

Summary
Osteoarthritis (OA) is a prevalent degenerative disease of the elderly. The NRF2 antioxidant system plays a critical role in maintaining redox balance. Mitoquinone (MitoQ) is a mitochondria-targeted antioxidant. This research aimed to determine whether MitoQ alleviated OA and the role of the NRF2/Parkin axis in MitoQ-mediated protective effects. In interleukin (IL)-1β-induced OA chondrocytes, MitoQ activated the NRF2 pathway, reducing extracellular matrix (ECM) degradation and inflammation. MitoQ also increased glutathione peroxidase 4 (GPX4) expression, leading to decreased levels of reactive oxygen species (ROS) and lipid ROS. Silencing NRF2 weakened MitoQ’s protective effects, while knockdown of Parkin upregulated the NRF2 pathway, inhibiting OA progression. Intra-articular injection of MitoQ mitigated cartilage destruction in destabilized medial meniscus (DMM)-induced OA mice. Our study demonstrates that MitoQ maintains cartilage homeostasis in vivo and in vitro through the NRF2/Parkin axis. We supplemented the negative feedback regulation mechanism between NRF2 and Parkin. These findings highlight the therapeutic potential of MitoQ for OA treatment.
Subject areas: Orthopedics, Molecular biology, Cell biology
Graphical abstract
Highlights
-
•
Mitoquinone alleviates osteoarthritis progress by promoting the expression of NRF2
-
•
NRF2 activates NRF2 antioxidant pathway and mitophagy and suppresses ferroptosis
-
•
Parkin regulates NRF2 expression through feedback regulation
Orthopedics; Molecular biology; Cell biology
Introduction
Osteoarthritis (OA), a prevalent degenerative disease, is a major factor of disability for the elderly.1 Since 1990, the age-standardized prevalence and incidence rate have increased by about 8%–10%.2 The aggravated damage of articular cartilage, subchondral bone, synovial tissue, and osteophyte formation are characteristics of OA.3 Although a few risk factors related to OA have been proposed, such as joint malalignment, aging, and high BMI, the pathogenesis of OA is still not clear.4,5,6 OA is a complicated process composed of oxidative stress, inflammation, metabolic factors, etc.7 The stiff and painful joint is characterized by oxidative stress, ferroptosis,8 and mitophagy9 in chondrocytes, synovial inflammation, degradation of cartilage extracellular matrix (ECM), osteophyte formation, and subchondral osteosclerosis.7 In addition, metabolic alterations, including changes in lipid metabolism, the enhancement of aerobic glycolysis, and mitochondrial dysfunction, play a role in OA.10
Nuclear factor erythroid2-related factor 2 (NRF2) regulated cellular redox homeostasis11 and initially offered protection against a variety of chronic diseases, involving neurodegenerative diseases and heart disease.12 Recently, a growing number of studies support that the NRF2 signaling pathway participates in OA progression. NRF2 antioxidant pathway attenuates oxidative stress and inflammation in chondrocytes.13,14,15,16,17,18 Thus, it is necessary to research the protection of NRF2 on chondrocytes.
As the only cellular component in cartilage, chondrocytes protect articular cartilage by maintaining ECM homeostasis.19 In the process of OA, chondrocyte injury plays a crucial role. Numerous studies have shown that chondrocyte injury is due to apoptosis, autophagic cell death, and cell necrosis. Ferroptosis, a new type of cell death, is first discovered and named by Dixon et al.20 Iron-dependent lipid peroxidation is the key characteristic of ferroptosis.21 Many researchers find that acyl-CoA synthetase long chain family member 4 (ACSL4) is a susceptible contributor and marker of ferroptosis.21,22 The antioxidant enzyme glutathione peroxidase 4 (GPX4) is a vital marker of ferroptosis.23 Inhibition of GPX4 brings about the accumulation of lipid reactive oxygen species (ROS) and eventually causes ferroptosis.17,23 Our previous research verified that ferroptosis produces a marked effect on the occurrence and progression of OA.8
Mitochondria have a variety of interrelated functions, supplying cell energy and producing various biosynthetic intermediates.24 Mitochondrial dysfunction is related to cell damage and broad age-related diseases, such as degenerative joint disease.25 During mitochondrial damage, mitophagy pathways in normal cells enhance cellular protective effects26 by removing dysfunctional mitochondria.27 Recently, numerous studies have demonstrated that mitophagy plays a role in degenerative joint diseases, including our previous one.9
To protect mitochondrion, researchers paid more attention to the research and development of mitochondria-targeted antioxidants. Mitoquinone (MitoQ), a mitochondria-targeted antioxidant, is more efficient than non-targeted antioxidants in preventing mitochondrial oxidative damage.28 MitoQ has protective functions on various oxidative damage-related diseases.29,30,31 However, the effects of MitoQ on the procession of OA are still unexplored. The evidence proposed that the NRF2 pathway and mitophagy partially mediated the protective function of MitoQ.32,33,34
Based on these results, we supposed that MitoQ delays OA progression by activating the NRF2/Parkin axis and attenuating the interleukin (IL)-1β-induced inflammatory response in chondrocytes. In vivo, we used the destabilization of the medial meniscus to induce post-traumatic OA in mice. Here, we aim to clarify whether MitoQ can protect chondrocytes and attenuate OA cartilage degeneration. Moreover, we explored the role of MitoQ in regulating the NRF2 antioxidant pathway, mitophagy, and ferroptosis.
Results
Cytotoxicity of MitoQ
Figure 1A showed the chemical structure of MitoQ. Firstly, the cytotoxicity of MitoQ was measured. After 24 h treatment, we used Cell Counting Kit-8 (CCK-8) to detect the toxicity of MitoQ on chondrocytes at concentrations of 0.125, 0.25, 0.5, 1, and 2 μM, with half maximal inhibitory concentration (IC50) values of 1.094 μM (Figure 1B). Accordingly, chondrocytes were treated with 0.125, 0.25, and 0.5 μM MitoQ in this experiment. Then, we used toluidine blue staining to measure the morphology of chondrocytes. Figure 1C showed that the chondrocytes exposed to IL-1β and MitoQ displayed less loss of coloration compared with the IL-1β group. Then, we used 5-ethynyl-2’-deoxyuridine (EdU) staining to assess the chondrocyte proliferation activity after being treated with MitoQ. We observed little difference in EdU-positive staining between the control group and chondrocytes exposed to MitoQ (0.25 and 0.5 μM) for 24 h (Figures 1D and 1E).
Figure 1.

Cytotoxicity of MitoQ
(A) Chemical structure of MitoQ.
(B) The CCK-8 results of MitoQ (0.125, 0.25, 0.5, 1, and 2 μM). IC50 = 1.094 μM.
(C–E) Toluidine blue staining on chondrocytes exposed to IL-1β and MitoQ (scale bar: 400 μm) (D and E) EdU staining on chondrocytes (scale bar: 200 μm). Data are represented as mean ± SD, ns = not significant, (n = 3).
MitoQ activated antioxidant system and mitophagy in chondrocytes
In our experiments, the levels of NRF2, heme oxygenase 1 (HO-1), and NAD(P)H:quinone oxidoreductase 1 (NQO1) were utilized as markers of the NRF2 antioxidant pathway, and the protein expression of Parkin and PINK1 was utilized as markers of mitophagy. In the study, the expression of NRF2 slightly increased in chondrocytes exposed to IL-1β. Compared with the control group, IL-1β promoted an increase in the expression and nuclear translocation of NRF2 in chondrocytes (Figure 2A). IL-1β increased HO-1 and NQO1 expression, and MitoQ further improved the level of NRF2, HO-1, and NQO1 (Figures 2B and 2C). Figure 2D showed that MitoQ inversed the decreased mRNA of Nrf2 in chondrocytes induced by IL-1β. The immunofluorescence results revealed that NRF2 was found largely in the cytoplasm of normal chondrocytes, while green-stained NRF2 was observed in the chondrocyte nucleus in the IL-1β group and MitoQ further promoted the nuclear localization of NRF2(Figure 2E).
