

Abstract
Hyper-IgM1 is a rare X-linked combined immunodeficiency caused by mutations in the CD40 ligand (CD40LG) gene with a median survival of 25 years, potentially treatable with in situ CD4+ T cell gene editing with Cas9 and a one-size-fits-most corrective donor template. Here, starting from our research-grade editing protocol, we pursued the development of a good manufacturing practice (GMP)-compliant, scalable process that allows for correction, selection and expansion of edited cells, using an integrase defective lentiviral vector as donor template. After systematic optimization of reagents and conditions we proved maintenance of stem and central memory phenotypes and expression and function of CD40LG in edited healthy donor and patient cells recapitulating the physiological CD40LG regulation. We then documented the preserved fitness of edited cells by xenotransplantation into immunodeficient mice. Finally, we transitioned to large-scale manufacturing, and developed a panel of quality control assays. Overall, our GMP-compliant process takes long-range gene editing one step closer to clinical application with a reassuring safety profile.
Keywords: IDLV, gene editing, Cas9, GMP, large-scale process, hyper-IgM1
Graphical abstract
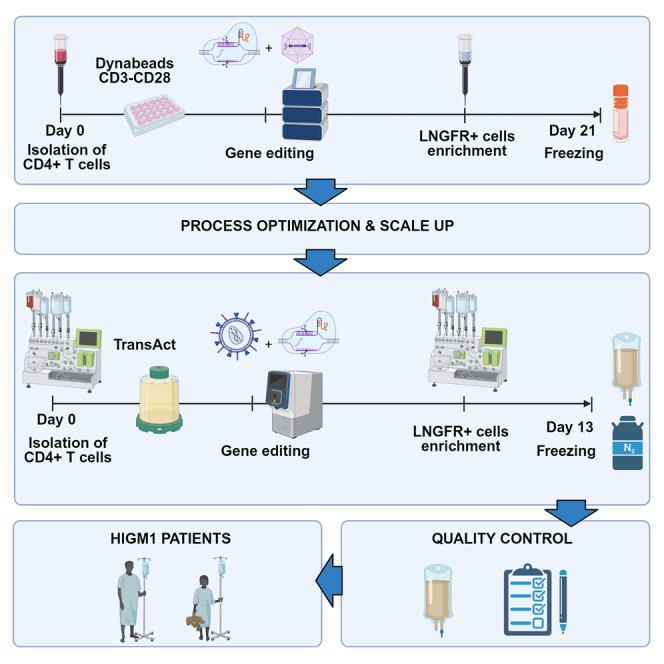
Naldini and colleagues developed a scalable, GMP-compliant manufacturing process for ex vivo, HDR gene editing of CD4 T cells for the treatment of hyper-IgM1. Cells are purified from leukapheresis with CliniMACS-Prodigy, activated with TransAct, transduced with a CD40LG-LNGFR integrase defective lentiviral vector, electroporated with MaxCyte-GTx, immunomagnetically selected by LNGFR, expanded and formulated.
Introduction
Mutations in the X-linked CD40 ligand (CD40LG) gene underlie hyper-IgM1 (HIGM1), a combined immunodeficiency characterized by low levels of IgG and IgA, normal or increased levels of IgM, and a median survival of 25 years.1,2,3 The phenotype results from impaired CD40L-CD40 crosstalk between CD4+ T cells and B cells, where it is required for class switch recombination, and myeloid cells, where it regulates immune activation.4,5 Restoration of physiological regulation of CD40LG expression is paramount, as constitutive expression or impaired regulation can result in lymphoproliferative or autoimmune disease.6,7 In situ gene correction strategies using a donor adenoviral-associated vector (AAV6) have been previously published by our group8 and others.9,10 Specifically, we have shown potential clinical benefit of correcting CD40L-defective CD4+ lymphocytes or hematopoietic stem cells, pasting into CD40LG intron 1 an AAV6-delivered donor template encoding for a splice acceptor sequence and the cDNA of CD40LG exons 2–5. In addition, we have shown that CD40L corrected CD4+ T cells were able to promote Ig class switch in vitro. Furthering our pursuit of treating HIGM1 patients with autologous gene-corrected CD4+ lymphocytes, we reasoned that infusion of a large number of corrected cells may be advantageous to potentially avoid lymphodepletion and treat frail patients with ongoing chronic infections, as destination therapy or bridge to hematopoietic stem cell transplantation. Therefore, we strove to develop a sustainable and scalable GMP process, suitable for a wide range of starting cell numbers—mirroring harvests from either pediatric or adult donors—and allowing enrichment and expansion of large numbers of edited cells. Here, we systematically tested and optimized manufacturing conditions, from selection of CD4+ cells to gene editing, enrichment of corrected cells, and expansion. Furthermore, we validated the functional rescue of CD40L and the fitness of the drug product into a xenograft model and built a custom quality control (QC) panel.
