

Abstract
From studies on postmortem anatomical descriptions of the uveal vascular bed, it was generally concluded that occlusion of PCA or its branches should not produce an ischemic lesion. However, in vivo studies have recorded that the PCAs and their branches, right down to the terminal choroidal arterioles, and the choriocapillaris, have a segmental distribution in the choroid, and that PCAs and choroidal arteries function as end-arteries. This explains the basis of the occurrence of isolated inflammatory, ischemic, metastatic, and degenerative choroidal lesions, which are usually localized. Thus, in vivo studies have completely revolutionized our concept of the uveal vascular bed in disease.
Subject terms: Pathogenesis, Outcomes research, Microscopy
Introduction
“Who does not know that every scientific accomplishment dislodges some deeply rooted error and that behind it is usually concealed injured pride, if not enraged interest?” Ramón y Cajal (1923) [1]
The eyeball contains two sets of vascular beds: (i) retinal and (2) uveal vascular beds. With the advent of ophthalmoscopy in 1851, the retinal vascular bed has always been primarily the centre of the interest clinically, with little interest in the background uveal vascular bed since it is not visible on ophthalmoscopy. So, there is a massive amount of literature on the retinal vascular bed, having been the primary focus of interest all along. As regards the uveal vascular beds, however, apart from bits and pieces reported over the years, there is no full composite review published in any scientific ophthalmic journal based on the latest scientific advances. The objective of this review is to provide a comprehensive account of: (A) the anatomy of the posterior ciliary arteries (PCAs), anterior ciliary arteries (ACAs), cilioretinal arteries and vortex veins (VVs), and (B) lesions produced by their occlusion and acute uveal ischemic lesions seen clinically.
Before the advent of fluorescein fundus angiography (FFA) in 1961, our understanding of the uveal (ciliary) vascular bed since 1700 was based primarily on postmortem cast studies. In 1964, some enigmatic observations inspired me to use FFA to explore this vascular bed comprehensively.
In my FFA studies on central retinal artery occlusion in the early1960s, I noticed that the optic nerve head (ONH) showed vascular filling but no filling of the retinal vasculature (Fig. 1) [2, 3]; this finding contradicted the then prevalent concept that the ONH was supplied by the central retinal artery. Thus, this new finding showed for the first time that the ONH was supplied by the PCA circulation and not by the central retinal artery. And FFA also unveiled the in vivo filling pattern of the choroid.
There were reports in the literature in which ophthalmoscopically seen choroidal infarcts (initially whitish, and then resolving to chorioretinal pigmented lesions) were erroneously diagnosed as retinal infarcts due to branch retinal artery occlusion.
Duke-Elder [4] in 1961 rightly commented: “The tendency for inflammatory and degenerative diseases of the choroid to show a considerable degree of selective localization, despite the fact that anatomically (postmortem casts) the vessels would appear to form a continuous network, has given rise to speculations regarding the anatomical isolation of specific choroidal areas”. As discussed below, fluorescein fundus angiographic studies provided the answer and confirmed Duke-Elder’s views.
It was observed that the findings of postmortem casts are not always supported by the in vivo studies of the uveal vascular bed. As discussed below, FFA studies amply showed that.
Fig. 1. Fluorescein fundus angiogram of an eye with central retinal artery occlusion.
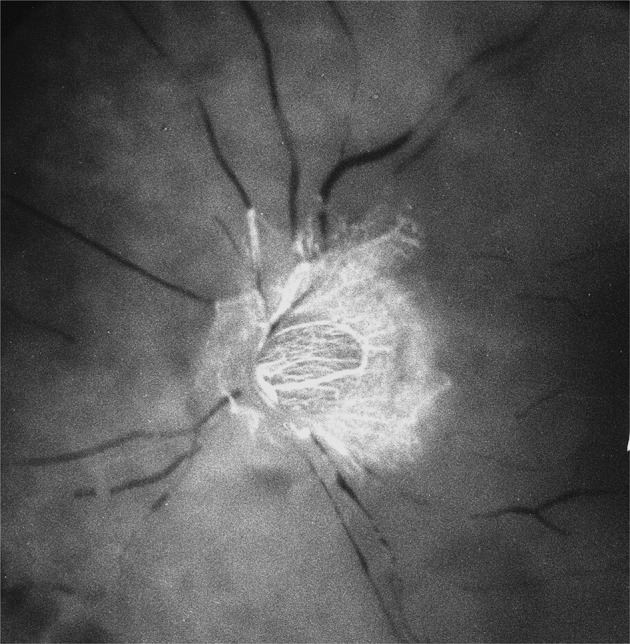
The optic disc shows filling from the PCA circulation.
Uveal vascular system is the largest and most important vascular system in the eye, and has a role in supplying almost every tissue in the eyeball. Following is a comprehensive, but very abbreviated, summary of the uveal vascular bed in health and disease, based on (A) the anatomy of the PCAs, ACAs, cilioretinal arteries and VVs, (After all, most importantly, basic sciences are the foundation of Medicine), and (B) the lesions produced by their occlusion and acute uveal ischemic lesions seen clinically.
Method of literature search
This review is based on bibliographies of my 36 research studies on the subject, published in peer review ophthalmic journals (mainly British and American, and a few European) since 1962; and an update of the literature by using the following “PubMed” search strategy.
[“Optic Disk”[Mesh]) AND ((“Blood Circulation”[Mesh]) OR (“Blood Vessels”[Mesh]))) OR ((“Uvea”[Mesh]) AND ((“Blood Circulation”[Mesh]) OR (“Blood Vessels”[Mesh])))) OR ((“ciliary processes”) AND ((“Blood Circulation”[Mesh]) OR (“Blood Vessels”[Mesh])))) OR ((“Choroid”[Mesh]) AND ((“Blood Circulation”[Mesh]) OR (“Blood Vessels”[Mesh])))) OR ((“Iris”[Mesh]) AND ((“Blood Circulation”[Mesh]) OR (“Blood Vessels”[Mesh])))) OR ((“Ciliary Body”[Mesh]) AND ((“Blood Circulation”[Mesh]) OR (“Blood Vessels”[Mesh]))) AND (“ocular circulation”)) OR (“uveal circulation”) Filters: from 1950–2022.]
Lesions Produced By PCA Occlusion
As discussed previously, the PCAs are the main source of blood supply to the choroid up to the equator and the ONH, the retinal pigment epithelium (RPE), the outer 130 µ of retina (and, when a cilioretinal artery is present, the entire thickness of the retina in that region), the ciliary body and the iris. Information on the vascular pattern of the choroid and of the PCAs was previously drawn entirely from postmortem cast studies, ever since the first description by Frederick Ruysch [5] in about 1700. Since then, extensive anatomical studies of the choroidal vascular bed have been conducted, mostly by studying casts prepared by the postmortem injection of a variety of materials [5–10] and by studying casts by scanning electron microscopy [11, 12] These studies formed the basis of the classical textbook anatomical description of the choroidal vasculature. According to most of these descriptions: (i) PCAs have no segmental distribution, (ii) they anastomose freely with one another as well as with the ACAs, (iii) there are inter-arterial and arteriovenous anastomoses in the choroid, and (iv) choriocapillaris form a freely communicating and an uninterrupted vascular bed in the entire choroid. From these postmortem anatomical descriptions, it was generally concluded that occlusion of PCA or one of its branches should not produce an ischemic lesion.
Experimental Pca Occlusion Studies
After the advent of fluorescein fundus angiography in the 1960s, I decided to investigate the enigma which Duke-Elder [4] described in 1961: “The tendency for inflammatory and degenerative diseases of the choroid to show a considerable degree of selective localization, despite the fact that anatomically (postmortem casts) the vessels would appear to form a continuous network, has given rise to speculations regarding the anatomical isolation of specific choroidal areas”.
In 1970, I investigated this in a comprehensive experimental study. I experimentally occluded the PCAs in rhesus monkeys (since the vascular pattern in rhesus monkeys is identical to that in the human) by fluorescein angiography to investigate: (1) in vivo circulation and distribution pattern of the PCAs and their branches, and (2) the types of ischemic lesions produced by cutting various PCAs and their branches. I proceeded by cutting one or another or all the PCAs at a time in 113 monkey eyes [13–17], one or the other SPCA in 87 eye [18], and a LPCA in 22 eyes [19]. That had never been done before, and provided wholly new information.
In Vivo Circulation And Distribution Pattern Of The Pcas And Their Branches
As discussed at length above, my in vivo fluorescein angiographic studies [13, 16, 18, 19] on the PCAs and their various branches provided much new information. This included information about the in vivo circulation and distribution pattern of the PCAs and their branches, right down to the terminal choroidal arterioles, and the choriocapillaris. They showed for the first time that the PCAs and their branches, right down to the terminal choroidal arterioles, and the choriocapillaris, have a segmental distribution in the choroid, and that the PCAs and choroidal arteries function as end-arteries. These findings have great clinical importance in explaining various clinically seen choroidal lesions. These fluorescein angiographic studies showed that the postmortem injection cast studies had misled us greatly for centuries.
Choroidal Ischemic Lesions Produced By Occlusion Of The Pcas
I investigated this by experimental occlusions of the PCAs in rhesus monkeys. This was the first comprehensive study of the choroidal ischemic lesions produced by PCA occlusion in a large number of eyes. It is necessary to describe: (1) the nature of these lesions, and (2) their evolution with time, in detail, because there was little information on this subject elsewhere in the ophthalmic literature, and the result was frequent misdiagnoses and management of these lesions. Following is a detailed account of the: (1) ophthalmoscopic, (2) fluorescein fundus angiographic, (3) histopathological findings of the choroidal ischemic lesions, and (4) their evolution, which were provided by an experimental study.
Ophthalmoscopic Apperances Of Choroidal Ischemic Lesions And Their Evolution
About I hour after occlusion of the PCA, ophthalmoscopic examination of the fundus showed no detectable lesions. When the eyes were examined 1-2 days after that, white patches were seen in the part of the fundus supplied by the occluded artery (Figs. 2, 3A). These patches assumed a large variety of shapes and sizes. Their size varied from a tiny spot to a large area of the fundus. They were irregular in shape, many being elongated with pointed ends, and frequently tended to be triangular and sectoral (Figs. 2, 3A). These patches were separated from one another by narrow strips of normal coloured fundus and sometimes by the big visible choroidal vessels. The white patches were infarcts, lying deep to the retina.
Fig. 2.
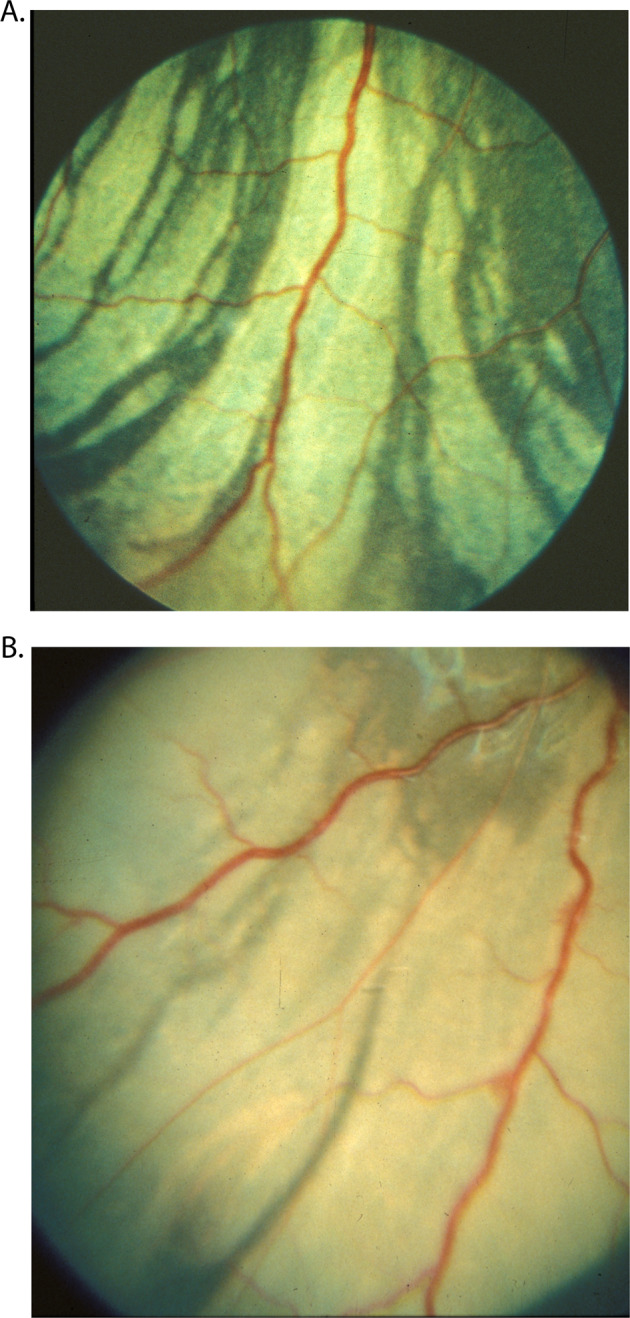
Fundus photographs (A, B) show white ischemic choroidal lesions inferotemporal region 2 days after the occlusion of lateral PCA in rhesus monkey eyes.
Fig. 3.
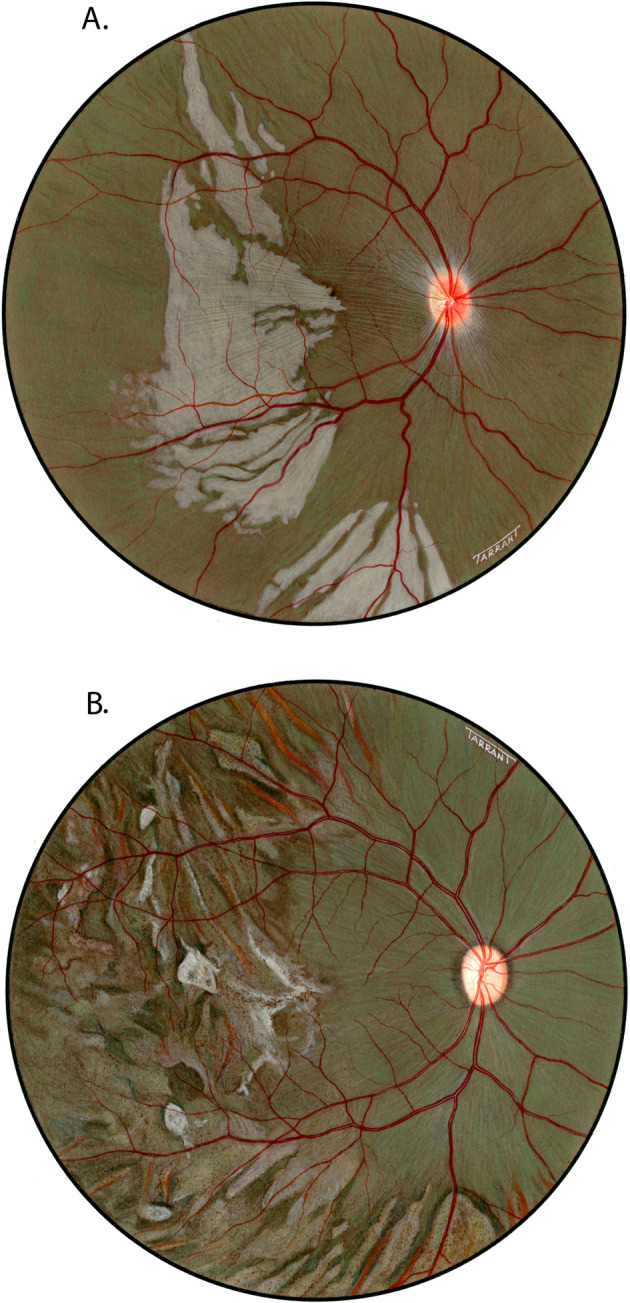
Fundus drawings of the right eye shows lesions after lateral PCA occlusion in rhesus monkey after (A) 5 and (B) 90 days of occlusion.
On follow-up, these lesions did not increase significantly in size. They, however, became more clearly defined over the following few days. At the beginning of the second week, they began to resolve. They became less white, assumed a granular appearance, becoming less dense and grey. After 2 to 3 weeks these lesions appeared as granular, greyish, depigmented scars in the fundus (Figs. 3B, 4). With the passage of time some of the patches tended to become ill-defined.
Fig. 4.
Fundus photograph (A), and (B) fluorescein fundus angiogram during the retinal arterial phase of the right eye of rhesus monkey, show lesions situated in superior nasal region, after occlusion of all the PCAs.
Another interesting change appeared towards the end of the second week. The grey lesions became surrounded by a narrow zone, like a dark-coloured halo, which was darker than the surrounding fundus (Figs. 3B, 4). These patches became more marked with time and their size increased, extending to involve the adjacent normal-looking fundus. This was a slowly progressive phenomenon, and the lesion was found to be still increasing in size when followed for more than 3 months but became stationary by about 5 to 7 months. Thus, the involved part of the fundus had large, irregular dark-brown areas, with steel-grey patches, in addition to the greyish-white granular depigmented patches scattered amongst them, and the pigmented areas encroaching upon the normal looking fundus.
In the depigmented areas, the main choroidal vessels were sometimes more conspicuous than in the normal fundus. Some of these patches, when associated with choroidal destruction, had a mottled, shining, greyish-white appearance, with prominently visible large choroidal vessels.
In addition to the characteristic fundus lesions seen in these eyes, a few showed the following pattern:
Instead of the ophthalmoscopically well-defined patches of lesions mentioned above, only a diffuse, scattered, irregular mottling of the pigment was visible in some areas. These patches were seen better on fluorescein angiography than by ophthalmoscopy.
In others no depigmented patches were seen, but large areas of dark brown discoloration of the fundus (like the haloes) were seen, usually situated in the region extending from the outer limits of the central part of the fundus (the central part includes the macular region, optic disc, peripapillary choroid, and adjacent areas) to the equator of the eye. Examination of the peripheral fundus showed that in the equatorial region of the affected part of the fundus there was a scalloped border between the posterior dark-coloured fundus and the anterior normal fundus. This indicated that the pigmentary disturbance in the involved region was diffuse and extensive and was associated with increased pigmentation of the fundus.
There were differences in the distribution of the fundus lesions in occlusion of the lateral, medial or all PCAs, as is evident from the following description.
After Lateral PCA Occlusion
The lesions involved the temporal part of the fundus in every eye (Fig. 3). In two eyes they also encroached upon the adjacent part of the inferior nasal sector. In no eye did the lesions involve the peripapillary choroid, or the area extending from the optic disc to the nasal margin of the macula. The macula was involved in three eyes. The temporal lesions were of extremely varied distribution but were usually situated in the area between the equator and the central region. In the macular region, particularly the region between the optic disc and the macula, horizontal retinal folds were frequently seen for the first 2 to 3 weeks.
After Medial PCA Occlusion
The lesions appeared in the nasal part of the fundus in one-half of the eyes, no lesion being seen in the rest. Their distribution was also very varied, and they were situated in the area between the equator and the central region of the fundus. In almost all cases the lesions seen in medial PCA occlusion were much smaller in size and more localized than those seen in lateral PCA occlusion.
After All PCA Occlusion
When the PCA supply to the eye was completely cut off, there were much more extensive lesions than in either of the previous groups. In this group, the lesions were seen in every eye and involved many parts of the fundus. In eyes in which a small SPCA escaped cutting, no fundus lesions were seen in the corresponding sector.
Fluorescein Fundus Angiographic Findings And Their Evolution
These ischemic choroidal lesions were also investigated by fluorescein fundus angiography.
Fluorescein Pattern at the Initial Stages of PCA Occlusion
During the transit of the dye through the eye, no background choroidal fluorescence was seen in the region of the white patches; these patches, however, showed late fluorescence a few minutes after the transit of the dye (Fig. 5A). The late fluorescence was due to staining of the necrotic tissue of the white patches by the fluorescein, which had leaked very slowly from the underlying choroid into the overlying tissue. This may be due to the following two mechanisms.
Late Filling of the Choroid in the Region of the Occluded PCA Soon after the Occlusion: The various mechanisms responsible for this were discussed in detail elsewhere [13]. Briefly, these include the following modes, singly or in combination:
Fig. 5. Fluorescein fundus angiograms of the macular region and the area temporal to it after lateral PCA occlusion 2 days after the occlusion in rhesus monkey.
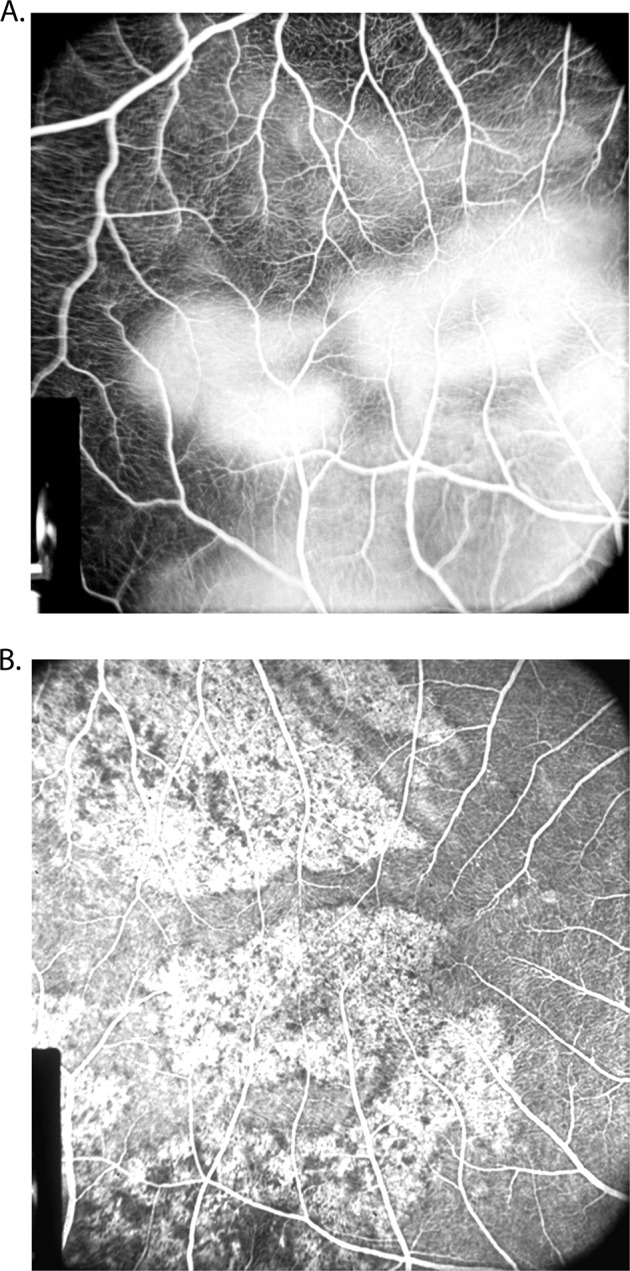
A Shows late fluorescence of the white patches. B The same after 26 days of occlusion shows unmasking of the choroidal fluorescence.
(a) Via Retrograde Choroidal Circulation from the Vortex Veins: The territory of the uveal tract drained by one vortex vein is usually supplied by three sets of arteries, medial and lateral PCAs, and the ACAs, as shown in Fig. 6. Thus, occlusion of the medial and/or lateral PCA reduces the blood pressure in the venous tributaries draining the non-filling sector of the choroid. Blood could regurgitate from the main stem of the vortex vein (which still has venous return from the anterior segment of the eye) into these empty tributaries and lead to retrograde filling of the choroidal vascular bed. This is further supported by the fact that these isolated filling areas appear during the late venous phase of the dye transit. The normal ocular pulsation, by acting as a pumping mechanism, would help in the filling and emptying of the vortex veins in this sector.
Fig. 6. Diagram shows route of retrograde flow of blood via the vortex vein into the part of choroid supplied by an occluded PCA (large arrow).
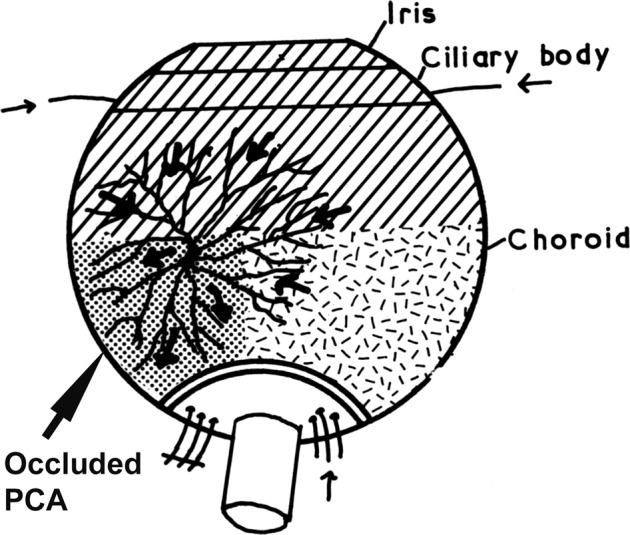
Small arrows indicate direction of flow of blood in various vessels.
(b) Via the Posterior Episcleral Arterial Plexus: Anastomoses on the surface of the posterior sclera between the PCAs, optic nerve sheath vessels and other vessels would enable dye or blood to reach the occluded vessel distal to the occlusion, as demonstrated in the silicone rubber injection studies [13]. The cutting of the PCAs some distance posterior to the sclera would leave these anastomoses intact. The choroidal filling through these anastomoses increased with the passage of time.
(c) Via the Retrolaminar Optic Nerve Pial Vascular Plexus: The peripapillary choroid anastomoses with the retrolaminar optic nerve pial vascular plexus (Fig. 7B). With normal blood supply in the pial plexus, isolated, small, patchy filling of the peripapillary choroid slowly starts from that source. This usually appeared around the venous phase. Fluorescence spreads from these patches to adjacent areas of the choroid.
Fig. 7. Schematic representation of blood supply of the optic nerve.

A arachnoid, C choroid, CRA central retinal artery, Col. Br. Collateral branches, CRV central retinal vein, CZ circle of Zinn and Haller, D dura, LC lamina cribrosa, OD optic disc, ON optic nerve, PCA posterior ciliary artery, PR prelaminar region, R retina, S sclera, SAS subarachnoid space.
2 Breakdown of Blood Retinal Barrier in the Retinal Pigment Epithelium (RPE): Normally the RPE cell layer exercises a blood-retinal-barrier which prevents diffusion of choroidal fluid into the retina. But, in the region of the infarcted RPE, the blood retinal barrier breaks down, as shown by our horseradish peroxidase tracer study, discussed below [17]. That results in the diffusion of fluorescein from the choroid into the infarcted RPE and staining of it.
