

Abstract
Background and Aims
Although type 2 innate lymphoid cells (ILC2s) were originally found to be liver-resident lymphocytes, the role and importance of ILC2 in liver injury remains poorly understood. In the current study, we sought to determine whether ILC2 is an important regulator of hepatic ischaemia/reperfusion injury (IRI).
Methods
ILC2-deficient mice (ICOS-T or NSG) and genetically modified ILC2s were used to investigate the role of ILC2s in murine hepatic IRI. Interactions between ILC2s and eosinophils or macrophages were studied in coculture. The role of human ILC2s was assessed in an immunocompromised mouse model of hepatic IRI.
Results
Administration of IL-33 prevented hepatic IRI in association with reduction of neutrophil infiltration and inflammatory mediators in the liver. IL-33-treated mice had elevated numbers of ILC2s, eosinophils, and regulatory T cells. Eosinophils, but not regulatory T cells, were required for IL-33-mediated hepatoprotection in IRI mice. Depletion of ILC2s substantially abolished the protective effect of IL-33 in hepatic IRI, indicating that ILC2s play critical roles in IL-33-mediated liver protection. Adoptive transfer of ex vivo-expanded ILC2s improved liver function and attenuated histologic damage in mice subjected to IRI. Mechanistic studies combining genetic and adoptive transfer approaches identified a protective role of ILC2s through promoting IL-13-dependent induction of anti-inflammatory macrophages and IL-5-dependent elevation of eosinophils in IRI. Furthermore, in vivo expansion of human ILC2s by IL-33 or transfer of ex vivo-expanded human ILC2s ameliorated hepatic IRI in an immunocompromised mouse model of hepatic IRI.
Conclusions
This study provides insight into the mechanisms of ILC2-mediated liver protection that could serve as therapeutic targets to treat acute liver injury.
Impact and Implications
We report that type 2 innate lymphoid cells (ILC2s) are important regulators in a mouse model of liver ischaemia/reperfusion injury (IRI). Through manipulation of macrophage and eosinophil phenotypes, ILC2s mitigate liver inflammation and injury during liver IRI. We propose that ILC2s have the potential to serve as a therapeutic tool for protecting against acute liver injury and lay the foundation for translation of ILC2 therapy to human liver disease.
Keywords: Innate lymphoid cells, Hepatic ischaemia/reperfusion injury, IL-33, Eosinophils, Macrophages
Graphical abstract
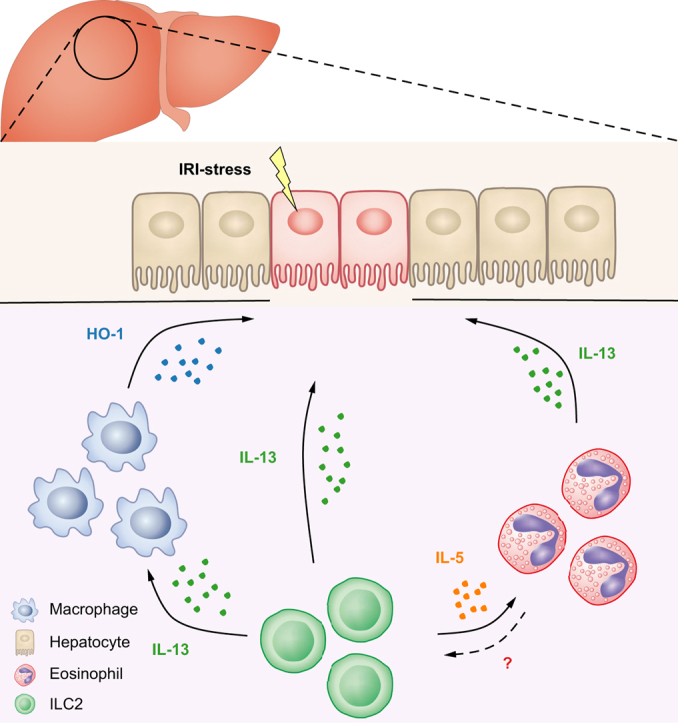
Highlights
-
•
ILC2s played critical roles in IL-33-mediated hepatoprotection in IRI mice.
-
•
IL-13 induction of anti-inflammatory Mφ and IL-5 elevation of eosinophils mediate the protective effect of ILC2s in vivo.
-
•
Human ILC2s protected against hepatic IRI in NSG mice, supporting their therapeutic potential in humans.
Introduction
Hepatic ischaemia/reperfusion injury (IRI) occurs in many clinical settings, such as liver surgery, trauma, and liver transplantation, and is often triggered by transient exposure of the liver to hypoxia followed by reperfusion with oxygenated blood.1 IRI can cause acute liver tissue damage resulting in significantly increased patient mortality and morbidity. In hepatic IRI, innate immune cells, such as neutrophils, macrophages, and natural killer (NK) cells, infiltrate the liver following reperfusion and produce inflammatory mediators, leading to hepatocyte death and necrosis, and ultimately organ failure.2,3
In recent years, a new group of innate immune cells, called innate lymphoid cells (ILCs) have gained much attention in inflammatory diseases.4,5 ILCs can be classified into three subgroups by their lineage-defining transcription factors and effector cytokines, termed ILC1, ILC2, and ILC3, which are distributed broadly in non-barrier organs, including the liver, kidney, and nervous system.6 Hepatic ILCs exert important functions in balancing inflammatory and reparative responses during tissue injury.7,8 In chronic hepatitis B, liver ILC1s have a pro-inflammatory function, whereas in acute liver injury induced by carbon tetrachloride, they are involved in the repair process and promote survival of damaged hepatocytes through ILC1-derived interferon-γ (IFN-γ).9,10 A larger number of ILC3s are present in the liver and play a profibrotic role in the carbon tetrachloride-induced liver fibrosis model.11 As ILC2 is the most abundant ILC subset in human and mouse liver tissue, its role under disease conditions has attracted increasing attention. Interestingly, ILC2s in the liver exhibit opposing effects in different hepatic injuries. One study showed that ILC2s protect against liver injury at the immune contraction stage of lymphocytic choriomeningitis virus infection-induced viral hepatitis.12 By contrast, liver-resident ILC2s aggravated inflammation and tissue damage in concanavalin A (ConA)-induced hepatitis.13 The underlying mechanism, however, is not clear. Meanwhile, IL-33, an ‘alarmin’ protein released from damaged hepatic parenchymal cells, can activate ILC2s in response to inflammation or damage.[14], [15], [16] Previous studies have shown that IL-33 was involved in a variety of acute liver diseases, but whether IL-33 promotes or inhibits disease progression in liver injury remains controversial. As a pathogenic factor, IL-33 released by necrotic hepatocytes promotes liver injury by neutrophil infiltration in acetaminophen-induced liver injury.16 By contrast, IL-33 has a protective role in hepatic IRI and Con A-induced acute hepatitis by inducing anti-apoptotic effects on hepatocytes and increasing liver regulatory T cells (Tregs), respectively.14,15 Given these contradictory results, further study is needed to better understand the importance of IL-33/ILC2-mediated immune responses in liver injury and to elucidate the mechanisms for these discrepancies.
In the current study, we uncovered the critical role of the IL-33–ILC2 pathway in protection against hepatic IRI. We revealed that ILC2s exert hepatoprotection against IRI by promoting IL-13-dependent induction of anti-inflammatory macrophages and IL-5-dependent elevation of eosinophils. Importantly, we demonstrated that ex vivo-expanded human ILC2s attenuated hepatic IRI in NSG mice. Therefore, manipulating IL-33–ILC2 responses may be a novel therapeutic strategy in the prevention and treatment of acute liver injury.
Materials and methods
Mice
BALB/c, C57BL/6 (CD45.2+), congenic C57BL/6 (CD45.1+), and NOD-scid IL2rγnull (NSG) mice were purchased from the Australian BioResources (Sydney, Australia). ICOS-T (Icosdtr/+ Cd4cre/+) mice and depletion of regulatory T cells (DEREG) (C57BL/6-Tg23.2Spar/Mmjax) mice were bred at Westmead Hospital Animal House and Australian BioResources (Sydney, Australia). For all studies, adult (8–12 weeks of age) male mice were used in accordance with the animal care and use protocol approved by the Animal Ethics Committee of Western Sydney Local Health District.
Hepatic IRI murine model and IL-33 administration
Partial hepatic ischaemia was induced as previously described.17 In brief, fasted mice were anaesthetised with a ketamine/xylazine mixture. After a midline laparotomy, mice were injected with heparin, and an atraumatic clip (Roboz, Gaithersburg, MD, USA) was placed across the portal vein and hepatic artery to interrupt blood supply to the left lateral/median lobes (70%) of the liver. The mice were placed on a heating pad to maintain body temperature at 37 °C and kept well hydrated with warm saline. After 60 min of partial hepatic ischaemia, the clip was removed to initiate reperfusion. Sham-operated mice underwent the same surgical procedure without vascular occlusion. Mice were euthanised at indicated time points after reperfusion to collect blood and tissues for further analysis.
