

Significance
Rhabdomyosarcoma is a lethal pediatric cancer for which new therapies are needed. In this study, we developed a high-throughput genetic screening method to identify genes that cause rhabdomyosarcoma cells to differentiate into normal muscle. We used this platform to discover the protein NF-Y as an important molecule that contributes to rhabdomyosarcoma biology. CRISPR-based genetic targeting of NF-Y converts rhabdomyosarcoma cells into differentiated muscle, and we reveal the mechanism by which this occurs. Since many forms of human sarcoma exhibit a defect in cell differentiation, the methodology described here might have broad relevance for the investigation of these tumors. In addition, our findings suggest that NF-Y could serve as a molecular target for the development of differentiation therapy in rhabdomyosarcoma.
Keywords: rhabdomyosarcoma, myo-differentiation, PAX3–FOXO1, NF-Y, muscle
Abstract
Recurrent chromosomal rearrangements found in rhabdomyosarcoma (RMS) produce the PAX3–FOXO1 fusion protein, which is an oncogenic driver and a dependency in this disease. One important function of PAX3–FOXO1 is to arrest myogenic differentiation, which is linked to the ability of RMS cells to gain an unlimited proliferation potential. Here, we developed a phenotypic screening strategy for identifying factors that collaborate with PAX3–FOXO1 to block myo-differentiation in RMS. Unlike most genes evaluated in our screen, we found that loss of any of the three subunits of the Nuclear Factor Y (NF-Y) complex leads to a myo-differentiation phenotype that resembles the effect of inactivating PAX3–FOXO1. While the transcriptomes of NF-Y- and PAX3–FOXO1-deficient RMS cells bear remarkable similarity to one another, we found that these two transcription factors occupy nonoverlapping sites along the genome: NF-Y preferentially occupies promoters, whereas PAX3–FOXO1 primarily binds to distal enhancers. By integrating multiple functional approaches, we map the PAX3 promoter as the point of intersection between these two regulators. We show that NF-Y occupies CCAAT motifs present upstream of PAX3 to function as a transcriptional activator of PAX3–FOXO1 expression in RMS. These findings reveal a critical upstream role of NF-Y in the oncogenic PAX3–FOXO1 pathway, highlighting how a broadly essential transcription factor can perform tumor-specific roles in governing cellular state.
Rhabdomyosarcoma (RMS) is an aggressive pediatric tumor composed of cells that resemble myoblasts of the skeletal muscle lineage (1, 2). However, the myoblast-like state of RMS is distinct from normal myogenesis because of a prominent defect in terminal differentiation (3–7). For example, RMS cells fail to express genes that encode the contractile apparatus, including myosin heavy and light chains, troponins, tropomyosins, myomesins, and actinins (8). Since terminal myo-differentiation is coupled with an arrest in cell proliferation, a long-standing interest exists in developing therapies that allow RMS cells to regain their potential for terminal muscle differentiation (5, 9–11).
One of the most common genetic alterations that initiate RMS is chromosomal translocations that produce the PAX3–FOXO1 fusion oncoprotein (12–15). The presence of PAX3–FOXO1, in combination with other mutations, in human cell culture systems (16–20) and animal models (21, 22) leads to the formation of RMS-like tumors, which recapitulate the block in myo-differentiation that defines this disease (23). In addition, multiple genetic approaches have demonstrated that RMS cells harboring PAX3–FOXO1 remain dependent on this oncoprotein to sustain tumor cell growth and viability (24–29). Inactivation of PAX3–FOXO1 triggers RMS myo-differentiation in association with upregulation of multiple terminal myogenic markers, including myosin heavy chain proteins (24, 30). Collectively, these findings implicate PAX3–FOXO1 as a powerful driver and a genetic dependency in RMS, and hence a compelling therapeutic target in this disease.
The importance of PAX3–FOXO1 in RMS has provided a strong rationale to understand the molecular details of its transcriptional function. PAX3–FOXO1 retains the N-terminal DNA-binding domains of Paired Box 3 (PAX3), which becomes fused to the potent C-terminal trans-activation domain of Forkhead Box O1 (FOXO1). During normal myogenesis, wild-type PAX3 is critical for lineage commitment and for the survival of progenitor cells (31). While wild-type PAX3 becomes attenuated as myoblasts undergo terminal differentiation (32), the gain-of-function attribute of PAX3–FOXO1 results in a sustained and dysregulated transcriptional output that blocks terminal differentiation (33, 34). To carry out its transcriptional function, PAX3–FOXO1 collaborates with several general chromatin regulators at distal enhancer DNA elements, including SMARCA4 (35) and CHD4 (36) chromatin remodeling ATPases and the bromodomain protein BRD4 (37). While much of our knowledge of PAX3–FOXO1 mechanisms has been obtained using biochemical and epigenomic methods, a high-throughput genetic screening strategy has yet to be applied to PAX3–FOXO1 in search of cooperating factors in an unbiased manner.
NF-Y (Nuclear Factor Y) is a heterotrimeric complex composed of NF-YA, NF-YB, and NF-YC, which performs a transcriptional activation function primarily at promoter regions in mammalian cells (38). A remarkable attribute of NF-Y is its use of a nucleosome-like mechanism of DNA wrapping via the histone fold domains of NF-YB and NF-YC, in conjunction with a sequence-specific DNA-binding domain of NF-YA, which together lead to tethering of NF-Y at CCAAT DNA sequences (39). The NF-Y complex is ubiquitously expressed and regulates hundreds of genes in proliferating cell types during development (40, 41), and its activity is commonly elevated in cancer (42). In addition, the CCAAT motif is enriched at the promoters of cell cycle and stemness genes (40, 43), suggesting that NF-Y has evolved to sustain self-renewal in proliferating stem and progenitor cells (44). NF-Y function can also be dynamically modulated during cell differentiation (45). For example, in the myoblast lineage NF-YA is silenced in differentiated muscle cells (38, 46–48); however, it is retained in satellite cells, where it coordinates regenerative myogenesis in response to injury (49). To our knowledge, the relevance of NF-Y in RMS has not been previously investigated.
In this study, we coupled a myo-differentiation reporter with pooled CRISPR screening to nominate proteins that cooperate with PAX3–FOXO1 in RMS. Using this approach, we identified NF-Y as a critical factor needed to block myo-differentiation in PAX3–FOXO1+ RMS. Unexpectedly, we found that the cooperation between PAX3–FOXO1 and NF-Y does not occur through a physical interaction between these two transcription factors but instead via a strict NF-Y requirement for the activity of the PAX3 promoter, which is a key cis regulatory element needed for PAX3–FOXO1 expression. Collectively, this work reveals NF-Y as a previously overlooked upstream regulator of the myo-differentiation blockade in RMS.
Results
Profiling the Myo-Differentiation Response Following CRISPR-Based Inactivation of PAX3–FOXO1 in RMS Cell Lines.
Here, we sought to perform an unbiased genetic screen that would nominate genes that function in the same genetic pathway as the PAX3–FOXO1 oncoprotein. Since myo-differentiation is known to occur following genetic targeting of PAX3–FOXO1 in human RMS cell lines (20, 25, 26), we pursued the identification of a myo-differentiation marker that could be adapted to fluorescence-activated cell sorting (FACS) and multiplexed CRISPR screening (50). To identify such a marker, we began by profiling the myo-differentiation phenotype after CRISPR-mediated targeting of PAX3–FOXO1 in RMS. Green fluorescent protein (GFP)-linked single guide RNAs (sgRNAs) targeting various exons of PAX3 or FOXO1 were transduced into a Cas9-expressing human RMS cell line RH4 (harboring an endogenous PAX3–FOXO1 translocation) via lentivirus (all sgRNA sequences used in this study are listed in Dataset S1). Using western blotting and competition-based proliferation assays, we found that sgRNAs targeting exons encoding the N-terminus of PAX3 or the C-terminus of FOXO1 (regions present in the fusion oncoprotein) led to a reduction of PAX3–FOXO1 protein and a robust decrease in cellular proliferation (Fig. 1 A and B). In contrast, targeting the C-terminus of PAX3 or the N-terminus of FOXO1 (absent on the oncogenic fusion protein) had no effect on cellular fitness (Fig. 1B). Similar observations were made in multiple PAX3–FOXO1+ RMS lines; however, targeting of PAX3 or FOXO1 in RMS cell lines lacking this chromosomal translocation had no significant impact on cell proliferation (SI Appendix, Fig. S1A). Quantification of RNA-seq reads mapping to WT PAX3, WT FOXO1, and the PAX3–FOXO1 fusion revealed that the WT PAX3 allele is not expressed in RH4 cells (SI Appendix, Fig. S1B), which suggests that the observed phenotype is solely a result of PAX3–FOXO1 loss and not an effect of cotargeting the fusion and WT alleles. These findings validated PAX3–FOXO1 as a dependency in RMS, in accord with prior findings (24–29).
Fig. 1.

CRISPR-based targeting of PAX3–FOXO1 in RMS cells triggers myo-differentiation. (A) Western blot analysis of RH4 PAX3–FOXO1+ RMS cells 7 d post lentiviral transduction (poi) with sgRNAs targeting PAX3–FOXO1 (sgPAX3–FOXO1) or negative control (sgRosa). Membranes were incubated with an anti-FOXO1 antibody, which recognizes both wild-type FOXO1 and PAX3–FOXO1 fusion. Anti-β-actin (ACTB) antibody was used as a loading control. (B) Competition-based proliferation assay in RH4 PAX3–FOXO1+ RMS cells stably expressing Cas9. The cells were lentivirally transduced with vectors encoding GFP and sgRNAs targeting different regions of PAX3 and FOXO1 (N-PAX3 and C-FOXO1 are present in wild-type genes and the PAX3–FOXO1 fusion; C-PAX3 present in wild-type PAX3 only; N-FOXO1 present in wild-type FOXO1 only). Percentage of GFP+ cells was tracked over time using flow cytometry. Data represent an average of GFP percentage normalized to day 3 poi. Error bars represent SD of all sgRNAs targeting a denoted region (N-PAX3 n = 9, C-PAX3 n = 3, N-FOXO1 n = 3, C-FOXO1 n = 2). (C) F-actin immunofluorescence staining of RH4 cells transduced with sgRosa or sgPAX3–FOXO1 analyzed 7 d poi. (D) Volcano plot depicting differentially expressed genes between sgPAX3–FOXO1 and sgRosa control in RH4 cells. RNA-seq was performed 7 d poi with sgRNAs (n = 2 biological replicates). Selected up-regulated myo-differentiation genes are indicated. (E) GSEA plot demonstrating strong correlation between genes up-regulated following PAX3–FOXO1 knockout and the hallmark myogenesis gene set. The color of the circle indicates normalized enrichment score (NES); the size of the circle indicates a false discovery rate-adjusted q-value (FDR q-val). (F) Top 10 GSEA signatures positively correlated with gene expression changes induced by the knockout of PAX3–FOXO1. (G) Western blot analysis of multiple myo-differentiation markers in RH4 PAX3–FOXO1+ RMS cells 7 d poi with sgRNAs targeting PAX3–FOXO1 or Rosa26 negative control. A representative ACTB loading control is shown.