Fluorescein Pattern at Later Stages of PCA Occlusion
As the white patches progressed to become thin, unmasking of the choroidal fluorescence became evident, indicating a thinning of the RPE (Fig. 5B). This unmasking became more prominent with the passage of time and was at its greatest when the lesions assumed the granular depigmented greyish-white appearance (Fig. 4). The marginal parts of the lesion showed a bright granular fluorescence, which appeared with the filling of the choroid. The central part of the lesion showed evidence of reduction of choroidal vessels and some granular fluorescence as well. In these patches the main choroidal arteries showed a distinct filling and unmasking. This fluorescence of the patches persisted only during the transit of dye through the choroid and faded off thereafter. There was no staining of the lesions after about 3 weeks.
The steel-grey patches in the haloes around the depigmented choroidal lesions usually slightly masked the choroidal fluorescence (Fig. 4). In the eyes showing no depigmented patches but large areas of dark brown discoloration, fluorescein fundus angiography showed no abnormality. Similarly, when such a discoloration was associated with typical depigmented lesions, no fluorescein angiographic abnormality corresponding to the dark brown patches was evident.
No other fluorescein angiographic abnormality was detected in the fundus. The findings suggested a degeneration of the RPE in the areas of the patches, with some destruction of small and medium-sized choroidal vessels in the central part of these lesions (Fig. 4B). The retina showed no abnormality. In areas with marked choroidal degeneration, apart from a few big choroidal vessels, no other choroidal vessels were seen (Fig. 8), except at their margins.
Fig. 8.
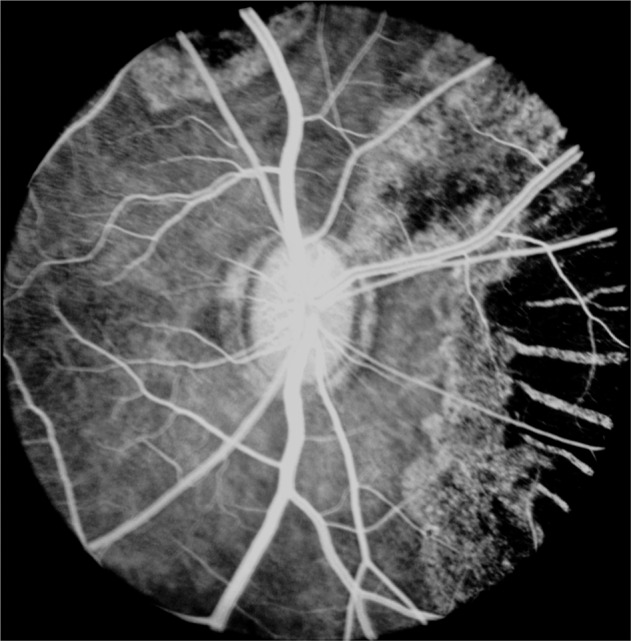
Fluorescein fundus angiogram 58 days after occlusion of all PCAs in rhesus monkey during retinal arteriovenous phase, shows marked destruction of choroid nasally with choroidal unmasking along the borders.
Progressive Filling of the Choroid Later on in PCA Occlusion
Fluorescein fundus angiography also showed that with time, the choroidal vascular bed filling in the region of the occluded PCA gradually improved, so that finally the choroidal vascular bed filled almost normally. The mechanism of that filling is discussed above. This fact is important clinically because normal filling of the choroidal vascular bed months later in eyes with PCA occlusion (e.g., in GCA) has been misinterpreted as showing that there had been no occlusion of the PCA—a common argument. I have seen that in eyes with PCA occlusion due to GCA. Therefore, complete occlusion of the PCA can only be demonstrated on fluorescein angiography within the first few days.
Histopathological Findings And Their Evolution
These are discussed in detail elsewhere [14]. Briefly, the various fundus lesions showed the following histological changes on light microscopy. These changes never involved the anterior part of the choroid supplied by the ACAs.
Retina
In eyes with white patches, necrotic material was present between Bruch’s membrane and the inner layers of the retina in the region of the patches. The necrotic material consisted of dead RPE, rods, cones, and to a variable degree the outer nuclear layer. The pigment granules of the RPE were irregularly scattered in the necrotic mass. The latter varied in thickness and structure, depending upon its age, and disappeared completely in older lesions.
In old lesions, the most common change observed was the loss of visual cells and outer nuclear layer in the lesion. A few nuclei of the outer nuclear layer might still be seen. Rarely, the inner nuclear layer was somewhat involved and disorganized, but not significantly.
Chorioretinal adhesions were frequently seen in ophthalmoscopically well-marked lesions. In these adhesions, the RPE and Bruch’s membrane were often absent or disorganized. In a few instances some big, pigmented cells were seen to have invaded the retina close to the choroid. No migration of the pigment into the deeper layers of the retina was seen.
Retinal Pigment Epithelium
This was involved significantly in most of these eyes. There were, however, instances where, with the retina showing absence of the visual cells and outer nuclear layer, no significantly demonstrable changes in the RPE could be seen. The reverse was also true.
The changes in the RPE varied from mild degeneration to complete disorganization or even to the complete absence of the RPE. Usually, the degeneration of the RPE was more widespread than the changes in the overlying retina. The normal RPE is thin, uniform in thickness, light brown, having regularly arranged cells and a very finely granular appearance. The changes in the RPE seen in the region of the white patches have already been mentioned under “retina”. In old lesions, the RPE might be represented by very dark (sometimes even black) clumps or granules. In others the epithelium might be irregular in thickness, with coarse granules. Some of these granules were discrete, rounded, of varying size, and black. At some places a few of these isolated granules were the sole evidence of the RPE. Big, round, pigmented cells were seen lying over Bruch’s membrane in some areas, or even invading the adjacent layers of the retina. These were grossly hypertrophied RPE cells. Sometimes localized hypertrophy of the RPE was seen around the area of the destruction of the RPE.
In eyes where the ophthalmoscope showed pigmented fundus lesions, e.g., brownish discoloration of the fundus - the only histological evidence was roughening and some thickening of the RPE.
Bruch’s Membrane
Usually, Bruch’s membrane showed no demonstrable changes in mild fundus lesions. Where chorioretinal adhesions were seen, the membrane was generally absent. A possible thickening and hypertrophy of Bruch’s membrane was seen in some places and frequently in areas close to chorioretinal adhesions. In some eyes, however, a normal Bruch’s membrane was present, even in the presence of the adhesions.
Choroid
Usually, no demonstrable change was seen in the choroid, except for some obliteration of the choriocapillaris. In some of the areas which showed evidence of absence of choroidal vessels on fluorescein fundus angiography, there was localized atrophy of the choroid. In the eyes irrigated with gluteraldehyde, normally no blood was seen in the choroidal vessels if adequate circulation was present, but in eyes with freshly occluded PCAs, the choroid was full of blood on the side of the occlusion.
Light And Electron Microscopy Study
In another study [17], evolution of histopathological changes was investigated by both light and electron microscopy. In this investigation, the horseradish peroxidase (HRP) tracer technique was also used to study the RPE blood-retinal barrier. Following were the findings of this study.
Twenty-Four hours after PCA occlusion
In the macular region the choriocapillaris was congested and showed extensive thrombosis. The RPE was focally necrotic, alternating with early focal reactive proliferation. Necrosis of photoreceptor inner and outer segments was evident and was associated with extensive focal pyknosis in the outer nuclear layer. The inner retina appeared unaffected. Leakage of HRP extended through the RPE into the subretinal space and passed the outer limiting membrane and outer nuclear layer to the outer plexiform layer. Regional variation of the ischemic changes was remarkable. Nasally, focal RPE necrosis was seen, associated with macrophage-like cells that were infiltrating into the sub-RPE space, and focal thinning and necrosis of the outer nuclear layer were also noted. However, only mild HRP leakage through the RPE was present at this site. Inferiorly, changes included scattered RPE pyknosis with focal necrosis in the overlying photoreceptor cells.
Two weeks after PCA occlusion
In the macula, the choriocapillaris revealed an extensive occlusion and loss of capillaries that was accompanied by a proliferation of pericytes. The remarkable feature was plaques of RPE fibrous metaplasia proliferating in multiple layers on Bruch’s membrane. The HRP tracer infiltrated the RPE plaque and extended into the subretinal space to the opposing outer limiting membrane. Some RPE cells showed marked vacuolation and liquefaction of the cytoplasm. Photoreceptor outer and inner segments were largely lost, and the outer limiting membrane approached the apical RPE surface. The outer nuclear layer was severely thinned, and pyknotic nuclei were observed. The inner retina overlying the RPE plaque appeared mildly gliotic. An extensive RPE plaque was also noted in the superior quadrant of the retina, but HRP leakage was only apparent within the RPE plaque and in the subretinal space. Beneath the plaque, the choriocapillaris exhibited marked replacement fibrosis and the capillaries were extensively reduced in number. The outer nuclear layer was almost completely lost at this site. In the temporal quadrant, the choriocapillaris was patent and there was a focal area of RPE depigmentation and proliferation, and a markedly thinned outer nuclear layer.
Three Months after PCA Occlusion
In the macula, there was patchy loss of choriocapillaris, and a RPE plaque had formed, and the outer nuclear layer was mildly reduced in thickness. The regional ischemic changes appeared to be milder than the necrotic changes that were noted in the previously described eyes. Scattered macrophages were observed in the outer plexiform layer. The HRP infiltrated the RPE plaque and extended into the subretinal space. Nasally, a large RPE plaque with focal thinning of the overlying outer nuclear layer and occasional macrophages in the outer plexiform layer were present, but no significant HRP leakage was detected in this area.
Thus, this study showed that the RPE reaction to ischaemia began with intracellular oedema and cell necrosis in the area of damage while, over time, adjacent RPE cells migrated to repopulate this region. Plaque formation as a response to ischaemia was noted as early as 1 week after PCA occlusion, but it was only seen in those areas with the most severe necrotic changes in the outer retina. The outer nuclear layer became thinned with focal photoreceptor cell death 24 h after vascular occlusion. Damage to the photoreceptor cell layer appeared irrevocable and obvious at 3 months, even overlying fairly normal repopulated RPE. Three months after the ischemic insult, no areas with complete RPE denudation were seen. It appeared that RPE cells could repopulate a previously denuded area within this period. The inner retina appeared largely unaffected, but rarefaction of axons in the outer plexiform layer was frequently noted.
Horseradish Peroxidase (Hrp) Tracer Findings
The breakdown of the blood-retinal barrier was evident 6 h after ischaemia when leakage of HRP from the choroid into the outer retina was observed. As in the studies by Uyama et al. [20], and Koshibu and ltotagawa [21], penetration of HRP into diseased RPE cells was noticed, while the tight junctions between these cells were still intact and prevented intercellular HRP movement into the outer retina. Continuous disruption of the chorioretinal blood barrier was noted until 1 month after ischaemia, but leakage of HRP through the RPE appeared to decrease after 1 week. Three months later, the chorioretinal blood barrier was mostly restored, with only minimal HRP leakage at the sites of severe ischaemia, whereas the loss of photoreceptor cells from the outer nuclear layer was irrevocable.
Conclusions Of Pca Occlusion
When various PCAs were cut, white patches of markedly varied shape, size, and distribution appeared in the parts of the fundus supplied by the occluded artery within 24 h. These patches stained during the late phase of fluorescein in angiography. After one week the patches began to change gradually, so that over a 2 to 3-week period they became greyish-white, granular, depigmented areas. During the evolution of the lesions, they became surrounded by deeply pigmented haloes, and the adjacent fundus showed similar pigmentation. In the greyish-white depigmented patches, intravenous fluorescein fundus angiography revealed degeneration of the RPE and partial destruction of the choroidal vasculature. The sites of these white patches corresponded to post-occlusion non-perfusion areas in the choroid. Histological studies of the fundus lesions revealed destruction of visual cells and the outer nuclear layer, with degeneration of the RPE and Bruch’s membrane, but with little change in the choroid. Later, it appeared that RPE cells could repopulate a previously denuded area within a 3-month period. The inner retina appeared largely unaffected, but rarefaction of axons in the outer plexiform layer was frequently noted. HRP study showed breakdown of the RPE blood retinal barrier initially, but 3 months later it was mostly restored.
Correlation Of Site Of Lesion With Choroidal Ischaemia
In most of the eyes, the site and extent of the lesions after PCA occlusion depended to a great extent upon the extent of the choroidal filling, via the routes discussed above and elsewhere [13]. The lesions usually appeared in areas devoid of choroidal filling after the occlusion, as revealed by non-perfusion of the area of the choroid during the transit of the dye in angiographic studies. In some, however, it was difficult to be definite about the exact area of non-perfusion in the choroid. On follow-up studies, a delayed filling of the choroid in the areas of the lesion was usually evident. It is, again, difficult to explain the occurrence of elongated pointed lesions with narrow intervening strips of normal-looking fundus.
Since the first publication about experimental choroidal ischemic lesions in PCA occlusion in monkeys, several similar studies—all but one in Japanese (which I cannot read)—have been reported with almost similar findings. The Japanese studies [22–24] mainly deal with ultrastructural changes of the RPE cells following experimental occlusion of the PCA in the rhesus monkey. Algvere [25] produced choroidal ischaemia in owl monkeys on occlusion of a vortex vein, associated with proliferation or degeneration of the RPE, frequently accompanied by photoreceptor degeneration and circumscribed retinal detachment.
Onh Ischemic Lesions Produced By The Pca Occlusion
Since AION is a commonly seen clinical entity, and there is a good deal of confusion and controversy on the aetiology and evolution of ischemic ONH lesions. The effect of experimental PCA occlusion on the ONH was investigated in 85 rhesus monkey eyes. The findings are discussed in detail elsewhere [15]. PCA occlusion produced AION. It is essential to describe in detail (1) the nature of these lesions and (2) their evolution with time, in eyes with occlusions of lateral, medial or all PCAs. As discussed above, the ONH supply by the various PCA can vary.
Following is a detailed account of the ophthalmoscopic, fluorescein fundus angiographic and histopathological findings of the ONH ischemic lesions and their evolution seen with various PCA occlusions.
Ophthalmoscopic And Fluorescein Fundus Angiographic Findings In The Onh
After Occlusion Of Lateral Pca (31 Eyes)
One Hour after Occlusion
Ophthalmoscopic examination of the fundus at this time showed slight oedema of the disc in 7 of 31 eyes. This represented AION. This oedema involved the lower part of the optic disc in 3 eyes, the temporal part in 2, the nasal part in one, and the whole optic disc in one. Eight of 31 eyes showed pallor of the disc, as compared to its pre-occlusion appearance, the pallor being usually more evident on the temporal part of the disc. It is pertinent to point out that there is wide interindividual variation in the supply by PCA to the ONH and that may be responsible for different sites of optic disc oedema in different eyes.
At this stage, fluorescein fundus angiographic studies revealed very faint or no fluorescence of the disc during the retinal arterial phase, and moderate fluorescence during the arteriovenous phase. The fluorescence was usually more marked on the nasal than the temporal part of the disc. This is interesting, because the pre-occlusion studies in these, and in normal eyes, mostly showed a more marked fluorescence on the temporal than the nasal part. Thus, occlusion of the lateral PCA reversed the fluorescence pattern in these discs. During the late phase, blurring of the disc margins was seen in the 7 eyes with optic disc oedema.
One to 2 Days after Occlusion
Out of the 12 follow-up eyes, 8 eyes were examined at this stage. Five of these showed a mild degree of oedema of the disc, involving either the whole (Fig. 9) or a part of the disc. Three of the discs with pallor one hour after PCA remained pale at this stage. Two eyes were normal.
Fig. 9.
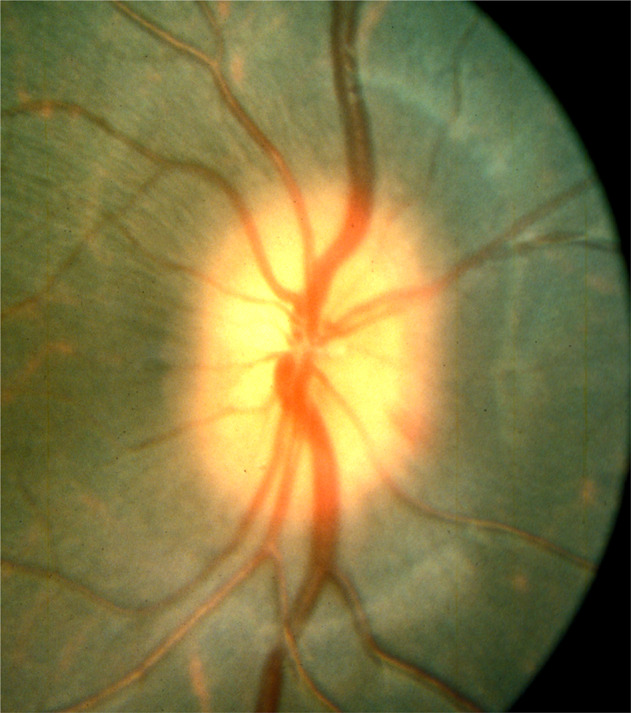
Fundus photograph shows oedema of the optic disc 2 days after occlusion of the lateral PCA and arteritic AION.
At this stage, fluorescein fundus angiographic studies revealed a slight fluorescence of the disc in the retinal arterial phase, largely uniform in distribution in the majority, which increased during the arteriovenous phase and again decreased in the venous phase. A mild degree of fluorescein leakage was seen in 5 eyes—usually along the temporal or lower margins.
One Week after Occlusion
There was no significant change.
After 3 to 4 Weeks’ PCA Occlusion
In the 3 eyes with oedema of the disc, which were followed for more than one week, the oedema subsided in about 3 to 4 weeks. Subsequently, these 3 eyes developed pallor, more marked on the temporal than the nasal part, and showed varying degrees of optic atrophy. None of these 3 eyes showed any immediate post-occlusion pallor of the disc, though two showed a degree of oedema. Fluorescein fundus angiography at the end of the follow-up revealed reduced fluorescence in the trophic discs.
After Occlusion Of Medial Pca (17 Eyes)
One Hour after PCA Occlusion
One disc was oedematous in its lower part. No hyperaemia was seen.
Five discs showed a mild degree of pallor.
On fluorescein fundus angiography, the discs showed faint to moderate fluorescence in the retinal arterial phase, with the temporal part usually more fluorescent than the nasal. Fluorescence increased in intensity during the arteriovenous phase and was reduced in the venous phase. In the disc showing oedema inferiorly, fluorescein leakage was seen in the same area.
Two Days after PCA Occlusion
Mild oedema of the disc was seen in 2 eyes involving the upper part in one, and the lower part in the other. Half of the discs showed a mild degree of hyperaemia.
One Week after PCA Occlusion
The oedema of the disc cleared.
Two months after PCA occlusion
One of the previously oedematous discs developed a moderate degree of optic atrophy with pallor, more marked in the nasal part than the temporal part (before the occlusion, the temporal part of the disc had been paler than the nasal part.
After Occlusion Of All Pca (37 Eyes)
One Hour after PCA Occlusion
The discs showed uniform pallor in about a quarter of the eyes involved. On fluorescein fundus angiography, during the retinal arterial phase, only a faint disc fluorescence was usually seen, with some discs showing no fluorescence at all. A uniform fluorescence of moderate to marked degree was seen during the arteriovenous phase. The late phase showed, in a third of these eyes, a leak of fluorescein with blurring of the lower border or the disc; the earliest filling of the peripapillary choroid occurred in the corresponding sector. This suggests that the fluorescein had leaked from the ischemic vessels on restoration of the peripapillary circulation.
Two Days after PCA Occlusion
A mild degree of oedema of the disc with fluorescein leakage was seen in most of these eyes; this usually subsided in about 2 weeks.
More Than 5 Weeks after PCA Occlusion
Of the 13 eyes followed for more than 5 weeks, 11 showed optic atrophy of variable extent. In 4 eyes with optic atrophy, there were enlarged collateral vessels on the disc, connecting the retinal vessels on the disc surface with the peripapillary choroid. These were on the temporal side in three, and on the nasal side in one. No such vessels were seen before occluding the PCAs in these eyes. Their exact significance is not known, because they were not seen in other eyes with or without optic atrophy.
These findings for the first showed ophthalmoscopic and fluorescein angiographic changes in AION and evolutionary changes after occlusion of the various PCAs.
Fluorescein fundus angiography immediately after occlusion of the PCAs mostly revealed, during the early part of the transit of the dye, reduced fluorescence of the part of the disc supplied by the occluded PCA, e.g., usually the temporal part in lateral PCA occlusion and the nasal part in medial PCA occlusion. This study (1) confirmed the role of the ciliary circulation in the blood supply of the ONH, and (2) provided more information about the origins of fluorescence of the optic disc during the different phases of the retinal transit of the dye in fluorescein fundus angiography.
During the retinal arterial phase: Before the retinal capillaries fill, the disc fluorescence represents the deep ciliary supply to the optic disc.
During the arteriovenous phase: When the retinal capillaries are completely filled, the fluorescence of the disc is mainly due to the retinal capillary fluorescence and very little to the deep ciliary fluorescence. The one eye in which the cilioretinal artery did not fill on occlusion of the lateral PCA demonstrated this very clearly, because the area where the retinal capillaries were missing was much less fluorescent than the area where they were full.
Histopathological Studies
In the optic discs showing ophthalmoscopic evidence of atrophy, histological examination revealed atrophic changes in the disc and the retrolaminar part supplied by the PCAs. In some eyes, the atrophic area had a well-marked border with the normal optic nerve. Clinically, in GCA there is always PCA occlusion, and that results in development of arteritic AION. Thus, the PCA occlusion produced arteritic AION. In arteritic AION, well-marked infarction of the ONH has also been reported [26–29] (Fig. 10). In other monkey eyes, the well-marked junction between the atrophic and normal regions was not always so well-defined, although the distribution of atrophic changes was similar. In some of these, the atrophic changes were more marked in the peripheral part of the optic nerve. There was a localized patch of optic atrophy in the peripheral part of the retrolaminar optic nerve on the occluded side in a significant number of these nerves.
Fig. 10. Photomicrograph of the optic nerve head and retrolaminar optic nerve of right eye of a patient with 4-week-old arteritic AION, shows a well-defined area of infarction.
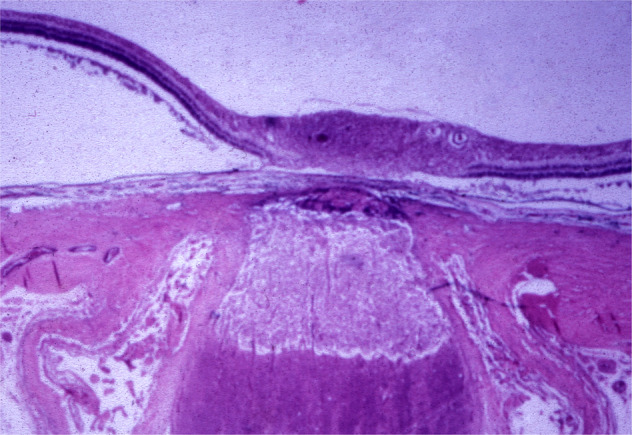
(Verhoeff’s modified elastic stain). (Reproduced from late Dr. J.F. Cullen).
Conclusion
Experimental occlusion of the various PCAs, during the initial phases produced optic disc oedema. The incidence of development of optic atrophy after 5 to 6 weeks varies in occlusion of various PCA in monkeys – it was seen in 16%, 60%, and 85% of the eyes after occlusion of the medial, lateral, and all PCAs respectively. These changes in the optic disc are typical of AION. Histopathological studies revealed involvement of the ONH and retrolaminar part of the optic nerve - the parts of the optic nerve supplied by the PCA circulation, as has been shown in arteritic AION eyes of patients with GCA [26–29]. Thus, this experimental study provided critical information about AION: its controversial pathogenesis, and the ophthalmoscopic, fluorescein angiographic and histopathological issues, particularly in arteritic AION.
Simultaneous Occlusion Of Pcas And Vortex Veins
My studies [14, 15] discussed above, dealing with experimental occlusion of various PCA in monkeys, showed patchy infarction of the RPE and outer parts of the retina and development of AION. The lesions were in the distribution of the occluded PCAs. One consistent, intriguing finding was the great variation in the extent of the ischemic lesions produced by identical experimental PCA occlusions, even within the same study. For example, the study [14] showed a notable variation in the extent of the lesions after occlusion of the lateral, medial, and all PCAs—not only between the three groups, but, much more importantly, between eyes in the same group. This study [14] suggested that the extent of the retinal infarcts after occlusion of the PCA depends largely on the extent of choroidal filling from other sources, because I found that the lesions usually appeared in areas devoid of choroidal filling on fluorescein fundus angiography immediately after the occlusion. One important source of choroidal filling was considered to be the vortex vein system [13]; as discussed above, the territory of the uveal tract drained by one vortex vein is usually supplied not only by the medial and lateral PCAs but also by a large number of the ACAs [13, 18]. Thus, occlusion of the PCAs cuts down the blood flow in the posterior tributaries of the vortex vein draining the non-perfused part of the choroid; however, the anterior tributaries of the vortex vein are still receiving blood from the uvea in front of the equator (mainly supplied by the ACAs). I [13, 18] postulated that in these eyes the blood would reflow from the main stem of the vortex vein into its empty posterior tributaries, causing retrograde filling of the choroid in the region of the occluded artery (Fig. 6). This was suggested by the fact that, in the occluded PCA territory, isolated areas of the choroid started to fill with fluorescein on angiography during the late venous phase) [13]. The normal ocular pulsation, by acting as a pumping mechanism, would help in the filling and emptying of the veins in the occluded territory. The high concentration of oxygen in the blood of the vortex veins should help substantially in preventing ischemic damage [13]. Occluding the vortex vein in such eyes should force much more blood into the occluded choroid than is seen normally and should protect the choroid and the tissues in the region of the occluded artery from ischemic damage.
To test this hypothesis, I conducted a study in 32 eyes of rhesus monkeys [16]. In 28 eyes, occlusion of all the PCAs was either performed alone (in 7 eyes) or accompanied simultaneously by occlusion of one vortex vein (in 11 eyes) or two vortex veins (in ten eyes). In addition, in 4 eyes lateral PCA occlusion was combined with occlusion of two vortex veins. All the eyes were investigated for acute ischemic lesions in the fundus-by-fundus photography and fluorescein fundus angiography, for outer retinal infarcts and AION; and in some of the histopathologic changes of the ischemic retina were examined by light and electron microscopy [17].
