

Significance
We report that specific HLA-DRB1*04 alleles are protective against Alzheimer’s dementia (AD), Parkinson’s disease (PD), and other neurodegenerative disorders. Further, we found that these HLA (Human Leukocyte Antigen) subtypes selectively bind a piece of Tau crucial to aggregation but only when it is acetylated (a-PHF6). This a-PHF6 piece is significant as it is a common posttranslational modification of Tau found in Alzheimer’s brains. Only when someone is HLA-DRB1*04:04 or HLA-DRB1*04:01 can PHF6 be presented as a T cell epitope to T cell receptors and mount a memory immune response against this pro-aggregation fragment. This immune response would protect against AD, PD, and neurodegeneration, explaining the HLA association. Vaccination with a-PHF6 in HLA-DRB1*04 individuals could have preventive effects.
Keywords: HLA, Alzheimer’s dementia, Parkinson’s disease, neurodegeneration, autoimmunity
Abstract
Across multiancestry groups, we analyzed Human Leukocyte Antigen (HLA) associations in over 176,000 individuals with Parkinson’s disease (PD) and Alzheimer’s disease (AD) versus controls. We demonstrate that the two diseases share the same protective association at the HLA locus. HLA-specific fine-mapping showed that hierarchical protective effects of HLA-DRB1*04 subtypes best accounted for the association, strongest with HLA-DRB1*04:04 and HLA-DRB1*04:07, and intermediary with HLA-DRB1*04:01 and HLA-DRB1*04:03. The same signal was associated with decreased neurofibrillary tangles in postmortem brains and was associated with reduced tau levels in cerebrospinal fluid and to a lower extent with increased Aβ42. Protective HLA-DRB1*04 subtypes strongly bound the aggregation-prone tau PHF6 sequence, however only when acetylated at a lysine (K311), a common posttranslational modification central to tau aggregation. An HLA-DRB1*04-mediated adaptive immune response decreases PD and AD risks, potentially by acting against tau, offering the possibility of therapeutic avenues.
Alzheimer’s disease (AD) and Parkinson’s disease (PD) are responsible for considerable morbidity and mortality. Pathophysiology involves the accumulation of tau (neurofibrillary tangles) and Amyloid-β-rich aggregates (amyloid plaques) in AD, and α-synuclein-rich aggregates (Lewy bodies) in PD, although copresence of these aggregates may occur. Consensus is growing that tau may also play a key role in PD (1–3).
Innate immune responses and microglial involvement have long been implicated in neurodegenerative diseases (4–6). More recently, a role for adaptive immunity in PD and AD has also been outlined through genetic (7–10) and immunological studies (11–15). Notably, comparisons of healthy controls and individuals with neurodegenerative diseases have shown that a complex polyclonal T cell response develops against α-synuclein, and tau (11–13). Whether or not these responses are epiphenomenal, contribute to, or protect against neurodegeneration is unknown.
A cornerstone of the adaptive immune response is the highly polymorphic HLA (Human Leukocyte Antigen) locus located on human chromosome 6. HLA genes encode a set of proteins that bind peptides derived from foreign or self-antigens, allowing recognition by T cells and subsequent coordination of immune responses. In this context, PD (8) and AD (10) have genome-wide associations in the HLA class II region, within a region containing HLA-DR and DQ, two tightly linked sets of genes. Depending on one’s specific HLA, individuals present distinct repertoires of bound peptides to CD4+ T cells, thus HLA establishes how the immune system sees and reacts to foreign and self-antigens in different individuals.
In PD genome-wide association studies (GWAS), the HLA signal was initially attributed to HLA-DRA, HLA-DRB1*15:01, and HLA-DRB5 (8), but recent studies indicate it marks HLA-DRB1*04 (16–18). In AD, the HLA signal remains uncharacterized (7, 9, 19) or was assigned to HLA-DRB1*15:01 (7). SI Appendix, Table S1 provides a list of previous studies reporting association results at the HLA locus in genetic association studies of AD and PD.
To better understand the involvement of HLA in these diseases, we first gathered genome-wide data available in PD and AD, refined the signal to the HLA subtype level through HIBAG imputation (20), and performed HLA haplotyping to disentangle causal alleles. HLA signals are best dissected across ancestry groups, due to the high variability of HLA alleles across ancestry, as first shown in narcolepsy (21). Therefore, we gathered a large and diverse set of participants including Europeans (10, 17), Asians (22–24), Latin Americans (25), and African Americans (26, 27) to perform fine-mapping of the HLA association in AD and PD. Second, we studied HLA association with pathological observations, finding the strongest protective associations with tau-associated biomarkers. Last, we tested the binding of HLA alleles associated with AD and PD, with epitopes from alpha-synuclein and tau, identifying potential epitopes mediating these effects.
Results
Multiancestry Local-GWAS of AD and PD and Colocalization at the HLA Locus.
In our AD local-GWAS, 110,927 cases and proxy-cases, and 423,415 controls were included in the meta-analysis (SI Appendix, Table S2). Separate analyses were carried out per ancestry and per dataset and meta-analyzed at the HLA locus, with the full GWAS of European and Japanese cohorts now published separately with publicly available summary statistics (10, 24). The most significant association with AD was at rs35472547 (odds ratio [OR] = 0.91; 95% CI, [0.89; 0.93]; P = 9.7 × 10−23, SI Appendix, Fig. S1). In our PD local-GWAS, 41,205 cases and proxy-cases and 474,597 controls were included in the meta-analysis (SI Appendix, Table S2). Summary statistics from three published studies (8, 22, 25) were included in this meta-analysis at the HLA locus. The most significant association with PD was at rs504594 (OR = 0.84; 95% CI, [0.80; 0.88]; P = 1.83 × 10−13, SI Appendix, Fig. S1). Colocalization analysis emphasized that the same HLA association signal is shared across AD and PD (posterior probability of colocalization, PP4 = 99.5%, Fig. 1), with rs601945 common to the two GWAS and lead variant in their combination. SI Appendix, Figs. S3 and S4 provide locus zoom plots per ancestry of the AD and PD local-GWAS.
Fig. 1.

Colocalization of the HLA locus signal in AD, PD, and ALS. PP4: posterior probability of colocalization.
Same Protective HLA Association in Other Neurodegenerative Diseases.
Amyotrophic lateral sclerosis (ALS) GWAS (9) recently reported a genome-wide significant protective association at the HLA locus. Our colocalization analysis shows that this association is shared with AD and PD (PP4 = 94.6%, and PP4 = 60.6%, respectively, Fig. 1 and SI Appendix, Fig. S1). The largest Lewy body dementia (LBD) GWAS (28) to date did not contain a genome-wide significant peak at the HLA locus. However, querying rs601945 in this GWAS summary statistics, we noted an association with decreased LBD risk close to nominal significance (OR = 0.91; [0.83; 1.01]; P = 0.07) and in the same range as observed in AD and PD, suggesting that a larger sample size may lead to a similarly shared protective association.
Fine-Mapping of the HLA Association—Allele-, Haplotype-, Amino Acid–Level Analyses—Highlights Association with HLA-DRB1*04 Alleles.
Our HLA-fine-mapping analysis includes a slightly different set of individuals than the local-GWAS. In the AD analysis, 121,411 cases and proxy-cases and 409,096 controls were included (SI Appendix, Table S3). In the PD analysis, 55,554 cases and proxy-cases and 1,454,443 controls were included (SI Appendix, Table S3). Briefly, all associations were tested under a dominant model as HLA effects are mostly dominant; the presence or absence of an allele allows for recognition of epitopes. All HLA associations from allele-, haplotype-, and amino acid–level analyses are reported in SI Appendix, Tables S4 and S5, respectively, for AD and PD, with key findings highlighted in Table 1 and details per cohort in Datasets S1–S5.
Table 1.
HLA-DRB1 alleles HLA-DRB1*04:04 and HLA-DRB1*04:01 are associated with a decreased risk of Parkinson’s and ADs
| HLA | PD | AD | PD + AD | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| FreqC | N | OR | P-val | FreqC | N | OR | P-val | OR | P-val | p-het | ||
| HLA Alleles | HLA-DRB1*04:01 | 0.196 | 1,484,656 | 0.92[0.89; 0.95] | 2.4E-08 | 0.191 | 486,478 | 0.93[0.9; 0.96] | 6.4E-07 | 0.92[0.91; 0.94] | 8.9E-14 | 0.56 |
| HLA-DRB1*04:02 | 0.019 | 1,474,730 | 0.92[0.85; 0.99] | 0.02 | 0.022 | 155,846 | 1.00[0.91; 1.10] | 0.99 | 0.95[0.89; 1.01] | 0.07 | 0.17 | |
| HLA-DRB1*04:03 | 0.012 | 980,868 | 0.89[0.81; 0.97] | 0.01 | 0.072 | 7,587 | 1.09[0.91; 1.30] | 0.34 | 0.93[0.85; 1.01] | 0.07 | 0.04 | |
| HLA-DRB1*04:04 | 0.074 | 1,475,574 | 0.84[0.80; 0.88] | 1.5E-11 | 0.073 | 476,236 | 0.86[0.82; 0.90] | 8.9E-12 | 0.85[0.82; 0.88] | 9.3E-22 | 0.60 | |
| HLA-DRB1*04:05 | 0.013 | 1,507,057 | 1.00[0.95; 1.05] | 0.86 | 0.026 | 169,080 | 0.98[0.91; 1.06] | 0.62 | 0.99[0.95; 1.03] | 0.68 | 0.75 | |
| HLA-DRB1*04:06 | 0.046 | 32,327 | 0.95[0.82; 1.09] | 0.46 | 0.058 | 7,587 | 0.95[0.78; 1.15] | 0.60 | 0.95[0.85; 1.06] | 0.37 | 0.99 | |
| HLA-DRB1*04:07 | 0.021 | 526,189 | 0.79[0.69; 0.91] | 7.3E-04 | 0.019 | 474,840 | 0.88[0.81; 0.96] | 4.5E-03 | 0.86[0.79; 0.92] | 2.7E-05 | 0.18 | |
| HLA-DRB1*04:10 | 0.041 | 4,853 | 0.91[0.67; 1.25] | 0.57 | 0.031 | 7,985 | 1.23[0.94; 1.59] | 0.13 | 1.08[0.89; 1.32] | 0.42 | 0.16 | |
| HLA-DRB1*01:01 | 0.181 | 1,485,033 | 1.05[1.01; 1.09] | 7.0E-03 | 0.186 | 487,120 | 1.07[1.04; 1.10] | 3.9E-07 | 1.06[1.04; 1.09] | 1.3E-08 | 0.44 | |