Using immunofluorescent imaging of filamentous actin (f-actin), we found that targeting of PAX3–FOXO1 led to a change in cellular morphology, with RMS cells becoming elongated and spindle-like (Fig. 1C). Using an RNA-seq analysis, we found that PAX3–FOXO1-deficient RMS cells upregulated several mRNAs encoding functional components of differentiated skeletal muscle (Fig. 1D; complete RNA-seq data for all conditions presented in this study can be found in Dataset S3). An unbiased Gene Set Enrichment Analysis (GSEA) (51) of these data further corroborated muscle differentiation being among the top transcriptional signatures induced upon PAX3–FOXO1 inactivation (Fig. 1 E and F). We validated the robust upregulation of myosin heavy chain (MYH), myomesin 3 (MYOM3), actinin alpha 2 (ACTN2), and myotilin (MYOT) by western blotting (Fig. 1G). Overall, these data validated a powerful myo-differentiation response upon targeting of PAX3–FOXO1 in RMS, which includes several protein markers of this phenotype that could be monitored for a genetic screen.
A FACS-Based Reporter Screen to Identify Genetic Perturbations that Trigger Myo-Differentiation in RMS.
We next evaluated the antibodies shown in Fig. 1G in a FACS-based CRISPR screening method to identify genetic knockouts that phenocopied the loss of PAX3–FOXO1 (Fig. 2A). In this strategy, Cas9+ RH4 cells were lentivirally transduced with a pooled sgRNA library, followed by glutaraldehyde fixation, detergent-based cell permeabilization, staining with fluorophore-linked antibodies that detect a myo-differentiation marker, and sorting of cell populations of varying fluorescence intensity. After the reversal of glutaraldehyde crosslinking, genomic DNA was prepared from each cell population and used for PCR amplification of the sgRNA cassette. Finally, deep sequencing was applied to each library to identify sgRNAs that became enriched in the marker-positive cell population. Through this multistep procedure, sgRNA-mediated knockouts that triggered a myo-differentiation response in RMS could be identified in a nonbiased manner.
Fig. 2.

Optimization of a FACS-based genetic screen to identify knockouts that cause myo-differentiation of rhabdomyosarcoma cells. (A) Cartoon representation of a myo-differentiation screening strategy. PAX3–FOXO1+ RMS cells stably expressing Cas9 are lentivirally transduced with a library of sgRNAs and enriched using blasticidin selection. Seven days poi, the pool of knockout cells is harvested, fixed, permeabilized, and stained. The cells are then sorted into myosin heavy chain (MYH) low (undifferentiated) and high (differentiated) pools using FACS. sgRNA cassette is then PCR amplified from the genomic DNA of cells in each pool, next-generation sequencing libraries are prepared, sequenced, and the abundance of sgRNAs in each pool is quantified. Cartoon created with BioRender.com. (B) Flow cytometry plots depicting myosin heavy chain staining of RH4 RMS cells transduced with sgRNAs targeting Rosa26, PAX3–FOXO1, MYC, CDK1, and MYOD1. Essentiality scores of these genes in RH4 cells is depicted in a heatmap based on DepMap CRISPR Avana Public 21Q1 data. (C) Small-scale pooled myo-differentiation test screen with a library of sgRNAs targeting PAX3–FOXO1 (sgPAX3-N n = 13 and sgFOXO1-C n = 6) and neutral controls (n = 78). (D) Negative selection and myo-differentiation exon scan of the PAX3–FOXO1 fusion. Top: schematic representation of the PAX3–FOXO1 fusion. The fusion junction and the annotated functional domains of the protein are indicated. PD—paired domain; OP—octapeptide motif; HD—homeodomain; TAD (truncated)—fragment of a transactivation domain of PAX3 (truncated in PAX3–FOXO1); FKHR (truncated)—fragment of a forkhead domain of FOXO1 (truncated in PAX3–FOXO1); KIX bdg—kinase-inducible domain interacting domain (KIX) binding domain; TAD—transactivation domain of FOXO1. Bottom: results of the PAX3–FOXO1 exon scan. RH4 PAX3–FOXO1+ RMS cells were lentivirally transduced with a library of all possible sgRNAs targeting all coding exons of PAX3 and FOXO1 (plus controls; see: Materials and Methods) and drug selected. For the negative selection screen, the cells were harvested at day 3 poi (starting pool of sgRNAs) and at multiple timepoints thereafter to track negative selection of sgRNAs. The data depict log2-transformed fold change of sgRNA abundance between day 3 and day 18 samples plotted along the length of the PAX3–FOXO1 fusion polypeptide chain. Pale purple dots—individual sgRNAs; dark purple line—a trendline depicting rolling average over 35 amino acid window. For the myo-differentiation screen, the abundance of sgRNAs was compared between the MYH high and MYH low pools following cell sorting of the cells harvested 7 d poi. Pale orange dots—individual sgRNAs; dark orange line—a trendline depicting rolling average over 35 amino acid window.
Among the antibodies we tested for intracellular FACS staining, we found that a monoclonal antibody recognizing MYH exhibited the strongest fluorescence increase following PAX3–FOXO1 inactivation (Fig. 2B). In this assay, we consistently observed between 10% and 30% of cells becoming MYH+ following fusion inactivation (SI Appendix, Fig. S2 A and B). CRISPR-based targeting of other essential genes in RMS (e.g., MYC and MYOD1) did not trigger MYH upregulation using this FACS-based assay (Fig. 2B and SI Appendix, Fig. S2C), which highlights the specificity of MYH upregulation as a reporter of PAX3–FOXO1 and not of other essential transcription factors.
We piloted the screening strategy in RH4 cells by evaluating a small test library composed of 19 sgRNAs targeting PAX3–FOXO1 mixed with 78 neutral sgRNAs. Using a deep sequencing analysis, we confirmed that PAX3–FOXO1-targeting sgRNAs became enriched relative to control sgRNAs in sorted MYH+ cells relative to the MYH- cell population (Fig. 2C). To further demonstrate the capabilities of this screening method, we performed a CRISPR exon scan of the entire length of PAX3–FOXO1, which is a method that can nominate functionally important domains based on the magnitude of sgRNA depletion or enrichment (52). We carried out exon scanning using the MYH reporter in parallel with performing a negative-selection “dropout” screen with the same sgRNA library. Using these two approaches, we observed a largely congruent pattern: the PAX3 and FOXO1 sgRNAs that became preferentially enriched in the myo-differentiation screen were also preferentially depleted in the negative selection screen. This included stronger effects for sgRNAs targeting the Paired Box DNA-binding domain of PAX3 and the known trans-activation domain of FOXO1 (Fig. 2D and SI Appendix, Fig. S3). In addition, both screens nominated functional hotspots of this fusion oncoprotein that lie outside of annotated domains (Fig. 2D), which we will investigate in future work. As expected, sgRNAs that targeted regions of PAX3-C and FOXO1-N absent on the fusion oncoprotein exhibited minimal effects in these two screens (SI Appendix, Fig. S3). Collectively, these control experiments supported the feasibility of coupling a FACS-based measurement of MYH expression with multiplexed CRISPR screening to identify genes that function upstream or downstream of PAX3–FOXO1.
We made attempts at applying a genome-wide CRISPR library (GeCKO) (53) consisting of pools of 65,383 sgRNAs to the MYH myo-differentiation reporter screen, but these experiments failed to produce reliable findings, as evidenced by the poor consistency among independent sgRNAs targeting the same gene and the failure to recover PAX3–FOXO1-targeting sgRNAs in the MYH+ gate. To overcome these technical issues, we next designed custom sgRNA libraries that would improve the quality of the screen results for the MYH reporter. Our design criteria included 10 sgRNAs per gene, which were designed to exclusively target exons encoding conserved protein domains to enrich for null mutations (52). To limit prohibitive cell sorting times, we split the sgRNAs into multiple sublibraries (10 libraries ranging from 817 to 1,188 sgRNAs) and limited the number of targeted genes to ~1,000, which were chosen based on their demonstrated essentiality in RMS (54), as well as having functions related to gene regulation and signal transduction (SI Appendix, Fig. S4 and Dataset S1).
The performance of these custom sgRNA libraries in the myo-differentiation screen was evident by the identification of PAX3–FOXO1 as the top hit and the behavior of negative controls (e.g., MYC and MYOD1) (Fig. 3A). In accord with our pilot experiments, we found that most of the genes evaluated in our screen behaved similarly to negative controls. Among the top hits in the screen identified after PAX3–FOXO1 were all three components of the NF-Y complex: NF-YC (#2), NF-YA (#3), and NF-YB (#6) (complete list of screened genes and their respective beta scores are listed in Dataset S2). We used western blotting to validate robust MYH upregulation following knockout of each NF-Y subunit in RH4 cells (Fig. 3B). Since prior work established that loss of any of the three NF-Y subunits displaces the complex from chromatin and abrogates its function (40), in subsequent experiments, we used sgRNAs targeting the NF-YC subunit (sgNFYC). Targeting NF-Y in two additional PAX3–FOXO1+ RMS cell lines also resulted in MYH upregulation (Fig. 3E). Importantly, no significant MYH upregulation was observed following NF-Y knockout in RMS cell lines that lacked the PAX3–FOXO1 translocation (Fig. 3D). In addition, immunofluorescence staining demonstrated remarkable similarity between the morphology of cells depleted of PAX3–FOXO1 or NF-Y (Fig. 3C). When compared to control cells, the two knockouts exhibited spindle-shaped, differentiated cell morphology marked by f-actin, TNNT3, and MYH staining (Fig. 3 C and D). These data suggest that inactivation of NF-Y in RMS triggers a myo-differentiation response that resembles the effects of targeting PAX3–FOXO1, thus validating our marker-based genetic screen as a means to reveal myo-differentiation regulators in this tumor context.