For IL-33 treatment, C57BL/6 (male, 8–12 weeks of age) mice were administered 0.4-μg mouse recombinant IL-33 (BioLegend, San Diego, CA, USA) i.p. daily for 5 consecutive days before IRI surgery. The dose and duration were selected according to previous published studies.[18], [19], [20] Control animals received PBS only. To deplete Tregs or ILC2, DEREG or ICOS-T mice were injected i.p. with diphtheria toxin (DT; 30 mg/kg, Sigma Aldrich, Castle Hill, NSW, Australia) at Days -5, -3, and -1 before ischaemia. Depletion of Tregs and ILC2s was verified using flow cytometry. For eosinophil depletion, mice were injected i.p. with anti-C–C motif chemokine receptor 3 (CCR3) antibody (350 μg/mouse, clone 6S2-19-4) or control IgG three times before ischaemia.21 Mice were humanely culled at the indicated time points. Blood and tissues were harvested for analysis. Serum alanine aminotransferase (ALT) levels were measured using an ALT kit (Thermo Fisher, North Ryde, NSW, Australia) according to the manufacturer’s instructions.
Murine ILC2 cell expansion and adoptive transfer to IRI mice
ILC2s were isolated from the liver of C57BL/6 or BALB/c mice treated with IL-33 daily for 5 consecutive days. ILC2s were cultured in Royal Park Memorial Institute (RPMI) 1640 medium, supplemented with 10% FBS, penicillin (100 U/ml), and streptomycin (100 μg/ml), plus IL-2 (20 ng/ml), IL-7 (20 ng/ml), and IL-33 (50 ng/ml) for 14 days. Cell-free supernatants were assessed for IL-5 and IL-13 cytokine production by ELISA.
For ILC2 treatment, 5 × 106 ILC2s were transferred into C57BL/6 mice by a single tail-vein injection 1 day before ischaemia. Mice received recombinant murine IL-13 (1 μg; BioLegend) via the lateral tail vein 1 day before ischaemia. M2 macrophage depletion was induced by administration of GW2580 (BioVision, Waltham, MA, USA) once daily at a dose of 160 mg/kg by oral gavage for 2 consecutive days before ischaemia. For eosinophil depletion, mice were injected i.p. anti-CCR3 antibody (350 μg/mouse, clone 6S2-19-4) or control IgG for 2 consecutive days before ischaemia. In parallel, 5-(and 6)-carboxyfluorescein diacetate succinimidyl ester (CFSE)-labelled ILC2s (5 × 106) were transfused into C57BL/6 mice 1 day before sham or IRI surgery. All mice were euthanised at Day 1 after IRI surgery. The distribution of CFSE-labelled ILC2s was analysed in liver sections by fluorescence microscopy. The number of transfused ILC2s was quantitated in 8–10 nonoverlapping high-power fields. For in vivo expansion of ILC2s, NSG mice were injected with 0.5 × 106 ILC2s isolated from BALB/c mice at Day -5 before ischaemia and were administered mouse recombinant IL-33 (0.4 μg/mouse) i.p. daily for 5 consecutive days. Mice were humanely culled at the indicated time point.
Human ILC2 cell expansion and adoptive transfer to NSG mice
Human ILC2s isolated from donor peripheral blood mononuclear cells were cultured in RPMI 1640 medium containing 100 U/ml penicillin–streptomycin, supplemented with 10% human AB serum, plus IL-2 (20 ng/ml), IL-7 (20 ng/ml), and IL-33 (50 ng/ml) for 14 days. Cell-free supernatants were assessed for IL-5 and IL-13 production by ELISA (R&D Systems, Minneapolis, MN, USA). For ILC2 treatment, 5 × 106 human ILC2s were transferred into NSG mice by a single tail-vein injection 1 day before hepatic ischaemia. In parallel, 0.5 × 106 human ILC2s were transfused into NSG mice 5 days before IRI surgery, and then human recombinant IL-33 (0.4 μg/mouse; BioLegend) was administered i.p. daily for 5 consecutive days. NSG mice were humanely culled at Day 1 after IRI surgery. Serum ALT levels were measured using an ALT kit (Thermo Fisher) according to the manufacturer’s instructions.
Cell suspension preparation
Spleen and liver draining lymph nodes were isolated, minced, and digested for 30 min at 37 °C in RPMI 1640 medium containing 1 mg/ml collagenase D (Roche, Basel, Switzerland) and 100 μg/ml DNase I (Roche). The digested cell suspension was then passed through a 70-μm cell strainer. Liver was perfused with saline before removal and digested with collagenase and DNase as previously described.22,23 The ischaemic lobes were cut into small pieces and digested in DMEM containing 1 mg/ml collagenase IV (Sigma Aldrich), and 100 μg/ml DNase I (Roche) for 30 min at 37 °C with intermittent agitation. Then, the mixture was dissociated using a gentle-MACS Dissociator (Miltenyi Biotec, Macquarie Park, NSW, Australia). The digested cell suspension was then filtered through a 70-μm cell strainer. F4/80+ macrophages were sorted from the liver by FACS. Sorted macrophages were used for real-time PCR analyses to detect macrophage phenotypes.
Flow cytometry and cell sorting
For FACS analysis of mouse cells, single-cell suspensions were stained with Fc block/anti-CD16/32 (2.4G2) and antibodies to CD45.2 (104), ST2 (RMST2-2), CD127 (A7R34), GATA3 (TWAJ), KLRG1 (2F1), CD90.2 (30-H12), and CD25 (PC61), as well as with antibodies to T cell, B cell, NK cell, monocyte/macrophage, dendritic cell, eosinophil, neutrophil, and erythroid cell lineages (referred to hereafter as ‘lin’): CD3 (145-2C11), CD5 (53-7.3), TCRβ (H57-597), TCRγδ (eBioGL3), CD19 (1D3), B220 (RA3-6B2), CD49b (DX5), CD11b (M1/70), CD11c (N418), FcεRIα (MAR-1), Gr-1 (RB6-8C5), and Ter-119. Other antibodies used in this study include CD4 (GK1.5), Foxp3 (FJK-16s), F4/80 (BM8), and Siglec-F (1RNM44N), as well as corresponding isotype controls, all purchased from eBioscience (North Ryde, NSW, Australia) or BioLegend. Cells were analysed on an LSRFortessa flow cytometer (BD Biosciences, Macquarie Park, NSW, AU). For intracellular cytokine analysis, liver leucocytes were pre-enriched by anti-CD45 microbeads (Miltenyi Biotec) and were then incubated with 500 ng/ml of ionomycin and 50 ng/ml of phorbol myristate acetate at 37 °C for 2 h. Brefeldin A (3 mg/ml) was then added to the wells and incubated for 3 h. Intracellular staining with antibodies against IL-13 (eBio13A) and IFN-γ (XMG1.2) was performed and analysed by flow cytometry. For FACS, single-cell suspensions were pregated on haematopoietic cells using anti-CD45.2 antibody; then, lineage markers were used to exclude immune cells, and DAPI was used to exclude dead cells. Mouse ILC2 cells (CD45+Lin-CD127+ST2+CD90+) were sorted using a FACSAria II (BD Biosciences). After sorting, cells were used for phenotypic and functional assays.
For FACS analysis or sorting of human ILC2s, single-cell suspensions were stained with antibodies to CD45 (HI30), CD127 (A019D5), CRTH2 (BM16), CD161 (HP-3G10), KLRG1 (13F12F2), and ST2 (HB12), as well as with antibodies to T cell, B cell, NK cell, monocyte/macrophage, dendritic cell, eosinophil, neutrophil, and erythroid cell lineages (referred to hereafter as ‘lin’): CD3 (UCHT1), TCRαβ (IP26), CD19 (HIB19), CD20 (2H7), CD14 (M5E2), CD16 (3G8), CD11b (ICRF44), CD11c (BU15), CD123 (6H6), CD56 (HCD56), and FcεRIα (AER-37), all purchased from BioLegend or BD Biosciences. Human ILC2 cells (CD45+Lin-CD127+CRTH2+CD161+) were sorted using a FACSAria II (BD Biosciences). After sorting, cells were used for phenotypic and functional assays. The transfused human ILC2s (CD45+CD127+CRTH2+) in the liver of NSG mice was analysed on an LSR Fortessa flow cytometer (BD Biosciences).
CRISPR-Cas9 transfection
ILC2s were transfected with clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated 9 (Cas9) plasmid or its control (Santa Cruz, CA, USA) in accordance with the manufacturer’s instructions. In brief, ILC2s were transfected with IL-5 (sc-421112), or IL-13 (sc-421086) CRISPR-Cas9 plasmid or its control (sc-418922) and incubated for 24 h. Media were replaced 24 h post transfection. Puromycin antibiotic (2 μg/ml) was added to allow for positive selection of transfected cells. IL-5 or IL-13 was measured in culture supernatant of ILC2 via ELISA.
