

Significance
A significant fraction of oncogenic proteins is localized intracellularly but could hardly be utilized for targeted cancer treatment by conventional targeting agents such as antibody-based therapeutics. Furthermore, most of these intracellular targets are “nonlethal,” in that inhibiting their activities is also insufficient for the treatment. Using intracellular nonlethal aldehyde dehydrogenase 1A1 as an example, we transform intracellular targets into targetable cell surface artificial markers for the “lethal” treatment of the cancer stem-like cells enabled by de novo design of selective cell labeling agents and click chemistry-mediated toxin delivery. This technique is transferable to other unique intracellular targets and holds promise for phenotypic identification of cancer intracellular markers both in vitro and in vivo.
Keywords: cancer targeting, metabolic glycoengineering, click chemistry, cancer stem cell, enzymes
Abstract
Inhibition of overexpressed enzymes is among the most promising approaches for targeted cancer treatment. However, many cancer-expressed enzymes are “nonlethal,” in that the inhibition of the enzymes’ activity is insufficient to kill cancer cells. Conventional antibody-based therapeutics can mediate efficient treatment by targeting extracellular nonlethal targets but can hardly target intracellular enzymes. Herein, we report a cancer targeting and treatment strategy to utilize intracellular nonlethal enzymes through a combination of selective cancer stem-like cell (CSC) labeling and Click chemistry-mediated drug delivery. A de novo designed compound, AAMCHO [N-(3,4,6-triacetyl- N-azidoacetylmannosamine)-cis-2-ethyl-3-formylacrylamideglycoside], selectively labeled cancer CSCs in vitro and in vivo through enzymatic oxidation by intracellular aldehyde dehydrogenase 1A1. Notably, azide labeling is more efficient in identifying tumorigenic cell populations than endogenous markers such as CD44. A dibenzocyclooctyne (DBCO)-toxin conjugate, DBCO-MMAE (Monomethylauristatin E), could next target the labeled CSCs in vivo via bioorthogonal Click reaction to achieve excellent anticancer efficacy against a series of tumor models, including orthotopic xenograft, drug-resistant tumor, and lung metastasis with low toxicity. A 5/7 complete remission was observed after single-cycle treatment of an advanced triple-negative breast cancer xenograft (~500 mm3).
Inhibition of overexpressed enzymes is among the most promising approaches for targeted cancer treatment. However, over 80% of cancer-expressed enzymes are “nonlethal,” in that inhibiting the enzymes’ activity is insufficient to kill cancer cells (1). Nonlethal proteins on the cell surface could be targeted by antibody-based therapeutics to kill the cancer cells (2), such as antibody-drug conjugates (ADCs) (3) or bispecific T cell engagers (4) via the engagement of toxins or immune effector cells, respectively. For example, Trop-2 is a widely expressed marker on the cancer cell surface, and direct inhibition of Trop-2 is insufficient to block tumor growth (5). In comparison, sacituzumab govitecan, an ADC for Trop-2, has shown promising clinical results in treating advanced triple-negative breast cancers (TNBCs) and was recently approved by the U.S. Food and Drug Administration (6). However, over 75% of human oncogenes will translate into intracellular markers (7), which cannot be targeted by antibody-based therapeutics due to their poor permeability. It is thus challenging but highly desired to develop strategies to target nonlethal intracellular proteins for anticancer treatment. To overcome this obstacle, using oncogenic intracellular aldehyde dehydrogenase 1A1 (ALDH1A1) as an example, we report the techniques for transforming intracellular nonlethal enzymes into targetable cell surface artificial markers for the “lethal” targeting of the cancer cells.
ALDH1A1 is an evolutionarily conserved intracellular enzyme that is generally overexpressed by tumorigenic cancer-stem-like cells (CSCs), a cancer cell subpopulation capable of self-renewal and differentiation, and contributes to drug resistance, metastasis, and relapse (8). As an oncogenic factor, ALDH1A1 plays important roles in the promotion of DNA repair and induction of drug resistance (9). Overexpression of ALDH1A1 in breast (10), ovarian (11), prostate (12), colon (13), and lung cancer CSCs (14) is correlated with a poor prognosis. However, little effort has been reported to target ALDH1A1 for in vivo CSC identification and cancer treatment due to several challenges. First, ALDH1A1 is an intracellular nonlethal enzyme, rendering direct inhibition of the enzyme activity insufficient to kill cancer cells effectively (15, 16). Second, humans have over 18 ALDH isoforms, many of which share similar substrate scope with ALDH1A1 (17). It is therefore challenging to selectively target ALDH1A1 while sparing other ALDH isoforms commonly found in normal tissues to avoid side effects. Cancer therapeutics that fail to ablate CSCs will inadvertently lead to the enrichment of CSCs in the tumor, contributing to aggressive and drug-resistant phenotypes during the relapse (18–20). Thus, there is a consensus that targeted therapeutics capable of ablating CSC could contribute to the more effective treatment of cancers (21). Extensive efforts have been made to identify CSC-specific markers for targeting (22). For example, CD44 is overexpressed by breast cancer, colon cancer, and glioblastoma CSCs but not by melanoma and lung cancer CSCs (23). CD34 can be used to identify and target stem-like cells in leukemia but not solid tumors (24). Furthermore, the low density (104 to 106 per cell) of such markers also limited the development of effective therapeutics (25).
Given the lack of an efficient approach to target CSCs, in this study, we leveraged an otherwise untargetable intracellular enzyme, ALDH1A1, to target CSCs. Specifically, we do novo design an unnatural azide-sugar N-(3,4,6-triacetyl-N-azidoacetylmannosamine)-cis-2-ethyl-3-formylacrylamide glycoside (AAMCHO) that can be selectively activated by ALDH1A1 and label CSCs with azide groups as an artificial CSC marker with high density through the endogenous metabolic pathway (Fig. 1). First, we verify that ALDH1A1-responsive AAMCHO can selectively label CSCs with azide groups in vitro and in vivo. More importantly, we demonstrated that AAMCHO-labeling is a more precise method to sort out cancer cells with higher self-renewal tendency and tumorigenic properties than conventional stem-like cell markers such as CD44. We next showed that AAMCHO coupled with the conjugation of dibenzocyclooctyne (DBCO)-drugs via bioorthogonal click chemistry in situ significantly improved antitumor efficacy against aggressive TNBCs, metastasis, and drug-resistant tumors in vivo. In an advanced-stage mice TNBC xenograft model in which treatment started at ~500 mm3 tumors in volume, 5 out of 7 mice exhibited complete remission. This platform presents an example of converting nonlethal enzymes into lethal targetable cell markers for cancer treatment and is transferable to other unique cancer intracellular enzymes.
Fig. 1.
