5,56,57,64 our phenotype clustering analysis implicated C7orf26 as a core integrator complex subunit, C1orf131 as a regulator of ribosome biogenesis, and AKIRIN2 in proteasome function. We also identified gene knockouts resulting in defects in mitotic function including unanticipated roles for the membrane-bound transporters AQP7 and ATP1A1 and cellular osmolarity in promoting accurate chromosome segregation. In addition, our work revealed roles for multiple gene expression regulators in controlling cell division, including the predicted transcription factor ZNF335, the DREAM complex (LIN52), the 3’ end mRNA processing complex (CLP1), and the minor spliceosome (RNPC3) in the expression of specific cell division components. These examples highlight the power of optical screening to identify co-functional genes across diverse biological pathways, with the potential for further discovery from the data published here. This work also creates a powerful data resource for researchers to explore and an extensive testbed for future analytical method development.
Our work highlights the power of combining CRISPR perturbations with rich, quantitatively-defined cell biological phenotypes extracted using image-based analyses to precisely identify co-functional genes across a wide range of cellular processes. As two proteins may act in a single biological pathway without displaying a direct interaction, strategies for defining co-functional genes based on functional perturbations and phenotypic measurements provide an important and highly scalable orthogonal approach to proteome-wide protein interaction studies.45 Recent related work has defined functional relationships based on their correlated fitness requirements in pooled CRISPR screens across more than 1000 cell lines17,21,46,51 and by analyzing single-cell transcriptional states using Perturb-seq.5 The precision and breadth of the clustering behaviors reported here highlights the ability of quantitative image-based phenotypic profiling to provide a similar scale of functional information, with both overlapping and distinct insights. For example, as gene co-essentiality analyses rely on substantial differences in fitness requirements across cell lines, gene clusters based on the DepMap dataset are excellent for identifying developmental signaling pathways such as TGF-Beta, Interferon, p53, and MHC signaling.46 These genes are not essential in most cell lines, and thus were largely excluded from our screening library or do not result in potent cellular phenotypes in HeLa cells. Within the essential gene landscape, we found that our approach excelled at defining gene clusters for pan-essential genes, such as ribosome components, which display clear similarities in cellular image-based phenotypes but do not show variable requirements across cell lines, thereby limiting their detectability based on co-essentiality analyses. Similarly, the Perturb-seq approach identified similar gene clusters to many of those in our analysis, including ribosome components and other protein complexes.5 However, in many cases, our analysis identified additional genes and achieved a more fine-grained resolution for these clusters, for example allowing us to effectively distinguish amongst core ribosome subunits and ribosome biogenesis factors (Figure S5C–D). Our approach was also highly successful at identifying gene clusters for morphological processes, including cytokinesis, nuclear transport, chromosome condensation, adhesion, and others, which may be harder to identify based on transcriptional changes. Finally, both the Wainberg et al.46 co-essentiality analysis and Replogle et al.5 were better able to identify clusters containing mitochondrial genes than our study, highlighting the potential value of including a specific cellular marker for mitochondrial function in future analyses. Overall, the flexibility of phenotype measurements and modest relative cost of image-based pooled CRISPR screens, together with the results reported here, further establish image-based pooled profiling screens as a robust strategy for defining gene networks and the functional contributions of human genes.
Image-based measurements have the unique ability to define cellular and morphological phenotypes across a broad range of time and length scales, and report on non-cell autonomous phenotypes that require spatial context. Arrayed image-based screens have been used successfully for diverse purposes,7,79–82 but these depend on extensive automation and are subject to multilevel batch effects that are difficult to control. In contrast, pooled image-based screens are comparatively simple to scale with intermixed controls providing a strong statistical basis for comparisons. Recent studies have used physical enrichment via FACS or other approaches to isolate individual cells with desired phenotypes using pooled microscopy screening,10–14 but this strategy is only feasible using a limited number of pre-defined phenotypes. In contrast, pooled image-based screens produce complex profile data and perturbation identity for each cell to enable multiple high-dimensional phenotypes to be directly associated with individual perturbations in a single experiment. Here, we efficiently leverage complex, multi-dimensional image-based phenotypes to yield dozens of functionally-relevant clusters of genes across diverse cellular processes at a scale an order of magnitude greater than any reported individual image-based pooled profiling screen.8,9 Together, this paper represents important advances through the data and initial insights produced, as well as the general strategy for exploring biological function at scale.
Limitations of the study
Although the image-based phenotypic profiling strategy described here is applicable to a wide range of phenotypes and cellular models, we chose to focus on the well-characterized, transformed, aneuploid cervical cancer HeLa cell line, which lacks a functional p53 pathway. As the gene targets analyzed in this study are pan-essential, we expect the majority of the observations in HeLa cells to be reflected across most cell lines. However, p53 status plays an important role in modulating the observed phenotypes for some cell cycle factors,27 suggesting that future work should prioritize extending this approach to non-transformed cell lines with intact p53 signaling. Here we also selected a time point (78 hrs) that maximized the observed penetrance of phenotypic defects based on pilot screens (Figure S1A), but we recognize that our results are time point-dependendent. At the later time points required to elicit phenotypes associated with eliminating stable proteins, increased cell death will reduce phenotypic differences between gene knockouts and rising fluorescence background in dying cells will degrade in situ sequencing signals. Finally, although we were struck by how powerfully four cellular stains revealed requirements for diverse cellular processes, the inclusion of additional cellular markers, such as a reporter for mitochondrial function or early-stage apoptosis, would likely provide further increases in phenotypic depth and insights.
In addition to the cell model and time point, the results are sensitive to the methods used to extract information from the cell images. We used previously-defined intensity and morphological metrics to extract information from each stain and each cell at a low computational cost. Although this approach proved exceptionally powerful, more information likely remains latent in the raw images. Other approaches, including training convolutional neural networks to learn cellular phenotype representations,83–85 are well-poised to make additional findings in this dataset. We also focused primarily on robust central tendency statistics, which do not capture biological information contained in the distribution of phenotypes across cells (apparent in Figures S2E, S3C, and S6G). However, sgRNA performance and other technical effects will also contribute to cell-level phenotype variation. Most sgRNAs targeting the same gene in our screens exhibit similar phenotypes (Figures S1E and S6G), but there is value in considering differences that may reflect biologically-important effects of targeting specific functional protein regions.
STAR★Methods
Resource availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Iain Cheeseman (icheese@wi.mit.edu).
Materials Availability
Plasmids generated in this study have been deposited to Addgene (additional details provided in the Key Resources Table).
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-gamma H2A.X (phospho S139) | Abcam | Cat#ab81299 |
| Anti-alpha-tubulin-FITC antibody | Sigma | Cat#F2168 |
| Goat anti-rabbit antibody disulfide-linked
to Alexa Fluor 594 (custom conjugation) |
Invitrogen; Thermo Fisher Scientific |
Cat#21212; Cat#A10270 |
| Rabbit anti-GFP | Cheeseman Lab | N/A |
| Mouse anti-gamma H2A.X (phospho S139) | Millipore | Cat#05636 |
| Rabbit anti c-Myc | Abcam | Cat#ab32072 |
| Rabbit anti-“Bonsai”/NDC80 | Schmidt et al., 201293 | N/A |
| HRP Anti-GAPDH antibody | Abcam | Cat#Ab185059 |
| Mouse Anti-alpha-tubulin | Sigma | Cat#T6199 |
| Rabbit anti-centrin | Backer et al., 201294 | N/A |
| Mouse anti-CENPA | Invitrogen | Cat#MA1–20832 |
| Chemicals, peptides, and recombinant proteins | ||
| Alexa Fluor Plus 750 Phalloidin | Thermo Fisher Scientific | Cat#A30105 |
| Critical commercial assays | ||
| RevertAid H minus Reverse Transcriptase | Thermo Fisher Scientific | Cat#EP0452 |
| Ribolock RNase inhibitor | Thermo Fisher Scientific | Cat#EO0384 |
| RNase H | Enzymatics | Cat#Y9220L |
| TaqIT DNA polymerase | Enzymatics | Cat#P7620L |
| Ampligase | Lucigen | Cat#A3210K |
| Phi29 DNA polymerase | Thermo Fisher Scientific | Cat#EP0091 |
| MiSeq Reagent Nano Kit v2 with PR2 buffer | Illumina | Cat#MS-103–1003 |
| MitoTracker Orange CMTMRos | Invitrogen | Cat#M7510 |
| Deposited data | ||
| Processed images and data | This paper | https://vesuvius.wi.mit.edu |
| Processed single-cell phenotype measurements | This paper | Harvard Dataverse https://doi.org/10.7910/DVN/VYKTI5 |
| Raw image data | This paper | BioImage Archive S-BIAD394 |
| RNA-sequencing data | This paper | Gene Expression Omnibus: GSE199901 |
| Experimental Models: Cell lines | ||
| All cell lines used in this study, see Table S5 | This study | N/A |
| Oligonucleotides | ||
| All oligonucleotides used in this study, see Table S1 | This study | N/A |
| Recombinant DNA | ||
| CROPseq-puro-v2 | Feldman et al., 20198 | #127458 (Addgene) |
| sgOPTI | Fulco et al., 201688 | #85681(Addgene) |
| pBABE H2B-mCherry (pKC96) | This paper | Undergoing submission (Addgene) |
| pBABE GFP-SPC24 (pKG422) | Gascoigne et al., 201187 | Undergoing submission (Addgene) |
| pBABE EGFP-LIN52 (pKC518) | This paper | Undergoing submission (Addgene) |
| pBABE EGFP-ZNF335 (pKC530) | This paper | Undergoing submission (Addgene) |
| Software and algorithms | ||
| General repository used for processing optical pooled screening data | Feldman et al., 202215 |
https://github.com/feldman4/OpticalPooledScreens (https://doi.org/10.5281/zenodo.5002684) |
| Application-specific tools for the screens presented in this paper | This paper |
https://github.com/lukebfunk/OpticalPooledScreens (https://doi.org/10.5281/zenodo.7155628) |
Data and Code Availability
RNA-sequencing data have been deposited at the Gene Expression Omnibus (GEO) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Processed images and data from the screen are available in the supplemental materials and through the companion web portal (https://vesuvius.wi.mit.edu). Full single-cell extracted image phenotype measurements from the primary screen have been deposited at the Harvard Dataverse and are publicly available as of the date of publication. DOIs are listed in the key resources table. Raw image data are being deposited at the BioImage Archive and are publicly available. Accession numbers are listed in the key resources table.
All original code has been deposited at GitHub and Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
Experimental model and subject details
Tissue Culture
HeLa and HEK293 cells were cultured in DMEM with sodium pyruvate and GlutaMAX (Life Technologies 10569044) or 2 mM L-glutamine supplemented with 10% heat-inactivated fetal bovine serum (Sigma F4135) and 100 U/mL penicillin-streptomycin (Thermo Fisher Scientific 15140122). Cells were screened routinely for mycoplasma contamination and validated based on the presence of appropriate cellular behaviors and markers.
Method details
Library design and cloning
The primary screen library of fitness-conferring genes was defined based on evidence from multiple published sources. First, we used data from the Broad Institute DepMap project to identify genes that are broadly fitness-conferring in a variety of cell lines.17,18,21 Specifically, we selected genes with a genetic dependency probability of >0.35 in at least 10% of the >600 tested cell lines, resulting in 3,991 selected genes. We subsequently chose 1,081 additional genes that had evidence of essentiality in at least 2 other published screens.16,19,20,22–24 CRISPR sgRNA sequences were selected from published libraries,24–26 with simultaneous optimization of sgRNA performance (e.g., on- and off-target efficiency) and minimization of 5’ sequence length required to demultiplex all sgRNAs during in situ sequencing. In total, we selected 20,445 sgRNA sequences, including 4 sgRNAs each for all but one gene target (3 sgRNAs targeting RGPD5) and 250 non-targeting control sgRNAs that lack targets in the human genome, with a minimum Levenshtein distance of 2 between the leading 11-nucleotide 5’ sequence for all possible pairs of sgRNAs (Table S1). Although most sgRNAs targeting the same gene display similar phenotypes (Figure S1E), multiple sgRNAs per gene are used to buffer any gene or protein domain-specific targeting effects or variable efficiency. We note that, for some groups of genes with high sequence homology, it is not possible to design distinct targeting sgRNAs for each gene. For groups of genes where the full lists of possible sgRNAs collected from previously published libraries were identical, a single set of 4 sgRNAs was chosen to target these genes collectively (specified as gene identifiers joined by “&” in the supplementary tables). Two sgRNAs per gene were selected for the 239 genes in the live cell screen based on performance in the fixed-cell screen, in addition to 50 non-targeting guides selected using the 5’ sequence optimization described above. Targeting and non-targeting sgRNA libraries were designed as separate subpools of synthesized oligo arrays (Agilent) and independently cloned into CROPseq-puro-v2 (Addgene #127458), which contains an optimized sgRNA scaffold as described previously,8,86 via Golden Gate assembly with Esp3I restriction sites.