The findings of these studies are discussed at length elsewhere [16, 17]. The retinal lesions were extensive with vortex vein occlusion. Histopathologic changes also suggested that simultaneous vortex vein occlusion had an ameliorative effect of on the damaging effects of PCA occlusion.
Conclusion
Occlusion of one or two vortex veins exercises a distinct protective influence against the acute ischemic lesions caused by PCA occlusion—an important new finding.
The study also demonstrated the reason for the notable interstudy and intrastudy variations in the incidence and extent of retinal infarcts and AION among the various previously reported experimental PCA occlusion studies: it was caused by unintentional and unnoticed cutting of some of the vortex veins in the orbit. This is because vortex veins frequently follow a long, devious, and elusive course in the orbital fat before joining the final draining vein. Thus, although, while doing the studies by orbitotomy, I believed on a routine inspection that I had not interfered with the vortex veins, further careful exploration of the veins into the orbit sometimes disclosed that the vein or veins had been cut inadvertently, while clearing the retrobulbar and orbital fat and other tissues, to expose the PCAs and optic nerve.
From these studies, it is reasonable to conclude that, although the PCAs and their subdivisions right down to the choriocapillaris have a segmental distribution, with no direct communication between adjacent segments [18], in an acute arterial occlusion, the various arterial segments do communicate via the vortex venous system, which probably acts as a partial shield against many acute ischemic insults in the choroid. Simoens [30] in a similar study in pigs, also showed that ligation of the vortex veins has a protective influence on the acute ischemic lesions provoked by transection of the posterior ciliary arteries.
Causes Of Occlusion Of The Pca And Choroidal Ischaemia
This can be due to systemic diseases or local vascular diseases.
Systemic Diseases
Giant cell arteritis
This is the most important systemic disease because it results in the development of arteritic AION with or without cilioretinal artery occlusion or central retinal artery occlusion (CRAO). These are seriously blinding conditions. There are multiple histopathological studies [26–29]. (Fig. 10), and fluorescein angiographic studies in these eyes [31, 32] (Fig. 11) showing occlusion of the PCA by GCA. Since the cilioretinal artery is a branch of the PCA, occlusion of the latter also results in occlusion of the former (Fig. 11). In eyes where the PCA and central retinal artery arise by a common stem from the ophthalmic artery, when the common trunk is occluded by GCA – such eyes then present with occlusion of both the PCA and CRAO.
Fig. 11.
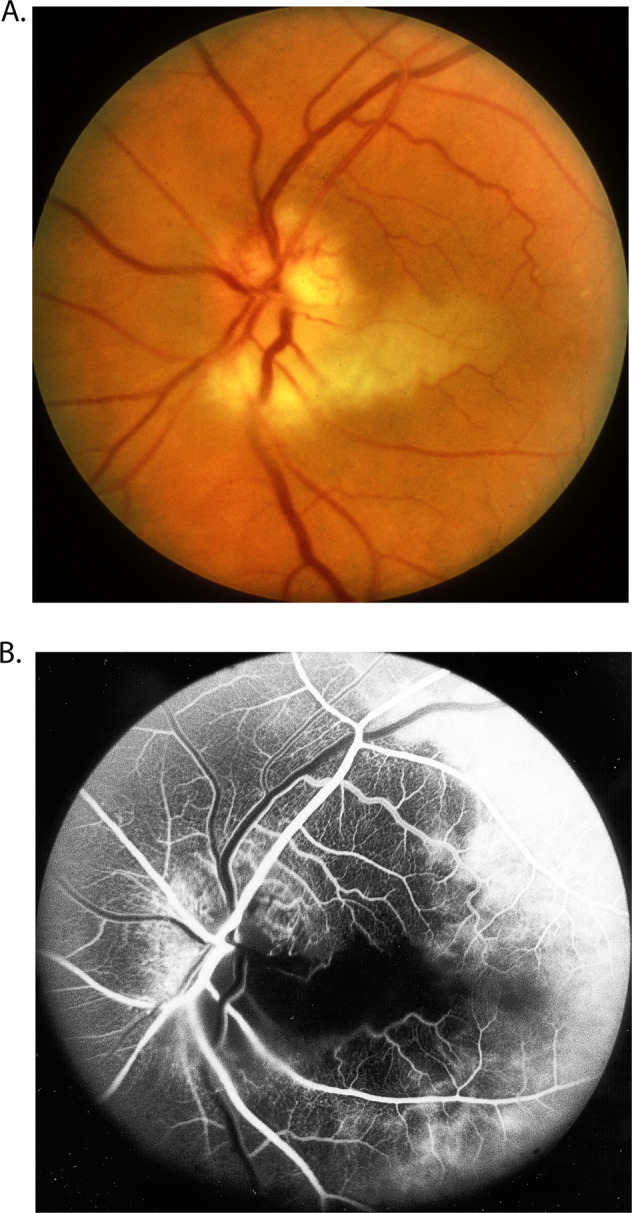
Fundus photograph (A) and fluorescein fundus angiogram (B), of left eye of a GCA patient with arteritic AION, and a cilioretinal artery occlusion. A Fundus photograph shows a classical appearance of arteritic AION, i.e., chalky white optic disc oedema with some hyperaemia. B Fluorescein fundus angiogram shows normal filling of the area supplied by the lateral PCA, but no filling of the area supplied by the medial PCA (including the entire optic disc, with no perception of light).
Malignant Arterial Hypertension
In this case, hypertensive choroidopathy develops, associated with choroidal ischaemia caused by choroidal arterial occlusion. I produced this experimentally in rhesus monkeys and that showed on ophthalmoscopy a variety of lesions of hypertensive choroidopathy due to choroidal ischaemia (see below) [33]. We also demonstrated by histopathological studies that choroidal vascular occlusive changes involving arteries, arterioles, and choriocapillaris occurred later [34]. In this category also falls toxaemia of pregnancy and chronic glomerulonephritis.
Carotid Artery Disease
This can cause acute choroidal ischemic lesions either due to atheromatous lesion (by producing emboli) or due to stenosis, or occlusion (by reducing the mean blood pressure in the choroid far below the IOP and producing a collapse of the choroidal arteries) or by their involvement by phycomycosis.
Autoimmune Diseases
These can also be associated with choroidal ischaemia, and include systemic lupus erythematosus, Goodpasture’s syndrome, periarteritis nodosa, scleroderma, and dermatomyositis. In patients with Birdshot Chorioretinopathy [35], however, the choroidal vascularity index is normal.
Hematologic Diseases
Acute choroidal ischemic lesions have been reported in these conditions, including polycythaemia, cryoglobulinemia, sickle cell disease, disseminated intra vascular coagulopathy, and thrombotic thrombocytopenic purpura.
Miscellaneous Conditions
These include haemorrhagic shock [3], Raynaud’s disease, cardiac valvular lesions, hyperlipidaemia, subclavian steal syndrome, polidocanol injection to a glabellar haemangioma, after autologous fat injection into the nasolabial fold, after subcutaneous silicone-oil injection for cosmetic purposes, and haemodialysis.
In these diseases there is involvement of either the PCAs (e.g., in GCA, embolism into carotid arteries) or the choroidal arteries or the terminal choroidal arterioles.
Local Vascular Disorders
The arterial supply to the choroid may be obstructed by local causes, including the following categories.
Due To Ocular Compression During Anaesthesia
A large number of case reports are available in the literature in which a patient had an eye compressed during anaesthesia either by the face mask or by the headrest (as in the sitting position used in neurosurgical procedures); in some cases, this situation was made worse by associated systemic arterial hypotension. Since in almost all cases there is associated CRAO as well, the retinal changes mask the underlying choroidal lesion, misleading earlier authors into considering that the visual loss and fundus lesions were due to CRAO, in spite of the unexplained associated chorioretinal pigmentary degeneration. Some of these cases were even included in the category of retinitis pigmentosa because of the close ophthalmoscopic resemblance between the two.
Photocoagulation Treatment of the Various Retinal and Fundus Lesions
These may inadvertently close the underlying choroidal arteries., as can surgery for retinal detachment.
Traumatic Lesions
Choroidal arteries or their parent trunks may be injured and produce choroidal infarcts.
Arteriosclerosis and/or Atherosclerosis of the PCAS or Their Subdivisions
In eyes of middle-aged and older patients, where no obvious cause can be found for the vascular occlusion, these may be responsible for choroidal ischaemia.
Vasculitis
This, could involve the choroidal arteries and produce secondary occlusion, and has been reported in herpes zoster ophthalmicus, orbital mucormycosis, and Wegener’s granulomatosis.
Miscellaneous Conditions
These include a variety of conditions, including onchocerciasis., effect of vitrectomy combined with lensectomy, after phacoemulsification, rheumatoid hyperviscosity syndrome.and sectioning of the posterior scleral rings as treatment for central retinal vein occlusion.
Investigations In Patients With Pca Occlusion Or Choroidal Ischaemia
From the above discussion, it is evident that these patients should have the following investigations to determine the cause and appropriate management.
In all patients 50 years and older with PCA occlusion, it is critical to rule out GCA, because that is an important ophthalmic ocular emergency; if it is missed, the result can be catastrophic visual loss in both eyes, which is preventable by immediate diagnosis and aggressive management with corticosteroid therapy. Apart from the human tragedy involved, this fact has medicolegal implications. All these patients must have immediate evaluation of erythrocyte sedimentation rate and C-reactive protein levels—both are helpful, but the latter is more reliable. Presence of systemic symptoms of GCA, arteritic AION, or associated cilioretinal artery occlusion or CRAO can be helpful. All these patients must also have fluorescein fundus angiography to detect the presence of PCA occlusion.
Since embolism is a common cause of arterial occlusions in the eye, all these patients need a complete evaluation, to find the source of embolism. The most common sources of emboli are the carotid artery and the heart. In the carotid artery, the most common source of embolus is a plaque, so that it is essential to find if there are any plaques; unfortunately, dealing with ocular arterial occlusion for more 50 years, I have found that most of the time evaluation is primarily focused on whether there is hemodynamically significant carotid artery stenosis, without attention being paid to the presence of plaques. The actual source of emboli may be missed.
Cardiac evaluation is equally essential, even if a carotid artery shows lesions, because I have found that at times a patient can have lesions in both places, and either of them can be the source of emboli. The most common source of emboli in the heart are the valves, although other lesions can also be causative. Transesophageal echocardiography is the best way to evaluate heart lesions.
Atherosclerosis is a common cause of arterial occlusion, so that it is essential to do a lipid evaluation. I have found that physicians commonly tend to look only at the total cholesterol level. I have seen many patients with perfectly normal cholesterol level but with high low-density lipoprotein, which is the most important parameter. The current guideline is to have low-density lipoprotein not more than 70 s mg/dL. Similarly, triglycerides should also be evaluated.
As discussed above, malignant arterial hypertension is an important cause of choroidal ischaemia. Therefore, patients must be evaluated for this, and managed appropriately.
As discussed above, there are many rare causes of PCA occlusion or choroidal ischaemia. In difficult cases one must be aware of all possibilities.
Clinical Features And Types Of Acute Choroidal Ischemic Lesions
The primary object of any experimental study is to give a better understanding of a clinical condition, the pathogenesis of which is not very evident in a clinical study. There are large numbers of reports in the literature of choroidal ischemic lesions due to a variety of causes. I hope that the above discussion of experimental studies will help to clarify the pathogenesis of choroidal ischaemia. The experimental studies reviewed above show that clinical features i.e., the appearance and types of acute choroid al ischemic lesions, depend upon the following factors:
Stage Of Evolution of the Lesions
As debated above, fresh lesions are homogeneously white (Figs. 2, 3A), but evolve into chorioretinal pigmentary degenerative lesions after two to three weeks (Figs. 3B, 4A). The fresh acute choroidal ischemic lesions have long been confused with retinal infarcts (due to retinal arterial occlusion) on routine ophthalmoscopy, because of their white colour; however, a resolved retinal infarct does not produce the pigmentary lesions seen in resolved choroidal infarcts.
Size of the Occluded Artery or Arteriole
Occlusion of a PCA or large choroidal arteries produces large triangular wedge-shaped lesions (Fig. 2), with the apex of the wedge towards the posterior pole and the base toward the periphery of the fundus. Amalric [36] called this “triangular syndrome”. Those due to occlusion of small arteries or arterioles usually produce geographical lesions of varying sizes, depending upon the size of the occluded vessel. Occlusion of a terminal choroidal arteriole supplying a single lobule of the choriocapillaris produces the so-called Elschnig’s spot or Siegrist’s streaks.
Severity of Ischaemia
This may vary from severe, producing complete infarction of the overlying tissue, to mild, producing only a physiological dysfunction (such as breakdown of the chorioretinal barrier of the RPE, often resulting in serous retinal detachment). Very mild choroidal ischaemia may produce no visual dysfunction and leave no scars, whereas more severe damage must produce visual dysfunction, scarring, and even loss of the eye.
There are many clinical reports in the literature dealing with acute choroidal ischaemia in patients. The earliest definition information I could find was that by Hepburn in 1912 [37]. He [37, 38] described sectoral fundus lesions in patients, starting with an ill-defined area of oedema, and later resolving to sectoral pigmentary changes (fine granular pigmentary changes in the RPE) with normal retinal vessels. In one of his cases the changes involved the lower half of the fundus. He rightly attributed these lesions to occlusion of the main PCA. Similarly, Amalric [36] in 1971 described triangular sector-shaped areas of pigmentary disturbance in the fundus in several conditions and ascribed these to PCA occlusion. Foulds et al. [39] also described similar cases. I described two cases in 1972 [14].
My experimental and clinical studies showed that acute choroidal ischemic lesions have polymorphic lesions. They represent a continuous spectrum of disease which, for descriptive purposes, can be divided into separate clinical entities, depending upon the size of the involved choroidal artery.
Acute Choroidal Ischemic Lesions Seen Clinically
The available information indicates that the following lesions belong to this group.
Elschnig’s Spot
This is a localized choroidal infarct, produced by occlusion of a terminal choroidal arteriole supplying a single choriocapillaris lobule [33, 40]. During the acute phase these are white spots (Fig. 12A) which later develop into typical lesions with a pigmented centre surrounded by a non-pigmented ring.
Fig. 12.
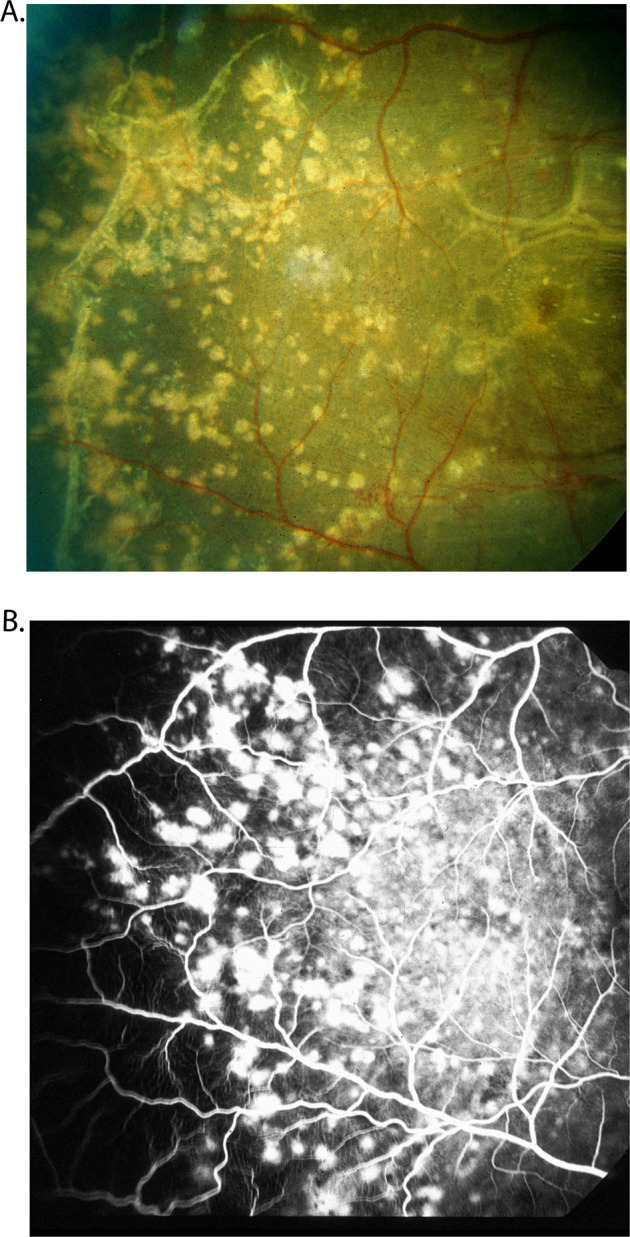
Fundus photograph (A) and fluorescein angiogram (during the late phase) (B) of right eye of a rhesus monkey with malignant arterial hypertension (day 78; blood pressure 190 mmHg). A It shows multiple acute focal retinal pigment epithelial lesions with serous retinal detachment (B). It shows fluorescein staining of the acute focal retinal pigment epithelial lesions.
Multifocal Acute Ischemic Choroidopathy
This entity has erroneously been called “acute posterior multifocal placoid pigment epitheliopathy”. Since Gass [41] described this clinical entity in 1968, many case reports have since been published. Based on my experimental and clinical studies in 1972 [14], I pointed out that this clinical entity is in fact an acute choroidal ischemic lesion, which produces focal areas of infarction of the overlying RPE and outer retinal layers. This was confirmed by Deutman [42]. The evidence supporting that concept is as follows:
-
(i)
Fluorescein angiography, during the early stages of the acute phase of the disease, shows typical nonperfusion of the underlying choriocapillaris in the areas of the lesions during the transit of the dye, with late staining of the lesions (Fig. 12B). In the past, non-fluorescence of the lesions was ascribed a masking effect by the white lesions, but this idea is not supported by my experimental and clinical studies on acute choroidal ischaemia. On resolution, the lesions are replaced by chorioretinal degenerative patches, and, in some cases, even by permanent destruction of the choriocapillaris. The ophthalmoscopic and angiographic appearance is classically that of acute choroidal ischaemia, as shown by the experimental studies discussed above. Serous retinal detachment reported in some of these cases is due to loss of the chorioretinal blood retinal barrier property of the RPE ischaemia, as shown by our HRP study [17].
-
(ii)
This condition may also be associated with other systemic vascular lesions, some of which are clearly of an acute focal ischemic type, e.g., in the cerebral cortex [43] and kidney [44]. The ocular lesions may be associated with erythema nodosum, uveitis, episcleritis or cerebral vasculitis, retinal vasculitis [45] suggesting vasculitis as the basic aetiology; however, the almost universal failure of the ocular lesions to respond to systemic corticosteroid therapy conflicts with an aetiology of simple vasculitis (but some vasculitides do not respond to corticosteroid therapy), and strongly favours an occlusive nature. That many of the lesions are embolic in nature cannot be ruled out.
Thus, the available evidence indicates that multifocal acute ischemic choroidopathy is due to focal occlusion of small choroidal arterioles. The associated optic disc swelling reported in some cases [45] probably represents AION due to focal ischaemia of the optic disc (secondary to peripapillary choroidal ischaemia). lt could be argued that most patients with multifocal acute ischemic choroidopathy have no detectable vascular aetiology; however, those who deal extensively with vascular disorders very frequently see patients who have a confirmed vascular lesion (e.g., embolism or vasculitis) in one organ, yet extensive investigation reveals no aetiology and no other vascular lesion in the body.
Geographic Choroidopathy
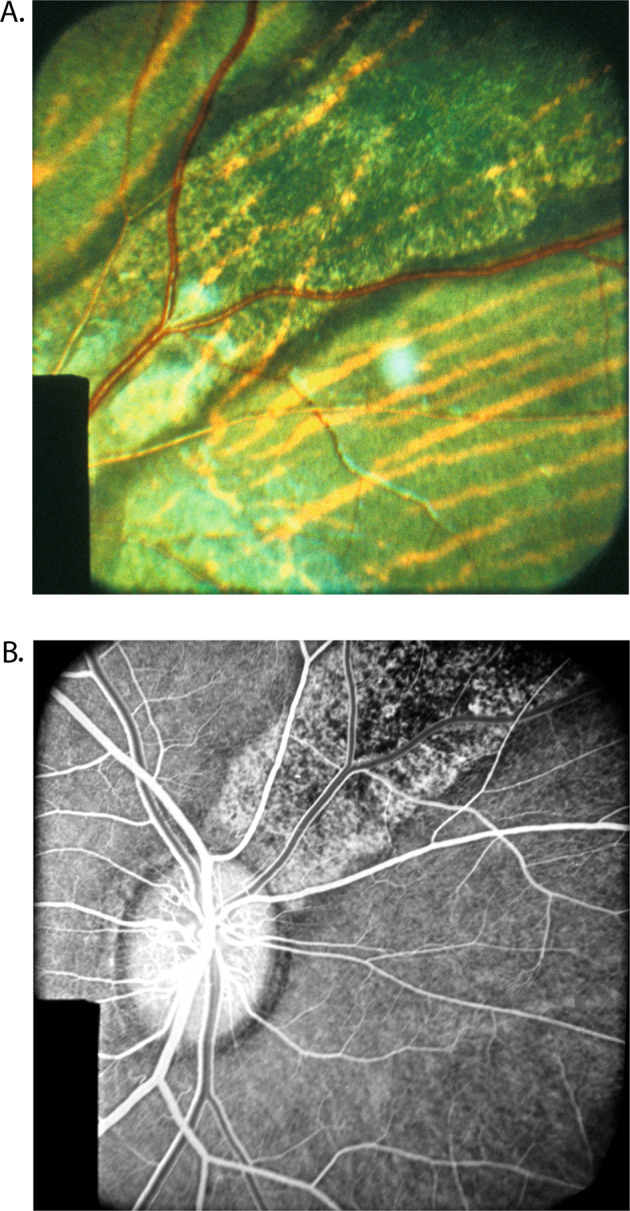
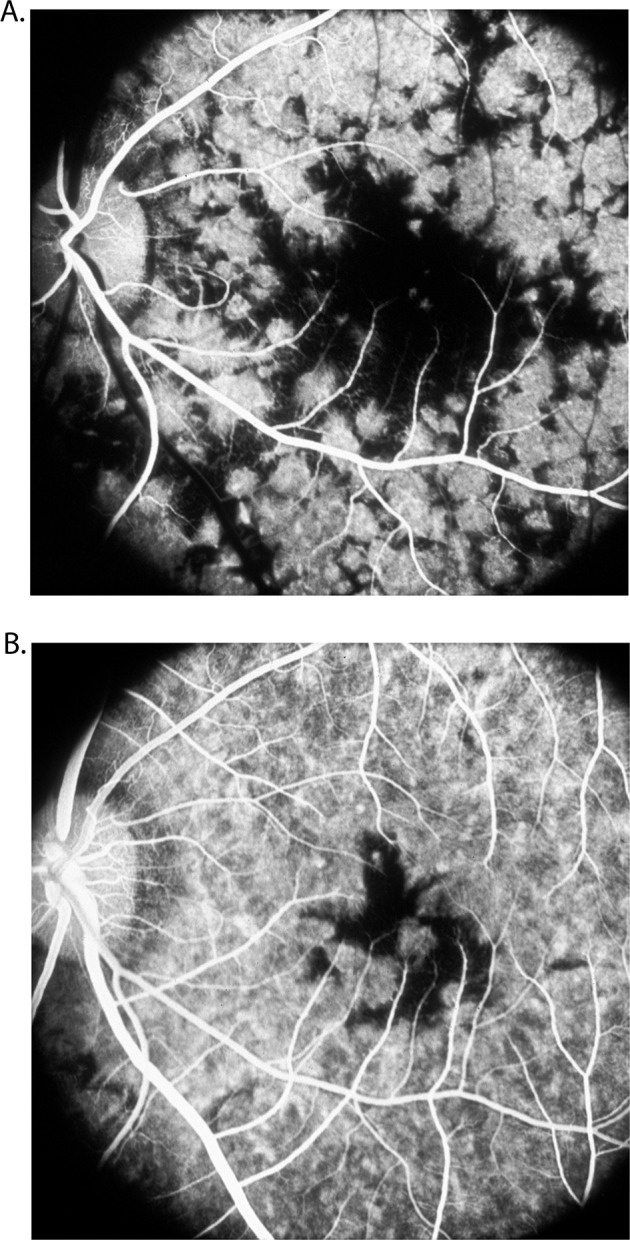
Hyvarinen et al. [46]. first pointed out that the fluorescein fundus angiographic pattern of this suggests an acute ischemic lesion of the choroid; an ischemic aetiology has also been suggested by others. The ophthalmoscopic, fluorescein fundus angiographic and evolution patterns of the lesions are typically those seen in focal acute choroidal ischaemia. However, others consider a vascular aetiology for this condition unlikely [47]. Studies have shown significant thinning of all vascular layers of the choroid; that further suggests a vascular aetiology for geographic choroidopathy (Fig. 13).
Fig. 13.