ILC2 coculture with macrophages or eosinophils
Macrophages (CD45+F4/80+CD11b+) isolated from the liver by flow cytometry were cultured in RPMI 1640 medium, supplemented with 10% FBS, penicillin (100 U/ml), and streptomycin (100 μg/ml), plus 10 ng/ml macrophage colony-stimulating factor for 2 days. CRISPR-Cas9 plasmid transfected ILC2s (ILC2-C or ILC2–IL-13; 4 × 105 cells/well) were cocultured with liver macrophages (2 × 105 cells/well) for 6 h. Macrophage phenotype was examined by quantitative PCR (qPCR). Eosinophils (CD45+CD11b+Siglec-F+) were freshly isolated from the bone marrow. CRISPR-Cas9 plasmid-transfected ILC2s (ILC2-C or ILC2–IL-5; 2 × 105 cells/well) were cocultured with eosinophils (2 × 105 cells/well) in fresh medium without recombinant mouse IL-5 for 24 h. IL-13 expression in eosinophils was examined by flow cytometry. ILC2s (2 × 105 cells/well) were cultured with IL-5 or eosinophils (2 × 105 cells/well) for 24 h. The expression of IL-13 in ILC2s was measured by qPCR.
Primary culture of hepatocytes and simulated ischaemia
Mouse hepatocytes were isolated from the liver by in situ collagenase perfusion through the portal vein.14 In brief, mice were anaesthetised with inhalation anaesthesia, and livers were perfused in situ with Liver Perfusion Medium (Life Technologies, Carlsbad, CA, USA) followed by Liver Digest Medium (Life Technologies). Then, the liver was minced and strained through a sterile 100-μm nylon mesh. Hepatocytes were separated by Percoll gradient centrifugation followed by low-speed centrifugation. Hepatocytes were cultured with complete William’s E Medium plus maintenance supplement (Thermo Fisher) on a collagen-coated plate. To simulate IRI in vitro, hepatocytes were cultured with serum-free DMEM/F12 medium in a modular incubator chamber (BioSpherix, Parish, NY, USA) gassed with 1% O2, 5% CO2, and 94% N2. After incubating under hypoxia for 60 min, cells were incubated under normoxic conditions with 95% air and 5% CO2. ILC2 cells were cocultured with ischaemic hepatocytes for 12 h. The medium and cells were collected for further analysis. Apoptosis of hepatocytes at 12 h after the coculture was measured by staining with 7-AAD and Annexin V following the manufacturer’s protocol (BD Biosciences).
ELISA of cytokines
IL-4, IL-5, and IL-13 levels in sera and culture supernatants were assayed using an ELISA kit (eBioscience). ELISA was performed according to the manufacturer’s protocol.
Quantitative PCR
Total RNA was isolated from tissue or cells using the RNeasy Mini Kit (Qiagen, Clayton, VIC, Australia) and then reverse-transcribed into cDNA using the First Strand cDNA Synthesis Kit (Invitrogen, Riverstone, NSW, Australia). Real-time PCR was performed on the CFX96 Touch Real-Time PCR Detection System (Bio-Rad, South Granville, NSW, Australia) using the SYBR mastermix (Invitrogen). The data were normalised to housekeeping gene expression and quantified using the 2-ΔΔCt method. The primer sequences of the target genes are shown in Table S1.
Histology and immunofluorescence
Liver sections (5 μm) were stained with H&E for necrotic area examination and Sirius Red for determination of collagen deposition. All slides were blindly quantified in 10–12 high-power fields, where the percent necrosis or fibrosis was calculated from the total area of the tissue section. The data were then averaged to calculate the necrotic area or fibrosis for each mouse. To avoid selection bias, the areas to be viewed for morphometric analysis were anatomically identical for each section and were positioned before microscopic visualisation.
For immunofluorescence staining of ILC2s, frozen sections were stained with rabbit anti-mouse CD127 (1140A), polyclonal goat anti-mouse GATA3 (R&D Systems), and rat anti-mouse CD3e (17A2) antibodies, and then incubated with the secondary antibodies, namely, AF488 donkey anti-rabbit IgG, AF546 donkey anti-goat IgG, and AF647 donkey anti-rat IgG. For immunofluorescence staining of haem oxygenase-1 (HO-1)+ macrophages, rat anti-mouse F4/80 (BM8) and polyclonal rabbit anti-mouse HO-1 (ADI-SPA-894; Enzo Life Sciences, Farmingdale, NY, USA) were used as the primary antibodies and AF546 goat anti-rat IgG and AF488 goat anti-rabbit IgG as the secondary antibodies. For immunofluorescence staining of eosinophils, rat anti-mouse Siglec-F (1RNM44N) was used as the primary antibody and AF546 goat anti-rat IgG as the secondary antibody. Control rat, rabbit, and goat IgG to primary antibodies were included in staining. The sections were viewed under an FV1000 microscope (Olympus, Macquarie Park, NSW, Australia). The numbers of ILC2s [CD3(-)CD127+GATA3+], M2 macrophages [F4/80+/HO-1+], and Siglec-F+ eosinophils were quantitated in 8–10 nonoverlapping high-power fields of the liver sections.
Statistics
Statistical tests included unpaired, two-tailed Student’s t test using Welch’s correction for unequal variances and one-way ANOVA with Tukey’s multiple comparison test. Statistical analyses were performed using Prism (version 8, GraphPad, San Diego, CA, USA). Results are expressed as the mean ± SEM. A p <0.05 was considered statistically significant.
Results
IL-33 protected against hepatic IRI
We recently demonstrated that short-term IL-33 administration attenuated renal IRI.18 To determine whether IL-33 could modulate acute liver injury, we treated C57BL/6 mice with recombinant mouse IL-33 (0.4 μg/mouse/day, i.p.) for 5 consecutive days before hepatic IRI (Fig. 1A). IL-33-treated mice developed much milder liver injury at 24 h compared with PBS-treated mice, as demonstrated by marked decrease in serum levels of ALT and areas of hepatocyte necrosis (Fig. 1B and C). Hepatic IRI in wild-type (WT) mice led to significantly increased recruitment of neutrophils compared with sham mice, whereas Gr-1+/CD11b+ neutrophil accumulation in liver was significantly reduced in IRI mice treated with IL-33 (Fig. 1D). Sterile inflammation is a hallmark of hepatic IRI.2 Therefore, we examined the inflammatory response in IRI mice with IL-33 treatment. IL-33 treatment significantly reduced the production of serum pro-inflammatory cytokines/chemokines, including tumour necrosis factor-α, IL-1β, IL-6, monocyte chemoattractant protein-1 (MCP-1), and CXC motif chemokine ligand 1 (CXCL1), and mRNA levels of these cytokines/chemokines in the livers compared with those of PBS-treated IRI mice (Fig. 1E and F). In addition, IL-33 treatment also enhanced serum levels of the Th2-associated cytokines IL-4, IL-5, and IL-13 and the expression of hepatic IL-4, IL-5, and IL-13 (Fig. 1G and H). Collectively, these data demonstrate that IL-33 markedly attenuated liver inflammation and acute liver injury in IRI mice.
Fig. 1.

IL-33 protected against liver injury in IRI mice.
(A) C57BL/6 mice were administered mouse recombinant IL-33 daily for 5 consecutive days before hepatic ischaemia. (B) Liver injury was assessed by serum levels of ALT. (C) Representative histological H&E staining images and statistics showing necrotic areas in livers from sham mice, IRI mice treated with vehicle (PBS), or IRI mice treated with IL-33 1 day after IRI. Bar = 200 μm. (D) The accumulation of neutrophils in livers from these mice was assessed by flow cytometry. (E and F) The production of pro-inflammatory cytokines/chemokines in the serum and mRNA expression in the livers from these mice were measured. (G and H) IL-4, IL-5, and IL-13 in the serum (G) and their mRNA expression in the livers (H) of these mice were measured. Data shown are the mean ± SEM (n = 4–6 per group). Statistical significance was assessed using a one-way ANOVA. ∗p <0.05, ∗∗p <0.01, ∗∗∗p <0.001. ALT, alanine aminotransferase; IRI, ischaemia/reperfusion injury; TNF-α, tumour necrosis factor-α.
IL-33 induced ILC2s, eosinophils, and Tregs in IRI mice
To better understand the hepatoprotective function of IL-33, we performed a detailed analysis of the cellular immune response milieu in IL-33-treated mice with IRI. IL-33-mediated ILC2 expansion has been demonstrated in multiple anatomical sites where they regulate inflammation and promote repair.[18], [19], [20] Flow cytometric analysis of leucocytes isolated from the liver of naïve C57BL/6 mice revealed a population of Lin-CD127+GATA3+ST2+ ILC2s that comprised around 60% of CD45+Lin-CD127+ ILCs (Fig. 2A). Lin-CD127+GATA3+ST2+ cells in liver expressed CD90, KLRG1, and CD25 (Fig. 2B), a phenotype of ILC2 similar to that of the mouse lung and kidney.22,24 IL-33-treated IRI mice exhibited a massive increase in GATA3+ST2+ ILC2 frequencies and numbers (40-fold) as compared with PBS-treated controls (Fig. 2C and D). However, we did not observe an increase of total ILCs or ILC2s in the peripheral blood or liver of IRI mice in comparison with sham mice (Fig. S1). Immunofluorescence staining for CD3, CD127, and GATA-3 clearly identified CD3-GATA-3+CD127+ ILC2s in the interstitial and intravascular compartments of the livers of sham and PBS-treated IRI mice and IL-33-treated IRI mice. Quantitative analyses confirmed a marked expansion of ILC2s in the liver of the IL-33-treated group (Fig. 2F and G). ILC2 accumulation was accompanied by a significant increase in IL-5 and IL-13 mRNA expression in the livers of IL-33-treated mice (Fig. 1G and H). We also observed a significant increase of eosinophils, which have been shown to promote tissue repair and resolution of inflammation (Fig. 2G).25,26 As compared with ILC2s and eosinophils, Treg populations in the liver and liver draining lymph nodes were only modestly increased in response to IL-33 treatment (Fig. 2H and I). Therefore, these data indicate that IL-33 elicits the ILC2, eosinophil, and Treg response in the liver, which may drive IL-33-mediated hepatoprotection.