| HLA-DQB1*03:02 | 0.191 | 1,501,065 | 0.91[0.88; 0.93] | 2.6E-14 | 0.190 | 510,130 | 0.89[0.86; 0.91] | 1.2E-19 | 0.90[0.88; 0.91] | 4.7E-32 | 0.23 | |
| HLA-DQA1*03:01 | 0.177 | 1,507,147 | 0.89[0.87; 0.91] | 2.5E-20 | 0.186 | 507,263 | 0.89[0.87; 0.91] | 1.8E-19 | 0.89[0.88; 0.91] | 3.E-37 | 0.79 | |
| HLA-DRB1*15:01 | 0.272 | 1,507,057 | 1.06[1.03; 1.10] | 5.0E-04 | 0.263 | 485,383 | 1.02[0.99; 1.04] | 0.13 | 1.03[1.01; 1.05] | 1.1E-03 | 0.054 | |
| HLA Haplotypes | DRB1*04:01~DQA1* 03:01~DQB1*03:02 | 0.088 | 1,473,386 | 0.95[0.92; 0.98] | 3.6E-03 | 0.092 | 342,115 | 0.91[0.87; 0.95] | 6.5E-06 | 0.93[0.91; 0.96] | 3.2E-07 | 0.10 |
| DRB1*04:01~DQA1* 03:03~DQB1*03:01 | 0.12 | 1,481,518 | 0.98[0.97; 0.99] | 5.2E-04 | 0.133 | 346,157 | 0.98[0.94; 1.02] | 0.36 | 0.98[0.97; 0.99] | 3.4E-04 | 0.86 | |
| DRB1*04:04~DQA1* 03:01~DQB1*03:02 | 0.077 | 1,475,734 | 0.84[0.80; 0.89] | 1.7E-10 | 0.086 | 344,760 | 0.85[0.81; 0.89] | 5.4E-11 | 0.85[0.82; 0.88] | 5.7E-20 | 0.78 | |
| DRB1*04:07~DQA1* 03:01~DQB1*03:02 | 0.198 | 1498 | 0.58[0.44; 0.76] | 6.1E-05 | 0.063 | 1,865 | 0.75[0.47; 1.21] | 0.23 | 0.62[0.49; 0.78] | 4.6E-05 | 0.36 | |
| DRB1*04:07~DQA1* 03:03~DQB1*03:01 | 0.021 | 524,845 | 0.87[0.73; 1.04] | 0.12 | 0.017 | 340,065 | 0.87[0.79; 0.97] | 8.0E-03 | 0.87[0.80; 0.95] | 2.1E-03 | 0.99 | |
| DRB1*04:05~DQA1* 03:03~DQB1*04:01 | 0.11 | 35,369 | 0.99[0.92; 1.06] | 0.76 | 0.21 | 4,149 | 0.97[0.83; 1.13] | 0.68 | 0.99[0.92; 1.05] | 0.65 | 0.81 | |
| DRB1*08:02~DQA1* 03:01~DQB1*03:02 | 0.026 | 5,602 | 1.10[0.76; 1.60] | 0.61 | 0.02 | 4,149 | 0.85[0.55; 1.31] | 0.46 | 0.99[0.74; 1.31] | 0.93 | 0.37 | |
| DRB1*01:01~DQA1* 01:01~DQB1*05:01 | 0.150 | 1,485,406 | 1.05[1.01; 1.09] | 0.02 | 0.222 | 351,276 | 1.08[1.05; 1.11] | 4.8E-07 | 1.07[1.04; 1.09] | 6.8E-08 | 0.27 | |
| Amino acid: | HLA-DRB1 13H/33H | 0.300 | 1,507,057 | 0.89[0.87; 0.91] | 9.3E-22 | 0.315 | 527,173 | 0.91[0.89; 0.93] | 1.7E-18 | 0.90[0.89; 0.92] | 2.0E-38 | 0.36 |
Effect sizes are reported as odds ratio (OR), with 95% CI, and significance (P-value). FreqC: frequency of carriers, N: number of individuals analyzed for a given allele/haplotype/amino acid, p-het: heterogeneity test P-value.
In the HLA-allele-level analysis, the most significant association in AD was observed for HLA-DQB1*03:02 (OR = 0.89[0.86; 0.91]; P = 1.2E-19), which was the second most significant allele in PD (OR = 0.91[0.88; 0.93]; P = 2.6E-14). In PD, the most significant association was observed for HLA-DQA1*03:01 (OR = 0.89[0.87; 0.91]; P = 2.3E-20), which was the second most significant allele in AD (OR = 0.89[0.87; 0.91]; P = 1.7E-18). In both AD and PD, HLA-DRB1*15:01 only had a marginal effect (Table 1). It is worth noting that HLA-DQA1*03:01 and DQB1*03:02 are in high linkage disequilibrium and typically found on haplotypes harboring HLA-DRB1*04 subtypes. Because HLA-DRB1*04 alleles are numerous and have more variations, each individually is less common than HLA-DQA1*03:01 and DQB1*03:02; thus, the strength of this association may reflect the sum of HLA-DRB1*04 subtype association.
This is particularly clear in the HLA-haplotype-level analysis, where haplotypes harboring HLA-DRB1*04 subtypes are most significant (Table 1 and SI Appendix, Tables S4 and S5). The two significant haplotypes in linkage with DQA1*03:03~DQB1*03:01 are the most interesting since they advocate against the causality of DQA1*03:01~DQB1*03:02 (Fig. 2). Finally, DQA1*03:01~DQB1 *03:02~DRB1*08:02, relatively common in East-Asian populations, does not show any association (Fig. 2), which also argues against DQA1*03:01~DQB1*03:02 causality. Overall, the data suggest hierarchical protective effects of HLA-DRB1*04 subtypes, with *04:04 and *04:07 having the strongest effects on disease protection than other subtypes (Table 1), followed by weaker effects of *04:01, *04:06, and *04:03, and no effect of *04:05.
Fig. 2.

Haplotypes harboring key HLA-DRB1*04 subtypes and/or HLA-DQB1*03:02. Effect sizes highlighted in red were nominally significant (P < 0.05).
Last, the HLA-amino acid–level analysis emphasized the pair of HLA-DRB1 amino acids H13 and H33 as the most significant amino acid changes associated with AD and PD risks (H13 association with AD: OR = 0.91[0.89; 0.93]; P = 1.7E-18; and with PD: OR = 0.89[0.87; 0.91]; P = 9.3E-22). This pair of amino acid changes is in complete linkage disequilibrium across ancestries and present on all HLA-DRB1*04 subtypes. It is worth emphasizing that the lead variant in the local-GWAS (rs601945) is also in high linkage disequilibrium with these amino acids (D′ close to 1 in Europeans and East-Asians, and R2 above 0.9 in Europeans, SI Appendix, Fig. S4). Importantly, no significant heterogeneity in effect sizes between AD and PD was observed (Table 1), as formally tested using heterogeneity tests at each allele, haplotype, and amino acid.
Conditional Analyses Suggest a Shared Association Signal at HLA across AD and PD.
A subset of the participants analyzed with HLA-fine-mapping was also available for conditional analyses using the lead variant (rs601945) and lead amino acid (HLA-DRB1 H13). In the AD analysis, 120,403 cases and proxy-cases and 408,720 controls were included (SI Appendix, Table S6). In the PD analysis, 41,515 cases and proxy-cases and 518,923 controls were included (SI Appendix, Table S6). Four conditional analyses were implemented i) local-GWAS conditioned on rs601945, ii) local-GWAS conditioned on HLA-DRB1 H13, iii) HLA-class II amino acid level conditioned on rs601945, and iv) HLA-class II amino acid level conditioned on H13 (SI Appendix, Tables S7 and S8). Overall, the main signal colocalized at the HLA locus across AD and PD disappeared after conditional analyses. Of note, however, whereas no other signals were observed in PD, two independent signals remained genome-wide significant in AD after conditional analysis (SI Appendix, Fig. S5). The first significant signal is between BTLN2 and HLA-DRA at lead variant rs3129841 (OR = 0.94[0.92; 0.96]; P = 1.0E-08), while the second is between HLA-DQB1 and HLA-DQA2 at lead variant rs9275222 (OR = 1.05[1.03; 1.07]; P = 6.6E-09). In the HLA-class II amino acid–level conditional analyses, no significant associations were observed in PD (SI Appendix, Table S8), while in AD there were several significant associations when conditioning on H13 (SI Appendix, Table S7). The two most significant ones were HLA-DRB1 amino acid changes, N37 (OR = 0.94[0.92; 0.96]; P = 6.0E-08) and H32 (OR = 0.94[0.92; 0.96]; P = 9.3E-08). However, given the location of the two AD peaks in the conditional analysis (SI Appendix, Fig. S5), it seems unlikely that these will be related to these amino acid changes, and other regulatory mechanisms should be explored. In conclusion, the HLA signal in AD is likely more complex than initially described. However, conditional analyses confirmed a shared signal across AD and PD, and this main common signal was the only one pursued in all additional analyses.
HLA-DRB1 H13/H33 Is Associated with Decreased Tau Braak Staging.
How could an HLA-DRB1*04 subtype-specific association be involved in AD? To investigate this question, we first used neuropathological information from 7,259 postmortem samples of European ancestry available through the Religious Orders Study and Memory and Aging Project (29) and the National Institute on Aging–AD Center cohorts 1 to 7 (30), looking at the effect of HLA-DRB1 on tau Braak staging and neuritic plaque density in pathological samples. As shown in Table 2, a significant association of HLA-DRB1 H13/H33, with decreased neurofibrillary tangles (β = −0.13, 95% CI, [−0.21; −0.05]; P = 0.001), but not neuritic plaque density (β = −0.04, 95% CI, [−0.10; 0.02]; P = 0.19), was observed, suggesting the involvement of tau. Due to linkage disequilibrium, the same associations were observed for rs601945, and by extension for HLA-DRB1*04 alleles (SI Appendix, Tables S9 and S10). Last, in a subset of autopsied individuals with either Lewy Body pathology, AD pathology, or both pathologies, we tested the association of rs601945 with each pathology group versus pathology-free controls. In this comparison, rs601945 was associated with reduced AD only pathology (OR = 0.81[0.69; 0.96], P = 0.01) and concordant protective effects in the Lewy Body only pathology group (OR = 0.84[0.69; 1.03], P = 0.09) and in the dual pathology group (OR = 0.81[0.59; 1.09], P = 0.16), though the last two associations were not significant due to smaller sample sizes in these groups (SI Appendix, Table S10).
Table 2.
HLA-DRB1 H13/H33 amino acid is associated with reduced tau and neurofibrillary tangles and to a lesser extent with reduced Amyloid-β or neuritic plaques, when testing their association with AD neuropathology and CSF biomarkers
| Phenotype | DRB1 H13 | ||||
|---|---|---|---|---|---|
| N | Freq | β [95% CI] | P val | ||
| All individuals | Tau Braak staging | 7,456 | 0.293 | −0.13[−0.21; −0.05] | 1.4E-03 |
| Neuritic plaques density | 5,876 | 0.292 | −0.04[−0.10; 0.02] | 0.19 | |
| total-tau in CSF | 5,289 | 0.232 | −0.11[−0.17; −0.05] | 5.5E-04 | |
| p-tau in CSF | 5,269 | 0.234 | −0.08[−0.14; −0.02] | 1.0E-02 | |
| Aβ42 in CSF | 5,368 | 0.232 | 0.08[0.01; 0.14] | 0.02 | |
| Cases only | Tau Braak staging | 5,126 | 0.283 | −0.07[−0.12; −0.01] | 0.02 |
| Neuritic plaques density | 4,124 | 0.289 | −0.01[−0.04; 0.02] | 0.39 | |
| total-tau in CSF | – | 0.228 | −0.14[−0.23; −0.06] | 8.2E-04 | |
| p-tau in CSF | – | 0.228 | −0.10[−0.19; −0.01] | 0.02 | |
| Aβ42 in CSF | – | 0.227 | 0.09[0.02; 0.16] | 1.0E-02 | |
| Age-at-AD-onset | 11,900 | 0.278 | 0.39[0.03; 0.76] | 0.03 | |
p-tau: phosphorylated tau, t-tau: total tau, N: number of individuals, MAF: minor allele frequency, OR: odds ratio, β: parameter estimate, CI: confidence interval. Braak: Tau Braak staging, Neur: Neuritic plaques density.