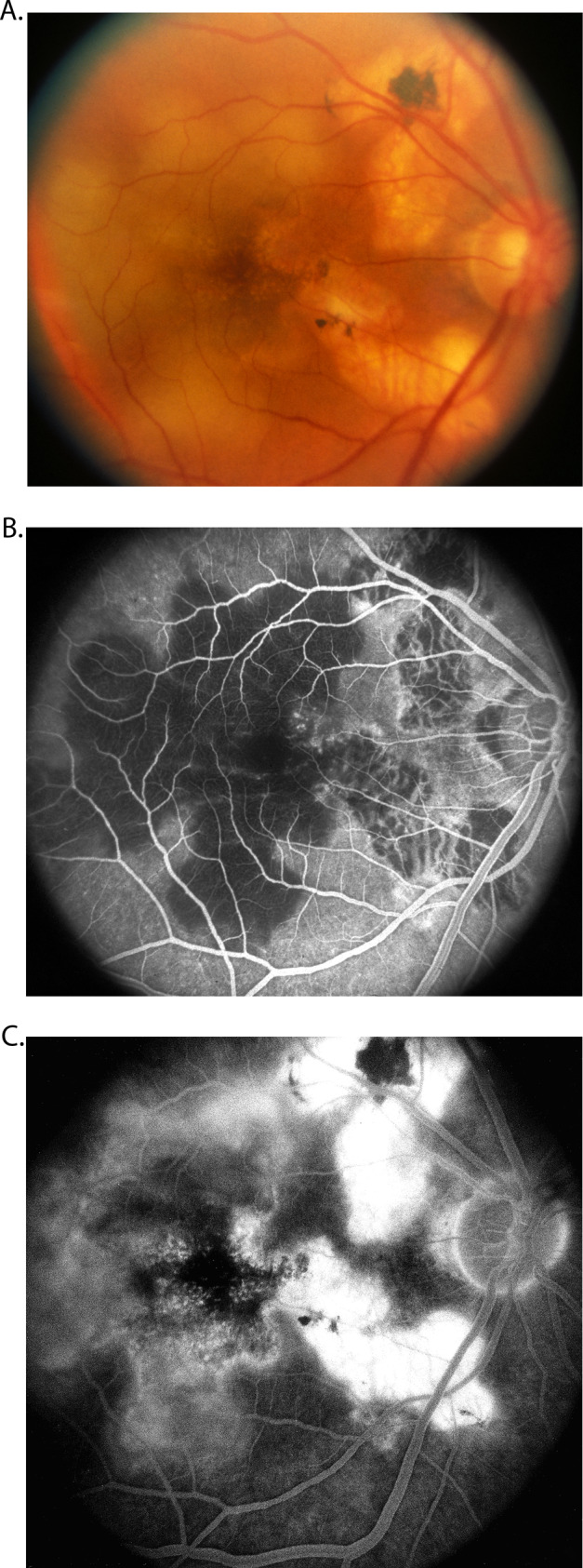
Fundus photograph (A) and fluorescein fundus angiograms (B, C) of right eye of a 57-year-old woman with geographic choroidopathy. A Fundus photograph shows acute and old lesions. B Angiogram during retinal arteriovenous phase shows no filling of the lesions and loss of choriocapillaris in the old, healed patches. C Angiogram during late phase shows fluorescein staining of lesions.
Conditions Associated with Serous Retinal Detachment
Ischemic dysfunction of the RPE (with or without demonstrable frank infarction) produces breakdown of the chorioretinal blood barrier. This is seen in conditions associated with acute onset of arterial hypertension, e.g., malignant hypertension [33], renal failure or insufficiency, eclampsia, Goodpasture’s disease and other allied disorders. In all these conditions, the choroidal arterioles show hyalinization and fibrinoid necrosis [34] and consequent choroidal ischaemia which produces ischaemia of the overlying RPE and consequent breakdown of the barrier and leakage of fluid under the retina. Once the hypertension is controlled, the retinal detachment settles spontaneously. Similar serous retinal detachment is seen in some cases of multifocal acute ischemic choroidopathy [48] and disseminated intra- vascular coagulopathy. The central serous choroidopathy (mistakenly called retinopathy) may also belong to this group. It is possible that some of the cases of Harada’s disease may fit into this category as well [48].
Post-operative Acute Choroidal Ischaemia
This develops during the post-operative period in some eyes subjected to closed-system intraocular surgery where the IOP is kept very high during surgery [49]. A number of these cases have been reported. The necrosis of the outer retina and RPE is due to acute choroidal ischaemia [14, 16]. To understand the mechanism of acute choroidal ischaemia in these eyes, one must take into consideration two important factors governing their intraocular blood:
Perfusion Pressure in the Intraocular Arteries
This is equal to the mean blood pressure in the vessels minus the IOP (Mean blood pressure = Diastolic blood pressure + 1 /3 of difference between systolic and diastolic blood pressures).
Presence or Absence of Autoregulation
The choroidal vascular bed has no autoregulatory control, so that its blood flow cannot adjust to the fall of the perfusion pressure and is reduced proportionately; in contrast, the retinal vascular bed can autoregulate its blood flow to a certain level and is thus less vulnerable to circulatory disorders in the event of a fall of perfusion pressure.
In vitrectomy or any other intraocular surgery in a closed eye, if the IOP is excessively raised for a prolonged period, the blood flow in the choroidal vascular bed would be seriously impaired during that time and this would produce acute choroidal ischaemia. If at the same time the systemic blood pressure falls (as in general anaesthesia or deep sedation), this would lower the perfusion pressure still further. The fact that some of these eyes may recover good visual function [50] indicates that the ischemic damage is not permanent, since normal blood flow is restored at the end of surgery, and the RPE and outer retina must recover function in due course. This contrasts with my experimental acute choroidal ischaemia produced in monkeys by permanent cauterization of the PCAs, in which case the RPE and the outer retina suffered permanent damage. This recovery of visual function in eyes subjected to transient choroidal ischaemia gives us important information about the degree of tolerance of the RPE and outer retina to transient ischaemia. This information is helpful in understanding some of the acute choroidal ischemic disorders.
Occlusion of the LPCA
Figure 14A shows the acute lesion in a patient, and Fig. 14B in another patient with later pigmentary degeneration in the involved region.
Fig. 14.
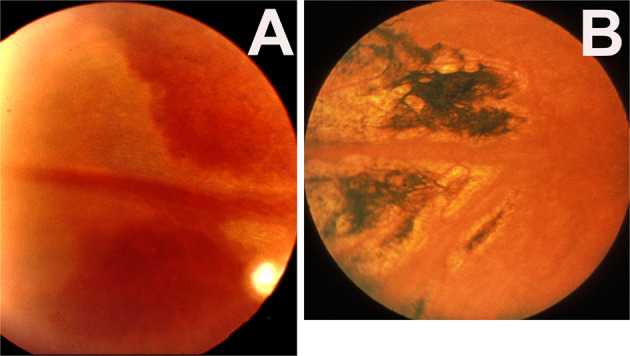
A Whitish choroidal ischemic lesion in the distribution of the temporal long PCA three days after the onset of severe posterior scleritis. B A triangular chorioretinal lesions situated in the area corresponding to the distribution of the resolved ischemic lesion of LPCA. (Reproduced by kind courtesy of late Dr. Pierre Amalric).
With further awareness of acute choroidal ischaemia, more hitherto obscure conditions may be found to fall in this group of diseases.
Macular Lesions Secondary To Choroidal Vascular Disorders
Choroidal vascular disorders can produce a variety of lesions in the macular region. These include the following.
Acute Choroidal Ischemic Lesions
These are seen in a variety of systemic diseases discussed above. These lesions present a typical clinical picture. They appear acutely. As discussed above, ophthalmoscopically the lesions are homogeneously white at first (because of infarction of the RPE and the overlying outer retinal layers) but after two to three weeks they resolve into chorioretinal pigmentary degenerative lesions [14, 16]. Their size and shape depend upon the size of the occluded choroidal artery. They are usually small, localized lesions in the distribution of a terminal or larger choroidal arteriole/artery. The small localized chorioretinal infarcts are equivalent to cotton wool spots (i.e. localized superficial retinal infarcts)—the former due to occlusion of terminal choroidal arterioles and the latter to occlusion of terminal retinal arterioles.
Acute choroidal ischemic lesions may present as a variety of distinct clinical entities; however, they represent a continuous spectrum of the disease, depending upon the size and number of the involved vessels and the severity of ischaemia. The various lesions are discussed above, and they include the following clinical entities: Elschnig’s spots, multifocal acute ischemic choroidopathy (so-called “acute posterior multifocal placoid pigment epitheliopathy”), and geographic choroidopathy and postoperative acute choroidal ischaemia.
Chronic Choroidal Ischemic Disorders
Age-related Macular Degeneration
In 1974, based on my fluorescein fundus angiography of submacular choroidal vascular bed in rhesus monkey [18], I discovered that all the temporal SPCAs enter the eyeball in the macular region and spread out to the periphery of the fundus to supply the temporal half of the choroid, as shown by fluorescein fundus angiography of a man (Fig. 15) [51]. It is, therefore, natural that most of the segments of the choroid supplied by the temporal SPCAs and their watershed zones meet in the submacular region [18] (Fig. 16). This was a consistent pattern in my studies. It is well-established that an area where numerous watershed zones meet is an area of comparatively poor vascularity and in the event of circulatory disorders, most vulnerable to ischaemia. Watershed zones are known to play an important role in ischemic disorders, for example, in ischemic infarcts of the brain and in AION [52]. Based on my findings about the location of multiple watershed zones in the submacular choroid and their importance, in 1974 I concluded: “The present studies, therefore, suggest that the macular region is especially vulnerable to chronic ischaemic disorders.” I further stated: “In the light of my findings, the frequent occurrence of senile macular degeneration (now called “age-related macular degeneration”) is not at all surprising. In senile macular degeneration, the well documented submacular choroidal neovascularization from the choroidal vascular bed may represent a response to chronic ischaemia.” [18] This concept was attacked immediately because it contradicted the conventional wisdom that the submacular choroid was the most vascular part of the choroid. As discussed above, this concept of increased vascularity was based on the misconception that histological and anatomical studies of submacular choroid show many arteries there. One has to keep in mind, however that all the temporal SPCAs enter the choroid in the macular region, as shown in a fluorescein fundus angiography of a man (Fig. 15), but they immediately run radially towards the equator of the eye to supply the rest of the choroid, and only the apical parts of the various segments supplied by the temporal SPCAs meet each other in the centre of the macular region (Fig. 16), and the macular region is not the only area supplied by these arteries.
Fig. 15. Fluorescein fundus angiogram of a normal human eye, showing the sites of entry of the SPCAs and their course in the choroid.
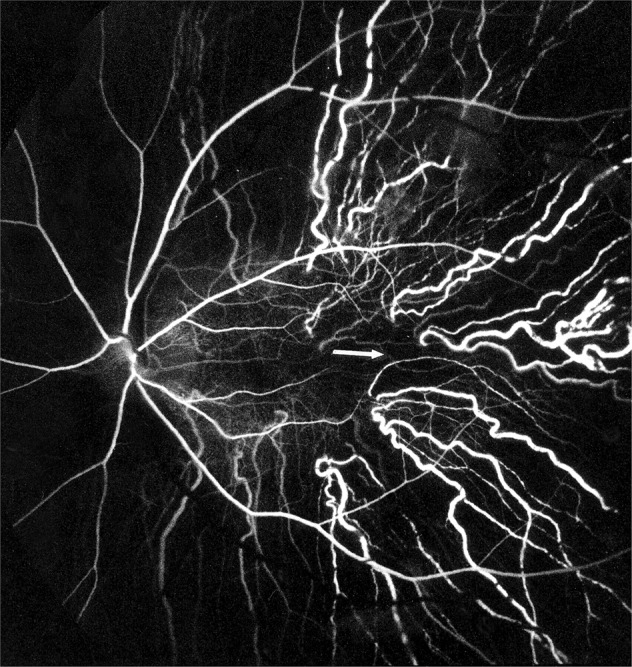
Arrow marks the center of the macular region. Note that no artery lies in the centre of the macular region. (Reproduced from late P. Amalric: Int. Ophthalmol. 1983;6:149-53).
Fig. 16.
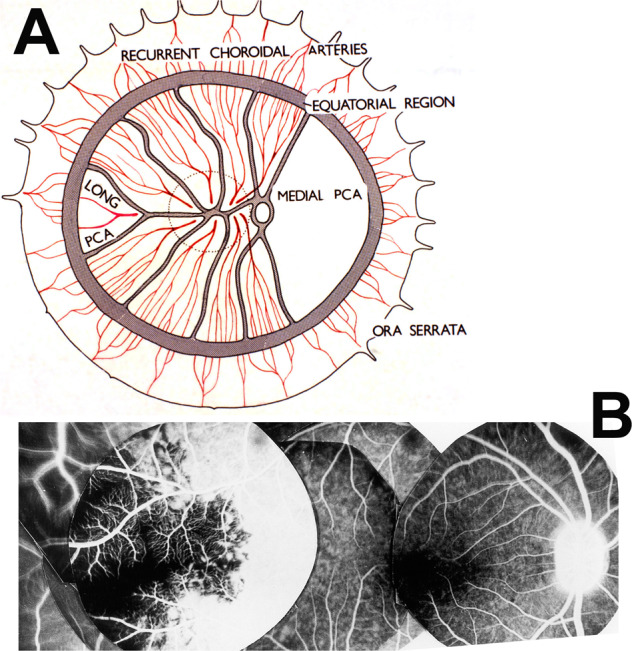
A Diagrammatic representation of the distribution by the various ciliary arteries in the choroid and watershed zones between them. The choroid posterior to the equator is supplied by the medial and lateral PCAs. In the area supplied by the lateral PCA are shown the segments supplied by the various short PCAs and the one by the long PCA, with the watershed zones between them (dotted circle in this area indicates the macular region). Recurrent choroidal arteries from the ACAs and the greater arterial circle of the iris supply in front of the equator. The watershed zone between the anterior and posterior choroidal arteries lies in the equatorial region. B Composite fluorescein fundus angiograms of rhesus monkey eye, after cutting the temporal long PCA, showing no filling of the choroid in the extreme temporal periphery, temporal to the macular region.
My original concept is further supported by the following evidence.
1. In fluorescein angiography of early age-related macular degeneration, I reported a filling delay of the watershed zones between the temporal SPCAs in the centre of the macular region [53], (Fig. 17. Chen et al. [54] also showed delayed choriocapillaris filling on fluorescein fundus angiography in age-related macular degeneration with decreased visual acuity. They concluded that chronic choroidal vascular compromise is an important cause of visual impairment.
Fig. 17.
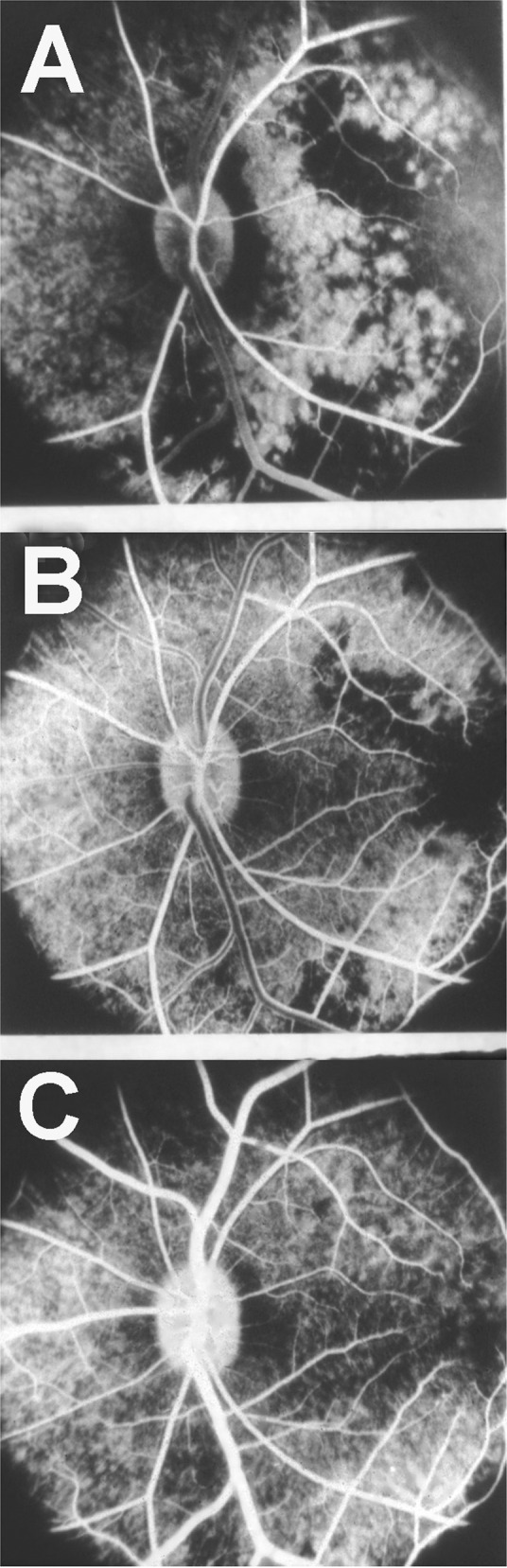
Fluorescein fundus angiograms in left eye of a rhesus monkey, after reduced perfusion pressure in the eye, 9.1 (A), 11.2 (B) and 17.2 (C) seconds after intravenous injection of fluorescein. Note slow filling of the choroid, particularly marked in the watershed zones between the various temporal short PCAs and the macula region in (A) and (B).
2. Ross and associates [55], on indocyanine green angiography, showed an increased incidence of choroidal watershed zone filling defects in patients with age-related macular degeneration and postulated that that may predispose them to choroidal neovascularization.
3. When I experimentally reduced perfusion pressure in the ocular vessels of rhesus monkey eyes, fluorescein fundus angiography revealed delay in the filling of the watershed zones in the macular region between the temporal SPCAs, and of the central macular choriocapillaris (Fig. 17).
4. My studies have shown that the watershed zones of the various vortex veins are also located in the submacular choroid (Fig. 18) [18].
Fig. 18. Diagrammatic representation of watershed zones between the four various vortex veins in rhesus monkeys.

X = fovea.
5. In studies of experimental malignant hypertension in rhesus monkeys, choroidal ischaemia was found to be an important manifestation [33]. Fluorescein fundus angiographic studies in these eyes during the acute phase revealed markedly delayed filling of the submacular choroid, particularly in its central part (Fig. 19), and there were choroidal ischemic lesions in the macular region which produced macular retinal detachment (Fig. 12A).
Fig. 19.
Fluorescein fundus angiograms of left eye of a rhesus monkey with malignant arterial hypertension (on day 42, with blood pressure 205 mmHg), during retinal arterial (A) and (B) late venous phases, show marked delay in filling of the submacular choroid.
The effect of the meeting of multiple watershed zones of the temporal short PCAs in the macular region, causing macular ischaemia under certain circumstances, can be explained by the following common example, illustrated in Fig. 20 and discussed previously [18]. Normally (Fig. 20A), the part of the lawn situated in the centre of the areas of multiple sprinklers would receive an adequate water supply from all of them combined; each would supply a segment of it (comparable to each temporal SPCA supplying a segment of the central part of the macular region) (Fig. 16). However, when the water pressure is reduced in the sprinklers, the territory irrigated by each sprinkler shrinks, leaving the central part inadequately watered or even dry (Fig. 20B), depending upon the extent of the fall of water pressure. The occurrence of this phenomenon in the macular region is suggested by the fluorescein angiograms in Fig. 17. These angiograms show a prolonged delay in the filling of the watershed zones in the macular region, and they very well illustrate the meeting of the various watershed zones in the macular region and their selective susceptibility to hypoperfusion.
Fig. 20. Two illustrations demonstrating effect of reducing water pressure in eight garden sprinklers watering a lawn.
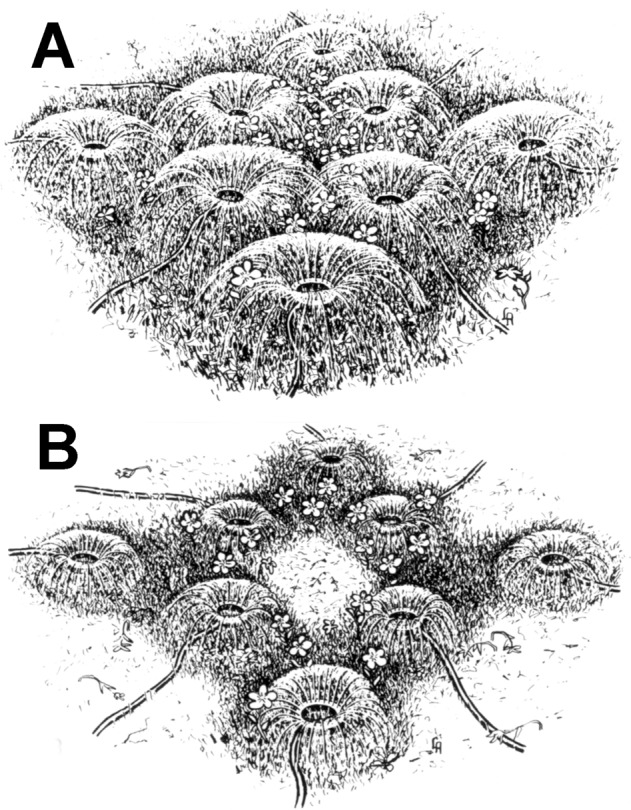
A Watering at normal water pressure with normal areas of supply by the various sprinklers. B Same areas supplied by sprinklers with reduced water pressure. Note shrinkage of the central and other areas when water supply is markedly reduced now.
There have been reports of selective localized senile atrophy of the choriocapillaris, and in some cases even of larger choroidal vessels, in the submacular choroid [56–59]. Grunwald and colleagues have measured the foveal choroidal blood flow in age-related macular degeneration in several studies [60]. In their latest study [60], they concluded that in age related macular degeneration “development and progression are associated with decreased choroidal circulatory parameters.”
The prevalence of age-related macular degeneration increases progressively with age. Some studies have shown an increased incidence of arterial hypertension and smoking in these patients. A reduction in perfusion pressure and blood flow can also be influenced by several systemic and local factors in this age group, and those further add to its multifactorial aetiology. In the light of all this information, it would seem logical to consider that the submacular choroid must be more vulnerable to ischemic disorders than other parts of the posterior choroid. The same phenomenon is seen in the equatorial choroidal region where there is watershed zone between the PCAs and ACAs (Figs. 16, 21).
Fig. 21. Reticular pigmentary degeneration in the equatorial region of a patient with age-related macular degeneration.
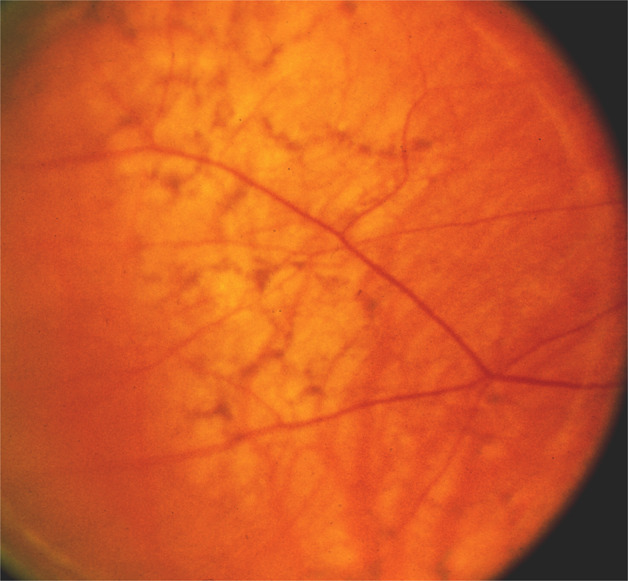
The pigment accumulates in lines forming irregular polygons.
Thus, all the evidence collectively provides support for my original hypothesis of 1974, that the submacular choroidal ischaemia plays a role in age-related macular degeneration. When Grunwald and colleagues [60] recently demonstrated decreased foveolar choroidal blood flow in eyes with age-related macular degeneration, I remarked, “It is gratifying to see that finally new investigations are confirming what I reported 40 years ago, but was ignored and forgotten for all these years.” [61].
Macular Lesions Associated With Subretinal Choroidal Neovascularization
This type of neovascularization is an important cause of macular diseases. The blood vessels arise from the choroidal vascular bed, and travel through defects in the Bruch’s membrane to reach the sub-pigment epithelial space and later the subretinal space. Thus, an eye with a break in the Bruch’s membrane from any cause is in danger of developing choroidal neovascularization. The neovascularization may lead to localized macular subretinal and/or sub-epithelial haemorrhages or serous detachment. Radial chorioretinal folds in the macular region may be an early sign of choroidal neovascularization. The choroidal neovascularization in the macular region has been reported in several conditions. These include: age-related macular degeneration, presumed histoplasmosis syndrome, myopic choroidal degeneration, choroidal rupture, angioid streaks, old chorioretinal scar of focal choroiditis, old chorioretinal scar of toxoplasmosis, macular dystrophies, e.g., vitelliform degeneration, macular drusen, hereditary haemorrhagic macular dystrophy, optic disc drusen, choroidal nevus and other choroidal tumours, geographic choroidopathy, rubella retinopathy, toxocara canis infection, photocoagulation scar, chronic uveitis, idiopathic subretinal neovascularization, and others.
Macular Lesions Associated With Subretinal Detachment
These are seen frequently and may be due to a definite or a presumed choroidal vascular lesion. These include the following:
Conditions Associated with Choroidal Neovascularization in the Macular Region
This probably constitutes the major cause of subretinal serous macular retinal detachment. The various conditions in which the neovascularization is found are listed above.
Conditions Associated with Breakdown of the Chorioretinal Blood Barrier
As discussed above, the normal RPE exercises a chorioretinal blood barrier, which prevents any leakage of the serous choroidal fluid into the retina [62]. Any lesion which produces damage to the RPE and consequent breakdown of this barrier must result in seepage of serous fluid under the retina and produce a serous retinal detachment. On fluorescein fundus angiography, fluorescent spots are usually seen at the level of the RPE during the acute phase of the disease process (Fig. 12B) but later the spots frequently develop RPE degeneration. This disorder is seen in several conditions, including the following:
Conditions Associated with Acute Onset of Arterial Hypertension
This is seen in malignant hypertension (Fig. 12A), renal failure or insufficiency, eclampsia, Goodpasture’s disease, and other allied disorders. In all these conditions, there is choroidal ischaemia which produces ischaemia of the overlying RPE and consequently breakdown of the barrier and leakage of fluid under the retina. Once the hypertension is controlled, the retinal detachment settles spontaneously.
Multifocal Acute Ischaemic Choroidopathy
A number of these cases have been reported with serous macular detachment [48]. The ischemic damage to the RPE breaks down the barrier.
Disseminated Intravascular Coagulopathy
The characteristic histopathological abnormalities consist of thrombotic occlusion of the choriocapillaris and adjacent choroidal arterioles and venules, limited to the submacular and peripapillary choroid. Damage to the RPE over the foci of occluded choriocapillaris causes breakdown of their barrier property and results in serous retinal detachment. Similar changes have been reported in thrombotic thrombocytopenic purpura.
Cavernous Haemangioma of the Choroid
There are many reports of this in the literature.
Other Miscellaneous Conditions
These include central serous choroidopathy (erroneously called central serous retinopathy), Harada’s disease (or Vogt-Koyanagi Harada syndrome), and sympathetic uveitis. The presence of fluorescein-leaking spots on angiography, at the level of the RPE, is well documented in these diseases, but the cause of localized RPE damage is still obscure. In Harada’s disease the evidence points more to an inflammatory aetiology as also seems to be the case in sympathetic uveitis; however, Harada’s disease could be a manifestation of choroidal ischaemia, and some authors feel that Harada’s disease (Vogt-Koyanagi-Harada syndrome) and multi focal acute ischemic choroidopathy represent the spectrum of a single disease entity [48]. In central serous choroidopathy the possibility of ischaemia (due to embolism or other causes) cannot be ruled out. There are many non-vascular causes of serous macular retinal detachment; they are beyond the scope of this article.