Fig. 2.

IL-33 induced ILC2s, eosinophils, and Tregs in the liver of IRI mice.
(A) Representative FACS analysis showing the gating strategy to identify CD45+Lin-CD127+ST2+GATA3+ ILC2 in mouse liver. (B) Histogram showing expression of CD90, KLRG1, and CD25 on liver ILC2 cells. Specific markers (red lines) and isotype controls (grey-filled areas) are shown. (C and D) Proportion and absolute number of ST2+GATA3+ ILC2 in the livers of sham, IRI + vehicle, or IRI + IL-33 mice. (E and F) Immunofluorescence staining for CD127 (green), GATA3 (red), and CD3 (magenta) in the liver sections of sham and IRI mice. The white arrowheads indicate ILC2s (CD3-CD127+GATA3+). Bar = 100 mm. (G) Proportion of Siglec-F+ eosinophils in the CD45+ leucocyte compartment from the liver were measured by flow cytometry. (H and I) Proportion of CD4+Foxp3+ Tregs in the CD4+ T-cell compartment from the liver (H) and liver draining lymph node (I) were measured by flow cytometry. Data shown are the mean ± SEM (n = 4–6 per group). Statistical significance was assessed using a one-way ANOVA. ∗∗∗p <0.001. FSC, forward scatter; hpf, high-power field; ICL2, type 2 innate lymphoid cell; IRI, ischaemia/reperfusion injury; SSC, side scatter; Treg, regulatory T cell.
Eosinophils, but not Tregs, are required for IL-33-mediated hepatoprotection in IRI mice
Eosinophils are known to be cytotoxic cells involved in host defence against parasitic infections and pathogenesis of allergic diseases.27 However, recent studies have shown that eosinophils also play a protective function during acute liver injury.25,26 To investigate the role of eosinophils in protection against hepatic IRI through delivery of IL-33, we depleted these cells by i.p. injection of an anti-CCR3 antibody as previously reported (Fig. 3A).21 Eosinophil depletion by anti-CCR3 antibody in IL-33-treated C57BL/6 mice was confirmed in the liver by flow cytometry and immunofluorescence staining (Fig. 3B and C). Eosinophil depletion worsened liver histological and functional injury in IRI mice with IL-33 treatment. However, the protective effect of IL-33 was only mildly reversed in eosinophil-depleted IRI mice (Fig. 3D–F). Next, we examined whether Tregs contributed to IL-33-mediated hepatoprotection in IRI mice. Transgenic DEREG mice were administered DT to selectively deplete Tregs during IL-33 administration (Fig. S2A). Tregs were effectively depleted from the liver of IRI mice (Fig. S2B and C). However, Treg depletion did not affect IL-33-mediated hepatoprotection in IRI, indicating that Tregs are unlikely to be important in IL-33-mediated protection against hepatic IRI (Fig. S2D–F).
Fig. 3.

Eosinophils were required for IL-33-mediated hepatoprotection in IRI mice.
(A) C57BL/6 mice were treated with mouse recombinant IL-33 daily for 5 consecutive days, as well as anti-CCR3 antibody or control rat IgG three times before hepatic IRI surgery. (B) Frequency of Siglec-F+ eosinophils in the CD45+ leucocyte compartment from the liver was assessed in control, IRI + vehicle, IRI + IL-33 + IgG, or IRI + IL-33 + anti-CCR3 mice. (C) Numbers of Siglec-F+ eosinophils in the liver section (number/hpf) were assessed by immunofluorescence staining. (D) Representative H&E-stained sections of livers from mice 1 day after IRI. Bar = 200 μm. (E and F) Liver necrosis areas and serum ALT levels were assessed in these mice. Data shown are the mean ± SEM (n = 4–6 per group). Statistical significance was assessed using a one-way ANOVA. ∗∗p <0.01, ∗∗∗p <0.001. ALT, alanine aminotransferase; CCR3, C–C motif chemokine receptor 3; hpf, high-power field; IRI, ischaemia/reperfusion injury.
ILC2s played critical roles in the hepatoprotective effect of IL-33 in IRI mice
ILC2s play important roles in tissue repair and immunoregulation.28,29 IL-33-mediated renoprotection in renal IRI is dependent on ILC2s. The potential contribution of ILC2 to IL-33-mediated protection of hepatic IRI was assessed in ICOS-T mice, in which administration of DT leads to selective depletion of ILC2s (Fig. 4A). Flow cytometric analyses confirmed that CD45+Lin-ST2+ ILC2s were effectively depleted from the liver of IRI mice with IL-33 treatment (Fig. 4B and C). Notably, the protective effect of IL-33 on liver histological and functional injury was significantly abolished in ILC2-depleted IRI mice, pointing towards a critical role for ILC2 in IL-33-mediated protection of liver injury following IRI (Fig. 4D–F). To further determine whether ILC2s are required for IL-33-mediated hepatoprotection, we injected IL-33 and ILC2s into NSG mice before IRI surgery; these NSG mice lack ILC2 as well as T cells, B cells, and functional NK cells (Fig. 4G). As expected, the proportion of ILC2s in the liver was not increased in response to daily doses of IL-33, but IL-33 induced a marked expansion of ILC2s in NSG mice reconstituted with ILC2s before IL-33 administration (Fig. 4H). In terms of outcome, IL-33-treated NSG mice were not protected from IRI-induced liver injury accompanied by no increase in the number of ILC2s in the liver. However, reconstitution with ILC2s in NSG mice (lacking ILC2s) restored the protective effect of IL-33 in hepatic IRI (Fig. 4I–K), suggesting that IL-33 prevents liver injury through induction of ILC2 expansion in NSG mice. We also found that eosinophils were significantly reduced in NSG mice compared with WT mice, and there was no increase of eosinophils in the livers of NSG mice treated with IL-33 and ILC2 (Fig. S3). These data identify ILC2s as key targets of IL-33 and further demonstrate that IL-33-expanded ILC2s play a major role in protecting against acute liver injury in IRI mice.
Fig. 4.

ILC2 played a key role in IL-33-mediated hepatoprotection in IRI mice.
(A) ICOS-T C57BL/6 mice were treated with mouse recombinant IL-33 daily for 5 consecutive days, as well as DT at Days -5, -3, and -1 before hepatic ischaemia. (B and C) Percentage of ST2+ ILC2 in the CD45+ leucocyte compartment from the livers of sham, IRI + vehicle, IRI + DT, IRI + IL-33, or IRI + IL-33 + DT mice. (D) Representative H&E-stained sections of livers from mice 1 day after IRI. Bar = 200 μm. (E and F) Liver necrosis areas and serum ALT levels were assessed in these mice. (G) NSG mice were injected with ILC2s isolated from BALB/c mice at Day -5 before ischaemia and were administered mouse recombinant IL-33 daily for 5 consecutive days. (H and I) Percentage of ST2+ ILC2 in the CD45+ leucocyte compartment from the livers of mice. (J) Representative H&E-stained sections of livers from mice 1 day after IRI. Bar = 200 μm. (K and L) Liver necrosis areas and serum ALT levels were assessed in these mice. Data shown are the mean ± SEM (n = 4–6 per group). Statistical significance was assessed using a one-way ANOVA. ∗∗∗p <0.001. ALT, alanine aminotransferase; DT, diphtheria toxin; ICL2, type 2 innate lymphoid cell; IRI, ischaemia/reperfusion injury; NSG, NOD-scid IL2rγnull.
ILC2s protected against hepatic IRI predominantly through IL-13 production and induction of anti-inflammatory macrophages
We then investigated the mechanism by which ILC2s protect mice against hepatic IRI in more detail. It is known that ILC2s produce IL-13, which has been shown to play a protective role in hepatic IRI.30 Therefore, we investigated whether IL-13 may confer the hepatoprotective effects of ILC2s during IRI. ILC2s isolated from livers after daily injections of IL-33 produced progressively increased amounts of IL-13 but not IFN-γ in vitro (Fig. 5A and B). Furthermore, ILC2s separated from C57BL/6 mice with IL-33 treatment were expanded in culture with IL-2/IL-7/IL-33 for 14 days (Fig. 5C). The cultured ILC2s maintained their expression of key markers, including ST2, GATA3, and CD90 (data now shown), and produced a large amount of IL-13 (Fig. 5D). To confirm the importance of IL-13 in ILC2-mediated protection of IRI, we deleted IL-13 in ILC2s using CRISPR-Cas9. ILC2s transfected with control empty vector produced a large amount of IL-13 in the supernatant, whereas ILC2s transfected with IL-13 CRISPR-Cas9 did not produce IL-13 (Fig. 5D). A greater number of transfused ILC2s were found in the IRI liver than in the sham liver, indicating that ILC2s tend to migrate to the damaged liver undergoing inflammatory response (Fig. S4). ILC2-treated hepatic IRI mice developed much milder liver injury than PBS-treated mice accompanied by a significant increase in IL-13 in the peripheral blood and liver (Fig. S5). IL-13 depletion in ILC2s markedly impaired their protective effect on hepatic IRI, suggesting a protective role for ILC2-derived IL-13 (Fig. 5E–H).