Figure 2.

MitoQ activated antioxidant system and mitophagy in chondrocytes
Chondrocytes were exposed to IL-1β and MitoQ (0.125,0.25, and 0.5 μM) for 24 h.
(A) The chondrocytes were treated with IL-1β for 1, 6, 12, 24, and 48 h, and then whole-cell lysates together with nuclear lysates were prepared for western blot.
(B, C) The levels of NRF2, HO-1, and NQO1.
(D) Nrf2 expression was examined by qRT-PCR.
(E–G) Nuclear translocation of Nrf2 was observed by immunofluorescence (scale bar: 50 μm) (F and G) The levels of Parkin, PINK1, and LC3 II.
(H) Parkin expression was examined by qRT-PCR.
(I) Chondrocytes were stained with Mitotracker (mitochondria marker, red) and Parkin (green) (scale bar: 25 μm). ∗p < 0.05 vs. the control group, ∗∗p < 0.01 vs. the control group, #p < 0.05 vs. the IL-1β treated group, ##p < 0.01 vs. the IL-1β treated group, Data are represented as mean ± SD.
Parkin RBR E3 ubiquitin protein ligase (PRKN)-dependent mitophagy is the main type of mitophagy.9 Next, we exposed chondrocytes to IL-1β to detect PRKN-dependent pathways expression. Compared with the control group, we observed that the levels of Parkin, PTEN induced putative kinase 1 (PINK1), and microtubule-associated protein 1 light chain 3 II (LC3 II) were decreased in chondrocytes exposed to IL-1β, while MitoQ partly reversed the decline (Figures 2F and 2G). We discovered via qRT-PCR that the expression of Parkin decreased in chondrocytes induced by IL-1β, while MitoQ promoted the mRNA of Parkin (Figure 2H). Co-localization of Parkin and Mitotracker in chondrocytes indicated that Parkin localized to the mitochondria following activation (Figure 2I). Therefore, MitoQ activated NRF2 antioxidant pathway and mitophagy in chondrocytes.
MitoQ alleviated the ECM degradation and inflammatory response in chondrocytes
In our experiments, the expressions of SRY-box transcription factor 9 (SOX9), collagen type II alpha 1 (COL2A1), matrix metalloproteinase 13 (MMP13), and MMP3 were used as ECM degradation markers, and inducible nitric oxide synthase (iNOS) was utilized as a marker of inflammation. Compared with the control group, the inflammatory biomarker iNOS was dramatically increased in chondrocytes exposed to IL-1β. MitoQ alleviated the increased expression of iNOS (Figures 3A and 3B). In chondrocytes exposed to IL-1β, western blot and qRT-PCR measured that the expression of COL2A1 and SOX9 reduced, while the production of MMP3 and MMP13 increased. (Figures 3C–3E). The immunofluorescence results of COL2A1 are consistent with western blots (Figure 3F). Compared to the control group, chondrocytes showed less EdU-positive staining when exposed to IL-1β for 24 h. Meanwhile, compared to the IL-1β group, chondrocytes exposed to IL-1β and MitoQ (0.5 μM) increased EdU-positive staining, which indicated that MitoQ recovered the declined chondrocyte proliferation activity induced by IL-1β (Figures 3G and 3H). These results indicated that MitoQ relieved the degradation of ECM and inflammation of chondrocytes.
Figure 3.

MitoQ alleviated the ECM degradation and inflammatory response in chondrocytes
(A and B) iNOS protein expression.
(C, D, and E) The levels of MMP3, MMP13, COL2A1, and SOX9 were detected by western blot and qRT-PCR.
(F) GPX4 expression was observed by immunofluorescence (scale bar: 100 μm).
(G and H) EdU staining on chondrocytes exposed to IL-1β and MitoQ (scale bar: 200 μm). ∗p < 0.05 vs. the control group, ∗∗p < 0.01 vs. the control group, #p < 0.05 vs. the IL-1β treated group, ##p < 0.01 vs. the IL-1β treated group, Data are represented as mean ± SD.
MitoQ alleviated ferroptosis in chondrocytes
In our study, we used the expression of GPX4 as the marker of ferroptosis. Western blot and immunofluorescence results showed that the expression of GPX4 decreased in chondrocytes exposed to IL-1β. Nevertheless, MitoQ recovered the GPX4 expression level (Figures 4A–4C). Moreover, MitoQ treatment could lighten the accumulation of ROS and lipid ROS in chondrocytes exposed to IL-1β (Figures 4D and 4E). These results indicated the inhibition of MitoQ on chondrocyte ferroptosis.
Figure 4.

MitoQ alleviated ferroptosis in chondrocytes
(A, B, and C) The expression of GPX4 was observed by western blot and immunofluorescence (scale bar: 50 μm).
(D) Intracellular ROS level (scale bar: 200 μm).
(E) Intracellular lipid ROS level (scale bar: 200 μm).
(F) Representative FerroOrange staining images of chondrocytes treated with Erastin and MitoQ (scale bar: 200 μm).
(G) The CCK-8 results of Erastin and MitoQ (0.25, 0.5 and 1 μM). ns = not significant, n = 5.
(H and I) EdU staining on chondrocytes exposed to Erastin and MitoQ (scale bar: 200 μm). ∗p < 0.05 vs. the control group, ∗∗p < 0.01 vs. the control group, #p < 0.05 vs. the IL-1β or Erastin treated group, ##p < 0.01 vs. the IL-1β or Erastin treated group, Data are represented as mean ± SD.
Furthermore, our previous studies have confirmed that IL-1β or Erastin-induced chondrocyte ferroptosis plays a role in the progression of OA.8,17 To verify the protective function of MitoQ, we used Erastin to induce ferroptosis in chondrocytes. FerroOrange staining revealed that Erastin increased intracellular iron levels in chondrocytes, promoting ferroptosis. On the other hand, MitoQ inhibited the levels of intracellular divalent iron (Figure 4F). After 24 or 48 h treatment, CCK-8 results indicated that MitoQ recovered the chondrocyte ferroptosis induced by Erastin (Figure 4G). Afterward, we used EdU staining to assess the chondrocyte proliferation activity after being treated with Erastin and MitoQ. Compared to the control group, chondrocytes observed less EdU-positive staining when exposed to Erastin for 24 h. Meanwhile, compared to the IL-1β group, chondrocytes exposed to Erastin and MitoQ (0.5 μM) increased EdU-positive staining (Figures 4H and 4I). These results indicated that MitoQ relieved the ferroptosis of chondrocytes.
NRF2 antioxidant pathway mediated the protection of MitoQ on chondrocytes
Furthermore, we knock down NRF2 with small interfering RNA (siRNA). As mentioned in the previous study, the high knockout efficiency was confirmed by western blotting.35 The results indicated that silencing NRF2 reduced the levels of NRF2, HO-1, and NQO1(Figures 5A and 5B). NRF2 knockdown also inversed the repression of MitoQ on the levels of iNOS, COX2, MMP13, and MMP3 (Figures 5C and 5D) and decreased the GPX4 expression level (Figures 5E and 5F). Of note, silencing NRF2 also reduced the expression of Parkin and PINK1 (Figures 5G and 5H). Together, these results suggested that the NRF2 antioxidant pathway participates in MitoQ-mediated inhibition of the ECM destruction, inflammation, and ferroptosis in chondrocytes exposed to IL-1β. And MitoQ promoted Parkin expression partially via NRF2. Therefore, we demonstrated that MitoQ could promote NRF2 expression, which in turn activated the NRF2 antioxidant pathway and mitophagy, inhibited ferroptosis, and ultimately led to increased anabolism and decreased catabolism in chondrocytes.