For expression of fusion proteins, H2B (pKC96) was amplified from a template retroviral construct87 and SPC24 (pKG422) from pJAG261 (gift from Jagesh Shah), while C7orf26 (NP_076972.2; pKC509) and LIN52 (Q52LA3.1; pKC518) were human codon-optimized and synthesized (Twist Biosciences). Gene fragments were ligated into an mCherry, GFP, or EGFP pBABE-based vector (Addgene #44432). ZNF335 (pKC530) was amplified from Synthetic construct Homo sapiens clone IMAGE:100066405 and ligated into a EGFP lentiviral vector. sgRNA constructs for individual inducible knockout cell lines were generated by primer annealing and ligation into sgOPTI88,89 (see Table S5). A control sgRNA with a single target site within the non-essential LBR gene (HS1)90 was used for comparison of all follow-up experiments.
Virus production, transduction and selection
Prior to lentiviral production of screening sgRNA libraries, the corresponding targeting and non-targeting plasmid pools were mixed (final non-targeting sgRNA pool fraction of 5% for the primary fixed-cell screen, 9.5% for the secondary live-cell screen). Lentiviral production and transduction were performed as described previously for libraries.8,15 Briefly, HEK293FT cells were seeded into 15-cm or multi-well plates at a density of 100,000 cells/cm2. After one day, cells were transfected with pMD2.G (Addgene #12259), psPAX2 (Addgene #12260), and a lentiviral transfer plasmid (2:3:4 ratio by mass) using Lipofectamine 3000 (Thermo Fisher Scientific L3000015). Viral supernatant was harvested 48 hours after transfection and filtered through 0.45 μm cellulose acetate filters (Corning 431220).
Retrovirus was generated by transfecting 2.5 μg VSVG packaging plasmid and 5 μg pBABE-based vectors containing H2B-mCherry, EGFP-C7orf26, EGFP-Lin52, GFP-SPC24 fusions or mNeonGreen into 4 million HEK293-GP cells in 300μl Buffer EC with 16 μl Enhancer and 60μl Effectene (Qiagen 301425) for transduction as described previously.91 Transduced cells were enriched by FACS (GFP-SPC24) or selected with 375 μg/ml hygromycin (Invitrogen).
Fluorescence microscopy
All screening datasets were acquired using a Nikon Ti-2 inverted epifluorescence microscope with automated stage control, hardware autofocus, and an Iris 9 sCMOS camera (Teledyne Photometrics). All hardware was controlled using NIS-Elements AR, and a CELESTA light engine (Lumencor) was used for fluorescence illumination. In situ sequencing cycles were acquired using a 10X 0.45 NA CFI Plan Apo Lambda objective (Nikon MRD00105) and 2×2 pixel binning with the following laser lines, filters, and exposure times for each channel: DAPI (408 nm laser excitation with 0.8% power, custom Chroma dual-band 408/473 dichroic and emission filter set, 50 ms exposure), Miseq G (545 nm laser with 30% power and Semrock FF01–543/3 excitation filter, Chroma T555LPXR dichroic filter, Chroma ET575/30 emission filter, 200 ms exposure), Miseq T (545 nm laser excitation with 30% power, Chroma T565LPXR dichroic filter, Semrock FF01–615/24 emission filter, 200 ms exposure), Miseq A (635 nm laser excitation with 30% power, Chroma ZET635RDC dichroic filter, Semrock FF01–680/42 emission filter, 200 ms), Miseq C (635 nm laser excitation with 30% power, Chroma ZET635RDC dichroic filter, Semrock FF01–732/68 emission filter, 200 ms exposure). Fixed-cell primary screen phenotype images were acquired using a 20X 0.75 NA CFI Plan Apo Lambda objective (Nikon MRD00205) using DAPI (as before), FITC (473 nm laser excitation, custom Chroma 408/473 filter set), Alexa Fluor 594 (same settings as MiSeq T), and Alexa Fluor 750 (750 nm laser excitation, Semrock FF765-Di01 dichroic filter, custom ET820/110 Chroma emission filter) fluorescence channels. For the live-cell secondary screen, timelapse phenotype images were acquired using the 20X objective lens, an mCherry fluorescence channel (same settings as MiSeq T), and a microscope enclosure with temperature and CO2 control along with passive humidification (Okolab H201).
Immunofluorescence images of single knockout cell lines were taken on the Deltavision Ultra (Cytiva) system using a 60x/1.42NA objective and deconvolution. For kinetochore component quantification, z-sections at 0.2 μm intervals were taken using a 100X/1.45NA objective. For time lapse imaging of individual inducible knockouts and EGFP fusion cell lines, we used a Nikon Eclipse microscope equipped with an ORCA-Fusion BT sCMOS camera (Hamamatsu) using a Plan Fluor 20X/0.5 NA (live cells) or 40x/1.3NA (EGFP) objective lens.
Fixed-cell optical pooled CRISPR screen
For the fixed cell screen, HeLa-TetR-Cas9 (A7) cells were transduced with the 20,445 sgRNA library in CROPseq-puro-v2 and selected with 2 μg/mL puromycin (Thermo Fisher Scientific A1113803) for 4 days. Cas9 expression was induced with 2 μg/mL doxycycline for 78 hours, and the cell library was seeded into eight 6-well glass-bottom plates (Cellvis P06–1.5H-N) at a density of 300,000 cells per well (~30,000 cells/cm2) 48 hours prior to fixation. Cells were fixed with 4% paraformaldehyde in PBS for 30 minutes, followed by in situ amplification as described previously.8,15 First, cells were permeabilized with 70% ethanol for 30 minutes. Cells were then carefully exchanged over six washes into PBS-T wash buffer (PBS + 0.05% Tween-20). Reverse transcription mix (1x RevertAid RT buffer, 250 μM dNTPs, 0.2 mg/mL BSA, 1 μM LNA-modified RT primer (oRT_CROPseq-v2), 0.8 U/μL Ribolock RNase inhibitor, and 4.8 U/μL RevertAid H minus reverse transcriptase) was added to the sample and incubated overnight at 37°C. Following reverse transcription, cells were washed 5 times with PBS-T and post-fixed with 3% paraformaldehyde and 0.1% glutaraldehyde for 30 minutes at room temperature. Cells were then washed with PBS-T 5 times before incubation in a padlock probe and gap-filling reaction mix (1x Ampligase buffer, 0.4 U/μL RNase H, 0.2 mg/mL BSA, 100 nM padlock probe (oPD_CROPseq-v2), 0.02 U/μL TaqIT polymerase, 0.5 U/μL Ampligase and 50 nM dNTPs) for 5 minutes at 37°C and 90 minutes at 45°C, and then washed 2 times with PBS-T. Circularized padlocks were then amplified using rolling circle amplification (1x Phi29 buffer, 250 μM dNTPs, 0.2 mg/mL BSA, 5% glycerol, and 1 U/μL Phi29 DNA polymerase) overnight at 30°C and washed 2 times with PBS-T. After rolling circle amplification, cells were stained with rabbit anti-gamma H2A.X (phospho S139) antibody (Abcam ab81299, 1:2000 dilution in PBS with 3% BSA) for 1 hour at room temperature. Cells were washed twice with PBS-T (PBS with 0.05% Tween-20), then stained with mouse anti-alpha-tubulin-FITC antibody (Sigma F2168, 1:500 dilution), goat anti-rabbit antibody disulfide-linked to Alexa Fluor 594 (Invitrogen 31212, Thermo Fisher Scientific A10270, custom conjugation; 1:500 dilution), and Alexa Fluor Plus 750 Phalloidin (Thermo Fisher Scientific A30105, 1:1000 dilution) in PBS with 3% BSA for 45 minutes at room temperature. After washing with PBS-T three times, well plates were replaced with 200 ng/mL DAPI in 2X SSC and imaged for cellular phenotypes using the microscope configuration described above with 4 z-slices at 1.5 μm intervals. Following phenotype imaging, Alexa Fluor 594 was cleaved from disulfide-linked antibodies by incubating cells in 50 mM TCEP in 2X SSC for 1 hour at room temperature, followed by three washes with PBS-T. Finally, 11 cycles of in situ sequencing-by-synthesis were performed as described previously.8,15 A sequencing primer was first hybridized for 30 minutes at 37°C (1 μM primer oSBS_CROPseq-v2 in 2X SSC). sgRNA sequences were read out in situ using sequencing-by-synthesis reagents from the Illumina MiSeq 500 cycle Nano kit (Illumina MS-103–1003). Samples were washed with incorporation buffer (Nano kit PR2) and incubated for 3 minutes in incorporation mix (Nano kit reagent 1) at 60°C on a flat-top thermocycler. Samples were then repeatedly washed with PR2 at 60°C (6 washes for 5 minutes each) and placed in 200 ng/mL DAPI in 2x SSC for fluorescence imaging. Following each cycle of imaging, samples were incubated for 6 minutes at 60°C in Illumina cleavage mix (Nano kit reagent 4), and thoroughly washed with PR2. In parallel with the optical pooled screen, cells expressing the same sgRNA library were induced with 1 μg/mL doxycycline, and then doxycycline media was refreshed every day for 2 more days. Cells were harvested on days 0 (pre-induction), 3, and 5 post-Cas9 induction and genomic DNA was extracted using PureLink (Invitrogen). sgRNA sequences were then PCR amplified using Q5 hotstart (NEB) with primers oDF344 and oDF112 before addition of index barcodes and sequencing on an Illumina HiSeq using sequencing primer oKC651. See Table S1 for sequence of all primers used.
Live-cell optical pooled CRISPR screen
HeLa-TetR-Cas9 cells expressing an H2B-mCherry fusion protein (cKC556) were transduced with the live-cell screening library of 526 sgRNA sequences in CROPseq-puro-v2. Cells were selected with 2 μg/mL puromycin (Thermo Fisher Scientific A1113803) for 3 days. Cas9 expression was induced with 2 μg/mL doxycycline for 48 (day 3 time course) or 72 hours (day 4 time course) prior to the beginning of live-cell imaging, and cells were seeded into 6-well glass-bottom plates (Cellvis P06–1.5H-N) at a density of 300,000 or 350,000 cells per well (~35,000 cells/cm2) 24 hours prior to imaging. Each time course was performed in three batches on separate days. Immediately before imaging, cells were washed once with PBS, and then replaced with imaging media consisting of phenol red-free DMEM with L-glutamine and HEPES (Thermo Fisher Scientific 21063029) supplemented with 10% heat-inactivated fetal bovine serum (Sigma F4135) and 100 U/mL penicillin-streptomycin (Thermo Fisher Scientific 15140122). Live-cell imaging was performed using the microscope configuration described above and 2 z-slices spaced at either 4 or 5 μm intervals. Cells were imaged for 24 hours at 10 minute time intervals, immediately fixed with 4% paraformaldehyde in PBS for 30 minutes, then processed through in situ amplification and sequencing-by-synthesis following the same protocol as the fixed-cell screen.
GFP immunoprecipitation and Mass-spectrometry
IP-MS experiments were performed as described previously.92 EGFP-C7orf26 and EGFP-LIN52 cells were mitotically enriched with 10μM STLC overnight, harvested and washed in PBS and resuspended 1:1 in 1X Lysis Buffer (50 mM HEPES, 1 mM EGTA, 1 mM MgCl2, 100 mM KCl, 10% glycerol, pH 7.4) then frozen in liquid nitrogen. Cells were thawed after addition of an equal volume of 1.5X lysis buffer supplemented with 0.075% Nonidet P-40, 1X Complete EDTA-free protease inhibitor cocktail (Roche), 1 mM phenylmethylsulfonyl fluoride, 20 mM beta-glycerophosphate, 1 mM sodium fluoride, and 0.4 mM sodium orthovanadate. Cells were then lysed by sonication and cleared by centrifugation. The supernatant was mixed with Protein A beads (Biorad) coupled to rabbit anti-GFP antibodies (Cheeseman lab) and rotated at 4°C for 1 hour. Beads were washed five times in wash buffer (50 mM HEPES, 1 mM EGTA, 1 mM MgCl2, 300 mM KCl, 10% glycerol, 0.05% NP-40, 1 mM dithiothreitol, 10 μg/mL leupeptin/pepstatin/chymostatin, pH 7.4). After a final rinse in wash buffer without detergent, bound protein was eluted with 100 mM glycine pH 2.6. Eluted proteins were precipitated by addition of 1/5th volume trichloroacetic acid at 4°C overnight. Precipitated proteins were reduced with TCEP, alkylated with iodoacetamide, and digested with mass-spectrometry grade trypsin (Promega) using S-Trap (Protifi) according to the manufacturer’s instructions. Peptides were separated by liquid chromatography and analyzed on an Orbitrap Elite mass spectrometer (Exploris 480, Thermo Fisher) with FAIMS Pro Interface (Thermo Fisher). Data were analyzed using Proteome Discoverer Software (Thermo Fisher).
Western Blotting
Cells expressing individual sgRNAs were induced in 1 μg/mL doxycycline for 2 to 5 days before lysis in Laemmli buffer and incubation at 95°C for 5 min. For mitotic samples, cells were harvested by mitotic shake off and, when necessary, after an overnight 10 μM STLC incubation. Samples were separated by SDS-PAGE and semi-dry transferred to nitrocellulose. Membranes were blocked for 30 min in blocking buffer (5% BSA for H2A.X; for all others, milk in TBS with 0.1% Tween-20) before incubation with primary antibodies: anti-phospho-H2A.X (Ser139, Millipore clone JBW301; 1:1000), anti c-Myc (Abcam, ab32072; 1:1000), anti-CENP-A (Clone 3–19, Invitrogen; 1:2000), or anti-“Bonsai”/NDC8093 (0.5 μg/mL). This was followed by HRP-conjugated secondary antibody (Kindle Biosciences) incubation at 1:1000 dilution. To detect GAPDH as a loading control, HRP-conjugated antibody (Abcam, ab185059) was applied at 1:20,000 dilution. Membranes were imaged with a KwikQuant Imager (Kindle Biosciences) and quantified using Image Studio software (LI-COR).
