

Abstract
Background
Limitations in trial design, accrual, and data reporting impact efficient and reliable drug evaluation in cancer clinical trials. These concerns have been recognized in neuro-oncology but have not been comprehensively evaluated. We conducted a semi-automated survey of adult interventional neuro-oncology trials, examining design, interventions, outcomes, and data availability trends.
Methods
Trials were selected programmatically from ClinicalTrials.gov using primary malignant central nervous system tumor classification terms. Regression analyses assessed design and accrual trends; effect size analysis utilized survival rates among trials investigating survival.
Results
Of 3038 reviewed trials, most trials reporting relevant information were nonblinded (92%), single group (65%), nonrandomized (51%), and studied glioblastomas (47%) or other gliomas. Basic design elements were reported by most trials, with reporting increasing over time (OR = 1.24, P < .00001). Trials assessing survival outcomes were estimated to assume large effect sizes of interventions when powering their designs. Forty-two percent of trials were completed; of these, 38% failed to meet their enrollment target, with worse accrual over time (R = −0.94, P < .00001) and for US versus non-US based trials (OR = 0.5, P < .00001). Twenty-eight percent of completed trials reported partial results, with greater reporting for US (34.6%) versus non-US based trials (9.3%, P < .00001). Efficacy signals were detected by 15%–23% of completed trials reporting survival outcomes.
Conclusion
Low randomization rates, underutilization of controls, and overestimation of effect size, particularly pronounced in early-phase trials, impede generalizability of results. Suboptimal designs may be driven by accrual challenges, underscoring the need for cooperative efforts and novel designs. The limited results reporting highlights the need to incentivize data reporting and harmonization.
Keywords: clinical trial design trends, leveraging of trial registry data, primary central nervous system malignancies, treatment effect size assumptions, trial accrual
Key Points.
Comprehensive analysis revealed severe overestimation of effect size and underenrollment in trials
Reporting of results data and publications in ClinicalTrials.gov remains sparse
Programmatic approaches can be effectively used to assess periodic trends
Importance of the Study.
Trial design limitations and underreporting of results are pervasive in oncology and contribute to the dismal success rates of cancer therapy trials. These limitations necessitate a better understanding of the challenges and trends, but past assessments of neuro-oncology trials have predominantly focused on glioblastoma, or other restricted trial subsets. In a comprehensive assessment of 3038 adult interventional neuro-oncology trials registered in ClinicalTrials.gov up to July 28, 2021, we found a landscape of severely underaccruing and underreporting trials, relying on large unsubstantiated treatment effect sizes to power trial designs. While these findings echo the results of past focused assessments, our study is the first to comprehensively evaluate assumptions at design and trends over time across the neuro-oncology landscape. Finally, our study is the first to follow a programmatic approach to leveraging the registry data, which is imperative for routine utilization of historical data in future trial designs.
Despite important advances in the molecular understanding of malignant primary central nervous system (CNS) tumors and efforts to translate these advances into clinical benefit, there has been limited progress in treatments across neuro-oncology.1,2 Therapeutic drug development in neuro-oncology is complicated by a unique set of challenges including vast inter and intratumoral heterogeneity, complex interactions of the tumor with the neuronal and tumor microenvironments, the existence of the blood-brain/tumor barrier which limits the penetrability of compounds, a distinct and isolated immunologic locale, and the inability of preclinical models to recapitulate this complexity.3 Beyond scientific challenges, past studies have pointed to shortcomings in clinical trial design (including a lack of randomization, absence of controls, overly restrictive eligibility criteria, and low accrual) as contributing to the translation failure of potentially promising therapies.4–7 These concerns are further exacerbated by the low incidence of malignant primary CNS cancers, which consist of over 100 different diagnostic groups and subtypes that all classify as rare cancers with a cumulative incidence of 7.08 per 100,000.8–11
Think tank discussions have highlighted the need to address design challenges through novel trial designs that leverage external patient-level data,3,12 but well-curated high-quality datasets do not exist for already conducted trials.13 Other higher-level data from past clinical trials can be accessed through trial registries, including ClinicalTrials.gov and the consolidated WHO ICTRP.14 Regulatory pushes, including the 2005 ICMJE mandate to register trials prior to publication and the FDAAA Final Rule mandate (effective 2017) to deposit design and key results on conducted trials, and efforts to curate the information into relational databases are making it possible to leverage such data.15,16 However, past assessments of neuro-oncology trials in these registries focused on glioblastoma or limited time frame subsets of trials,4,12,17 and relied on manual curation of the literature which is not feasible for periodic assessments. We therefore aimed to conduct a comprehensive semi-automated assessment of the landscape of neuro-oncology trials registered in ClinicalTrials.gov taking advantage of the programmatic infrastructure of the Aggregate Analysis of ClinicalTrials.gov (AACT) database as a first necessary step in periodically leveraging of historical information. The scope of our survey included past, ongoing, and planned trials assessing interventions on any malignant CNS tumor diagnoses; we evaluated trial design characteristics, estimated treatment effect size assumptions implied by the trial designs, overviewed the extent of data availability, and summarized reported efficacy and toxicity of interventions.