Figure 5.

NRF2 antioxidant pathway mediated the protection of MitoQ on chondrocytes
After silencing NRF2, chondrocytes were exposed to IL-1β and MitoQ for 24 h.
(A and B) The chondrocytes whole-cell lysates together with nuclear lysates were prepared for western blot. The levels of NRF2, HO-1, and NQO1.
(C and D) The levels of iNOS, COX2, MMP13, and MMP3.
(E and F) The GPX4 expression.
(G and H) The levels of Parkin and PINK1. ∗p < 0.05 vs. the IL-1β + negative control group, ∗∗p < 0.01 vs. the IL-1β + negative control group, #p < 0.05 vs. the IL-1β + MitoQ + negative control group, ##p < 0.01 vs. the IL-1β + MitoQ + negative control group, Data are represented as mean ± SD.
Parkin regulated the expression of the NRF2 antioxidant pathway through negative feedback
Previous results have shown that NRF2 promoted the expression of Parkin.36,37 Knockdown of Parkin with siRNA revealed the connection between NRF2 and Parkin. The western blot and qRT-PCR results verified the si-Parkin effect (Figures 6A–6C). Then chondrocytes were exposed to IL-1β with negative control or si-Parkin. The results revealed that silencing Parkin decreased the increased iNOS and MMP3 levels in chondrocytes exposed to IL-1β and partially recovered the level of COL2A1 (Figures 6D and 6E). Similarly, MitoQ reversed the inhibition of IL-1β on Col2a1 (Figure 6F). Meanwhile, the western blot and qRT-PCR results indicated that silencing Parkin promoted GPX4 expression (Figures 6G and 6H). These results revealed that the knockdown of Parkin repressed ECM degradation and ferroptosis induced by IL-1β. To explore the molecular mechanisms behind this, we measured the level of the NRF2 pathway. Interestingly, the results indicated that the knockdown of Parkin improved Nrf2 expression and the levels of NRF2, HO-1, and NQO1 (Figures 6I and 6J), which was not consistent with our expectations. Thus, we supposed that Parkin might regulate the NRF2 antioxidant pathway through negative feedback. The decreased expression of NRF2 reduced Parkin, while the decreased Parkin levels promoted the expression of NRF2, and the increased expression of NRF2 in turn promotes GPX4 expression. To further verify this view, Parkin knockdown chondrocytes were exposed to IL-1β and MitoQ. Western blot indicated that MitoQ increased the levels of NRF2, HO-1, and NQO1, and silencing Parkin further promoted the expression of NRF2 antioxidant pathway (Figures 6K and 6L). Figures 6M and 6N revealed that the knockdown of Parkin enhanced the beneficial effect of MitoQ. In short, these results indicated that Parkin regulates the NRF2 antioxidant pathway through negative feedback.
Figure 6.

Parkin regulated the expression of the NRF2 antioxidant pathway through negative feedback
After Parkin knockdown, chondrocytes were exposed to IL-1β and MitoQ for 24 h.
(A) The level of Parkin.
(B and C) The levels of Parkin and PINK1.
(D and E) The protein levels of iNOS, MMP3, and COL2A1.
(F) The mRNA level of Col2a1.
(G and H) The GPX4 level was measured by western blot and qRT-PCR.
(I and J) The protein levels of NRF2, HO-1, and NQO1, and the mRNA level of Nrf2.
(K and L) The levels of NRF2, HO-1, and NQO1 treated by MitoQ with negative control or si-Parkin.
(M and N) The expression of iNOS, MMP13, and COL2A1. ∗p < 0.05 vs. the IL-1β + negative control group, ∗∗p < 0.01 vs. the IL-1β + negative control group, #p < 0.05 vs. the IL-1β + negative control or IL-1β + MitoQ + negative control group, ##p < 0.01 vs. the IL-1β + negative control or IL-1β + MitoQ + negative control group, Data are represented as mean ± SD.
MitoQ reduced cartilage degeneration induced by DMM
To further investigate whether MitoQ could attenuate OA in vivo, destabilized medial meniscus (DMM) was used for mouse post-traumatic OA modeling. Safranin O/fast green staining revealed that injection of MitoQ into the joint lightened cartilage degeneration (Figure 7A). Moreover, Osteoarthritis Research Society International (OARSI) scores were used to estimate OA. Compared to the DMM group, the OARSI score of DMM + MitoQ (0.1 or 1 mg/kg) group was lower (Figure 7B). Immunohistochemical analysis revealed that the expression of COL2A1, NRF2, HO-1, GPX4, and Parkin decreased in the DMM group compared to the sham group, while the number of MMP13-positive cells significantly increased. Conversely, intra-articular injection of MitoQ significantly increased the expression of COL2A1, NRF2, HO-1, GPX4, and Parkin, while decreasing the number of MMP13-positive cells when compared to the DMM group (Figures 7C–7H). More specifically, the 0.1 mg/kg MitoQ group displayed a significant increase in PINK1 expression relative to the DMM group. However, the 1 mg/kg MitoQ group did not exhibit a significant increase in the number of PINK1-positive cells, suggesting that a concentration of 0.1 mg/kg MitoQ for intra-articular injection may be a more favorable choice. In conclusion, MitoQ reduced cartilage degeneration induced by DMM.
Figure 7.

MitoQ attenuated DMM-induced cartilage degeneration
After DMM surgery, MitoQ (0.1 mg/kg and 1 mg/kg) or vehicle was intra-articularly injected into mice’ knee joints twice a week for 8 weeks.
(A and B) Safranin O staining/fast green was used to measure cartilage degeneration. The OARSI scores were used to assess the progression of OA. Scale bar: 200 μm and 100 μm (The sham and the DMM +0.1 mg/kg MitoQ group (n = 8), the DMM and the DMM +1 mg/kg MitoQ group (n = 9)).
(C–H) Immunohistochemistry staining was used to measure COL2A1, NRF2, HO-1, GPX4, Parkin, and PINK1 levels. Quantification of COL2A1-, MMP13-, NRF2- HO-1-, GPX4-, Parkin-, and PINK1-positive staining cells in vivo (scale bar: 100 μm). ∗p < 0.05 vs. the sham group, #p < 0.05 vs. the DMM group, ##p < 0.01 vs. the DMM group, n = 7 or 8. Data are represented as mean ± SD.
Discussion
OA, a common degenerative disease, is related to various factors. ECM homeostasis is important for cartilage health. Disorder of cartilage homeostasis is a key link in the development of OA.38 Oxidative stress and inflammation are tightly associated with the progression of OA.39 IL-1β can destroy the dynamic balance of cartilage by causing inflammatory reactions and ferroptosis of chondrocytes. The redox state of chondrocytes is mainly regulated by NRF2, and the NRF2-mediated cell protection effects are critical to cartilage homeostasis.40
MitoQ has protective functions on many oxidative damage-related diseases.29,30,31 The evidence suggests that the NRF2 signaling pathway and mitophagy partially mediate the protection of MitoQ.32,33,34 In this research, we used OA models in vitro and in vivo to explore the protective function of MitoQ on OA and its mechanism. IL-1β can trigger a strong inflammatory reaction, promote the degradation of ECM,41,42 cause ferroptosis of chondrocytes,8 and affect the normal function of mitophagy.9,43 The results revealed that IL-1β could enhance the inflammatory, ferroptosis, mitophagy, and catabolic processes while inhibiting the anabolism of chondrocytes, which is identical to previous research results. We found that MitoQ treatment attenuated IL-1β-induced degradation of the ECM, accumulation of oxidative stress, inflammation response, and ferroptosis in chondrocytes. Moreover, silencing NRF2 partly eliminated the influence of MitoQ on reducing catabolism and inflammation and promoting anabolism and mitophagy. Our results confirmed that MitoQ alleviated OA progress by activating the NRF2 pathway and mitophagy and suppressed oxidative stress, inflammation, and ferroptosis in chondrocytes.