Arrayed imaging experiments with inducible knockout cell lines
Inducible knockout cell lines for immunofluorescence were seeded on poly-L-lysine (Sigma-Aldrich) coated coverslips and fixed in PHEM with 4% formaldehyde for 10 min at 37°C (microtubule staining) or ice cold methanol. Coverslips were washed with PBS, permeabilized with 0.2% Triton X-100 in PBS, and blocked in Abdil buffer (20 mM Tris-HCl, 150 mM NaCl, 0.1% Triton X-100, 3% bovine serum albumin, 0.1% NaN3, pH 7.5). Anti-alpha-tubulin (DM1A, Sigma; 1:3000 dilution), anti-Centrin94 (1μg/mL), anti-CENP-A (Clone 3–19, Invitrogen; 1:1000 dilution) and anti- “Bonsai”/NDC8093 (1 μg/mL) antibodies in Abdil buffer were used for primary staining. Cy2- and Cy5-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories) were diluted 1:500 with 1 μg/mL Hoechst-33342 (Sigma-Aldrich) in Abdil for subsequent staining. Slides were mounted with ProLong Gold Antifade (Invitrogen) prior to imaging using the microscope configuration described above.
For quantifications of Ndc80 and CENP-A kinetochore stain intensity, sections of cells were maximum intensity projected and cropped in Fiji.95 Integrated fluorescence intensity of mitotic kinetochores was measured with a custom pipeline in CellProfiler.96 The median intensity of a 5-pixel wide region surrounding each kinetochore was used to background subtract each measurement.
For live analysis of individual knockout and RNPC3 rescue cell lines, cells were induced with 1 μg/mL doxycycline for 3 days, refreshing doxycycline media each day. On day 3 or 4 post-Cas9 induction, cells were moved to CO2-independent media (Gibco) supplemented with 10% FBS, 100 U/mL penicillin and streptomycin, and 2 mM L-glutamine before imaging using the microscope configuration described above in 12-well polymer-bottomed plates (Cellvis). For the hyperosmotic stress experiments, polyethylene Glycol (PEG) 300 (TCI) was applied to the media at the indicated concentrations (w/v%) and incubated for 6 h prior to imaging. For imaging of active mitochondria with intact membrane potential in Figure S5F, MitoTracker Orange CMTMRos (Invitrogen) was applied at 25 nM for 30 min before imaging. For MitoTracker image analysis, nuclei were segmented using the CellPose segmentation algorithm97 with a Hoechst stain, then a cytoplasmic ring was defined by morphologically dilating the nuclei by 10 pixels. The stain was then quantified by taking the mean intensity within the ring area, normalized by the mean intensity of the nucleus for each cell.
RNA-sequencing
Inducible knockout cells were seeded in 1 μg/mL doxycycline, and doxycycline media was refreshed each day for 3 days before harvest of a mitotically-enriched cell population by shake-off on day 5. Control and ZNF335 knockout cells were additionally treated with 10 μM STLC for 12 h prior to harvest. Cells were washed in PBS before snap-freezing pellets of 500,000 cells in liquid nitrogen. RNA was purified using TRIzol reagent (Life Technologies) according to manufacturer’s instructions. 2ug of purified total RNA was mixed with 0.7 pg and 1.3 pg polyadenylated Nano luciferase and Firefly luciferase spike-in mRNA, respectively. KAPA mRNA HyperPrep kit with poly(A) selection was used to prepare libraries. Libraries were sequenced with the Illumina NovaSeq 6000 platform, 100×100 bp paired-end reads.
Reverse transcription and qPCR
Total RNA was purified as described for RNA-sequencing. 1 μg of total RNA was used in a cDNA synthesis reaction with the Maxima First Strand cDNA synthesis kit (Thermo Scientific) according to the manufacturer’s protocol. The cDNA was subjected to qPCR using the PowerUP SYBR Green Master Mix (Thermo Fisher) according to the manufacturer’s protocol or end point PCR using 2x Q5 polymerase mix (NEB). For qPCR, a standard curve was used for quantitative assessment of mRNA levels and normalized to GAPDH mRNA. See key resource table for sequences of qPCR primers used.
Quantification and statistical analysis
Screening image analysis
In situ sequencing spots were identified and barcode sequences extracted using our previously described workflow.8,15,98 Briefly, sequencing reads were detected by applying a Laplacian-of-Gaussian linear filter (kernel width σ = 1 pixel) with sequencing spots identified by finding the local minima of the per-pixel standard deviation over sequencing cycles. A linear transformation was estimated and applied to the extracted per-channel spot intensities to correct for fluorescence cross-talk between sequencing channels. Finally, each base was called according to the sequencing channel with maximum corrected intensity in a given cycle of sequencing. In addition, phenotype images were acquired at a higher magnification than in situ sequencing images, and thus the datasets were computationally aligned to match cell identities. This alignment was completed by computing the Delaunay triangulation of nuclei centroids for each phenotype and sequencing image tile, and then computationally comparing triangulations between images from the two datasets to find matching tiles and cell identities. Overall, approximately 60% of all segmented cells were included in the final dataset (Figure S1C), with remaining cells unused due to no in situ sequencing spots matching a designed sgRNA sequence, sequencing reads mapping to multiple gene targets, or an inability to match cell identities between the sequencing and phenotype images.
Phenotype images from both screens were first maximum intensity projected to compress z-slices into a single plane, and then a retrospective flat-field correction was applied to reduce effects from uneven illumination.99 Nuclei were semantically segmented by applying a local intensity threshold to the DAPI channel and then performing morphological operations to remove aberrant holes and particles. Individual nucleus instances were then segmented using the watershed algorithm. In the fixed-cell phenotype data, semantic segmentation of cytoplasmic foreground was achieved by thresholding a Gaussian-filtered copy of the phalloidin (actin) channel (sigma of 3 pixels), followed by morphological operations. Cell instances were identified by applying the watershed algorithm with nuclear segmentations as seeds. Phenotype parameters were extracted from nuclear and cellular segmentations for each channel by implementing image features derived from CellProfiler,96 scikit-image,100 and mahotas101 as Python functions operating on scikit-image RegionProperties objects (Table S2). Image segmentation, phenotype feature extraction, and in situ sequencing analysis were performed in parallel on a per-image tile basis using the Snakemake workflow manager.102
Fixed-cell screen phenotype analysis
After aligning the phenotype and sequencing datasets, a subset of features were transformed to approximate normal distributions (Table S2). All features for each cell were then normalized using the median and median absolute deviation of the population of cells carrying non-targeting sgRNAs within the same well (robust z-score). This internal control procedure was used to reduce batch effects between wells and plates that may be caused by intensity differences or cell density effects. Mitotic and interphase cells were identified using a support vector classifier (scikit-learn103 svm.SVC implementation, default parameters) trained with 2,514 annotated cells on a subset of 182 features that demonstrated the highest average difference between annotated mitotic and interphase cells (Figure S1D; Table S2). Cell-level measurements were then re-normalized from the raw data as before, but within interphase and mitotic cells separately.
Summary phenotype measurements were computed for each gene target by taking the median of z-scored parameters for all cells targeted by a single sgRNA sequence, then aggregating to the gene level by taking the median across sgRNAs targeting the same gene. Raw p-values for a subset of summary parameters were computed by comparing gene scores to null distributions of corresponding bootstrapped summary scores from cells expressing non-targeting sgRNAs, a nonparametric approach that does not depend on strong assumptions concerning the underlying distributions of the data. Separate null distributions were defined for each gene target by first performing 100,000 cell sampling repetitions to produce a distribution of bootstrapped non-targeting sgRNA scores for each cell sample size of the targeting sgRNAs. Cell sample sizes for each gene are reported in Table S2. These guide-level null distributions were then correspondingly sampled 100,000 times for each group of sgRNAs targeting the same gene and aggregated to produce gene-level null distributions with matched cell and guide sample sizes. The Benjamini-Hochberg procedure was applied to obtain the reported FDR q-values. As indicated in the figure legends, an FDR threshold of 0.05 was used for defining significance for all parameters. This process was modified for the mean nuclear 𝛾H2AX intensity measurements (Figure 1D), as the non-targeting cells do not provide an adequate null population for this phenotype given the lack of Cas9-induced DNA breaks. In this case, bootstrapped null distributions were generated by sampling from all cells expressing any targeting sgRNA, as indicated in the figure legend, resulting in the center of the volcano plot being offset from 0 as robust z-scores (x-axis) are calculated relative to the non-targeting sgRNA cell population. This approach, combined with an effect size threshold at the 2.5 and 97.5 percentiles of the non-targeting sgRNA scores, resulted in a conservative identification of mean nuclear 𝛾H2AX intensity phenotypes beyond baseline DNA damage resulting from Cas9 nuclease activity.
For the high-dimensional analysis, features with a Pearson correlation greater than 0.9 with another feature were iteratively excluded, and additional features with low variance or only a few unique discrete values across the dataset were removed. This resulted in a set of 475 features for the interphase dataset and 884 features for the mitotic dataset, selected independently from the full list of 1,084 extracted features (Table S2). Further feature redundancies were reduced by applying principal component analysis (PCA) and retaining the components that explain 95% of the variance in the datasets (103 components for the interphase data, 530 components for the mitotic data). The PHATE manifold learning and visualization algorithm43 was then used to produce two-dimensional representations of the phenotypic landscape of gene targets from the PCA-projected feature profiles (default parameters except n_pca=None). For a summary score of phenotype strength, the PHATE potential distance matrix was used to calculate the average distances between each gene phenotype profile and non-targeting control sgRNAs profiles, which were then min-max normalized between 0 and 1. To cluster knockout phenotypes, the diffusion operator local affinity graph produced as an intermediate of PHATE was supplied as input to the Leiden algorithm, a standard and well-developed clustering approach that optimizes cluster modularity.44 The Leiden resolution parameter was chosen by analyzing the robustness of clustering solutions to the subsampling of gene-level data with varying resolution (robustness plateaued at resolution = 10 for the interphase profiles, resolution = 9 for the mitotic profiles). Cluster annotations in Fig. 3B represent manually-identified groups of Leiden clusters corresponding to broad functional categories, as indicated in the legend. In cases where hierarchical sub-clustering is used as a straightforward approach to identify phenotype relationships between individual gene targets (e.g., Figure 4C), this was performed using average linkage (UPGMA) of the Pearson correlation between PCA-projected phenotype profiles. For visualization of differences between phenotype profiles and corresponding clusters in the presented heatmaps (Figures 3D and 3F, etc.), we first selected clusters to highlight based on the presence of known functional groups of genes, then iteratively selected a minimal set of phenotype parameters that together discriminated the various clusters. Features with clear explanation were prioritized to enable interpretability, resulting in 16 interphase and 16 mitotic phenotype features (Table S2). Feature values in all heatmaps are presented as z-scores from the distribution of non-targeting sgRNAs, visualized on a symmetric log scale (linear between −1 and 1). In parallel with the computational phenotype analysis, two individuals independently scored mitotic phenotypes from the primary screen by visually inspecting montages of mitotic cells from each gene target and assigning a phenotype severity score from 1 to 9 (Figures S6A–B). During this process, the scoring individuals were blinded to the gene identities associated with each montage of cells.
Comparisons to external data
To compare our interphase phenotype profile similarities to existing datasets of co-functional genes (Figures S4H and S5A–D), we used the CORUM 3.0 core set of protein complexes,47 BioPlex 3.0 HEK293T interactions,45 co-essential gene pairs defined in Wainberg et al.,46 KEGG pathways (accessed March 30, 2022),48 STRING v11.5 protein links,49 and data from the recent large-scale Perturb-seq screen.5 In all analyses of KEGG pathways, pathways categorized under “Organismal Systems,” “Human Diseases,” or “Drug Development” were excluded. For CORUM and KEGG, all possible pairs of genes within the same complex were considered as co-functional. For the Perturb-seq cluster comparison, we included both the annotated genes associated with each cluster as well as “nearest neighbor” genes listed in the supplemental data of Replogle et al.5 In each dataset, all present annotated co-functional gene pairs were included for Figure S4H, and correlations were computed between phenotype profiles in the same form as the input to PHATE (first 103 principal components). For cluster enrichment of CORUM complexes, complexes were included that contained at least 3 genes from the screening library with at least 2/3 of the full complex represented (Table S3). Cluster enrichment for KEGG and CORUM annotations for Figure S5A and Table S3 was determined using Fisher’s exact test for each annotation and cluster, with the background set as all genes targeted in the screen. The Benjamini-Hochberg procedure was applied across tested annotation sets for each cluster individually, with an FDR cut-off of 0.05. Enrichment of BioPlex, co-essentiality, and STRING associations within each cluster was determined using a permutation test. 10,000 repetitions of cluster label permutations were performed to generate null distributions of intra-cluster associations, and a p-value threshold of 0.05 was applied to determine significance.