Materials and Methods
Selecting and Obtaining Study Data
ClinicalTrials.gov registry was accessed programmatically on July 28, 2021 using R (RPostgreSQL library) through AACT.18–21 Adult neuro-oncology trials were selected by searching for terms derived from WHO-classified malignant CNS tumors (excluding nonmalignant ICDO-O codes) in their title, description, or study design (Supplementary Table S1)10; observational or expanded access trials, and trials with a maximum participant age under 19 were excluded. General oncology trials for comparative analyses were identified by searching for all Medical Subject Headings (MeSH) terms under “Neoplasm by Site” (Supplementary Table S2). Trial characteristics and results data obtained included conditions studied, interventions, year of initiation, number of arms, enrollment (anticipated and actual), primary purpose, intervention model, randomization, blinding, use of a data monitoring committee (DMC), efficacy outcomes, adverse events, and primary sponsors. Enrollment was augmented programmatically with data from the ClinicalTrials.gov webpage. Reported actual enrollment values that exceeded anticipated enrollment were assumed to reflect true anticipated enrollment. Basket trials with 6000 or more participants were excluded. Trial site locations were used to determine whether the trial was US or non-US based.
Trials assessing overall survival (OS), progression free survival (PFS), and tumor response were identified by searching for relevant terms among planned and collected outcomes data. Five-year OS rates (OS-5) and average incidence by CNS tumor histology classification were obtained from the 2020 CBTRUS Report.9 The median OS-5 for malignant CNS tumors (MOS-5) was used to define 3 survival categories: lowest survival (OS-5 < MOS-5), moderate survival (MOS-5 < OS-5 < 2 times MOS-5), and highest survival (OS-5 > 2 times MOS-5). Trials were categorized based on conditions studied; unmatched conditions were considered of moderate survival (Supplementary Table S3). The total incidence (rate per 100,000) of a given trial’s target diagnoses was computed as the sum of the incidence of individual diagnoses studied, accounting for diagnoses overlaps as in the case of glioma (Supplementary Table S4) and not exceeding the incidence of all malignant CNS tumors (7.08 per 100,000). Trials were placed into 3 incidence categories: lowest (incidence smaller than one-third of 7.08), moderate (incidence between one-third and two-thirds of 7.08), and highest (incidence larger than two-thirds of 7.08).
Interventions and Mechanisms of Action
MeSH terms of trial interventions were mapped to compound names from the Drug Repurposing Hub to obtain mechanisms of action.22
Data Reporting Analysis
Logistic regression was used to assess associations of trial characteristics (phase, primary sponsor class, primary purpose, blinding, intervention model, DMC use, randomization, year of initiation) with trial data reporting (basic design, enrollment, results, publication). Stratified subgroup differences were assessed using chi-squared tests.
Feasibility and Accrual
Differences between anticipated and actual enrollment of completed trials were assessed using paired Wilcoxon tests with a one-sided alternative hypothesis that anticipated enrollment is greater than actual enrollment, stratified by phase, survival category, incidence category, or location, and False Discovery Rate (FDR) multiple hypothesis correction. Pearson correlation was computed on the proportion of trials meeting (100%, or 90% of) their enrollment target versus initiation year. Logistic regression was used to assess whether the likelihood of a trial’s meeting its enrollment target depended on year of initiation, survival or incidence category, and location.
Treatment Effect Size Assumption Analysis
The sample sizes of trials with primary study outcomes of OS, PFS, or tumor response rate were used to estimate the minimum treatment effect sizes (ES) that they were powered to evaluate (with one-sided hypothesis of treatment-associated benefit, at α = 0.05 and power = 80%).
For trials evaluating OS, the minimum detectable effect sizes were estimated with respect to the control event (death) rate based on OS-5 rates of indicated conditions.9 Trials targeting multiple conditions were assessed based on the lowest survival condition. Trials evaluating PFS were similarly assessed using 5-year progression free survival (PFS-5) rates, estimated as 50%, 75%, and 80% of their corresponding OS-5 rates for the lowest, moderate, and highest survival categories, respectively.23 For trials evaluating tumor response rate, we estimated the minimum ES of increase in response rate that trials were powered to assess, from a baseline response rate of 10%.24 Trial follow-up periods were assumed to equal twice the median overall survival of the relevant diagnoses.
Survival-assessing trials were assumed to have 2 arms, with enrollment of single-arm trials doubled in the analysis to allow for use of a historical control arm. Enrollment for response-assessing trials was assumed equally distributed among the arms, as in Bugin et al.25
Efficacy and Toxicity Outcomes
The efficacy, toxicity, and other results reported for completed trials were mapped to the arm level using full or partial matches and synonyms in arm descriptions; arms mentioning control/placebo-related terms were labeled as control, and the remainder as experimental. Hazard ratios (HR) and median months of survival were obtained for trials assessing OS and PFS. Objective response rate (ORR) was obtained or calculated from available data.
Log HR and computed standard errors from OS and PFS were pooled using an inverse variance model with random effects using the R metafor library.26 One-sided Wilcoxon tests were conducted to compare the median months of OS, median months of PFS, and ORR in the (best performing) experimental arms versus those of control arms, with the alternative hypothesis of treatment-associated benefit. When pooling data by survival category, trials studying multiple conditions were included for each relevant category.
Adverse event (AE) terms and counts of patients affected and at-risk for each AE were extracted for completed trials. Risk ratios (RR) were calculated for the most frequently reported individual AEs, and for totals of serious AEs, other AEs, and all-cause mortality from trials with matched control and experimental arms. Risk ratios were pooled using an inverse variance model with random effects using the R metafor library.
Linear regression was used to assess the association between treatment efficacy and toxicity in experimental arms, weighted by total actual enrollment, with consideration of interaction between toxicity covariates. The model power was assessed using the R pwr library.27
Supplementary Information contains further details on search terms, histology categories and survival rates, and histology categories and incidence.