HLA-DRB1 H13/H33 Is Associated with Decreased Tau in CSF and Increased Age-at-Onset.
The analysis of cerebrospinal fluid (CSF) Aβ42 and tau levels in 8,074 subjects of European ancestry independently confirmed this observation (Table 2 and SI Appendix, Table S10). In CSF, HLA-DRB1 H13/H33 was significantly associated with lower levels of phosphorylated- and total-tau (for total tau, β = −0.11, 95% CI, [−0.17; −0.05]; P = 0.0006), but the association with increased Aβ42 levels was less significant (β = −0.08, 95% CI, [−0.14; −0.02]; P = 0.02). Interestingly, HLA-DRB1 H13/H33 was also associated with older age-at-onset in AD (β = 0.39, 95% CI, [0.03; 0.76]; P = 0.03), as also previously reported in PD (31), further supporting a protective role.
In Vitro Test of HLA-DRB1*04 Subtypes Binding to Tau and Alpha-Synuclein Peptides Emphasizes the Differential Binding of HLA-DRB1* 04 to Acetylated PHF6 Tau.
Based on these results, we hypothesized that an HLA-DRB1*04-restricted adaptive immune response directed against tau may be protective in AD. To test this hypothesis, we screened the entire tau protein for HLA-RB1*04 subtype-specific binding, using the highly protective HLA-DRB1*04:04 subtype, the moderately protective HLA-DRB1*04:01 subtype, and the neutral HLA-DRB1*04:05 subtype (Fig. 3). Because tau is extensively modified posttranslationally, all most frequent posttranslational modified (PTM) changes (32) were also included, totaling 448 peptides (SI Appendix, Table S11). As a control, alpha-synuclein, another extensively PTM protein involved in PD was also tested (SI Appendix, Fig. S6) for a total of 62 peptides (SI Appendix, Table S12).
Fig. 3.
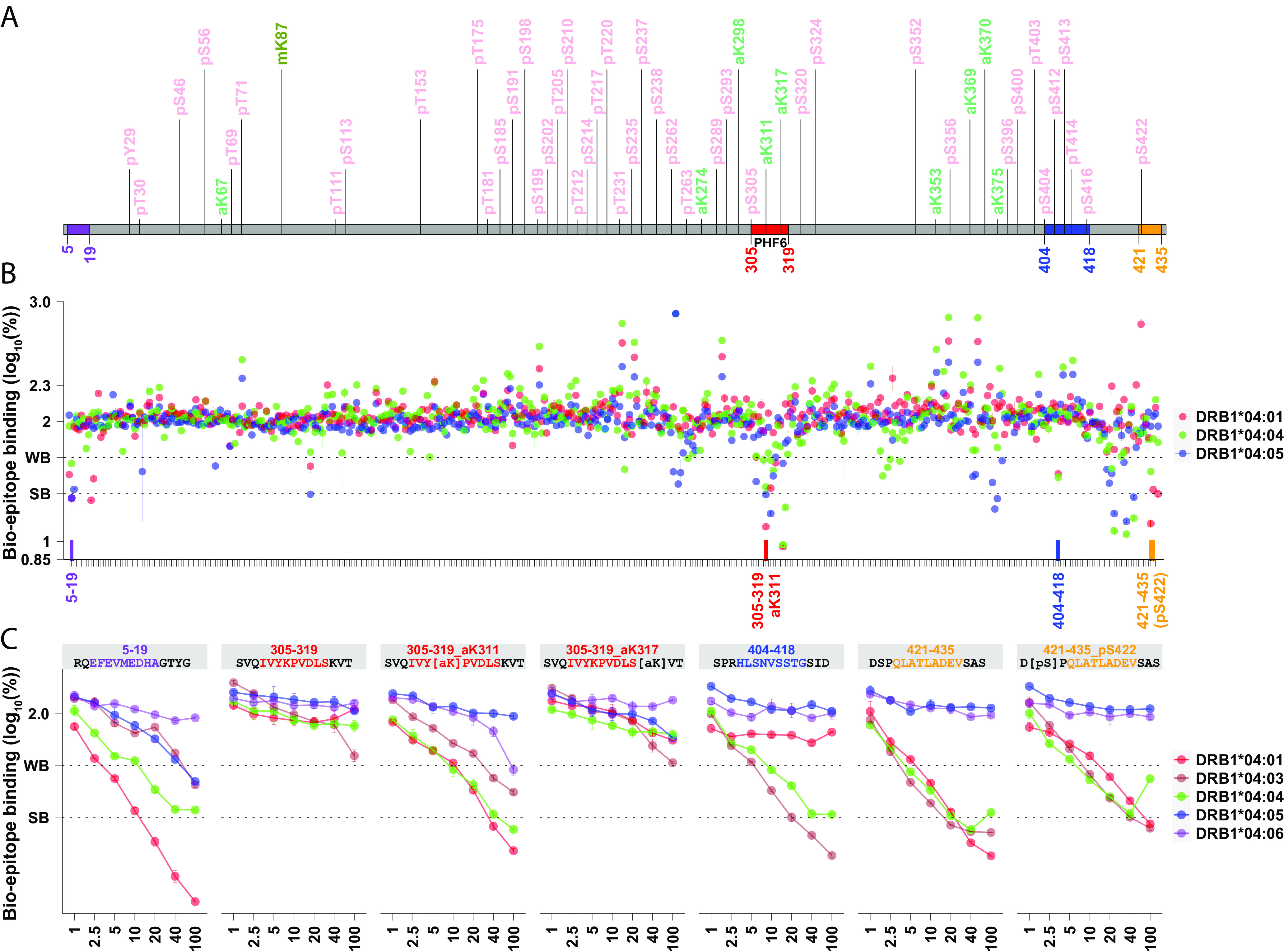
The pro-aggregation PHF6 region of tau binds HLA-DRB1*04 subtypes only when acetylated at K311. Fifteen-mer peptides (800 µM) encompassing the entirety of all tau isoforms (schematized in panel A), overlapping across 11 residues, were screened for HLA-DRB1*04:01, HLA-DRB1*04:04 and HLA-DRB1*04:05 binding (Methods), with and without common PTMs as reported by Wesseling et al. (32). Four regions (labeled in purple, red, blue, orange, panel B) containing strong HLA-DRB1*04 binders (log displacement <1 to 1.4, below 25% of baseline control) were further tested at various concentrations (panel C), showing three promising regions where binding was stronger with HLA-DRB1*04:04/ HLA-DRB1*04:01, intermediary with HLA-DRB1*04:03 and absent or weak with HLA-DRB1*04:05 and HLA-DRB1*04:06, a pattern similar to GWAS risk (Table 1). Among these candidate regions, PHF6 306VQIVY(acetylK)PVDLSK317 is the only one that strongly binds HLA-DRB1*04:01, HLA-DRB1*04:03, and HLA-DRB1*04:04 and only when posttranslationally modified at K311. This segment is well known to seed tau aggregation, and this process is increased in the presence of acetylK311. Outcompeting a biotinylated epitope (known binder) 75% and 50% is considered as strong binding (SB) and weak binding (WB), respectively. Predicted binding cores are highlighted correspondingly.
Among tested peptides (SI Appendix, Tables S11 and S12), only a few peptides strongly bound HLA-DRB1*04 subtypes, with PHF6 in tau (R3/wt; 306VQIVY(acetylK)PVDLSKV318) standing as the main candidate (Fig. 3). Most interestingly, PHF6 sequences only bind HLA-DRB1*04 when K311 is acetylated (titration repeated 3 times, SI Appendix, Fig. S8). Two-way repeated measure ANOVA comparing HLA binding to unacetylated versus acetylated PHF6 was significant for HLA-DRB1*04:01, and HLA-DRB1*04:04 (pDRB1*04:01 = 3.58 × 10−8, pDRB1*04:04 = 1.19 × 10−4, SI Appendix, Table S13). Additionally, acetylated sequences have significantly less affinity for HLA-DRB1*04:05 versus other subtypes (HLA-DRB1*04:04= HLA-DRB1*04:01> HLA-DRB1* 04:03> HLA-DRB1*04:06> HLA-DRB1*04:05), the same hierarchy observed in the case/control association results, with cores predicted to bind most strongly HLA-DRB1*04:04 (SI Appendix, Fig. S7) (33, 34). Additional PTMs in the area, such as acetylated K317, do not alter binding, although the presence of phosphoserine at S305, another frequent PTM, slightly reduces PHF6 binding (Fig. 3). Other peptides were found to bind HLA-DRB1*04 in both tau (Fig. 3) and α-synuclein (SI Appendix, Fig. S6), but in no other case was a PTM necessary for epitope binding.
In Silico Predictions Confirm HLA-DRB1*04 Subtypes as the Unique Common HLA-DRB1 Alleles Binding to PHF6 Tau.
In-silico predictions confirm that HLA-DRB1*04-associated subtypes are the only frequent HLA-DRB1 and DQ subtypes with predicted high affinity for the PHF6 (SI Appendix, Fig. S9), likely explaining why HLA-DRB1*04, and no other subtypes, mediate this effect. In-silico predicted binding to the PHF6 motif was also observed with accessory gene HLA-DRB4*01 (SI Appendix, Fig. S9). This PHF6/HLA-DRB4*01 strong binding was confirmed in vitro (SI Appendix, Fig. S10), but the absence of AD or PD association with HLA-DRB1*07:01 and HLA-DRB1*09:01, whose haplotypes are in linkage with HLA-DRB4*01, ruled out its involvement (SI Appendix, Table S14). Last, an absence of predicted binding to PHF6 for DQA1*01:01~DQB1*05:01 or DQA1*03:01~DQB1*03:02 was notable (SI Appendix, Fig. S9) and we confirmed this prediction by testing binding in vitro SI Appendix, Fig. S10.
Discussion
Here, we show that HLA HLA-DRB1*04 protects against AD, PD, and probably ALS, three prototypical neurodegenerative diseases and that HLA-DRB1*04 selectively binds the K311-acetylated epitope of the PHF6 sequence of the microtubule-associated protein tau (35), an important region in the mediation of tau aggregates. Presence of HLA-DRB1*04 was also associated with lower CSF tau and fewer neurofibrillary tangles in AD subjects. Although tau aggregation is unlikely to be the sole contributor to neurodegeneration in all these diseases, it may exacerbate pathology in the presence of other aggregates, as has been proposed previously (36, 37).