For the CORUM correspondence analysis (Figure S5B), a subset of minimal CORUM complexes were selected that contained limited overlap with other complexes (defined as containing at least 3 genes from the screening library with at least 2/3 of the full complex represented, and removing the largest complexes that share more than 10% of gene-pairs with smaller CORUM complexes; see Table S3), resulting in 9,781 gene pairs included across 292 annotated complexes. Precision and recall of correspondence were defined as indicated in Figure S5B, with the Leiden clustering resolution parameter varied between 0.1 and 1000 to generate curves. When restricting this comparison to varying overall phenotype strengths, the indicated quantiles were used as thresholds on the interphase PHATE mean potential distance to non-targeting sgRNAs (as also presented in Fig. S4G), only evaluating pairs of genes where both members met the given threshold. For comparisons to the DepMap-based co-essentiality clusters46 and the Perturb-seq clusters,5 due to the nature of the available date we conducted qualitative comparisons of the genes present in each cluster.
For the comparison to MitoCheck, all genes identified in at least one of the four main phenotype categories (“mitotic arrest/delay,” “binuclear,” “polylobed,” and “grape”) were considered as exhibiting mitotic phenotypes. These genes were compared to those in our fixed-cell screen demonstrating strong image-based mitotic phenotype profiles (using the mean PHATE potential distance to non-targeting sgRNAs with mitotic profiles analogously to Figure 5A, selecting gene targets above the 95th percentile of non-targeting sgRNAs) or significantly altered mitotic index (P<0.05 by permutation test with 10,000 permutations of sgRNA-gene assignments; Figure S6C; Table S3).
Live-cell screen phenotype analysis
Following nuclear segmentation of the time lapse data, cells were tracked across frames using the TrackMate implementation of the linear assignment problem approach to tracking.104,105 The cost of linking nuclei in consecutive frames was set as the squared distance between centroids, with maximum linking distance set to 60 pixels (~18 μm). Track gaps up to 2 frames were allowed, in addition to track merges and splits. Tracked cell lineages that did not last for the full 24 hour time-course were excluded from analysis.
The sgRNA assigned to each tracked cell lineage in the phenotype data was determined by matching cell identities between the in situ sequencing images and the final time point of the time course. Similar to the fixed-cell screen, individual cell feature measurements were normalized using the median and median absolute deviation of the non-targeting control cell population from the same well and time point to reduce batch effects and correct for temporal intensity variations. Interphase, mitotic, and apoptotic cells were classified using a support vector machine (scikit-learn svm.SVC, linear kernel) with 2,514 annotated cells using 81 features selected from the full set of 116 extracted features by iteratively removing features with Pearson correlation >0.9 with another feature (Figure S6E; Table S4). However, due to the difficulty of separating mitotic and apoptotic cells based on H2B-mCherry fluorescence alone, these categories were later combined into a single, broad “mitotic” bin. Cell division events were defined as a contiguous sequence of at least 2 frames of mitotic-classified cells immediately followed by a split in the track into 2 daughter cells. Also included as cell division events were continuous sequences of mitotic cells that start in the first frame or reach the end of the acquired time course, if the observed mitotic duration was at least as long as the average mitotic duration of non-targeting control cells in the same well. Mitotic duration was measured as the time difference between the first and last frame of the cell division event. The fraction of cells entering mitosis was calculated as the fraction of tracked lineages containing at least one cell division event as defined above. Both measurements were aggregated to the gene level by taking the average of sgRNAs targeting the same gene. Since many genes exhibited a stronger phenotype at either the Day 3 or Day 4 time point, likely due to differences in protein depletion timing, the strongest phenotype was selected for plotting in Figure 6B by selecting the time course with the highest absolute difference in mitotic duration compared to the mean of non-targeting sgRNAs. Sample sizes for each gene are reported in Table S4.
RNA-sequencing analysis of inducible knockout cell lines
RNA-sequencing reads were trimmed to remove any poly(A) sequences using Cutadapt106 (v3.7) with the parameters “--minimumlength 1 -a A{25}”. Reads were mapped to the human genome (Gencode v25) using STAR107 v2.7.1a with the parameters “--runMode alignReads --outFilterMultimapNmax 1 --outFilterType BySJout --outSAMattributes All --outSAMtype BAM SortedByCoordinate”. Aligned reads were quantified using htseq-count108 (0.11.0). A read cutoff of at least 20 reads for each gene sample was applied before further analysis. Differential expression analysis was performed using DESeq2.109 Differentially-expressed genes are defined as log2 effect size > 0.5 and FDR < 0.01. For the Clp1 RNA-seq data, the relative abundance of spike-in mRNAs was used as the sizeFactor for DESeq2 instead of median normalization due to 3’ end processing defects resulting in global mRNA downregulation. The Minor Intron Database110 (v1.2) was used to reference minor intron containing genes.
ShinyGO111 v0.75 was used to identify enriched GO terms from the GO Biological Process database. Enrichment analysis was performed within the downregulated genes from LIN52 knockout cells; genes that surpassed the 20 read cutoff and did not show differential expression were used as the background set.
Clp1 and RNPC3 meta plots were generated using the deepTools v3.5 package.112 The aligned reads were converted to RPKM-normalized coverage using bamCoverage with the parameters “--outFileFormat bigwig --normalizeUsing RPKM --binSize 1”. For analysis of transcription termination, 500bp upstream of the transcription termination site from longest isoform per gene was used for the annotation file for meta plots and the following parameters were used for computeMatrix “--binSize 1 --regionBodyLength 300 --downstream 1000” to generate a matrix of coverages. For analysis of minor introns, the following parameters were used for computeMatrix “--binSize 1 --regionBodyLength 100 --upstream 100 --downstream 100” with the minor intron annotation file from Minor Intron Database v1.2.110 Normalized coverage per bin was obtained using plotProfile with the parameter “--averageType mean” and the average coverage of two biological replicates was plotted. For minor intron analysis, the RPKM-normalized coverage in all bins was further normalized to the summed flanking exon coverage to correct for the decreased mRNA abundance of minor intron containing genes in RNPC3 knockouts.
Supplementary Material
Figure S1. Optimization of image-based pooled screening for essential gene function, Related to Figure 1. (A) Scatter plot showing the results from trial screens of 400 gene targets. This compares the fraction of mitotic cells with visually-identified phenotypic defects for established cell division factors at 3 and 4 days post-Cas9 induction. Overall, mitotic phenotypes were more commonly observed at the earlier time point. (B) Scatter plot showing mean change in abundance within the 20,445 sgRNA primary screen library at 3 and 5 days post-Cas9 induction, both time points relative to pre-induction (day 0). N=2 screen replicates were performed, averaged across sgRNAs targeting the same gene. Orange indicates non-targeting control sgRNAs. Many gene targets begin to drop out of the population at day 5 due to fitness defects. Based on these data and from (A), 78 hours post-Cas9 induction was chosen as the fixation time point for our image-based screen to maximize observable phenotypes. (C) Boxplot demonstrating in situ sequencing quality in our fixed-cell image-based pooled screen. Sequencing quality was consistent across the eight imaging plates, with the majority of imaging tiles exceeding 50% of cells with sequencing reads that uniquely match a single sgRNA sequence from the library. N = 1,665 or 1,998 imaging tiles in each plate column. Whiskers extend to 1.5 times the interquartile range. (D) Confusion matrix demonstrating performance of the support vector classifier in distinguishing interphase and mitotic cells, 5-fold cross-validation with N=2,514 manually annotated cell images. (E) Histograms indicating the distribution of phenotype similarities (correlation of phenotype profiles after principal component projection) between sgRNAs targeting the same gene (blue) or between sgRNAs targeting different genes (orange). Overall, sgRNAs targeting the same gene display more similar phenotypes to than sgRNAs targeting different genes.
Figure S2. Analysis of interphase nuclear phenotypes, Related to Figure 1. (A) Cumulative distribution plots of mean nuclear γH2AX intensity (DNA damage phenotype) sgRNA scores, with sgRNAs grouped by number and location of target sites. Non-targeting control sgRNAs and sgRNAs targeting a single genomic locus (blue) include the vast majority of sgRNAs in the library and displayed minimal DNA damage on average in the screen. In contrast, sgRNAs with increasing numbers of target sites (orange, green) tend to display stronger DNA damage phenotypes, in particular when the target sites are spread across multiple chromosomes (dotted lines). Genomic target sites are defined as the total number of cutting frequency determination (CFD) bin 1 matches (see Doench et al.25). *P<10−9 by 1-sided Mann-Whitney U test. (B) Bar graph indicating over-representation of KEGG pathways among gene targets exhibiting decreased or increased nuclear γH2AX mean intensity. *FDR<0.05 (C) Bar graph of over-representation analysis results as in (B) among gene targets with decreased or increased nuclear DNA (DAPI) integrated intensity. *FDR<0.05 (D) Scatter plot showing summary gene scores (see STAR Methods) for integrated nuclear DNA (DAPI) intensity compared to nuclear area. DNA content is relatively constant across gene targets exhibiting a range of nuclear areas, although a subset demonstrates increased nuclear area and DNA. Summary DAPI scores are plotted on a symmetric log scale (linear between −1 and 1) and labeled genes are colored by functional category. (E) Bivariate histograms of integrated nuclear DNA intensity and mean nuclear γH2AX intensity, displaying single-cell distributions for all cells expressing non-targeting sgRNAs (top left) and selected gene targets. Knockouts of genes that regulate chromosome segregation or cytokinesis result in more cells with increased DNA content, but only modest increases in γH2AX intensity. Histogram bins containing less than 1 in 104 of the total cells for a given gene target are not displayed.
Figure S3. Analysis of interphase cytoskeletal and morphological phenotypes, Related to Figure 2. (A) Scatter plot comparing mean cellular actin intensity summary scores between interphase and mitotic cell populations, indicating factors that robustly affect actin structures throughout the cell cycle. (B) Scatter plot indicating summary gene scores (see STAR Methods) for mean cellular actin intensity compared to cell area. A subset of gene knockouts display increased actin staining together with decreased cell area due to disrupted cellular adhesion. Labeled genes are colored by functional category. (C) Bivariate histograms of mean cellular actin intensity and cellular area, displaying single-cell distributions for all cells expressing non-targeting sgRNAs (left) and selected gene targets. Knockouts of genes that regulate cellular adhesion (e.g., ITGAV) show a distribution of cells shifted toward lower cellular area and correspondingly increased mean actin intensity. Histogram bins containing less than 1 in 104 total cells for a given gene target are not displayed. (D) Scatter plot showing summary gene scores for mean cellular tubulin intensity compared to cell area as in (B). Similar to actin, a subset of gene knockouts display increased tubulin staining in combination with decreased cell area. (E) Volcano plot for interphase cell area across gene targets in the screen, showing a wide range of decreased (magenta) and increased (green) cell areas (FDR<0.05). Raw P-values were computed by comparing gene targets to a bootstrapped null distribution of cells expressing non-targeting sgRNAs (see STAR Methods), with false discovery rate (FDR) estimated using the Benjamini-Hochberg procedure. (F) Bar graph of KEGG pathway over-representation analysis for gene targets that result in decreased or increased cell area. *FDR<0.05. (G) Example images and cumulative distributions of individual inducible knockout cell lines validating increased cell area for DTL and DONSON knockouts. *P<10−10 by Mann-Whitney U test relative to control sgRNA, N>3,700 cells per gene target. Scale bar, 10 μm.
Figure S4. Interphase phenotypes enable detailed clustering of specific functional categories, Related to Figure 3. Two-dimensional PHATE representations of interphase phenotype clusters and corresponding heat maps of a manually-selected subset of phenotype parameters as in Figures 3C–F for genes involved in (A) protein degradation (including the recently-characterized gene AKIRIN2), (B) RNA processing, (C) DNA replication and DNA damage, (D) cell cycle processes, (E) actin cytoskeleton and cellular adhesion, and (F) vesicle trafficking and related processes. The right plot in (C) demonstrates that most genes targeted by sgRNAs with excess target sites show related DNA damage phenotypes. Numbers indicate individual interphase cluster identities. All genes from selected clusters are listed below each heatmap. Parameters are presented as z-scores from the distribution of non-targeting sgRNAs, visualized on a consistent symmetric log scale (linear between −1 and 1). (G) Bivariate histogram showing the joint distribution of image-based interphase phenotype strength relative to non-targeting control sgRNAs computed by PHATE together with the strength of knockout fitness effect in the screening cell line (mean change in sgRNA abundance within the library after 5 days of Cas9 induction, N=2 screen replicates averaged across sgRNAs targeting the same gene, data from Figure S1B). >90% of gene targets exhibit a measurable interphase phenotype in the image-based screen (phenotype strength greater than the 95th percentile of non-targeting sgRNAs). Of the remaining 407 genes, only 55 demonstrate a meaningful fitness effect in the tested cell line (log2 fold change abundance less than the 5th percentile of non-targeting sgRNAs). Labeled genes are those that display a fitness effect and no interphase phenotype, but do show a measurable mitotic phenotype using the same method (not displayed). (H) Distributions of the Pearson correlation between image-based interphase phenotype profiles from the primary screen for all gene pairs (gray) or gene pairs annotated as co-functional in the labeled external databases (orange), indicating increased phenotype similarity between known co-functional genes across the full dataset. Note that this phenotype similarity is weaker among KEGG pathways, likely due to the broad pathway definitions and inclusion of many environmental response pathways not critical at the basal growth conditions tested in our screen.