All analyses were conducted using R version 3.6.0.18
Ethics Statement
The study uses preexisting publicly available data downloaded from ClinicalTrials.gov (www.clinicaltrials.gov) and Drug Repurposing Hub (https://www.broadinstitute.org/drug-repurposing-hub).
Results
Landscape of Neuro-Oncology Trials
We surveyed the landscape of adult interventional neuro-oncology trials by retrieving and analyzing data from the ClinicalTrials.gov registry. We identified 3038 registered trials with start dates between 1966 and 2025 that studied interventions for WHO-classified malignant primary CNS tumor diagnoses, and excluded pediatric-targeting studies. Trials were most commonly phase 2 (43.1%), completed (41.9%), nonblinded (92.3%), single group (64.9%), and nonrandomized (51.0%), with a primary purpose of treatment (85.8%) and a single primary outcome (67.3%) (Figure 1A; Table 1).
Figure 1.
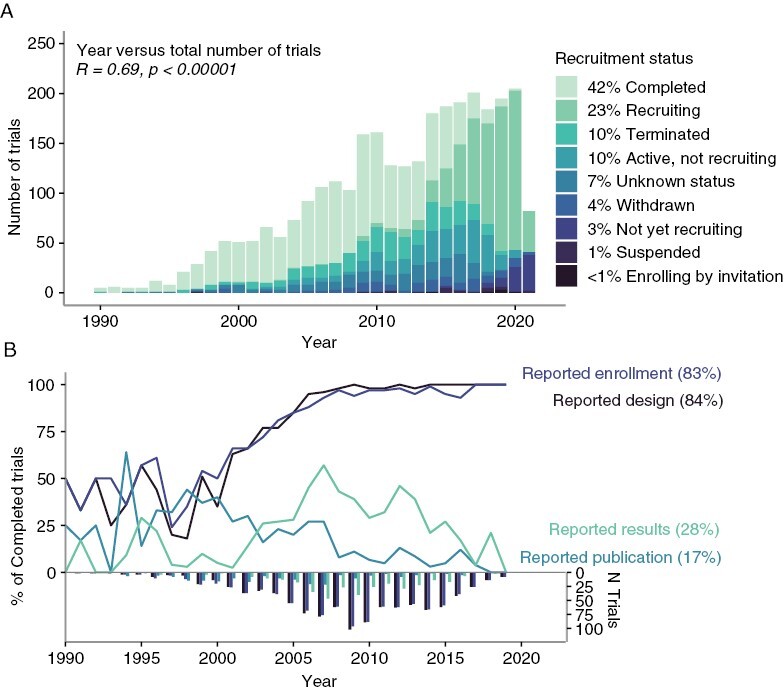
Overview of trial landscape. (A) Number and trial status and (B) Data reporting over years. (The plots exclude depiction of 5 pre-1990 trials).
Table 1.
Trial Characteristics of Interventional Studies of Primary Malignant CNS Tumors Registered on ClinicalTrials.gov
| Number of Trials (%) | ||
|---|---|---|
| Phase (n = 2626) * | Early Phase 1 | 96 (3.7%) |
| Phase 1 | 760 (28.9%) | |
| Phase 1/Phase 2 | 377 (14.4%) | |
| Phase 2 | 1131 (43.1%) | |
| Phase 2/Phase 3 | 40 (1.5%) | |
| Phase 3 | 186 (7.1%) | |
| Phase 4 | 36 (1.4%) | |
| Recruitment status (n = 3038) | Completed | 1271 (41.9%) |
| Recruiting | 688 (22.7%) | |
| Terminated | 316 (10.4%) | |
| Active, not recruiting | 304 (10.0%) | |
| Unknown status | 223 (7.3%) | |
| Withdrawn | 124 (4.1%) | |
| Not yet recruiting | 83 (2.7%) | |
| Suspended | 23 (0.8%) | |
| Enrolling by invitation | 6 (0.2%) | |
| Primary purpose (n = 3019) | Treatment | 2590 (85.8%) |
| Diagnostic | 210 (7.0%) | |
| Supportive care | 93 (3.1%) | |
| Other | 38 (1.3%) | |
| Basic science | 34 (1.1%) | |
| Prevention | 30 (1.0%) | |
| Health services research | 12 (0.4%) | |
| Device feasibility | 7 (0.2%) | |
| Screening | 5 (0.2%) | |
| Has data monitoring committee (n = 2381) | TRUE | 1495 (62.8%) |
| FALSE | 886 (37.2%) | |
| FDA regulated drug or device (n = 1133) | TRUE | 718 (63.4%) |
| FALSE | 415 (36.6%) | |
| Results reported (n = 3038) | FALSE | 2514 (82.8%) |
| TRUE | 524 (17.3%) | |
| Single center study (n = 3038) | TRUE | 1654 (54.4%) |
| FALSE | 1384 (45.6%) | |
| One or more trial site in the United States (n = 2876) | TRUE | 2004 (69.7%) |
| FALSE | 872 (30.3%) | |
| Number of primary outcomes planned (n = 2823) | 1 | 1901 (67.3%) |
| 2 | 542 (19.2%) | |
| 3 | 175 (6.2%) | |
| 4 | 89 (3.2%) | |
| >=5 | 116 (4.7%) | |
| Blinding (n = 2838) | None (Open label) | 2618 (92.3%) |
| Single | 78 (2.8%) | |
| Double | 62 (2.2%) | |
| Triple | 41 (1.4%) | |
| Quadruple | 39 (1.4%) | |
| Intervention model (n = 2785) | Single-group assignment | 1806 (64.9%) |
| Parallel assignment | 824 (29.6%) | |
| Sequential assignment | 114 (4.1%) | |
| Crossover assignment | 36 (1.3%) | |
| Factorial assignment | 5 (0.2%) | |
| Randomization (n = 1287) | Nonrandomized | 656 (51.0%) |
| Randomized | 631 (49.0%) | |
| Intervention type (n = 3038) ** | Drug | 2333 (51.0%) |
| Radiation | 547 (12.0%) | |
| Biological | 478 (10.5%) | |
| Procedure | 457 (10.0%) | |
| Other | 453 (9.9%) | |
| Device | 173 (3.8%) | |
| Behavioral | 50 (1.1%) | |