Fig. 3.
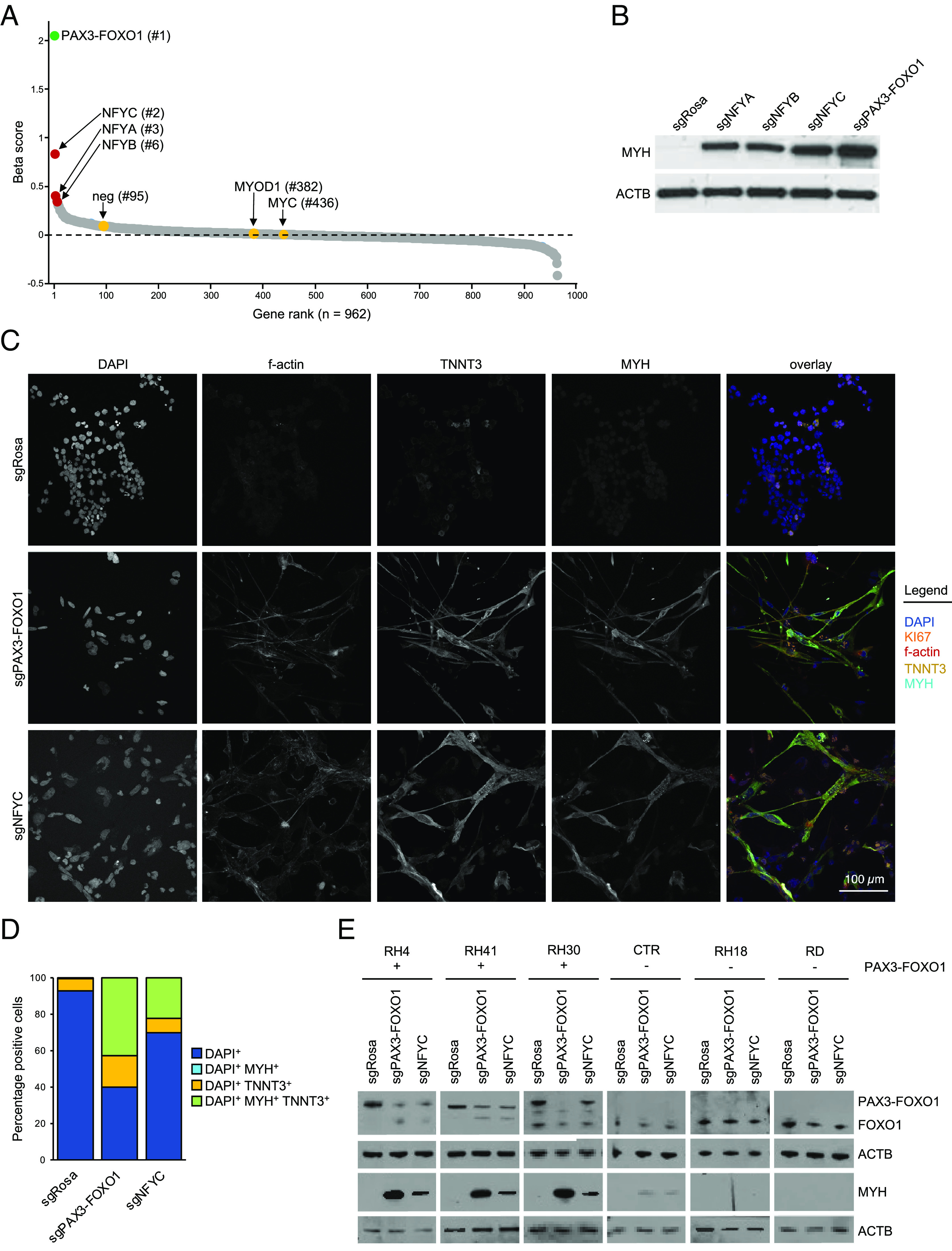
Loss of the NF-Y complex myo-differentiates PAX3–FOXO1+ RMS. (A) Results of the myo-differentiation screen in rhabdomyosarcoma. The dots represent beta scores based on all sgRNAs targeting each gene. Positive values indicate sgRNA enrichment in the MYH high pool of cells. (B) Western blot validation of the screening results. RH4 PAX3–FOXO1+ RMS cells were transduced with sgRNAs targeting PAX3–FOXO1 or either of the three subunits of the NF-Y complex (sgNFYA, sgNFYB, or sgNFYC). The membranes were stained with anti-MYH antibodies. ACTB was used as a loading control. (C) Immunofluorescence analysis of the control cells and cells depleted of PAX3–FOXO1 or NF-Y. The cells were harvested 7 d poi and stained with DAPI (visualizes cell nuclei), KI67 (Proliferation-Related Ki-67 Antigen; proliferation marker), and multiple myo-differentiation markers (f-actin—filamentous actin; TNNT3—Troponin T3, Fast Skeletal Type; MYH—myosin heavy chain). (D) Quantification of immunofluorescence staining. (E) Western blot analysis of gene knockouts in a panel of PAX3–FOXO1+ and PAX3–FOXO1− RMS cell lines. In PAX3–FOXO1− lines, sgPAX3–FOXO1 targets wild-type PAX3. The membranes were stained with an anti-FOXO1 antibody (recognizes wild-type FOXO1 and PAX3–FOXO1) to validate fusion status, and anti-MYH antibodies, to assess myo-differentiation in response to gene knockout. Corresponding ACTB loading controls are shown. Analysis was performed 7 d poi.
To further deepen our comparative analysis of the effects of targeting PAX3–FOXO1 and NF-Y, we performed bulk RNA-seq analysis in RH4 cells. GSEA revealed prominent enrichment of myo-differentiation gene signatures as the top-ranking gene sets that became altered following PAX3–FOXO1 and NF–YC knockout (Fig. 4 A–C; compare with Fig. 1 D–F). An RNA-seq scatterplot analysis revealed a striking similarity in the global transcriptome response incurred following these two knockouts (Fig. 4D). This pattern was also observed upon transcriptome profiling of NF-YC-depleted RH41 and RH30 cells (Fig. 4E). While NF-Y subunits are expressed to similar levels in fusion + and − RMS cell lines and tumors (SI Appendix, Fig. S5C) and are essential for the proliferation of PAX3–FOXO1+ and − RMS, as well as cancers of other lineages (SI Appendix, Fig. S5 A and B), the myogenic differentiation response following NF-Y loss was most evident in PAX3–FOXO1+ cells, as judged by the GSEA statistics and ranking of the “hallmark myogenesis” and other differentiation-related signatures, with little evidence of a myo-differentiation response in PAX3–FOXO1− RMS cells (CTR, RH18, and RD) (Fig. 4E and SI Appendix, Fig. S5D).
Fig. 4.

NF-Y-deficient RMS cells resemble cells depleted of PAX3–FOXO1. (A) Top 10 GSEA signatures positively correlated with gene expression changes induced by knockout of NF-Y and their corresponding rank and enrichment in PAX3–FOXO1 knockout. Bulk RNA-seq was performed 7 d poi with sgRNAs (n = 2 biological replicates). The color of the circle indicates normalized enrichment score (NES); the size of the circle indicates a false discovery rate-adjusted q-value (FDR q-val). (B) Volcano plot depicting differentially expressed genes between sgNFYC and sgRosa control in RH4 cells. Selected up-regulated myo-differentiation genes are indicated. The same genes are highlighted in Fig. 1D. (C) GSEA plot demonstrates strong correlation between genes up-regulated following NF-Y loss and the hallmark myogenesis gene set. (D) RNA-seq analysis comparing mRNA changes after PAX3–FOXO1 and NFYC knockout compared with Rosa26 negative control in RH4 cells. Each dot represents the log2-transformed fold change for every protein-coding gene. Linear trendline, Pearson correlation coefficient (Pearson r) and P-value (p-val) are indicated. (E) Analysis of the hallmark myogenesis gene signature following knockout of PAX3–FOXO1 or NF-Y in a panel of PAX3–FOXO1+ and PAX3–FOXO1− RMS cell lines, and an engineered myoblast line (Dbt-P3F1), where the PAX3–FOXO1 fusion is expressed from a viral promoter in a doxycycline-inducible manner. In PAX3–FOXO1- lines, sgPAX3–FOXO1 targets wild-type PAX3. The analysis was performed on differentially expressed genes identified through bulk RNA-seq performed 7 d poi with sgRNAs (n = 2 biological replicates, except RH30 and Dbt-P3F1, where n = 1). A rank of the hallmark myogenesis gene signature is plotted in relation to a total of 21,599 signatures queried. The color of the circle indicates normalized enrichment score (NES); the size of the circle indicates a false discovery rate-adjusted q-value (FDR q-val). (F) T-distributed Stochastic Neighbor Embedding (t-SNE) projections of a pseudotime trajectory analysis computed with Palantir. Ordering of the cells in individual knockout conditions along the trajectory is indicated. Equal cell numbers are plotted for each condition in all figures. Two branches largely absent in the sgRosa control condition emerge following PAX3–FOXO1 and NF-Y loss. Single-cell RNA-seq was performed on RH4 PAX3–FOXO1 fusion-positive cells 7 d poi with sgRNAs targeting PAX3–FOXO1, NFYC or ROSA26 safe locus. (G) Cell cycle phase analysis. Cell state was assigned to each cell based on generic cell-cycle scores computed for genes reported to be up-regulated during S and G2/M phases and plotted on the pseudotime projection. Bar chart displays cell cycle distribution for each knockout condition. (H) T-SNE projections depicting GSEA scores for the muscle: fast muscle and vasculature: smooth muscle signatures from the Tabula Sapiens collection of gene signatures.