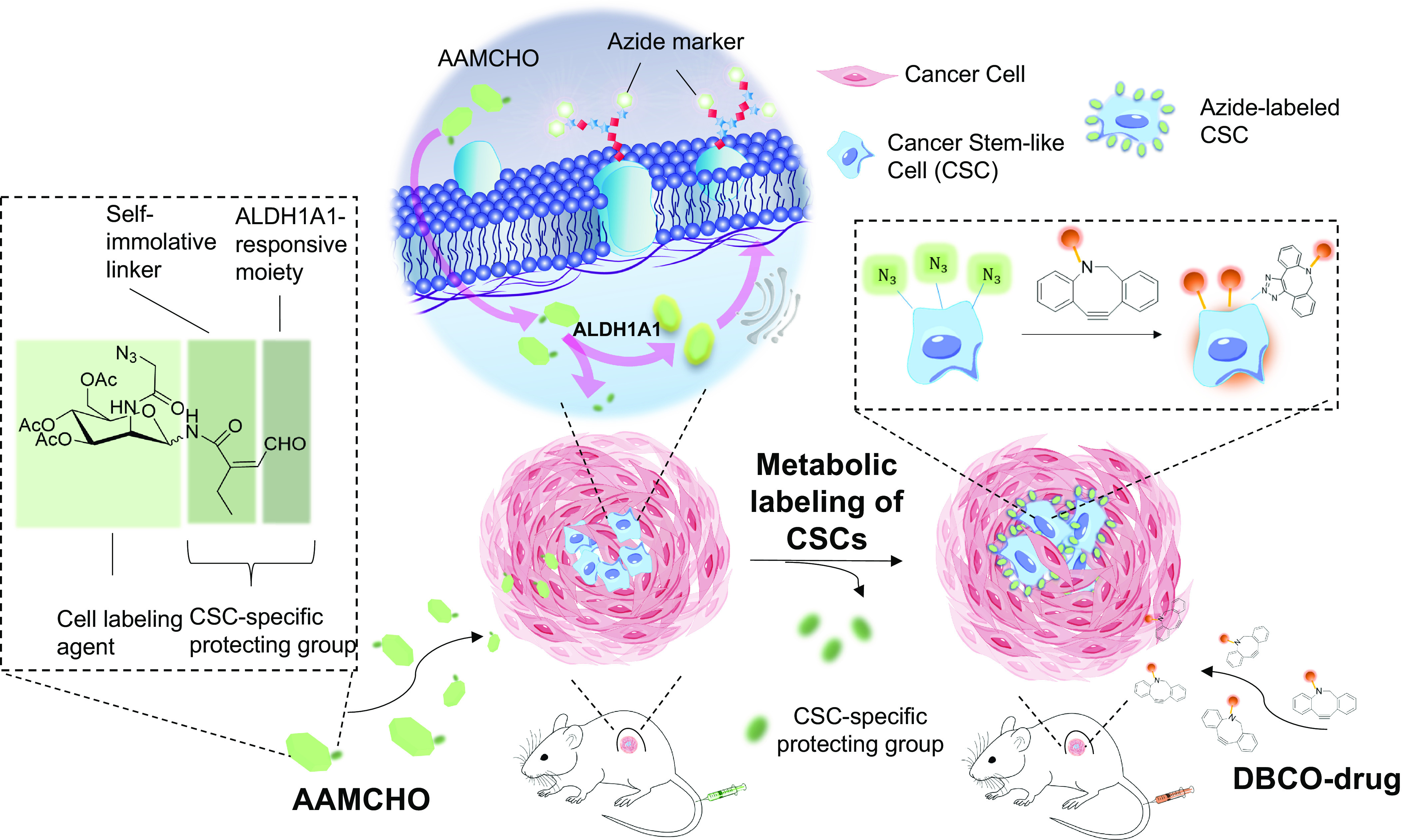
Schematic illustration of metabolic labeling of CSCs with azide groups and subsequent targeting of therapeutics via bioorthogonal click chemistry. AAMCHO, which is composed of azide–mannose, a CSC-specific protecting group, and with a self-immolating linker, can preferentially label CSCs with azide groups in vitro and in vivo. These azide-labeled CSCs can then mediate targeted conjugation of DBCO-drugs via click chemistry.
Results
Design, Synthesis, and Characterization of ALDH1A1-Responsive AAMCHO.
ALDH1A1 catalyzes the oxidization of alkene-conjugated aldehyde, as exemplified by retinaldehyde, 50 to 100 times faster than other ALDH isoforms, such as mitochondrial aldehyde dehydrogenase-2 (ALDH2) (26, 27). Given the lack of prodrug decaging design specifically for ALDH1A1 and challenges to avoid cross-reactivity with other ALDH isoforms, we combined the ALDH1A1 oxidation of alkene-conjugated aldehyde with a self-immolative moiety, maleic monoamide (Fig. 2A) (28). Using this moiety, we rationally designed caged azide-sugar, AAMCHO, as a cell labeling agent that is stable in ambient biological environment but reactivable in the presence of ALDH1A1.
Fig. 2.

Design and characterizations of ALDH1A1-responsive cell labeling agent, AAMCHO. (A) Metabolic glycoengineering process of ALDH1A1-responsive AAMCHO. Upon entering the cell, AAMCHO can be hydrolyzed into AM-CHO by esterase and then be degraded into AM-COOH in the presence of ALDH1A1, followed by the release of 2-ethyl maleic anhydride to yield AM-NH2. The hydrolysis of AM-NH2 will yield AM-OH, which can then be converted to azide-sialic acids, conjugated to proteins, and express azide groups on the cell surface. (B) Degradation kinetics of AAMCHO in the presence of ALDH1A1. (C) AAMCHO degradation after incubation with various ALDH isoforms for 30 min. Statistical analysis was conducted to compare with the control group. (D) AAMCHO degradation after incubation with various analytes for 30 min. Analytes were tested at a concentration of 100 μM. Statistical analysis was conducted to compare with the control group. (E) AAMCHO label cells with azide group in an ALDH1A1-dependent manner. Azide-labeled cells can be probed with DBCO-Cy5 via click chemistry. (F) Mean Cy5 fluorescence intensity of K562 cells treated with azide-sugars for 3 d in the presence or absence of ALDH1A1 siRNA. Azide groups were probed with DBCO-Cy5. (G) Western blot analysis of K562 cells treated with azide-sugars for 3 d. Disulfiram was used as an inhibitor of ALDH1A1. Azide-modified glycoproteins were biotinylated by incubating with DBCO-PEG4-biotin and then detected by streptavidin-horseradish peroxidase conjugate. All the numerical data are presented as mean ± SD (0.01 < *P ≤ 0.05; 0.001 < **P ≤ 0.01; 0.0001 < ***P ≤ 0.001, ****P ≤ 0.0001).
The synthetic route of AAMCHO is shown in SI Appendix, Scheme S1. The cell labeling process of AAMCHO is illustrated in Fig. 2A. AAMCHO first hydrolyzes into AM-CHO by intracellular esterase upon cell internalization (step 1); enzymatic conversion of the aldehyde group to the carboxylate group by intracellular ALDH1A1 (step 2) enables the resultant maleic monoamide to spontaneously self-cyclize and releases C1 amine-substituted azido-sugar (AM-NH2) with an anhydride by-product (step 3) (28). The unstable aminal group of AM-NH2 then undergoes rapid hydrolysis to afford metabolically active sugar, AM (step 4). AM could be further processed through a series of cellular biosynthetic pathways (steps 6 to 9) to express the azide group on the cell surface glycoproteins. In contrast, in cells without ALDH1A1, C1-caged AM-CHO could not undergo hemiacetal-aldehyde isomerization through step 6′ and express azide groups on the cell surface (SI Appendix, Fig. S1) (29). We first verified the selectivity of ALDH1A1-catalyzed reaction of the aldehyde moiety with a fluorogenic probe, Naph-CHO, which shares the same ALDH1A1-responsive moiety as AAMCHO (SI Appendix, Figs. S2 and S3). Naph-CHO could release the fluorescent product Naph-NH2 ex vivo (SI Appendix, Fig. S2) or label ALDH1A1Hi K562 human chronic myeloid leukemia cells efficiently (SI Appendix, Fig. S3), which was diminished by the addition of ALDH1A1 inhibitor N, N-diethylamino-benzaldehyde (DEAB), or disulfiram (30).
To elucidate the release mechanism of AAMCHO, we incubated AAMCHO with recombinant ALDH1A1. AAMCHO was rapidly degraded in the presence of ALDH1A1. The formation of AM and 2-ethyl maleic anhydride could be detected and quantified by High-Performance Liquid Chromatography (HPLC)-UV and LC-MS, respectively (Fig. 2B and SI Appendix, Fig. S4). ALDH1A1 inhibitor DEAB could significantly slow down the degradation of AAMCHO (Fig. 2C), while incubation with other ALDH isoforms including ALDH2, ALDH1A3, ALDH4A1, and ALDH5A1 resulted in limited degradation of AAMCHO (Fig. 2C). The potential cross-reactivity of AAMCHO toward common biological molecules including oxidants and thiols was also studied, and none of the tested molecules resulted in significant degradation of AAMCHO (Fig. 2D).