Tau, like other proteins involved in neurodegeneration, is highly posttranslationally modified (PTM) through e.g. phosphorylation and acetylation, phenomena that likely predispose tau to aggregation (32). In autoimmune diseases, PTMs frequently form part of culprit autoantigens, contributing to reduced self-tolerance as they are not presented in the thymus for negative selection (38). This likely explains the strong polyclonal T cell response observed in controls and AD/PD patients against tau, β-amyloid, and α-synuclein (11, 13, 39). A similarly broad B cell response against tau and α-synuclein is also reported in controls and patients (14, 15). As mentioned above, prior work has outlined strong polyclonal CD4+ T cell responses against α-synuclein, β-amyloid, and tau peptides when presented by various HLA subtypes in both cases and controls, thus it is unclear if these responses are effective in limiting disease or are a simple bystander effect (11).
A recent study in humans showed presence of CD4+ and CD8+ T cells in the CSF of PD and AD patients (40), suggesting CD8+ T cell–mediated clearing of amyloid plaques, or a response contributing to neuronal damage. Similarly, Wang et al. recently showed that a microglia-mediated T cell infiltration drives neurodegeneration in a mouse model with tau aggregates; the nefarious adaptive immune response was not observed in mice with amyloid deposition (41). These data suggest that T and B cell responses against tau are common and robust. In our case, we hypothesize that a specific HLA-DRB1*04 restricted response, unlike other adaptive immune responses that may be pathological, targets a particular tau epitope important for the pathological conformation of tau and/or the spreading of aggregates, resulting in a protective effect (SI Appendix, Fig. S7). Interestingly, this may be critical to AD, PD, ALS, and other neurodegenerative diseases but not to 4R Tauopathies such as CBD and PSP, given that no large HLA signal has been reported in small sample GWAS of CBD and PSP (42). In line with this hypothesis, in CBD and PSP, acetyl-K311 may not be involved as polyclonal anti-acetyl-K311 antibodies do not recognize the associated tau aggregates (unlike in Pick’s disease and AD, which are 3R or mixed 3/4R pathologies) (43). Further, CryoEM observations suggest that acetyl-K311 is critical to the formation of helical filaments of AD (mix of 3R/4R tau) (44), but not of tau fibrils of CBD (4R tau) (44).
The fact that HLA-DRB1*04 only binds acetylated forms of PHF6 also supports the involvement of this epitope in the protective effect of HLA-DRB1*04 in AD. With K317 located nearby, the K311 PTM of PHF6 is the most differentiating tau PTM found in AD versus control brains. Further, K311 acetylation has been shown by multiple investigators to promote aggregation of PHF6 in vivo (45), in vitro (46) and in silico (43), while K311 carbamylation is inhibitory (47). Crystallography studies have also shown that acetylated PHF6 dominates in the formation of long fibrils as in neurofibrillary tangles of AD (44, 48). PHF6 is also present in all other known forms of tau aggregates identified to date by cryoEM (49–51). It is also needed for all RT-Quick assays of tau (52). Finally, HLA-DRB1*04-associated subtypes are the only frequent HLA-DRB1 and DQ subtypes with predicted high affinity for this epitope (SI Appendix, Fig. S9), likely explaining why HLA-DRB1*04, and no other subtypes, mediate this effect. An absence of binding for DQA1*01:01~DQB1*05:01 or DQA1*03:01~DQB1*03:02 is for example notable and reported in SI Appendix, Fig. S10. Acetylation at the K311 tau residue may be mediated by SIRT1 and/or HDAC6, current therapeutic targets in AD (45, 53). Similarly, recent evidence suggests that reducing acetylated tau is neuroprotective in brain injury (54).
K311 is not only acetylated, but also methylated, ubiquitinated (32, 55), or succinylated (56), and the epitope is trafficked to the NLRP3 inflammasome of microglial cells (57), where HLA class II presentation of tau fragments by HLA-DRB1*04 to T cells is also likely to occur (58). The involvement of microglial cells in antigen processing and presentation is also suggested by various GWAS association signals observed in AD (4) and PD (5). Although the ubiquitinated K311 epitope is unlikely to bind HLA-DRB1*04 due to steric hindrance at P4, ubiquitination at K317 at P10 could further modulate HLA-DRB1*04 binding and the effect of K311 succinylation or methylation on DRB1*04 binding is unknown. Additional experiments exploring HLA-DRB1*04 subtype binding of PTM segments of PHF6 in various combinations will be needed to further this line of investigation.
Overall, our results indicate that an HLA-DRB1*04-subtype-specific adaptive immune response is protective against both AD and PD. The association with PD is more unexpected, but is in line with recent experiments implicating tau in human PD and in α-synuclein animal models of the disease (1–3, 59). For example, a large inversion that includes the MAPT gene encoding tau, is associated with PD (8, 60) and to a lesser extent with AD (10). These and associated polymorphisms are known to modulate tau levels (61), although other effects could be involved as the genetic inversion affects multiple genes and has pleiotropic effects (62, 63). Finally, tau has been involved in multiple other neurodegenerative diseases (37) and in chronic traumatic encephalopathy as well, suggesting it could be a cofactor in multiple brain diseases (64).
Although it is impossible to exclude the involvement of proteins other than tau in the HLA-DRB1*04 observed effect, CD4+ T cell reactivity toward PHF6 fragments containing the acetylated K311 epitope of tau is a strong candidate for mediating most of the effect (Fig. 4). In vitro assays did not yield strong candidates for DRB1*04-mediated effects in alpha-synuclein (for example within the aggregation prone region), although we did identify a strong binding in the C terminus, whose truncation (65) or phosphorylation (66) could additionally modulate alpha-synuclein aggregation. Our results also open the possibility that targeting tau epitopes containing acetylated-K311 through chimeric antigen receptor T cells or antibodies could have therapeutic values. Further, vaccination with acetylated PHF6-like epitopes could reduce disease progression in HLA-DRB1*04 individuals (~20 to 30% of the population across ancestries). It is noteworthy that antibodies, although not targeting acetylated-K311 per se, but adjacent regions within PHF6, were shown to reduce CSF tau and tau pathology in animal models (67) and are being tested as a means of preventing autosomal dominant forms of AD (68).
Fig. 4.
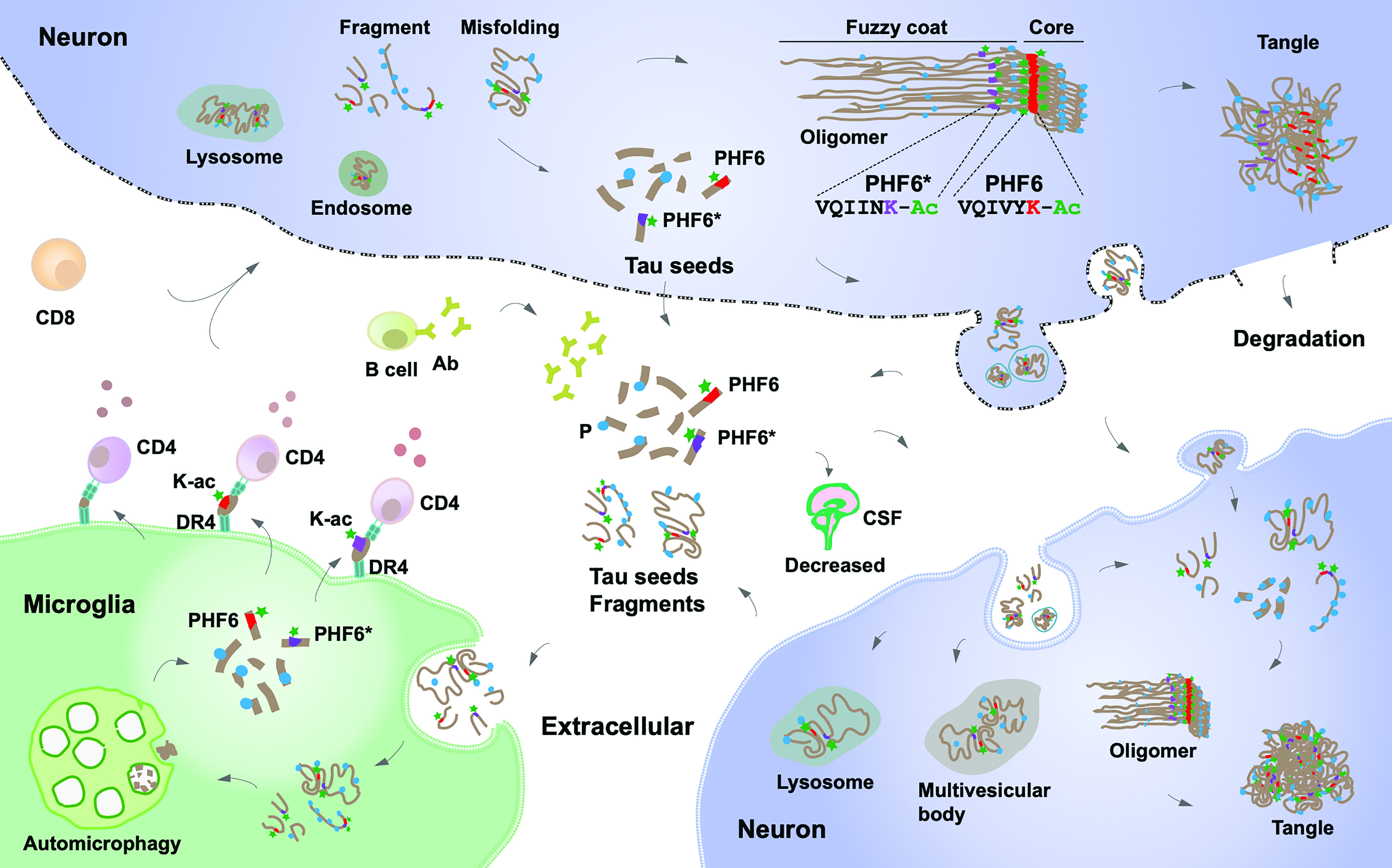
Immune clearance of Acetylated PHF6 tau sequences may reduce neurodegeneration in AD and PD. Pathological tau seeds, soluble tau fragments, or misfolded tau present in the extracellular space are taken up and phagocytosed by activated microglia where it is processed. In addition to autophagy, resulting tau peptide fragments, notably acetylated lysine (K-ac) 311 PHF6 are bound to HLA-DRB1*04:01 or HLA-DRB1*04:04 and the resulting HLA-peptide complexes presented by microglial cells (or other antigen presenting cells) to CD4+ T helper cells. CD4+ T cells trigger beneficial downstream immune responses perhaps involving CD8+ T and antibody producing B cells. These responses limit propagation of misfolded tau and reduce neuropathology, also explaining reduced CSF tau in HLA-DRB1*04 individuals.
Methods
Samples.