Fig. 5.

ILC2s protected against hepatic IRI through IL-13 production and induction of anti-inflammatory macrophages.
(A and B) Intracellular staining shows ILC2s harvested from livers at the specified time points after daily injections of IL-33 produce high levels of IL-13 and minimal amounts of IFN-γ. (A) Contains representative flow dot plots, and (B) shows percent of ILC2s expressing IL-13. (C) Liver ILC2 were isolated from C57BL/6 mice treated with or without IL-33 and cultured with IL-2, IL-7, and IL-33 for 14 days. The number of ILC2s was calculated at Days 3, 7, and 14. (D) ILC2s were transfected with control (ILC2-C) or IL-13 CRISPR-Cas9 (ILC2–IL-13). IL-13 was measured in culture supernatant of ILC2-C and ILC2–IL-13 via ELISA. (E) C57BL/6 mice were treated with transfected ILC2 1 day before ischaemia and with GW2580 daily for 3 consecutive days before ischaemia. (F) Representative H&E-stained sections of livers from mice 1 day after IRI. Bar = 200 μm. (G and H) Liver necrosis areas and serum ALT levels were assessed in these mice. (I) The mRNA expression of MR, arginase, HO-1, and FIZZ1 was quantified by qPCR in F4/80+ liver macrophages. (J and K) The numbers of HO-1+F4/80+ macrophages were assessed by immunofluorescence staining in liver sections. Bar = 100 μm. Data shown are the mean ± SEM (n = 4–6 per group). Statistical significance was assessed using Student’s t test or ANOVA. ∗∗∗p <0.001. ALT, alanine aminotransferase; HO-1, haem oxygenase-1; hpf, high-power field; ICL2, type 2 innate lymphoid cell; IFN-γ, interferon-γ; IRI, ischaemia/reperfusion injury; qPCR, quantitative PCR; MR, mannose receptor; WT, wild-type.
Macrophages play a critical role in the pathophysiology of liver IRI.3,31 Liver macrophages are very plastic and display variable functions in different hepatic microenvironments.32 GW2580, a cellular feline McDonough sarcoma kinase inhibitor, has been used to selectively deplete anti-inflammatory (M2) macrophages in vivo.33,34 Here, we found that M2 macrophage depletion by administration of GW2580 partially reversed the protective function of transfused ILC2s in IRI mice, suggesting that liver M2 macrophages play an important role in the protective function of ILC2s during hepatic IRI (Fig. 5F–H). Moreover, adoptive transfer of ILC2s, but not IL-13-deleted ILC2s, elevated the expression of M2 macrophage markers (mannose receptor [MR], arginase, HO-1, Retnla resistin like alpha (FIZZ1), and IL-10) in liver macrophages (Fig. 5I). Immunofluorescence staining of HO-1+ macrophages in liver sections confirmed their increased accumulation in mice treated with ILC2s, which did not occur when the mice were treated with IL-13-deleted ILC2s (Fig. 5J and K). In addition, confirming our previous findings, liver macrophages, when cocultured with ILC2s, had increased expression of M2 macrophage markers, and the expression of these markers was reduced when cocultured with IL-13-deleted ILC2s (Fig. S6). ILC2s or IL-13 alone exhibits a mild protection in macrophage-depleted IRI mice, further confirming the importance of macrophages in ILC2-mediated hepatoprotection (Fig. S7). Therefore, IL-13 mediates the protective effects of ILC2s, and the IL-13/M2 macrophage axis is necessary in promoting the protective function of ILC2s, leading to our hypothesis that IL-13 produced by ILC2s induces M2 macrophages, which protect against liver injury. Of note, apoptosis of ischaemic hepatocytes was significantly reduced when cocultured with ILC2s, but not with IL-13-deleted ILC2s, indicating the direct protective role of ILC2-derived IL-13 in ischaemic hepatocyte damage (Fig. S8).
ILC2 protected against hepatic IRI partially through IL-5-dependent activation of eosinophils
To further study whether eosinophils are required for the protective function of ILC2s in IRI mice, we performed eosinophil depletion in WT mice by administration of anti-CCR3 antibody. ILC2s were adoptively transferred into eosinophil-depleted WT mice that were then subjected to liver IRI (Fig. 6A). We observed a significant increase of eosinophils in WT mice treated with ILC2s, which was prevented by injection of anti-CCR3 antibody (Fig. 6B). Eosinophil depletion partially impaired the protective effect of ILC2s in hepatic IRI mice, as demonstrated by the significant increase in ALT and hepatocyte necrosis (Fig. 6C–E). We also observed a significant increase of neutrophils in eosinophil-depleted mice (Fig. S9). IL-5 was significantly increased in the peripheral blood and liver of IRI mice treated with ILC2s (Fig. S10). To determine whether the increase of eosinophils and reduced liver IRI in mice were related to ILC2-derived IL-5, we deleted IL-5 in ILC2 using CRISPR-Cas9 (Fig. 6F). IL-5 depletion in ILC2s was confirmed in the supernatant by ELISA (Fig. 6F). IL-5 depletion in ILC2s impaired eosinophil accumulation in the liver (Fig. 6G) and partially reversed ILC2s-mediated protection in hepatic IRI, evident from increased values of ALT, and worsened histology, suggesting a protective role for ILC2-derived IL-5 (Fig. 6H–J). In vitro, eosinophils that were freshly isolated from the bone marrow, when cocultured with ILC2, had increased expression of IL-13, and the expression of IL-13 in eosinophils was reduced when cocultured with IL-5-deleted ILC2s (Fig. 6K and L). However, the expression of IL-13 in ILC2s was not increased when cocultured with IL-5 or eosinophils (Fig. S11). Taken together, these data show that the beneficial effects of ILC2 treatment in hepatic IRI can be partially attributed to IL-5-dependent accumulation and activation of eosinophils.
Fig. 6.

ILC2 protected against hepatic IRI partially through activation of eosinophils.
(A) C57BL/6 mice were treated with anti-CCR3 or control IgG at indicated time points followed by administration of ILC2s 1 day before ischaemia. (B) Percentage of Siglec-F+ eosinophils in the CD45+ leucocyte compartment from the livers of mice. (C–E) Liver necrosis areas and serum ALT levels were assessed 1 day after IRI. Bar = 200 μm. (F) ILC2s were transfected with control (ILC2-C) or IL-5 CRISPR-Cas9 (ILC2–IL-5). IL-5 was measured in culture supernatant via ELISA. (G) C57BL/6 mice were treated with transfected ILC2s (ILC2-C or ILC2–IL-5) 1 day before ischaemia and euthanised 1 day after IRI. Percentage of Siglec-F+ liver eosinophils in the CD45+ leucocyte compartment was measured by flow cytometry. (H–J) Liver necrosis areas and serum ALT levels were assessed 1 day after IRI. Bar = 200 μm. (K and L) Eosinophils were cultured with ILC2-C or ILC2–IL-5 for 24 h. The expression of IL-13 on eosinophils was measured by flow cytometry and qPCR. Data shown are the mean ± SEM (n = 4–6 per group). Statistical significance was assessed using Student’s t test or ANOVA. ∗∗p <0.01, ∗∗∗p <0.001. ALT, alanine aminotransferase; CCR3, C–C motif chemokine receptor 3; ICL2, type 2 innate lymphoid cell; IRI, ischaemia/reperfusion injury; MFI, mean fluorescence intensity; qPCR, quantitative PCR.
ILC2 were protective post IRI
For future translation, it is important to assess whether ILC2s are protective after liver injury has been initiated. ILC2s were administered 6 h after hepatic IRI surgery (Fig. S12A). The accumulation of infused ILC2s in the liver was only maintained at a high level for up to 7 days and significantly reduced by Day 14, indicating that the ILC2 increase was transient in the liver (Fig. S12B). As expected, ILC2 treatment attenuated IRI-induced liver injury, evident from decreased values of ALT and areas of hepatocyte necrosis at different time points after IRI (Fig. S12C and D). The effect of ILC2s on liver fibrosis was also assessed from Week 1 to Week 8 after IRI. Histological staining with Sirius Red revealed that ILC2 treatment significantly reduced liver fibrosis in the post-ischaemic liver of IRI mice (Fig. S12E and F). Treatment with ILC2 also reduced gene expression of fibrogenic markers α-smooth muscle actin and collagen-1α1 (Fig. S12G).