AAMCHO-Mediated Metabolic Labeling of ALDH1A1Hi Cells In Vitro.
We next studied whether AAMCHO can label cancer cells in an ALDH1Al-dependent manner in vitro. ALDH1A1Hi K562 cancer cells were treated with AAMCHO for 3 d, followed by detecting cell-surface azide groups with DBCO-Cy5 through azide-DBCO Click reaction (Fig. 2E). Ac-N-AAM with C1 caged by a nondegradable moiety was used as a negative control (Fig. 2E and SI Appendix, Scheme S2). AAMCHO and Ac-N-AAM showed minimal cytotoxicity against K562 cells (SI Appendix, Fig. S5). Compared to cells treated with Ac-N-AAM, cells treated with AAMCHO or Ac4ManAz showed significantly enhanced Cy5 fluorescence intensity (SI Appendix, Fig. S6 A and B), indicating the successful labeling of cells with azide groups. The addition of ALDH1A1 siRNA that knocks down ALDH1A1 in K562 cells resulted in a significantly reduced azide labeling (Fig. 2F and SI Appendix, Fig. S6C), substantiating the ALDH1A1-dependent labeling activity of AAMCHO. Western blot analysis also confirmed successful azide labeling of glycoproteins by AAMCHO, and the labeling efficiency was significantly reduced by disulfiram, a clinically relevant ALDH1A1 inhibitor (Fig. 2G) (31). We then evaluated the labeling of K562 by AAMCHO under hypoxia conditions, which is common in the microenvironment of advanced-stage tumor that is often resistant to chemotherapeutics (32). It has also been proved that an increased population of cells overexpress ALDH1A1 under hypoxic conditions (33). After being incubated with AAMCHO under hypoxic or normoxic conditions for 72 h, the azide-positive population of K562 cells increased from 2% in the normoxia group to 18% in the hypoxia group (SI Appendix, Fig. S6 D and E). Notably, most azide-positive cells were CD34-positive, a stem-like cell marker for K562 cells (SI Appendix, Fig. S6 D and E) (24). In contrast, the impact of the hypoxic condition on the labeling activity of a nonselective sugar, Ac4ManAz, was negligible (SI Appendix, Fig. S6F). The addition of ALDH1A1 siRNA significantly decreased the labeling effect of AAMCHO of K562 cells under hypoxic conditions (SI Appendix, Fig. S6G). HEK293T, an ALDH1A1-negative cell line (34), showed minimal azide labeling by AAMCHO (SI Appendix, Fig. S7 A and B). These experiments demonstrated the high selectivity of the ALDH1A1-dependent cell labeling of AAMCHO.
AAMCHO-Mediated Metabolic Selective Labeling of CSCs In Vitro.
We next studied whether AAMCHO can metabolically label cancer stem-like cells of tumor spheroids with azide in vitro. We first tested AAMCHO-mediated metabolic labeling of MDA-MB-231 TNBC cells under hypoxia conditions in single-layer culture, where the azide signals were well colocalized with CD44+, a marker for stem-like cells (Fig. 3A). We then enriched the CSCs population of MDA-MB-231 by growing cells in mammosphere in low-serum medium on nonadherent plates (35, 36). As expected, the ALDH1A1 expression of MDA-MB-231 cells was up-regulated in the spheroids than that in single-layer culture (SI Appendix, Fig. S8A). AAMCHO could selectively label CSC spheres with azides compared to non-CSC spheres (Fig. 3B), while nonselective sugar, Ac4ManAz, did not show any labeling selectivity toward CSC spheres (Fig. 3B). Furthermore, over 85% of CD44+ stem-like cells are labeled with azide groups, while only ~1% of azide-labeled cells are CD44- populations (SI Appendix, Fig. S8C). These results indicate that AAMCHO can selectively label cancer cells with established stem-like markers. To verify whether the cell labeling of AAMCHO reflects the differentiation trend of the CSCs, CSC spheres were differentiated by incubating with 10% FBS for a different time. It has been reported that supplementing FBS into the spheroid culture can induce the differentiation of CSCs thus lowering the “stemness” of the cells (33). At 0, 2, 6, 12, and 24 h post-FBS addition, AAMCHO was added, and the cells were incubated for another 48 h, followed by the detection of cell-surface azide groups via DBCO-Cy5. Confocal Laser Scanning Microscopy (CLSM) imaging showed a significantly enhanced Cy5 signal, i.e., azide expression, for CSC spheres without FBS induction in comparison to spheres inducted with FBS for 24 h (Fig. 3C). Quantitative analysis confirmed that the level of azide expression decreased to baseline level after FBS induction of differentiation (Fig. 3 D and E). The azide-labeled cells were positive for CD44, indicating their stem-like phenotypes (SI Appendix, Fig. S8B).
Fig. 3.

AAMCHO labels CSCs in vitro, and the labeled cell populations exhibit stemness. (A) CSLM image of MDA-MB-231 cells treated with azide-sugars for 72 h and stained with DBCO-Cy5 and Fluorescein Isothiocyanate (FITC) conjugated anti-CD44. Cell nuclei were stained with Hoechst 33342 (blue). (Scale bar, 20 μm.) (B) Representative flow cytometry histograms of CSCs or non-CSCs of MDA-MBA-231 cells treated with azide-sugars for 48 h and probed with DBCO-Cy5. The schematics of CSC sphere formation and differentiation are shown. (C–E) MDA-MB-231 CSC sphere was formed under serum-free conditions. CSC sphere will be differentiated by FBS. Spheres were incubated with AAMCHO for 48 h and then probed with DBCO-Cy5. (C) CLSM images of MDA-MB-231 CSC spheres before and after differentiation. (Scale bar, 50 μm.) (D) Representative Cy5 fluorescence histograms of MDA-MB-231 spheres at a series of differentiation times. (E) Quantification of azide labeling of MDA-MBA-231 spheres by AAMCHO. Statistical comparison was conducted between the AAMCHO group and the Ac-N-AAM group at each time point. (F–H) Single-cell suspension was isolated from MDA-MB-231 xenograft, labeled with AAMCHO + DBCO-Cy5 and FITC conjugated CD44 antibody. Three populations were sorted and characterized by in vitro sphere-forming assay or inoculated subcutaneously into mice for tumorigenesis test. (F) Schematics of the experimental design. (G) Sphere-forming assays of different populations. Spheres were counted on day 7 (n = 6). (H) Tumorigenesis test: tumor volume of MDA-MB-231 xenografts derived from different populations over time. 10 k cells were injected subcutaneously into the flank. Statistical comparison on day 27 is shown. All the numerical data are presented as mean ± SD (0.01 <*P ≤ 0.05; 0.001 <**P ≤ 0.01; 0.0001 <***P ≤ 0.001, ****P ≤ 0.0001).