Zhang et al. reported that, in intestinal epithelial cells, MitoQ plays an antioxidant and anti-inflammatory role in sepsis-associated intestinal barrier damage by promoting the NRF2 pathway.44 For further verification, si-NRF2 was used. The influence of MitoQ on promoting chondrocyte anabolism and mitophagy, and reducing catabolism, inflammation, and ferroptosis, is partially eliminated by NRF2 knockdown. This finding suggested that MitoQ enhanced NRF2 expression to combat oxidative stress, inflammation, and ferroptosis in chondrocytes.
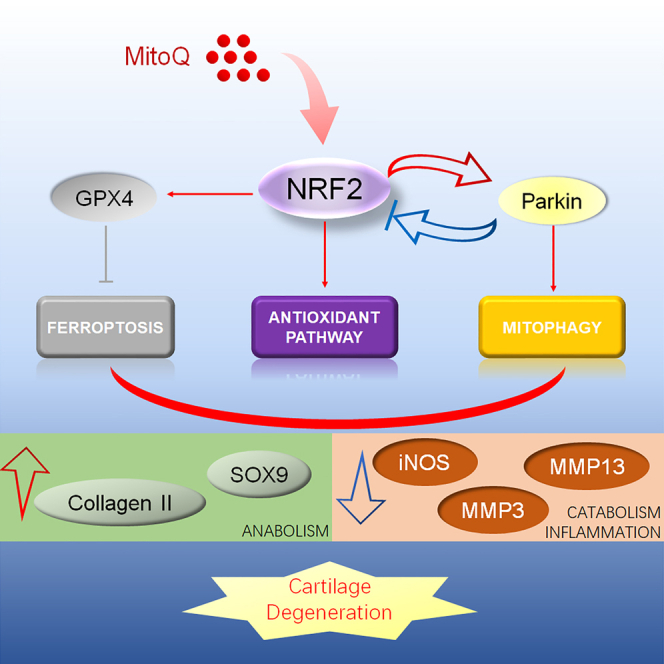
Our research group has reported that mitophagy is tightly associated with OA.9 Therefore, we further studied the function of Parkin in the protection of MitoQ against OA. Recent research demonstrated that MitoQ regulates Parkin expression via NRF2/Keap1 in tubular cells. MitoQ suppresses oxidative stress and apoptosis and activates mitophagy partly via NRF2.32 However, the regulation mechanism between NRF2 and Parkin has not been profoundly researched. In addition, Yun et al. and colleagues discovered that the silencing of PINK1 reversed the effects of ELAV like RNA binding protein 1 (ELAVL1) inhibition on GPX4, suggesting that PINK1 may be involved in regulating the expression of GPX4.45 Yaguang et al. proposed that mitochondrial autophagy may contribute to the degradation of GPX4.46 However, there is currently no report on the mutual regulation mechanism between PARKIN and GPX4. In our study, we discovered that MitoQ reversed the decreased expression of Parkin and PINK1 in chondrocytes exposed to IL-1β. The results were in line with previous studies. Si-Parkin was used and we observed the changes of various indicators through western blot. Surprisingly, silencing Parkin did not reduce the protective effect of MitoQ on OA as expected, but further improved the protective effect on chondrocytes. So we observed the changes in the NRF2 pathway and found that the knockdown of Parkin would promote the expression of the NRF2 pathway. These results sustain our hypothesis that Parkin could promote NRF2 expression through feedback regulation, and the increased expression of NRF2 in turn promotes GPX4 expression, thereby protecting chondrocytes. The results after adding MitoQ further verified this view. In short, by silencing NRF2 and Parkin, respectively, we supplemented the negative feedback regulation mechanism between NRF2 and Parkin (Figure 8). It provides a possible regulatory mechanism for further research.
Figure 8.

Possible regulatory mechanism of MitoQ’s anti-OA function
MitoQ relieved cartilage degeneration by activating the antioxidant system and mitophagy and inhibiting ferroptosis via the NRF2/Parkin axis. Parkin regulated the expression of the NRF2 antioxidant pathway through negative feedback.
To sum up, our research results confirmed the anti-OA function of MitoQ at the cell level and animal level. Additionally, we also found that MitoQ could partially activate NRF2 expression, thereby promoting the NRF2 pathway and mitophagy, inhibiting ferroptosis, and reducing inflammation and ECM degradation. We supplemented the negative feedback regulation between NRF2 and Parkin and provided possible regulatory mechanisms for other studies. These findings indicate MitoQ bid fair has therapeutic effects on OA.
Limitations of the study
There were some limitations in this research that needed further study. First, the specific mechanisms of the negative feedback regulation between NRF2 and Parkin remained unclear. However, further experiment on the mechanisms was an ongoing process. We put forward a hypothesis that Parkin, as an E3 ubiquitin ligase, downregulated the level of NRF2 by the ubiquitin pathway. Thus, silencing Parkin decreased the ubiquitination of NRF2, then increased NRF2 level, and protected chondrocytes from OA. Second, we identified the protective effects of MitoQ on mouse OA. However, the experiments on the protection of MitoQ on other large mammals needed to be further studied.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| GAPDH | Proteintech Group, Wuhan, Hubei, China | 60004-1-Ig; RRID: AB_2107436 |
| Collagen II | Proteintech Group, Wuhan, Hubei, China | 15943-1-AP; RRID: AB_2881147 |
| NRF2 | Proteintech Group, Wuhan, Hubei, China | 16396-1-AP; RRID: AB_2782956 |
| HO-1 | Proteintech Group, Wuhan, Hubei, China | 10701-1-AP; RRID: AB_2118685 |
| Parkin | Proteintech Group, Wuhan, Hubei, China | 66674-1-Ig; RRID: AB_2882028 |
| PINK1 | Proteintech Group, Wuhan, Hubei, China | 23274-1-AP; RRID: AB_2879244 |
| NQO1 | Abcam, Cambridge, UK | #ab80588; RRID: AB_1603750 |
| MMP13 | Abcam, Cambridge, UK | #ab39012; RRID: AB_776416 |
| SOX9 | Abcam, Cambridge, UK | #ab185966; RRID: AB_2728660 |
| GPX4 | Abcam, Cambridge, UK | #ab125066; RRID: AB_10973901 |
| MMP3 | Boster, Wuhan, Hubei, China | #BM4074 |
| iNOS | Cell Signaling Technology, Beverly, MA, USA | #13120; RRID: AB_2687529 |
| LC3 | Cell Signaling Technology, Beverly, MA, USA | #4108; RRID: AB_2137703 |
| COX2 | Cell Signaling Technology, Beverly, MA, USA | #12882; RRID: AB_2571729 |
| HRP Conjugated AffiniPure Goat Anti-mouse IgG | Boster, Wuhan, Hubei, China | BA1050 |
| HRP Conjugated AffiniPure Goat Anti-rabbit IgG | Boster, Wuhan, Hubei, China | BA1054 |
| FITC Conjugated AffiniPure Goat Anti-rabbit IgG | Boster, Wuhan, Hubei, China | BA1105 |
| FITC Conjugated AffiniPure Goat Anti-mouse IgG | Boster, Wuhan, Hubei, China | BA1101 |
| Chemicals, peptides, and recombinant proteins | ||
| Mitoquinone mesylate | Selleckchem, Houston, TX, USA | S8978 |
| Erastin | Selleckchem, Houston, TX, USA | S7242 |
| Recombinant mouse IL-1β protein | R&D Systems, Minneapolis, MN, USA | NP_032387 |
| Trypsin | Biosharp Life Sciences, Hefei, Anhui, China | BL527A |
| Collagenase II | Biosharp Life Sciences, Hefei, Anhui, China | BS164 |
| 4% Paraformaldehyde | Biosharp Life Sciences, Hefei, Anhui, China | BL539A |