| Diagnostic test | 30 (0.7%) | |
| Genetic | 24 (0.5%) | |
| Dietary supplement | 20 (0.4%) | |
| Combination product | 9 (0.2%) | |
| Top 30 conditions (n = 3038) *** | Glioblastoma | 1415 (47.4%) |
| Glioma | 1296 (43.4%) | |
| Astrocytoma | 384 (12.9%) | |
| Germ cell neoplasm | 253 (8.5%) | |
| Gliosarcoma | 242 (8.1%) | |
| Oligodendroglioma | 172 (5.8%) | |
| Medulloblastoma | 169 (5.7%) | |
| Central nervous system lymphoma | 147 (4.9%) | |
| Neuroectodermal neoplasm | 146 (4.9%) | |
| Ependymoma | 142 (4.8%) | |
| Meningioma | 152 (4.2%) | |
| Primitive neuro-ectodermal tumors (PNET) | 100 (3.4%) | |
| Pituitary neoplasm | 81 (2.71) | |
| Oligoastrocytoma | 78 (2.6%) | |
| Diffuse intrinsic pontine glioma (DIPG) | 64 (2.1%) | |
| Paraganglioma | 53 (1.8%) | |
| Nerve sheath neoplasm | 50 (1.7%) | |
| Teratoma | 46 (1.5%) | |
| Chordoma | 43 (1.4%) | |
| Germinoma | 43 (1.4%) | |
| Craniopharyngioma | 38 (1.3%) | |
| Pineoblastoma | 29 (1.0%) | |
| Choriocarcinoma | 21 (0.7%) | |
| Hemangiopericytoma | 21 (0.7%) | |
| Choroid plexus neoplasm | 19 (0.6%) | |
| Yolk sac neoplasm | 14 (0.5%) | |
| Xanthoastrocytoma | 10 (0.3%) | |
| Embryonal carcinoma | 9 (0.3%) | |
| Ganglioglioma | 8 (0.3%) | |
| Gliomatosis | 8 (0.3%) | |
| Top 10 trial site locations (n = 2876) | United States | 2004 (67.1%) |
| France | 256 (8.6%) | |
| Canada | 232 (7.8%) | |
| Germany | 181 (6.1%) | |
| China | 156 (5.2%) | |
| United Kingdom | 143 (4.8%) | |
| Netherlands | 131 (4.4%) | |
| Italy | 127 (4.3%) | |
| Spain | 126 (4.2%) | |
| Switzerland | 120 (4.0%) |
* n indicates the total number of trials reporting each trial characteristic listed;
** there may be multiple interventions reported per trial;
*** excluded top nonspecific categories such as “CNS neoplasms” and “brain neoplasms.”
Most (60.1%) trials studied a combination of multiple conditions, with a median of 2 per trial (inter-quartile range [IQR]: 1–3; Supplementary Table S5). Most frequently studied diagnoses were gliomas: glioblastoma (47.4%), nonspecified glioma (43.4%), astrocytoma (12.9%), gliosarcoma (8.1%), oligodendroglioma (5.8%), and ependymoma (4.8%). Excluding gliomas, the most frequently studied diagnoses included germ cell neoplasms (8.5%), medulloblastoma (5.7%), central nervous system lymphoma (4.9%), neuroectodermal neoplasm (4.9%), and meningioma (4.2%). Other neoplasms were studied by fewer than 4% of trials. North America hosted the largest number of trials (United States 67.1%, Canada 7.8%), followed by Europe (France 8.6%, Germany 6.1%, United Kingdom 4.8%, Netherlands 4.4%, Italy 4.3%, Spain 4.2%, Switzerland 4.0%), and Asia (China [5.2%]), with remaining regions or countries hosting fewer than 4% of trials (Table 1).
The majority of phase 1 (60.3%, n = 35) and phase 2 (64.8%, n = 463) trials were single-arm studies. Of phase 1 and phase 2 single-arm trials reporting efficacy primary outcomes, OS was reported in 28.6% and 20.1%, PFS in 54.3% and 39.7%, and tumor response in 42.9% and 51.2% of trials, respectively; more than one primary efficacy outcome was reported by 20% of phase 1 and 9.5% of phase 2 single-arm studies (Supplementary Tables S6 and S7). Among phase 3 trials reporting sufficient information (n = 86), only one reported single-arm design, with the primary outcome of tumor response. No significant change over time was found in the prevalence of OS, PFS, or ORR as primary endpoints for single-arm trials.