We next performed single-cell RNA-seq (scRNA-seq) to evaluate the differentiation trajectories of RH4 cells following NFY-C and PAX3–FOXO1 inactivation. Using the Palantir algorithm (55), we observed two distinct pseudotime branches that emerged following knockout of either PAX3–FOXO1 or NF-YC (Fig. 4F). By applying GSEA, we found that these two paths reflect distinct muscle differentiation signatures (striated muscle versus smooth muscle/senescence) coupled with proliferation arrest (Fig. 4 G and H and SI Appendix, Figs. S6 A–C and S7 A and B and Dataset S7). While PAX3–FOXO1 knockout cells reached a more terminal stage of differentiation than NF-Y knockout cells at this timepoint (day 7), the overall transcriptome similarity at the single cell level further validates a strong linkage between these transcriptional regulators in RMS.
NF-Y and PAX3–FOXO1 Occupy Largely Distinct Genomic Loci.
The findings above led us to hypothesize that NF-Y and PAX3–FOXO1 might bind to one another in a physical complex that coregulates differentiation-related genes in RMS cells. However, our coimmunoprecipitation experiments failed to reveal evidence of an association between these two factors in RMS cell lysates. In addition, we evaluated the genome-wide chromatin occupancy pattern of NF-Y in RH4 cells using Cleavage Under Targets & Release Using Nuclease (CUT&RUN) and compared it to PAX3–FOXO1 chromatin occupancy in the same cell line (56) (Dataset S4). We found that NF-Y and PAX3–FOXO1 occupied almost entirely non-overlapping sites (Fig. 5 A and B). Consistent with prior findings (40, 43), we detected NF-Y primarily at active gene promoters containing its cognate CCAAT motif, whereas PAX3–FOXO1 was found predominantly at active enhancers distant from promoters (Fig. 5 A and C and SI Appendix, Fig. S8 A–C). Together, these findings suggest that NF-Y does not collaborate with PAX3–FOXO1 via a shared transcriptional complex.
Fig. 5.

Genome-wide analysis of chromatin association of NF-Y and PAX3–FOXO1. (A) Heatmaps and profiles visualizing enrichment of PAX3–FOXO1, H3K27ac (active enhancer mark), H3K4me3 (active promoter mark), and NF-Y (NF-YB and NF-YC subunits) in RH4 PAX3–FOXO1+ RMS cells. Elements are categorized into two classes: PAX3–FOXO1 bound (n = 968) and NF-Y bound (n = 3,074). The rows depict 4-kb regions, centered on PAX3–FOXO1 or NF-Y peaks and ranked by the respective signal intensities. For PAX3–FOXO1, H3K27ac and H3K4me3, color shading corresponds to chromatin immunoprecipitation followed by sequencing (ChIP-seq) read counts; for NF-YB and NF-YC, color shading corresponds to CUT&RUN read counts. (B) Venn diagram depicting an overlap between NF-Y peaks and PAX3–FOXO1 peaks. (C) Classification and quantification of genomic elements bound by NF-Y and PAX3–FOXO1. (D) View of the SIX1 locus, representing NF-Y binding at the promoter, PAX3–FOXO1 binding at a distal enhancer, and interaction of the two sites on chromatin loops in 3D. H3K27ac marks and RNA expression of genes within the region is indicated. Bottom: 4C-seq validation of the 3D interaction between NF-Y bound promoter and PAX3–FOXO1 bound distal enhancer in sgRosa and sgNFYC cells analyzed 7 d post lentiviral transduction. Viewpoint is indicated. Plotted 4C-seq signal represents an average of two biological replicates.
While NF-Y-bound promoters are often tens to hundreds of kilobases away from the nearest PAX3–FOXO1-bound enhancers along the linear genome (Fig. 5D and SI Appendix, Fig. S9B), we considered the possibility that NF-Y cooperates with PAX3–FOXO1 via long-range enhancer–promoter interactions in 3D space. Using an absolute quantification of chromatin architecture (AQuA–HiChIP) analysis in RH4 cells (56, 57), we identified ~200 3D contacts between NF-Y-occupied promoters and PAX3–FOXO1-occupied enhancers (SI Appendix, Fig. S9A and Dataset S5). To validate these findings, we performed chromosome conformation capture combined with high-throughput sequencing (4C-seq), which confirmed contact between selected NF-Y- and PAX3–FOXO1-occupied elements at the SIX1, PEG3, ERRFI1, and JUN loci (Fig. 5D and SI Appendix, Fig. S9C). However, we found that these enhancer-promoter interactions did not occur in an NF-Y-dependent manner (Fig. 5D and SI Appendix, Fig. S9C). In addition, we were unable to identify robust correlations between the presence of looped interactions and changes in gene expression following knockout of either of the two transcription factors (SI Appendix, Fig. S9A). Collectively, these findings suggested that NF-Y and PAX3–FOXO1 block myo-differentiation via binding and direct activation of distinct genomic targets.
NF-Y Regulates RMS Myo-Differentiation by Activating the PAX3 Promoter to Maintain PAX3–FOXO1 Expression.
We next attempted to pinpoint the critical target genes of NF-Y that regulate RMS myo-differentiation. For this purpose, we integrated several epigenomic and genetic screening datasets to find 1) essential genes in RMS [derived from Cancer Dependency Map database (54, 58)] that are 2) directly occupied by NF-Y, and 3) significantly down-regulated at the RNA level at an early timepoint following NF-Y knockout, and 4) scored in our reporter screen as triggering myo-differentiation when inactivated (Dataset S6). Remarkably, the only NF-Y target gene that satisfied each of these criteria was PAX3–FOXO1 itself (Fig. 6A). Using qRT-PCR analysis and western blotting of endogenous PAX3–FOXO1 mRNA and protein, respectively, we confirmed that knockout of NF-Y triggered a rapid and robust downregulation of the fusion oncoprotein (Fig. 6 B and C). The similar magnitude of PAX3–FOXO1 mRNA and protein reduction following NF-Y knockout was suggestive of transcriptional regulation. Consistent with this, our CUT&RUN analysis revealed NF-Y occupancy spanning two CCAAT motifs within the PAX3 promoter, located 69 and 114 base pairs upstream of the transcriptional start site (TSS) (Fig. 6D and Dataset S8). These findings led us to consider whether NF-Y-mediated activation of the PAX3 promoter might explain how this complex regulates myo-differentiation in RMS.
Fig. 6.
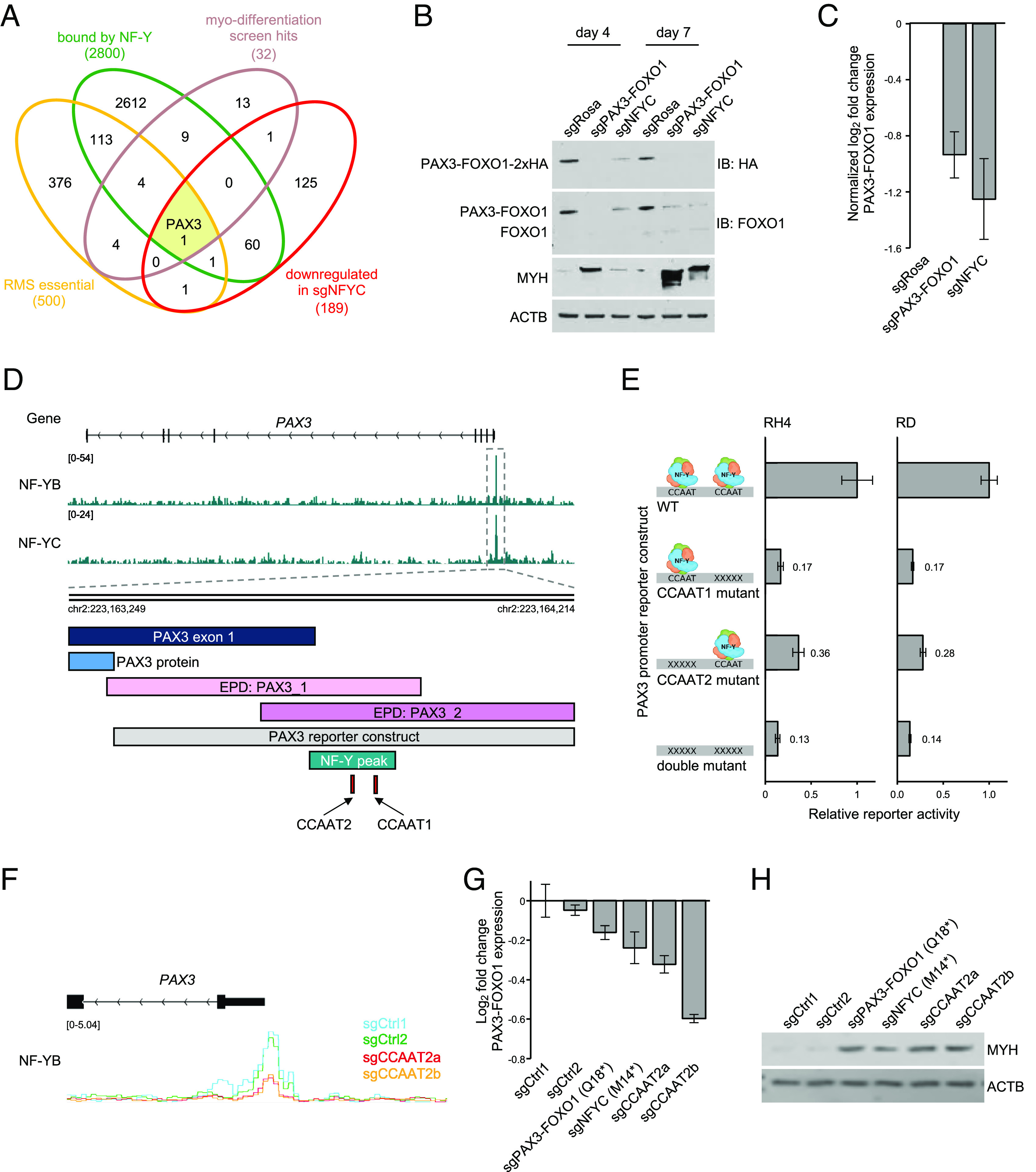
NF-Y complex binds to the PAX3 promoter and activates transcription of PAX3–FOXO1. (A) Venn diagram depicting an overlap between genes bound by NF-Y (based on CUT&RUN), genes essential in PAX3–FOXO1+ RMS (based on DepMap CRISPR Avana Public 21Q1 data; top 500 genes based on average CRISPR score in screened PAX3–FOXO1+ RMS cell lines), genes down-regulated following loss of NF-Y (based on bulk RNA-seq in RH4 cells 4 d poi; genes with log2-transformed fold change < −0.5 and adjusted P-value < 0.05; n = 2 and gene hits from the myo-differentiation screen (beta score > 0.16). (B) Western blot analysis of engineered RH4 cells, in which HA-tag is knocked in at the C-terminus of endogenous PAX3–FOXO1. The cells were lentivirally transduced with sgRNAs targeting PAX3–FOXO1, NFYC, or ROSA26, drug selected, and analyzed at day 4 and day 7 poi. Protein levels of PAX3–FOXO1 were visualized by staining with anti-FOXO1 and anti-HA antibodies. Differentiation state was assessed by MYH staining. ACTB was used as a loading control. (C) qRT-PCR analysis of PAX3–FOXO1 transcript levels following lentiviral transduction with sgRosa, sgPAX3–FOXO1, and sgNFYC. The values represent log2-transformed fold change of PAX3–FOXO1 expression normalized to sgRosa (n = 2 biological replicates, three technical replicates each). (D) The architecture of the PAX3 promoter, retained in the PAX3–FOXO1 fusion, depicting the NF-Y CUT&RUN peak. The two promoter annotations retrieved from the Eukaryotic Promoter Database (accession numbers PAX3_1 and PAX3_2), as well as the promoter sequence assessed in luciferase reporter assays in Fig. 6E are indicated. Not depicted on the figure is CCDC140 long noncoding RNA, encoded on the +DNA strand, which partially overlaps the promoter of PAX3. (E) Luciferase reporter assays. RH4 PAX3–FOXO1+ RMS cell line and RD PAX3–FOXO1− RMS cell line transduced with either wildtype (WT) or CCAAT NF-Y binding motif mutant PAX3 promoter constructs driving the expression of firefly luciferase and constitutively expressing renilla