Results
Optimization of a small- and medium-scale manufacturing process for gene editing of CD4+ T cells
Starting from our previously published research-grade gene editing process, we break down the decision points to be addressed for the process development in Figure 1A. While not strictly required for gene correction, we included the low-affinity nerve growth factor receptor (LNGFR) selector (Figure 1B) in the corrective donor template. As LNGFR is expressed only upon correct integration of the corrective cassette, through an internal ribosome entry site (IRES) sequence, it allows for in vitro enrichment of edited cells and better reconstitution of CD40L expression.8 Selection of edited cells was also shown to be beneficial in terms of safety as it supported purging of cells suffering large deletion at the edited locus (to be reported elsewhere). Moreover, enrichment by LNGFR favored standardization of product specifics in terms of proportion of corrected cells present in the cell product.
Figure 1.

Small- and medium-scale manufacturing of edited CD4+ T cells
(A) Flow chart with the decision points for manufacturing process development. For each step, corresponding panels are encircled in blue. (B) Gene editing strategy: the corrective cassette is integrated by homologous recombination (red lines) in the first intron of the CD40LG gene (dark gray). HA-L, homology arm left; SA, splice acceptor; HA-R, homology arm right. (C–E) Comparison of CD4+ selection by single-antibody-positive selection vs. negative selection by antibody cocktail in terms of editing efficiency (C), viability (D), proliferation rate (E), expression of activation markers (F), and phenotype (mean ± SD) (G). (H–J) Impact of human serum (HS) and IL-2 supplementation at different time points on HDR efficiency (H), cell growth (I), and phenotype (J) (mean ± SD), with AAV6 donor template. (K) Impact of transduction enhancers cyclosporine H (CsH)14 and Lentiboost on editing efficiency, alone or in combination (Combo) targeting the AAVS1 locus with a GFP reporter IDLV. (L) Impact of timing of transduction on editing efficiency, assessed by LNGFR expression in CD40LG locus with IDLV donor template (B). (M and N) Relative performance of different Maxcyte GTx electroporation protocols in terms of cell viability (M) and HDR efficiency (N). Kruskal-Wallis test with Dunn’s multiple comparisons. Exp T2-T5 correspond to increased voltage settings. Exp, expanded. (O) Immunomagnetic enrichment of LNGFR+ cells at different days after isolation and editing. (P) Fold growth at D6, D13, and cumulative growth of healthy donor (HD) cells. D6/D2 is the cell fold growth measured at day 6, 4 days after gene editing, before the cell selection step. D13/D6 is the fold growth obtained at the end of the process (from day 6 to day 13), D13/D2 is the total fold growth from the day of the electroporation to the end of the process considering the loss of cells after the selection step. (Q) Schematic representation of the optimized small- and medium-scale protocol. Cells were immunomagnetically selected from buffy coats or peripheral blood samples using CD4 microbeads and activated in G-Rex 24-6-6M on day 0; transduced with IDLV and electroporated with Cas9 RNP using MaxCyte GTx on day 2; on day 6 LNGFR+ cells were enriched and seeded for cell expansion; and, at day 13, cells were frozen. AAV6 donor was used in (C–J). IDLV donor was used in the remaining panels. Empty dots indicate HD cells; full dots indicate patient cells. Editing efficiency, phenotype, and activation markers were assessed by flow cytometry. UT, untreated; GE, gene edited; TSCM, T stem cell memory; CM, central memory; EM, effector memory; TEMRA, terminal differentiated; MOI, multiplicity of infection; MW, multiwell. Percentage of live cells was assessed by trypan blue exclusion.
As research-grade CD4+ negative selection was not translatable to clinical-grade manufacturing, we assessed the performance of single-antibody immunomagnetic positive selection, reaching, with an AAV6 donor, comparable results in terms of bona fide HDR editing efficiency by LNGFR expression, viability, growth, and immunophenotype (Figures 1C–1G and S1A). For activation and culture, use of TransAct (Figure S1B) and 0.5% human serum albumin (HSA) before editing, together with addition of IL-2 and human serum after editing, were beneficial in terms of editing efficiency, with no impact on growth and phenotype (Figures 1H–1J).
After initial experiments we favored integrase defective lentiviral vector (IDLV) over AAV6 as template delivery vehicle, given lower cyto- and geno-toxicity concerns observed in a different setting.11 While editing efficiency with IDLV was lower (20% vs. 57%, see Figure S1C), potency was comparable (Figure S1D).
Optimal editing efficiency in G-Rex was achieved by delivering IDLV at day 2, before electroporation, together with LentiBoost, a commercial poloxamer that enhances lentiviral transduction12 (Figures 1K, 1L, and S1E). Of note, neither cyclosporine H (CsH), an inhibitor of lentiviral restriction, nor supplementation with deoxyribonucleotids13 significantly improved editing efficiency (Figure S1F), as observed instead in other settings.14
Moving from research-grade electroporation with Lonza Nucleofector to clinical-grade Maxcyte GTx, we compared different electroporation protocols (expanded protocol T2 to T5, from lowest to highest energy delivery), striking a balance between toxicity and editing efficiency (Figures 1M and 1N), ultimately choosing “expanded protocol T3” (Exp T3). Immunomagnetic enrichment by LNGFR beads allowed formulating a nearly pure product (Figure 1O). We noticed an increased performance of the enrichment step from day 5 to day 7, possibly due to progressive dilution of residual CD4 microbeads used for initial cell purification (Figures 1C–1G), and correspondingly reduced LNGFR-independent cell binding to the enrichment column. In a large series of experiments in which enrichment was performed at day 6, we observed 21-fold growth in cell numbers in the 4 days following gene editing, up to LNGFR selection, and an additional 16-fold growth in the subsequent 7 days of expansion, with a cumulative growth of 46-fold, which also incorporates cell loss during selection (Figure 1P). The scheme of this first small and medium process is illustrated in Figure 1Q.