To study whether AAMCHO-labeled cells exhibit self-renewal properties and tumorigenicity of stem-like cancer cells, we sorted an azide+ subpopulation (top 15% azide-positive cells) and an azide- subpopulation of CSC spheres for sphere-forming assay (SI Appendix, Fig. S8D) (37). Azide+ subpopulations formed approximately 20-fold more spheres than azide- subpopulations (SI Appendix, Fig. S8E). The tumorigenic properties of azide+ and azide− subpopulations were further tested in vivo (SI Appendix, Fig. S8F). 10k isolated azide+ or azide− MDA-MB-231 cells after treatment with AAMCHO were injected into the mammary fat pat of athymic nude mice, and the orthotopic tumor growth was monitored. Compared with the azide− subpopulation and unsorted cells, the azide+ subpopulation resulted in an increased tumor growth rate (SI Appendix, Fig. S8F). To further evaluate the stem-like properties of AAMCHO-labeled cancer cells derived from growing tumors, TNBC cells were isolated from xenograft breast cancer of mice, and three distinct populations (CD44+/azide−, CD44−/azide+, and CD44+/azide+) were sorted (Fig. 3F). In the standard ex vivo sphere-forming assay, CD44+/azide+ cells formed fourfold more spheres than CD44+/azide− counterparts. Notably, CD44−/azide+ populations also formed considerable numbers of spheres (Fig. 3G and SI Appendix, Fig. S8 G and H), suggesting that the azide labeling better reflected the self-renewal property of the cell population than the widely used CD44 marker. In vivo xenograft study verified the high tumorigenic properties of azide-labeled cancer cells by AAMCHO (Fig. 3H).
AAMCHO-Mediated Labeling of CSCs In Vivo.
We next tested whether AAMCHO enabled metabolic labeling of CSCs in vivo. MDA-MB-231 xenografts in athymic nude mice were established via subcutaneous inoculation of CSC and non-CSC spheres into the left and right flanks, respectively (Fig. 4A). CSC spheres yielded larger tumors than non-CSC spheres, as expected (Fig. 4B). From day 7 post tumor inoculation, AAMCHO or Phosphate-Buffered Saline (PBS) or Ac4ManAz was intravenously injected once daily for 3 d, followed by intravenously injection of DBCO-Cy5 on day 10 for the detection of azide-labeled cells (Fig. 4A). At 48 h postinjection of DBCO-Cy5, the inoculated CSCs showed much higher Cy5 fluorescence signal than non-CSCs (Fig. 4C). Ex vivo fluorescence imaging of CSC tumors showed a 2.5-fold increase in Cy5 fluorescence intensity compared to non-CSC tumors (Fig. 4D). The CD44+ cells isolated from CSC tumors showed a higher Cy5 fluorescence intensity than CD44− cells in the AAMCHO group, while CD44+ and CD44− cells in the Ac4ManAz group showed similar Cy5 fluorescence intensity, demonstrating the selective labeling of stem-like cells by AAMCHO (Fig. 4E). To further evaluate the labeling selectivity of AAMCHO, at 24 h postinjection of three doses of AAMCHO or Ac4ManAz, the tissue-bounded azide sialic acid was extracted and quantified by HPLC after derivatization (SI Appendix, Fig. S16A). AAMCHO preferentially labeled CSC tumors compared to non-CSC tumors and other tissues, whereas nonselective Ac4ManAz labeled CSC and non-CSC tumors with azide groups similarly along with evident off-target labeling to other tissues (Fig. 4F).
Fig. 4.

AAMCHO can metabolically label CSCs in vivo for bioorthogonal click chemistry-mediated targeting. (A–F) Mice were subcutaneously inoculated with CSC or non-CSC on the left and right flank, respectively, followed by intravenous injection of AAMCHO on days 7 to 9. DBCO-Cy5 was intravenously injected on day 10. (A) Timeframe of the study. (B) Tumor volume on day 7. (C) Representative in vivo fluorescence image of mice at 24 h postinjection of DBCO-Cy5. The Cy5 fluorescence signal was overlaid over the CT background. (D) Mean Cy5 fluorescence intensity of tumor tissues at 24 h postinjection of DBCO-Cy5. (E) Mean Cy5 fluorescence intensity of CD44- or CD44+ subpopulations, as analyzed by flow cytometry. (F) Quantification of azide-labeled sialic acids (Neu5NAz) in different tissues. (G) CLSM image of MDA-MB-231 tumorsphere after a 3-d labeling with AAMCHO and 1-h incubation with DBCO-Cy5 and FITC conjugated anti-CD44. (Scale bar, 50 μm.) (H) Penetration profiles of DBCO-Cy5 and FITC conjugated anti-CD44 in tumorsphere, fluorescence intensity extracted and averaged from 25 radii. (I–K) After MDA-MB-231 Xenograft reached ~50 mm3, AAMCHO was intravenously injected on days 0, 1, and 2, followed by intravenous injection of DBCO-Cy5 and FITC conjugated anti-CD44 on day 3. Tumors were sectioned on day 5. (I) Schematics of the experimental design. (J) Representative CLSM image of the tumor tissue. The blood vessels were stained with platelet endothelial cell adhesion molecule 1 (PECAM-1) antibody and CFL-555 secondary antibody (yellow). The cell nuclei were stained with Hoechst 33342. (Scale bar, 50 μm.) (K) Microdistribution of DBCO-Cy5 or FITC conjugated anti-CD44 in tumor regions with varied distances from blood vessels. All the numerical data are presented as mean ± SD (0.01 <*P ≤ 0.05; 0.001 <**P ≤ 0.01; 0.0001 <***P ≤ 0.001, ****P ≤ 0.0001).
Tissue penetration is a critical attribute affecting the effectiveness of anticancer therapeutics in vivo. The aberrant vasculatures in the dense tumor microenvironment around CSCs make it hard for large molecules, such as antibodies, to penetrate as deeply as small molecules (38). We compared the penetration depth of DBCO-Cy5 and anti-CD44 antibodies toward azide-labeled CSCs. In AAMCHO-treated tumor spheres, DBCO-Cy5 showed a deeper penetration than FITC-conjugated anti-CD44 (Fig. 4 G and H and SI Appendix, Fig. S9). Compared to antibodies which are only located at the barriers of the tumorspheres, the DBCO-Cy5 signal is a better indicator of the heterogeneous distribution of CSC populations in the inner area of tumorspheres. To study the in vivo microdistribution, AAMCHO was intravenously injected into nude mice bearing orthotopic MDA-MB-231 tumor once daily for 3 d, and FITC-conjugated anti-CD44 and DBCO-Cy5 were intravenously injected into mice 24 h after the last AAMCHO injection (Fig. 4I). At 24 h postinjection, FITC-conjugated anti-CD44 showed a heterogeneous, perivascular distribution, with minimal penetration to areas distant from blood vessels (Fig. 4J and SI Appendix, Fig. S10), while DBCO-Cy5 existed in a broader area within the tumor microenvironment, including areas distant from blood vessels (Fig. 4K).
Combination of AAMCHO with DBCO-MMAE (Monomethyl Auristatin E) Conjugate for Cancer Treatment In Vitro and In Vivo.
We next studied whether AAMCHO coupled with a DBCO-drug conjugate can enhance antitumor efficacy in vivo via CSC targeting. MMAE is one of the most used toxins in ADCs (39). Here, we designed and synthesized DBCO-MMAE as a trigger-responsive prodrug to target AAMCHO-labeled cancer cells. The synthetic route of DBCO-MMAE is shown in SI Appendix, Schemes S3 and S4. DBCO-MMAE has a cleavable linker, which can release MMAE in response to β-glucuronidase, an enzyme expressed in cancer cells (Fig. 5A and SI Appendix, Fig. S11 A and B) (40). The IC50 value of DBCO-MMAE against MDA-MB-231 cells is 10.5 nM, which is 20-fold less potent than the pristine MMAE (0.56 nM, SI Appendix, Fig. S11C). AAMCHO combination with DBCO-MMAE resulted in improved destruction of MDA-MB-231 tumorspheres compared to DBCO-MMAE alone in vitro (Fig. 5B). To study whether targeted delivery of DBCO-MMAE to azide-labeled cancer cells by AAMCHO injection can improve the antitumor efficacy against TNBC tumors, athymic nude mice bearing orthotopic human breast MDA-MB-231 tumors were dividing into four groups: i) AAMCHO + DBCO-MMAE, ii) DBCO-MMAE, iii) MMAE, and iv) PBS (Fig. 5C). MMAE (0.25 mg/kg) was intraperitoneally injected in group (iii) at its Maximum Tolerated Dose (MTD), while a 32 times higher dose of DBCO-MMAE (8 mg/kg, MTD ~20 mg/kg) was systemically administrated in groups (i) and (ii). AAMCHO + DBCO-MMAE showed improved efficacy compared to DBCO-MMAE or MMAE alone (Fig. 5 D and E and SI Appendix, Fig. S12).