Human ILC2s reduced hepatic IRI in NSG mice
In the light of potential therapeutic applications, we next evaluated whether human ILC2s could be used to prevent hepatic IRI in NSG mice. Human ILC2s separated from peripheral blood mononuclear cells were effectively expanded in in vitro culture with IL-2/IL-7/IL-33 for 14 days (Fig. 7A), a method that has been established in our group.18 The ex vivo-expanded human ILC2s maintained their expression of key markers, including prostaglandin D2 receptor 2 (CRTH2), interleukin 1 receptor-like 1 (ST2), and killer cell lectin like receptor G1 (KRLG1) (Fig. 7B), and produced large amounts of IL-5 and IL-13 (Fig. 7C). The in vivo function of ex vivo-expanded human ILC2s or human ILC2s expanded in vivo by administration of human recombinant IL-33 was examined in NSG mice with hepatic IRI (Fig. 7D and E). The transfused human ILC2 cells were identified in livers of NSG mice with IRI by flow cytometry (Fig. 7F). There was a greater percentage of human ILC2s in the CD45+ leucocyte compartment from the livers of NSG mice that received short-term human IL-33 treatment (Fig. 7G). Human IL-5 and IL-13 were significantly increased in the peripheral blood of IRI mice transfused with human ILC2s (Fig. S13). Treatment with either ex vivo-expanded human ILC2s or in vivo IL-33-expanded human ILC2s significantly attenuated liver injury in NSG mice with IRI, indicating that human ILC2s can effectively prevent IRI. Human IL-33/ILC2-treated mice (20% of human ILC2s) developed much milder liver injury compared with human ILC2-treated mice (10% of human ILC2s), suggesting a dose-dependent protective effect of ILC2s in hepatic IRI (Fig. 7H–J). In addition, macrophages isolated from the liver of NSG IRI mice with human ILC2 or IL-33/ILC2 treatment had enhanced expression of M2 macrophage markers, including MR, arginase, HO-1, and FIZZ1 (Fig. 7K). These findings further supported our hypothesis that human ILC2s can effectively prevent hepatic IRI.
Fig. 7.

Human ILC2s reduced hepatic IRI in NSG mice.
(A) Human ILC2 were isolated from peripheral blood mononuclear cells and cultured with IL-2, IL-7, and IL-33 for 14 days. The number of ILC2s was calculated at indicated time points. (B) Histogram showing expression of CRTH2, ST2, and KLRG1 on human ILC2 cells. Specific markers (red lines) and isotype controls (grey-filled areas) are shown. (C) IL-5 and IL-13 were measured in culture supernatant via ELISA. (D) NSG mice were treated with human ILC2 (5 × 106) 1 day before IRI. (E) NSG mice were given with human ILC2s (0.5 × 106) at Day -5 before ischaemia followed by administration of human recombinant IL-33 daily for 5 consecutive days. (F) Representative FACS analysis showing the gating strategy to identify human CD45+CD127+CRTH2+ ILC2 in the liver of NSG mice. (G) Percentage of CD127+CRTH2+ ILC2s in the CD45+ leucocyte compartment from the livers were measured 1 day after IRI. (H–J) Liver necrosis areas and serum ALT levels were assessed 1 day after IRI. (K) The mRNA expression of MR, arginase, HO-1, and FIZZ1 was quantified by qPCR in F4/80+ liver macrophages. Data shown are the mean ± SEM (n = 4–8 per group). Statistical significance was assessed using Student’s t test or ANOVA. ∗p <0.05, ∗∗p <0.01, ∗∗∗p <0.001. ALT, alanine aminotransferase; HO-1, haem oxygenase-1; ICL2, type 2 innate lymphoid cell; IRI, ischaemia/reperfusion injury; MR, mannose receptor; NSG, NOD-scid IL2rγnull; qPCR, quantitative PCR.
Discussion
The role of IL-33/ILC2s in liver injury has been examined in very few studies with conflicting results. A study of ConA-induced hepatitis in mice demonstrated that ILC2 depletion by an anti-CD90.2 antibody reduced liver injury, suggesting a pathologic role of ILC2s in this context.13 However, this finding is contradictory to the protective role of the IL-33/ST2 axis in ConA-induced hepatitis.15 Two other studies showed that ILC2s protect against liver injury in adenovirus- and lymphocytic choriomeningitis virus-induced viral hepatitis.12,35 Our experiments using genetic models of ILC2 deficiency (ICOS-T or NSG mice) or adoptive transfer of genetically modified ILC2s (IL-5- or IL-13-deficient ILC2s) revealed a critical protective role for ILC2s and their underlying mechanisms in murine hepatic IRI. Furthermore, our data also showed that ex vivo-expanded human ILC2s protected against hepatic IRI in NSG mice, supporting their therapeutic potential in humans.
Our earlier studies showed that exogenous IL-33, administered before reperfusion, induced the expansion of ILC2s and subsequently prevented renal IRI via induction of anti-inflammatory macrophages.18 Anti-inflammatory macrophages have been shown to play a HO-1 pathway-dependent protective role during ischaemic-induced liver injury.36,37 Based on these studies, we hypothesised that ILC2-derived IL-13 may alleviate hepatic IRI through modulating macrophage phenotype. Our experiments using adoptive transfer of IL-13-/- ILC2s and depletion of macrophages showed that the ILC2 effect, at least in large part, can be attributed to production of IL-13 and induction of M2 macrophages. These results are supported by recent work demonstrating that ILC2s appear to regulate the polarisation of macrophages to alleviate hepatic IRI after IL-33 administration.38 Although this study did not investigate the underlying mechanisms, we have demonstrated a critical role for ILC2–IL-13–macrophage pathways in mediating this protection. Moreover, our work suggests that IL-13 may possess an additional protective role in other cells. We demonstrated that ILC2-derived IL-13 acted directly on ischaemic hepatocytes to prevent apoptosis, which is consistent with previous work showing that endogenous IL-13 protected hepatocytes from hydrogen peroxide-induced cytotoxicity.30 We also demonstrated decreased neutrophil infiltration in IL-33-treated IRI mice, which could attribute to reduction of pro-inflammatory cytokines/chemokines in the peripheral blood and liver. IL-13 has been shown to suppress neutrophil accumulation in an injured liver by regulating neutrophil transendothelial migration,12,30 suggesting that ILC2-derived IL-13 may contribute to this reduction in our model.
There is emerging evidence supporting the finding that the eosinophil is an important regulator of local immunity and tissue repair rather than a simple proinflammatory cell type.25,26,39 Indeed, recent studies have shown that eosinophils play a protective role during acute liver injury and acute lung injury.25,26,40 Here, we demonstrated that eosinophils are required for both IL-33- and ILC2-mediated hepatoprotection in hepatic IRI. ILC2s are important regulators of eosinophil recruitment.41 A previous study has shown that ILC2s promote the persistence of airway eosinophilia in patients with severe asthma through localised production of type 2 cytokine IL-5.42 To better understand how ILC2s work and whether they regulate eosinophil phenotypes via secretion of IL-5, we went on to analyse the interaction between ILC2s and eosinophils in vivo and in vitro. Of note, our data revealed that ILC2s produced the high levels of IL-5, promoting eosinophil accumulation and leading to protection against hepatic IRI. Our finding that eosinophils accumulate within 24 h after ILC2 injection strongly suggests that ILC2s drive eosinophil recruitment to the liver. In vitro, we further demonstrated that ILC2s activate eosinophils via IL-5, evident from upregulation of IL-13 expression on eosinophils. Eosinophil-derived IL-13 could further attenuate hepatic IRI by inhibiting neutrophil infiltration.26 Eosinophils are a major cellular source of IL-4. One study found that the eosinophil–ILC2 interplay resulted in the activation of ILC2s in an IL-4-dependent manner.43 Therefore, the crosstalk between ILC2s and eosinophils might be critical to enhance the protective effect of ILC2s in hepatic IRI. Although the role of ILC2 in promoting eosinophil accumulation has been reported,41,42 our study is the first to demonstrate that ILC2s protect against liver IRI through acting on eosinophils and initiate their accumulation and activation via production of IL-5.