Figure S5. Validation of interphase phenotypes and clusters, Related to Figures 3 and 4. (A) (A) Pie chart showing the fraction of genes present in clusters enriched for at least 2 of the comparison datasets (blue), or in clusters enriched for a single dataset. (B) Precision and recall of correspondence between our interphase phenotype clustering results and CORUM protein complex annotations across clustering resolution. A subset of 292 minimal CORUM complexes was used for comparison (see Methods), and comparisons were restricted to varying overall interphase phenotype strength as indicated. Error bars indicate the standard deviation of 10 clustering iterations with different random seeds. (C) Venn diagrams representing a qualitative comparison of similar clustering behaviors for co-functional genes that comprise specific protein complexes identified based on our interphase phenotype analysis and from the large-scale Perturb-seq screen performed by Replogle et al.5 (D) Venn diagrams as in (C), demonstrating the improved ability to identify and sub-categorize ribosome-related genes in our dataset in comparison to Replogle et al.5 (E) Interphase phenotype clustering identified a relationship between mitochondrial function and KRAS/BRAF signaling in cluster 149. (F) Example images and cumulative distributions of mitochondrial membrane potential dependent staining (MitoTracker) for control and knockout cells for KRAS, BRAF, and two co-clustering mitochondrial factors. Each knockout demonstrates disrupted mitochondrial activity as compared to control cells, supporting the phenotypic association of KRAS and BRAF with mitochondrial factors in the screen. *P<10−10 by Mann-Whitney U test relative to control sgRNA, N>1,400 cells per gene target. (G) Western blot MYC protein expression following knockout of several genes from cluster 121 (see Figures 4A–B). (H) Interphase phenotype clustering suggests a co-functional role of the RNA binding protein HNRNPD with METTL3 and METTL14, which form m6A modifications on RNA.
Figure S6. Fixed-cell mitotic phenotype analysis and pooled live cell screen, Related to Figures 5 and 6. (A-B) Box plots demonstrating strong agreement between manual phenotype scores by two individuals (blinded to gene labels) and computational phenotype strength (mean potential distance to non-targeting sgRNAs from mitotic PHATE analysis, normalized between 0 and 1). Whiskers extend to 1.5 times the interquartile range. (C) Comparison of mitotic phenotype genes identified in our fixed-cell screen to mitotic phenotypes found by MitoCheck.7 Mitotic phenotypes in the fixed-cell screen were defined as overall phenotype strength greater than the 95th percentile of non-targeting sgRNAs or significantly altered mitotic index (sgRNA permutation test, P<0.05). Bottom, bar graphs indicating KEGG pathway enrichment for all pathways enriched in any of the indicated gene sets. For the gene sets corresponding to the Venn diagram, the background for enrichment analysis was the intersection of all genes present in both screens. For the set of mitotic hit genes identified in MitoCheck that were not present in our screen (far right), the background for enrichment was all genes screened in MitoCheck not present in our screen. *FDR<0.05. (D) Example image indicating nuclear localization of GFP-tagged ZNF335. Scale bar, 10 μm. (E) Confusion matrix demonstrating performance of the support vector classifier in distinguishing interphase, mitotic, and apoptotic cells from the live-cell screen. 5-fold cross-validation with N=2,514 manually annotated cell images. Due to the relative difficulty of differentiating mitotic and apoptotic cells from H2B-mCherry fluorescence alone, the mitotic and apoptotic classes were combined after inference (cross-validation precision and recall after combining classes indicated by brackets). (F) Histograms of the total cell divisions observed across both day 3 and day 4 time courses for each gene target. (G) Cumulative distributions of mitotic duration for individual sgRNAs (blue, orange) compared to all mitotic events of non-targeting cells (gray) across both day 3 (left) and day 4 (right) post-Cas9 induction time course experiment for known cell division control genes included in the screen.
Figure S7. Targeted analysis of mitotic phenotypes, Related to Figures 6 and 7. (A) Immunofluorescence images of individual cell lines stably expressing a single sgRNA targeting each gene of interest to confirm live-cell pooled screen phenotypes and enable visualization at higher resolution across a single population. Images are deconvolved maximum intensity projections of fixed cells stained for microtubules (anti-alpha-tubulin) and DNA (Hoechst). Scale bar, 10 μm. (B) Bar plot showing kinetochore-localized intensity of the outer kinetochore microtubule-binding protein NDC80 in inducible knockout cell lines for all 29 genes pursued from the live-cell screen, along with CENP-A and HJURP controls. Each data point represents the median kinetochore signal of one experiment for >10 cells per gene target. Values are normalized relative to negative control cells from the same experiment. *P<0.01 by two-tailed independent T-test relative to negative control cells. Error bars indicate SD. (C) Bar plot of kinetochore-localized intensity for the inner kinetochore centromere-specific histone CENP-A in inducible knockout cell lines; experiment design as in (B). *P<0.01 by two-tailed independent T-test relative to control cells. Error bars indicate SD. (D) Western blot of CENP-A and NDC80 total protein levels for a subset of inducible gene knockouts. (E) Left, Fluorescence images of human cells expressing GFP-LIN52, indicating LIN52 nuclear localization in interphase cells and non-specific localization in mitotic cells. Scale bar, 10 μm. Right, Mass spectrometry analysis of an GFP-LIN52 immunoprecipitation from mitotically-enriched cells relative to controls, indicating that LIN52 associates with a subset of expected factors, but not the entire DREAM complex. (F) Images from time-lapse fluorescence imaging of individual knockout cell lines expressing H2B-mCherry, demonstrating similar mitotic phenotypes for LIN52, LIN9, and LIN54 knockouts. Scale bar, 10 μm. (G) Bar plot showing kinetochore-localized intensity for the inner kinetochore centromere-specific histone CENP-A in inducible knockout cell lines of LIN52-associated genes. LIN52, LIN9, and LIN54 each demonstrate a significant decrease in CENP-A kinetochore localization. *P<0.01 by two-tailed independent T-test relative to control sgRNA. (H) Metagene analysis of transcription termination in CLP1 knockouts. The increased density of reads downstream of annotated transcription termination sites suggests a defect in 3’ end mRNA processing. (I) RT-PCR validation of SPC24 minor intron retention, showing an intron 4-containing SPC24 amplicon after RNPC3 KO that is not present in control samples.
Table S1. Screening library sgRNA sequences and additional oligonucleotides. Related to Figures 1–7. (A) Library CRISPR sgRNA sequences and (B) additional oligonucleotides used in this study.
Table S2. Primary fixed-cell screen results. Related to Figures 1–5. (A) Summary results and (B) extracted image features for the primary fixed-cell screen.
Table S3. Comparison of primary screen results to external datasets. Related to Figures 1–5. (A) Comparison of genes with mitotic phenotypes in the primary screen with those identified in MitoCheck.7 (B) Summary of interphase phenotype cluster enrichment for comparison annotation datasets. (C) List of KEGG pathways and corresponding enriched clusters. (D) KEGG enrichment result details. (E) List of CORUM complexes and corresponding enriched clusters. (F) CORUM enrichment result details. (G) Wainberg et al.,46 BioPlex, and STRING enrichment result details.
Table S4. Secondary live-cell screen results. Related to Figure 6. (A) Summary results and (B) extracted image features for the secondary live-cell screen.
Movie S1. Example time lapse montages from the secondary live cell screen, Related to Figure 6.
Movie S2. Example time lapse images of individual knockout cell lines, Related to Figure 6.
Acknowledgments:
We thank Andy Nutter-Upham and Scott McCallum for creating the Vesuvius web portal, members of the Blainey and Cheeseman labs, Bingbing Yuan and Heather Keys for their support and input, and Dave Bartel, Kara McKinley, Anne Carpenter, Anna Le, Russell Walton, and Robert Majovski for comments on the manuscript. We thank the Broad Genomic Perturbation Platform for providing CFD scores for our custom sgRNA library, the Whitehead Institute Genome Technology Core for performing the RNA-seq library prep and sequencing, and Celeste Diaz and Julia Bauman in the lab of J.T. Neal at the Broad Institute for assistance in developing custom antibody conjugations. The HeLa cell line was used in this research. Henrietta Lacks, and the HeLa cell line that was established from her tumor cells without her knowledge or consent in 1951, have made significant contributions to scientific progress and advances in human health. We are grateful to Lacks, now deceased, and to the Lacks family for their contributions to biomedical research.
Funding:
This work was supported by grants from the National Institutes of Health (HG009283 and HG006193 to PCB and R35GM126930 to IMC) and support from the Gordon and Betty Moore Foundation to IMC. LF was supported by a National Defense Science and Engineering Graduate Fellowship. JL was supported in part by a Natural Sciences and Engineering Research Council fellowship.
Declaration of interests:
PCB is a consultant to and/or equity holder in companies in the life sciences industries including 10X Genomics, GALT, Celsius Therapeutics, Next Generation Diagnostics, Cache DNA, Concerto Biosciences, and Stately. PCB’s laboratory receives research funding from Calico Life Sciences and Merck for work related to genetic screening. The Broad Institute and MIT have filed U.S. patent applications on work described here and may seek to license the technology.
References
- 1.Wang T, Wei JJ, Sabatini DM, and Lander ES (2014). Genetic Screens in Human Cells Using the CRISPR-Cas9 System. Science 343, 80–84. 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Condon KJ, Orozco JM, Adelmann CH, Spinelli JB, van der Helm PW, Roberts JM, Kunchok T, and Sabatini DM (2021). Genome-wide CRISPR screens reveal multitiered mechanisms through which mTORC1 senses mitochondrial dysfunction. Proc. Natl. Acad. Sci. U. S. A 118, e2022120118. 10.1073/pnas.2022120118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nieuwenhuis J, Adamopoulos A, Bleijerveld OB, Mazouzi A, Stickel E, Celie P, Altelaar M, Knipscheer P, Perrakis A, Blomen VA, et al. (2017). Vasohibins encode tubulin detyrosinating activity. Science 358, 1453–1456. 10.1126/science.aao5676. [DOI] [PubMed] [Google Scholar]
- 4.Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, Marjanovic ND, Dionne D, Burks T, Raychowdhury R, et al. (2016). Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 167, 1853–1866.e17. 10.1016/j.cell.2016.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Replogle JM, Saunders RA, Pogson AN, Hussmann JA, Lenail A, Guna A, Mascibroda L, Wagner EJ, Adelman K, Lithwick-Yanai G, et al. (2022). Mapping information-rich genotype-phenotype landscapes with genome-scale Perturb-seq. Cell 185, 2559–2575.e28. 10.1016/j.cell.2022.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Datlinger P, Rendeiro AF, Schmidl C, Krausgruber T, Traxler P, Klughammer J, Schuster LC, Kuchler A, Alpar D, and Bock C (2017). Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 14, 297–301. 10.1038/nmeth.4177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neumann B, Walter T, Hériché J-K, Bulkescher J, Erfle H, Conrad C, Rogers P, Poser I, Held M, Liebel U, et al. (2010). Phenotypic profiling of the human genome by time-lapse microscopy reveals cell division genes. Nature 464, 721–727. 10.1038/nature08869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feldman D, Singh A, Schmid-Burgk JL, Carlson RJ, Mezger A, Garrity AJ, Zhang F, and Blainey PC (2019). Optical Pooled Screens in Human Cells. Cell 179, 787–799.e17. 10.1016/j.cell.2019.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang C, Lu T, Emanuel G, Babcock HP, and Zhuang X (2019). Imaging-based pooled CRISPR screening reveals regulators of lncRNA localization. Proc. Natl. Acad. Sci 116, 10842–10851. 10.1073/pnas.1903808116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hasle N, Cooke A, Srivatsan S, Huang H, Stephany JJ, Krieger Z, Jackson D, Tang W, Pendyala S, Monnat RJ Jr., et al. (2020). High-throughput, microscope-based sorting to dissect cellular heterogeneity. Mol. Syst. Biol 16, e9442. 10.15252/msb.20209442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kanfer G, Sarraf SA, Maman Y, Baldwin H, Dominguez-Martin E, Johnson KR, Ward ME, Kampmann M, Lippincott-Schwartz J, and Youle RJ (2021). Image-based pooled whole-genome CRISPRi screening for subcellular phenotypes. J. Cell Biol 220, e202006180. 10.1083/jcb.202006180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schraivogel D, Kuhn TM, Rauscher B, Rodríguez-Martínez M, Paulsen M, Owsley K, Middlebrook A, Tischer C, Ramasz B, Ordoñez-Rueda D, et al. (2022). High-speed fluorescence image–enabled cell sorting. Science 375, 315–320. 10.1126/science.abj3013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wheeler EC, Vu AQ, Einstein JM, DiSalvo M, Ahmed N, Van Nostrand EL, Shishkin AA, Jin W, Allbritton NL, and Yeo GW (2020). Pooled CRISPR screens with imaging on microraft arrays reveals stress granule-regulatory factors. Nat. Methods 17, 636–642. 10.1038/s41592-020-0826-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yan X, Stuurman N, Ribeiro SA, Tanenbaum ME, Horlbeck MA, Liem CR, Jost M, Weissman JS, and Vale RD (2021). High-content imaging-based pooled CRISPR screens in mammalian cells. J. Cell Biol 220, e202008158. 10.1083/jcb.202008158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feldman D, Funk L, Le A, Carlson RJ, Leiken MD, Tsai F, Soong B, Singh A, and Blainey PC (2022). Pooled genetic perturbation screens with image-based phenotypes. Nat. Protoc 17, 476–512. 10.1038/s41596-021-00653-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Blomen VA, Májek P, Jae LT, Bigenzahn JW, Nieuwenhuis J, Staring J, Sacco R, Diemen F.R. van, Olk N, Stukalov A, et al. (2015). Gene essentiality and synthetic lethality in haploid human cells. Science 350, 1092–1096. 10.1126/science.aac7557. [DOI] [PubMed] [Google Scholar]