Process validation with patient cells
We addressed potential concerns of reproducibility of our process with patient-derived cells, reasoning that disease-specific characteristics could potentially be relevant. Indeed, we confirmed the previously described differences between HIGM1 patients and healthy donors (HDs)15,16 in terms of white blood cell counts (Figure 2A) and CD4+ subset distribution (Table S1), highlighting an increased level of naive T cells (Figure 2B).
Figure 2.

Confirmatory experiments on patient cells
(A) White blood cell (WBC) count from patient blood samples. Dotted lines indicate the normal range of WBC in HDs. Full dots indicate the same patient analyzed at different time points. (B) Naive, TSCM, CM, EM, and TEMRA distribution in CD4+ cell-positive fraction (mean ± SEM). (C) Editing efficiency at day 5–6 by LNGFR expression and at day 13 by LNGFR expression and ddPCR (median ± IQR). Green bars, Pt; blue bars, HD. (D–F) Composition, phenotype distribution, and expression of exhaustion markers in patients and HD cells, (mean ± SEM). (G) Fold growth at D6, D13, and cumulative growth of patient and control cells. # indicates sample shipped overnight in suboptimal conditions. (H) Relative MFI of CD40L in LNGFR+ cells (median ± IQR). (I) Relative binding to CD40 in LNGFR+ cells (median ± IQR). (J) Gating strategy for expression of LNGFR in CD40L-expressing cells upon stimulation. Pt, patient; HD, healthy donor.
Despite these differences, and potentially other ones, related to previous patient medication that we could not account for, patient cells were edited with similar or better efficiency (Figure 2C), maintaining CD4+ cell purity until the end of the process (Figure 2D) and resulting in a higher proportion of T stem cell memory (TSCM) cells (Figure 2E).15 Analysis of activation/exhaustion markers showed very few cells double positive and none triple or quadruple positive for the markers analyzed (Figure 2F), suggesting that the optimized culture conditions and the high proliferation rates did not drive cell exhaustion.17,18 However, we observed a different kinetic of cell growth compared with HDs: patient cells displayed a higher initial proliferation rate, followed by a lower one, resulting in an overall slightly lower process yield (Figure 2G), possibly reflecting the aforementioned higher proportion of naive cells (Figures 2A and 2B). In addition, in lieu of the previously developed IgG class switch assay8 we developed three reproducible potency assays that documented the potency of corrected patient cells in terms of regulated CD40LG expression, binding to CD40, and downstream signal transduction (Figures 2H–2I). By correcting patient cells we were also able to experimentally demonstrate that cells expressing CD40L upon correction also expressed LNGFR, thus confirming that the latter is stringently coupled to correction of CD40LG (Figure 2J).
Engraftment of gene-edited cells upon xenotransplantation
The fitness of the edited cells was proven upon xenotransplantation into non-irradiated NSG mice and compared with that of untreated or mock electroporated cells from two different HDs undergoing the same culture process. The study was carried out in certified GLP conditions, as detailed in the materials and methods; a schematic representation of the experiment is reported in Figure 3A. Engraftment in peripheral blood was similar across groups (Figure 3B), with preserved human CD4+ purity in the human CD45+ subset (Figure 3C) and no expansion of other cell subtypes. Engrafted cells repopulated mice spleen as expected, with little but detectable engraftment in the bone marrow (Figure 3D). Editing efficiency in terms of both LNGFR expression and molecular analysis was stable in the peripheral blood of treated animals (Figures 3E and 3F) and could also be documented in bone marrow and spleen (Figure 3G). Infused cells showed progressive maturation toward effector phenotype, which was similar to their untreated or mock-electroporated counterparts (Figure 3H). In terms of fitness of infused cells, we observed spleen repopulation by human cells (Figures 3I and S2A; Table S2); comparable levels of graft-versus-host disease (GvHD) were observed, even despite the absence of CD8 T cells, across skin, lung, and liver of the treated animals (Figures 3J and S2A; Tables S3–S5), with mild impact on body weight (Figure S2B).
Figure 3.