Participants or their caregivers provided written informed consent in the original studies. The current study protocol was granted an exemption by the Stanford University institutional review board because the analyses were carried out on deidentified, off-the-shelf data; therefore, additional informed consent was not required.
The AD samples included in the HLA fine-mapping analysis are part of the following datasets with phenotype, genotype ascertainment, and quality control described elsewhere (7, 10, 23, 24, 26, 69–71): the European AD BioBank (EADB), The Genome Research @ Fundació ACE project (GR@ACE), Genetic and Environmental Risk in AD/Defining Genetic, Polygenic and Environmental Risk for AD Consortium (GERAD), the European AD Initiative (EADI), the Norwegian DemGene (DemGene), the Bonn study (Bonn), the Copenhagen City Heart Study (CCHS), the AD Genetics Consortium (ADGC), the Alzheimer Disease Sequencing Project (ADSP), the UK Biobank, the Gwangju Alzheimer’s and Related Dementias (GARD) study, the Japanese Genetic Study Consortium for AD from Niigata University and National Center for Geriatrics and Gerontology (NCGG). The PD samples included in the HLA fine-mapping analysis are part of the following datasets for which the phenotyping, genotyping, and quality control have been described elsewhere (8, 16, 17, 22, 25, 72): International PD Genomics Consortium (IPDGC) NeuroX dataset, McGill University (McGill), National Institute of Neurological Disorders and Stroke (NINDS) Genome-Wide genotyping in PD, NeuroGenetics Research Consortium (NGRC), Oslo PD Study (Oslo), Parkinson’s Progression Markers Initiative (PPMI), Autopsy-Confirmed Parkinson Disease GWAS Consortium (APDGC), the UK Biobank, East Asians samples from Japan, China, Singapore, Taiwan, and Hong-Kong (EastAsians-PD), 23andMe, and the Latin American Research Consortium on the Genetics of PD (LARGE-PD).
The samples assessed for AD and Lewy-body neuropathology included genetic data from the Rush Religious Orders Study and Memory and Aging Project (70) and from the AD Center cohorts 1 to 7 parts of the ADGC (7), and neuropathological assessment followed procedures described, respectively, in ref. 29 and in the National Alzheimer’s Coordinating Center (NACC) postmortem evaluation protocol (30). The samples with CSF biomarkers included in the analysis for which phenotype, genotype ascertainment, and quality control is described elsewhere (10, 73), are mostly part of the EADB. The remaining of this dataset includes samples originating from the Gothenburg H70 Birth Cohort studies and clinical AD samples from Sweden.
Genome-Wide Association at the HLA Locus and Colocalization between AD and PD.
Given the known signal at HLA in AD GWAS (7, 10) and PD GWAS (8), we aimed at refining the signal at the HLA locus using a multiancestry meta-analysis design. We considered a region, ± 1MB around HLA-DRB1, on chromosome 6 from base pair positions (hg38) 31578952 to 33589718. For the PD local-GWAS at the HLA locus, we meta-analyzed the summary statistics from the largest available GWAS to date in European ancestry (8) (distributed without 23andMe), with the Latino-Amerindian GWAS from Kunkle et al. (25) and the Asian GWAS from Kang et al. (22). For the AD local-GWAS at the HLA locus, we meta-analyzed the summary statistics the largest available GWAS to date in European ancestry (10) (which did not include their Stage 2), with the Korean/Japanese GWAS from (24), with in-house local-GWAS at the HLA locus on ADSP and ADGC data carried out by ancestry in European, Latino-Amerindian, African individuals, analyzed with a linear-mixed model as implemented in GENESIS (74) (see Imputation and Statistical Analysis of HLA Alleles, Haplotypes, and Amino Acids section) adjusted for 6 PCs and sex. All meta-analyses were implemented with a fixed-effect inverse variance weighted design implemented in METAL (75). Colocalization between the AD, PD, and ALS, HLA signals in these GWAS was assessed using the Bayesian model implemented in coloc (76) using default priors. We report the posterior probability of colocalization (PP4) between two GWAS associations in Fig. 2.
Imputation and Statistical Analysis of HLA Alleles, Haplotypes, and Amino Acids.
Two-field resolution alleles of HLA-A, HLA-B, HLA-C class I genes, and HLA-DPB1, HLA-DQA1, HLA-DQB1, and HLA-DRB1 class II genes were imputed using R package HIBAG v1.22 (20) for the following dataset: EADB, GR@ACE, GERAD, EADI, DemGene, Bonn, CCHS, UK Biobank, IPDGC, NINDS, NGRC, McGill, Oslo, PPMI, APDGC, LARGE-PD, ADSP, ADGC, GARD, NCGG. When available, training sets specific to ancestry (European, East Asian, Latino, African) and genotyping array were used, either available through HIBAG (20) or trained in-house as previously described (17).
In the allele-level analyses, alleles with an imputation posterior probability lower than 0.5 were considered as undetermined as recommended by HIBAG developers. Each allele was considered as a variant and analyzed under a dominant model; SI Appendix, Supplementary Methods provide details on the analysis per cohort.
In the haplotype-level analyses, only individuals with nonmissing allele genotypes were included in the haplotype-level analysis. Three-locus HLA class I or class II haplotypes were determined using the haplo.em function from the R haplo.stats package. Only haplotypes with posterior probability >0.5 and a carrier frequency of >1% were included in the analysis. Each haplotype was considered as a variant and analyzed under a dominant model; SI Appendix, Supplementary Methods provide details on the analysis per cohort.
In the amino acid–level analyses, HIBAG (20) was used to convert P-coded alleles to amino acid sequences for exon 2, 3 of HLA class I genes, and exon 2 of class II genes. Each amino acid was considered as a variant and analyzed under a dominant model; SI Appendix, Supplementary Methods provide details on the analysis per cohort.
For the East Asians-PD and 23andMe cohorts, the HLA alleles, haplotypes, and amino acids statistics were derived from GWAS summary statistics data using the DISH software (77) as described in ref. 16.
The allele-, haplotype-, amino acid–level analyses were, respectively, meta-analyzed separately between the two neurodegenerative diseases, and across diseases, using a fixed-effect inverse variance weighted design implemented in METAL (75).
Conditional Analyses.
A posteriori conditional analyses were implemented to condition the AD and PD GWAS, on the lead variant common to the two GWAS (rs601945), and separately on the lead amino acid HLA-DRB1 H13 (in complete linkage disequilibrium with H33). The set of conditional analyses were run the amino acid–level analysis, first conditioning on rs601945 and then conditioning on H13. As for the other analyses, these analyses were run across multiple centers and meta-analyzed with METAL (75).
Tau Peptide Binding.
Competition binding studies were conducted as previously described (78). In brief, tau peptides at different concentrations were incubated with DRB1*04:01, DRB1*04:03, DRB1*04:04, DRB1*04:05, or DRB1*04:06 (from the Emory University NIH core tetramer facility http://tetramer.yerkes.emory.edu/support/faq) for 3 d at 37 °C together with biotinylated GAD or EBV (Bio-GAD, EBV). The reaction was quenched by adding neutralization buffer and then transferred into anti-DR antibody precoated on a 96-well plate. DELFIA® time-resolved fluorescence (TRF) intensity was detected using a Tecan SPARK after adding Europium (Eu)-labeled streptavidin. Nonspecific binding was removed through an extensive wash. Each peptide was duplicated. Competitor tau peptide with Eu TRF intensity that was lower than 25% and 25 to 50% of Bio-GAD or EBV epitope alone was considered a strong binder and a weak binder, respectively. Binding for tau peptides significant for an HLA-DRB1*04 allele (Fig. 2) was repeated three times. Two-way repeated measures ANOVA was used to assess the significance of the differential binding of acetylated vs. unacetylated PHF6 (305-SVQIVY[acetylK]PVDLSKVT-319) across molar ratios.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (DOCX)
Acknowledgments
This work was supported by the Michael J. Fox Foundation grant MJFF-020161 (E.M., Z.G.-O.), NIH and National Institute of Aging grants AG060747 (M.D.G.), AG066206 (Z.H.), AG066515 (Z.H., M.D.G.), the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie (grant agreement No. 890650, Y.L.G.), the Alzheimer’s Association (AARF-20-683984, M.E.B.), and the Iqbal Farrukh and Asad Jamal Fund, a grant from the EU Joint Programme—Neurodegenerative Disease Research (European Alzheimer DNA BioBank, EADB; JPND), the Japan Agency for Medical Research and Development JP21dk0207045 (T.I.), JP21dk020704 (K.O., S.N.), JP21km040550 (K.O.), the Einstein Center for Neurosciences in Berlin (S.M.Y.), the Swedish Research Council (#2018-02532, H.Z.), the European Research Council (#681712, H.Z.), and the Swedish State Support for Clinical Research (#ALFGBG-720931, H.Z.). Inserm UMR1167 is also funded by the Inserm, Institut Pasteur de Lille, Lille Métropole Communauté Urbaine, and the French government’s LABEX DISTALZ program (development of innovative strategies for a transdisciplinary approach to AD). Additional funders of individual investigators and institutions who contributed to data collection and genotyping are provided in SI Appendix.
Author contributions
Y.L.G., A.A., J.-C.L., M.D.G., and E.M. designed research; Y.L.G., G.L., A.A., V.D., D.D., Y.J.J., L.L., and J.Z. performed research; Y.L.G., G.L., A.A., V.D., I.J., E.Y., A.N., I.d.R., T.P.L., A.M., C.B., M.M.L., K.P., T.M., B.G.-B., T.N., F.K., W.S., V.A., B.A., M.E.B., L.B., A.B., B.B., M.J.B., P.C., J.C., A.D., S.D., J.-F.D., M.D., C.D., E. During, E. Düzel, D.G., G.G.-R., J.M.G.-A., P.G.-G., V.G., O.G., C.G., E.G., O.H., L.H., S.H.-H., H.H., J.H., D.J., S.K., T.K., K.H.L., C.M., P.M., S. Mehrabian, A.d.M., M.B., P.M., S. Moebus, F.M., B.N., G.N., S.N., B.G.N., G.P., J. Papma, L.P., F.P., P.P., O.P., Y.A.L.P., G.P.-R., J. Popp, L.M.P., R.P., J.P.-T., I.R., L.M.R., S.R.-H., E.R.-R., O.A.R., J.L.R., D.R., N. Scarmeas, P.S., N. Scherbaum, A.S., D.S., I.S., V.S., G.S., A.S., J.v.S., R.S.-V., E.-K.T., T.T., C.T., J.Q.T., L.T., M.V., F.V., M.W., J.W., H.Z., K.B., Z.H., J.W., P.A., F.J., P.G.K., O.A.A., C.V.D., M.T., P.S.J., R.F.-S., K.S., T.T., A.Z., M.I., Y.O., G.R., M.H., J.G., K.O., R.S., J.N.F., W.v.d.F., T.I., A. Ramirez, I.M., A. Ruiz, Z.G.-O., EADB, G.s.g., D.c., DemGene, EADI, GERAD, A.P.D.G.c., J.-C.L., M.D.G., and E.M. contributed new reagents/analytic tools; Y.L.G., G.L., A.A., V.D., I.J., E.Y., A.N., I.d.R., T.P.L., A.M., C.B., M.M.L., T.M., H.P., B.G.-B., T.N., F.K., S.D.T., V.S., W.S., H.Z., K.B., Z.H., J.W., P.A., F.J., P.G.K., O.A.A., C.V.D., M.T., P.S.-J., R.F.-S., K.S., T.T., A.Z., M.I., Y.O., G.R., M.H., J.G., K.O., R.S., J.N.F., W.v.d.F., T.I., A. Ramirez, I.M., A. Ruiz, Z.G.-O., J.-C.L., M.D.G., and E.M. analyzed data; and Y.L.G., G.L., A.A., S.M.Y., J.-C.L., M.D.G., and E.M. wrote the paper. Data used in the preparation of this article were obtained from the EABD, GR@ACE, DEGESCO, DemGene, EADI, GERAD, and Asian Parkinson’s Disease Genetics consortia. As such, the investigators within these consortia contributed to the design and implementation of their respective consortium and/or provided data but did not participate in analysis or writing of this report.