Fig. 5.

AAMCHO-mediated CSC labeling, coupled with DBCO-MMAE, improves antitumor efficacy against orthotopic triple-negative breast cancer and metastasis. (A) Structure and triggered release mechanism of DBCO-MMAE. (B) MDA-MB-231 CSC spheres were incubated with AAMCHO or PBS for 72 h, followed by 24-h incubation with DBCO-MMAE or MMAE. The spheres were incubated for 6 d. Shown are the mean diameter and counts of spheres (n = 3). (C–E) MDA-MB-231 TNBC orthotopic tumors were established by injection of MDA-MB-231 cells into the mammary fat pat of athymic nude mice. When the tumor reached ~100 mm3, AAMCHO was intravenously injected on days 0, 1, and 2, followed by intravenous injection of DBCO-MMAE (8 mg/kg) on day 3. In the MMAE group, MMAE (0.25 mg/kg, MTD) was intraperitoneally injected on day 3 (n = 7). (C) Timeframe of the study. (D) Average tumor volume of each group over the course of the efficacy study. Statistical comparisons on day 28 are shown. (E) Kaplan–Meier plots for all groups. Loss of mice was a result of treatment-related death or euthanasia after the predetermined endpoint was reached. (F–I) 4T1 lung metastases were established in BALB/c mice by i.v. injection of luciferase-engineered 4T1 cells (day 0), and mice were randomly divided into five groups. AAMCHO was i.v. injected once daily on days 2, 3, and 4. DBCO-MMAE (8 mg/kg, i.v.) or MMAE (0.25 mg/kg i.p.) was injected on day 5. Tumor growth was monitored by bioluminescence imaging on days 4, 9, and 13. (F) Timeframe of the study. (G) Representative bioluminescence images of mice on days 4, 9, and 13. (H) Integrated bioluminescence intensity of mice on days 9 and 13. Statistical analysis on day 13 is shown. (I) Average metastatic tumor nodule counts for each group. All the numerical data are presented as mean ± SD (0.01 <*P ≤ 0.05; 0.001 <**P ≤ 0.01; 0.0001 <***P ≤ 0.001, ****P ≤ 0.0001).
Metastases are responsible for the majority of cancer deaths and overall low survival from various types of cancers (41). CSCs are the major cell population that results in the formation of early micrometastasis in distant organs during epithelial–mesenchymal transitions (41). We next tested whether metastatic lesions could be metabolically labeled by AAMCHO (SI Appendix, Fig. S13A). The 4T1 metastatic lung metastatic model was established in BALB/c mice by i.v. injection of 4T1 cells. After administration of AAMCHO intravenously once daily for 3 d, mice were intravenously administrated with DBCO-Cy5 1 d after the last AAMCHO injection to detect azide-labeled cells. At 48 h postinjection, an enhanced Cy5 fluorescence signal in metastatic nodes of lung parenchyma was observed, indicating successful azide labeling of the micrometastases (SI Appendix, Fig. S13B). In contrast, minimal fluorescence was seen in other tissues (SI Appendix, Fig. S13C). Cy5+ cells inside the lung were well colocalized with CD44+ cells with a ~90% colocalization factor (SI Appendix, Fig. S13D), substantiating the successful azide labeling of CSCs. We next investigated whether AAMCHO coupled with DBCO-MMAE could inhibit the growth of 4T1 metastases. BALB/c mice were intravenously injected with luciferase-expressing 4T1 cells on day 0, followed by intravenous injection of AAMCHO once daily for 3 d. Mice were administrated with DBCO-MMAE or MMAE systemically on day 5 (Fig. 5F). All the drug treatment groups resulted in reduced bioluminescent signals compared to the untreated group (Fig. 5G). Compared to DBCO-MMAE, AAMCHO + DBCO-MMAE resulted in a markedly weaker tumor signal (Fig. 5 G and H). Tumor nodule counts of dissected lungs further validated the fewer metastases in the AAMCHO + DBCO-MMAE group compared to the DBCO-MMAE group (23.2 ± 12.0 compared to 89.8 ± 35.9) and decreased percentage of tumor surface area (5.7% ± 4.6% compared to 35.7% ± 11.1%) (Fig. 5I and SI Appendix, Fig. S14). It is noteworthy that DBCO-MMAE, used at a much higher dose (4 mg/kg equivalent MMAE), exhibited lower toxicity than MMAE (0.25 mg/kg), especially for the spleen and liver, as revealed by histology analysis (SI Appendix, Fig. S15). These results demonstrated that AAMCHO-mediated labeling of tumors followed by targeted conjugation of DBCO-MMAE could significantly improve the antitumor efficacy with low toxicity.
To push the limit of the CSC targeting anticancer therapy, we tested the efficacy of an advanced-stage mice tumor model where tumors reached ~500 mm3 in volume and used the DBCO-MMAE at its maximum tolerable dose (20 mg/kg). AAMCHO was intravenously injected on days 0, 1, and 2, followed by an intravenous injection of DBCO-MMAE (20 mg/kg) on day 3 (Fig. 6A). AAMCHO + DBCO-MMAE exhibited superior efficacy against the large immune-compromised TNBC tumors after single-cycle treatment (Fig. 6 B and C), with 5 out of 7 tumors developing complete remission (Fig. 6G) which is not observed in other groups (Fig. 6 D–F) (42–44).
Fig. 6.

AAMCHO + DBCO-MMAE exhibit enhanced anticancer efficacy against late-stage and drug-resistant tumors. (A–D) MDA-MB-231 TNBC orthotopic tumors were established by injection of MDA-MB-231 cells into the mammary fat pat of athymic nude mice. When the tumor reached ~500 mm3, AAMCHO was intravenously injected on days 0, 1, and 2, followed by an intravenous injection of DBCO-MMAE (20 mg/kg) on day 3. In the MMAE group, MMAE (0.25 mg/kg) was intraperitoneally injected on day 3. (A) Timeframe of the study. (B) Average tumor volume of mice from each group over the course of the efficacy study (n = 7). Statistical comparisons on day 18 are shown. (C) Kaplan–Meier plots for all groups. (D–G) Individual tumor volume curve for each group. (H) ALDH1A1 expression in drug-resistant cancer cell lines A549-R and LS174T-R. Regular A549 and LS174T cells were used as controls. (I) Representative flow cytometry plot of the azide group and CD44 expression of LS174T-R cells labeled with AAMCHO. (J) Quantification of azide–sialic acids in LS174T-R and regular LS174T cells after labeling with AAMCHO. (K–M) Multidrug-resistant LS174T-R xenograft tumors were established by inoculation of LS174T-R cells into the flank of athymic nude mice. When the tumor reached ~200 mm3, AAMCHO was intravenously injected on days 0, 1, and 2, followed by an intravenous injection of DBCO-MMAE (8 mg/kg) on day 3. (K) Timeframe of the study. (L) Average tumor volume of mice from each group over the course of the efficacy study (n = 7). Statistical comparisons on day 16 are shown. (M) Kaplan–Meier plots for all groups. All the numerical data are presented as mean ± SD (0.01 <*P ≤ 0.05; 0.001 <**P ≤ 0.01; 0.0001 <***P ≤ 0.001, ****P ≤ 0.0001).