Xenotransplantation of edited cells
(A) Scheme of experiment: NSG mice (n = 5/group) were transplanted with 20E+06 cells/mouse isolated from two HDs and left untreated (CI 1–2), edited (TI 1–2), or mock electroporated (Mock 2). Infused cells were manipulated as described in Figure 1A. Mice were followed by serial bleeding until experiment termination (day 60) when organs were collected for cytofluorimetric, molecular, and histopathological analysis. (B) Percentage of hCD45+ cells in human graft evaluated in peripheral blood (PB) of NSG mice over time (mean ± SD). (C) Percentage of hCD3+CD4+ cells in human graft evaluated in peripheral blood (PB) of NSG mice over time (mean ± SD). (D) Percentage of human CD45+ cells in bone marrow (BM) and spleen (SPL) of NSG mice at termination (day 60), mean ± SD. n = 5 mice/group with the exception of the TI1 group in which two mice were excluded due to failure in human cell engraftment and premature death. (E–G) Percentage of LNGFR+ cells analyzed by FACS (mean ± SD) (E) and proportion of edited alleles by ddPCR (mean ± SD) in PB over time (F) and in BM and SPL (G) at the end of experiment at day 60. (H) Phenotype of CD4+ cells in PB over time (10–30 days post injection [d.p.i.]). ∗Population in which only 1 or 2 out of 5 samples were above the low limit of quantification of the method (3+ cells/μL). Phenotype of n = 5 mice/group with the exception of the TI1 group in which two mice were excluded due to failure in human cell engraftment and premature death. (I) Heatmap indicating the spleen repopulation by engrafted cells. n = number of mice with none, minimal, mild to moderate, and marked mononuclear infiltration. (J) Heatmap indicating GvHD damage in skin (back and ear), lung, and liver. n = number of mice with none, minimal, mild to moderate, and marked GvH reaction. CI1-2, control item 1–2, untreated cells; TI1-2, test item 1–2, edited cells; Mock, mock electroporation.
Scale up of the manufacturing process
Based on these encouraging results, we scaled up the manufacturing process to the full scale (up to 500 × 106 electroporated cells at day 2), as summarized in Figure 4A. Briefly, we took advantage of CliniMACS Prodigy for CD4+ selection and LNGFR+ enrichment, and GMP-compliant R-1000 and R-20K process assemblies for the MaxCyte GTx electroporator. Cells were kept in a closed system with the sole exception of cell wash and centrifugation steps. Hereby, we were able to recover 77% of CD4+ cells on the day of selection (day 0), and 69% on day 2, before gene editing (Figure 4B). On day 6 we enriched our cell product with high LNGFR purity and an average recovery of 85% (n = 3 LNGFR selection runs) (Figures 4B and 4C). For cell culture and expansion we exploited the G-Rex bioreactor, which was scalable from G-Rex 6M to G-Rex 500M-CS, yielding up to 20 × 109 cells (Figure 4D). Overall, CD4+ purity was maintained until the end of the process with no expansion of CD8+ T cells (Figure 4E), and the proportion of TSCM and central memory (CM) was comparable with that obtained with the medium-scale process (Figure 4F); no exhaustion was evident by surface marking (Figure 4G), and potency was in line with medium-scale experiments (Figures 4H–4J). Thus, we were able to transition from a laboratory-grade protocol to a manufacturing process suitable for clinical-grade product formulation with substantial improvements in the growth and purity of the edited cells while preserving the most relevant TSCM and CM fractions.
Figure 4.

Large-scale manufacturing process
(A) Overview of the process. CD4+ cells were immunomagnetically selected from leukapheresis using CliniMACS Prodigy and activated in G-REX 100 CS on day 0, transduced with IDLV, and electroporated with Cas9 RNP using MaxCyte GTx on day 2, enriched for LNGFR expression on day 6, and frozen after 7 days of expansion. (B) Recovery of CD4+ T cells after CD4+ immunomagnetic selection with CliniMACS Prodigy (day 0), after cell activation (day 2), and recovery of LNGFR+ cells after LNGFR immunomagnetic selection (day 6), (mean ± SD). L/F, large/full scale. (C) Purity of HDR edited cells by LNGFR expression at the day of selection (day 6) and at the end of the process (day 13) using G-Rex 6M (medium scale) and G-Rex 100-CS (large scale), and G-Rex 500-CS (full scale) (mean ± SD). (D) Large- and full-scale process yield compared with the historical mean ± 1 SD of the medium-scale processes (dotted lines) (mean ± SD). (E–G) Composition (E), phenotype (F), and expression of exhaustion markers (G) at the end of the process (mean ± SD). (H–J) Potency assessed by CD40L expression (H) and binding to CD40 (F) (median ± IQR), and CD40 downstream signal transduction by SEAP colorimetric assay (J) in healthy donor cells (mean ± SD). LNGFR and CD40LG expression, phenotype, and binding to CD40 were assessed by flow cytometry. MS, medium scale; LS, large scale; FS, full scale.
Critical quality attributes
Given the absence of comparable gene therapy products in clinical use, we devised a suitable QC panel addressing the specific characteristics of the manufacturing process (Figure 5A). For identity characterization, beyond previously described assays, we propose to assess clonal diversity by Vbeta repertoire analysis (Figure 5B), whereby we did not detect skewing after manipulation. In terms of process-related impurities, residual Cas9 was not detected (Figure 5C). No cytokine-independent growth was observed (Figure 5D); median residual p24 was 47 pg/mL (range 13–138, n = 6), corresponding to a total of 7,050 pg per batch (range 3,680–13,800), with an estimated depletion of >99.9% at the end of the process (Figures 5E and 5F). Detailed genomic integrity analysis will be reported in a separate manuscript.