Miscellaneous Conditions Associated With Macular Lesions
There are several other macular conditions in which the choroidal vascular bed plays either a definite or a presumable role. These include the following:
Infectious Disease
Toxocara canis and cysticercosis organisms may travel via the choroidal circulation. The organisms may lodge in the macular region to produce toxocara canis infection and submacular disciform lesion and later a chorioretinal-anastomosis when a scar is formed, and in cysticercosis infection a subretinal cyst formation. Presumed histoplasmic lesions in the macular region may be due to a previous infection by Histoplasma Capsulatum.
Traumatic Lesions
A blunt ocular trauma may cause choroidal rupture and consequent subretinal haemorrhage in the macular region and later chorioretinal scar on resolution of the haemorrhage.
Heredodystrophic Macular Diseases
In a number of these diseases, there is either definite evidence of associated choroidal vascular disorders or evidence suggestive of such disorders (without definite proof). The conditions include the following:
Sorsby’s central areolar choroidal sclerosis and allied disorders
In these conditions there is a defective perfusion of the macular choriocapillaris and later even disappearance of some of the bigger choroidal vessels. Some authors have suggested an ischemic basis for them [63].
Primary familial amyloidosis
The most striking site of amyloid deposition in this condition is in the choroid, and the whole choriocapillaris bed may be completely obliterated by the amyloid deposits [64].
Doyne’s honeycomb dystrophy
The lesions correspond to the arterial parts (i.e., the central parts) of the choriocapillaris lobules (Fig. 22A) and suggest that they may possibly be due to a disorder of the arterial segments of the choriocapillaris.
Fig. 22. Fluorescein fundus angiograms of a rhesus monkey eye at the posterior pole.
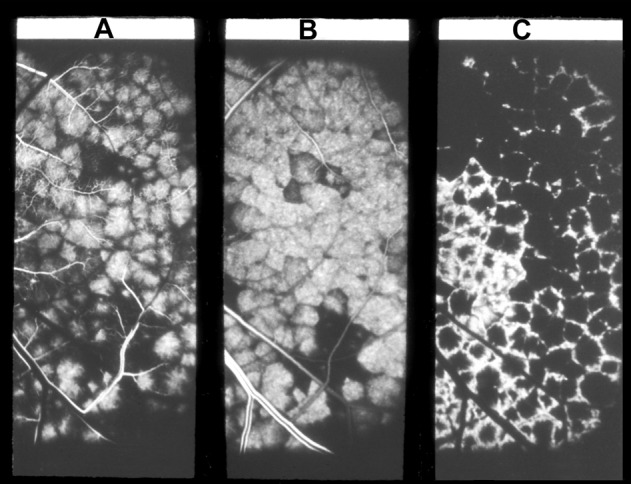
A The early arterial filling phase of the choriocapillaris, showing each lobule of the choriocapillaris (supplied by the terminal choroidal arteriole) forming a big fluorescent spot. Each spot is surrounded by a polygonal unfilled zone, producing a mosaic pattern in the choriocapillaris. B Peak arterial filling phase. Note the extraordinarily well-defined mosaic pattern, with each unit of the mosaic an independent entity, and the isolated nonfilling or slow filling of some of the lobules is clearly visible. It suggests that there is no communication between adjacent lobules. C Venous phase of the choriocapillaris filling, showing a honeycomb pattern; the fluorescent pattern is reverse of that seen in A., i.e., the fluorescent areas are nonfluorescent and vice versa.
Fundus flavimaculatus
The shape and distribution of the “fish tail” lesions in this condition resemble venous segments of the choriocapillaris lobules and suggest that this may be a disorder of the venous part of the choriocapillaris lobules (Fig. 22C). A vascular basis for the flecked retina syndrome is further suggested by the occurrence in the mid- and far-periphery of the fundus of chorioretinal lesions like those seen in choroidal ischaemia. However, the inheritance patterns seen in this syndrome are hard to explain on a vascular basis. A detailed description of each disease entity is beyond the scope of this discussion but can be found in detailed publications on the different disorders and in various recent publications.
Lesions Produced By Long Pca Occlusion
The LPCAs supply the anterior segment of the eye. There are many studies on the effects of experimental occlusion of the LPCAs on the anterior segment of the eye and the IOP. Most were carried out in rabbits; the production of ocular hypotony, phthisis bulbi, iris neovascularization, cataract and anterior segment necrosis has been recorded in rabbit. Interference with the LPCAs has been attributed to the occurrence of anterior segment ischaemia following diathermy, scleral buckling, and some other procedures for retinal detachment surgery [29].
In view of this, I investigated the effect of cutting a LPCA. This was done in two studies:
In 22 eyes of rhesus monkeys, one LPCA alone was cut [19].
In 85 eyes of the rhesus monkey one or the other main PCA (48 eyes) or all of them (37 eyes) were cut [13, 14].
In all cases, the anterior segment remained normal.
Retinal detachment surgery involving cutting of one or more rectus muscles, with extensive diathermy applications to the uveal tract, including the LPCAs, and encircling procedures, have been known to produce anterior segment necrosis. In such cases the occlusion of the LPCAs has been considered erroneously to be the major factor in the production of anterior segment ischaemia. The major factor in these cases has been the wrong concept created by the rabbit experiments. In rabbits, the major blood supply to the anterior segment of the eye is by the LPCA [65], but that is not the case in primates or humans, The observations in primates of occlusion of the LPCAs indicate that this by itself plays no significant role in anterior segment ischaemia in man, although it may act as a minor contributing factor in combination with interruption of the ACAs, and interference with venous drainage [66] from the anterior uveal tract due to liberal diathermy and indentation procedures in retinal detachment surgery. As shown above, this is further demonstrated by the absence of any lesions on follow-up in the anterior segment of eyes with occlusion of one or both LPCAs.
Lesions Produced By Anterior Ciliary Artery Occlusion
Anterior Segment Ischaemia
Anterior segment ischaemia is not a common disorder, but it can result in serious visual loss. In this condition, there is usually ischaemia of the iris and ciliary body, but it may involve the cornea and sclera. The blood supply of the anterior segment is discussed above.
Schmidt [67] gave the first clinical description of anterior segment ischaemia in 1874. Crock [68] reviewed the history of anterior segment ischaemia in 1967. The following account is based on his review. Clinical and pathological features were described by Harbitz [69] in 1926. Leinfelder and Black [70] in 1941 described it on the experimental transposition of the extra-ocular muscles in monkeys. Skipper and Flint [71] in 1952 described symptoms and signs in 18 cases. Ask-Upmark [72] in 1954, described atrophy of the iris, cataract, and corneal opacities in Takayasu’s arteritis. Knox [73] reviewed the literature in 1965. Wilson and Irvine [74] first reported it after retinal detachment surgery in 1954.
Causes Of Anterior Segment Ischaemia
This can be associated with a variety of clinical conditions. There are many reports, mostly anecdotal or based on only a few cases, of the development of anterior segment ischaemia with ocular ischemic syndrome [73, 75], and the literature on the subject was reviewed in our study on ocular ischaemic syndrome [75] Other conditions include surgery involving cutting recti, for the treatment of strabismus [70, 76] or retinal detachment [68, 76], GCA [32], following cyclocryotherapy [77], sickle cell hemoglobinopathy [78], chronic myelogenous leukaemia, chronic lymphocytic leukaemia, Takayasu’s arteritis [72], occlusion of a PCA, Fuchs’ heterochromic cyclitis, in angle-closure glaucoma [79], after intravitreal silicone injection, quinine toxicity, chloramine mustard, herpes zoster [69], rubella [68], irradiation for malignancies around the orbit and the globe [68], and after perfusion of intracranial malignancies with nitrogen mustard.
Anterior segment ischaemia is well-known to develop after the cutting of various rectus muscles for the treatment of strabismus [70, 76, 80, 81] or in retinal detachment [68, 76, 81] after cutting one or more rectus muscles, with extensive diathermy applications to the uveal tract, including the LPCAs, and encircling procedures. The anterior segment is supplied by various ACAs in all the rectus muscles and by the LPCAs. I investigated, by experimental and clinical studies, the effects of cutting various ACAs or long PCAs on the development of anterior segment ischaemia.
Experimental And Clinical Studies
Anterior Segment Ischaemia After Recession Of Various Recti
There is little comprehensive information on this important topic in the literature. To investigate this, we carried out recession of various recti (in various combinations) in 40 normal adult cynomolgus monkey [76], and in 33 human eyes [80]. The various combinations of recession were combined to evaluate their effects on the anterior segment; such combinations may be considered desirable in clinical practice but several of them had not been by ophthalmologists for fear of producing anterior segment ischaemia. These procedures were done as primary, secondary, tertiary and fourth (in one eye) procedures (each separated by several days, weeks or months), ultimately in monkeys involving all four rectus muscles in the eyes. The successive surgeries were always separated by a time interval that enabled the eye to recover normal filling of the iris vessels on fluorescein angiography, so that preoperative iris angiography in every case showed normal circulation in the iris.
Tenotomy of the recti cuts the ACAs; but the anterior segment is also supplied by the LPCAs; therefore, in some monkey eyes the LPCAs were also cut, alone or in combination with different recti, to find the effect. The anterior segment was evaluated by slit lamp examination and fluorescein iris angiography. The findings are discussed in detail elsewhere [76, 80].
This study showed that when all four recti were recessed simultaneously, as a primary procedure, the anterior segment changes were the most severe and the eyes were left with permanent lesions (e.g., cataracts in five of six eyes, atrophic iris in all, corneal vascularization, and opacity in two). However, when the simultaneous recession of the four recti was a secondary procedure (preceded by a simultaneous recession of both vertical recti and one horizontal rectus, about 2–5 months earlier), the anterior segment changes were comparatively much fewer and milder (e.g., corneal or lens changes did not develop in any of the six eyes, and iris atrophy did not even develop in two of the eyes). When the simultaneous recession of the four recti was a tertiary procedure, the anterior segment changes were once again fewer and milder than after the primary procedure. This indicates that simultaneous recession of all four recti as a secondary or tertiary procedure (after a previous recession of three to four muscles, 2–5 months earlier) did not produce serious permanent ocular complications.
Simultaneous recession of one vertical and one horizontal rectus (or both horizontal or both vertical recti), as a primary or secondary procedure, almost invariably produced no changes in the anterior segment (apart from angiographic filling defects in the iris) despite the fact that in all of these eyes, after the second procedure, all four recti had been recessed in two stages. As an exception to this, however, one eye, with simultaneous recession of the vertical recti as a primary procedure, showed a moderate anterior segment ischaemia during the immediate postoperative period.
Anterior Segment Ischaemia After Simultaneous Occlusion Of Both Lpcas And Recession Of The Horizontal Recti
When with this was a primary procedure, evidence of anterior segment ischaemia during the acute phase was very common, and later iris neovascularization developed in four of the 5 eyes, corneal neovascularization in two, iris atrophy in one, cataract in one, and prolonged hypotony in three. The anterior segment changes were thus much more severe in this group than after the recession of the corresponding recti alone or of occlusion of the PCAs alone, indicating a compounding effect of the two procedures. So, the risk of producing anterior segment ischaemia with this procedure is high and the eye may be left with permanent serious complications.
The findings of our experimental [76] and human [80] strabismus surgery studies also show that iris angiography is a very useful test to evaluate ischemic changes in the anterior segment because it shows, apart from the presence of filling defects in the iris, fluorescein leakage from the iris vessels, which is an important sign of ischaemia.
Conclusions
1. The nasal and temporal sectors (especially the former) have a dual blood supply-primary from the ACAs of the horizontal recti and subsidiary from the LPCAs; the former source plays a far more important role than the latter. Because most of the temporal perforating arteries are branches of the ACAs of the vertical recti [82], they constitute the major source of blood supply to the temporal sector. When recession of the medial rectus was combined with that of one of the vertical recti, the nasal iris filling defects were more frequent and more extensive than those seen after recession of the medial rectus alone. This suggests that even in the nasal region the ACAs of the vertical recti play some role in the blood supply.
2. The superior and inferior sectors are mainly supplied by the ACAs of the vertical recti, with no LPCAs in those regions. Cutting the vertical ACAs usually produces an abnormal filling defect in those areas, and the defect mostly extends to involve a part of the adjacent temporal sector also, because the ACAs of the vertical recti also generally contribute to the temporal region [82]. This study indicates that the ACAs of the vertical recti play a crucial role (more important than either the ACAs of the horizontal recti or the LPCAs) in the blood supply to the anterior segment.
3. The contribution by the LPCAs to the anterior segment is usually not very important. This study, as well as my experimental studies on the occlusion of the LPCAs or parent trunks of the LPCAs (i.e., the main PCAs [13, 14, 16, 19]), have clearly shown that that occlusion alone does not produce any anterior segment ischaemia. This was further confirmed by my patients with GCA in whom fluorescein fundus angiography showed complete occlusion of the main PCAs, without any evidence of anterior segment ischaemia. On the other hand, in seven eyes, with normal PCA circulation but simultaneous recession of all four recti as a primary procedure, immediate postoperative iris fluorescein angiography showed some filling of the nasal quadrant in four and of a small temporal sector in two, and no filling of the iris at all-in-one eye, with severe anterior segment ischaemia developing in all eyes. The latter indicates that the LPCAs, even when they contribute to the blood supply of the anterior segment, do not protect it from acute ischaemia in the event of occlusion of the ACAs. The evidence of this experimental study also indicates a frequent additive effect of occlusion of LPCAs with the occlusion of the horizontal ACAs, as in retinal detachment surgery.
4. As shown above, there is a marked disparity between the postmortem anatomic cast studies and in vivo occlusion studies,
5. Studies [31] established that there is marked interindividual variation in the distribution by the PCAs in humans, and this experimental study suggests that the same may be true of the distribution by the ACAs and LPCAs. This would explain the interindividual variation in the response after recession of all the four recti as well as in other groups. It seems that either certain sectors of the iris have more than one source of blood supply with interindividual variation, or the various arterial systems are independent and are connected to each other through collaterals, the situation, number, and efficacy of which determine the type of iris filling defect and the degree of iris ischaemia.
Physiological Anatomy Of The Blood Supply Of The Iris
The experimental [76] and human [80] strabismus studies suggest that the usual pattern of the circulation of the iris in man is as follows:
1. The nasal half of the iris seems to be supplied essentially by the medial LPCA, with the medial ACAs (from the medial rectus) playing only a subsidiary role.
2. It is not possible be definite what role the lateral LPCA plays in the supply of the iris. The findings in the limited number of patients of our series seem to suggest that the lateral LPCA has only a minor role in the supply of a small temporal sector of the iris. However, a large series and further investigation are required to obtain any definite information on the subject.
3. The lateral rectus ACA seems to supply only a small temporal sector of the iris, most probably along with the lateral long PCA.
4. Since no superior and inferior LPCAs exist in any animal species, including man, the superior and inferior ACAs seem to assume the major role in the supply of superior temporal and inferior temporal regions of the iris, respectively.
Role of the Major Arterial Circle of the Iris in Establishing Collateral Circulation
It could be argued that the major arterial circle of the iris should be able to compensate for the loss of one or more of the ACAs. In some of the eyes with vertical rectus tenotomy, angiograms of the limbal vessels were available; most of them showed filling defects in the corresponding limbal vessels as well, because that region is supplied by the episcleral branches of the ACAs. In these eyes the filling of the limbal and iris vessels was usually related, suggesting a possible mechanism of delayed filling of the iris vessels in the region of a cut vertical rectus. That is, the limbal vessels fill through free anastomoses with the adjacent vessels and this produces a retrograde filling of the remaining distal segment of the cut ACAs via their episcleral branches; the blood then flows via the perforating branches to fill the iris vessels. It is therefore possible that extensive extraocular muscle surgery associated with destruction of the episcleral and limbal vessels in the corresponding region may render the eye susceptible to anterior segment ischaemia.
Since the filling delays after tenotomy of the vertical recti seem to persist even months after the tenotomy, it seems that no appreciable collateral supply is established by the greater arterial circle of the iris. In some eyes a part of the major arterial circle may lie on the peripheral part of the iris [83, 84] instead of in the ciliary body.
These findings suggest that the greater arterial circle of the iris plays only a minor part or perhaps none, in filling the affected iris segment. Our knowledge about the anatomy of the greater arterial circle of the iris in man is essentially based on anatomical descriptions. Moreover, to judge from the in vivo fluorescein angiographic studies of the choroid, the morbid morphological vascular pattern does not always correspond with the in vivo circulatory pattern. Also, the iris angiographic findings of my study pose new questions about the true morphology, distribution, and haemodynamics of the greater arterial circle of the iris.
Signs Of Anterior Segment Ischaemia
Anterior segment ischaemia produces a whole range of signs. The early signs in the anterior chamber consist of aqueous flare, even exudation and haemorrhages. IOP may be low. There may be exudate deposits on the anterior surface of the lens. Minor changes can be missed without slit lamp examination. Advanced changes include corneal oedema, folds in Descemet’s membrane, keratic precipitates on corneal endothelium, corneal neovascularization, iris neovascularization, iris atrophy and cataract. The eye with the development of iris and angle neovascularization may develop neovascular glaucoma. The pupil may be affected and may show segmental palsy and/or transillumination defect. The anterior segment may become dystrophic, and the eye may develop phthisis bulbi. If the ophthalmologist is not aware of the clinical entity of anterior segment ischaemia, these clinical signs may be misdiagnosed. For example, initial changes in the anterior chamber and the presence of keratic precipitates may be confused with anterior uveitis or, if seen after retinal detachment surgery, with infection.
Investigations In Anterior Segment Ischaemia
Most important is to be aware of this clinical entity. As discussed above, it may be associated with a variety of systemic conditions, the most important of which is GCA. I have seen rare cases where this was entirely missed, resulting in complete blindness. Therefore, it is essential to rule out GCA.
Anterior segment ischaemia is common in association with ocular ischemic syndrome, in which severe carotid artery stenosis or occlusion is the cause; if that is missed, there is a risk of marked visual loss, stroke or myocardial ischemic complications, because a common cause of death in severe carotid artery disease is myocardial infarction. Therefore, a detailed evaluation of the carotid arteries is essential. If ocular ischemic syndrome is suspected, fluorescein fundus angiography is helpful because it shows poor filling of the iris, choroidal and retinal vasculature.
A thorough slit lamp examination is essential looking for the early signs of anterior segment ischaemia in the anterior chamber: aqueous flare, even exudation and haemorrhages. IOP may be low. Fluorescein dye given intravenously leaks profusely into the anterior chamber.
Management Of Anterior Segment Ischaemia
It is essential to be aware of this clinical entity and various conditions discussed above with which it can be associated. As discussed above, early signs of anterior segment ischaemia have been misdiagnosed as anterior uveitis or infection. In the case of strabismus surgery, as shown above, a certain combination of tenotomies is more likely to produce anterior segment ischaemia; it would be advisable to avoid those combinations as initial procedures. In retinal detachment surgery, once again, prophylactic measures are helpful to prevent development of anterior segment ischaemia. If signs of anterior segment ischaemia are detected following retinal detachment surgery and there is an encircling band, it may be helpful to release that. Apart from that, there is no definite treatment modality for anterior segment ischaemia so far.
Lesions produced by cilioretinal artery occlusion
The anatomy of the cilioretinal arteries is discussed above.
Classification Of Cilioretinal Artery Occlusion
From the point of view of aetiology, clinical features, and visual outcome, cilioretinal artery occlusion can be classified into the following three types:
1. Non-arteritic cilioretinal artery occlusion alone.
2. Arteritic cilioretinal artery occlusion associated with GCA.
3. Nonarteritic cilioretinal artery occlusion associated with central retinal vein occlusion or hemi-central retinal vein occlusion.
Of the three types of cilioretinal artery occlusion, those associated with GCA and those with central retinal vein occlusion or hemi-central retinal vein occlusion require detailed discussion about their pathogeneses, which are controversial.
Non-Arteritic Cilioretinal Artery Occlusion Alone
In a study [85] of 212 eyes with branch retinal arteriolar occlusion, there were 11 eyes with non-arteritic cilioretinal artery occlusion. It was located superiorly in 5, inferiorly in 2, and in the macular region in 4. There were 9 males and one female, with age range of 27 to 87 (mean 53.5 ± 18.3 SD) years. It was almost always due to embolism.
Associated Systemic Conditions
The most common cause, as with branch retinal arteriolar occlusion, is embolism. It can be caused by emboli from the heart or the carotid artery. In the literature there are reports of its association with a variety of other conditions; those do not necessarily imply a cause-and-effect relationship. These conditions include the following: atherosclerotic carotid artery, carotid artery dissection, coronary catheterization, subacute bacterial endocarditis, fibromuscular dysplasia, Behçet’s disease, polycythaemia vera, sickle cell trait, postoperative, following scleral reinforcement surgery, following intranasal cocaine insufflations, systemic lupus erythematosus, antiphospholipid syndrome, ulcerative colitis, embolization of an artery to an intracranial meningioma, laser in situ keratomileusis, posterior scleritis, Fabry’s disease, migraine, and oral contraceptives.
In a study [86] of 10 patients with non-arteritic cilioretinal artery occlusion, there was ipsilateral carotid artery stenosis (16–49%) in 3 and plaques in 3, stroke in 3, arterial hypertension in 4, coronary artery disease in 2, cardiac valvular disease (aortic/mitral valve replacement in 2, calcific mitral valve in 1), and smoking in 7.
Arteritic Cilioretinal Artery Occlusion Associated With Gca
In GCA, arteritic cilioretinal artery occlusion is almost always associated with arteritic AION. This clinical entity was first described in 1974 [87]. There are multiple published reports of it in various GCA publications and there are a few more anecdotal reports in the literature.
In the study [85] of 212 eyes with branch retinal arteriolar occlusion, there were 12 eyes with arteritic cilioretinal artery occlusion—in 11 of them it was associated with arteritic AION. In the one where cilioretinal artery occlusion was not associated with AION, the occluded PCA did not supply the ONH. In the 11 patients (12 eyes) there was temporal artery biopsy confirmed GCA. There were 7 females and 4 males, with age range 57 to 79 (mean 69.4 ± 6.8 SD) years.
Pathogenesis
This is an extremely important type of cilioretinal artery occlusion. As Kearns [88] rightly stressed, GCA “ranks as the prime medical emergency in ophthalmology, there being no other disease in which the prevention of blindness depends so much on prompt recognition and early treatment.” It can result in massive bilateral visual loss if not detected and treated urgently—the sooner high dose steroid therapy is instituted, the smaller is the risk of any further visual loss.
The PCA supplies the ONH as well as the cilioretinal artery [89]. GCA has a selective tendency to involve the PCA [32], resulting in its occlusion, which in turn results in simultaneous development of both arteritic AION and cilioretinal artery occlusion [88, 32]. The arteritic AION causes massive visual loss in these eyes.
The cilioretinal artery occlusion in GCA is sometime erroneously diagnosed as “branch retinal artery occlusion”, but the so-called “branch retinal arteries”, are in fact arterioles, and GCA is a disease of the medium-sized and large arteries and not of the arterioles [32]. I have seen patients with cilioretinal artery occlusion diagnosed by ophthalmologists as ordinary branch retinal arteriolar occlusion and left untreated, resulting in catastrophic and unnecessary visual loss in both eyes. These eyes present with the following classical, diagnostic clinical picture. A combination of chalky white optic disc oedema, retinal infarct in the region of the occluded cilioretinal artery occlusion and the presence of PCA occlusion on fluorescein angiography is diagnostic of GCA as the cause (Fig. 11). In all patients ≥ 50 years who have cilioretinal artery occlusion, it is essential to rule out GCA and associated PCA occlusion by fluorescein fundus angiography.
Cilioretinal Artery Occlusion Associated With Central Retinal Vein Occlusion Or Hemi-Central Retinal Vein Occlusion
This clinical entity was first described by Oosterhuis [90] in 1968 and later by me [91] and McLeod and Ring [92]. Since then, a large number of reports have been published, mostly anecdotal in nature or based on small series, except for one recent report based on 38 eyes [93]. In this condition, there is cilioretinal artery occlusion secondary to central retinal vein occlusion or hemi-central retinal vein occlusion.
This clinical entity is important and requires detailed discussion. I investigated this in 38 eyes in which cilioretinal artery occlusion was associated with central retinal vein occlusion or hemi- central retinal vein occlusion[93]. A full account is given elsewhere [93]. In summary, of the 38 eyes with cilioretinal artery occlusion in this study, 30 had nonischaemic central retinal vein occlusion, 5 had ischemic central retinal vein occlusion and 3 nonischaemic hemi-central retinal vein occlusion. The patients with nonischaemic central retinal vein occlusion were significantly younger (mean 45.3 + 16.0 years) than those with ischemic central retinal vein occlusion (72.3 + 9.2 years; P = 0.001) and nonischaemic hemi-central retinal vein occlusion (64.7 + 7.5 years; P = 0.018). Stoffelns [94], in his retrospective study of 31 patients, reported a mean patient age of 49 years, range 25–71 years.
Initially the ophthalmoscopic and fluorescein angiographic findings are like those seen in central retinal vein occlusion and hemi-central retinal vein occlusion, except that all these eyes have retinal infarct in the distribution of the cilioretinal artery. The size and site of the occluded cilioretinal artery varies widely. The occluded cilioretinal arteries are usually temporal in location, and sometimes nasal. There may be three cilioretinal arteries, and all of them get occluded.
Fluorescein angiography typically showed only a transient hemodynamic block in the cilioretinal artery [93] and not the typical organic occlusion seen in other types of cilioretinal artery occlusion. The fluorescein fundus angiograms show a variable degree of filling of the cilioretinal arteries.
Associated Systemic Conditions
Associated systemic conditions in this case are primarily those of central retinal vein occlusion. In my the study [93] of this condition in 38 patients, there were differences in the associated systemic conditions between the 3 types of retinal vein occlusion. Among 30 patients with non-ischemic central retinal vein occlusion, there was arterial hypertension in 17%, ischemic heart disease in 7% and smoking in 40%, and none had diabetes mellitus or stroke. In 5 patients with ischemic central retinal vein occlusion, there was arterial hypertension in 3, ischemic heart disease in 2, smoking in 1, and no diabetes mellitus or stroke. One of 3 patients with hemi-central retinal vein occlusion had a stroke and 2 were smokers, but none had arterial hypertension, diabetes mellitus or ischemic heart disease. This is, no doubt, too small a sample to provide fully reliable information.