Cancer stem-like cells play a critical role in developing resistance to chemotherapy. Multidrug-resistant (MDR) LS174T colon cancer cell and A549 lung cancer cell, namely LS174T-R and A549-R, were developed following an established method (45). An enhanced expression of ALDH1A1 was observed for these MDR cell lines (Fig. 6H), which was translated into a higher azide-labeling efficiency by AAMCHO (Fig. 6 I and J and SI Appendix, Fig. S16 A and B). To study whether targeted delivery of DBCO-MMAE can improve the antitumor efficacy against MDR tumors, athymic nude mice bearing LS174T-R tumors were divided into four groups: AAMCHO + DBCO-MMAE, DBCO-MMAE, oxaliplatin (first-line chemodrug for colon cancer) (46), and PBS (Fig. 6K). Compared to oxaliplatin or DBCO-MMAE alone, AAMCHO + DBCO-MMAE resulted in a much slower tumor growth (Fig. 6L). This targeted therapy significantly improved the median survival of animals by 90.9% compared to the PBS group and 50.0% compared to DBCO-MMAE alone (Fig. 6M and SI Appendix, Fig. S17 A–C). Furthermore, the expression of stemness-related factors, Oct-4 and Sox-2, was also reduced upon treatment with AAMCHO + DBCO-MMAE (SI Appendix, Fig. S17 D and E).
Discussion
Cancer cells overexpress various unique enzymes than normal tissue, which have provided an extensive reservoir of biomarkers and targets for the in vivo identification and targeted treatment of cancers, respectively (1). However, many oncogenic enzymes are nonlethal and localize intracellularly, as exemplified by ALDH1A1, which poses a challenge to develop targeted cancer therapies aiming at these enzymes. Here, we developed an ALDH1A1-responsive azide-sugar (AAMCHO) that can be activated in the presence of ALDH1A1 and thus selectively label cancer stem-like ALDH1A1Hi cells. We rationally designed an ALDH1A1-responsive moiety that can convert oxidative enzymatic activity into a self-immolative signal to release active cell labeling agents. The unique design also ensured low cross-reactivity to other ALDH isoforms (Fig. 2 C and D). This approach can convert an untargetable nonlethal enzymatic activity into targetable cellular surface artificial receptors that can attract lethal DBCO-toxins via in vivo click chemistry. Compared to enzyme inhibitors that are only therapeutically effective for lethal enzyme target, or antibody-mediated therapeutics that only target extracellular and membrane-bound targets, our strategy has demonstrated a platform to take advantage of the intracellular nonlethal enzymes for cancer treatment and expandable to crucial but otherwise untargetable oncogenic enzymes other than ALDH1A1.
Metabolic glycoengineering of unnatural sugars provides a facile approach to introduce chemical tags (e.g., azide groups) to cell-surface glycoproteins and glycolipids (47–54). Recent studies demonstrated the feasibility of using this approach for targeting TNBC, infectious disease, and in vivo imaging (55–57). However, the challenge lies in the selective labeling of specific cell populations that are responsible for tumorigenesis and progression. Herein, AAMCHO achieves selective labeling of cancer stem-like cells by ALDH1A1-enzymatic activation. In addition, the overexpression of sialic acids and overall increased metabolic activities of cancer cells may add up to the enhanced selectivity of metabolic labeling in vitro and in vivo (55). Limited densities of endogenous cancer markers on the cell surface hindered the efficacy of targeted therapies such ADCs (58). By taking advantage of the amplification effect of enzymatic reaction and metabolic glycoengineering, the AAMCHO-mediated metabolic labeling process can introduce up to 108 azide groups per cell, significantly higher than typical protein receptors (104 to 106 per cell) (SI Appendix, Fig. S16) (58). Furthermore, small-molecule azido-sugar and DBCO-toxins exhibit superior tissue penetration compared to large-molecule antibodies. These advantages add up to the CSC targeting efficiency in vivo. Compared to the MMAE treatment alone, AAMCHO-mediated azide labeling followed by DBCO-MMAE administration resulted in significantly improved antitumor efficacy and reduced toxicity in multiple tumor models (Figs. 5 and 6). This is consistent with the therapeutic effects on preventing cancer relapse, metastatic dissemination, and reverting chemo drug resistance by other CSCs or CSC niches targeted therapeutics, such as inhibiting the key CSC signaling pathways and differentiation therapy (21). Notably, due to disparities in the MTD, a higher dosage of DBCO-MMAE can be applied compared to MMAE alone, resulting in enhanced targeted accumulation of warhead in CSC niches. Furthermore, since DBCO-MMAE contains enzymatic-responsive moiety, which can release cell membrane permeable MMAE upon conjugation and internalization into CSCs, the neighboring cancer cells within the tumor microenvironment may also been affected through the “bystander effect,” which could add up to tumor regression after CSC-targeted therapy. However, although we used DBCO-MMAE as a model DBCO-toxins to evaluate the targeting and cytotoxic effect against CSCs in vitro and in vivo, further efforts on modulation of DBCO-toxins, especially reducing the nonspecific cytotoxicity before release compared to other caged prodrugs (59), are necessary to achieve a better therapeutic index and translational potential. Given the feasibility of modular design of DBCO-conjugates with different payloads such as cytotoxins, radioisotopes, and immune-modulatory agents, this CSC labeling techniques can be extended for the development other cancer targeted therapies, including chemotherapies, radiotherapies, and immunotherapies.
Targeted delivery of therapeutic agents to CSCs is deemed critical for overcoming drug resistance and preventing cancer metastases (21). However, the development of the CSC targeting method is hindered mainly by the lack of characteristic biomarkers, low biomarker abundance, and tumor microenvironment heterogeneity. Identifying unique markers for CSCs is in great demand for developing efficacious CSC-targeted therapies (22). Instead of relying on endogenous cell-surface receptors, our approach takes advantage of nonlethal ALDH1A1 enzymatic activities to achieve CSC targeting. More importantly, our result shows that the AAMCHO labeling is a more precise marker to reflect the self-renewal and tumorigenic properties of the cancer cells compared to conventional stem-like cell markers such as CD44. Thus, this technique also provides a platform for elucidating the role of CSCs in tumor initiation, development, and invasion.
Conclusion
To conclude, we developed an ALDH1A1-activatable sugar precursor, AAMCHO, for metabolic labeling of CSCs in vitro and in vivo and further developed click chemistry-based CSC-targeted therapies. AAMCHO can metabolically label cells in the presence of ALDH1A1 that is overexpressed by CSCs. We further showed that AAMCHO could metabolically label CSCs with azide groups in vivo for subsequently targeted conjugation of DBCO-drugs via efficient click chemistry. We also demonstrated that small-molecule DBCO-drugs exhibited improved penetration in tumor tissues with the help of the sugar labeling compared to antibodies, which could improve the targeting of CSCs that are often distant far from vasculatures inside tumor. Using cytotoxin MMAE as an example, we demonstrated that AAMCHO, coupled with DBCO-MMAE conjugate, showed significantly improved antitumor efficacy against orthotopic TNBC xenograft, syngeneic TNBC lung metastasis, advanced-stage MDA-MB-231 TNBC, and drug-resistant LS174T-R, compared to nontargeted therapies. Notably, compared to the widely used CSC marker CD44, AAMCHO-labeling is more efficient to identify aggressive cancer populations in vitro and in vivo. This CSC labeling and targeting technology holds tremendous potential for the development of potent cancer-targeted therapies, including chemotherapies, radiation therapies, and immunotherapies against various types of cancers.
Methods
Detailed descriptions of the experimental procedures are provided in SI Appendix.
K562 Labeling with AAMCHO in Hypoxia Conditions.