Figure 5.

Critical quality attributes
(A) Overview of the quality control panel. The panel includes identity purity and potency assays, the identification of process-related impurities and safety tests. Blue boxes include routine characterization, either with release specifications or for information only (FIO), while red boxes include non-routine characterization of the cell product. (B) Cytofluorimetric analysis of the VBeta repertoire at day 0 or on the drug substance at day 13, n = 2 HDs. Each color represents VBeta sub-families, pink area shows uncovered repertoire (mean ± SD). (C) Residual Cas9 on the cell product at different time points during the manufacturing. (D) Growth factor-dependent growth assay. Cells from DP were seeded in presence or in absence of human cytokines and cell growth was measured by counting at different time points (mean ± SD). (E–F) Quantification of p24 concentration (E) and the amount of total p24 (F) to determine residual IDLV from the supernatant of cells before the formulation (median ± range).
Discussion
Here, starting from a research-grade protocol8 we have developed a GMP-compliant, large-scale, IDLV- and Cas9-based HDR long-range editing process suitable for clinical use, while shortening its length from the original 21 days down to 13 days. To our knowledge, this strategy is unique in its design. Our process preserves CD4+ T cell purity, therapeutically relevant phenotypes, with in vivo documented fitness and function, reproducible potency, while also expanding the cells ≥45-fold. In our systematic testing of the raw materials and conditions we found that the most significant improvements in terms of cell growth and expansion came from culture conditions, and especially from stimulating the cells with a high dose of TransAct, in the absence of serum in the closed G-Rex system. The process is expected to yield at least 1 × 1010 cells per patient in a single run, which is in the range of doses used in other T cell therapies.19,21
T cells are a prime candidate for clinical application of HDR gene editing, as the risk of transformation stemming from unexpected genomic rearrangements is thought to be low in terminally differentiated cells, even when using AAV6 as donor template. In the pursuit of a common template delivery platform for both T cell and HSPC gene editing we favored IDLV for safety benefits observed in the latter context. While IDLV was significantly less efficient than AAV6 for template delivery, editing efficiency could be partially rescued by the use of LentiBoost. Inclusion of the LNGFR selector in the corrective cassette allowed for the formulation of a highly enriched product. In the hemizygous condition of CD40LG, enrichment of the desired outcome may also be expected to be beneficial in terms of genotoxicity, purging out cells that have undergone large deletions or other complex rearrangements that compromise regulation of the gene. In absence of alternative clinically compliant systems, RNP delivery by electroporation required balancing efficiency with the significant electroporation toxicity, which plateaued around protocol Exp T3. Around 30% cell loss had to be accounted for at this voltage level. In the future, less toxic delivery tools, such as lipid nanoparticles, may allow mitigating the cell death due to electroporation.22
In vivo assessment of functionality and fitness of edited cells is somewhat challenging given the intrinsic dynamics of human CD4+ cells upon xenotransplantation, resulting from clonal expansion and GvHD. Indeed, in absence of alternative options, we took advantage of the model to show that gene editing did not detectably alter the functionality of CD4+ cells, which were able to engraft and give rise to GvHD similarly to their unedited counterparts, while maintaining the expression of LNGFR in the long term. In principle, robust engraftment is an advantage of gene editing CD4+ T cells over HSPCs, whose repopulation capacity is instead usually hampered upon long-range gene editing.11,20 In terms of QC, we developed assays to address product-specific potency, and theoretical immunogenicity concerns for residual Cas9, to meet recent indications by regulatory authorities.
An intrinsic limitation of this process is its complexity, stemming from the necessity of delivering all the editing components and enriching for edited cells; this may reasonably be expected to impact on the non-trivial costs of manufacturing. Still, some efficiencies may be gained from the process scalability, which allows to save critical reagents by reducing the scale (e.g., in case of pediatric patients).
Inter-donor variability, while present, was overall manageable. Conversely, we observed consistent differences between HD and patient cells. The latter were edited with similar or even higher efficiency but displayed a slightly different proliferation kinetic. While patient samples may be difficult to collect in large numbers for the sake of process development, we reckon that assessment of the process performance with real-world patient cells provides valuable information for tailoring of its conditions. Large-scale runs with patient cells may be warranted in the future.
In summary, here we report that the feasibility of large-scale clinical-grade manufacturing of gene-corrected CD4+ T cells supports clinical translation with a favorable risk-benefit profile of one of the first applications of long-range HDR-mediated gene editing. Moreover, most of our findings may be portable to other T cell gene therapy platforms.