Data Reporting Trends and Associated Trial Characteristics
We assessed the degree to which trials reported design elements, results, and publications in the registry. Basic study design information reported included phase (86.5%), purpose (99.4%), intervention model (91.7%), blinding (93.4%), randomization (42.4%), number of arms (87.0%), and planned outcomes (92.9%). Conditions studied and eligibilities were reported by 100% of trials; however, the latter were reported as unstructured free text, not amenable to routine automated analysis. Excluding withdrawn and terminated trials, information available on completed trials (n = 1271) included basic design (including phase, intervention model, blinding, and randomization) for 84.0%, enrollment for 83.5%, results for 28.0%, and PubMed ID of a publication associated with study results for 17.5% of trials (Figure 1B). Baseline patient counts by trial arm, and subgroups of sex and age were provided by 28.0% of completed trials; counts by race/ethnicity were reported by 13.6%, and all other subgroup-level data (including performance status, diagnostic subgroups, and prior treatments) were reported by <4% (Supplementary Table S8). Few (n = 168) completed trials reported arms labels and efficacy results; even fewer reported subgroup-level counts, which additionally lacked explicit connections to results data. Further discordance in data reporting was apparent as we considered more detailed questions, including arm-level analyses (Supplementary Figure S1).
Studies located in the United States were significantly more likely to report results than those located elsewhere (34.6% and 9.3%, chi-squared = 68.89, P < .00001), with no location-dependent significant differences in design, enrollment, and publication reporting. Results reporting in neuro-oncology was similar to that of general oncology (26.8% (4198/15,663), chi-squared = 0.81, P = .36770), but greater than that of all completed interventional studies in the registry (23.9% (39,376/164,974), chi-squared = 11.67, P = .00064); publication reporting was significantly greater in neuro-oncology compared to both general oncology (14.3% (2232/15,663), chi-squared = 9.56, P = .00200) and all studies (9.8% (16,105/164,974), chi-squared = 83.67, P < .00001).
We evaluated whether the likelihood of data reporting was associated with specific trial characteristics, including primary sponsor, phase, primary purpose, intervention model, DMC use, randomization, blinding, and year. Trials were more likely to report basic design characteristics in the registry over time (OR = 1.24, P-adj < .00001). Phase 1 trials (vs. phase 3) were significantly less likely to report results (OR = 0.07, P-adj = .01997). The likelihood of trials to report a results publication significantly decreased each year (OR = 0.87, P-adj = .01997). No other significant associations were found (Supplementary Table S9).
Enrollment and Accrual Challenges Across Primary CNS Malignancies
As a first indicator of how successfully completed trials were conducted, we investigated whether they met their anticipated enrollment target, and factors associated with higher accrual rates. Among completed trials reporting sufficient data, 37.9% failed to reach their enrollment target, and the median actual enrollment was lower than the median anticipated enrollment (35 [IQR: 19–68] < 45 [IQR: 25–81], P < .00001); 33.2% failed to meet at least 90% of their enrollment target, and the proportion of trials initiated in a given year meeting this threshold dropped over time for both US (R= −0.9, P < .00001) and non-US studies (R = −0.5, P = .026) (Figure 2A). Accounting for the negative association with initiation year, US-based trials were half as likely to meet enrollment goals compared to non-US based trials (OR = 0.50, P < .00001), with no significant dependence on the trial size (Supplementary Figure S11).
Figure 2.
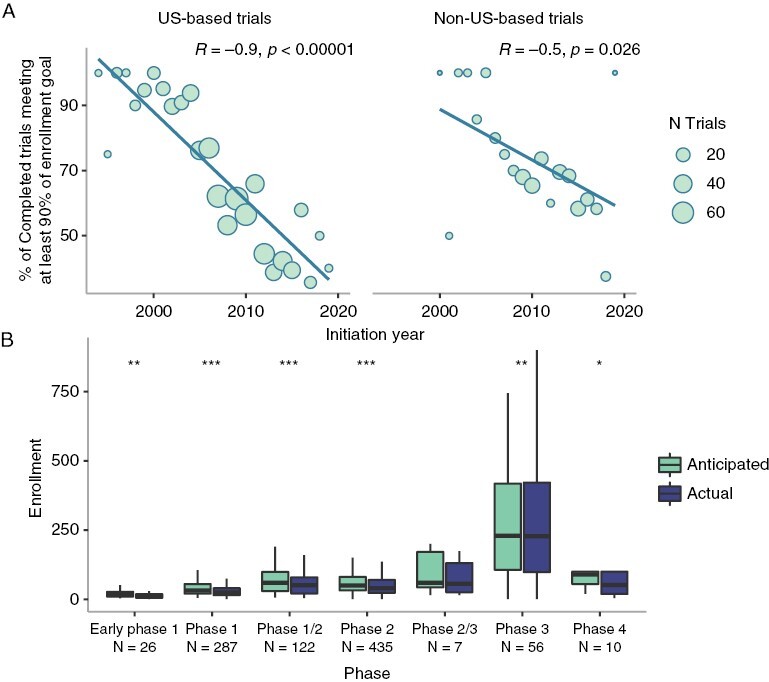
Enrollment trends. (A) Proportion of completed trials meeting at least 90% of their target enrollment stratified by US versus non-US location. (B) Comparison of target and actual enrollment for completed trials, by phase. Significance levels for P-adj: 0 < *** ≤ .001 ≤ ** ≤ .01 ≤ * ≤ .05.
When stratified by trial phase, actual enrollment was significantly below anticipated enrollment for all phase trials except phase 2/phase 3 (Figure 2B). We assessed whether differences existed based on disease prognosis or total incidence, hypothesizing that underenrollment may reflect reluctance or inability of patients to participate in trials due to their prognosis or rarity of the tumors; however, significant underenrollment pervaded all grouped prognostic and incidence levels (Supplementary Figures S2, S3C and F). Stratifying trials by conditions studied, actual enrollment was significantly lower than anticipated enrollment for 40.5% (15/37) of individual conditions considered (Supplementary Table S10).