luciferase. The relative activity of the promoter was quantified 48 h poi by measuring the luciferase signal. The data represent an average of firefly luciferase signal normalized to the average of renilla luciferase signal for each sample and then normalized to the wild-type PAX3 promoter activity. Error bars represent SEM; n = 3 biological replicates, three technical replicates each. (F) CUT&RUN NF-YB signal tracks at the PAX3 promoter in RH4 cells. The cells constitutively expressing the two components of the split cytosine base editor were lentiviral transduced with sgRNAs inducing C to T conversion at two control sites (sgCtrl1 and sgCtrl2) or in the second CCAAT NF-Y binding motif (sgCCAAT2_a and sgCCAAT2_b) within the PAX3 promoter. Transduced cells were drug selected, and base editing was induced by rapamycin addition. The cells were harvested 8 d post rapamycin addition. Base editing at the CCAAT motif was confirmed by Sanger trace deconvolution using EditR (see: SI Appendix, Fig. S10E). The plotted CUT&RUN signal was normalized to counts per million reads mapped (CPM). (G) qRT-PCR analysis on base edited cells, assessing transcript levels of PAX3–FOXO1. The numbers represent log2-transformed fold change values normalized to negative control. Error bars represent SD; n = 1 biological replicate in three technical replicates. (H) Western blot on the CCAAT motif mutant cells, showing their myo-differentiation status with MYH staining. ACTB was used as a loading control. The same negative control guides were used as above. sgRNAs that introduce premature stop codon into PAX3–FOXO1 (sgPAX3–FOXO1(Q18*)) and NF-YC (sgNFYC(M14*)) were used as positive controls for myo-differentiation.
To begin assessing the role of NF-Y as an activator of the PAX3 promoter, we cloned wild-type and CCAAT-mutant PAX3 promoter sequences into luciferase reporter constructs, which were transduced into RMS cells. Luciferase measurements revealed that mutation of each of the two CCAAT motifs was sufficient to abrogate promoter activity, suggesting that the PAX3 promoter is an NF-Y-dependent cis-regulatory element (Fig. 6E). These findings led us to consider whether expression of PAX3–FOXO1 from a nonnative promoter that lacks CCAAT motifs would rescue the myo-differentiation response observed following NF-Y knockout. For this purpose, we used a transformed human myoblast cells engineered to express PAX3–FOXO1 under the control of a tetracycline response element and a minimal cytomegalovirus (CMV) promoter (referred to as Dbt-P3F1) (59). In our experiments, Dbt-P3F1 cells were continuously cultured in the presence of doxycycline to maintain PAX3–FOXO1 expression. As expected, CRISPR-mediated targeting of PAX3–FOXO1 in Dbt-P3F1 cells resulted in myo-differentiation as assayed by bulk RNA-seq (Fig. 4E). However, the myo-differentiation response to NF-YC knockout was largely attenuated in this cell system (Fig. 4E), which supports a hypothesis in which NF-Y regulates RMS myo-differentiation through transcriptional control of the endogenous PAX3–FOXO1 locus.
Finally, we employed inducible base editing technology to evaluate whether point mutations of CCAAT motifs within the endogenous PAX3 promoter of RH4 cells would be sufficient to trigger myo-differentiation (SI Appendix, Fig. S10 A, B, and D). This base editing tool is a modification of the BE4max system (60), in which the cytosine deaminase is split into two polypeptide halves fused to FKBP12-FRB dimerization domains. Transient exposure to low concentrations of rapamycin triggers dimerization to reconstitute deaminase activity, thereby generating targeted C to T conversions while mitigating off-target mutagenesis (61). Of the two CCAAT sequences present within the PAX3 promoter, the −113 motif was located near a PAM motif that allowed for sgRNA targeting and potential for base editing. After establishing the efficiency of the rapamycin-induced cytosine deamination in RH4 cells (SI Appendix, Fig. S10C), we expressed two sgRNAs targeting the PAX3 −114 bp CCAAT motif and validated successful cytosine to thymine conversion within the targeting window and at adjacent bases by Sanger sequencing (SI Appendix, Fig. S10E) (62). Using CUT&RUN analysis, we confirmed that the two sgRNAs targeting the distal CCAAT motif resulted in loss of NF-Y occupancy at the endogenous PAX3 promoter (Fig. 6F), which was associated with diminished PAX3–FOXO1 expression (Fig. 6G). Remarkably, these base edits were sufficient to cause a potent MYH upregulation, with effects that were comparable to targeting PAX3–FOXO1 and NFYC with point mutations that introduce premature stop codons (Fig. 6H). Collectively, these experiments support that a key function of NF-Y in blocking RMS myo-differentiation is via activation of the PAX3 promoter to promote PAX3–FOXO1 expression.
Discussion
In this study, we describe a genetic screening strategy for revealing genes that cause a robust myo-differentiation phenotype when inactivated in RMS. We optimized this method using knockout of the PAX3–FOXO1 oncoprotein as a positive control, which is known to cause a robust myo-differentiation phenotype in RMS (24, 26, 30). Using this approach, we identified a role for the NF-Y complex as a key regulator of myo-differentiation in PAX3–FOXO1+ RMS. Importantly, this function of NF-Y is less apparent in RMS cells lacking the PAX3–FOXO1 oncoprotein. Using an integrative functional approach, we accounted for this differentiation phenotype by showing that NF-Y activates PAX3–FOXO1 expression by binding to CCAAT motifs found within the PAX3 promoter. Since impaired mesenchymal differentiation is a hallmark property of many soft tissue sarcomas, we anticipate that our phenotypic screening strategy will have broader utility in the study of oncogenic transcriptional programs in these aggressive tumors.
From a technical perspective, our study outlines several parameters that required optimization before obtaining high-quality screening results using the myo-differentiation MYH reporter. One limitation of our approach is that it required generation of custom high-depth sgRNA libraries, which only allowed us to probe ~1,000 genes in a multiplexed format, which collectively required ~70 h of FACS to implement. Due to this restriction, we prioritized genes that had been previously identified in genome-wide screens as impacting RMS cell fitness (54, 63) and were known to have epigenetic and signaling functions, which we estimate covers only ~30% of the essential genome in RMS. Nevertheless, our screening effort thus far emphasizes the rarity of observing a robust myo-differentiation response in RMS following a genetic knockout, as the overwhelming majority of genes did not incur MYH upregulation when inactivated. Notably, targeting the known PAX3–FOXO1 coactivators BRD4, SMARCA4, and CHD4 failed to elicit a robust myo-differentiation response. This is to be expected, as all three factors are likely to exert a general function in transcriptional activation, being required for both the direct output of PAX3–FOXO1 and for the activation of MYH and other terminal differentiation genes. In future work, an alternative marker could be employed (e.g., a direct transcriptional target of PAX3–FOXO1) to screen for additional coactivators that support PAX3–FOXO1 activity.
The observation of robust myo-differentiation in RMS following the NF-Y knockout was unexpected for several reasons. First, the NF-Y complex is required for the proliferation of nearly all cancer cell lines, which is likely due to an enrichment of CCAAT motifs in the promoters of genes that promote the cell cycle (42, 64). However, we have noticed that the promoters of terminal myo-differentiation genes (e.g., MYH2) generally lack CCAAT motifs when compared to other gene categories. This is consistent with diminished NF-Y function (via transcriptional silencing of the NF-YA subunit) in terminally differentiated mouse myotubes (65). These data provide further evidence that NF-Y is dispensable for terminal differentiation of muscle cells and might explain why NF-Y scored in our MYH reporter screen, whereas the general transcriptional coactivators (e.g., BRD4) did not.