Material and methods
Culturing and gene editing of CD4+ T cells with IDLV
For small- (1–2 × 106 cells at day 2) and medium-scale (5–10 × 106 cells at day 2) experiments CD4+ T cells were isolated from buffy coats of HDs with StraightFrom Buffy Coat CD4 Microbeads or from patient peripheral blood samples with StraightFrom Whole Blood CD4 Microbeads human (both Miltenyi Biotec). CD4+ T cells were cultured in X-Vivo 15 (Lonza) supplemented with 0.5% HSA (Baxalta), 1% Pen/Strep (Lonza), 100 IU/mL IL-7, and 200 IU/mL IL-15 (both from Miltenyi Biotec) and stimulated with T cell TransAct 40 μL/106 cells (Miltenyi Biotec) at 2 × 106 cells/mL. On day 2, unless stated otherwise, cells were transduced at 1 × 106 cells/mL with CD40LG-IRES_LNGFR-IDLV at an MOI of 40, and after 8 ± 1 h were washed with at least 1 volume of DPBS followed by a second wash with the indicated electroporation buffer (EB), resuspended in the indicated EB, and electroporated with SpyFi high fidelity Cas9 (Aldevron) complexed with a single guide RNA targeting the first intron of CD40LG. In preliminary experiments, recombinant AAV6, delivered after electroporation, instead of IDLV was used for donor template delivery, as indicated.
After electroporation, cells were cultured in X-Vivo 15 (Lonza) supplemented with 5% human serum (Pan Biotech), 1% Pen/Strep (Lonza), 100 IU/mL IL-7, 200 IU/mL IL-15, and 50 IU/mL IL-2 (all from Miltenyi Biotec). On day 6, unless stated otherwise, cells were enriched by custom-made immunomagnetic CD271 Microbeads (Mitenyi Biotec) and subsequently expanded until days 13–14 in the same culture medium. Cells were then frozen in saline solution supplemented with 7% HSA and 5% DMSO (Sigma) with KryoPlaner560-16 Software v.5.26 (Planer, Sunbury-on-Thames, UK) and stored in nitrogen vapors.
Multiwell plates (24-well) were used for small-scale experiments, while G-Rex 6-6M cell culture systems (Wilson Wolf) were used for medium-scale (5–10 × 106 cells at day 2) experiments. Characterization of corrected cells is reported in the supplemental information.
Large- and full-scale GMP-compliant manufacturing process
For large-scale experiments (100–500 × 106 cells at day 2) CD4+ T cells were isolated from male HD leukaphereses with the Enrichment Protocol 2.0 of CliniMACS Prodigy (Miltenyi) and CliniMACS CD4 GMP Microbeads, activated, and cultured in G-Rex 100CS or G-Rex 500CS (Wilson Wolf). Cell activation and culture conditions were identical to those indicated before. Electroporation was performed using R-1000 (100 × 106 cells) or R-20K process assembly (500 × 106 cells) using a MaxCyte GTx electroporator after a first wash with at least 1 volume of DPBS, followed by a second wash with Opti-MEM, resuspended in Opti-MEM at a concentration of 100–150 × 106 cells/mL. On day 6, LNGFR+ cells were isolated using the Enrichment Protocol 2.0 of CliniMACS Prodigy (Miltenyi) and CD271 Microbeads (Miltenyi). Cells were seeded in a G-Rex bioreactor for cell expansion and on day 13 frozen in CryoMACS bags (Miltenyi) in saline solution supplemented with 7% HSA, 5% DMSO (Sigma) using KryoPlaner560-16 Software v.5.26 (Planer), then stored in nitrogen vapors.
Xenotransplantation in NSG mice
Edited (test item), untreated (control item), and mock-electroporated (Mock) human cells derived from two different HDs were infused into male non-irradiated NSG mice (n = 5 mice/group) at a dose of 20 × 106 cells/mouse at day 0. Animals were monitored daily for clinical signs while body weight was recorded weekly until the end of study (day 60). Blood samples were collected at different time points to determine composition and phenotype of transplanted cells by cytofluorimetric analysis as well as the proportion of edited alleles by ddPCR. At the end of the experiment, mice were euthanized and organs were collected for cytofluorimetric and molecular analysis (bone marrow and spleen) and for histopathological evaluation (bone marrow, spleen, brain, gut, liver, skin, ear, lungs). According to very low percentage of hCD45+ cells retrieved in blood samples collected in the first 20 days after cell infusion, molecular analysis was performed only on samples collected from day 30 on. All the mice were maintained in specific pathogen-free conditions, and all animal procedures were designed and performed with the approval of the Animal Care and Use Committee of the San Raffaele Hospital (IACUC no. 1167) and communicated to the Ministry of Health and local authorities according to Italian law.
Human subject research
Informed consent for biological sample collection and anonymized biological sample/data sharing for HIGM1 patients were obtained by the referring physician according to local research protocols, and reviewed and approved by local ethics committees or institutional review boards. Buffy coats were obtained as anonymized residues of blood donations, used upon signature of specific institutional informed consent for blood product donation by healthy blood donors. Whole blood samples were obtained according to Tiget Clinical Protocol TIGET09: “Collection of biological material for the study of blood cells and their microenvironment and for the development of new therapeutic approaches for genetic diseases and tumors.” The experiments conformed to the principles set out in the WMA Declaration of Helsinki and the Department of Health and Human Services Belmont Report.
Data and code availability
For original data please contact asperti.claudia@hsr.it. Additional methods may be found in a data supplement available with the online version of this article.