Competing interests
A patent application was filed by G.L. and E.M. on this discovery. Y.L.G. and reviewer F.C. shared the same institution.
Footnotes
Reviewers: F.C., Institut du Cerveau; and M.G., MRC Laboratory of Molecular Biology.
Contributor Information
Yann Le Guen, Email: yleguen@stanford.edu.
Emmanuel Mignot, Email: mignot@stanford.edu.
Collaborators: Céline Bellenguez, Fahri Küçükali, Iris Jansen, Victor Andrade, Sonia Moreno-Grau, Najaf Amin, Benjamin Grenier-Boley, Rafael Campos-Martin, Peter A. Holmans, Anne Boland, Luca Kleineidam, Vincent Damotte, Sven J. van der Lee, Teemu Kuulasmaa, Itziar de Rojas, Amber Yaqub, Ivana Prokic, Marcos R Costa, Julien Chapuis, Shahzad Ahmad, Vilmantas Giedraitis, Dag Aarsland, Pablo Garcia-Gonzalez, Carla Abdelnour, Emilio Alarcón-Martín, Daniel Alcolea, Montserrat Alegret, Ignacio Alvarez, Victoria Álvarez, Nicola J. Armstrong, Tsolaki Anthoula, Ildebrando Appollonio, Marina Arcaro, Silvana Archetti, Alfonso Arias Pastor, Beatrice Arosio, Lavinia Athanasiu, Henri Bailly, Nerisa Banaj, Miquel Baquero, Ana Belén Pastor, Luisa Benussi, Claudine Berr, Céline Besse, Valentina Bessi, Giuliano Binetti, Alessandra Bizarro, Rafael Blesa, Mercè Boada, Barbara Borroni, Silvia Boschi, Paola Bossù, Geir Bråthen, Catherine Bresner, Henry Brodaty, Keeley J. Brookes, Luis Ignacio Brusco, Dolores Buiza-Rueda, Katharina BÛrger, Vanessa Burholt, Miguel Calero, Geneviève Chene, Ángel Carracedo, Roberta Cecchetti, Laura Cervera-Carles, Camille Charbonnier, Caterina Chillotti, Simona Ciccone, Jurgen A.H.R. Claassen, Jordi Clarimon, Christopher Clark, Elisa Conti, Anaïs Corma-Gómez, Emanuele Costantini, Carlo Custodero, Delphine Daian, Maria Carolina Dalmasso, Antonio Daniele, Efthimios Dardiotis, Jean-François Dartigues, Peter Paul de Deyn, Stéphanie Debette, Jürgen Deckert, Teodoro del Ser, Nicola Denning, Martin Dichgans, Janine Diehl-Schmid, Mónica Diez-Fairen, Paolo Dionigi Rossi, Srdjan Djurovic, Emmanuelle Duron, Emrah Düzel, Carole Dufouil, Valentina Escott-Price, Ana Espinosa, Michael Ewers, Marta Fernández-Fuertes, Catarina B Ferreira, Evelyn Ferri, Bertrand Fin, Peter Fischer, Tormod Fladby, Klaus Fließbach, Juan Fortea, Silvia Fostinelli, Nick C. Fox, Emlio Franco-Macías, María J. Bullido, Ana Frank-García, Lutz Froelich, Daniela Galimberti, Jose Maria García-Alberca, Pablo García-González, Sebastian Garcia-Madrona, Guillermo Garcia-Ribas, Roberta Ghidoni, Ina Giegling, Giaccone Giorgio, Oliver Goldhardt, Antonio González-Pérez, Caroline Graff, Giulia Grande, Emma Green, Timo Grimmer, Edna Grünblatt, Tamar Guetta-Baranes, Annakaisa Haapasalo, Georgios Hadjigeorgiou, Harald Hampel, Olivier Hanon, John Hardy, Annette M. Hartmann, Lucrezia Hausner, Janet Harwood, Stefanie Heilmann-Heimbach, Seppo Helisalmi, Michael T. Heneka, Isabel Hernández, Martin J. Herrmann, Per Hoffmann, Clive Holmes, Henne Holstege, Raquel Huerto Vilas, Marc Hulsman, Charlotte Johansson, Lena Kilander, Anne Kinhult Ståhlbom, Miia Kivipelto, Anne Koivisto, Johannes Kornhuber, Mary H. Kosmidis, Carmen Lage, Erika J Laukka, Alessandra Lauria, Jenni Lehtisalo, Ondrej Lerch, Alberto Lleó, Adolfo Lopez de Munain, Malin Löwemark, Lauren Luckcuck, Juan Macías, Catherine A. MacLeod, Wolfgang Maier, Francesca Mangialasche, Spallazzi Marco, Marta Marquié, Rachel Marshall, Angel Martín Montes, Carmen Martínez Rodríguez, Carlo Masullo, Simon Mead, Patrizia Mecocci, Miguel Medina, Alun Meggy, Shima Mehrabian, Silvia Mendoza, Manuel Menéndez-González, Pablo Mir, Susanne Moebus, Merel Mol, Laura Molina-Porcel, Laura Montrreal, Laura Morelli, Fermin Moreno, Kevin Morgan, Markus M Möthen, Carolina Muchnik, Benedetta Nacmias, Tiia Ngandu, Gael Nicolas, Børge G. Nordestgaard, Robert Olaso, Adelina Orellana, Michela Orsini, Gemma Ortega, Alessandro Padovani, Caffarra Paolo, Goran Papenberg, Lucilla Parnetti, Pau Pastor, Alba Pérez-Cordón, Jordi Pérez-Tur, Pierre Pericard, Oliver Peters, Yolande A.L. Pijnenburg, Juan A Pineda, Gerard Piñol-Ripoll, Claudia Pisanu, Thomas Polak, Julius Popp, Danielle Posthuma, Josef Priller, Raquel Puerta, Olivier Quenez, Inés Quintela, Jesper Qvist Thomassen, Alberto Rábano, Innocenzo Rainero, Inez Ramakers, Luis M Real, Marcel J.T. Reinders, Steffi Riedel-Heller, Peter Riederer, Natalia Roberto, Eloy Rodriguez-Rodriguez, Arvid Rongve, Irene Rosas Allende, Maitée Rosende-Roca, Jose Luis Royo, Elisa Rubino, Dan Rujescu, María Eugenia Sáez, Paraskevi Sakka, Ingvild Saltvedt, Ángela Sanabria, María Bernal Sánchez-Arjona, Florentino Sanchez-Garcia, Pascual Sánchez Juan, Raquel Sánchez-Valle, Sigrid B Sando, Michela Scamosci, Nikolaos Scarmeas, Elio Scarpini, Philip Scheltens, Norbert Scherbaum, Martin Scherer, Matthias Schmid, Anja Schneider, Jonathan M. Schott, Geir Selbæk, Davide Seripa, Alexey A Shadrin, Olivia Skrobot, Hilkka Soininen, Vincenzo Solfrizzi, Alina Solomon, Sandro Sorbi, Oscar Sotolongo-Grau, Gianfranco Spalletta, Annika Spottke, Alessio Squassina, Eystein Stordal, Juan Pablo Tartan, Lluís Tárraga, Niccolo Tesí, Anbupalam Thalamuthu, Tegos Thomas, Latchezar Traykov, Lucio Tremolizzo, Anne Tybjærg-Hansen, Andre Uitterlinden, Abbe Ullgren, Ingun Ulstein, Sergi Valero, Aad van der Lugt, Jasper Van Dongen, Jeroen van Rooij, John van Swieten, Rik Vandenberghe, Frans Verhey, Jean-Sébastien Vidal, Jonathan Vogelgsang, Martin Vyhnalek, Michael Wagner, David Wallon, Leonie Weinhold, Jens Wiltfang, Gill Windle, Bob Woods, Mary Yannakoulia, Miren Zulaica, Jan Laczo, Vaclav Matoska, Maria Serpente, Francesca Assogna, Fabrizio Piras, Federica Piras, Valentina Ciullo, Jacob Shofany, Carlo Ferrarese, Simona Andreoni, Gessica Sala, Chiara Paola Zoia, Maria Del Zompo, Alberto Benussi, Patrizia Bastiani, Mari Takalo, Teemu Natunen, Tiina Laatikainen, Jaakko Tuomilehto, Riitta Antikainen, Timo Strandberg, Jaana Lindström, Markku Peltonen, Richard Abraham, Ammar Al-Chalabi, Nicholas J. Bass, Carol Brayne, Kristelle S. Brown, John Collinge, David Craig, Pangiotis Deloukas, Nick Fox, Amy Gerrish, Michael Gill, Rhian Gwilliam, John Hardy, Denise Harold, Paul Hollingworth, Jarret A. Johnston, Lesley Jones, Brian Lawlor, Gill Livingston, Simon Lovestone, Michelle Lupton, Aoibhinn Lynch, David Mann, Bernadette McGuinness, Andrew McQuillin, Michael C. O’Donovan, Michael J. Owen, Peter Passmore, John F Powell, Petra Proitsi, Martin Rossor, Christopher E. Shaw, A. David Smith, Hugh Gurling, Stephen Todd, Catherine Mummery, Nathalie Ryan, Giordano Lacidogna, Ad Adarmes-Gómez, Ana Mauleón, Ana Pancho, Anna Gailhajenet, Asunción Lafuente, D Macias-García, Elvira Martín, Esther Pelejà, F Carrillo, Isabel Sastre Merlín, L Garrote-Espina, Liliana Vargas, M Carrion-Claro, M Marín, Ma Labrador, Mar