It is important to emphasize that NF-Y performs at least two essential functions in RMS cells: 1) activation of promoters of genes involved in cell cycle and metabolism (66), which are functions carried out in all proliferating cells, and 2) activation of PAX3–FOXO1 expression, a role that is RMS-specific. The PAX3 promoter is expressed in a highly tissue-specific manner, which is distinct from the ubiquitous expression pattern of NF-Y. Mechanistically, NF-Y is not a strong activator per se but rather synergizes with other transcription factors which bind near CCAAT motifs (67). This suggests that while NF-Y is required for activation of the PAX3 promoter, it is unlikely to be sufficient; additional tissue-specific factors are likely cooperating with NF-Y at this cis-element that await identification (Dataset S8). Nevertheless, our study emphasizes the importance of promoter-encoded cancer vulnerabilities of oncogenes; in a prior study, we identified ZFP64 as an activator of MLL-fusion oncoproteins by binding to the MLL promoter (68). These findings reinforce the profound sensitivity of RMS cells to undergo myo-differentiation following partial reduction in PAX3–FOXO1 expression.
While we have demonstrated that NF-Y regulates myo-differentiation in RMS in part through transcriptional regulation of PAX3–FOXO1, we cannot exclude additional contributions from its other target genes. For example, SIX1 is activated by NF-Y and PAX3–FOXO1 and has previously been described as a master regulator of RMS differentiation (69). At this locus, we observe long-range contact between the NF-Y-bound promoter and PAX3–FOXO1-bound distal enhancer, and this gene becomes down-regulated upon knockout of each factor. However, this 3D contact does not appear to be maintained by the presence of NF-Y but could instead be mediated by other factors (e.g., CTCF). Nevertheless, we cannot exclude the possibility that regulation of SIX1, as well as other genes with shared NF-Y and PAX3–FOXO1 occupancy, also contribute to the differentiation phenotype observed upon NF-Y inactivation.
While the cell-of-origin of RMS is currently unclear, a prevailing view in the field is that rhabdomyosarcoma reflects hijacked developmental programs of skeletal muscle progenitor cells (4, 70). However, our scRNA-seq profiling of RMS cells following PAX3–FOXO1 and NF-Y knockout revealed two distinct pseudotime branches. The cells occupying branch 1 express smooth muscle and senesence genes (e.g., TAGLN, MYL6, and CDKN2A), while cells falling within branch 2 resemble striated muscle, expressing either skeletal (e.g., MYL1 and MYBPC1) or cardiac (e.g., ACTC1, TNNT2, and MYL4) genes. While it remains to be validated whether these states represent distinct myogenic lineages, our observation suggests that myo-differentiation is not a simple return to a cell-of-origin state but instead is a more stochastic transition of RMS cells along multiple lineage trajectories. The relationship between multilineage differentiation and the RMS cell-of-origin warrants further investigation, but these responses are possibly influenced by the level of PAX3–FOXO1 expression in these different RMS contexts.
Our findings justify consideration of NF-Y as a target for the development of differentiation therapies in RMS. The approach of reprogramming cancer cells into differentiated cells has been successfully implemented in other malignancies, most prominently in PML-RARα fusion-driven acute promyelocytic leukemia (71). Active efforts are underway to design inhibitors of NF-Y, which primarily focus on disrupting NF-Y heterotrimer formation (72) or NF-Y:DNA binding (73, 74). A major challenge for targeting NF-Y will be in avoiding toxicity, since this complex activates genes involved in cell cycle and metabolic pathways in normal proliferating tissues. This issue is further reinforced by the broad NF-Y dependency observed across diverse cancer types. However, rhabdomyosarcoma cells are highly sensitive to perturbations of PAX3–FOXO1, with acute reductions resulting in myo-differentiation, which might be an irreversible state. Hence, a window of opportunity could exist in which transient inhibition of NF-Y differentiates RMS cells, while having minimal effects on healthy tissues, a possibility that warrants further exploration.
Materials and Methods
MYH Staining for Flow Cytometry and Cell Sorting.
Cas9-expressing RMS cells were lentivirally transduced with sgRNAs and drug selected. Cells have not been passaged throughout the duration of the experiment in order to enrich for differentiated cells. On day 7 post-transduction, the media was aspirated, the cells were trypsinized, resuspended in media, and counted using Invitrogen Countess Automated Cell Counter. All knockout conditions were processed in parallel to minimize variability in staining. The cells were pelleted by centrifugation and washed in PBS. All centrifugation steps were performed at 1,500 rpm at 4 °C. To fix, cell pellets were dry vortexed to minimize fixation in clumps and resuspended in 0.05% glutaraldehyde (Sigma, G6257-10X10ML) in PBS (200 µL per 1 million cells) while being constantly agitated. Resuspended cells were incubated in glutaraldehyde for 10 min at room temperature (RT) with periodic gentle vortexing. Cells were then pelleted by centrifugation for 2 min and washed three times with 0.1% PBS/BSA (all washes are performed with ~1 mL 0.1% PBS/BSA per 1 M cells). Following the last wash, the pellets were dry vortexed and resuspended in 0.1% Triton X-100 (Thermo Fisher Scientific, A16046.AP) in 0.1% PBS/BSA (100 µL per 1 M cells) to permeabilize. Cells were incubated for 5 min at RT, pelleted, and washed once with 0.1% PBS/BSA. Fixed and permeabilized cells were aliquoted into different staining conditions. For fully stained samples, the cells were resuspended in MYH staining solution (for 1 M cells: 7.2 µL anti-MYH antibody (DSHB, MF20) + 12.8 µL 0.1% PBS/BSA). For unstained, cells are resuspended in 0.1% PBS/BSA (20 µL per 1 M cells). Cells were stained overnight at 4 °C with constant agitation. The following day, the cells were washed three times with 0.1% PBS/BSA and stained with a secondary antibody (for 1 M cells: 1.5 µL BV421 Rat Anti-Mouse Igκ Light Chain (BD Biosciences, 562888) + 18.5 µL 0.1% PBS/BSA) or 0.1% PBS/BSA for unstained controls (20 µL per 1 M cells). Cells were stained for 3 h at RT with constant agitation. Cells were then washed twice with 0.1% PBS/BSA and resuspended in 0.1% PBS/BSA for flow cytometry or cell sorting.
Flow Cytometry.
Stained cell samples were passed through 35-µm strainers to break up clumps and analyzed on a BD LSRFortessa flow cytometer (BD Biosciences). Data analysis was performed using the FlowJo software (TreeStar).
Myo-Differentiation CRISPR Screens.
Cas9-expressing RH4 cells were infected at low multiplicity of infection (MOI) (~0.3) with lentivirus carrying a library of sgRNAs. Sufficient numbers of cells were used to achieve at least 1,000× coverage of the library. Control cells were infected with sgRosa negative control and sgPAX3–FOXO1 positive control sgRNAs. Media was changed 12 h post-transduction. Puromycin selection was introduced 48 hrs post-transduction to enrich for infected cells. Media was refreshed at day 5 and cells have not been passaged throughout the duration of the experiment to prevent loss of myo-differentiated cells. Knockout cells were harvested 7 d post-transduction and stained for MYH as described above. Cells infected with the library were diluted at 50 million cells per mL to maximize sorting speed and passed through 35-µm strainers. Cells were sorted on BD FACS ARIA-SORP instrument recording FSC, SSC, and BV421 fluorescence (405 nm laser, 450/50 bandpass filter). Control cells were used to set PMT voltages and draw gates (SSC-A vs. FSC-A to gate out debris, SSC-W vs. SSC-H and FSC-W vs. FSC-H to gate on single cells, SSC-A vs. BV421-A to distinguish MYH high and MYH low populations). Sufficient number of cells was sorted to achieve ~1,000× sgRNA representation within the MYH high gate. Harvested cells were pelleted, washed with PBS, and resuspended in DNA extraction buffer (10 mM Tris-HCL (pH8) + 150 mM NaCl + 10 mM ethylenediaminetetraacetic acid (EDTA)) with 0.1% SDS and 80 µg proteinase K. Digestion was performed O/N at 56 °C. Following lysis, gDNA was harvested using the phenol-chlorophorm method. Precipitation was done in 100% ethanol + 30 µL 5 M sodium acetate at −80 °C O/N followed by centrifugation for 30 min at 4 °C. Libraries were constructed through a two-step PCR; first, sgRNA cassette was amplified with the following primers:
forward:
TCTTGTGGAAAGGACGAAACACCG;
reverse:
TCTACTATTCTTTCCCCTGCACTGT (cycling conditions: 10 min at 95 °C; 25 cycles of 30 s at 95 °C, 45 s at 60 °C, 30 s at 72 °C; 7 min at 72 °C). The 242 bp fragments were gel purified using the QIAquick Gel Extraction Kit (Qiagen, 28706X4) and used as a template for the second PCR to incorporate sequencing adapters and barcode the samples. The following primers were used for the second PCR:
forward:
AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACGCTCTTCCGATCTNHNNNNATCACGGTGGAAAGGACGAAACACC;
reverse:
CAAGCAGAAGACGGCATACGAGATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTCTGTTTCCAGCATAGCTCTTAAAC; cycling conditions as above but for eight cycles total. The libraries (~190 bp) were gel purified and their quality was assessed on Agilent 2100 Bioanalyzer using the Agilent DNA 1000 Assay (Agilent, 5067-1504). Libraries were quantified using Qubit dsDNA BR Assay (Thermo Fisher Scientific, Q32850) and KAPA Library Quantification Kit (KK4824), diluted, pooled at equimolar concentrations, and sequenced on the Illumina NextSeq500 platform using the single end SE75 bp mode of sequencing.
Negative Selection CRISPR Screens.
Cas9-espressing RH4 RMS cells were infected with the exon scan library at low MOI. Sufficient numbers of cells were used to achieve at least 1,000× coverage of the library. Cells were harvested at day 3 post-transduction to assess the initial representation of the library and drug selection was applied to enrich for infected cells. Cells were then harvested at day 18 posttransduction to assess negative selection of individual sgRNAs. Sufficient numbers of cells were harvested at each timepoint to maintain at least 1,000× representation of each sgRNA. Genomic DNA extraction, library preparation, and sequencing were performed as described above.