Acknowledgments
We wish to thank all patients; the following key opinion leaders, internal and external collaborators: Pietro Genovese (Harvard Medical School), Harry Malech (National Institutes of Health), Suk See DeRavin (National Institutes of Health), Isabelle Meyts (University Hospitals Leuven), Arjan C. Lankester (Leiden University Medical Center), Andrew R. Gennery (Newcastle University), Sandro Plebani (Università di Brescia), Pere Soler Palacín (Vall d’Hebron Hospital), Luigi D. Notarangelo (National Institutes of Health), and Alessandro Aiuti (Università Vita-Salute San Raffaele) for guidance; Akiva Zablocki and the Hyper IgM foundation for patient referral; present and past SR-TIGET members including Tiziana Plati, Mariasole Vespasiano, Maria Carmina Castiello, Martina Fiumara, Paola Albertini and the SR-TIGET GLP team, and San Raffaele Flow Cytometry facility (FRACTAL) for technical support; the GeneSpire team for support. Some schematics were created using BioRender.com. This work was supported by Fondazione Telethon ETS (TIGET Core Grant support to L.N.), E-Rare-3-JTC 2017 to L.N., and GeneSpire S.r.l., a startup company aiming to develop ex vivo gene editing in genetic diseases.
Author contributions
D.C. and C.A. designed the study, performed research, interpreted data, and wrote the manuscript. E.R. performed gene editing experiments. S.P. performed potency and quality control experiments supervised by A.V., S.F. and V.V. contributed to experimental design and data interpretation. A.J. performed initial gene editing experiments. L.A. performed pilot in vivo experiments. F.C. and F.S. performed in vivo experiments and histopathological analyses, respectively. L.S.S. produced IDLV batches. E.M. performed p24 quantification. L.I.G.-G., V.L., A.F., and F.F. provided patient-derived cells. M.R. and L.N. supervised research, coordinated the work and revised the manuscript.
Declaration of interests
L.N., C.A., M.R., V.V., A.V., S.F., S.P., D.C., and A.J. are inventors of patent applications owned by Ospedale San Raffaele S.r.l. and Fondazione Telethon ETS, including one patent application on CD40LG gene editing. L.N. is founder, quota holder, and consultant of GeneSpire S.r.l.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtm.2023.08.020.
Contributor Information
Marina Radrizzani, Email: radrizzani.marina@hsr.it.
Luigi Naldini, Email: naldini.luigi@hsr.it.
Supplemental information
References
- 1.de la Morena M.T., Leonard D., Torgerson T.R., Cabral-Marques O., Slatter M., Aghamohammadi A., Chandra S., Murguia-Favela L., Bonilla F.A., Kanariou M., et al. Long-term outcomes of 176 patients with X-linked hyper-IgM syndrome treated with or without hematopoietic cell transplantation. J. Allergy Clin. Immunol. 2017;139:1282–1292. doi: 10.1016/j.jaci.2016.07.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ferrua F., Galimberti S., Courteille V., Slatter M.A., Booth C., Moshous D., Neven B., Blanche S., Cavazzana M., Laberko A., et al. Hematopoietic stem cell transplantation for CD40 ligand deficiency: Results from an EBMT/ESID-IEWP-SCETIDE-PIDTC study. J. Allergy Clin. Immunol. 2019;143:2238–2253. doi: 10.1016/j.jaci.2018.12.1010. [DOI] [PubMed] [Google Scholar]
- 3.Qamar N., Fuleihan R.L. The hyper IgM syndromes. Clin. Rev. Allergy Immunol. 2014;46:120–130. doi: 10.1007/s12016-013-8378-7. [DOI] [PubMed] [Google Scholar]
- 4.Díaz Á., González-Alayón I., Pérez-Torrado V., Suárez-Martins M. CD40-CD154: A perspective from type 2 immunity. Semin. Immunol. 2021;53 doi: 10.1016/j.smim.2021.101528. [DOI] [PubMed] [Google Scholar]
- 5.Ledbetter J.A., Shu G., Gallagher M., Clark E.A. Augmentation of normal and malignant B cell proliferation by monoclonal antibody to the B cell-specific antigen BP50 (CDW40) J. Immunol. 1987;138:788–794. [PubMed] [Google Scholar]
- 6.Brown M.P., Topham D.J., Sangster M.Y., Zhao J., Flynn K.J., Surman S.L., Woodland D.L., Doherty P.C., Farr A.G., Pattengale P.K., Brenner M.K. Thymic lymphoproliferative disease after successful correction of CD40 ligand deficiency by gene transfer in mice. Nat. Med. 1998;4:1253–1260. doi: 10.1038/3233. [DOI] [PubMed] [Google Scholar]
- 7.Sacco M.G., Ungari M., Catò E.M., Villa A., Strina D., Notarangelo L.D., Jonkers J., Zecca L., Facchetti F., Vezzoni P. Lymphoid abnormalities in CD40 ligand transgenic mice suggest the need for tight regulation in gene therapy approaches to hyper immunoglobulin M (IgM) syndrome. Cancer Gene Ther. 2000;7:1299–1306. doi: 10.1038/sj.cgt.7700232. [DOI] [PubMed] [Google Scholar]