- 17.Dempster JM, Rossen J, Kazachkova M, Pan J, Kugener G, Root DE, and Tsherniak A (2019). Extracting Biological Insights from the Project Achilles Genome-Scale CRISPR Screens in Cancer Cell Lines. bioRxiv, 720243. 10.1101/720243. [DOI] [Google Scholar]
- 18.DepMap Broad Institute DepMap 19Q3 Public. figshare. Dataset doi: 10.6084/m9.figshare.9201770.v2. [DOI] [Google Scholar]
- 19.Hart T, Chandrashekhar M, Aregger M, Steinhart Z, Brown KR, MacLeod G, Mis M, Zimmermann M, Fradet-Turcotte A, Sun S, et al. (2015). High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 163, 1515–1526. 10.1016/j.cell.2015.11.015. [DOI] [PubMed] [Google Scholar]
- 20.Horlbeck MA, Gilbert LA, Villalta JE, Adamson B, Pak RA, Chen Y, Fields AP, Park CY, Corn JE, Kampmann M, et al. (2016). Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. eLife 5, e19760. 10.7554/eLife.19760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, et al. (2017). Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells. Nat. Genet 49, 1779–1784. 10.1038/ng.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tzelepis K, Koike-Yusa H, De Braekeleer E, Li Y, Metzakopian E, Dovey OM, Mupo A, Grinkevich V, Li M, Mazan M, et al. (2016). A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Rep. 17, 1193–1205. 10.1016/j.celrep.2016.09.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, and Sabatini DM (2015). Identification and characterization of essential genes in the human genome. Science 350, 1096–1101. 10.1126/science.aac7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang T, Yu H, Hughes NW, Liu B, Kendirli A, Klein K, Chen WW, Lander ES, and Sabatini DM (2017). Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell 168, 890–903.e15. 10.1016/j.cell.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol 34, 184–191. 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hart T, Tong AHY, Chan K, Van Leeuwen J, Seetharaman A, Aregger M, Chandrashekhar M, Hustedt N, Seth S, Noonan A, et al. (2017). Evaluation and Design of Genome-Wide CRISPR/SpCas9 Knockout Screens. G3 Bethesda Md 7, 2719–2727. 10.1534/g3.117.041277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McKinley KL, and Cheeseman IM (2017). Large-Scale Analysis of CRISPR/Cas9 Cell-Cycle Knockouts Reveals the Diversity of p53-Dependent Responses to Cell-Cycle Defects. Dev. Cell 40, 405–420.e2. 10.1016/j.devcel.2017.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sancar A, Lindsey-Boltz LA, Unsal-Kaçmaz K, and Linn S (2004). Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem 73, 39–85. 10.1146/annurev.biochem.73.011303.073723. [DOI] [PubMed] [Google Scholar]
- 29.Bell SP, and Dutta A (2002). DNA Replication in Eukaryotic Cells. Annu. Rev. Biochem 71, 333–374. 10.1146/annurev.biochem.71.110601.135425. [DOI] [PubMed] [Google Scholar]
- 30.Carmena M, Wheelock M, Funabiki H, and Earnshaw WC (2012). The chromosomal passenger complex (CPC): from easy rider to the godfather of mitosis. Nat. Rev. Mol. Cell Biol 13, 789–803. 10.1038/nrm3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pollard TD, and O’Shaughnessy B (2019). Molecular Mechanism of Cytokinesis. Annu. Rev. Biochem 88, 661–689. 10.1146/annurev-biochem-062917-012530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lara-Gonzalez P, Pines J, and Desai A (2021). Spindle assembly checkpoint activation and silencing at kinetochores. Semin. Cell Dev. Biol 117, 86–98. 10.1016/j.semcdb.2021.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pederiva C, Böhm S, Julner A, and Farnebo M (2016). Splicing controls the ubiquitin response during DNA double-strand break repair. Cell Death Differ. 23, 1648–1657. 10.1038/cdd.2016.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shi R, Hou W, Wang Z-Q, and Xu X (2021). Biogenesis of Iron–Sulfur Clusters and Their Role in DNA Metabolism. Front. Cell Dev. Biol 9, 2676. 10.3389/fcell.2021.735678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Villa F, Fujisawa R, Ainsworth J, Nishimura K, Lie‐A‐Ling M, Lacaud G, and Labib KP (2021). CUL2LRR1, TRAIP and p97 control CMG helicase disassembly in the mammalian cell cycle. EMBO Rep. 22, e52164. 10.15252/embr.202052164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goodson HV, and Jonasson EM (2018). Microtubules and Microtubule-Associated Proteins. Cold Spring Harb. Perspect. Biol 10, a022608. 10.1101/cshperspect.a022608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pollard TD (2016). Actin and Actin-Binding Proteins. Cold Spring Harb. Perspect. Biol 8, a018226. 10.1101/cshperspect.a018226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li L, Zhang W, Liu Y, Liu X, Cai L, Kang J, Zhang Y, Chen W, Dong C, Zhang Y, et al. (2020). The CRL3BTBD9 E3 ubiquitin ligase complex targets TNFAIP1 for degradation to suppress cancer cell migration. Signal Transduct. Target. Ther 5, 1–9. 10.1038/s41392-020-0140-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rodríguez-Pérez F, Manford AG, Pogson A, Ingersoll AJ, Martínez-González B, and Rape M (2021). Ubiquitin-dependent remodeling of the actin cytoskeleton drives cell fusion. Dev. Cell 56, 588–601.e9. 10.1016/j.devcel.2021.01.016. [DOI] [PubMed] [Google Scholar]
- 40.Zyss D, Ebrahimi H, and Gergely F (2011). Casein kinase I delta controls centrosome positioning during T cell activation. J. Cell Biol 195, 781–797. 10.1083/jcb.201106025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Collart MA (2016). The Ccr4-Not complex is a key regulator of eukaryotic gene expression. WIREs RNA 7, 438–454. 10.1002/wrna.1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cantwell H, and Nurse P (2019). Unravelling nuclear size control. Curr. Genet 65, 1281–1285. 10.1007/s00294-019-00999-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Moon KR, van Dijk D, Wang Z, Gigante S, Burkhardt DB, Chen WS, Yim K, Elzen A. van den, Hirn MJ, Coifman RR, et al. (2019). Visualizing structure and transitions in high-dimensional biological data. Nat. Biotechnol 37, 1482–1492. 10.1038/s41587-019-0336-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Traag VA, Waltman L, and van Eck NJ (2019). From Louvain to Leiden: guaranteeing well-connected communities. Sci. Rep 9, 5233. 10.1038/s41598-019-41695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huttlin EL, Bruckner RJ, Navarrete-Perea J, Cannon JR, Baltier K, Gebreab F, Gygi MP, Thornock A, Zarraga G, Tam S, et al. (2021). Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. Cell 184, 3022–3040.e28. 10.1016/j.cell.2021.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wainberg M, Kamber RA, Balsubramani A, Meyers RM, Sinnott-Armstrong N, Hornburg D, Jiang L, Chan J, Jian R, Gu M, et al. (2021). A genome-wide atlas of co-essential modules assigns function to uncharacterized genes. Nat. Genet 53, 638–649. 10.1038/s41588-021-00840-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Giurgiu M, Reinhard J, Brauner B, Dunger-Kaltenbach I, Fobo G, Frishman G, Montrone C, and Ruepp A (2019). CORUM: the comprehensive resource of mammalian protein complexes—2019. Nucleic Acids Res. 47, D559–D563. 10.1093/nar/gky973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kanehisa M, and Goto S (2000). KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 28, 27–30. 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.von Mering C, Jensen LJ, Snel B, Hooper SD, Krupp M, Foglierini M, Jouffre N, Huynen MA, and Bork P (2005). STRING: known and predicted protein–protein associations, integrated and transferred across organisms. Nucleic Acids Res. 33, D433–D437. 10.1093/nar/gki005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cho NH, Cheveralls KC, Brunner A-D, Kim K, Michaelis AC, Raghavan P, Kobayashi H, Savy L, Li JY, Canaj H, et al. (2022). OpenCell: Endogenous tagging for the cartography of human cellular organization. Science 375, eabi6983. 10.1126/science.abi6983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pan J, Meyers RM, Michel BC, Mashtalir N, Sizemore AE, Wells JN, Cassel SH, Vazquez F, Weir BA, Hahn WC, et al. (2018). Interrogation of Mammalian Protein Complex Structure, Function, and Membership Using Genome-Scale Fitness Screens. Cell Syst. 6, 555–568.e7. 10.1016/j.cels.2018.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, Frederick DT, Hurley AD, Nellore A, Kung AL, et al. (2013). Oncogenic BRAF Regulates Oxidative Metabolism via PGC1α and MITF. Cancer Cell 23, 302–315. 10.1016/j.ccr.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liou G-Y, Döppler H, DelGiorno KE, Zhang L, Leitges M, Crawford HC, Murphy MP, and Storz P (2016). Mutant KRas-Induced Mitochondrial Oxidative Stress in Acinar Cells Upregulates EGFR Signaling to Drive Formation of Pancreatic Precancerous Lesions. Cell Rep. 14, 2325–2336. 10.1016/j.celrep.2016.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GRS, and Chandel NS (2010). Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci 107, 8788–8793. 10.1073/pnas.1003428107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cao Z, Budinich KA, Huang H, Ren D, Lu B, Zhang Z, Chen Q, Zhou Y, Huang Y-H, Alikarami, F., et al. (2021). ZMYND8-regulated IRF8 transcription axis is an acute myeloid leukemia dependency. Mol. Cell 81, 3604–3622.e10. 10.1016/j.molcel.2021.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Singh S, Vanden Broeck A, Miller L, Chaker-Margot M, and Klinge S (2021). Nucleolar maturation of the human small subunit processome. Science 373, eabj5338. 10.1126/science.abj5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.de Almeida M, Hinterndorfer M, Brunner H, Grishkovskaya I, Singh K, Schleiffer A, Jude J, Deswal S, Kalis R, Vunjak M, et al. (2021). AKIRIN2 controls the nuclear import of proteasomes in vertebrates. Nature, 1–6. 10.1038/s41586-021-04035-8. [DOI] [PubMed] [Google Scholar]
- 58.Song H, Feng X, Zhang H, Luo Y, Huang J, Lin M, Jin J, Ding X, Wu S, Huang H, et al. (2019). METTL3 and ALKBH5 oppositely regulate m6A modification of TFEB mRNA, which dictates the fate of hypoxia/reoxygenation-treated cardiomyocytes. Autophagy 15, 1419–1437. 10.1080/15548627.2019.1586246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Baillat D, Hakimi M-A, Näär AM, Shilatifard A, Cooch N, and Shiekhattar R (2005). Integrator, a Multiprotein Mediator of Small Nuclear RNA Processing, Associates with the C-Terminal Repeat of RNA Polymerase II. Cell 123, 265–276. 10.1016/j.cell.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 60.Jodoin JN, Sitaram P, Albrecht TR, May SB, Shboul M, Lee E, Reversade B, Wagner EJ, and Lee LA (2013). Nuclear-localized Asunder regulates cytoplasmic dynein localization via its role in the integrator complex. Mol. Biol. Cell 24, 2954–2965. 10.1091/mbc.E13-05-0254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boeing S, Williamson L, Encheva V, Gori I, Saunders RE, Instrell R, Aygün O, Rodriguez-Martinez M, Weems JC, Kelly GP, et al. (2016). Multiomic Analysis of the UV-Induced DNA Damage Response. Cell Rep. 15, 1597–1610. 10.1016/j.celrep.2016.04.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Malovannaya A, Li Y, Bulynko Y, Jung SY, Wang Y, Lanz RB, O’Malley BW, and Qin J (2010). Streamlined analysis schema for high-throughput identification of endogenous protein complexes. Proc. Natl. Acad. Sci 107, 2431–2436. 10.1073/pnas.0912599106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sabath K, Stäubli ML, Marti S, Leitner A, Moes M, and Jonas S (2020). INTS10–INTS13–INTS14 form a functional module of Integrator that binds nucleic acids and the cleavage module. Nat. Commun 11, 3422. 10.1038/s41467-020-17232-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Pan J, Kwon JJ, Talamas JA, Borah AA, Vazquez F, Boehm JS, Tsherniak A, Zitnik M, McFarland JM, and Hahn WC (2022). Sparse dictionary learning recovers pleiotropy from human cell fitness screens. Cell Syst. 13, 286–303. 10.1016/j.cels.2021.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yang YJ, Baltus AE, Mathew RS, Murphy EA, Evrony GD, Gonzalez DM, Wang EP, Marshall-Walker CA, Barry BJ, Murn J, et al. (2012). Microcephaly gene links trithorax and REST/NRSF to control neural stem cell proliferation and differentiation. Cell 151, 1097–1112. 10.1016/j.cell.2012.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jayaraman D, Bae B-I, and Walsh CA (2018). The Genetics of Primary Microcephaly. Annu. Rev. Genomics Hum. Genet 19, 177–200. 10.1146/annurev-genom-083117-021441. [DOI] [PubMed] [Google Scholar]