| DMEM/F12 medium | Hyclone, Logan, UT, USA | SH30023.01 |
| Fetal bovine serum Excellent | NEWZERUM LIMITED, Upper Riccarton, Christchurch, New Zealand | E500 |
| RNA isolation kit | Omega Bio-tek, USA | R6834-01 |
| cDNA synthesis kit | Yeasen, Shanghai, China | 11141ES60 |
| Mitotracker Deep Red FM | Yeasen, Shanghai, China | 40743ES50 |
| RT-qPCR kit | Yeasen, Shanghai, China | 11201es08 |
| ROS assay kit | Beyotime, Shanghai, China | S0033 |
| Nuclear and Cytoplasmic Protein Extraction Kit | Beyotime, Shanghai, China | P0027 |
| C11-BODIPY Lipid Peroxidation Sensor | Thermo Fisher, Waltham, MA | D3861 |
| RIPA lysis buffer | Boster, Wuhan, Hubei, China | AR0102 |
| Protease inhibitors | Boster, Wuhan, Hubei, China | AR1182 |
| Phosphatase inhibitors | Boster, Wuhan, Hubei, China | AR1183 |
| DAPI reagent | Boster, Wuhan, Hubei, China | AR1176 |
| Cell counting kit-8 (CCK-8) | Boster, Wuhan, Hubei, China | AR1160 |
| FerroOrange | DOJINDO, ShangHai, China | F374 |
| Safranin O Stain Solution | Solarbio Life Sciences, Beijing, China | G1067 |
| Toluidine Blue Stain Solution | Solarbio Life Sciences, Beijing, China | G3660 |
| PVDF membranes | Immobilon, USA | ISEQ00010 |
| SDS-PAGE gels | BioRad, Hercules, CA, USA | 4561036 |
| Experimental models: Organisms/strains | ||
| C57BL/6J mouse | GemPharmatech, Nanjing, China | Strain NO.N000013 |
| Oligonucleotides | ||
| Primer for RT-qPCR, see below table | This paper | N/A |
| Software and algorithms | ||
| ImageJ Version 1.52i | National Institutes of Health | https://www.nih.gov/ |
| GraphPad Prism Version 8 | GraphPad Software | https://www.graphpad.com/ |
| Adobe Photoshop CC Version 19.0 | Adobe | https://www.adobe.com/ |
Primer sequence used in the RT-qPCR experiment
| Sequence | |
|---|---|
| Mmp3 | Forward: 5′-GAAACGGGACAAGTCTGTGGAG-3′ |
| Reverse: 5′-ATGAAAATGAAGGGTCTTCCGGTCC-3′ | |
| Mmp13 | Forward: 5′-GCTGGACTCCCTGTTG-3′ |
| Reverse: 5′-TCGGAGCCTGTCAACT-3′ | |
| Col2a1 | Forward: 5′-GGGAATGTCCTCTGCGATGAC-3′ |
| Reverse: 5′-GAAGGGGA TCTCGGGGTTG-3′ | |
| Gapdh | Forward: 5′-AACATCAAATGGGGTGAGGCC-3′ |
| Reverse: 5′-GTTGTCATGGATGACCTTGGC-3′ | |
| Gpx4 | Forward: 5′-GATGGAGCCCATTCCTGAACC-3′ |
| Reverse: 5′-CCCTGTACTTATCCAGGCAGA-3′ | |
| Nrf2 | Forward: 5′-ACCAAGGGGCACCATATAAAAG-3′ |
| Reverse: 5′-CTTCGCCGAGTTGCACTCA-3′ | |
| Parkin | Forward: 5′-TCCTTCGTCCACTGTTTCACA-3′ |
| Reverse: 5′-GGGCATTGCTCTCAGTCACAT -3′ |
Resource availability
Lead contact
Further information and requests for reagents and resources should be directed to and will be fulfilled by the lead contact, Fengjing Guo (guofjdoc@163.com).
Materials availability
The study did not generate new unique reagents.
Experimental model and study participant details
Animals and the OA models
The Laboratory Animal Center (Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China) approved all animal experimental procedures. (TJH-202112022). Based on the impact of estrogen levels on the development of osteoarthritis in female mice, we conducted our experiments using male mice. 8-week-aged male C57BL/6J mice were used to establish the OA model. The mice were provided by The Experimental Animal Center of Tongji Hospital. According to the previously published protocol,47 1% pentobarbital (35 mg/kg) was injected into the mouse abdominal cavity, then the destabilized medial meniscus (DMM) surgical procedure was performed on the left knee of mice. Thirty-five mice were randomly divided into four groups, the sham group (n = 8), the DMM group (n = 9), the DMM +MitoQ (0.1 mg/kg) group (n = 9), and the DMM + MitoQ (1 mg/kg) group (n = 9). The DMM group and the sham group were intra-articular injected with 10 μL of the vehicle. The DMM +MitoQ group received MitoQ (0.1 or 1 mg/kg) dissolved in 10 μL of a solution twice a week for 8 weeks. After 8 weeks, the joint tissues of each left knee were collected.
Isolation and culture of murine chondrocytes
The Laboratory Animal Center (Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China) approved all animal experimental procedures. C57BL/6J mice (3–5 days old) knee joints were used to separate chondrocytes using a previously described procedure.48 Cartilage was taken out from joints and sheared, then digested with trypsin and collagenase at 37°C. Afterward, DME/F12 medium containing 10% FBS was used to culture chondrocytes. In this study, we use Chondrocytes in the first and second passages.
Method details
Cell counting Kit-8
The assay kit was provided by Boster, China. In brief, 96-well plates were used to culture chondrocytes for 24 h. Subsequently, chondrocytes were exposed to MitoQ (0.125, 0. 25, 0. 5, 1 and 2 μM). After 24 h, CCK-8 solution was used to plates (100 μL per well). Then, the 96-well plates get away from light for 1 h. Measure the absorbance at 450 nm with a microplate reader (Leica Microsystems, Wetzlar, Germany).
Toluidine blue staining
In brief, chondrocytes were transferred to 6-well plate and exposed to IL-1β and MitoQ. After 24 h, chondrocytes were fixed by paraformaldehyde for 10 min. Next, chondrocytes were stained with toluidine blue for 5 min, then chondrocytes were washed with PBS. Morphological characteristics of chondrocytes examined by microscope (Evos Fl Auto, Life Technologies, USA).
EdU staining
The EdU staining kit was purchased from RiboBio (Guangzhou, China). In brief, 10 μmol/L EdU was used to chondrocytes for 2 h. Chondrocytes were fixed with paraformaldehyde. After that, 2 mg/mL glycine was added to chondrocytes. Then, 0.5% Triton X-100 was used for chondrocyte permeabilization. After washing with PBS, a staining solution was added to chondrocytes for 30 min. Then, chondrocytes were washed with 0.5% Triton X-100. Finally, chondrocytes were incubated with Hoechst. Images of the staining were observed by microscope. We repeated the EdU staining three times and selected the same field of vision each time. Then, we divided the numbers of EdU-positive cells by the total numbers of cells present in selected areas.