IRI is a common clinically significant problem in many different organ systems, including the liver, kidney, brain, heart, and lung. In response to IRI, numerous studies have indeed described how ILC2s become licensed to protect against injury in various organs through suppressing the onset of immune responses or enhancing tissue regeneration.18,22,38,44 We recently demonstrated for the first time that the ILC2-activating cytokine IL-25, IL-33, or ex vivo-expanded ILC2s could reduce tubular cell injury and improve kidney function in renal IRI.18,22 In the context of hepatic IRI, and consistent with our results, other groups have shown that ILC2s play a protective role by modulating macrophage polarisation.38 Furthermore, ILC2s demonstrated a direct and protective role in the recovery of experimental myocardial infarction,44 a condition similar to IRI, via ILC2-derived IL-5 and reparative heart macrophages. Until now, the direct role of ILC2-derived IL-5 and IL-13 (and downstream pathways) was not clear. Our study clearly identified two main signature pathways of ILC2-mediated protection in hepatic IRI: ILC2/IL-13/macrophage and ILC2/IL-5/eosinophil. To date, therapies targeting individual mechanisms of IRI pathology (e.g. N-acetylcysteine for reactive oxygen species [ROS]45 or prostaglandin E1 as a hepatoprotective agent46) have been largely ineffective. Successful therapies to alleviate IRI must therefore act on many of the mechanisms by which IRI causes organ rejection, parenchymal cell death, ROS, inflammatory cytokine induction, and immune activation. As intrinsic mediators of protective and regenerative responses in many tissues, ILC2s possess the ability to significantly reduce IRI by acting on parenchymal and immune cell populations alike. Herein, we have shown that ILC2s reduce necrotic area, inflammatory cytokine expression, and influx of ROS-producing neutrophil populations while shifting the hepatic milieu towards an anti-inflammatory phenotype. Moreover, we have shown that human ILC2s can be expanded in vitro, facilitating their use as an autologous cell therapy for patients undergoing liver transplant or resection. Although no human ILC-based cell therapy studies have been conducted to date, adoptive cell transfers have been conducted using other innate cell populations such as NK cells,47 supporting their feasibility for clinical use. The clinical utility of ILC2s is supported by our data herein, demonstrating that adoptive transfer of human ILC2s ameliorated liver injury in NSG mice with hepatic IRI, which is consistent with our previous study showing the protective function of human ILC2s in humanised mice with renal IRI.18
A concern regarding ILC2s as a therapeutic approach is that they may cause organ fibrosis.48,49 Hepatic resident ILC2s have been shown to mediate chronic liver fibrosis via IL-4Rα and STAT6-dependent signalling pathways.48 Our data demonstrated that adoptive transfer of ILC2s post hepatic IRI reduced the development of liver fibrosis, which could be explained by ILC2s attenuating early liver inflammation and injury, and subsequently reducing fibrosis in the late stage of IRI. Importantly, the number of transfused ILC2s dropped significantly by 14 days post cell transfer, suggesting that they do not persist long-term within the hepatic microenvironment. Therefore, our data suggest that ILC2 therapy will not exacerbate the development of fibrosis following chronic insults.
The present study uncovered a highly protective function of ILC2s in hepatic IRI and provides novel insights into the molecular and cellular mechanisms underlying ILC2-medicated hepatoprotection. Specifically, the data revealed that IL-13-dependent induction of anti-inflammatory macrophages and IL-5-dependent elevation of eosinophils mediated the protective effect of ILC2s. Furthermore, human ILC2 effectively reduced liver injury in an immunocompromised mouse model of hepatic IRI. These findings suggest that strategies to promote ILC2 recruitment and/or adoptive transfer of ILC2s may represent a novel cell-based therapeutic approach to improve the outcomes of liver surgery and transplantation.
Financial support
This work was supported by the National Health and Medical Research Council of Australia (NHMRC; grants 1146156, 2008347, and 1195437), the National Natural Science Foundation of China (Nos. 81770721 and 81671752), and the Natural Science Foundation of Anhui Province (2208085MH207 and 2022AH040189).
Authors’ contributions
Designed and supervised the study: QC, YW, DCHH. Performed animal experiments and in vitro experiments, and analysed data: QC, RW, YW, ZN, TC, JC, FA, CZ, VWSL, QH. Contributed to analysis of IRI models: QC, RW, YW, ZN, VWSL, SR, YMW, QH. Provided reagents and expertise: MRS, SR, VWSL, SJ, GZ. Participated in discussions, provided intellectual input, and wrote the paper: QC, RW, MRS, SR, GZ, SIA, JG, YW and DCHH.
Data availability statement
Data are available on reasonable request to the corresponding author.
Conflicts of interest
The authors declare that no conflicts of interest exist.
Please refer to the accompanying ICMJE disclosure forms for further details.
Acknowledgements
We thank Andrew McKenzie for generating and providing ICOS-T mice. We also thank Maggie Wang, Edwin Lau, Suat Dervish, and Hong Yu from Westmead Core Facilities for technical assistance.
Footnotes
Author names in bold designate shared co-first authorship
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jhepr.2023.100837.
Contributor Information
Qi Cao, Email: qi.cao@sydney.edu.au.
David C.H. Harris, Email: david.harris@sydney.edu.au.
Supplementary data
The following are the supplementary data to this article.
References
- 1.Zhai Y., Petrowsky H., Hong J.C., Busuttil R.W., Kupiec-Weglinski J.W. Ischaemia–reperfusion injury in liver transplantation – from bench to bedside. Nat Rev Gastroenterol Hepatol. 2013;10:79–89. doi: 10.1038/nrgastro.2012.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hirao H., Nakamura K., Kupiec-Weglinski J.W. Liver ischaemia-reperfusion injury: a new understanding of the role of innate immunity. Nat Rev Gastroenterol Hepatol. 2022;19:239–256. doi: 10.1038/s41575-021-00549-8. [DOI] [PubMed] [Google Scholar]
- 3.Krenkel O., Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol. 2017;17:306–321. doi: 10.1038/nri.2017.11. [DOI] [PubMed] [Google Scholar]
- 4.Eberl G., Colonna M., Di Santo J.P., McKenzie A.N.J. Innate lymphoid cells. Innate lymphoid cells: a new paradigm in immunology. Science. 2015;348:aaa6566. doi: 10.1126/science.aaa6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sonnenberg G.F., Artis D. Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat Med. 2015;21:698–708. doi: 10.1038/nm.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vivier E., Artis D., Colonna M., Diefenbach A., Di Santo J.P., Eberl G., et al. Innate lymphoid cells: 10 years on. Cell. 2018;174:1054–1066. doi: 10.1016/j.cell.2018.07.017. [DOI] [PubMed] [Google Scholar]
- 7.Shen Y., Li J., Wang S.Q., Jang W. Ambiguous roles of innate lymphoid cells in chronic development of liver diseases. World J Gastroenterol. 2018;24:1962–1977. doi: 10.3748/wjg.v24.i18.1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu M., Zhang C. The role of innate lymphoid cells in immune-mediated liver diseases. Front Immunol. 2017;8:695. doi: 10.3389/fimmu.2017.00695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang Z., Tang T., Wei X., Yang S., Tian Z. Type 1 innate lymphoid cells contribute to the pathogenesis of chronic hepatitis B. Innate Immun. 2015;21:665–673. doi: 10.1177/1753425915586074. [DOI] [PubMed] [Google Scholar]
- 10.Nabekura T., Riggan L., Hildreth A.D., O'Sullivan T.E., Akira Shibuya A. Type 1 innate lymphoid cells protect mice from acute liver injury via interferon-gamma secretion for upregulating Bcl-xL expression in hepatocytes. Immunity. 2020;52:96–108.e109. doi: 10.1016/j.immuni.2019.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang S., Li J., Wu S., Cheng L., Shen Y., Wei Ma W., et al. Type 3 innate lymphoid cell: a new player in liver fibrosis progression. Clin Sci (Lond) 2018;132:2565–2582. doi: 10.1042/CS20180482. [DOI] [PubMed] [Google Scholar]
- 12.Liang Y., Yi P., Yuan D.M.K., Jie Z., Kwota Z., Soong L., et al. IL-33 induces immunosuppressive neutrophils via a type 2 innate lymphoid cell/IL-13/STAT6 axis and protects the liver against injury in LCMV infection-induced viral hepatitis. Cell Mol Immunol. 2019;16:126–137. doi: 10.1038/cmi.2017.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Neumann K., Karimi K., Meiners J., Voetlause R., Steinmann S., Dammermann W., et al. A proinflammatory role of type 2 innate lymphoid cells in murine immune-mediated hepatitis. J Immunol. 2017;198:128–137. doi: 10.4049/jimmunol.1600418. [DOI] [PubMed] [Google Scholar]
- 14.Sakai N., Van Sweringen H.L., Quillin R.C., Schuster R., Blanchard J., Burns J.M., et al. Interleukin-33 is hepatoprotective during liver ischemia/reperfusion in mice. Hepatology. 2012;56:1468–1478. doi: 10.1002/hep.25768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Volarevic V., Mitrovic M., Milovanovic M., Zelen I., Nikolic I., Mitrovic S., et al. Protective role of IL-33/ST2 axis in Con A-induced hepatitis. J Hepatol. 2012;56:26–33. doi: 10.1016/j.jhep.2011.03.022. [DOI] [PubMed] [Google Scholar]