Pathogenesis
Several hypotheses have been put forward to explain the simultaneous development of cilioretinal artery occlusion and central retinal vein occlusion; that has resulted in a controversy on the subject. McLeod and Ring [92], based on a study of 11 eyes, and McLeod [95], based on a series of presumptions and speculations postulated a somewhat confusing hypothesis to explain it. However, based on my comprehensive studies in the subject, I find the hypothesis postulated by McLeod inadequate to explain the pathogenesis of development of cilioretinal artery occlusion with central retinal vein occlusion and hemi-central retinal vein occlusion. Glacet-Bernard et al. [96] stated that the possibility of primary occlusion of the cilioretinal artery must be considered in these eyes. It has also been postulated that optic disc oedema caused by central retinal vein occlusion can cause cilioretinal artery occlusion, but optic disc oedema cannot cause cilioretinal artery occlusion. Some have even attributed cilioretinal artery occlusion with central retinal vein occlusion to embolism.
To comprehend the pathogenesis of this clinical entity logically, one must understand the factors that influence the ocular blood flow.
1. Blood flow in general depends upon the intraluminal perfusion pressure (Perfusion pressure = arterial pressure minus venous pressure). Therefore, any of the factors that either reduce the arterial pressure or increase the venous pressure, or a combination of both, result in reduced perfusion pressure, and consequently decreased blood flow or even no circulation.
2. Eyes with an additional cilioretinal artery differ from eyes with the central retinal artery as the only source of blood supply, in their retinal arterial and venous drainage systems.
(a) The venous drainage from the entire retina is by the central retinal vein, irrespective of the numbers and sources of the arteries that supply the retina.
(b) In eyes with a cilioretinal artery, the arterial supply to the retina is obviously from two independent sources - the central retinal artery is the major source, and the cilioretinal artery usually supplies only a small part of the retina, though its distribution can vary widely.
(c) The central retinal artery and cilioretinal artery belong to two very different arterial systems, with different physiological properties.
(d) The central retinal artery has efficient blood flow autoregulation, so that when there is a fall in perfusion pressure in the retinal arterial bed caused by a rise in the retinal venous pressure, the autoregulatory mechanism in the retinal arterial bed causes a rise in its pressure, to try to maintain retinal circulation.
(e) By contrast, since the cilioretinal artery usually belongs to the choroidal vascular system.
(f) The following two entirely different mechanisms are working in the cilioretinal artery circulation in eyes with central retinal vein occlusion: (i) the choroidal vascular bed has no or poor autoregulation, and (ii) in eyes with central retinal vein occlusion, there is no vortex venous obstruction. Therefore, a sudden onset of central retinal vein occlusion increases the retinal capillary intraluminal pressure, and that in turn decreases the perfusion pressure, and that then triggers in the autoregulatory mechanism in the central retinal artery to maintain its blood flow; by contrast, no such compensatory mechanism exists in the choroid or the SPCAs supplying the choroid. Moreover, studies have shown that the perfusion pressure in the choroidal vascular bed is normally lower than that in the central retinal artery [97]. Thus, the theory proposed by McLeod and Ring [92] that in these eyes there is partial obstruction of their PCAs, and that this represents “a spectrum of ocular vascular lesions intermediate between acute central retinal vein occlusion and acute ischaemic optic neuropathy” is not valid; moreover, contrary to what they stated, there is no evidence of AION in these eyes.
In the light of the above facts, then, let us consider the hemodynamic situation in eyes that have a cilioretinal artery and develop central retinal vein occlusion. Sudden occlusion of the central retinal vein results in a marked rise of intraluminal pressure in the entire retinal capillary bed; when that intraluminal pressure rises above that in the cilioretinal artery, the result is a hemodynamic stasis in the cilioretinal artery. In many of these eyes, angiography performed during the early acute phase provides information about the in vivo dynamics of blood flow in the eye. During the early stages of the transit of the dye, the cilioretinal artery usually fills up to the optic disc, since the ONH is supplied mainly by the PCA circulation [89]. During systole, the cilioretinal artery often fills for a variable length from the optic disc into the retina but during diastole, the filling retracts to the optic disc, resulting in an oscillating blood column in the cilioretinal artery, moving back and forth from the optic disc for a variable distance into the retina; I have often seen this when observing the fundus while doing fluorescein fundus angiography in these eyes—it presents a fascinating phenomenon. Thus, due to combination of these factors, the cilioretinal artery occlusion in these eyes is simply a hemodynamic stasis and is not due to embolism or thrombosis. The hemodynamic stasis is invariably transient, lasting from a few hours to several days, depending upon how severe the retinal venous stasis is and how rapidly the collateral circulation is established by the central retinal vein through its multiple tributaries in the optic nerve. Therefore, in the optic nerve, the further anterior the site of occlusion in the central retinal vein, the fewer tributaries are available, and the longer it takes to re-establish the circulation—and vice versa. As soon as those tributaries establish collateral circulation, there is a fall of intraluminal pressure in the retinal capillary bed to below that of the blood pressure in the cilioretinal artery, resulting in restoration of retinal circulation in the distribution of the cilioretinal artery. Hence, if a patient with central retinal vein occlusion is seen for the first time many days after the onset of cilioretinal artery occlusion, a retinal infarct is present ophthalmoscopically, but on angiography the cilioretinal artery fills normally; that can result in confusion and mistaken diagnosis of the cause of retinal infarct. This may be the reason why occlusion of the cilioretinal artery has mistakenly been attributed to embolism. In hemi-central retinal vein occlusion, when one of the two trunks of the central retinal vein is occluded [98], the above mechanism applies only to the segment of the retina drained by the occluded trunk.
Patients with central retinal vein occlusion associated with cilioretinal artery occlusion often have two complaints:
1. Visual loss is often first discovered on waking up from sleep, or in the morning on first opportunity to use fine central vision. To comprehend the reason for that, one must consider the important phenomenon of nocturnal arterial hypotension (see below).
2. In the study of cilioretinal artery occlusion and central retinal vein occlusion [93], 38% of patients gave a definite history of episode(s) of transient visual blurring before the onset of constant blurred vision. This is most probably also due to a transient fall in systemic blood pressure during waking hours, for whatever reason (e.g., orthostatic hypotension), resulting in a transient hemodynamic stasis in the cilioretinal artery. Naturally the question arises—why did only 38% of the patients experience that, and not all? Whether a patient gets these episodes of transient blurring before developing cilioretinal artery occlusion depends upon the difference between the intraluminal pressures in the retinal capillary bed and in the cilioretinal artery (see above). There can be two scenarios. (i) If the difference between the two pressures is very small, even a transient, mild fall of systemic blood pressure is enough to precipitate such an episode, e.g., with orthostatic hypotension. (ii) But if that difference is substantial, it would require, proportionately, a much greater fall of systemic blood pressure (e.g., with marked nocturnal arterial hypotension); in the latter case, these episodes may be occurring during sleep, but the patient is not aware of them.
Nocturnal Arterial Hypotension
A fall of blood pressure during sleep is a well-established physiological phenomenon. We investigated that by doing 24-hour ambulatory blood pressure monitoring in more than 700 patients, whose blood pressure was recorded every 10 min during waking hours and every 20 min during sleep. In that study [99], I found that during sleep systolic blood pressure falls by 34.8 ± 1.2% and diastolic by 44.0 ± 1.3% from the daytime pressures [100]. This fall is aggravated by overmedication with blood pressure lowering medication, particularly when given in the evening or at bedtime. There were two other important relevant findings of the study.
(1) The most impressive finding in 24-hour ambulatory blood pressure monitoring was that systemic arterial blood pressure is the most volatile parameter in the human body, and is greatly influenced, instantaneously, by emotional factors. This is particularly true of a patient who has just suffered visual loss and is emotionally upset. In view of that, arterial hypertension discovered in central retinal vein occlusion patients at the time of their diagnosis may be of one of the three types: (a) genuine, (b) temporary arterial hypertension due to emotional upset at sudden visual loss, or (c) “white coat hypertension”. Unfortunately, it is not unusual that a patient newly diagnosed with central retinal vein occlusion and found to have transient arterial hypertension in his ophthalmologist’s office may be treated aggressively by a physician who may not realize that the patient may not have genuine arterial hypertension. That action has the potential of precipitating or aggravating the visual loss in eyes with central retinal vein occlusion which have cilioretinal artery (see below), and also can convert non-ischemic central retinal vein occlusion to ischemic central retinal vein occlusion.
(2) Daytime blood pressure rarely has any relationship to the night-time blood pressure. Since blood pressure is routinely evaluated during the daytime, the physician almost invariably has no information about the blood pressure during sleep.
Thus, an understanding of nocturnal arterial hypotension has important implications both for comprehension of the mechanism of development of cilioretinal artery occlusion with central retinal vein occlusion and for management of these patients (see below).
In eyes with central retinal vein occlusion and cilioretinal artery, the following sequence of events takes place: a fall of systemic blood pressure during the night → secondary fall in the cilioretinal artery blood pressure (without any appreciable change in the intraluminal pressure in the retinal capillary bed caused by central retinal vein occlusion) → hemodynamic block in the cilioretinal artery during sleep → no retinal circulation in its distribution for several sleeping hours → retinal infarct in the distribution of the cilioretinal artery. Thus, a marked fall of blood pressure during sleep may play an important role in the development of cilioretinal artery occlusion in these patients.
As discussed above, there are eyes with a cilioretinal artery, which do not develop cilioretinal artery occlusion with central retinal vein occlusion. I have seen the following two types of cases.
1. Many patients, when first seen many days or weeks after the onset of visual blurring, showed no evidence of infarction or any significant delay in filling of the cilioretinal artery. In these eyes, obviously, the intraluminal pressure in the retinal capillary bed was never high enough to interfere with cilioretinal artery filling. This may be due to slow development of central retinal vein occlusion, which allows time for collaterals to develop in the optic nerve, so that the intraluminal pressure in the retinal capillaries never goes high enough to cause hemodynamic stasis in the cilioretinal artery. The other possible explanation is that the cilioretinal artery is a direct branch of the PCA and not a part of the choroidal vascular bed, so that it has the same intraluminal pressure as the central retinal artery—both arising from the ophthalmic artery.
2. I have also seen an occasional patient who presented soon after having developed transient visual obscuration, with markedly engorged retinal veins and a rare or no retinal haemorrhage. In these eyes, angiography revealed markedly delayed filling of the cilioretinal artery but no occlusion. This indicates that in these eyes, just the intraluminal pressure in the retinal capillary bed is high enough to cause delayed filling of the cilioretinal artery, but it is not high enough yet to produce a complete hemodynamic block and infarction, although enough to produce transient visual obscuration. This would indicate that delayed filling of the cilioretinal artery occurs much earlier than development of complete hemodynamic block and retinal infarction. Most likely, the retinal infarction develops in these eyes when nocturnal arterial hypotension during sleep causes intraluminal pressure in the cilioretinal artery to fall below the critical level, resulting in a hemodynamic block in the cilioretinal artery lasting many hours.
To understand why some eyes with central retinal vein occlusion associated with cilioretinal artery occlusion develop permanent visual loss in the distribution of cilioretinal artery while others have only temporary loss, it is essential to consider retinal tolerance time to acute ischaemia [101]. The severity of visual loss and the recovery of retinal function depend upon the duration and severity of retinal ischaemia in the area of retina supplied by the cilioretinal artery. If acute retinal ischaemia lasts for ≤97 min in these eyes, that causes no irreversible retinal damage and the retina recovers its function fully, but after that, the longer the acute ischaemia, the greater the irreversible ischemic damage. If the acute retinal ischaemia lasts for about 4 h, there will be no recovery of visual function.
Investigations In Patients With Cilioretinal Artery Occlusion
From the above discussion, it is evident that patients with cilioretinal artery occlusion should have the following investigations to find out the cause and to prevent or reduce the risk of any further visual problems.
1. In all patients 50 years and older with cilioretinal artery occlusion, it is absolutely essential to rule out GCA because that is an ocular emergency; if it is missed, that can result in catastrophic visual loss in both eyes, which is preventable by immediate diagnosis and aggressive management with corticosteroid therapy; this fact has medicolegal implication, apart from being a human tragedy. All these patients must have immediate evaluation of erythrocyte sedimentation rate and C-reactive protein levels estimated—the latter of the two is more reliable. The presence of systemic symptoms of GCA, when found, can be helpful. All these patients must also have fluorescein fundus angiography because the presence of associated PCA occlusion is diagnostic in these cases (Fig. 11).
2. In eyes with non-arteritic cilioretinal artery occlusion, since embolism is the most common cause, all these patients must have a complete evaluation to find the source of embolism. The most common sources of emboli are the carotid artery and the heart. In the carotid artery the most common source of embolus is a plaque, so that it is essential to find out if there are any plaques; unfortunately, I have found that evaluation usually is primarily focused on whether there is hemodynamically significant carotid artery stenosis, without paying any attention to presence of plaques. That can cause the actual source of emboli to be missed.
Cardiac evaluation is equally essential, even if the carotid artery shows lesions, because I have found that at times a patient can have lesions in both places and either of them can be the source of emboli. The most common source in the heart is the valves, although other lesions can also be causal. Transesophageal echocardiography is the best way to evaluate heart lesions.
3. Systemic evaluation for arterial hypertension, diabetes mellitus, hyperlipidaemia and other abnormalities is also indicated.
Clinical Findings
As discussed above, cilioretinal artery occlusion is of three types. In the literature there is no such distinction when describing clinical features in these cases; therefore, it is difficult to compare others’ findings with my own. For example, Stoffelns [94], in a retrospective study of 31 eyes of 31 patients lumped all cilioretinal arteries together.
Symptoms Of Cilioretinal Artery Occlusion
Typically, there is a sudden loss of vision in the involved segment of the eye. The visual loss may be preceded by transient visual obscurations [102].
In a study [85], episode/s of amaurosis before permanent visual loss in arteritic cilioretinal artery occlusion was seen 3 of the 12 eyes. This is due to a transient fall of perfusion pressure in the ciliary circulation caused by thrombosis of the PCAs. The transient fall of perfusion pressure may be due to a fall of systemic blood pressure and/or rise of IOP (usually caused by stooping or massage of the eye).
In the study [93] of cilioretinal artery occlusion associated with central retinal vein occlusion or hemi- central retinal vein occlusion, at least one third of the 38 eyes had a definite history of episode(s) of transient visual blurring before the onset of constant blurred vision. The duration of amaurosis fugax in the non-ischemic CRVO group varied from about 15 to 30 (23.7 ± 7.5) minutes. During the episode, the patients experienced seeing a purple or lavender haze and occasionally orange spots. Some patients experienced multiple episodes. This is due to a recurrent fall of perfusion pressure in the cilioretinal artery from recurrent transient fall of blood pressure (as in orthostatic hypotension). In this type of cilioretinal artery occlusion, the sudden onset of visual blurring was discovered more often in the morning or forenoon, usually when the patient first tried to use fine vision, compared to other times of the day, and in two cases with non-ischemic CRVO after a syncopal episode.
In non-arteritic cilioretinal artery occlusion amaurosis fugax is due to transient migrating emboli. Among 11 eyes with non-arteritic cilioretinal artery occlusion [85], only 1 experienced transient visual loss before permanent visual loss, due to transient migration of an embolus.
Visual Function In Cilioretinal Artery Occlusion
Visual Acuity And Visual Fields
Visual function varied with the type of cilioretinal artery occlusion, as is evident from the following.
Arteritic Cilioretinal Artery Occlusion Associated with GCA
In a study [85] initial deterioration of visual acuity was primarily due to associated arteritic AION in all but one case; in the latter visual acuity was 20/70. In the remaining 11 eyes, initial visual acuity was 6/6in 1, 6/7.5 in 2, 6/12 in 1, counting fingers in 2, hand motion in 1, light perception in 2 and no light perception in 2. All were treated with systemic corticosteroids without any visual change on follow-up. The visual field defect was primarily caused by arteritic anterior/posterior ION.
Cilioretinal Artery Occlusion Associated with Central Retinal Vein Occlusion or Hemi- Central Retinal Vein Occlusion
This is discussed in detail in elsewhere [93].
Non-Arteritic Cilioretinal Artery Occlusion Alone
In the study [93] of 11 eyes, (5 superior, 2 inferior, and 4 macular), 3 eyes presented with worse than 6/12 visual acuity, but all 3 improved during follow-up. All 11 eyes had a central visual defect at their initial visit (6 eyes with centrocecal scotoma, 3 with central scotoma, 1 with superior central altitudinal defect, and 1 with inferior central altitudinal defect). A peripheral visual field defect was seen in 3 eyes with cilioretinal arteries of large size (1 inferior altitudinal, 1 superior altitudinal, and 1 superior nasal). Of the 9 eyes with follow-up, the central field improved in 4 eyes, worsened in 1, and remained stable in 4. Only one of the 3 eyes with peripheral field defect had follow-up, and it remained stable. All 8 eyes with normal peripheral field remained normal during the follow-up period.
Anterior Segment Of The Eye
In cilioretinal artery occlusion there are no abnormalities attributable to it in the anterior segment. The IOP is normal.
Ophthalmoscopic Findings In Cilioretinal Artery Occlusion
The findings again depend upon the type of cilioretinal artery occlusion.
Non-Arteritic Cilioretinal Artery Occlusion Alone
In the study [85] of 11 eyes (5 superior, 2 inferior, and 4 macular) of this type. Seven were seen within a week after onset and the others after 2, 4 and 15 weeks and unknown in one. Retinal infarct in the region of supply of the cilioretinal artery was present in those seen up to about 4 weeks, after which the retina looked normal. The retinal veins were normal. The optic disc showed pallor in the involved region of the retina. The retinal arterioles looked normal. In one eye, seen 7 days after the onset of a visual problem, there was a visible embolus in the cilioretinal artery.
Arteritic Cilioretinal Artery Occlusion
In the study [85], in this type, there were 11 eyes in this group. In these eyes, apart from retinal infarction in the involved region, there was also optic disc oedema, which is mostly a chalky white swelling—a diagnostic appearance of arteritic AION (Fig. 11A).
Non-Arteritic CLRAO Associated with Central/Hemi-Central Retinal Vein Occlusion
In the study [93], in this group, there were 38 eyes. In these eyes there were fundus findings of central/hemi-central retinal vein occlusion, with retinal infarct in the region of the involved cilioretinal arteriole.
In this type and in non-arteritic cilioretinal artery occlusion, if the infarcted retina touches or passes through the fovea, initially the visual acuity drops markedly, but once the retinal infarct resolves, there is usually marked improvement. The same is true of the visual field defect. In Stoffeln’s [94] retrospective study of 31 eyes with cilioretinal artery occlusion, he found that all occlusions were located temporally and involved some portion of the fovea (2 had cholesterol emboli).
Fluorescein Fundus Angiographic Findings In Cilioretinal Artery Occlusion
The findings depend, again, upon the type of cilioretinal artery occlusion.
Non-Arteritic Cilioretinal Artery Occlusion Alone
The findings are like those in permanent branch retinal arterioles.
Arteritic Cilioretinal Artery Occlusion Associated with GCA
Apart from non-filling of the involved cilioretinal artery, typically there is also no filling of the choroid in the distribution of the occluded PCA; that is the diagnostic feature of this type of cilioretinal artery occlusion.
Cilioretinal Artery Occlusion Associated with Central/Hemi-Central Retinal Vein Occlusion
Fluorescein fundus angiography provides useful information in these eyes. Normally, the cilioretinal artery starts to fill just before the central retinal artery at the optic disc, though in some eyes the cilioretinal and central retinal arteries start to fill at the same time. However, in eyes with central retinal vein occlusion associated with cilioretinal artery occlusion, the filling pattern seen in the cilioretinal artery depends upon the time lapse between the onset of visual symptoms and fluorescein angiography.
(1)When the eyes are seen shortly after the onset, as discussed above, they show the classical oscillating blood column in the cilioretinal artery, i.e., the artery fills for a variable distance from the optic disc during systole, but the filling is retracted to the optic disc during diastole. This aspect can only be appreciated on a dynamic view and may be missed on routine fluorescein fundus angiograms.
(2)When the eyes are seen a few days after the onset of symptoms, however, the cilioretinal artery starts to fill—the shorter the time interval, the longer it takes the artery to fill; this filling can cause confusion about the cause of the retinal infarct in these eyes. The time the cilioretinal arteriole takes to fill on angiography depends upon: (a) the severity of retinal venous stasis caused by central retinal vein occlusion—the more marked the stasis, the longer it takes to fill, (b) the speed with which the venous collaterals developed in the optic nerve, and (c) the time lapse between the onset of visual symptoms and the first clinic visit (and angiography), which can vary widely among patients. The same applies to cilioretinal artery occlusion in hemi-central retinal vein occlusion.
Management Of Cilioretinal Artery Occlusion
As discussed above, cilioretinal artery occlusion is of three types, and treatment depends upon the type of occlusion, as is evident from the following.
Arteritic Cilioretinal Artery Occlusion Associated With Gca
From the point of view of management of any cilioretinal artery occlusion, this is the most important type. GCA has the potential to produce massive bilateral visual loss. It is well established that early and intensive corticosteroid therapy is the treatment of choice for GCA to prevent any further visual loss, although it does not improve visual outcome which has already been lost. Steroid therapy in GCA is discussed at length elsewhere [103].
Non-Arteritic Cilioretinal Artery Occlusion Alone
In this condition there is no known beneficial treatment. The most important action in management of non-arteritic cilioretinal artery occlusion is to undertake a complete evaluation, to find the source of embolism, since embolism is the most common cause. The carotid artery and the heart are, as discussed above, the most common sources.
Cilioretinal Artery Occlusion Associated With Central/Hemi-Central Retinal Vein Occlusion
From the management point of view, for patients who present with any of the symptoms or findings of this type of cilioretinal artery occlusion, it is essential to evaluate and regulate their blood pressure–lowering medication, to prevent further visual loss. Our above discussed 24-hour ambulatory blood pressure monitoring studies in about 700 patients has shown that patients who are overmedicated with blood pressure–lowering drugs or take those drugs in the evening or at bedtime are highly susceptible to marked nocturnal arterial hypotension [100] and consequent visual loss, as discussed above.
It is a common practice to prescribe aspirin or anticoagulants for eyes with central/hemi-central retinal vein occlusion. Studies [104] have shown these have a detrimental effect on visual outcome, by increasing retinal haemorrhages. Therefore, it is not advisable to prescribe these drugs in these cases.
Lesions Produced By Vortex Vein Occlusion
Retinal vein occlusion is a well-known clinical entity, but there is no corresponding detailed information about the clinical features of vortex vein occlusion, in man or in primates. There are some old studies dealing with experimental occlusion of vortex veins designed primarily to produce glaucoma, one study designed to produce retinal detachment, and others to study histopathological changes. All the earlier studies were carried out in rabbits and other animals.
Vortex venous occlusion is of great clinical interest in context of glaucoma, anterior uveitis, necrosis of the anterior segment, choroidal and intraocular haemorrhages and retinal detachment surgery. These are important clinical conditions. Thus, vortex venous occlusion requires a comprehensive information discussed here.
To investigate comprehensively clinical features of vortex vein occlusion, I [66] conducted an experimental study of vortex vein occlusion in 40 eyes of rhesus monkeys. All eyes had these examinations: external ocular, IOP, fundus, fluorescein fundus angiographic and histopathological features, or silicone rubber perfusion. Various vortex veins were occluded in different combinations. The findings of the study are discussed in detail elsewhere; [66] following are the main features.
In this study, the maximum brunt of the vortex vein occlusion was borne by the anterior segment of the eye—most marked by the vessels in the ciliary processes.
Ciliary Processes And Ciliary Body Findings
Histological examination of the eyes, soon after the occlusion of the vortex veins, revealed marked vascular congestion and dilated vessels in the ciliary processes (Fig. 23A–C); some eyes even showed the presence of thin-walled blood cysts on the ciliary processes. The area affected depended upon the vortex vein occluded, because each vein had a segmental distribution in the ciliary processes as well. When all the veins were occluded, the fine blood vessels under the ciliary epithelium ruptured in most of the eyes within a period of from a few minutes to about 3 h after occlusion. This produced bleeding into the posterior chamber which escaped via the pupil and led to a hyphaema in the anterior chamber. Histological examination of some of these eyes revealed ciliary epithelial cysts projecting into the posterior chamber. When three vortex veins were occluded, such a bleeding was seen in four of nine eyes, (or in four of five eyes followed-up for more than 24 h), while it was seen in two of twelve eyes on occluding two vortex veins, but in none after occlusion of one vortex vein. In eyes with occlusion of three or two vortex veins, the hyphaema was not only less common but also less marked than after occlusion of four veins. The hyphaema cleared in the former eyes in a week and in the latter in 2 to 5 weeks.
Fig. 23.
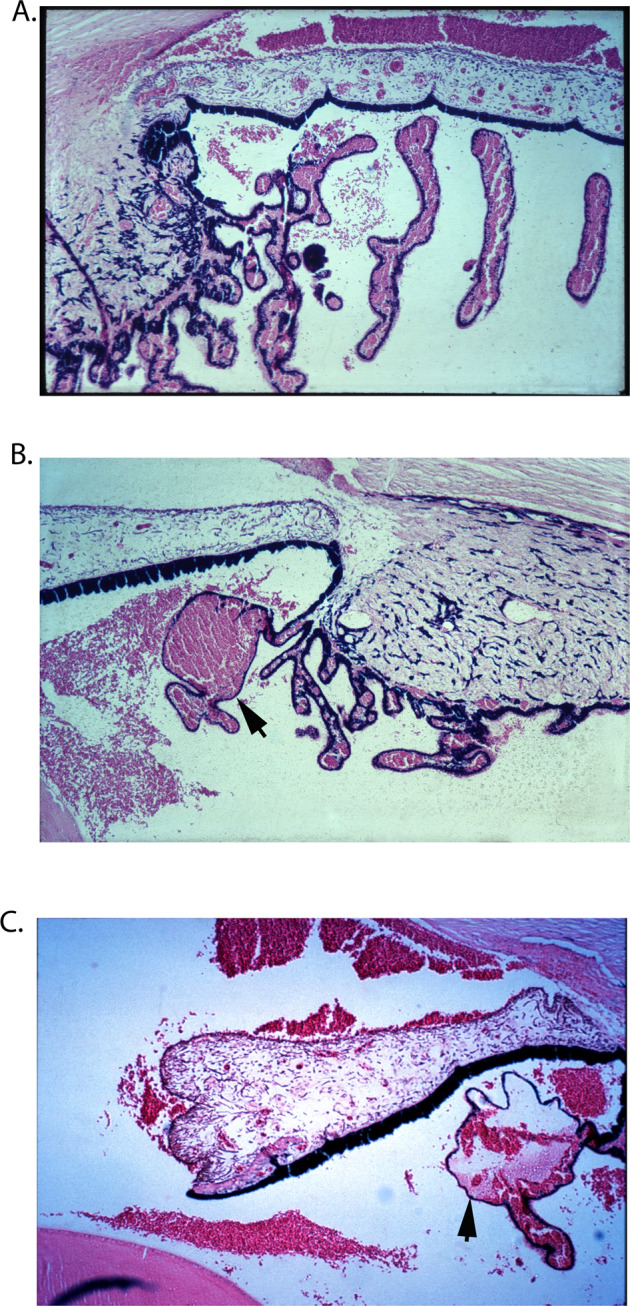
A–C These show markedly engorged vessels in ciliary processes, ciliary body and iris, with haemorrhages in the posterior and anterior chambers.