Quantitative real-time PCR (qRT-PCR)
The expression of related genes was quantified using qRT-PCR. A total RNA extraction kit was purchased from OMEGA, Britain. A first Strand cDNA Synthesis Kit and SYBR Green Real-time PCR Master Mix were purchased from Yeasen, China. Sequences of the primers for the genes of interest are as follows: MMP3 (F) 5′-GAAACGGGACAAGTCTGTGGAG-3′, (R) 5′-ATGAAAATGAAGGGTCTTCCGG TCC-3′. MMP13 (F) 5′-GCTGGACTCCCTGTTG-3′, (R) 5′-TCGGAGCCTGT CAA CT-3′. COL2A1 (F) 5′-GGGAATGTCCTCTGC GATGAC-3′, (R) 5′-GAAGGGGA TCTCGGGGTTG-3′. GAPDH (F) 5′-AACATCAAATGGGGTGAGGCC-3′, (R): 5′-GTTGTCATGGATGACCTTGGC-3′. GPX4 (F) 5′- GATGGAGCCCATTCCTGAAC C-3′, (R) 5′-CCCTGTACTTATCCAGGCAGA-3′. NRF2 (F) 5′-ACCAAGGGGCAC CATATAAAAG-3′, (R) 5′-CTTCGCCGAGTTGCACTCA-3′. Parkin (F) 5′-TCCTTC GTCCACTGTTTCACA -3′, (R) 5′-GGGCATTGCTCTCAGTCACAT -3′.
Western blotting
Chondrocytes were seeded into 6-well plates and cultured for 48 h. The total cellular protein was extracted as before.49The nuclear protein was extracted according to instructions of nuclear and cytoplasmic extraction reagents kit (Beyotime, Beijing, China, p0027). The protein concentrations were determined by Bradford protein-binding assay. Briefly, RIPA lysis buffer (Boster, China, AR0102) was added to chondrocytes for 15 min. Next, the solution was centrifuged at 12,000 rpm and 4°C for 30 min. We used 12% SDS-PAGE gels to separate protein samples. Then, samples were transferred to PVDF membranes (Millipore, Billerica, MA) and blocked by 5% skim milk for 1 h, then incubated with primary antibodies (1:1000) at 4°C for 12 to 16 h. GAPDH (#60004-1-Ig), Collagen Type II (#15943-1-AP), NRF2 (#16396-1-AP), HO-1 (#10701-1-AP), Parkin (#66674-1-Ig) and PINK1 (#23274-1-AP) were purchased from the Proteintech Group (Wuhan, China). Antibodies against NQO1 (#ab80588), MMP13 (#ab39012), SOX9 (#ab185966), and GPX4 (UK, ab125066, diluted 1:5,000) were purchased from Abcam (Cambridge, UK). Antibodies against MMP3 (#BM4074 1:500) were bought from Boster (Wuhan, China). Antibodies against COX2 (#12882), iNOS (#sc-7271), and LC3 (#4108) were purchased from Cell Signaling Technology Inc. (Beverly, MA, USA). Next, TBST was used to wash membranes. Then, membranes were incubated with a horseradish peroxidase-conjugated secondary antibody (1:5000) for 1 h. A chemiluminescent reagent (Boster, China) was added to observe protein bands, and we obtained images with a Bio-Rad scanner (Hercules, CA, USA).
Reactive oxygen species (ROS) and lipid ROS
In brief, we used six well plates to culture chondrocytes. After 24 h, PBS was used to wash chondrocytes. Then chondrocytes were incubated with C11-BODIPY or DCFH-DA for 1 h away from light. Next, PBS was used to wash chondrocytes. Finally, a fluorescence microscope was used to observe images.
FerroOrange staining
In brief, we used six well plates to culture chondrocytes. After 24 h, HBSS was used to wash chondrocytes. Then chondrocytes were incubated with 1 mol/L FerroOrange (dojingo) in HBSS for exactly 30 min at 37°C, 21% oxygen and 5% CO2 and imaged immediately. Finally, a fluorescence microscope was used to observe images.
Immunofluorescence microscopy
We used six-well plates to culture chondrocytes. In brief, chondrocytes were fixed with paraformaldehyde. Next, 0.5% Triton X-100 was used for chondrocyte permeabilization. Then normal goat serum was used to block chondrocytes for 1.5 h. Next, chondrocytes were incubated with primary antibodies against Parkin, GPX4 and COL2A1 staining for 12 to 16 h at 4°C. Then, PBS was used to wash chondrocytes and FITC-conjugated goat anti-rabbit secondary antibody was added to chondrocytes protected from light. Lastly, chondrocytes were stained with DAPI and washed by PBST. A fluorescence microscope was used to observe images.
Co-localization of Parkin and Mitotracker staining was performed. Chondrocytes were treated and subsequently incubated with 200 nmol/L Mitotracker Deep Red FM at 37°C for 30 min. Following this, chondrocytes were washed with PBS. The staining procedure for Parkin was carried out as described earlier.
siRNA
RiboBio (Guangzhou, China) chemically synthesized siRNA targeting mouse NRF2 and Parkin genes and transfected it into chondrocytes. Lipofectamine 3000 was purchased from Thermo Fisher, UT, USA. The Parkin siRNA sequences are as follows: sense strand 5′-GCTTTTTCATCTACTGCAA-3′. The NRF2 siRNA sequences are as follows: sense strand 5′-CGACAGACCCTCCATCTA-3′.
Histological and immunohistochemical staining
We collected left knee joints. Then joints were fixed with paraformaldehyde for 3 days. Knee joint sections were embedded in paraffin wax and cut into thin slices. Next, the specimens were stained with Safranin O/Fast green. The development of OA was assessed by histopathologists using the OARSI histopathology scoring system in a blinded manner.50 After they were deparaffinized and rehydrated, BSA containing 0.1% Triton X-100 was used to block the specimens for 1 h. The specimens were incubated with anti-NRF2, anti-MMP13, anti-COL2A1, or anti-Parkin antibodies, then added secondary antibody, colored with DAB. Finally, the specimens were counterstained with hematoxylin. A fluorescence microscope was used to observe images.
Quantification and statistical analysis
Graph Pad Prism 8.0 (San Diego, CA, USA) was used to analyze all of the data. Data are expressed as the mean ± SD for independent experiments. We used the Student’s t test to analyze between two groups. One-way analysis of variance followed by post hoc comparison with Bonferroni’s test. p-values <0.05 were considered significant. Statistical details can be found in the figure legends and results.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Nos. 82172498).
Author contributions
Each author has contributed sufficiently to the work to take public responsibility for the appropriate parts of this project. Planning and design of the study: LH, KS, and FG. The revision work of the paper has been completed by GW. Data collection: analysis and interpretation: LH, XZ, FL, JX, ZG, KS, GW, HL, and YH. Drafting the article: LH and KS. All authors critically read the manuscript.
Declaration of interests
The authors declare no competing interests.
Inclusion and diversity
We support inclusive, diverse, and equitable conduct of research.
Published: August 15, 2023
Contributor Information
Kai Sun, Email: 1085844308@qq.com.
Fengjing Guo, Email: guofjdoc@163.com.