Treatment Effect Size Assumptions and Power of Trials Evaluating Efficacy Outcomes
Underenrollment has direct implications on the statistical power of the trials to assess their anticipated treatment effect, so we were interested in estimating the tolerance of trial designs toward underenrollment. One challenge encountered as we set out to do so was determining lenient yet realistic criteria against which to assess the power of the trials. Literature search of landmark neuro-oncology trials revealed wide ranges of assumptions on anticipated treatment effect sizes and narrowing these down was further complicated by the heterogeneity of the trials’ assessed diagnoses and planned outcomes. Thus instead, for a given trial we estimated the minimum treatment effect size (ES) that the trial would be sufficiently powered to assess (at α = 0.05 and power = 80%). For most lenient analysis, survival-assessing trials studying multiple diagnoses were evaluated using the worst-prognosis diagnosis studied, which, for a given trial size, would power detection of the smallest treatment effect size. Single-arm survival-assessing trials were assumed to assess their outcomes against external controls, doubling their power for the given size. For tumor response outcomes we assumed a minimum baseline response rate of 10%. Finally, no α adjustment was assumed for trials assessing multiple outcomes.
Figure 3 summarizes the estimated minimum ES among trials with anticipated enrollment data and primary outcomes of OS (n = 348), PFS (n = 548), and tumor response (n = 477). The estimates reveal that most trials assessing survival outcomes were designed with assumptions of large ES (median minimum ES of 43%, 48%, and 60% for low, moderate, and high survival categories for OS; median minimum ES of 48%, 46%, and 53% for low, moderate and high survival categories for PFS) and insufficient power to detect smaller treatment-associated benefits. Trials assessing tumor response rates were overall more realistic in their ES assumptions, designed to detect smaller minimum ES (median minimum ES of 14% for each of low, moderate, and high survival categories). Sensitivity analysis using a higher baseline ORR up to 30% provided more stringent estimations (yielding higher minimum detectable ES) as expected.
Figure 3.
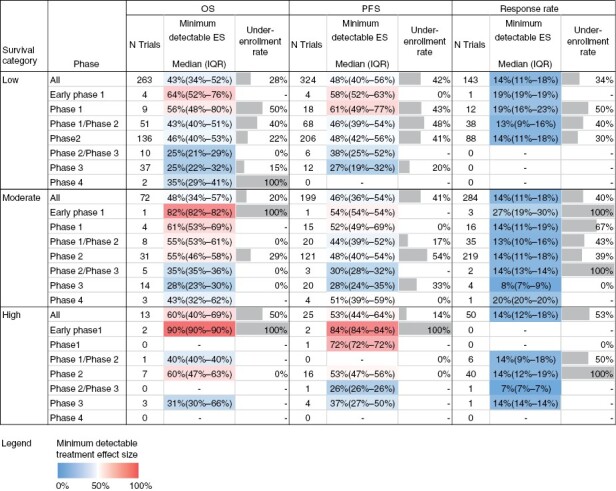
Effect size assumption analysis. The minimum ES that each trial is sufficiently powered to assess was computed for trials reporting anticipated enrollment and primary endpoints of OS, PFS, or ORR. For OS/PFS calculations, trials studying multiple diagnoses were assessed based on the diagnosis with poorest prognosis to allow for most lenient assessment. Therefore, survival information for 100% of low survival category trials was based on Glioblastoma; for moderate survival category trials, survival information was based on survival for Glioma for 56.2%, Neuroectodermal Neoplasm for 9.0%, Astrocytoma for 7.7%, Meningioma for 7.7%, CNS Lymphoma for 7.2%, and other diagnoses for <7% of the trials; for high survival category trials, survival information was based on Germ Cell Neoplasm for 46.6%, Pituitary Neoplasm for 24.6%, Medulloblastoma for 16.7%, Ependymoma for 8.5%, Craniopharyngioma for 5.6%, and other diagnoses for <1% of trials. The percentage of trials that failed to meet their target enrollment was computed for completed trials reporting anticipated and actual enrollment.
Stratification by phase revealed that phase 3 trials of the low-survival category, which comprises of all glioblastoma trials, were powered to assess more sensitive ES down to a median of 25% and 27% for OS and PFS outcomes, respectively, while phase 2 studies of this category were designed to detect median minimum ES of 46% and 48%, respectively. Similar trends persisted across moderate and high prognostic categories. Tumor response outcomes in phase 3 trials were also designed to detect smaller treatment effects compared to early phases, where data existed.
We also tabulated the percentage of completed trials of a given prognostic category and phase that failed to reach their enrollment target (also shown in Figure 3). As anticipated, underenrollment led to an increase in the minimum ES the trials could assess across all categories considered (Supplementary Table S12).
Efficacy and Toxicity Outcomes and Correlations
We also sought to assess whether the results of completed trials could provide insights into the efficacy and toxicity of the interventions and feasibly provide the basis for the establishment of data-driven historical controls. Among completed trials reporting results (n = 356), 30.3%, 27.8%, and 22.8% reported efficacy results on OS, PFS, and tumor response (with sufficient information to extract ORR), respectively (Supplementary Figure S1). Median data collection timeframes varied by outcome. Trials studying OS and PFS had median collection timeframes of 28.0 months (IQR: 24.0–52.0) and 24.0 months (IQR: 12.0–48.0), and ORR had the shortest median time of 15.0 months (IQR: 2.8–24.0). However, as subgroup-level counts were reported by few trials (28.0% of completed trials reporting counts by age/sex, and fewer than 4% reporting other subgroup-level counts) it was not feasible to account for any subgroup confounders in the analyses below.