- 67.Gheiratmand L, Coyaud E, Gupta GD, Laurent EM, Hasegan M, Prosser SL, Gonçalves J, Raught B, and Pelletier L (2019). Spatial and proteomic profiling reveals centrosome-independent features of centriolar satellites. EMBO J. 38, e101109. 10.15252/embj.2018101109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Goshima G, Wollman R, Goodwin SS, Zhang N, Scholey JM, Vale RD, and Stuurman N (2007). Genes Required for Mitotic Spindle Assembly in Drosophila S2 Cells. Science 316, 417–421. 10.1126/science.1141314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Taïeb HM, Garske DS, Contzen J, Gossen M, Bertinetti L, Robinson T, and Cipitria A (2021). Osmotic pressure modulates single cell cycle dynamics inducing reversible growth arrest and reactivation of human metastatic cells. Sci. Rep 11, 13455. 10.1038/s41598-021-92054-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cheeseman IM (2014). The kinetochore. Cold Spring Harb. Perspect. Biol 6, a015826. 10.1101/cshperspect.a015826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Esterlechner J, Reichert N, Iltzsche F, Krause M, Finkernagel F, and Gaubatz S (2013). LIN9, a Subunit of the DREAM Complex, Regulates Mitotic Gene Expression and Proliferation of Embryonic Stem Cells. PLOS ONE 8, e62882. 10.1371/journal.pone.0062882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fischer M, and Müller GA (2017). Cell cycle transcription control: DREAM/MuvB and RB-E2F complexes. Crit. Rev. Biochem. Mol. Biol 52, 638–662. 10.1080/10409238.2017.1360836. [DOI] [PubMed] [Google Scholar]
- 73.Ghazy MA, Gordon JMB, Lee SD, Singh BN, Bohm A, Hampsey M, and Moore C (2012). The interaction of Pcf11 and Clp1 is needed for mRNA 3′-end formation and is modulated by amino acids in the ATP-binding site. Nucleic Acids Res. 40, 1214–1225. 10.1093/nar/gkr801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Austenaa LMI, Piccolo V, Russo M, Prosperini E, Polletti S, Polizzese D, Ghisletti S, Barozzi I, Diaferia GR, and Natoli G (2021). A first exon termination checkpoint preferentially suppresses extragenic transcription. Nat. Struct. Mol. Biol 28, 337–346. 10.1038/s41594-021-00572-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Estell C, Davidson L, Steketee PC, Monier A, and West S (2021). ZC3H4 restricts non-coding transcription in human cells. eLife 10, e67305. 10.7554/eLife.67305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kamieniarz-Gdula K, Gdula MR, Panser K, Nojima T, Monks J, Wiśniewski JR, Riepsaame J, Brockdorff N, Pauli A, and Proudfoot NJ (2019). Selective Roles of Vertebrate PCF11 in Premature and Full-Length Transcript Termination. Mol. Cell 74, 158–172.e9. 10.1016/j.molcel.2019.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Verma B, Akinyi MV, Norppa AJ, and Frilander MJ (2018). Minor spliceosome and disease. Semin. Cell Dev. Biol 79, 103–112. 10.1016/j.semcdb.2017.09.036. [DOI] [PubMed] [Google Scholar]
- 78.de Wolf B, Oghabian A, Akinyi MV, Hanks S, Tromer EC, van Hooff JJE, van Voorthuijsen L, van Rooijen LE, Verbeeren J, Uijttewaal ECH, et al. (2021). Chromosomal instability by mutations in the novel minor spliceosome component CENATAC. EMBO J. 40, e106536. 10.15252/embj.2020106536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chandrasekaran SN, Ceulemans H, Boyd JD, and Carpenter AE (2021). Image-based profiling for drug discovery: due for a machine-learning upgrade? Nat. Rev. Drug Discov 20, 145–159. 10.1038/s41573-020-00117-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cox MJ, Jaensch S, Van de Waeter J, Cougnaud L, Seynaeve D, Benalla S, Koo SJ, Van Den Wyngaert I, Neefs J-M, Malkov D, et al. (2020). Tales of 1,008 small molecules: phenomic profiling through live-cell imaging in a panel of reporter cell lines. Sci. Rep 10, 13262. 10.1038/s41598-020-69354-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kang J, Hsu C-H, Wu Q, Liu S, Coster AD, Posner BA, Altschuler SJ, and Wu LF (2016). Improving drug discovery with high-content phenotypic screens by systematic selection of reporter cell lines. Nat. Biotechnol 34, 70–77. 10.1038/nbt.3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rohban MH, Singh S, Wu X, Berthet JB, Bray M-A, Shrestha Y, Varelas X, Boehm JS, and Carpenter AE (2017). Systematic morphological profiling of human gene and allele function via Cell Painting. eLife 6, e24060. 10.7554/eLife.24060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Caicedo JC, McQuin C, Goodman A, Singh S, and Carpenter AE (2018). Weakly Supervised Learning of Single-Cell Feature Embeddings. In 2018 IEEE/CVF Conference on Computer Vision and Pattern Recognition, pp. 9309–9318. 10.1109/CVPR.2018.00970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kobayashi H, Cheveralls KC, Leonetti MD, and Royer LA (2022). Self-supervised deep learning encodes high-resolution features of protein subcellular localization. Nat. Methods 19, 995–1003. 10.1038/s41592-022-01541-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lu AX, Kraus OZ, Cooper S, and Moses AM (2019). Learning unsupervised feature representations for single cell microscopy images with paired cell inpainting. PLOS Comput. Biol 15, e1007348. 10.1371/journal.pcbi.1007348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Dang Y, Jia G, Choi J, Ma H, Anaya E, Ye C, Shankar P, and Wu H (2015). Optimizing sgRNA structure to improve CRISPR-Cas9 knockout efficiency. Genome Biol. 16, 280. 10.1186/s13059-015-0846-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gascoigne KE, Takeuchi K, Suzuki A, Hori T, Fukagawa T, and Cheeseman IM (2011). Induced Ectopic Kinetochore Assembly Bypasses the Requirement for CENP-A Nucleosomes. Cell 145, 410–422. 10.1016/j.cell.2011.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fulco CP, Munschauer M, Anyoha R, Munson G, Grossman SR, Perez EM, Kane M, Cleary B, Lander ES, and Engreitz JM (2016). Systematic mapping of functional enhancer–promoter connections with CRISPR interference. Science 354, 769–773. 10.1126/science.aag2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.McKinley KL, Sekulic N, Guo LY, Tsinman T, Black BE, and Cheeseman IM (2015). The CENP-L-N Complex Forms a Critical Node in an Integrated Meshwork of Interactions at the Centromere-Kinetochore Interface. Mol. Cell 60, 886–898. 10.1016/j.molcel.2015.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.van den Berg J, G. Manjón A, Kielbassa K, Feringa FM, Freire R, and Medema RH (2018). A limited number of double-strand DNA breaks is sufficient to delay cell cycle progression. Nucleic Acids Res. 46, 10132–10144. 10.1093/nar/gky786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Morgenstern JP, and Land H (1990). Advanced mammalian gene transfer: high titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 18, 3587–3596. 10.1093/nar/18.12.3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cheeseman IM, and Desai A (2005). A Combined Approach for the Localization and Tandem Affinity Purification of Protein Complexes from Metazoans. Sci. STKE 2005, pl1–pl1. 10.1126/stke.2662005pl1. [DOI] [PubMed] [Google Scholar]
- 93.Schmidt JC, Arthanari H, Boeszoermenyi A, Dashkevich NM, Wilson-Kubalek EM, Monnier N, Markus M, Oberer M, Milligan RA, Bathe M, et al. (2012). The Kinetochore-Bound Ska1 Complex Tracks Depolymerizing Microtubules and Binds to Curved Protofilaments. Dev. Cell 23, 968–980. 10.1016/j.devcel.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Backer CB, Gutzman JH, Pearson CG, and Cheeseman IM (2012). CSAP localizes to polyglutamylated microtubules and promotes proper cilia function and zebrafish development. Mol. Biol. Cell 23, 2122–2130. 10.1091/mbc.E11-11-0931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.McQuin C, Goodman A, Chernyshev V, Kamentsky L, Cimini BA, Karhohs KW, Doan M, Ding L, Rafelski SM, Thirstrup D, et al. (2018). CellProfiler 3.0: Next-generation image processing for biology. PLOS Biol. 16, e2005970. 10.1371/journal.pbio.2005970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Stringer C, Wang T, Michaelos M, and Pachitariu M (2021). Cellpose: a generalist algorithm for cellular segmentation. Nat. Methods 18, 100–106. 10.1038/s41592-020-01018-x. [DOI] [PubMed] [Google Scholar]
- 98.Feldman D, and Funk L (2021). Pooled genetic perturbation screens with image-based phenotypes, OpticalPooledScreens. 10.5281/zenodo.5002684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Singh S, Bray M-A, Jones T, and Carpenter A (2014). Pipeline for illumination correction of images for high-throughput microscopy. J. Microsc 256, 231–236. 10.1111/jmi.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Walt S. van der, Schönberger JL, Nunez-Iglesias J, Boulogne F, Warner JD, Yager N, Gouillart E, and Yu T (2014). scikit-image: image processing in Python. PeerJ 2, e453. 10.7717/peerj.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Coelho LP (2013). Mahotas: Open source software for scriptable computer vision. J. Open Res. Softw 1, e3. 10.5334/jors.ac. [DOI] [Google Scholar]
- 102.Köster J, and Rahmann S (2012). Snakemake—a scalable bioinformatics workflow engine. Bioinformatics 28, 2520–2522. 10.1093/bioinformatics/bts480. [DOI] [PubMed] [Google Scholar]
- 103.Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, et al. (2011). Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res 12, 2825–2830. [Google Scholar]
- 104.Jaqaman K, Loerke D, Mettlen M, Kuwata H, Grinstein S, Schmid SL, and Danuser G (2008). Robust single-particle tracking in live-cell time-lapse sequences. Nat. Methods 5, 695–702. 10.1038/nmeth.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Tinevez J-Y, Perry N, Schindelin J, Hoopes GM, Reynolds GD, Laplantine E, Bednarek SY, Shorte SL, and Eliceiri KW (2017). TrackMate: An open and extensible platform for single-particle tracking. Methods 115, 80–90. 10.1016/j.ymeth.2016.09.016. [DOI] [PubMed] [Google Scholar]
- 106.Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12. 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 107.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Anders S, Pyl PT, and Huber W (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Olthof AM, Hyatt KC, and Kanadia RN (2019). Minor intron splicing revisited: identification of new minor intron-containing genes and tissue-dependent retention and alternative splicing of minor introns. BMC Genomics 20, 686. 10.1186/s12864-019-6046-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ge SX, Jung D, and Yao R (2020). ShinyGO: a graphical gene-set enrichment tool for animals and plants. Bioinformatics 36, 2628–2629. 10.1093/bioinformatics/btz931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ramírez F, Dündar F, Diehl S, Grüning BA, and Manke T (2014). deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–W191. 10.1093/nar/gku365. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Optimization of image-based pooled screening for essential gene function, Related to Figure 1. (A) Scatter plot showing the results from trial screens of 400 gene targets. This compares the fraction of mitotic cells with visually-identified phenotypic defects for established cell division factors at 3 and 4 days post-Cas9 induction. Overall, mitotic phenotypes were more commonly observed at the earlier time point. (B) Scatter plot showing mean change in abundance within the 20,445 sgRNA primary screen library at 3 and 5 days post-Cas9 induction, both time points relative to pre-induction (day 0). N=2 screen replicates were performed, averaged across sgRNAs targeting the same gene. Orange indicates non-targeting control sgRNAs. Many gene targets begin to drop out of the population at day 5 due to fitness defects. Based on these data and from (A), 78 hours post-Cas9 induction was chosen as the fixation time point for our image-based screen to maximize observable phenotypes. (C) Boxplot demonstrating in situ sequencing quality in our fixed-cell image-based pooled screen. Sequencing quality was consistent across the eight imaging plates, with the majority of imaging tiles exceeding 50% of cells with sequencing reads that uniquely match a single sgRNA sequence from the library. N = 1,665 or 1,998 imaging tiles in each plate column. Whiskers extend to 1.5 times the interquartile range. (D) Confusion matrix demonstrating performance of the support vector classifier in distinguishing interphase and mitotic cells, 5-fold cross-validation with N=2,514 manually annotated cell images. (E) Histograms indicating the distribution of phenotype similarities (correlation of phenotype profiles after principal component projection) between sgRNAs targeting the same gene (blue) or between sgRNAs targeting different genes (orange). Overall, sgRNAs targeting the same gene display more similar phenotypes to than sgRNAs targeting different genes.