In all the eyes of this series, immediately after occlusion, and before the appearance of any hyphaema, thick protein-rich fluid extended from the ciliary processes in the sector drained by the occluded vortex vein; this exudate coated the anterior surface of the lens. In the anterior chamber, the protein-rich aqueous resulted in the formation of a gelatinous cobweb-like deposit in its dependent part (Fig. 24). It would thus appear that a marked rise in blood pressure in the capillaries of the ciliary processes results in the outpouring of protein-rich aqueous. This was due to the breakdown of the blood-aqueous barrier. Soon after the injection of intravenous fluorescein, profuse leakage of fluorescein, from the involved sector only was seen; this was at first localized to the involved sector of the pupil and anterior chamber but progressively became more prominent and diffuse (Fig. 24). The blood, protein-rich aqueous, and fluorescein from the posterior chamber passed through the pupil on to the anterior lens surface and anterior chamber, but none was seen in the vitreous or on the posterior lens surface. The vitreous in all eyes was clear. On follow-up, the protein-rich aqueous cleared with the return to normal of the uveal circulation.
Fig. 24. Fluorescein photographs of anterior segment of an eye 21 h after occlusion of superior temporal and nasal vortex veins in a rhesus monkey (about 10 to 15 min after intravenous injection of fluorescein).
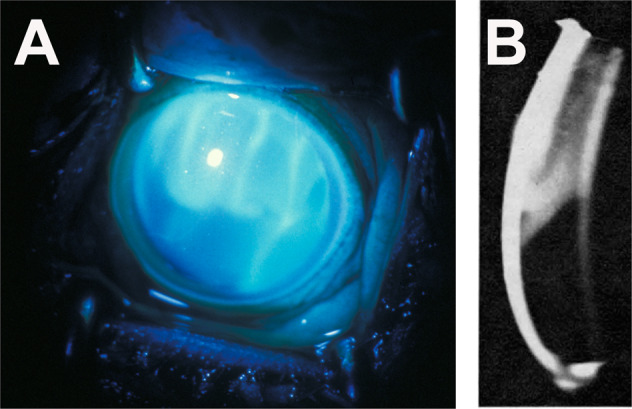
A Ordinary view shows fluorescein stained proteinous leakage in upper part of anterior chamber, as well seepage of fluorescein from posterior chamber into anterior chamber. B Slit-lamp photograph shows proteinous exudate in the upper of the anterior chamber, corresponding to that see in A.
Depth Of Anterior Chamber Findings
The iris-lens diaphragm moved forward significantly in eyes with occlusion of all or three vortex veins, because there was an increase in the volume of the posterior compartment of the eye, due to marked venous congestion in the choroidal vascular bed. This resulted in a shallow anterior chamber—shallower after occlusion of all the vortex veins than after three veins. On follow-up, in three of six eyes with all the vortex veins occluded, the anterior chamber was found to be absent after the hyphaema had cleared and it never reformed, while in the remaining three eye the depth of the anterior chamber became nearly normal after the hyphaema had cleared. In eyes with occlusion of one or two vortex veins, no significant change in the depth of the anterior chamber was noticed.
Intraocular Pressure Findings
As mentioned above, vortex vein occlusion was originally designed to produce raised IOP and glaucoma. In my series [66], eyes with occlusion of all or three vortex veins showed a marked rise in the IOP immediately after occlusion - about 40 to 60 mmHg after occlusion of all the vortex veins, and about 30 to 40 mmHg after occlusion of three vortex veins; but no significant rise in IOP was recorded after occlusion of two or one vortex vein. This rise in pressure was not related to hyphaema, because the IOP rose as much in eyes without as those with hyphaema. Similarly, the protein-rich aqueous seemed to play no role in the rise in IOP, because the eyes with occlusion of one or two vortex veins had a large amount of gelatinous deposit in the anterior chamber without any rise in the IOP. In the eyes with elevated IOP, the angle of the anterior chamber was very narrow, and it could have been blocked in most of them by a forward shift in the iris-lens diaphragm and engorgement of the ciliary body and iris, because of marked venous congestion in the entire uveal tissue.
On the day after the occlusion, the IOP was either the same as or even higher than the immediate post-occlusion pressure. After one week a fall in the IOP had set in, so that it returned to near normal in eyes with occlusion of all the vortex veins and to unrecordable low levels in eyes with occlusion of three vortex veins; the eyes with occlusion of all the veins IOP then dropped to an unrecordable level in 2 weeks. Having reached the lowest level, the IOP started to recover; this occurred within 2 weeks after occlusion of three vortex veins. In eyes with occlusion of all the vortex veins, however, the recovery of the IOP depended upon the state of the anterior chamber, i.e., if the anterior chamber was absent the eye had permanent marked hypotony, but if the anterior chamber was present the recovery started after an interval of more than 2 weeks. The IOP in these eyes did not recover completely but remained subnormal during the period of follow-up. Histological studies showed evidence of atrophy of the ciliary processes and ciliary body in these eyes.
Iris Findings
The iris was markedly hyperaemic soon after the occlusion of all or three vortex veins (Fig. 23C). With occlusion of one to three vortex veins, there was sectoral hyperaemia of the iris corresponding to the occluded vortex veins. Other authors reported similar findings [105].
On follow-up, three eyes with all the veins occluded and an absent anterior chamber had anterior and posterior synechiae with iris atrophy and iris neovascularization: the clinical appearance resembling that of an eye with long-standing chronic anterior uveitis or anterior segment ischaemia. In one of the eyes with three vortex veins occluded, posterior synechiae and sectoral iris atrophy were present. Koster [106] also found these changes in eyes after occlusion of all the vortex veins in rabbits.
Lens Findings
In all the eyes the lens, soon after occlusion of the various vortex veins, had a proteinous deposit on its anterior surface (Fig. 24). During a follow-up period of up to 4 weeks, the deposit cleared without leaving any lens abnormalities in any eye except those with all the vortex veins occluded. During follow-up, all the eyes with all the vortex veins occluded showed pigment deposit on the anterior lens surface; in three of these eyes with an absent anterior chamber and permanent ocular hypotony, the lens became completely cataractous within 5 weeks. The changes appeared at first in the anterior cortex of the lens and later resulted in a complete cataract 14, 19, 38, and 90 days after the occlusion. The cataractous change is evidently secondary to impaired nutrition due to degeneration of the ciliary epithelium and ciliary body, as shown by histological studies.
Corneal Findings
Epithelial oedema was seen when the IOP was very high, in eyes with all or three vortex veins occlusion. On follow-up, stromal clouding and vascularization by radial vessels from the limbus was seen in three eyes with all the vortex veins occlusion. Two of these had no anterior chamber. Koster [106] also noticed a similar corneal vascularization after occlusion of all the vortex veins and likened it to that of interstitial keratitis, with a gradually progressive pannus from the limbus towards the centre. I feel that the corneal changes are secondary to the changes in the IOP and anterior chamber.
The degenerative changes seen in the lens and anterior segment of the eye in these monkeys (particularly in those with occlusion of all the vortex veins) very much resemble those seen in patients with long-standing anterior uveitis or anterior segment ischaemia. Almost all the pathological changes in the anterior segment of the monkey eyes depend upon the changes in the anterior chamber (e.g., protein-rich aqueous, hyphaema, shallow or absent anterior chamber), which in turn are secondary to marked disturbances of the uveal circulation associated with the changes in the ciliary processes and the forward displacement of the iris-lens diaphragm, by engorgement of the posterior uveal vascular bed increasing the volume of the posterior segment.
Choroidal Circulation Findings
On intravenous fluorescein fundus angiography, the choroidal vascular bed normally starts to fill during the pre-retinal arterial phase and the filling is complete by the retinal arterial or early arteriovenous phase. When all the vortex veins in an eye were occluded, however, the choroidal bed showed no filling at all until the arteriovenous phase Even then the choroidal filling was extremely sluggish and patchy, so that complete filling did not occur even by the end of the retinal venous phase; the temporal part of the choroid filled earlier than the nasal part, and the territory of one vortex vein could fill before that of another. This delay in the filling of the choroidal vascular bed is primarily due to back pressure in the uveal vascular bed because of complete blockage of its outlet; the raised IOP is also a contributory factor to some extent [97, 107].
On occluding three vortex veins, angiography showed normal filling of a well-defined quadrant of the choroid corresponding to the unoccluded vein (Fig. 25). The choroid drained by the occluded vortex veins started to fill slowly during the late retinal arterial phase and the filling was not complete till the late retinal venous phase. Similarly, eyes with occlusion of two or one vortex veins showed in the choroidal well-defined quadrantic normal filling in the unoccluded segment, and a very sluggish filling in the occluded segment. The watershed zones between the various vortex veins were well-defined and were the last to fill (Figs. 18, 25). The fact that the choriocapillaris in the watershed zones did not fill until very late, while the choriocapillaris on either side of that filled completely, further confirms the segmental nature of the choriocapillaris. Thus, each vortex vein has a well-defined segmental distribution in the choroid, with little communication with the adjacent vein. A similar sectoral distribution of the vortex veins has also been recorded by other workers in rabbits and dogs. Nishikawa et al. [108]. on Indocyanine green angiography in 5 Japanese macaque monkeys showed a delay in filling of the choroidal arteries in the field of the occluded vortex veins, and the choroidal veins were filled retrogradely in a pulsatile manner. Histology showed that the choroidal veins in the occluded field were engorged with red blood cells.
Fig. 25.
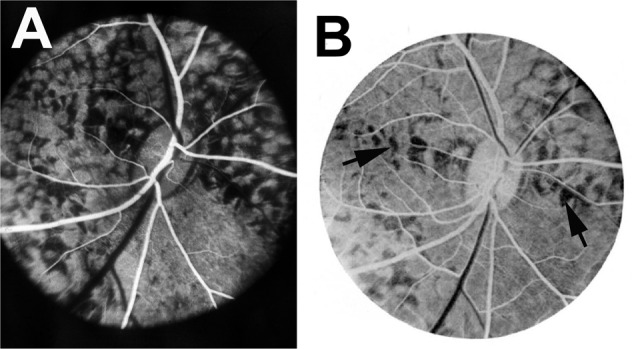
A, B Fluorescein fundus angiograms of right eye 1 h after occlusion of superior temporal, superior nasal, and inferior temporal vortex veins. (A) During retinal arterial phase, shows normal filling of only inferior nasal quadrant of the choroid. B During retinal arteriovenous phase, shows unfilled watershed zone (arrows) and complete filling of inferior nasal quadrant of choroid, with incomplete choroidal filling in other three quadrants.
On follow-up, the choroidal perfusion in the region of the occluded vortex veins was restored to normal in 1 week after the occlusion of one or two vortex veins, and in 2 weeks after the occlusion of three veins. It was not possible to determine the time taken for such a restoration of choroidal perfusion after occlusion of all the vortex veins because hyphaema lasted for a few weeks and later there were cataractous changes in half of the cases; however, it was normal when seen after about 3 months. The exact mode of this restoration of the vortex vein outflow is not known. Koster [106], in his studies of occlusion of all the vortex veins in rabbits, found after injection of Berlin blue gelatin, that the occluded vortex veins established small collateral vessels at their point of exit from the sclera; the collateral vessels went forwards and backwards to the adjacent muscles. He also recorded the ·presence of enlarged collateral vessels between the choroidal and retinal vessels around the optic nerve but did not find any collaterals between the ciliary body and the overlying episcleral plexus or muscles. My silicone rubber perfusion studies were not conclusive in this respect, except that in some of the eyes, after silicone injection, the normal stem of the vortex vein outside the globe was seen to have re-formed in spite of its having been cut with a cautery at the time of occlusion. I found no other collateral of any significance between the vortex veins and the extraocular venous channels. Takahashi and Kishi [109] performed wide-angle indocyanine green angiography in eyes with rhegmatogenous retinal detachment that had been treated with scleral buckling and cryopexy and had developed vortex veins occlusion after that. In these eyes, angiography 3 months to more than one year after the surgery, revealed the development of new venous drainage routes which were connected to the intact vortex veins.
Fundus Findings
Immediately after occlusion of the vortex veins no lesions were visible in the posterior part of the fundus. The equatorial and peripheral parts of the fundus could not usually be properly examined. Histological studies immediately after the occlusion showed a markedly engorged choroidal bed and even choroidal and suprachoroidal haemorrhages in the region of the occluded vortex veins (Fig. 26).
Fig. 26. Photomicrograph of eye about 1 to 2 h after occlusion of all vortex veins, shows extensive choroidal and suprachoroidal haemorrhages.
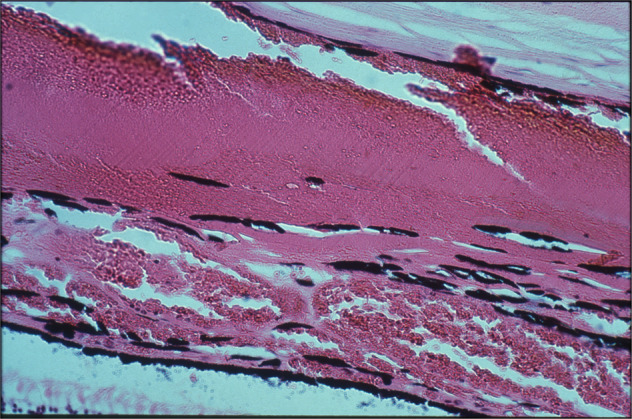
Haematoxylin and eosin.
On follow-up, patches of chorioretinal atrophy were seen near the equator, in the region of the occluded vortex vein (in one eye each with occlusion of four, two, and one vortex vein, and two eyes with three vortex veins occluded). In some of the other eyes such lesions could have been missed because of the difficulty in examining the peripheral part of the fundus in these eyes. Degenerative changes in the RPE have also been reported in rabbits after vortex vein occlusion [66]. On angiography, these lesions show marked destruction of the choroidal vascular bed. I feel that the chorioretinal lesions are secondary to the choroidal haemorrhages seen in these eyes.
Sachsenweger and Lukoff [110] described the occurrence of a flat, circumscribed or mould-like retinal detachment after the occlusion of two or three vortex veins in dogs and a total retinal detachment after the occlusion of four vortex veins. The latter group also had intrachoroidal and subretinal haemorrhage. Koster [106] found histological evidence of retinal detachment in some eyes after occlusion of two to four vortex veins in rabbits and a total detachment 4 months after occlusion of four vortex veins. All these workers reported the presence of subretinal transudate in their retinal detachments, which was considered to be due to venous congestion by Sachsenweger and Lukoff [110]. Similarly, de Faria and Silva [111] thought that extensive retinal detachment with choroidal tumours in the posterior part of the choroid were secondary to compression of the vortex veins and associated transudation. Klien [112] suggested that central serous retinopathy was due to stasis in the vortex veins, because on histopathological examination of three eyes with central serous retinopathy she found congestion and stasis in the vortex vein bed and in the other orbital and retinal venous beds. Recent fluorescein fundus angiographic studies of the eyes of patients with central serous retinopathy have lent no support to Klien’s [112] view. In my studies, no retinal detachment was seen ophthalmoscopically in any of my animals; nor did histology reveal any retinal detachment and associated subretinal transudation. Where all the vortex veins were occluded and the eye had become markedly hypotonic and degenerate with a cataract, histological examination revealed retinal detachment; such a detachment was part of a generalized degeneration and disorganization of the eye. Of the remaining eyes, if a retinal detachment was detected histologically, it was an artefact and not a true detachment and there was no associated subretinal transudation. In my study no clinical or histological evidence of choroidal detachment was seen in any eye. I feel that most of the retinal detachments reported by Koster [106] were histological artefacts.
Histological examination of some discs with all or three vortex veins occluded showed some evidence of cupping and atrophy of the optic disc. Ophthalmoscopically, no definite optic atrophy or cupping could be detected in eyes with raised IOP. Koster [106] and Sachsenweger and Lukoff [110] reported the presence of a hyperaemic optic disc and distended retinal veins soon after the occlusion of all the vortex veins when the IOP was very high. I feel that the fundus changes described by these authors were secondary to the marked ocular hypertension. Koster [106] found these discs to become pale eventually, with gliosis of the optic nerve.
Our studies [16, 17] showed that in occlusion of the PCA, simultaneous occlusion of one or two vortex veins exercised a distinct protective influence against the acute ischemic lesions. Similarly, a study by Simoens [30] reported ligation of the lateral vortex veins has a protective influence on the acute ischemic lesions caused by cutting the lateral PCAs. Bonnet [113] found that occlusion of two vortex veins in eyes with senile pigment epithelial detachment did not provide any benefit.
Conclusions
From my study [66], one can derive the following conclusions.
1. The vortex veins do not communicate freely with one another but have a well-defined distribution in the entire length of the uveal tract, each vein usually draining a quadrant of the uvea. Because of this segmental distribution, and the segmental distribution of the PCAs and of the choriocapillaris, occlusion of a vortex vein produces marked venous congestion and sluggish circulation in the affected segment of the uveal tract only. The circulation is restored to normal in 1 to 2 weeks or longer depending upon the number of vortex veins occluded.
2. No significant fundus lesions are seen, other than, on follow-up, chorioretinal degenerative lesions near the equator in the region of the occluded vortex vein, even if only one vein was occluded.
3. The ciliary processes bear the main brunt of the vortex vein occlusion because of marked engorgement of subepithelial capillaries in the occluded sector. On occlusion of the vortex veins, instead of the normal aqueous production, marked exudation of protein-rich aqueous and profuse leakage of fluorescein are seen from the sector drained by the occluded vortex vein. This produces a gelatinous deposit on the anterior surface of the lens and in the anterior chamber. On occlusion of two, three or all the vortex veins, in addition to the protein-rich aqueous, haemorrhages due to rupture of the subepithelial capillaries produce hyphaema - the more vortex veins are occluded, the more common this consequence is. On follow-up, atrophy of the ciliary processes develops, the extent of involvement depending upon the number of vortex veins occluded.
4. On occlusion of three or all vortex veins, forward movement of the iris-lens diaphragm leads to a shallow anterior chamber. On follow-up, the anterior chamber was permanently absent in half of the eyes with occlusion of all the vortex veins.
5. On occlusion of three or all the vortex veins, there is a sudden rise of IOP. However, 1 to 2 weeks after occlusion there is marked ocular hypotony. The IOP improved slowly after that but was never found to have recovered to normal during the period of follow-up of my study.
6. Immediately after the occlusion, the iris shows sectoral hyperaemia corresponding to the occluded vortex vein, which is most marked after the occlusion of three or all vortex veins.
7. Occlusion of all the vortex veins later resulted in iris atrophy with anterior and posterior synechiae, cataractous lens, and vascularized cornea in half of the eyes on follow-up. Almost all these eyes had an absent anterior chamber and permanent ocular hypotony.
8. Clinically, vortex vein occlusion can mimic non-granulomatous anterior uveitis. In some of the cases of vortex vein occlusion, particularly those with occlusion of three or all the vortex veins, the syndrome of anterior segment ischaemia could be present because of the production of marked disturbances in the uveal circulation in them.
9. Interference with the circulation in the vortex veins after retinal detachment surgery seems to play an important role in producing some of the major complications of surgery, e.g., glaucoma, “anterior uveitis”, necrosis of the anterior segment, and choroidal and intraocular haemorrhages.
Clinical Significance
The finding in the anterior chamber after vortex vein occlusion of thick flare or thick gelatinous deposit and hyperaemia of the iris mimics perfectly the syndrome diagnosed in patients as non-granulomatous iritis of sudden onset. It is probable that some patients with the latter diagnosis might, in fact, be cases of occlusion of one vortex vein or more. Intravenous fluorescein injection can be a useful test in such cases to diagnose and determine whether vortex vein occlusion has occurred. Similarly, some of the patients with obscure hyphaema of sudden onset could belong to this category.
Retinal Detachment Surgery Complications and Vortex Vein Occlusion
The important role played by interference with the circulation of the vortex veins in some of the major complications following retinal detachment surgery has not previously been adequately dealt with in the literature. Of particular interest in this context are glaucoma, anterior uveitis, necrosis of the anterior segment, and choroidal and intraocular haemorrhages. Most of these complications are usually consequent on the encircling procedure and are aggravated by the application of diathermy or cryopexy. The encircling band would, by invagination and compression, not only reduce the volume of the posterior compartment of the eye (and hence raise the IOP) but also compress and occlude the choroidal vessels (particularly the thin-walled veins). This must produce marked venous congestion in the anterior part of the uveal tract (i.e., the iris, ciliary body, and anterior part of the choroid). Since (a) the exit of the vortex veins lies posterior to the encircling band, (b) there is no free communication between the ACAs and PCAs, and (c) the venous drainage from the part supplied by the ACAs is provided almost entirely by the vortex veins, the amount of venous congestion produced depends upon the degree of strangulation produced by the band and the area involved (e.g. the more posterior the encircling band, the greater will be the area involved). The venous congestion causes marked engorgement of the big thin-walled choroidal veins and also of the loops of blood vessels in the ciliary processes. The ciliary body may also be swollen because of this venous congestion. All these changes increase the volume of the posterior compartment of the eye, and at the same time the capacity of the posterior compartment is decreased by the encircling band: therefore, the iris-lens diaphragm is pushed forward, producing a shallow anterior chamber and raised IOP. This will be particularly marked in eyes where the anterior chamber is shallow and has a narrow angle normally.
Marked venous engorgement in the choroid in these cases leads to choroidal and even vitreous haemorrhages. Congestion of the vessels in the ciliary processes leads to the production of protein-rich aqueous which, in turn, causes signs resembling those of anterior uveitis, e.g., flare in the anterior chamber, and even the deposition of gelatinous material in the anterior chamber in severe cases (which may be confused with endophthalmitis). I feel that the so-called uveitis seen after retinal detachment surgery is, in fact, a part of this syndrome rather than a true uveitis. Intravenous injection of fluorescein soon after detachment surgery in such cases should produce immediate and marked leakage of the dye from the posterior into the anterior chamber (Fig. 24), indicating uveal venous congestion.
Microscopic hyphaema can occur and in severe cases frank hyphaema may be present, depending upon the degree of venous congestion in the ciliary processes. In very marked venous stasis of the anterior uveal tract, anterior segment necrosis would supervene.
When a sectoral scleral indentation is made to produce a buckle in one sector and is accompanied by the application of diathermy or cryopexy, this must interfere with the outflow of one or more vortex veins and thus produce changes like those seen after occlusion of one or more of the vortex veins—in mild cases simulating the signs of anterior uveitis.
Thus, in retinal detachment surgery, interference with the venous drainage from the anterior half or more of the uveal tract can give rise to many serious complications. As a safety precaution, it is advisable that in all these cases the following investigations should be carried out immediately after the operation:
(i) Slit-lamp examination of the anterior chamber for protein-rich aqueous and depth of the anterior chamber.
(ii) Intravenous fluorescein test for leakage of fluorescein into the anterior chamber from the posterior chamber, 15 min after the injection. A large amount of fluorescein seeping into the anterior chamber from the posterior chamber via the pupil (Fig. 24) indicates embarrassment of the anterior uveal venous drainage.
(iii) Estimation of the IOP: A very high IOP, even if it is below the leveI required to produce pulsation of the central retinal artery on the optic disc, can interfere with the blood supply of the optic nerve and, in older people, may even produce AION with permanent total or partial visual loss.
Some workers have speculated that interference with the choroidal venous drainage from the macular area may be responsible for the production of subretinal haemorrhage in disciform macular degeneration. As mentioned above, the meeting point of the distribution of the four vortex veins is situated slightly nasal to the foveal region, so that the choroidal veins tend to radiate away from the foveal area (or, to describe the picture in another way, they meet one another close to the fovea). It is very tempting to postulate under these circumstances that a sudden occlusion of one of the vortex veins in patients with unhealthy vessels could initiate choroidal haemorrhage in the macular region. However, in my study, occlusion of one or more vortex veins did not produce any macular pathology. In one eye (not included in this series) occlusion of the superior temporal vortex vein combined with occlusion of one of the many SPCAs in that region produced a subretinal macular haemorrhage, originating from the choriocapillaris and clinically resembling the subretinal haemorrhage seen in macular degeneration. However, on repeating the experiment in six more eyes, no such haemorrhage was seen! It is therefore not productive to base speculations as to the effects of vortex vein occlusion in old and arteriosclerotic human eyes on results obtained from the eye of a young, healthy monkeys. With the evidence so far available, I find no relationship between age-related macular degeneration and vortex vein occlusion.
Author contributions
SSH was responsible for the writing and editing of the final manuscript. SBH provided feedback on the manuscript.
Funding
Supported by research grants from the British Medical Research Council, EY-1151, EY-1576, EY 3330 and RR-59 from the U.S. National Institutes of Health, in part by unrestricted grants from Research to Prevent Blindness, Inc., New York.
Competing interests
The authors declare no competing interests.
Footnotes
Due to Dr. Hayreh’s death, some errors and omissions may exist in the article as published.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ramon Y, Cajal S. Recollections of My Life. Santiago Ramon Y Cajal. Translated by EH Craigie from the third Spanish edition (1923). Philadelphia: American Philosophical Society; 1937; p. 564-5.
- 2.Hayreh SS. Blood supply of the optic nerve. In: Shaffer RN, Glaucoma Research Conference. Am J Ophthalmol. 1966;61:564. [Google Scholar]
- 3.Hayreh SS. Blood supply of the optic nerve head and its role in optic atrophy, glaucoma and oedema of the optic disc. Br J Ophthalmol. 1969;53:721–48. doi: 10.1136/bjo.53.11.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Duke-Elder S, Wybar KC. System of Ophthalmology, Vol. 2: The anatomy of the visual system. London: Kimpton; 1961; p. 345,351, 358,69
- 5.Wedel C, Ruysch F. Epistola anatomica, problematica ad Fredericum Ruyschium. De oculorum tunicis. Amstelaedami: Janssonio-Waesbergios; 1737.