There was no statistically significant treatment-associated improvement between control and experimental arms of studies per median OS (16.4 months [IQR: 13.6–18.9] vs. 12.4 months [IQR: 8.6–17.9]), median PFS (6.2 months [IQR: 4.1–7.4] vs. 7 months [IQR: 3.0–10.6]), and ORR (6% [IQR: 4.4%–11%] vs. 11% [IQR: 0%–24%]) (Figure 4). Stratifying studies by survival categories revealed a significant treatment-associated improvement in median PFS for trials studying cancers with moderate survival (median PFS of 1.1 [IQR: 0.6–1.7] and 7.8 [IQR: 3.9–11.8]; P = .0057) although this finding is likely an artifact of only two data points being available for the control side. Sensitivity analysis by collection timeframe (less than vs. greater than or equal to 6 months) revealed no new statistical significance (Supplementary Figure S4).
Figure 4.
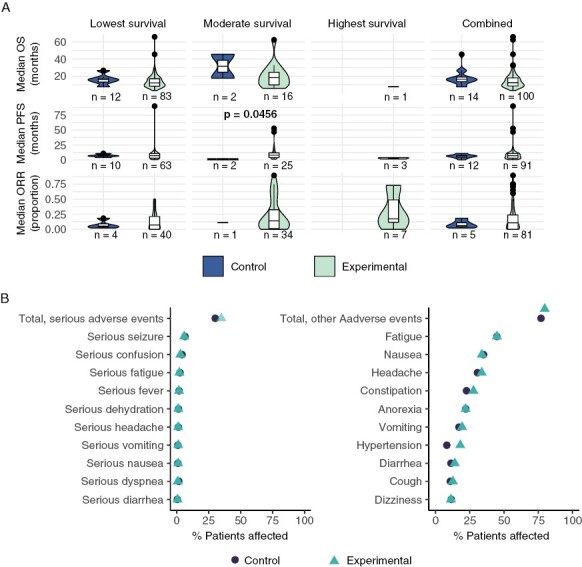
Overview of reported efficacy and toxicity. (A) Median responses were compared for experimental and control arms using one-sided Wilcoxon test for completed trials reporting response in either or both arms. One comparison is significant after FDR correction. (B) Proportion of patients experiencing serious and other AEs among trials reporting both experimental and control arm toxicity data.
The HR of OS and PFS endpoints were reported by 4.7% and 2.7% of applicable trials, respectively. Of these, 3 trials (15.0%) studying OS and 3 trials (23.1%) studying PFS reported HR indicating statistically significant survival benefit; pooled HR for all trials reporting OS revealed no significant treatment-associated survival benefit overall (HR: 0.89, P = .05263), whereas pooled HR for trials reporting PFS (HR: 0.84, P = .00622) indicated a statistically significant benefit, with both results limited by small sample size (Supplementary Figure S5A and B).
The total number of serious (SAE) and other (nonserious) adverse events (AEs) were reported by 21.5% of completed trials; individual serious and other adverse events were reported by 18.6% and 22.9%, respectively (Figure 4B). Extracting RR from these trials, 7.6% and 3.8% reported a significantly greater risk of total SAE and total other AE with experimental therapy (RR > 1), but no significance was found when pooled across trials (Supplementary Figure S5C–E). One trial reported a significantly lower risk of total other AE with experimental therapy (RR < 1). No trials reported significant treatment-associated differences in total mortality.
Toxicity and efficacy correlations may inform underlying treatment mechanisms and identify predictors of response and pharmacokinetics in drug discovery; thus, we assessed the feasibility of such analyses within the present framework of data availability.28–30 We evaluated correlations between efficacy (median OS, median PFS, and ORR) and total toxicity (rate of total SAE and total AE) in experimental arms, and noted a significant negative correlation between median PFS in experimental arms and total SAE (PFS: β = −18.9, P = .001), but no other significant correlations (Supplementary Table S13). Assessing correlations between efficacy and the top three most frequently reported AEs in experimental arms, we noted that OS was positively associated with serious nausea (OS: β = 436.5, P = .013), and both OS and PFS were negatively associated with fatigue (OS: β = −44.9, P < .0010; PFS: β = −40.5, P < .0010; Supplementary Table S14). The registry lacked sufficient data for the inclusion of controlled values in the regression, and our analysis was limited by small sample size.
Landscape and Redundancies of Interventions and Mechanisms of Action
Finally, we surveyed the landscape of drugs and biological therapies, assessing their predominant mechanisms of action and trends over time. DNA alkylating agents were the most common class of tested drugs (34% of interventions), followed by topoisomerase inhibitors (11%), DNA inhibitors (8%), DNA synthesis inhibitors (6%), and tubulin polymerization inhibitors (5%); the cumulative use of these classes of therapeutics decreased or plateaued over time, with novel therapies increasing in use (Figure 5; Supplementary Figure S6). Further associations between targeted mechanism of action and the efficacy or toxicity of interventions were not considered due to limited data and efficacy signals.
Figure 5.