Buendia, María Dolores Alonso, Marina Guitart, Mariona Moreno, Marta Ibarria, Mt Periñán, Nuria Aguilera, P Gómez-Garre, Pilar Cañabate, R Escuela, R Pineda-Sánchez, R Vigo-Ortega, S Jesús, Silvia Preckler, Silvia Rodrigo-Herrero, Susana Diego, Alessandro Vacca, Fausto Roveta, Nicola Salvadori, Elena Chipi, Henning Boecker, Christoph Laske, Robert Perneczky, Costas Anastasiou, Daniel Janowitz, Rainer Malik, Anna Anastasiou, Kayenat Parveen, Carmen Lage, Sara López-García, Anna Antonell, Kalina Yonkova Mihova, Diyana Belezhanska, Heike Weber, Silvia Kochen, Patricia Solis, Nancy Medel, Julieta Lisso, Zulma Sevillano, Daniel G Politis, Valeria Cores, Carolina Cuesta, Cecilia Ortiz, Juan Ignacio Bacha, Mario Rios, Aldo Saenz, Mariana Sanchez Abalos, Eduardo Kohler, Dana Lis Palacio, Ignacio Etchepareborda, Matias Kohler, Gisela Novack, Federico Ariel Prestia, Pablo Galeano, Eduardo M. Castaño, Sandra Germani, Carlos Reyes Toso, Matias Rojo, Carlos Ingino, Carlos Mangone, Sebastiaan Engelborghs, Tagliavini Fabrizio, Sune Fallgaard Nielsen, Lucia Farotti, Chiara Fenoglio, Geert Jan Biessels, Seth Love, Patrick G. Kehoe, Florence Pasquier, Christine Van Broeckhoven, David C. Rubinsztein, Stefan Teipel, Nathalie Fievet, Vincent Deramecourt, Charlotte Forsell, Håkan Thonberg, Maria Bjerke, Ellen De Roeck, María Teresa Martínez-Larrad, Natividad Olivar, Mohsen Ghanbari, Perminder Sachdev, Karen Mather, Frank Jessen, M. Arfan Ikram, Alexandre de Mendonça, Jakub Hort, Tsolaki Magda, Philippe Amouyel, Julie Williams, Ruth Frikke-Schmidt, Jordi Clarimon, Jean-François Deleuze, Giacomina Rossi, Ole A. Andreassen, Martin Ingelsson, Mikko Hiltunen, Kristel Sleegers, Cornelia M. van Duijn, Rebecca Sims, Wiesje M. van der Flier, Agustín Ruiz, Alfredo Ramirez, Jean-Charles Lambert, N Aguilera, E Alarcon, M Alegret, M Boada, M Buendia, A Cano, P Cañabate, A Carracedo, A Corbatón-Anchuelo, I de Rojas, S Diego, A Espinosa, A Gailhajenet, P García-González, M Guitart, A González-Péerez, M Ibarria, A Lafuente, J Macias, O Maroñas, E Martín, MT Martínez, M Marquié, L Montrreal, S Moreno-Grau, M Moreno, R. Nuñez-Llaves, C Olivé, A Orellana, G Ortega, A Pancho, E Pelejà, A Pérez-Cordon, JA Pineda, R Puerta, S Preckler, I Quintela, LM Real, M Rosende-Roca, A Ruiz, ME Sáez, A Sanabria, M Serrano-Rios, O Sotolongo-Grau, L Táarraga, S Valero, L Vargas, AD Adarmes-Gómez, E Alarcón-Martín, MD Alonso, I Álvarez, V Álvarez, G Amer-Ferrer, M Antequera, C Antúnez, M Baquero, M Bernal, R Blesa, M Boada, D Buiza-Rueda, MJ Bullido, JA Burguera, M Calero, F Carrillo, M Carrión-Claro, MJ Casajeros, J Clarimón, JM Cruz-Gamero, MM de Pancorbo, I de Rojas, T del Ser, M Diez-Fairen, R Escuela, L Garrote-Espina, J Fortea, E Franco, A Frank-García, JM García-Alberca, S Garcia Madrona, G Garcia-Ribas, P Gómez-Garre, S Hevilla, S Jesús, MA Labrador Espinosa, C Lage, A Legaz, A Lleóo, A Lóopez de Munáin, S López-García, D Macias-García, S Manzanares, M Marín, J Marín-Muñoz, T Marín, M Marquié, A Martín Montes, B Martínez, C Martínez, V Martínez, A Martínez-Lage Álvarez, M Medina, M Mendioroz Iriarte, M Menéndez-González, P Mir, L Montrreal, A Orellana, P Pastor, J Péerez Tur, T Periñán-Tocino, R Pineda-Sánchez, G Piñol Ripoll, A Ráabano, D Real de Asúa, S Rodrigo, E Rodríguez-Rodríguez, JL Royo, A Ruiz, R Sanchez del Valle Díaz, P Sánchez-Juan, I Sastre, O Sotolongo-Grau, S Valero, MP Vicente, R Vigo-Ortega, L Vivancos, Alexey A Shadrin, Shahram Bahrami, Arvid Rongve, Geir Bråthen, Ingunn Bosnes, Eystein Stordal, Lavinia Athanasiu, Per Selnes, Ingvild Saltvedt, Sigrid B. Sando, Sverre Bergh, Ingun Ulstein, Srdjan Djurovic, Tormod Fladby, Dag Aarsland, Geir Selbæk, Ole A. Andreassen, Céline Bellenguez, Benjamin Grenier-Boley, Jacques Epelbaum, David Wallon, Didier Hannequin, Florence Pasquier, Claudine Berr, Jean-Francois Dartigues, Dominique campion, Christophe Tzourio, Vincent Dermecourt, Nathalie Fievet, Olivier Hanon, Carole Dufouil, Alexis Brice, Bruno Dubois, Karen Ritchie, Phillippe Amouyel, Jean-Charles Lambert, Denise Harold, Paul Hollingworth, Rebecca Sims, Amy Gerrish, Nicola Denning, Amy Williams, Charlene Thomas, Alun Meggy, Rachel Marshall, Chloe Davies, Lauren Luckcuck, William Nash, Kimberley Dowzell, Atahualpa Castillo Morales, Mateus Bernardo-Harrington, Patrick Kehoe, Per Hoffmann, Seth Love, James Turton, Jenny Lord, Kristelle Brown, Kevin Morgan, Emma Vardy, Elizabeth Fisher, Jason D. Warren, Jonathan M. Schott, Martin Rossor, Natalie S. Ryan, Nick C. Fox, Rita Guerreiro, Simon Mead, James Uphill, John Collinge, Michelle Lupton, Ammar Al-Chalabi, Christopher E. Shaw, Nick Bass, Richard Abraham, Reinhard Heun, Heike Kölsch, Britta Schürmann, Frank Jessen, Wolfgang Maier, ndré Lacour, Christine Herold, Simon Lovestone, Bernadette McGuinness, David Craig, Janet A. Johnston, Michael Gill, Peter Passmore, Stephen Todd, John Powell, Petra Proitsi, Yogen Patel, Angela Hodges, Tim Becker, A. David Smith, Donald Warden, Gordon Wilcock, Robert Clarke, Aoibhinn Lynch, Brian Lawlor, Michael Gill, Andrew McQuillin, Gill Livingston, John Hardy, David C. Rubinsztein, Carol Brayne, Rhian Gwilliam, Panagiotis Deloukas, Yoav Ben-Shlomo, David Mann, Nigel M. Hooper, Stuart Pickering-Brown, Clive Holmes, Rebecca Sussams, Nick Warner, Anthony Bayer, Andrew B. Singleton, Annette M Hartmann, Dan Rujescu, Ina Giegling, Harald Hampel, Martin Dichgans, Isabella Heuser, Dmitriy Drichel, Norman Klopp, Markus M. Nöthen, Manuel Mayhaus, Matthias Riemenschneider, Sabrina Pinchler, Thomas Feulner, Wei Gu, van Hendrik den Bussche, Martin Scherer, Jens Wiltfang, Johannes Kornhuber, Michael Hüll, Lutz Frölich, H-Erich Wichmann, Karl-Heinz Jöckel, Susanne Moebus, Steffi Riedel-Heller, John Kauwe, John Morris, Kevin Mayo, Magda Tsolaki, Michael O’Donovan, Lesley Jones, Michael Owen, Valentina Escott-Price, Alfredo Ramirez, Peter Holmans, Julie Williams, Jia Nee Foo, Elaine Guo Yan Chew, Sun Ju Chung, Rong Peng, Yinxia Chao, Louis CS Tan, Moses Tandiono, Michelle M Lian, Ebonne Y Ng, Kumar-M. Prakash, Wing-Lok Au, Wee-Yang Meah, Shi Qi Mok, Azlina Ahmad Annuar, Anne YY Chan, Ling Chen, Yongping Chen, Beom S Jeon, Lulu Jiang, Jia Lun Lim, Juei-Jueng Lin, Chunfeng Liu, Chengjie Mao, Vincent Mok, Zhong Pei, Hui-Fang Shang, Chang-He Shi, Kyuyoung Song, Ai Huey Tan, Yih-Ru Wu, Yu-ming Xu, Renshi Xu, Yaping Yan, Jing Yang, Bao Rong Zhang, Woon-Puay Koh, Shen-Yang Lim, Chiea Chuen Khor, Jianjun Liu, and Eng-King Tan
Data, Materials, and Software Availability
All HLA -alleles, -haplotypes, -amino-acid levels associations derived from this study are available per cohort in Datasets S1–S5, as well as the list of tau and alpha-synuclein peptides that were tested for binding.