CRISPR Screening Data Analysis.
Quality of the sequencing data was assessed using FastQC. Demultiplexing was performed in the command line, using the -grep function. The reads were mapped to the reference sgRNA library and counted using MAGeCK (75) count function. For myo-differentiation screens, comparison between samples and calculation of the beta scores for each gene was done using MAGeCK-MLE (using the mageck test and mageck mle commands (75)). For the test library and exon scans, MAGeCK-derived sgRNA counts were normalized across timepoints and fold changes (for negative selection: day 3 vs. day 8; for MYH screens: MYH high vs. MYH low) were calculated.
For additional experimental information, please see SI Appendix, Materials and Methods.
Supplementary Material
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (XLSX)
Dataset S08 (XLSX)
Acknowledgments
We would like thank Fred Barr (National Institutes of Health) for providing the immortalized myoblast cells engineered to express PAX3–FOXO1. Our study was supported by the Cold Spring Harbor Laboratory Next Generation Sequencing, Single Cell Genomics, Flow Cytometry, Microscopy, and Animal Imaging Core Facilities. This work was supported by Cold Spring Harbor Laboratory NCI Cancer Center Support grant P30-CA045508. Additional funding was provided to C.R.V. by the Pershing Square Sohn Cancer Research Alliance, NIH grants P01CA013106 and CA245859, the Edward and Martha Gerry Fellowship and The Miles Levin Impact Award, as well as support from the Christina Renna Foundation, The Mary Ruchalski Foundation, Friends of T.J. Foundation, Michelle Paternoster Foundation, Summer’s Way Foundation, Clark Gillies Foundation, Daniela Conte Foundation, and Maddie’s Promise Foundation. We received research funding from Boehringer-Ingelheim and Treeline Biosciences.
Author contributions
M.W.S., D.S., M.W.V., J.B.P., K.C., D.A.T., G.A.B., and C.R.V. designed research; M.W.S., D.S., M.W.V., J.B.P., L.M.N.d.A., K.C., R.U., G.C., B.N., J.P.M., and A.D. performed research; M.W.S., O.E.D., D.R., S.W.H., K.R.S., R.M., R.M.K., and J.S. contributed new reagents/analytic tools; M.W.S., D.S., M.W.V., J.B.P., O.E.D., B.G., and J.K. analyzed data; and M.W.S. and C.R.V. wrote the paper.
Competing interests
C.R.V. has received consulting fees from Flare Therapeutics, Roivant Sciences, and C4 Therapeutics; has served on the advisory boards of KSQ Therapeutics, Syros Pharmaceuticals and Treeline Biosciences; and owns stock from Treeline Biosciences. R.M.K. and J.S. through the University of Pennsylvania have filed a patent application on small-molecule controllable base editors.
Footnotes
This article is a PNAS Direct Submission.
Data, Materials, and Software Availability
The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) as a part of the GSE227603 SuperSeries and is composed of the following SubSeries: RNA-seq - GSE227594 (76), CRISPR screens - GSE227595 (77), CUT&RUN - GSE227596 (78), scRNA-seq - GSE227597 (79), and 4C-seq - GSE227600 (80). In addition, the following publicly-available datasets were used in this study: ChIP-seq - GSE116344 (81), AQuA-HiChIP - GSE120770 (82), and RNA-seq on RMS tumor samples (83). All other data are included in the article and/or supporting information.
Supporting Information
References
- 1.Parham D. M., Pathologic classification of rhabdomyosarcomas and correlations with molecular studies. Mod. Pathol. 14, 506–514 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Skapek S. X., et al. , Rhabdomyosarcoma. Nat. Rev. Primer 5, 1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Epstein J. A., Lam P., Jepeal L., Maas R. L., Shapiro D. N., Pax3 inhibits myogenic differentiation of cultured myoblast cells. J. Biol. Chem. 270, 11719–11722 (1995). [DOI] [PubMed] [Google Scholar]
- 4.Charytonowicz E., Cordon-Cardo C., Matushansky I., Ziman M., Alveolar rhabdomyosarcoma: Is the cell of origin a mesenchymal stem cell? Cancer Lett. 279, 126–136 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Saab R., Spunt S. L., Skapek S. X., Myogenesis and Rhabdomyosarcoma: The Jekyll and Hyde of Skeletal Muscle (Elsevier Inc., 2011). [DOI] [PubMed] [Google Scholar]
- 6.Keller C., Guttridge D. C., Mechanisms of impaired differentiation in rhabdomyosarcoma. FEBS J. 280, 4323–4334 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu P. Y., Guttridge D. C., Dysregulated Myogenesis in Rhabdomyosarcoma (Elsevier Inc., 2018). [DOI] [PubMed] [Google Scholar]
- 8.Schaaf G. J., et al. , Full transcriptome analysis of rhabdomyosarcoma, normal and fetal skeletal muscle: Statistical comparison of multiple SAGE libraries. FASEB J. 19, 1–26 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Wachtel M., Schäfer B. W., PAX3-FOXO1: Zooming in on an “undruggable” target. Semin. Cancer Biol. 50, 115–123 (2018). [DOI] [PubMed] [Google Scholar]
- 10.Hettmer S., et al. , Rhabdomyosarcoma: Current challenges and their implications for developing therapies. Cold Spring Harb. Perspect. Med. 4, a025650 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barlow J. W., et al. , Differentiation of rhabdomyosarcoma cell lines using retinoic acid. Pediatr. Blood Cancer 47, 773–784 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Barr F. G., et al. , Rearrangement of the PAX3 paired box gene in the paediatric solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 3, 113–117 (1993). [DOI] [PubMed] [Google Scholar]
- 13.Shapiro D. N., Sublett J. E., Li B., Downing J. R., Naeve C. W., Fusion of PAX3 to a member of the forkhead family of transcription factors in human alveolar rhabdomyosarcoma. Cancer Res. 53, 5108–5112 (1993). [PubMed] [Google Scholar]
- 14.Galili N., et al. , Fusion of a fork head domain gene to PAX3 in the solid tumour alveolar rhabdomyosarcoma. Nat. Genet. 5, 230–235 (1993). [DOI] [PubMed] [Google Scholar]
- 15.Linardic C. M., PAX3-FOXO1 fusion gene in rhabdomyosarcoma. Cancer Lett. 270, 10–18 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scheidler S., Fredericks W. J., Rauscher F. J., Barr F. G., Vogt P. K., The hybrid PAX3-FKHR fusion protein of alveolar rhabdomyosarcoma transforms fibroblasts in culture. Proc. Natl. Acad. Sci. U. S. A. 93, 9805–9809 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lam P. Y. P., Sublett J. E., Hollenbach A. D., Roussel M. F., The oncogenic potential of the Pax3-FKHR fusion protein requires the Pax3 homeodomain recognition helix but not the Pax3 paired-box DNA binding domain. Mol. Cell. Biol. 19, 594–601 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia S. J., Barr F. G., Analysis of the transforming and growth suppressive activities of the PAX3-FKHR oncoprotein. Oncogene 23, 6864–6871 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Linardic C. M., et al. , The PAX3-FKHR fusion gene of rhabdomyosarcoma cooperates with loss of p16INK4A to promote bypass of cellular senescence. Cancer Res. 67, 6691–6699 (2007). [DOI] [PubMed] [Google Scholar]
- 20.Xia S. J., Holder D. D., Pawel B. R., Zhang C., Barr F. G., High expression of the PAX3-FKHR oncoprotein is required to promote tumorigenesis of human myoblasts. Am. J. Pathol. 175, 2600–8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Keller C., et al. , Alveolar rhabdomyosarcomas in conditional Pax3: Fkhr mice: Cooperativity of Ink4a/ARF and Trp53 loss of function. Genes Dev. 18, 2614–2626 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keller C., Hansen M. S., Coffin C. M., Capecchi M. R., Pax3: Fkhr interferes with embryonic Pax3 and Pax7 function: Implications for alveolar rhabdomyosarcoma cell of origin. Genes Dev. 18, 2608–2613 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davicioni E., et al. , Identification of a PAX-FKHR gene expression signature that defines molecular classes and determines the prognosis of alveolar rhabdomyosarcomas. Cancer Res. 66, 6936–6946 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Zhang S., et al. , PAX3-FOXO1 coordinates enhancer architecture, eRNA transcription, and RNA polymerase pause release at select gene targets. Mol. Cell, (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kikuchi K., et al. , Effects of PAX3-FKHR on malignant phenotypes in alveolar rhabdomyosarcoma. Biochem. Biophys. Res. Commun. 365, 568–574 (2008). [DOI] [PubMed] [Google Scholar]
- 26.Ebauer M., Wachtel M., Niggli F. K., Schäfer B. W., Comparative expression profiling identifies an in vivo target gene signature with TFAP2B as a mediator of the survival function of PAX3/FKHR. Oncogene 26, 7267–7281 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Bernasconi M., Remppis A., Fredericks W. J., Rauscher F. J., Schäfer B. W., Induction of apoptosis in rhabdomyosarcoma cells through down-regulation of PAX proteins. Proc. Natl. Acad. Sci. U.S.A. 93, 13164–13169 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fredericks W. J., Ayyanathan K., Herlyn M., Friedman J. R., Rauscher F. J., An engineered PAX3-KRAB transcriptional repressor inhibits the malignant phenotype of alveolar rhabdomyosarcoma cells harboring the endogenous PAX3-FKHR oncogene. Mol. Cell. Biol. 20, 5019–5031 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ayyanathan K., et al. , Hormone-dependent tumor regression in vivo by an inducible transcriptional repressor directed at the PAX3-FKHR oncogene. Cancer Res. 60, 5803–5814 (2000). [PubMed] [Google Scholar]
- 30.Nguyen T. H., Barr F. G., Therapeutic approaches targeting PAX3-FOXO1 and its regulatory and transcriptional pathways in rhabdomyosarcoma. Molecules 23, 2798 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Collins C. A., et al. , Integrated functions of Pax3 and Pax7 in the regulation of proliferation, cell size and myogenic differentiation. PLOS ONE 4, e4475 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Relaix F., et al. , Pax3 and Pax7 have distinct and overlapping functions in adult muscle progenitor cells. J. Cell Biol. 172, 91–102 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davis R. J., Barr F. G., Fusion genes resulting from alternative chromosomal translocations are overexpressed by gene-specific mechanisms in alveolar rhabdomyosarcoma. Proc. Natl. Acad. Sci. U.S.A. 94, 8047–8051 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Barr F. G., et al. , In vivo amplification of the PAX3-FKHR and PAX7-FKHR fusion genes in alveolar rhabdomyosarcoma. Hum. Mol. Genet. 5, 15–21 (1996). [DOI] [PubMed] [Google Scholar]