- 16.Yazdani H.O., Chen H.W., Tohme S., Tai S., van der Windt D.J., Loughran P., et al. IL-33 exacerbates liver sterile inflammation by amplifying neutrophil extracellular trap formation. J Hepatol. 2018;68:130–139. doi: 10.1016/j.jhep.2017.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abe Y., Hines I.N., Zibari G., Pavlick K., Gray L., Kitagawa Y., et al. Mouse model of liver ischemia and reperfusion injury: method for studying reactive oxygen and nitrogen metabolites in vivo. Free Radic Biol Med. 2009;46:1–7. doi: 10.1016/j.freeradbiomed.2008.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao Q., Wang Y., Niu Z., Wang C., Wang R., Zhang Z., et al. Potentiating tissue-resident type 2 innate lymphoid cells by IL-33 to prevent renal ischemia-reperfusion injury. J Am Soc Nephrol. 2018;29:961–976. doi: 10.1681/ASN.2017070774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gadani S.P., Smirnov I., Smith A.T., Overall C.C., Kipnis J. Characterization of meningeal type 2 innate lymphocytes and their response to CNS injury. J Exp Med. 2017;214:285–296. doi: 10.1084/jem.20161982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rak G.D., Osborne L.C., Siracusa M.C., Kim B.S., Wang K., Bayat A., et al. IL-33-dependent group 2 innate lymphoid cells promote cutaneous wound healing. J Invest Dermatol. 2016;136:487–496. doi: 10.1038/JID.2015.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O'Connell A.E., Hess J.A., Santiago G.A., Nolan T.J., Lok J.B., Lee J.J., et al. Major basic protein from eosinophils and myeloperoxidase from neutrophils are required for protective immunity to Strongyloides stercoralis in mice. Infect Immun. 2011;79:2770–2778. doi: 10.1128/IAI.00931-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang Q., Niu Z., Tan J., Yang J., Liu Y., Ma H., et al. IL-25 elicits innate lymphoid cells and multipotent progenitor type 2 cells that reduce renal ischemic/reperfusion injury. J Am Soc Nephrol. 2015;26:2199–2211. doi: 10.1681/ASN.2014050479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cao Q., Wang Y., Wang X.M., Lu J., Lee V.W.S., Ye Q., et al. Renal F4/80+ CD11c+ mononuclear phagocytes display phenotypic and functional characteristics of macrophages in health and in adriamycin nephropathy. J Am Soc Nephrol. 2015;26:349–363. doi: 10.1681/ASN.2013121336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Monticelli L.A., Sonnenberg G.F., Abt M.C., Alenghat T., Ziegler C.G.K., Doering T.A., et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol. 2011;12:1045–1054. doi: 10.1031/ni.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu L., Yang Y., Wen Y., Jeong J.M., Emontzpohl C., Atkins C.L., et al. Hepatic recruitment of eosinophils and their protective function during acute liver injury. J Hepatol. 2022;77:344–352. doi: 10.1016/j.jhep.2022.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang Y., Yang Y., Wang M., Wang S., Jeong J.M., Xu L., et al. Eosinophils attenuate hepatic ischemia–reperfusion injury in mice through ST2-dependent IL-13 production. Sci Transl Med. 2021;13 doi: 10.1126/scitranslmed.abb6576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klion A.D., Nutman T.B. The role of eosinophils in host defense against helminth parasites. J Allergy Clin Immunol. 2004;113:30–37. doi: 10.1016/j.jaci.2003.10.050. [DOI] [PubMed] [Google Scholar]
- 28.Brestoff J.R., Kim B.S., Saenz S.A., Stine R.R., Monticelli L.A., Sonnenberg G.F., et al. Group 2 innate lymphoid cells promote beiging of white adipose tissue and limit obesity. Nature. 2015;519:242–246. doi: 10.1038/nature14115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang Q., Ma X., Wang Y., Niu Z., Wang R., Yang F., et al. IL-10 producing type 2 innate lymphoid cells prolong islet allograft survival. EMBO Mol Med. 2020;12 doi: 10.15252/emmm.202012305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kato A., Okaya T., Lentsch A.B. Endogenous IL-13 protects hepatocytes and vascular endothelial cells during ischemia/reperfusion injury. Hepatology. 2003;37:304–312. doi: 10.1053/jhep.2003.50075. [DOI] [PubMed] [Google Scholar]
- 31.Zhang M., Nakamura K., Kageyama S., Lawal A.O., Gong K.W., Bhetraratana M., et al. Myeloid HO-1 modulates macrophage polarization and protects against ischemia-reperfusion injury. JCI Insight. 2018;3 doi: 10.1172/jci.insight.120596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tacke F., Zimmermann H.W. Macrophage heterogeneity in liver injury and fibrosis. J Hepatol. 2014;60:1090–1096. doi: 10.1016/j.jhep.2013.12.025. [DOI] [PubMed] [Google Scholar]
- 33.Pyonteck S.M., Akkari L., Schuhmacher A.J., Bowman R.L., Sevenich L., Quail D.F., et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19:1264–1272. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Klinkert K., Whelan D., Clover A.J.P., Leblond A.L., Kumar A.H.S., Caplice N.M. Selective M2 macrophage depletion leads to prolonged inflammation in surgical wounds. Eur Surg Res. 2017;58:109–120. doi: 10.1159/000451078. [DOI] [PubMed] [Google Scholar]
- 35.Liang Y., Jie Z., Hou L., Aguilar-Valenzuela R., Vu D., Soong L., et al. IL-33 induces nuocytes and modulates liver injury in viral hepatitis. J Immunol. 2013;190:5666–5675. doi: 10.4049/jimmunol.1300117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ke B., Shen X.D., Gao F., Ji H., Qiao B., Zhai Y., et al. Adoptive transfer of ex vivo HO-1 modified bone marrow-derived macrophages prevents liver ischemia and reperfusion injury. Mol Ther. 2010;18:1019–1025. doi: 10.1038/mt.2009.285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakamura K., Zhang M., Kageyama S., Ke B., Fujii T., Sosa R., et al. Macrophage heme oxygenase-1-SIRT1-p53 axis regulates sterile inflammation in liver ischemia-reperfusion injury. J Hepatol. 2017;67:1232–1242. doi: 10.1016/j.jhep.2017.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang H.M., Chen X.J., Li S.P., Zhang J.M., Sun J., Zhou L.X., et al. ILC2s expanded by exogenous IL-33 regulate CD45+CD11b+F4/80high macrophage polarization to alleviate hepatic ischemia-reperfusion injury. Front Immunol. 2022;13 doi: 10.3389/fimmu.2022.869365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takeda K., Shiraishi Y., Ashino S., Han J., Jia Y., Wang M., et al. Eosinophils contribute to the resolution of lung-allergic responses following repeated allergen challenge. J Allergy Clin Immunol. 2015;135:451–460. doi: 10.1016/j.jaci.2014.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krishack P.A., Hollinger M.K., Kuzel T.G., Decker T.S., Louviere T.J., Hrusch C.L., et al. IL-33-mediated eosinophilia protects against acute lung injury. Am J Respir Cell Mol Biol. 2021;64:569–578. doi: 10.1165/rcmb.2020-0166OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nussbaum J.C., Van Dyken S.J., von Moltke J., Cheng L.E., Mohapatra A., Molofsky A.B., et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature. 2013;502:245–248. doi: 10.1038/nature12526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Smith S.G., Chen R., Kjarsgaard M., Huang C., Oliveria J.P., O'Byrne P.M., et al. Increased numbers of activated group 2 innate lymphoid cells in the airways of patients with severe asthma and persistent airway eosinophilia. J Allergy Clin Immunol. 2016;137:75–86.e78. doi: 10.1016/j.jaci.2015.05.037. [DOI] [PubMed] [Google Scholar]
- 43.Bal S.M., Bernink J.H., Nagasawa M., Groot J., Shikhagaie M.M., Golebski K., et al. IL-1β, IL-4 and IL-12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nat Immunol. 2016;17:636–645. doi: 10.1038/ni.3444. [DOI] [PubMed] [Google Scholar]
- 44.Yu X., Newland S.A., Zhao T.X., Lu Y., Sage A.S., Sun Y., et al. Innate lymphoid cells promote recovery of ventricular function after myocardial infarction. J Am Coll Cardiol. 2021;78:1127–1142. doi: 10.1016/j.jacc.2021.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thirunavayakalathil M.A., Varghese C.T., Bharathan V.K., Chandran B., Nair K., Mallick S., et al. Double-blind placebo-controlled randomized trial of N-acetylcysteine infusion following live donor liver transplantation. Hepatol Int. 2020;14:1075–1082. doi: 10.1007/s12072-020-10109-y. [DOI] [PubMed] [Google Scholar]
- 46.Bharathan V.K., Chandran B., Gopalakrishnan U., Varghese C.T., Menon R.N., Balakrishnan D., et al. Perioperative prostaglandin e1 infusion in living donor liver transplantation: a double-blind, placebo-controlled randomized trial. Liver Transpl. 2016;22:1067–1074. doi: 10.1002/lt.24479. [DOI] [PubMed] [Google Scholar]
- 47.Romee R., Rosario M., Berrien-Elliott M.M., Wagner J.A., Jewell B.A., Schappe T., et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. 2016;8 doi: 10.1126/scitranslmed.aaf2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McHedlidze T., Waldner M., Zopf S., Walker J., Rankin A.L., Schuchmann M., et al. Interleukin-33-dependent innate lymphoid cells mediate hepatic fibrosis. Immunity. 2013;39:357–371. doi: 10.1016/j.immuni.2013.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li D., Guabiraba R., Besnard A.G., Komai-Koma M., Jabir M.S., Zhang L., et al. IL-33 promotes ST2-dependent lung fibrosis by the induction of alternatively activated macrophages and innate lymphoid cells in mice. J Allergy Clin Immunol. 2014;134:1422–1432.e1411. doi: 10.1016/j.jaci.2014.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available on reasonable request to the corresponding author.