Supporting Information
References
- 1.Lei P., et al. , Tau protein: Relevance to Parkinson’s disease. Int. J. Biochem. Cell Biol. 42, 1775–1778 (2010). [DOI] [PubMed] [Google Scholar]
- 2.Pan L., et al. , Tau accelerates α-synuclein aggregation and spreading in Parkinson’s disease. Brain 145, 3454–3471 (2022). [DOI] [PubMed] [Google Scholar]
- 3.Vermilyea S. C., et al. , Loss of tau expression attenuates neurodegeneration associated with α-synucleinopathy. Transl. Neurodegener. 11, 34 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li Y., et al. , Genomics of Alzheimer’s disease implicates the innate and adaptive immune systems. Cell Mol. Life Sci. 78, 7397–7426 (2021), 10.1007/s00018-021-03986-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Andersen M. S., et al. , Heritability enrichment implicates microglia in Parkinson’s disease pathogenesis. Ann. Neurol. 89, 942–951 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Keren-Shaul H., et al. , A unique microglia type associated with restricting development of Alzheimer’s disease. Cell 169, 1276–1290.e17 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Kunkle B. W., et al. , Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing. Nat. Genet. 51, 414–430 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nalls M. A., et al. , Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 18, 1091–1102 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Rheenen W., et al. , Common and rare variant association analyses in amyotrophic lateral sclerosis identify 15 risk loci with distinct genetic architectures and neuron-specific biology. Nat. Genet. 53, 1636–1648 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bellenguez C., et al. , New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat. Genet. 54, 412–436 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dhanwani R., et al. , T cell responses to neural autoantigens are similar in Alzheimer’s disease patients and age-matched healthy controls. Front. Neurosci. 14, 874 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sulzer D., et al. , T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 546, 656–661 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lindestam Arlehamn C. S., et al. , α-Synuclein-specific T cell reactivity is associated with preclinical and early Parkinson’s disease. Nat. Commun. 11, 1875 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kuhn I., et al. , Serum titers of autoantibodies against α-synuclein and tau in child- and adulthood. J. Neuroimmunol. 315, 33–39 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Hromadkova L., Ovsepian S. V., Tau-reactive endogenous antibodies: Origin, functionality, and implications for the pathophysiology of Alzheimer’s disease. J. Immunol. Res. 2019, 7406810 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Naito T., et al. , Trans-ethnic fine-mapping of the major histocompatibility complex region linked to Parkinson’s disease. Mov. Disord. 36, 1805–1814 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yu E., et al. , Fine mapping of the HLA locus in Parkinson’s disease in Europeans. NPJ Parkinsons Dis. 7, 84 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hollenbach J. A., et al. , A specific amino acid motif of HLA-DRB1 mediates risk and interacts with smoking history in Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A. 116, 7419–7424 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wightman D. P., et al. , A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet. 53, 1276–1282 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng X., et al. , HIBAG—HLA genotype imputation with attribute bagging. Pharmacogenomics J. 14, 192–200 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsuki K., et al. , DQ (rather than DR) gene marks susceptibility to narcolepsy. Lancet 339, 1052 (1992). [DOI] [PubMed] [Google Scholar]
- 22.Foo J. N., et al. , Identification of risk loci for Parkinson disease in asians and comparison of risk between Asians and Europeans: A genome-wide association study. JAMA Neurol. 77, 746–754 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kang S., et al. , Potential novel genes for late-onset Alzheimer’s disease in East-Asian descent identified by APOE-stratified genome-wide association study. J. Alzheimers Dis. 82, 1451–1460 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shigemizu D., et al. , Ethnic and trans-ethnic genome-wide association studies identify new loci influencing Japanese Alzheimer’s disease risk. Transl. Psychiatry 11, 151 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Loesch D. P., et al. , Characterizing the genetic architecture of Parkinson’s disease in latinos. Ann. Neurol. 90, 353–365 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kunkle B. W., et al. , Novel Alzheimer disease risk loci and pathways in African American individuals using the African genome resources panel: A meta-analysis. JAMA Neurol. 78, 102 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jun G. R., et al. , Transethnic genome-wide scan identifies novel Alzheimer’s disease loci. Alzheimers Dement. 13, 727–738 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chia R., et al. , Genome sequencing analysis identifies new loci associated with Lewy body dementia and provides insights into its genetic architecture. Nat. Genet. 53, 294–303 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schneider J. A., et al. , Cognitive impairment, decline and fluctuations in older community-dwelling subjects with Lewy bodies. Brain 135, 3005–3014 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Besser L. M., et al. , The revised national Alzheimer’s coordinating center’s neuropathology form-available data and new analyses. J. Neuropathol. Exp. Neurol. 77, 717–726 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Blauwendraat C., et al. , Parkinson’s disease age at onset genome-wide association study: Defining heritability, genetic loci, and α-synuclein mechanisms. Mov. Disord. 34, 866–875 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wesseling H., et al. , Tau PTM profiles identify patient heterogeneity and stages of Alzheimer’s disease. Cell 183, 1699–1713.e13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Scally S. W., et al. , A molecular basis for the association of the HLA-DRB1 locus, citrullination, and rheumatoid arthritis. J. Exp. Med. 210, 2569–2582 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ting Y. T., et al. , The interplay between citrullination and HLA-DRB1 polymorphism in shaping peptide binding hierarchies in rheumatoid arthritis. J. Biol. Chem. 293, 3236–3251 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.von Bergen M., et al. , Assembly of τ protein into Alzheimer paired helical filaments depends on a local sequence motif (306VQIVYK311) forming β structure. Proc. Natl. Acad. Sci. U.S.A. 97, 5129–5134 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Giasson B. I., Lee V. M.-Y., Trojanowski J. Q., Interactions of amyloidogenic proteins. Neuromol. Med. 4, 49–58 (2003). [DOI] [PubMed] [Google Scholar]
- 37.Sengupta U., Kayed R., Amyloid β, Tau, and α-Synuclein aggregates in the pathogenesis, prognosis, and therapeutics for neurodegenerative diseases. Prog. Neurobiol. 214, 102270 (2022). [DOI] [PubMed] [Google Scholar]
- 38.Raposo B., et al. , T cells specific for post-translational modifications escape intrathymic tolerance induction. Nat. Commun. 9, 353 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singhania A., et al. , The TCR repertoire of α-synuclein-specific T cells in Parkinson’s disease is surprisingly diverse. Sci. Rep. 11, 302 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gate D., et al. , Clonally expanded CD8 T cells patrol the cerebrospinal fluid in Alzheimer’s disease. Nature 577, 399–404 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen X., et al. , Microglia-mediated T cell infiltration drives neurodegeneration in tauopathy. Nature 615, 668–677 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jabbari E., et al. , Genetic determinants of survival in progressive supranuclear palsy: A genome-wide association study. Lancet Neurol. 20, 107–116 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kim Y.-C., Jeong B.-H., In silico evaluation of acetylation mimics in the 27 lysine residues of human tau protein. Curr. Alzheimer Res. 16, 379–387 (2019). [DOI] [PubMed] [Google Scholar]
- 44.Arakhamia T., et al. , Posttranslational modifications mediate the structural diversity of tauopathy strains. Cell 180, 633–644.e12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Trzeciakiewicz H., et al. , An HDAC6-dependent surveillance mechanism suppresses tau-mediated neurodegeneration and cognitive decline. Nat. Commun. 11, 5522 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ganguly P., et al. , Tau assembly: The dominant role of PHF6 (VQIVYK) in microtubule binding region repeat R3. J. Phys. Chem. B. 119, 4582–4593 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guru KrishnaKumar V., Baweja L., Ralhan K., Gupta S., Carbamylation promotes amyloidogenesis and induces structural changes in Tau-core hexapeptide fibrils. Biochim. Biophys. Acta Gen. Subj. 1862, 2590–2604 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Vaquer-Alicea J., Diamond M. I., Joachimiak L. A., Tau strains shape disease. Acta Neuropathol. 142, 57–71 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Scheres S. H., Zhang W., Falcon B., Goedert M., Cryo-EM structures of tau filaments. Curr. Opin. Struct. Biol. 64, 17–25 (2020). [DOI] [PubMed] [Google Scholar]
- 50.Lövestam S., et al. , Assembly of recombinant tau into filaments identical to those of Alzheimer’s disease and chronic traumatic encephalopathy. Elife 11, e76494 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goedert M., Cryo-EM structures of τ filaments from human brain. Essays Biochem. 65, 949–959 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Metrick M. A., et al. , A single ultrasensitive assay for detection and discrimination of tau aggregates of Alzheimer and Pick diseases. Acta Neuropathol. Commun. 8, 22 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Park S., Lee J. H., Jeon J. H., Lee M. J., Degradation or aggregation: The ramifications of post-translational modifications on tau. BMB Rep. 51, 265–273 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shin M.-K., et al. , Reducing acetylated tau is neuroprotective in brain injury. Cell 184, 2715–2732.e23 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kontaxi C., Piccardo P., Gill A. C., Lysine-directed post-translational modifications of tau protein in Alzheimer’s disease and related tauopathies. Front. Mol. Biosci. 4, 56 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Acosta D. M., Mancinelli C., Bracken C., Eliezer D., Post-translational modifications within tau paired helical filament nucleating motifs perturb microtubule interactions and oligomer formation. J. Biol. Chem. 298, 101442 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Panda C., et al. , Aggregated Tau-PHF6 (VQIVYK) potentiates NLRP3 inflammasome expression and autophagy in human microglial cells. Cells 10, 1652 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Carmona-Abellan M., et al. , Microglia is associated with p-Tau aggregates in the olfactory bulb of patients with neurodegenerative diseases. Neurol. Sci. 42, 1473–1482 (2021). [DOI] [PubMed] [Google Scholar]
- 59.Henderson M. X., Sengupta M., Trojanowski J. Q., Lee V. M. Y., Alzheimer’s disease tau is a prominent pathology in LRRK2 Parkinson’s disease. Acta Neuropathol. Commun. 7, 183 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Simón-Sánchez J., et al. , Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 41, 1308–1312 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sarnowski C., et al. , Meta-analysis of genome-wide association studies identifies ancestry-specific associations underlying circulating total tau levels. Commun. Biol. 5, 336 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Koks S., Pfaff A. L., Bubb V. J., Quinn J. P., Transcript variants of genes involved in neurodegeneration are differentially regulated by the APOE and MAPT haplotypes. Genes (Basel) 12, 423 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Leveille E., Ross O. A., Gan-Or Z., Tau and MAPT genetics in tauopathies and synucleinopathies. Parkinsonism Relat. Disord. 90, 142–154 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Holper S., Watson R., Yassi N., Tau as a biomarker of neurodegeneration. Int. J. Mol. Sci. 23, 7307 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Spillantini M. G., Crowther R. A., Jakes R., Hasegawa M., Goedert M., α-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc. Natl. Acad. Sci. U.S.A. 95, 6469–6473 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ghanem S. S., et al. , α-Synuclein phosphorylation at serine 129 occurs after initial protein deposition and inhibits seeded fibril formation and toxicity. Proc. Natl. Acad. Sci. U.S.A. 119, e2109617119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sankaranarayanan S., et al. , Passive immunization with phospho-tau antibodies reduces tau pathology and functional deficits in two distinct mouse tauopathy models. PLoS One 10, e0125614 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Šimić G., et al. , Tau protein hyperphosphorylation and aggregation in Alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomolecules 6, 6 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Beecham G. W., et al. , The Alzheimer’s Disease sequencing project: Study design and sample selection. Neurol. Genet. 3, e194 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bennett D. A., et al. , Overview and findings from the rush memory and aging project. Curr. Alzheimer Res. 9, 646–663 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Le Guen Y., et al. , Association of African ancestry-specific APOE missense variant R145C with risk of Alzheimer disease. JAMA 329, 551–560 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Satake W., et al. , Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet. 41, 1303–1307 (2009). [DOI] [PubMed] [Google Scholar]
- 73.Najar J., et al. , Polygenic risk scores for Alzheimer’s disease are related to dementia risk in APOE ɛ4 negatives. Alzheimer’s Dementia (Amst). 13, e12142 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gogarten S. M., et al. , Genetic association testing using the GENESIS R/Bioconductor package. Bioinformatics 35, 5346–5348 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Willer C. J., Li Y., Abecasis G. R., METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics 26, 2190–2191 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Giambartolomei C., et al. , Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genetics 10, e1004383 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lim J., Bae S.-C., Kim K., Understanding HLA associations from SNP summary association statistics. Sci. Rep. 9, 1337 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Luo G., et al. , Autoimmunity to hypocretin and molecular mimicry to flu in type 1 narcolepsy. Proc. Natl. Acad. Sci. U.S.A. 115, E12323–E12332 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (DOCX)
Data Availability Statement
All HLA -alleles, -haplotypes, -amino-acid levels associations derived from this study are available per cohort in Datasets S1–S5, as well as the list of tau and alpha-synuclein peptides that were tested for binding.