- 35.Laubscher D., et al. , BAF complexes drive proliferation and block myogenic differentiation in fusion-positive rhabdomyosarcoma. Nat. Commun. 12, 1–16 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marques J. G., et al. , NuRD subunit CHD4 regulates super-enhancer accessibility in rhabdomyosarcoma and represents a general tumor dependency. eLife 9, e54993 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gryder B. E., et al. , PAX3-FOXO1 establishes myogenic super enhancers and confers BET bromodomain vulnerability. Cancer Discov. 7, 884–899 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maity S. N., NF-Y (CBF) regulation in specific cell types and mouse models. Biochim. Biophys. Acta 1860, 598–603 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nardini M., et al. , Sequence-specific transcription factor NF-Y Displays histone-like DNA binding and H2B-like ubiquitination. Cell 152, 132–143 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Oldfield A. J., et al. , Histone-fold domain protein NF-Y promotes chromatin accessibility for cell type-specific master transcription factors. Mol. Cell 55, 708–722 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu N., et al. , Transcription factor competition at the γ-globin promoters controls hemoglobin switching. Nat. Genet. 53, 511–520 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gurtner A., Manni I., Piaggio G., NF-Y in cancer: Impact on cell transformation of a gene essential for proliferation. Biochim. Biophys. Acta BBA - Gene Regul. Mech. 1860, 604–616 (2017). [DOI] [PubMed] [Google Scholar]
- 43.Gurtner A., Manni I., Piaggio G., NF-Y in cancer: Impact on cell transformation of a gene essential for proliferation. Biochim. Biophys. Acta 1860, 604–616 (2017). [DOI] [PubMed] [Google Scholar]
- 44.Dolfini D., Minuzzo M., Pavesi G., Mantovani R., The Short Isoform of NF-YA Belongs to the Embryonic Stem Cell Transcription Factor Circuitry. Stem Cells 30, 2450–2459 (2012). [DOI] [PubMed] [Google Scholar]
- 45.Marziali G., et al. , The activity of the CCAAT-box binding factor NF-Y is modulated through the regulated expression of its A subunit during monocyte to macrophage differentiation: Regulation of tissue-specific genes through a ubiquitous transcription factor. Blood 93, 519–526 (1999). [PubMed] [Google Scholar]
- 46.Gurtner A., et al. , Requirement for down-regulation of the CCAAT-binding activity of the NF-Y transcription factor during skeletal muscle differentiation. Mol. Biol. Cell 14, 2706–2715 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gurtner A., et al. , NF-Y dependent epigenetic modifications discriminate between proliferating and postmitotic tissue. PLoS One 3, e2047 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Farina A., et al. , Down-regulation of cyclin B1 gene transcription in terminally differentiated skeletal muscle cells is associated with loss of functional CCAAT-binding NF-Y complex. Oncogene 18, 2818–2827 (1999). [DOI] [PubMed] [Google Scholar]
- 49.Rigillo G., et al. , The transcription factor NF-Y participates to stem cell fate decision and regeneration in adult skeletal muscle. Nat. Commun. 12, 1–17 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bock C., et al. , High-content CRISPR screening. Nat. Rev. Methods Primer 2, 1–23 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Subramanian A., et al. , Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shi J., et al. , Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat. Biotechnol. 33, 661–667 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shalem O., et al. , Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dharia N. V., et al. , A first-generation pediatric cancer dependency map. Nat. Genet. 53, 529–538 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Setty M., et al. , Characterization of cell fate probabilities in single-cell data with Palantir. Nat. Biotechnol. 37, 451–460 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gryder B. E., et al. , Histone hyperacetylation disrupts core gene regulatory architecture in rhabdomyosarcoma. Nat. Genet. 51, 1714–1722 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gryder B. E., Khan J., Stanton B. Z., Measurement of differential chromatin interactions with absolute quantification of architecture (AQuA-HiChIP). Nat. Protoc. 15, 1209–1236 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Behan F. M., et al. , Prioritization of cancer therapeutic targets using CRISPR-Cas9 screens. Nature 568, 511–516 (2019). [DOI] [PubMed] [Google Scholar]
- 59.Pandey P. R., et al. , PAX3–FOXO1 is essential for tumour initiation and maintenance but not recurrence in a human myoblast model of rhabdomyosarcoma. J. Pathol. 241, 626–637 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Koblan L. W., et al. , Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol. 36, 843–846 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Berríos K. N., et al. , Controllable genome editing with split-engineered base editors. Nat. Chem. Biol. 17, 1262–1270 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kluesner M. G., et al. , EditR: A Method to quantify base editing from Sanger sequencing. CRISPR J. 1, 239–250 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsherniak A., et al. , Defining a cancer dependency map. Cell 170, 564–576.e16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mantovani R., The molecular biology of the CCAAT-binding factor NF-Y. Gene 239, 15–27 (1999). [DOI] [PubMed] [Google Scholar]
- 65.Goeman F., et al. , Molecular imaging of nuclear factor-Y transcriptional activity maps proliferation sites in live animals. Mol. Biol. Cell 23, 1467–1474 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Benatti P., et al. , NF-Y activates genes of metabolic pathways altered in cancer cells. Oncotarget 7, 1633–1650 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dolfini D., Zambelli F., Pedrazzoli M., Mantovani R., Pavesi G., A high definition look at the NF-Y regulome reveals genome-wide associations with selected transcription factors. Nucleic Acids Res. 44, 4684–4702 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lu B., et al. , A transcription factor addiction in leukemia imposed by the MLL promoter sequence. Cancer Cell 34, 970–981.e8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hsu J. Y., et al. , SIX1 reprograms myogenic transcription factors to maintain the rhabdomyosarcoma undifferentiated state. Cell Rep. 38, 110323 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Abraham J., et al. , Lineage of origin in rhabdomyosarcoma informs pharmacological response. Genes Dev. 28, 1578–1591 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Stubbins R. J., Karsan A., Differentiation therapy for myeloid malignancies: Beyond cytotoxicity. Blood Cancer J. 11, 1–9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jeganathan S., et al. , Constrained peptides with fine-tuned flexibility inhibit NF-Y transcription factor assembly. Angew. Chem. Int. Ed Engl. 58, 17351–17358 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Nardone V., et al. , Structural basis of inhibition of the pioneer transcription factor NF-Y by suramin. Cells 29, 2370 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kotecha M., et al. , Inhibition of DNA binding of the NF-Y transcription factor by the pyrrolobenzodiazepine-polyamide conjugate GWL-78. Mol. Cancer Ther. 7, 1319–1329 (2008). [DOI] [PubMed] [Google Scholar]
- 75.Li W., et al. , MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sroka M. W., Vakoc C. R., Myo-differentiation reporter screen reveals NF-Y as an activator of PAX3-FOXO1 in rhabdomyosarcoma [RNA-Seq]. GEO repository. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE227594. Deposited 17 March 2023. [DOI] [PMC free article] [PubMed]
- 77.Sroka M. W., Skopelitis D., Vakoc C. R., Myo-differentiation reporter screen reveals NF-Y as an activator of PAX3-FOXO1 in rhabdomyosarcoma [CRISPR screens]. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE227595. Deposited 17 March 2023. [DOI] [PMC free article] [PubMed]
- 78.Sroka M. W., Gryder B., Vakoc C. R., Myo-differentiation reporter screen reveals NF-Y as an activator of PAX3-FOXO1 in rhabdomyosarcoma [Cut & Run]. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE227596. Deposited 17 March 2023. [DOI] [PMC free article] [PubMed]
- 79.Sroka M. W., Preall J. B., Vakoc C. R., Myo-differentiation reporter screen reveals NF-Y as an activator of PAX3-FOXO1 in rhabdomyosarcoma [scRNA-Seq]. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE227597. Deposited 17 March 2023. [DOI] [PMC free article] [PubMed]
- 80.Sroka M. W., Vermunt M. W., Blobel G. A., Vakoc C. R., Myo-differentiation reporter screen reveals NF-Y as an activator of PAX3-FOXO1 in rhabdomyosarcoma [4C-seq]. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE227600. Deposited 17 March 2023. [DOI] [PMC free article] [PubMed]
- 81.Gryder B. E., Wen X., Khan J., Selective Disruption of Core Regulatory Transcription [ChIP-seq]. GEO repository. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE116344. Accessed 20 May 2020.
- 82.Gryder B. E., Wen X., Khan J., Selective Disruption of Core Regulatory Transcription [AQuA-HiChIP]. GEO repository. https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE120770. Accessed 9 December 2020.
- 83.Khan J., Oncogenomics Database, National Cancer Institute. https://omics-oncogenomics.ccr.cancer.gov/cgi-bin/JK. Accessed 1 May 2023.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Dataset S01 (XLSX)
Dataset S02 (XLSX)
Dataset S03 (XLSX)
Dataset S04 (XLSX)
Dataset S05 (XLSX)
Dataset S06 (XLSX)
Dataset S07 (XLSX)
Dataset S08 (XLSX)
Data Availability Statement
The data reported in this paper have been deposited in the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/) as a part of the GSE227603 SuperSeries and is composed of the following SubSeries: RNA-seq - GSE227594 (76), CRISPR screens - GSE227595 (77), CUT&RUN - GSE227596 (78), scRNA-seq - GSE227597 (79), and 4C-seq - GSE227600 (80). In addition, the following publicly-available datasets were used in this study: ChIP-seq - GSE116344 (81), AQuA-HiChIP - GSE120770 (82), and RNA-seq on RMS tumor samples (83). All other data are included in the article and/or supporting information.