Data availability statement
All data reported in this paper will be shared by the lead contact upon reasonable request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- 1.Cai X., Yuan S., Zeng Y., Wang C., Yu N., Ding C. New Trends in Pharmacological Treatments for Osteoarthritis. Front. Pharmacol. 2021;12 doi: 10.3389/fphar.2021.645842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peat G., Thomas M.J. Osteoarthritis year in review 2020: epidemiology & therapy. Osteoarthritis Cartilage. 2021;29:180–189. doi: 10.1016/j.joca.2020.10.007. [DOI] [PubMed] [Google Scholar]
- 3.Glyn-Jones S., Palmer A.J.R., Agricola R., Price A.J., Vincent T.L., Weinans H., Carr A.J. Osteoarthritis. Lancet. 2015;386:376–387. doi: 10.1016/S0140-6736(14)60802-3. [DOI] [PubMed] [Google Scholar]
- 4.Liu M., Jin F., Yao X., Zhu Z. Disease burden of osteoarthritis of the knee and hip due to a high body mass index in China and the USA: 1990-2019 findings from the global burden of disease study 2019. BMC Musculoskelet. Disord. 2022;23:63. doi: 10.1186/s12891-022-05027-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mobasheri A. Osteoarthritis year 2012 in review: biomarkers. Osteoarthritis Cartilage. 2012;20:1451–1464. doi: 10.1016/j.joca.2012.07.009. [DOI] [PubMed] [Google Scholar]
- 6.Rai M.F., Sandell L.J. Inflammatory mediators: tracing links between obesity and osteoarthritis. Crit. Rev. Eukaryot. Gene Expr. 2011;21:131–142. doi: 10.1615/critreveukargeneexpr.v21.i2.30. [DOI] [PubMed] [Google Scholar]
- 7.Ansari M.Y., Ahmad N., Haqqi T.M. Oxidative stress and inflammation in osteoarthritis pathogenesis: Role of polyphenols. Biomed. Pharmacother. 2020;129 doi: 10.1016/j.biopha.2020.110452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yao X., Sun K., Yu S., Luo J., Guo J., Lin J., Wang G., Guo Z., Ye Y., Guo F. Chondrocyte ferroptosis contribute to the progression of osteoarthritis. J. Orthop. Translat. 2021;27:33–43. doi: 10.1016/j.jot.2020.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun K., Jing X., Guo J., Yao X., Guo F. Mitophagy in degenerative joint diseases. Autophagy. 2021;17:2082–2092. doi: 10.1080/15548627.2020.1822097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng L., Zhang Z., Sheng P., Mobasheri A. The role of metabolism in chondrocyte dysfunction and the progression of osteoarthritis. Ageing Res. Rev. 2021;66 doi: 10.1016/j.arr.2020.101249. [DOI] [PubMed] [Google Scholar]
- 11.Cuadrado A., Rojo A.I., Wells G., Hayes J.D., Cousin S.P., Rumsey W.L., Attucks O.C., Franklin S., Levonen A.L., Kensler T.W., Dinkova-Kostova A.T. Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat. Rev. Drug Discov. 2019;18:295–317. doi: 10.1038/s41573-018-0008-x. [DOI] [PubMed] [Google Scholar]
- 12.Kumar H., Kim I.S., More S.V., Kim B.W., Choi D.K. Natural product-derived pharmacological modulators of Nrf2/ARE pathway for chronic diseases. Nat. Prod. Rep. 2014;31:109–139. doi: 10.1039/c3np70065h. [DOI] [PubMed] [Google Scholar]
- 13.Peng Y.J., Lu J.W., Lee C.H., Lee H.S., Chu Y.H., Ho Y.J., Liu F.C., Huang C.J., Wu C.C., Wang C.C. Cardamonin Attenuates Inflammation and Oxidative Stress in Interleukin-1beta-Stimulated Osteoarthritis Chondrocyte through the Nrf2 Pathway. Antioxidants. 2021;10 doi: 10.3390/antiox10060862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marchev A.S., Dimitrova P.A., Burns A.J., Kostov R.V., Dinkova-Kostova A.T., Georgiev M.I. Oxidative stress and chronic inflammation in osteoarthritis: can NRF2 counteract these partners in crime? Ann. N. Y. Acad. Sci. 2017;1401:114–135. doi: 10.1111/nyas.13407. [DOI] [PubMed] [Google Scholar]
- 15.Chen Z., Zhong H., Wei J., Lin S., Zong Z., Gong F., Huang X., Sun J., Li P., Lin H., et al. Inhibition of Nrf2/HO-1 signaling leads to increased activation of the NLRP3 inflammasome in osteoarthritis. Arthritis Res. Ther. 2019;21:300. doi: 10.1186/s13075-019-2085-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun K., Luo J., Jing X., Guo J., Yao X., Hao X., Ye Y., Liang S., Lin J., Wang G., Guo F. Astaxanthin protects against osteoarthritis via Nrf2: a guardian of cartilage homeostasis. Aging (Albany NY) 2019;11:10513–10531. doi: 10.18632/aging.102474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo Z., Lin J., Sun K., Guo J., Yao X., Wang G., Hou L., Xu J., Guo J., Guo F. Deferoxamine Alleviates Osteoarthritis by Inhibiting Chondrocyte Ferroptosis and Activating the Nrf2 Pathway. Front. Pharmacol. 2022;13 doi: 10.3389/fphar.2022.791376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun K., Luo J., Jing X., Xiang W., Guo J., Yao X., Liang S., Guo F., Xu T. Hyperoside ameliorates the progression of osteoarthritis: An in vitro and in vivo study. Phytomedicine. 2021;80 doi: 10.1016/j.phymed.2020.153387. [DOI] [PubMed] [Google Scholar]
- 19.Sandell L.J., Aigner T. Articular cartilage and changes in arthritis. An introduction: cell biology of osteoarthritis. Arthritis Res. 2001;3:107–113. doi: 10.1186/ar148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dixon S.J., Lemberg K.M., Lamprecht M.R., Skouta R., Zaitsev E.M., Gleason C.E., Patel D.N., Bauer A.J., Cantley A.M., Yang W.S., et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wei X., Yi X., Zhu X.H., Jiang D.S. Posttranslational Modifications in Ferroptosis. Oxid. Med. Cell. Longev. 2020;2020 doi: 10.1155/2020/8832043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuan H., Li X., Zhang X., Kang R., Tang D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 2016;478:1338–1343. doi: 10.1016/j.bbrc.2016.08.124. [DOI] [PubMed] [Google Scholar]
- 23.Imai H., Matsuoka M., Kumagai T., Sakamoto T., Koumura T. Lipid Peroxidation-Dependent Cell Death Regulated by GPx4 and Ferroptosis. Curr. Top. Microbiol. Immunol. 2017;403:143–170. doi: 10.1007/82_2016_508. [DOI] [PubMed] [Google Scholar]
- 24.Nunnari J., Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giorgi C., Marchi S., Simoes I.C.M., Ren Z., Morciano G., Perrone M., Patalas-Krawczyk P., Borchard S., Jędrak P., Pierzynowska K., et al. Mitochondria and Reactive Oxygen Species in Aging and Age-Related Diseases. Int. Rev. Cell Mol. Biol. 2018;340:209–344. doi: 10.1016/bs.ircmb.2018.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Belousov D.M., Mikhaylenko E.V., Somasundaram S.G., Kirkland C.E., Aliev G. The Dawn of Mitophagy: What Do We Know by Now? Curr. Neuropharmacol. 2021;19:170–192. doi: 10.2174/1570159X18666200522202319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palikaras K., Lionaki E., Tavernarakis N. Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell Biol. 2018;20:1013–1022. doi: 10.1038/s41556-018-0176-2. [DOI] [PubMed] [Google Scholar]
- 28.Rodriguez-Cuenca S., Cochemé H.M., Logan A., Abakumova I., Prime T.A., Rose C., Vidal-Puig A., Smith A.C., Rubinsztein D.C., Fearnley I.M., et al. Consequences of long-term oral administration of the mitochondria-targeted antioxidant MitoQ to wild-type mice. Free Radic. Biol. Med. 2010;48:161–172. doi: 10.1016/j.freeradbiomed.2009.10.039. [DOI] [PubMed] [Google Scholar]