- 8.Vavassori V., Mercuri E., Marcovecchio G.E., Castiello M.C., Schiroli G., Albano L., Margulies C., Buquicchio F., Fontana E., Beretta S., et al. Modeling, optimization, and comparable efficacy of T cell and hematopoietic stem cell gene editing for treating hyper-IgM syndrome. EMBO Mol. Med. 2021;13 doi: 10.15252/emmm.202013545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuo C.Y., Long J.D., Campo-Fernandez B., de Oliveira S., Cooper A.R., Romero Z., Hoban M.D., Joglekar A.v., Lill G.R., Kaufman M.L., et al. Site-Specific Gene Editing of Human Hematopoietic Stem Cells for X-Linked Hyper-IgM Syndrome. Cell Rep. 2018;23:2606–2616. doi: 10.1016/j.celrep.2018.04.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hubbard N., Hagin D., Sommer K., Song Y., Khan I., Clough C., Ochs H.D., Rawlings D.J., Scharenberg A.M., Torgerson T.R. Targeted gene editing restores regulated CD40L function in X-linked hyper-IgM syndrome. Blood. 2016;127:2513–2522. doi: 10.1182/blood-2015-11-683235. [DOI] [PubMed] [Google Scholar]
- 11.Ferrari S., Jacob A., Cesana D., Laugel M., Beretta S., Varesi A., Unali G., Conti A., Canarutto D., Albano L., et al. Choice of template delivery mitigates the genotoxic risk and adverse impact of editing in human hematopoietic stem cells. Cell Stem Cell. 2022;29:1428–1444.e9. doi: 10.1016/j.stem.2022.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schott J.W., León-Rico D., Ferreira C.B., Buckland K.F., Santilli G., Armant M.A., Schambach A., Cavazza A., Thrasher A.J. Enhancing Lentiviral and Alpharetroviral Transduction of Human Hematopoietic Stem Cells for Clinical Application. Mol. Ther. Methods Clin. Dev. 2019;14:134–147. doi: 10.1016/j.omtm.2019.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lahouassa H., Daddacha W., Hofmann H., Ayinde D., Logue E.C., Dragin L., Bloch N., Maudet C., Bertrand M., Gramberg T., et al. SAMHD1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 2012;13:223–228. doi: 10.1038/ni.2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petrillo C., Thorne L.G., Unali G., Schiroli G., Giordano A.M.S., Piras F., Cuccovillo I., Petit S.J., Ahsan F., Noursadeghi M., et al. Cyclosporine H Overcomes Innate Immune Restrictions to Improve Lentiviral Transduction and Gene Editing In Human Hematopoietic Stem Cells. Cell Stem Cell. 2018;23:820–832.e9. doi: 10.1016/j.stem.2018.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jain A., Atkinson T.P., Lipsky P.E., Slater J.E., Nelson D.L., Strober W. Defects of T-cell effector function and post-thymic maturation in X- linked hyper-IgM syndrome. J. Clin. Invest. 1999;103:1151–1158. doi: 10.1172/JCI5891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Levy J., Espanol-Boren T., Thomas C., Fischer A., Tovo P., Bordigoni P., Resnick I., Fasth A., Baer M., Gomez L., et al. Clinical spectrum of X-linked hyper-IgM syndrome. J. Pediatr. 1997;131:47–54. doi: 10.1016/S0022-3476(97)70123-9. [DOI] [PubMed] [Google Scholar]
- 17.Wherry E.J., Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015;15:486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Attanasio J., Wherry E.J. Costimulatory and Coinhibitory Receptor Pathways in Infectious Disease. Immunity. 2016;44:1052–1068. doi: 10.1016/j.immuni.2016.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tebas P., Stein D., Tang W.W., Frank I., Wang S.Q., Lee G., Spratt S.K., Surosky R.T., Giedlin M.A., Nichol G., et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 2014;370:901–910. doi: 10.1056/NEJMoa1300662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ferrari S., Valeri E., Conti A., Scala S., Aprile A., Di Micco R., Kajaste-Rudnitski A., Montini E., Ferrari G., Aiuti A., Naldini L. Genetic engineering meets hematopoietic stem cell biology for next-generation gene therapy. Cell Stem Cell. 2023;30:549–570. doi: 10.1016/j.stem.2023.04.014. [DOI] [PubMed] [Google Scholar]
- 21.Nagarsheth N.B., Norberg S.M., Sinkoe A.L., Adhikary S., Meyer T.J., Lack J.B., Warner A.C., Schweitzer C., Doran S.L., Korrapati S., et al. TCR-engineered T cells targeting E7 for patients with metastatic HPV-associated epithelial cancers. Nat. Med. 2021;27:419–425. doi: 10.1038/s41591-020-01225-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vavassori V., Ferrari S., Beretta S., Asperti C., Albano L., Annoni A., Gaddoni C., Varesi A., Soldi M., Cuomo A., et al. Lipid Nanoparticles Allow Efficient and Harmless Ex Vivo Gene Editing of Human Hematopoietic Cells. Blood. 2023 doi: 10.1182/blood.2022019333. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
For original data please contact asperti.claudia@hsr.it. Additional methods may be found in a data supplement available with the online version of this article.