Figure S2. Analysis of interphase nuclear phenotypes, Related to Figure 1. (A) Cumulative distribution plots of mean nuclear γH2AX intensity (DNA damage phenotype) sgRNA scores, with sgRNAs grouped by number and location of target sites. Non-targeting control sgRNAs and sgRNAs targeting a single genomic locus (blue) include the vast majority of sgRNAs in the library and displayed minimal DNA damage on average in the screen. In contrast, sgRNAs with increasing numbers of target sites (orange, green) tend to display stronger DNA damage phenotypes, in particular when the target sites are spread across multiple chromosomes (dotted lines). Genomic target sites are defined as the total number of cutting frequency determination (CFD) bin 1 matches (see Doench et al.25). *P<10−9 by 1-sided Mann-Whitney U test. (B) Bar graph indicating over-representation of KEGG pathways among gene targets exhibiting decreased or increased nuclear γH2AX mean intensity. *FDR<0.05 (C) Bar graph of over-representation analysis results as in (B) among gene targets with decreased or increased nuclear DNA (DAPI) integrated intensity. *FDR<0.05 (D) Scatter plot showing summary gene scores (see STAR Methods) for integrated nuclear DNA (DAPI) intensity compared to nuclear area. DNA content is relatively constant across gene targets exhibiting a range of nuclear areas, although a subset demonstrates increased nuclear area and DNA. Summary DAPI scores are plotted on a symmetric log scale (linear between −1 and 1) and labeled genes are colored by functional category. (E) Bivariate histograms of integrated nuclear DNA intensity and mean nuclear γH2AX intensity, displaying single-cell distributions for all cells expressing non-targeting sgRNAs (top left) and selected gene targets. Knockouts of genes that regulate chromosome segregation or cytokinesis result in more cells with increased DNA content, but only modest increases in γH2AX intensity. Histogram bins containing less than 1 in 104 of the total cells for a given gene target are not displayed.
Figure S3. Analysis of interphase cytoskeletal and morphological phenotypes, Related to Figure 2. (A) Scatter plot comparing mean cellular actin intensity summary scores between interphase and mitotic cell populations, indicating factors that robustly affect actin structures throughout the cell cycle. (B) Scatter plot indicating summary gene scores (see STAR Methods) for mean cellular actin intensity compared to cell area. A subset of gene knockouts display increased actin staining together with decreased cell area due to disrupted cellular adhesion. Labeled genes are colored by functional category. (C) Bivariate histograms of mean cellular actin intensity and cellular area, displaying single-cell distributions for all cells expressing non-targeting sgRNAs (left) and selected gene targets. Knockouts of genes that regulate cellular adhesion (e.g., ITGAV) show a distribution of cells shifted toward lower cellular area and correspondingly increased mean actin intensity. Histogram bins containing less than 1 in 104 total cells for a given gene target are not displayed. (D) Scatter plot showing summary gene scores for mean cellular tubulin intensity compared to cell area as in (B). Similar to actin, a subset of gene knockouts display increased tubulin staining in combination with decreased cell area. (E) Volcano plot for interphase cell area across gene targets in the screen, showing a wide range of decreased (magenta) and increased (green) cell areas (FDR<0.05). Raw P-values were computed by comparing gene targets to a bootstrapped null distribution of cells expressing non-targeting sgRNAs (see STAR Methods), with false discovery rate (FDR) estimated using the Benjamini-Hochberg procedure. (F) Bar graph of KEGG pathway over-representation analysis for gene targets that result in decreased or increased cell area. *FDR<0.05. (G) Example images and cumulative distributions of individual inducible knockout cell lines validating increased cell area for DTL and DONSON knockouts. *P<10−10 by Mann-Whitney U test relative to control sgRNA, N>3,700 cells per gene target. Scale bar, 10 μm.
Figure S4. Interphase phenotypes enable detailed clustering of specific functional categories, Related to Figure 3. Two-dimensional PHATE representations of interphase phenotype clusters and corresponding heat maps of a manually-selected subset of phenotype parameters as in Figures 3C–F for genes involved in (A) protein degradation (including the recently-characterized gene AKIRIN2), (B) RNA processing, (C) DNA replication and DNA damage, (D) cell cycle processes, (E) actin cytoskeleton and cellular adhesion, and (F) vesicle trafficking and related processes. The right plot in (C) demonstrates that most genes targeted by sgRNAs with excess target sites show related DNA damage phenotypes. Numbers indicate individual interphase cluster identities. All genes from selected clusters are listed below each heatmap. Parameters are presented as z-scores from the distribution of non-targeting sgRNAs, visualized on a consistent symmetric log scale (linear between −1 and 1). (G) Bivariate histogram showing the joint distribution of image-based interphase phenotype strength relative to non-targeting control sgRNAs computed by PHATE together with the strength of knockout fitness effect in the screening cell line (mean change in sgRNA abundance within the library after 5 days of Cas9 induction, N=2 screen replicates averaged across sgRNAs targeting the same gene, data from Figure S1B). >90% of gene targets exhibit a measurable interphase phenotype in the image-based screen (phenotype strength greater than the 95th percentile of non-targeting sgRNAs). Of the remaining 407 genes, only 55 demonstrate a meaningful fitness effect in the tested cell line (log2 fold change abundance less than the 5th percentile of non-targeting sgRNAs). Labeled genes are those that display a fitness effect and no interphase phenotype, but do show a measurable mitotic phenotype using the same method (not displayed). (H) Distributions of the Pearson correlation between image-based interphase phenotype profiles from the primary screen for all gene pairs (gray) or gene pairs annotated as co-functional in the labeled external databases (orange), indicating increased phenotype similarity between known co-functional genes across the full dataset. Note that this phenotype similarity is weaker among KEGG pathways, likely due to the broad pathway definitions and inclusion of many environmental response pathways not critical at the basal growth conditions tested in our screen.
Figure S5. Validation of interphase phenotypes and clusters, Related to Figures 3 and 4. (A) (A) Pie chart showing the fraction of genes present in clusters enriched for at least 2 of the comparison datasets (blue), or in clusters enriched for a single dataset. (B) Precision and recall of correspondence between our interphase phenotype clustering results and CORUM protein complex annotations across clustering resolution. A subset of 292 minimal CORUM complexes was used for comparison (see Methods), and comparisons were restricted to varying overall interphase phenotype strength as indicated. Error bars indicate the standard deviation of 10 clustering iterations with different random seeds. (C) Venn diagrams representing a qualitative comparison of similar clustering behaviors for co-functional genes that comprise specific protein complexes identified based on our interphase phenotype analysis and from the large-scale Perturb-seq screen performed by Replogle et al.5 (D) Venn diagrams as in (C), demonstrating the improved ability to identify and sub-categorize ribosome-related genes in our dataset in comparison to Replogle et al.5 (E) Interphase phenotype clustering identified a relationship between mitochondrial function and KRAS/BRAF signaling in cluster 149. (F) Example images and cumulative distributions of mitochondrial membrane potential dependent staining (MitoTracker) for control and knockout cells for KRAS, BRAF, and two co-clustering mitochondrial factors. Each knockout demonstrates disrupted mitochondrial activity as compared to control cells, supporting the phenotypic association of KRAS and BRAF with mitochondrial factors in the screen. *P<10−10 by Mann-Whitney U test relative to control sgRNA, N>1,400 cells per gene target. (G) Western blot MYC protein expression following knockout of several genes from cluster 121 (see Figures 4A–B). (H) Interphase phenotype clustering suggests a co-functional role of the RNA binding protein HNRNPD with METTL3 and METTL14, which form m6A modifications on RNA.
Figure S6. Fixed-cell mitotic phenotype analysis and pooled live cell screen, Related to Figures 5 and 6. (A-B) Box plots demonstrating strong agreement between manual phenotype scores by two individuals (blinded to gene labels) and computational phenotype strength (mean potential distance to non-targeting sgRNAs from mitotic PHATE analysis, normalized between 0 and 1). Whiskers extend to 1.5 times the interquartile range. (C) Comparison of mitotic phenotype genes identified in our fixed-cell screen to mitotic phenotypes found by MitoCheck.7 Mitotic phenotypes in the fixed-cell screen were defined as overall phenotype strength greater than the 95th percentile of non-targeting sgRNAs or significantly altered mitotic index (sgRNA permutation test, P<0.05). Bottom, bar graphs indicating KEGG pathway enrichment for all pathways enriched in any of the indicated gene sets. For the gene sets corresponding to the Venn diagram, the background for enrichment analysis was the intersection of all genes present in both screens. For the set of mitotic hit genes identified in MitoCheck that were not present in our screen (far right), the background for enrichment was all genes screened in MitoCheck not present in our screen. *FDR<0.05. (D) Example image indicating nuclear localization of GFP-tagged ZNF335. Scale bar, 10 μm. (E) Confusion matrix demonstrating performance of the support vector classifier in distinguishing interphase, mitotic, and apoptotic cells from the live-cell screen. 5-fold cross-validation with N=2,514 manually annotated cell images. Due to the relative difficulty of differentiating mitotic and apoptotic cells from H2B-mCherry fluorescence alone, the mitotic and apoptotic classes were combined after inference (cross-validation precision and recall after combining classes indicated by brackets). (F) Histograms of the total cell divisions observed across both day 3 and day 4 time courses for each gene target. (G) Cumulative distributions of mitotic duration for individual sgRNAs (blue, orange) compared to all mitotic events of non-targeting cells (gray) across both day 3 (left) and day 4 (right) post-Cas9 induction time course experiment for known cell division control genes included in the screen.
Figure S7. Targeted analysis of mitotic phenotypes, Related to Figures 6 and 7. (A) Immunofluorescence images of individual cell lines stably expressing a single sgRNA targeting each gene of interest to confirm live-cell pooled screen phenotypes and enable visualization at higher resolution across a single population. Images are deconvolved maximum intensity projections of fixed cells stained for microtubules (anti-alpha-tubulin) and DNA (Hoechst). Scale bar, 10 μm. (B) Bar plot showing kinetochore-localized intensity of the outer kinetochore microtubule-binding protein NDC80 in inducible knockout cell lines for all 29 genes pursued from the live-cell screen, along with CENP-A and HJURP controls. Each data point represents the median kinetochore signal of one experiment for >10 cells per gene target. Values are normalized relative to negative control cells from the same experiment. *P<0.01 by two-tailed independent T-test relative to negative control cells. Error bars indicate SD. (C) Bar plot of kinetochore-localized intensity for the inner kinetochore centromere-specific histone CENP-A in inducible knockout cell lines; experiment design as in (B). *P<0.01 by two-tailed independent T-test relative to control cells. Error bars indicate SD. (D) Western blot of CENP-A and NDC80 total protein levels for a subset of inducible gene knockouts. (E) Left, Fluorescence images of human cells expressing GFP-LIN52, indicating LIN52 nuclear localization in interphase cells and non-specific localization in mitotic cells. Scale bar, 10 μm. Right, Mass spectrometry analysis of an GFP-LIN52 immunoprecipitation from mitotically-enriched cells relative to controls, indicating that LIN52 associates with a subset of expected factors, but not the entire DREAM complex. (F) Images from time-lapse fluorescence imaging of individual knockout cell lines expressing H2B-mCherry, demonstrating similar mitotic phenotypes for LIN52, LIN9, and LIN54 knockouts. Scale bar, 10 μm. (G) Bar plot showing kinetochore-localized intensity for the inner kinetochore centromere-specific histone CENP-A in inducible knockout cell lines of LIN52-associated genes. LIN52, LIN9, and LIN54 each demonstrate a significant decrease in CENP-A kinetochore localization. *P<0.01 by two-tailed independent T-test relative to control sgRNA. (H) Metagene analysis of transcription termination in CLP1 knockouts. The increased density of reads downstream of annotated transcription termination sites suggests a defect in 3’ end mRNA processing. (I) RT-PCR validation of SPC24 minor intron retention, showing an intron 4-containing SPC24 amplicon after RNPC3 KO that is not present in control samples.
Table S1. Screening library sgRNA sequences and additional oligonucleotides. Related to Figures 1–7. (A) Library CRISPR sgRNA sequences and (B) additional oligonucleotides used in this study.
Table S2. Primary fixed-cell screen results. Related to Figures 1–5. (A) Summary results and (B) extracted image features for the primary fixed-cell screen.
Table S3. Comparison of primary screen results to external datasets. Related to Figures 1–5. (A) Comparison of genes with mitotic phenotypes in the primary screen with those identified in MitoCheck.7 (B) Summary of interphase phenotype cluster enrichment for comparison annotation datasets. (C) List of KEGG pathways and corresponding enriched clusters. (D) KEGG enrichment result details. (E) List of CORUM complexes and corresponding enriched clusters. (F) CORUM enrichment result details. (G) Wainberg et al.,46 BioPlex, and STRING enrichment result details.
Table S4. Secondary live-cell screen results. Related to Figure 6. (A) Summary results and (B) extracted image features for the secondary live-cell screen.
Movie S1. Example time lapse montages from the secondary live cell screen, Related to Figure 6.
Movie S2. Example time lapse images of individual knockout cell lines, Related to Figure 6.
Data Availability Statement
RNA-sequencing data have been deposited at the Gene Expression Omnibus (GEO) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Processed images and data from the screen are available in the supplemental materials and through the companion web portal (https://vesuvius.wi.mit.edu). Full single-cell extracted image phenotype measurements from the primary screen have been deposited at the Harvard Dataverse and are publicly available as of the date of publication. DOIs are listed in the key resources table. Raw image data are being deposited at the BioImage Archive and are publicly available. Accession numbers are listed in the key resources table.
All original code has been deposited at GitHub and Zenodo and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.