- 29.Mukhopadhyay P., Horváth B., Zsengellėr Z., Bátkai S., Cao Z., Kechrid M., Holovac E., Erdėlyi K., Tanchian G., Liaudet L., et al. Mitochondrial reactive oxygen species generation triggers inflammatory response and tissue injury associated with hepatic ischemia-reperfusion: therapeutic potential of mitochondrially targeted antioxidants. Free Radic. Biol. Med. 2012;53:1123–1138. doi: 10.1016/j.freeradbiomed.2012.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Graham D., Huynh N.N., Hamilton C.A., Beattie E., Smith R.A.J., Cochemé H.M., Murphy M.P., Dominiczak A.F. Mitochondria-targeted antioxidant MitoQ10 improves endothelial function and attenuates cardiac hypertrophy. Hypertension. 2009;54:322–328. doi: 10.1161/HYPERTENSIONAHA.109.130351. [DOI] [PubMed] [Google Scholar]
- 31.Rehman H., Liu Q., Krishnasamy Y., Shi Z., Ramshesh V.K., Haque K., Schnellmann R.G., Murphy M.P., Lemasters J.J., Rockey D.C., Zhong Z. The mitochondria-targeted antioxidant MitoQ attenuates liver fibrosis in mice. Int. J. Physiol. Pathophysiol. Pharmacol. 2016;8:14–27. [PMC free article] [PubMed] [Google Scholar]
- 32.Xiao L., Xu X., Zhang F., Wang M., Xu Y., Tang D., Wang J., Qin Y., Liu Y., Tang C., et al. The mitochondria-targeted antioxidant MitoQ ameliorated tubular injury mediated by mitophagy in diabetic kidney disease via Nrf2/PINK1. Redox Biol. 2017;11:297–311. doi: 10.1016/j.redox.2016.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Snow B.J., Rolfe F.L., Lockhart M.M., Frampton C.M., O'Sullivan J.D., Fung V., Smith R.A.J., Murphy M.P., Taylor K.M., Protect Study Group A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson's disease. Mov. Disord. 2010;25:1670–1674. doi: 10.1002/mds.23148. [DOI] [PubMed] [Google Scholar]
- 34.Xi Y., Feng D., Tao K., Wang R., Shi Y., Qin H., Murphy M.P., Yang Q., Zhao G. MitoQ protects dopaminergic neurons in a 6-OHDA induced PD model by enhancing Mfn2-dependent mitochondrial fusion via activation of PGC-1alpha. Biochim. Biophys. Acta Mol. Basis Dis. 2018;1864:2859–2870. doi: 10.1016/j.bbadis.2018.05.018. [DOI] [PubMed] [Google Scholar]
- 35.Yao X., Zhang Y., Hao J., Duan H.Q., Zhao C.X., Sun C., Li B., Fan B.Y., Wang X., Li W.X., et al. Deferoxamine promotes recovery of traumatic spinal cord injury by inhibiting ferroptosis. Neural Regen. Res. 2019;14:532–541. doi: 10.4103/1673-5374.245480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang L., Liu S., Li J., Tian Y., Xue Y., Liu X. Parkin and Nrf2 prevent oxidative stress-induced apoptosis in intervertebral endplate chondrocytes via inducing mitophagy and anti-oxidant defenses. Life Sci. 2020;243 doi: 10.1016/j.lfs.2019.117244. [DOI] [PubMed] [Google Scholar]
- 37.Gumeni S., Papanagnou E.D., Manola M.S., Trougakos I.P. Nrf2 activation induces mitophagy and reverses Parkin/Pink1 knock down-mediated neuronal and muscle degeneration phenotypes. Cell Death Dis. 2021;12:671. doi: 10.1038/s41419-021-03952-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mobasheri A., Rayman M.P., Gualillo O., Sellam J., van der Kraan P., Fearon U. The role of metabolism in the pathogenesis of osteoarthritis. Nat. Rev. Rheumatol. 2017;13:302–311. doi: 10.1038/nrrheum.2017.50. [DOI] [PubMed] [Google Scholar]
- 39.Sellam J., Berenbaum F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat. Rev. Rheumatol. 2010;6:625–635. doi: 10.1038/nrrheum.2010.159. [DOI] [PubMed] [Google Scholar]
- 40.Khan N.M., Ahmad I., Haqqi T.M. Nrf2/ARE pathway attenuates oxidative and apoptotic response in human osteoarthritis chondrocytes by activating ERK1/2/ELK1-P70S6K-P90RSK signaling axis. Free Radic. Biol. Med. 2018;116:159–171. doi: 10.1016/j.freeradbiomed.2018.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang X., Xi Y., Mao Z., Chu X., Zhang R., Ma X., Ni B., Cheng H., You H. Vanillic acid attenuates cartilage degeneration by regulating the MAPK and PI3K/AKT/NF-kappaB pathways. Eur. J. Pharmacol. 2019;859 doi: 10.1016/j.ejphar.2019.172481. [DOI] [PubMed] [Google Scholar]
- 42.Rousset F., Hazane-Puch F., Pinosa C., Nguyen M.V.C., Grange L., Soldini A., Rubens-Duval B., Dupuy C., Morel F., Lardy B. IL-1beta mediates MMP secretion and IL-1beta neosynthesis via upregulation of p22(phox) and NOX4 activity in human articular chondrocytes. Osteoarthritis Cartilage. 2015;23:1972–1980. doi: 10.1016/j.joca.2015.02.167. [DOI] [PubMed] [Google Scholar]
- 43.Zhou R., Yazdi A.S., Menu P., Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 44.Zhang S., Zhou Q., Li Y., Zhang Y., Wu Y. MitoQ Modulates Lipopolysaccharide-Induced Intestinal Barrier Dysfunction via Regulating Nrf2 Signaling. Mediators Inflamm. 2020;2020 doi: 10.1155/2020/3276148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Du Y., Zhang R., Zhang G., Wu H., Zhan S., Bu N. Downregulation of ELAVL1 attenuates ferroptosis-induced neuronal impairment in rats with cerebral ischemia/reperfusion via reducing DNMT3B-dependent PINK1 methylation. Metab. Brain Dis. 2022;37:2763–2775. doi: 10.1007/s11011-022-01080-8. [DOI] [PubMed] [Google Scholar]
- 46.Bi Y., Liu S., Qin X., Abudureyimu M., Wang L., Zou R., Ajoolabady A., Zhang W., Peng H., Ren J., Zhang Y. FUNDC1 interacts with GPx4 to govern hepatic ferroptosis and fibrotic injury through a mitophagy-dependent manner. J. Adv. Res. 2023 doi: 10.1016/j.jare.2023.02.012. In Press. [DOI] [PubMed] [Google Scholar]
- 47.Ma H.L., Blanchet T.J., Peluso D., Hopkins B., Morris E.A., Glasson S.S. Osteoarthritis severity is sex dependent in a surgical mouse model. Osteoarthritis Cartilage. 2007;15:695–700. doi: 10.1016/j.joca.2006.11.005. [DOI] [PubMed] [Google Scholar]
- 48.Liang S., Lv Z.T., Zhang J.M., Wang Y.T., Dong Y.H., Wang Z.G., Chen K., Cheng P., Yang Q., Guo F.J., et al. Necrostatin-1 Attenuates Trauma-Induced Mouse Osteoarthritis and IL-1beta Induced Apoptosis via HMGB1/TLR4/SDF-1 in Primary Mouse Chondrocytes. Front. Pharmacol. 2018;9:1378. doi: 10.3389/fphar.2018.01378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yao X., Jing X., Guo J., Sun K., Deng Y., Zhang Y., Guo F., Ye Y. Icariin Protects Bone Marrow Mesenchymal Stem Cells Against Iron Overload Induced Dysfunction Through Mitochondrial Fusion and Fission, PI3K/AKT/mTOR and MAPK Pathways. Front. Pharmacol. 2019;10:163. doi: 10.3389/fphar.2019.00163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Glasson S.S., Chambers M.G., Van Den Berg W.B., Little C.B. The OARSI histopathology initiative - recommendations for histological assessments of osteoarthritis in the mouse. Osteoarthritis Cartilage. 2010;18:S17–S23. doi: 10.1016/j.joca.2010.05.025. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon reasonable request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.