Using a hypoxic incubator, K562 cells were grown in full IMDM media with 1% O2, 5% CO2, and 94% N2 for 72 h. As a control, cells were grown in a standard normoxia incubator supplied with 5% CO2, while coincubated with 50 μM AAMCHO, or Ac-N-AAM, or Ac4ManAz as controls. After 48 h, the cells were collected by centrifugation and stained with 20 μM DBCO-Cy5 for 1 h. The cells were then spun down and resuspended in PBS. Anti-CD34-FITC and anti-CD38-VioBlue were added and incubated for 20 min on ice before subjecting to flow cytometry analysis.
Tumorsphere Labeling.
The tumorsphere was labeled by AAMCHO or Ac4ManAz for 48 h after being treated with FBS for 0 h or 36 h. Single-cell suspensions were collected via trypsin digesting and pipetting and incubated with 50 μM DBCO-Cy5 for 30 min prior to flow cytometry analysis. For cell sorting, the isolated cells from either the sphere culture or xenograft tumor were counterstained with 1 μg/mL FITC-conjugated anti-CD44 for 30 min. Sorting was performed using FACSAria II (BD Biosciences).
Quantification of Azide–Sialic Acid in Cell or Tissue Lysate.
The quantification of conjugated azide–monosaccharide was based on an established method using Cu(I) catalyzed click reaction with coumarin–alkyne. The tissue or cell lysate was homogenized with a mechanical disruptor and then disrupted by an ultrasonicator with lysis buffer (50 mM Tris–HCl and 1% SDS, pH 7.4). The protein concentration of each sample was quantified with the standard BCA protocol. Ninety microliters of lysate was mixed with 10 μL pure acetic acid while heated at 80 °C for 3 h. Then, the mixture was centrifuged at 10,000 rpm for 5 min. The supernatant was mixed with a freshly prepared reaction kit containing 7-Ethynylcoumarin, THPTA, CuSO4, and ascorbic acid. The reaction mixture was shaken at 37 °C overnight and detected by HPLC equipped with a fluorescence detector (λExt: 328 nm, λEmi: 415 nm).
Antitumor Efficacy Study.
MDA-MB-231 tumors were established in 6-wk-old female athymic nude mice by orthotopic injection of MDA-MB-231 cells [1.5 × 106 cells in Hank's Balanced Salt Solution/Matrigel (50 μL, 1/1, v/v)] into the fat pad under the mammary gland. When the tumors reached ~100 mm3, mice were randomly divided into four groups (group 1: AAMCHO + DBCO-MMAE; group 2: DBCO-MMAE; and group 3: MMAE; group 4: PBS). AAMCHO (60 mg/kg) was i.v. injected to group 1 mice once daily for three consecutive days (days 0, 1, and 2). DBCO-MMAE (8 mg/kg) was i.v. injected on day 3. For group 3, i.p. injection of MMAE (0.25 mg/kg, maximum tolerance dose) was conducted at day 3. Each mouse’s tumor volume and body weight were measured twice a week. In a separate study for anticancer efficacy against the late-stage tumor, when the tumors reached ~500 mm3, mice were randomly divided into four groups (group 1: AAMCHO + DBCO-MMAE; group 2: DBCO-MMAE; group 3: MMAE; and group 4: PBS; n = 7 or 8). AAMCHO (60 mg/kg) was i.v. injected to group 1 mice once daily for three consecutive days (days 0, 1, and 2). DBCO-MMAE (20 mg/kg) was i.v. injected on day 3.
Supplementary Material
Appendix 01 (PDF)
Acknowledgments
We thank Dr. Barbara Pilas and the Flow Cytometry Facility at University of Illinois at Urbana-Champaign for cell sorting and data analysis. We thank Karen Doty and the Histology Laboratory at the College of Veterinary Medicine of University of Illinois at Urbana-Champaign for tissue sample preparation and analysis. We would like to acknowledge the financial support from the NIH (R01 CA207584 and R01 EB025651). Iria Pharma acknowledges funding from the NIH (R43 CA235929).
Author contributions
Y. Bo and J. Cheng designed research; Y. Bo, J.Z., K.C., Y.W., Y.F., W.L., Y.J., J.R., C.A., S.H.G., L.M.L., and Y. Bao performed research; K.C. contributed new reagents/analytic tools; Y. Bo, J.Z., S.H.K., G.W.L., Y. Bao, J. Chan, H.W., and J. Cheng analyzed data; and Y. Bo, K.C., Y. Bao, H.W., and J. Cheng wrote the paper.
Competing interests
Y. Bo and J. Cheng have applied through the University of Illinois at Urbana-Champaign for a patent on this technology.
Footnotes
This article is a PNAS Direct Submission.
Contributor Information
Hua Wang, Email: huawang3@illinois.edu.
Jianjun Cheng, Email: chengjianjun@westlake.edu.cn.
Data, Materials, and Software Availability
All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Dang C. V., Reddy E. P., Shokat K. M., Soucek L., Drugging the ‘undruggable’cancer targets. Nat. Rev. Cancer 17, 502–508 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reichert J. M., Valge-Archer V. E., Development trends for monoclonal antibody cancer therapeutics. Nat. Rev. Drug Discovery 6, 349–356 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Drago J. Z., Modi S., Chandarlapaty S., Unlocking the potential of antibody–drug conjugates for cancer therapy. Nat. Rev. Clin. Oncol. 18, 327–344 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huehls A. M., Coupet T. A., Sentman C. L., Bispecific T-cell engagers for cancer immunotherapy. Immunol. Cell Biol. 93, 290–296 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zaman S., Jadid H., Denson A. C., Gray J. E., Targeting Trop-2 in solid tumors: Future prospects. Onco Targets Ther. 12, 1781 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldenberg D. M., Stein R., Sharkey R. M., The emergence of trophoblast cell-surface antigen 2 (TROP-2) as a novel cancer target. Oncotarget 9, 28989 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Uhlén M., et al. , Tissue-based map of the human proteome. Science 347, 1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Dagogo-Jack I., Shaw A. T., Tumour heterogeneity and resistance to cancer therapies. Nat. Rev. Clin. Oncol. 15, 81 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Ginestier C., et al. , ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem. Cell 1, 555–567 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Y., et al. , ALDH1A1 expression correlates with clinicopathologic features and poor prognosis of breast cancer patients: A systematic review and meta-analysis. BMC Cancer 14, 444 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Landen C. N., et al. , Targeting aldehyde dehydrogenase cancer stem cells in ovarian cancer. Mol. Cancer Therapeut. 9, 3186–3199 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li T., et al. , ALDH1A1 is a marker for malignant prostate stem cells and predictor of prostate cancer patients’ outcome. Lab. Invest. 90, 234 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kahlert C., et al. , Expression analysis of aldehyde dehydrogenase 1A1 (ALDH1A1) in colon and rectal cancer in association with prognosis and response to chemotherapy. Ann. Surg. Oncol. 19, 4193–4201 (2012). [DOI] [PubMed] [Google Scholar]
- 14.Huang C.-P., et al. , ALDH-positive lung cancer stem cells confer resistance to epidermal growth factor receptor tyrosine kinase inhibitors. Cancer Lett. 328, 144–151 (2013). [DOI] [PubMed] [Google Scholar]
- 15.Yang S.-M., et al. , Discovery of orally bioavailable, quinoline-based aldehyde dehydrogenase 1A1 (ALDH1A1) inhibitors with potent cellular activity. J. Med. Chem. 61, 4883–4903 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chefetz I., et al. , A pan-ALDH1A inhibitor induces necroptosis in ovarian cancer stem-like cells. Cell Rep. 26, 3061–3075.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vasiliou V., Nebert D. W., Analysis and update of the human aldehyde dehydrogenase (ALDH) gene family. Hum. Genom. 2, 1–6 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pallini R., et al. , Expression of the stem cell marker CD133 in recurrent glioblastoma and its value for prognosis. Cancer 117, 162–174 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Kreso A., et al. , Variable clonal repopulation dynamics influence chemotherapy response in colorectal cancer. Science 339, 543–548 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kurtova A. V., et al. , Blocking PGE 2-induced tumour repopulation abrogates bladder cancer chemoresistance. Nature 517, 209 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Batlle E., Clevers H., Cancer stem cells revisited. Nat. Med. 23, 1124 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Pattabiraman D. R., Weinberg R. A., Tackling the cancer stem cells—What challenges do they pose? Nat. Rev. Drug Discovery 13, 497 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zöller M., CD44: Can a cancer-initiating cell profit from an abundantly expressed molecule? Nat. Rev. Cancer 11, 254–267 (2011). [DOI] [PubMed] [Google Scholar]