- 6.Olver JM. Functional anatomy of the choroidal circulation: methyl methacrylate casting of human choroid. Eye. 1990;4:262–72. doi: 10.1038/eye.1990.38. [DOI] [PubMed] [Google Scholar]
- 7.Ashton N. Observations on the choroidal circulation. Br J Ophthalmol. 1952;36:465–81. doi: 10.1136/bjo.36.9.465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vilstrup G. Studies on the choroid circulation [Thesis (doctoral)]. Copenhagen: Ejnar Munksgaard; 1952.
- 9.Weiter JJ, Ernest JT. Anatomy of the choroidal vasculature. Am J Ophthalmol. 1974;78:583–90. doi: 10.1016/S0002-9394(14)76294-4. [DOI] [PubMed] [Google Scholar]
- 10.Wybar KC. A study of the choroidal circulation of the eye in man. J Anat. 1954;88:94–98. [PMC free article] [PubMed] [Google Scholar]
- 11.Shimizu K, Ujiie K Structure of ocular vessels. New York: Igaku-Shoin; 1978; p. 54,70-9, 108-24.
- 12.Wybar KC. Vascular anatomy of the choroid in relation to selective localization of ocular disease. Br J Ophthalmol. 1954;38:513–17. doi: 10.1136/bjo.38.9.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayreh SS, Baines JA. Occlusion of the posterior ciliary artery. I. Effects on choroidal circulation. Br J Ophthalmol. 1972;56:719–35. doi: 10.1136/bjo.56.10.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hayreh SS, Baines JA. Occlusion of the posterior ciliary artery. II. Chorio-retinal lesions Br J Ophthalmol Br J Ophthalmol. 1972;56:736–53. doi: 10.1136/bjo.56.10.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hayreh SS, Baines JA. Occlusion of the posterior ciliary artery. III. Eff Opt nerve head Br J Ophthalmol. 1972;56:754–64. doi: 10.1136/bjo.56.10.754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hayreh SS, Chopdar A. Occlusion of the posterior ciliary artery. V. Protective influence of simultaneous vortex vein occlusion. Arch Ophthalmol. 1982;100:1481–91. doi: 10.1001/archopht.1982.01030040459019. [DOI] [PubMed] [Google Scholar]
- 17.Loeffler KU, Hayreh SS, Tso MOM. The effects of simultaneous occlusion of the posterior ciliary artery and vortex veins: a histopathologic study. Arch Ophthalmol. 1994;112:674–82. doi: 10.1001/archopht.1994.01090170118033. [DOI] [PubMed] [Google Scholar]
- 18.Hayreh SS. Segmental nature of the choroidal vasculature. Br J Ophthalmol. 1975;59:631–48. doi: 10.1136/bjo.59.11.631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hayreh SS. The Long posterior ciliary arteries. Graefes Arch Clin Exp Ophthalmol. 1974;192:197–213. doi: 10.1007/BF00416866. [DOI] [PubMed] [Google Scholar]
- 20.Uyama M, Ohkuma H, Miki K, Koshibu A, Uraguchi K, Itotagawa S. [Pathology of choroidal circulatory disturbance. Part 3. Experimental choroidal circulatory disturbance, histopathological study of retino-choroidal lesion (author’s transl)]. Nihon Ganka Gakkai Zasshi. 1980;84:1924–46. [PubMed]
- 21.Koshibu A, Itotagawa S. [Disruption and repair of blood-retinal barrier at the retinal pigment epithelium after experimentally produced choroidal circulatory disturbance (author’s transl)] Nihon Ganka Gakkai Zasshi. 1980;84:721–35. [PubMed] [Google Scholar]
- 22.Ueno S, Tsukahara I. [Ultrastructural observations of the retinal pigment epithelial cells following experimental occlusion of the posterior ciliary artery in the rhesus monkey. I. Early changes of the damaged retinal pigment epithelial cells] Nihon Ganka Gakkai Zasshi. 1976;80:572–84. [PubMed] [Google Scholar]
- 23.Ueno S, Harayama K, Tsukahara I. [Ultrastructural observations of the retinal pigment epithelial cells following experimental occlusion of the posterior ciliary artery in the rhesus monkey. II. Mid-phase changes of the damaged retinal pigment epithelial cells] Nihon Ganka Gakkai Zasshi. 1977;81:728–42. [PubMed] [Google Scholar]
- 24.Ueno S, Ohta M, Tsukahara I. [Ultrastructual observations of the retinal pigment epithelial cells following experimental occlusion of the posterior ciliary artery in the rhesus monkey. III. Late changes of the damaged retinal pigment epithelial cells] Nihon Ganka Gakkai Zasshi. 1977;81:1801–13. [PubMed] [Google Scholar]
- 25.Algvere P. Retinal detachment and pathology following experimental embolization of choroidal and retinal circulation. Albrecht von Graefes Arch Klin Exp Ophthalmol. 1976;201:123–34. doi: 10.1007/BF00410063. [DOI] [PubMed] [Google Scholar]
- 26.Cogan DG. Neurology of the visual system. Springfield, IL: Charles C. Thomas; 1966. p. 137, 173, 185–8.
- 27.Henkind P, Charles NC, Pearson J. Histopathology of ischemic optic neuropathy. Am J Ophthalmol. 1970;69:78–90. doi: 10.1016/0002-9394(70)91859-3. [DOI] [PubMed] [Google Scholar]
- 28.MacMichael IM, Cullen JF. Pathology of ischaemic optic neuropathy. In: Cant JS, editor. Proceedings, 2nd William Mackenzie Symposium on the optic nerve. London: Kimpton; 1972. p.108–16.
- 29.Hinzpeter EN, Naumann G. Ischemic papilledema in giant-cell arteritis. Mucopolysaccharide deposition with normal intraocular pressure. Arch Ophthalmol. 1976;94:624–8. doi: 10.1001/archopht.1976.03910030306011. [DOI] [PubMed] [Google Scholar]
- 30.Simoens P. [In vivo and in vitro study of experimental occlusion of choroidal and retinal blood vessels in the miniature pig] Verh K Acad Geneeskd Belg. 1993;55:319–75. [PubMed] [Google Scholar]
- 31.Hayreh SS. Inter-individual variation in blood supply of the optic nerve head. Its importance in various ischemic disorders of the optic nerve head, and glaucoma, low-tension glaucoma and allied disorders. Doc Ophthalmol. 1985;59:217–46. doi: 10.1007/BF00159262. [DOI] [PubMed] [Google Scholar]
- 32.Hayreh SS, Podhajsky PA, Zimmerman B. Ocular manifestations of giant cell arteritis. Am J Ophthalmol. 1998;125:509–20. doi: 10.1016/S0002-9394(99)80192-5. [DOI] [PubMed] [Google Scholar]
- 33.Hayreh SS, Servais GE, Virdi PS. Fundus lesions in malignant hypertension. VI. Hypertensive choroidopathy. Ophthalmology. 1986;93:1383–400. doi: 10.1016/S0161-6420(86)33554-1. [DOI] [PubMed] [Google Scholar]
- 34.Kishi S, Tso MO, Hayreh SS. Fundus Lesions in Malignant Hypertension I. A Pathologic Study of Experimental Hypertensive Choroidopathy. Arch Ophthalmol. 1985;103:1189–97. doi: 10.1001/archopht.1985.01050080101029. [DOI] [PubMed] [Google Scholar]
- 35.Bousquet E, Khandelwal N, Séminel M, Mehanna C, Salah S, Eymard P, et al. Choroidal Structural Changes in Patients with Birdshot Chorioretinopathy. Ocul Immunol Inflamm. 2021;29:346–51. doi: 10.1080/09273948.2019.1681472. [DOI] [PubMed] [Google Scholar]
- 36.Amalric P. Acute choroidal ischaemia. Trans Ophthalmol Soc U K. 1971;91:305–22. [PubMed] [Google Scholar]
- 37.Hepburn ML. Inflammatory and vascular diseases of the choroid. Trans Ophthalmol Soc U K. 1912;32:361–86. [Google Scholar]
- 38.Hepburn ML. The Doyne Memorial Lencture: The role played by the pigment and visual fields in the diagnosis of diseases of the fundus. Trans Ophthalmol Soc U K. 1935;55:434–77. [Google Scholar]
- 39.Foulds WS, Lee WR, Taylor WO. Clinical and pathological aspects of choroidal ischaemia. Trans Ophthalmol Soc U K. 1971;91:323–41. [PubMed] [Google Scholar]
- 40.Hayreh SS. The choriocapillaris. Albrecht Von Graefes Arch Klin Exp Ophthalmol. 1974;192:165–79. doi: 10.1007/BF00416864. [DOI] [PubMed] [Google Scholar]
- 41.Gass JD. Acute posterior multifocal placoid pigment epitheliopathy. Arch Ophthalmol. 1968;80:177–85. doi: 10.1001/archopht.1968.00980050179005. [DOI] [PubMed] [Google Scholar]
- 42.Deutman AF, Lion F. Choriocapillaris nonperfusion in acute multifocal placoid pigment epitheliopathy. Am J Ophthalmol. 1977;84:652–7. doi: 10.1016/0002-9394(77)90380-4. [DOI] [PubMed] [Google Scholar]
- 43.Tsang BK-T, Chauhan DS, Haward R, Whiteman I, Frayne J, McLean C. Fatal Ischemic Stroke Complicating Acute Multifocal Placoid Pigment Epitheliopathy: Histopathological Findings. J Neuro-Ophthalmol. 2014;34:10–15. doi: 10.1097/WNO.0b013e318294a4b0. [DOI] [PubMed] [Google Scholar]
- 44.Priluck IA, Robertson DM, Buettner H. Acute posterior multifocal placoid pigment epitheliopathy. Urinary findings. Arch Ophthalmol. 1981;99:1560–2. doi: 10.1001/archopht.1981.03930020434004. [DOI] [PubMed] [Google Scholar]
- 45.Kirkham TH, Ffytche TJ, Sanders MD. Placoid pigment epitheliopathy with retinal vasculitis and papillitis. Br J Ophthalmol. 1972;56:875–80. doi: 10.1136/bjo.56.12.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hyvarinen L, Maumenee AE, George T, Weinstein GW. Fluorescein angiography of the choriocapillaris. Am J Ophthalmol. 1969;67:653–66. doi: 10.1016/S0002-9394(69)90987-8. [DOI] [PubMed] [Google Scholar]
- 47.Hamilton AM, Bird AC. Geographical choroidopathy. Br J Ophthalmol. 1974;58:784–97. doi: 10.1136/bjo.58.9.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wright BE, Bird AC, Hamilton AM. Placoid pigment epitheliopathy and Harada’s disease. Br J Ophthalmol. 1978;62:609–21. doi: 10.1136/bjo.62.9.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hayreh SS. Choroidal ischemiaischaemia. Retina. 1982;2:191–2. doi: 10.1097/00006982-198200230-00012. [DOI] [PubMed] [Google Scholar]
- 50.Hayreh SS. Choroidal ischemiaischaemia after extra capsular cataract extraction by phacoemulsification. Retina. 1982;2:191–3. doi: 10.1097/00006982-198200230-00012. [DOI] [PubMed] [Google Scholar]
- 51.Amalric P. The choriocapillaris in the macular area. Clinical and angiographic study. Int Ophthalmol. 1983;6:149–53. doi: 10.1007/BF00127643. [DOI] [PubMed] [Google Scholar]
- 52.Hayreh SS. Ischemic optic neuropathy. Prog Retin Eye Res. 2009;28:34–62. doi: 10.1016/j.preteyeres.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 53.Hayreh SS. In vivo choroidal circulation and its watershed zones. Eye. 1990;4:273–89. doi: 10.1038/eye.1990.39. [DOI] [PubMed] [Google Scholar]
- 54.Chen JC, Fitzke FW, Pauliekhoff D, Bird AC. Poor choroidal perfusion is a cause of visual morbidity in age-related macular degeneration [abstract] Investig Ophthalmol Vis Sci. 1989;30:153. [Google Scholar]
- 55.Ross RD, Barofsky JM, Cohen G, Baber WB, Palao SW, Gitter KA. Presumed macular choroidal watershed vascular filling, choroidal neovascularization, and systemic vascular disease in patients with age-related macular degeneration. Am J Ophthalmol. 1998;125:71–80. doi: 10.1016/S0002-9394(99)80237-2. [DOI] [PubMed] [Google Scholar]
- 56.Sarks SH. Ageing and degeneration in the macular region: a clinico-pathological study. Br J Ophthalmol. 1976;60:324–41. doi: 10.1136/bjo.60.5.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kornzweig AL. Changes in the choriocapillaris associated with senile macular degeneration. Ann Ophthalmol. 1977;9:753–6. [PubMed] [Google Scholar]
- 58.Corvi F, Tiosano L, Corradetti G, Nittala MG, et al. Choriocapillaris flow deficits as a risk factor for progression of age-related macular degeneration. Retina. 2021;41:686–93. doi: 10.1097/IAE.0000000000002990. [DOI] [PubMed] [Google Scholar]
- 59.Nesper PL, Ong JX, Fawzi AA. Exploring the Relationship Between Multilayered Choroidal Neovascularization and Choriocapillaris Flow Deficits in AMD. Investig Ophthalmol Vis Sci. 2021;62:12. doi: 10.1167/iovs.62.3.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu W, Grunwald JE, Metelitsina TI, DuPont JC, Ying GS, Martin ER, et al. Association of risk factors for choroidal neovascularization in age-related macular degeneration with decreased foveolar choroidal circulation. Am J Ophthalmol. 2010;150:40–7. doi: 10.1016/j.ajo.2010.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hayreh SS. Submacular choroidal vascular bed watershed zones and their clinical importance. Am J Ophthalmol. 2010;150:940–1. doi: 10.1016/j.ajo.2010.08.011. [DOI] [PubMed] [Google Scholar]
- 62.Morrison JC, Van Buskirk EM. Microanatomy and modulation of the ciliary vasculature. Trans Ophthalmol Soc U K. 1986;105:131–9. [PubMed] [Google Scholar]
- 63.Weiter J, Fine BS. A histologic study of regional choroidal dystrophy. Am J Ophthalmol. 1977;83:741–50. doi: 10.1016/0002-9394(77)90143-X. [DOI] [PubMed] [Google Scholar]
- 64.Paton D, Duke JR. Primary familial amyloidosis. Ocular manifestations with histopathologic observations. Am J Ophthalmol. 1966;61:736–47. doi: 10.1016/0002-9394(66)91211-6. [DOI] [PubMed] [Google Scholar]
- 65.Prince JH. The rabbit in eye research. Springfield, Illinois: Charles C. Thomas; 1964; p. 531, 539–40.
- 66.Hayreh SS, Baines JA. Occlusion of the vortex veins. An experimental study. Br J Ophthalmol. 1973;57:217–38. doi: 10.1136/bjo.57.4.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schmidt H. Beitrag zur Kenntniss der Embolie der Arteria Centralis Retinae. Albrecht Von Graefes Arch Klin Exp Ophthalmol. 1874;20:287–307. doi: 10.1007/BF01694160. [DOI] [Google Scholar]
- 68.Crock G. Clinical syndromes of anterior segment ischaemia. Trans Ophthalmol Soc U K. 1967;87:513–33. [PubMed] [Google Scholar]
- 69.Harbitz F. Bilateral carotid arteritis. Arch Pathol Lab Med. 1926;1:499–510. [Google Scholar]
- 70.Leinfelder PJ, Black NM. Experimental Transposition of the Extraocular Muscles in Monkeys. Am J Ophthalmol. 1941;24:1115–9. doi: 10.1016/S0002-9394(41)90512-3. [DOI] [Google Scholar]
- 71.Skipper E, Flint FJ. Symmetrical arterial occlusion of upper extremities head, and neck: a rare syndrome. Br Med J. 1952;2:9–14. doi: 10.1136/bmj.2.4774.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ask-Upmark E. On the pulseless disease outside of Japan. Acta Med Scand. 1954;149:161–78. doi: 10.1111/j.0954-6820.1954.tb11424.x. [DOI] [PubMed] [Google Scholar]
- 73.Knox DL. Ischemic ocular inflammation. Am J Ophthalmol. 1965;60:995–1002. doi: 10.1016/0002-9394(65)92806-0. [DOI] [PubMed] [Google Scholar]
- 74.Wilson WA, Irvine SR. Pathologic changes following disruption of blood supply to iris and ciliary body. Trans Am Acad Ophthalmol Otolaryngol. 1955;59:501–2. [PubMed] [Google Scholar]
- 75.Mizener JB, Podhajsky P, Hayreh SS. Ocular Ischemic Syndrome. Ophthalmology. 1997;104:859–64. doi: 10.1016/S0161-6420(97)30221-8. [DOI] [PubMed] [Google Scholar]
- 76.Virdi PS, Hayreh SS. Anterior segment ischemiaischaemia after recession of various recti. An experimental study. Ophthalmology. 1977;94:1258–71. doi: 10.1016/S0161-6420(87)80009-X. [DOI] [PubMed] [Google Scholar]
- 77.Krupin T, Johnson MF, Becker B. Anterior segment ischemiaischaemia after cyclocryotherapy. Am J Ophthalmol. 1977;84:426–8. doi: 10.1016/0002-9394(77)90688-2. [DOI] [PubMed] [Google Scholar]
- 78.Ryan SJ, Goldberg MF. Anterior segment ischemiaischaemia following scleral buckling in sickle cell hemoglobinopathy. Am J Ophthalmol. 1971;72:35–50. doi: 10.1016/0002-9394(71)91588-1. [DOI] [PubMed] [Google Scholar]
- 79.Friedman AH, Bloch R, Henkind P. Hypopyon and iris necrosis in angle-closure glaucoma. Report of two cases. Br J Ophthalmol. 1972;56:632–5. doi: 10.1136/bjo.56.8.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hayreh SS, Scott WE. Fluorescein iris angiography. II. Disturbances in iris circulation following strabismus operation on the various recti. Arch Ophthalmol. 1978;96:1390–1400. doi: 10.1001/archopht.1978.03910060144010. [DOI] [PubMed] [Google Scholar]
- 81.Amalric P. The angiography of the anterior segment of the eye. In: Shimizu K, editor. Proceedings of the International Symposium of Fluorescein Angiography, Tokyo, 1972. Tokyo: Igaku Shoin Ltd; 1974. p. 281–318.
- 82.Morrison JC, Van, Buskirk EM. Anterior collateral circulation in the primate eye. Ophthalmology. 1983;90:707–15. doi: 10.1016/S0161-6420(83)34501-2. [DOI] [PubMed] [Google Scholar]
- 83.Iovino C, Peiretti E, Braghiroli M, Tatti F, et al. Imaging of iris vasculature: current limitations and future perspective. Eye. 2022;36:930–40. doi: 10.1038/s41433-021-01809-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Henkind P. Angle Vessels in Normal Eyes. A Gonioscopic Evaluation and Anatomic Correlation. Br J Ophthalmol. 1964;48:551–7. doi: 10.1136/bjo.48.10.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hayreh SS, Podhajsky PA, Zimmerman MB. Branch retinal artery occlusion natural history of visual outcome. Ophthalmology. 2009;16:1188–94. doi: 10.1016/j.ophtha.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hayreh SS, Podhajsky PA, Zimmerman MB. Retinal artery occlusion: associated systemic and ophthalmic abnormalities. Ophthalmology. 2009;116:1928–36. doi: 10.1016/j.ophtha.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hayreh SS. Anterior ischaemic optic neuropathy. II. Fundus on ophthalmoscopy and fluorescein angiography. Br J Ophthalmol. 1974;58:964–80. doi: 10.1136/bjo.58.12.964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Kearns TP. Collagen and rheumatic diseases: ophthalmic aspects. In: Mausolf FA, editor. The Eye and Systemic Disease. St. Louis: Mosby; 1975. p. 105–8.
- 89.Hayreh SS. The blood supply of the optic nerve head and the evaluation of it: myth and reality. Prog Retin Eye Res. 2001;20:563–93. doi: 10.1016/S1350-9462(01)00004-0. [DOI] [PubMed] [Google Scholar]
- 90.Oosterhuis JA Fluorescein fundus angiography in retinal venous occlusion. In: Henkes HE, editor. Perspectives in ophthalmology. Amsterdam: Excerpta Medica; 1968. p. 29–47.
- 91.Hayreh SS. Pathogenesis of occlusion of the central retinal vessels. Am J Ophthalmol. 1971;72:998–1011. doi: 10.1016/0002-9394(71)91706-5. [DOI] [PubMed] [Google Scholar]
- 92.McLeod D, Ring CP. Cilio-retinal infarction after retinal vein occlusion. Br J Ophthalmol. 1976;60:419–27. doi: 10.1136/bjo.60.6.419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hayreh SS, Fraterrigo L, Jonas J. Central retinal vein occlusion associated with cilioretinal artery occlusion. Retina. 2008;28:581–94. doi: 10.1097/IAE.0b013e31815ec29b. [DOI] [PubMed] [Google Scholar]
- 94.Stoffelns BM. [Isolated cilioretinal artery occlusion—clinical findings and outcome in 31 cases] Klin Monbl Augenheilkd. 2012;229:338–42. doi: 10.1055/s-0031-1299402. [DOI] [PubMed] [Google Scholar]
- 95.McLeod D. Central retinal vein occlusion with cilioretinal infarction from branch flow exclusion and choroidal arterial steal. Retina. 2009;29:1381–95. doi: 10.1097/IAE.0b013e3181b85f41. [DOI] [PubMed] [Google Scholar]
- 96.Glacet-Bernard A, Gaudric A, Touboul C, Coscas G. [Occlusion of the central retinal vein with occlusion of a cilioretinal artery: apropos of 7 cases] J Fr Ophtalmol. 1987;10:269–77. [PubMed] [Google Scholar]
- 97.Hayreh SS, Revie IH, Edwards J. Vasogenic origin of visual field defects and optic nerve changes in glaucoma. Br J Ophthalmol. 1970;54:461–72. doi: 10.1136/bjo.54.7.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hayreh SS, Hayreh MS. Hemi-central retinal vein occulsion. Pathogenesis, clinical features, and natural history. Arch Ophthalmol. 1980;98:1600–09. doi: 10.1001/archopht.1980.01020040452011. [DOI] [PubMed] [Google Scholar]
- 99.Hayreh SS, Zimmerman MB, Podhajsky P, Alward WLM. Nocturnal arterial hypotension and its role in optic nerve head and ocular ischemic disorders. Am J Ophthalmol. 1994;117:603–24. doi: 10.1016/S0002-9394(14)70067-4. [DOI] [PubMed] [Google Scholar]
- 100.Hayreh SS, Podhajsky P, Zimmerman MB. Role of nocturnal arterial hypotension in optic nerve head ischemic disorders. Ophthalmologica. 1999;213:76–96. doi: 10.1159/000027399. [DOI] [PubMed] [Google Scholar]
- 101.Hayreh SS, Zimmerman MB, Kimura A, Sanon A. Central retinal artery occlusion. Retinal survival time. Exp Eye Res. 2004;78:723–36. doi: 10.1016/S0014-4835(03)00214-8. [DOI] [PubMed] [Google Scholar]
- 102.Hayreh SS, Zimmerman MB. Amaurosis fugax in ocular vascular occlusive disorders: Prevalence and pathogeneses. Retina. 2014;34:115–22. doi: 10.1097/IAE.0b013e31829234b5. [DOI] [PubMed] [Google Scholar]
- 103.Hayreh SS, Zimmerman B. Management of giant cell arteritis. Our 27-year clinical study: new light on old controversies. Ophthalmologica. 2003;217:239–59. doi: 10.1159/000070631. [DOI] [PubMed] [Google Scholar]
- 104.Hayreh SS, Podhajsky PA, Zimmerman MB. Central and Hemicentral Retinal Vein Occlusion Role of Anti-Platelet Aggregation Agents and Anticoagulants. Ophthalmology. 2011;118:1603–11. doi: 10.1016/j.ophtha.2011.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Leber T. Studien über den Flüssigkeitswechsel im Auge. Albrecht Von Graefes Arch Ophthalmol. 1873;19:87–195. doi: 10.1007/BF01720618. [DOI] [Google Scholar]
- 106.Koster W. [Contributions to the theory of glaucoma] Beiträge zur Lehre vom Glaukom. Albrecht Von Graefes Arch Ophthalmol. 1895;41:30–112. doi: 10.1007/BF01693633. [DOI] [Google Scholar]
- 107.Hayreh SS. Blood supply of the optic nerve head and its role in optic atrophy, glaucoma, and oedema of the optic disc. Br J Ophthalmol. 1969;53:721–48. doi: 10.1136/bjo.53.11.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nishikawa M, Matsunaga H, Takahashi K, Matsumura M. Indocyanine green angiography in experimental choroidal circulatory disturbance. Ophthalmic Res. 2009;41:53–8. doi: 10.1159/000165916. [DOI] [PubMed] [Google Scholar]
- 109.Takahashi K, Kishi S. Remodeling of choroidal venous drainage after vortex vein occlusion following scleral buckling for retinal detachment. Am J Ophthalmol. 2000;129:191–8. doi: 10.1016/S0002-9394(99)00425-0. [DOI] [PubMed] [Google Scholar]
- 110.Sachsenweger R, Lukoff L. [Animal experimental studies on the sequelae of surgical occlusion of the vortex veins] Klinische Monatsblatter fur Augenheilkd und fur augenarztliche Fortbild. 1959;134:364–73. [PubMed] [Google Scholar]
- 111.de Faria E, Silva D. [Cancer of the choroid and retinal detachment] Cancer de la choroide et décollement de la rétine. Annales d’oculistique. 1950;183:400–7. [PubMed] [Google Scholar]
- 112.Klien BA. Macular and extramacular serous chorieretinopathy. With remarks upon the role of an extrabulbar mechanism in its pathogenesis. Am J Ophthalmol. 1961;51:231–42. doi: 10.1016/0002-9394(61)91943-2. [DOI] [PubMed] [Google Scholar]
- 113.Bonnet M. [Surgical occlusion of 2 vortex veins in the treatment of decompensated senile macular degeneration. A longer-term evaluation] J Fr Ophtalmol. 1985;8:779–83. [PubMed] [Google Scholar]