- 24.Gerber J. M., et al. , A clinically relevant population of leukemic CD34+ CD38− cells in acute myeloid leukemia. Blood J. Am. Soc. Hematol. 119, 3571–3577 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Junttila M. R., et al. , Targeting LGR5+ cells with an antibody-drug conjugate for the treatment of colon cancer. Sci. Transl. Med. 7, 314ra186 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Klyosov A. A., Kinetics and specificity of human liver aldehyde dehydrogenases toward aliphatic, aromatic, and fused polycyclic aldehydes. Biochemistry 35, 4457–4467 (1996). [DOI] [PubMed] [Google Scholar]
- 27.Wang M.-F., Han C.-L., Yin S.-J., Substrate specificity of human and yeast aldehyde dehydrogenases. Chem. Biol. Interact. 178, 36–39 (2009). [DOI] [PubMed] [Google Scholar]
- 28.Rozema D. B., Ekena K., Lewis D. L., Loomis A. G., Wolff J. A., Endosomolysis by masking of a membrane-active agent (EMMA) for cytoplasmic release of macromolecules. Bioconjug. Chem. 14, 51–57 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Trott O., Olson A. J., AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Russo J. E., Hilton J., Characterization of cytosolic aldehyde dehydrogenase from cyclophosphamide resistant L1210 cells. Cancer Res. 48, 2963–2968 (1988). [PubMed] [Google Scholar]
- 31.Liu P., et al. , Cytotoxic effect of disulfiram/copper on human glioblastoma cell lines and ALDH-positive cancer-stem-like cells. Br. J. Cancer 107, 1488–1497 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Samanta D., Gilkes D. M., Chaturvedi P., Xiang L., Semenza G. L., Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc. Natl. Acad. Sci. U.S.A. 111, E5429–E5438 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Anorma C., et al. , Surveillance of cancer stem cell plasticity using an isoform-selective fluorescent probe for aldehyde dehydrogenase 1A1. ACS Central Sci. 4, 1045–1055 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thul P. J., et al. , A subcellular map of the human proteome. Science 356, eaal3321 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Eirew P., et al. , A method for quantifying normal human mammary epithelial stem cells with in vivo regenerative ability. Nat. Med. 14, 1384 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Shaw F. L., et al. , A detailed mammosphere assay protocol for the quantification of breast stem cell activity. J. Mammary Gland Biol. Neoplasia. 17, 111–117 (2012). [DOI] [PubMed] [Google Scholar]
- 37.Dontu G., et al. , In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 17, 1253–1270 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Das B., et al. , Hypoxia enhances tumor stemness by increasing the invasive and tumorigenic side population fraction. Stem Cells 26, 1818–1830 (2008). [DOI] [PubMed] [Google Scholar]
- 39.Fishman W. H., Anlyan A., Comparison of the β-glucuronidase activity of normal, tumor, and lymph node tissues of surgical patients. Science 106, 66–67 (1947). [DOI] [PubMed] [Google Scholar]
- 40.Younes A., et al. , Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N Engl. J. Med. 363, 1812–1821 (2010). [DOI] [PubMed] [Google Scholar]
- 41.Malanchi I., et al. , Interactions between cancer stem cells and their niche govern metastatic colonization. Nature 481, 85–89 (2012). [DOI] [PubMed] [Google Scholar]
- 42.Sengupta P., et al. , Cholesterol-tethered platinum II-based supramolecular nanoparticle increases antitumor efficacy and reduces nephrotoxicity. Proc. Natl. Acad. Sci. U.S.A. 109, 11294–11299 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Roncato F., et al. , Improvement and extension of anti-EGFR targeting in breast cancer therapy by integration with the Avidin-Nucleic-Acid-Nano-Assemblies. Nat. Commun. 9, 4070 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bitonti A. J., et al. , Regression of human breast tumor xenografts in response to (E)-2′-deoxy-2′-(fluoromethylene) cytidine, an inhibitor of ribonucleoside diphosphate reductase. Cancer Res. 54, 1485–1490 (1994). [PubMed] [Google Scholar]
- 45.Jiang D., Sui M., Zhong W., Huang Y., Fan W., Different administration strategies with paclitaxel induce distinct phenotypes of multidrug resistance in breast cancer cells. Cancer Lett. 335, 404–411 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.André T., et al. , Oxaliplatin, fluorouracil, and leucovorin as adjuvant treatment for colon cancer. N Engl. J. Med. 350, 2343–2351 (2004). [DOI] [PubMed] [Google Scholar]
- 47.Xie R., et al. , In vivo metabolic labeling of sialoglycans in the mouse brain by using a liposome-assisted bioorthogonal reporter strategy. Proc. Natl. Acad. Sci. U.S.A. 113, 5173–5178 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun Y., et al. , Mechanistic investigation and multiplexing of liposome-assisted metabolic glycan labeling. J. Am. Chem. Soc. 140, 3592–3602 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang H., et al. , Metabolic labeling and targeted modulation of dendritic cells. Nat. Mater. 19, 1244–1252 (2020), 10.1038/s41563-020-0680-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chang P. V., Dube D. H., Sletten E. M., Bertozzi C. R., A strategy for the selective imaging of glycans using caged metabolic precursors. J. Am. Chem. Soc. 132, 9516–9518 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Laughlin S. T., Bertozzi C. R., Metabolic labeling of glycans with azido sugars and subsequent glycan-profiling and visualization via Staudinger ligation. Nat. Protoc. 2, 2930–2944 (2007). [DOI] [PubMed] [Google Scholar]
- 52.Prescher J. A., Dube D. H., Bertozzi C. R., Chemical remodelling of cell surfaces in living animals. Nature 430, 873–877 (2004). [DOI] [PubMed] [Google Scholar]
- 53.Xie R., Hong S., Feng L., Rong J., Chen X., Cell-selective metabolic glycan labeling based on ligand-targeted liposomes. J. Am. Chem. Soc. 134, 9914–9917 (2012). [DOI] [PubMed] [Google Scholar]
- 54.Fan X., et al. , Cell-type-specific labeling and profiling of glycans in living mice. Nat. Chem. Biol. 18, 625–633 (2022). [DOI] [PubMed] [Google Scholar]
- 55.Wang H., et al. , Selective in vivo metabolic cell-labeling-mediated cancer targeting. Nat. Chem. Biol. 13, 415–424 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang H., et al. , Targeted ultrasound-assisted cancer-selective chemical labeling and subsequent cancer imaging using click chemistry. Angew. Chem. Int. Ed. Engl. 55, 5452–5456 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bo Y., et al. , Targeting infected host cells in vivo via responsive azido-sugar mediated metabolic cell labeling followed by click reaction. Biomaterials 238, 119843 (2020). [DOI] [PubMed] [Google Scholar]
- 58.Anastas J. N., Moon R. T., WNT signalling pathways as therapeutic targets in cancer. Nat. Rev. Cancer 13, 11–26 (2013). [DOI] [PubMed] [Google Scholar]
- 59.Wu K., et al. , Click activated protodrugs against cancer increase the therapeutic potential of chemotherapy through local capture and activation. Chem. Sci. 12, 1259–1271 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Data Availability Statement
All study data are included in the article and/or SI Appendix.