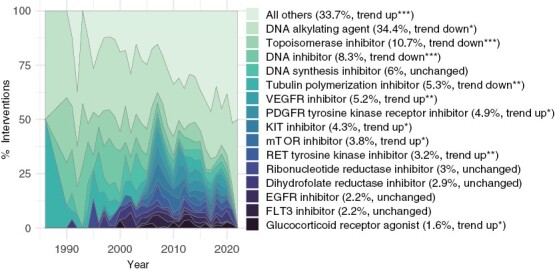
Trends of Intervention Mechanisms of Action. Proportion of most common mechanisms of action among interventions over years. Significance levels for P-adj: 0 < *** ≤ .001 ≤ ** ≤ .01 ≤ * ≤ .05.
Discussion
Understanding the landscape of past neuro-oncology trials is an important step in identifying opportunities to correct past shortcomings in future studies. Our assessment of neuro-oncology trials registered in ClinicalTrials.gov revealed that the majority of trials were nonrandomized, single-group studies, in agreement with, and expanding upon past findings in glioblastoma trials.4 Furthermore, in a first comprehensive assessment of treatment effect size assumptions, we found that studies evaluating survival benefit largely overestimated the expected treatment effects to power studies. Most early-phase studies were estimated to rely on median effect sizes of 40%–63%, across prognostic categories. Phase 3 studies were powered to detect smaller effect sizes but even those relied on median effect sizes of over 25%. Additionally, the heterogeneity in assumed effect sizes across studies signifies poor consensus on expected treatment effect during trial design. Trials assessing tumor response were designed with more conservative effect size expectations, likely due to smaller sample size requirements compared to survival analyses and ability to utilize single-arm designs. However, response assessment is not without challenges: concerns of objectivity and the need to make assumptions regarding baseline control-arm response rates for single-arm design point to the need to establish disease-specific standards for radiographic assessments,31 standard-of-care efficacy outcomes, and clinically meaningful effect size targets.
Challenges in accrual can dictate the observed design limitations.7 Indeed, we found that 38% of completed trials failed to meet their enrollment threshold. Underenrollment pervaded across prognostic and incidence subgroups, with significant underenrollment impacting more than 40% of diagnoses, broadly recapitulating concerns previously identified in glioblastoma trials.4 US-based studies were more likely to face accrual failure than non-US counterparts. Our recent work illustrating geographic and socio-economic barriers to the US trial infrastructure, and prior reports suggesting more stringent eligibility criteria among US trials recapitulate the need to address these challenges in the United States.32,33 Nonetheless, the persistent and worsening underenrollment remains a concern for all studies, highlighting the need to promote higher-powered, multi-site, wider-access clinical studies and international collaborations rather than disjointed and isolated efforts.3,7 Novel trial designs can also help overcome some design limitations, allowing, for example, for flexible testing of multiple hypotheses as data is gathered and disease-agnostic testing of targeted therapies.34–37
The identified limitations at the design and accrual levels may be responsible for non-generalizable results. For example, it has previously been reported that only 9% of glioblastoma therapies with successful phase 2 evaluation led to positive phase 3 results.38 We found low success rates across treatment types, conditions, and phases, with only 15% of trials that reported a hazard ratio showing treatment-associated OS benefit; the true rate of success is likely lower due to potential positive reporting bias. Similarly, only 8% of trials reported greater treatment-associated risk of serious AEs, testifying to the underreporting of harm in clinical studies that bias drug evaluation.39 Redundancy in mechanisms of actions of tested interventions may also contribute to the high failure rates, although encouragingly, the use of less-common interventions, including targeted therapies, increased over time.
Our findings reflect the overall state of data availability and quality in clinical research. Less than one-third of completed neuro-oncology trials reported any form of results, with 17% reporting relevant publications, in agreement with past evaluations of glioblastoma trials.4 Even fewer trials reported standardized efficacy and toxicity information. Efficacy data were often limited to summary statistics; AEs were reported by cumulative incidence by only 1 in 5 trials and individual incidence by a smaller proportion, all with levels of severity being grouped into serious and other, which do not align with the standardization efforts of Common Terminology Criteria for Adverse Events (CTCAE).40 The relatively higher reporting rates of US-based studies compared to their counterparts may echo the impact of ClincalTrials.gov reporting mandates, emphasizing the importance and efficacy of standardized guidelines. Interestingly, the results reporting rate in neuro-oncology was comparable to that of broader oncology and greater than that of broader clinical research. Considering previous findings that oncology publication rates are lower than in other diseases likely due to lower positive trial rates, the greater results reporting in oncology may reflect encouraging trends toward sharing negative data in registries and overcoming publication bias.41
The paucity and low quality of data led to several limitations of our study. We sought to maximize automated methods within the framework of readily available data, making several simplifying assumptions in search and analytical parameters attempting to balance the usability of low-quality data with the accurate depiction of the heterogenous trials landscape. The low data quality and restrictions of automated searches also present challenges in disambiguating when findings reflect data reporting versus underlying design issues, such as in the case of phase 1 trials identified (and manually verified) to report efficacy measures among their primary outcomes, in apparent contradiction to the traditional phase 1 focus on safety. We assumed minimum fixed tumor response rates and estimated PFS from OS data for our effect size analysis from published trials; we assessed trials by prognostic and incidence categories, since trials studying multiple diagnoses did not report data at the diagnosis granularity. These decisions may have muted nuanced differences across individual conditions and trial designs. Furthermore, without a framework for patient-level or meaningful clinical and molecular subgroup summaries, we could not provide outcome data that met the gold standard of historical efficacy data for external controls.34
The exercise of seeking to maximize insights from the registry led us to identify several key avenues that may incentivize data reporting and utilization needed to improve the state of the practice. First, demonstrating the value of resources resulting from high-quality data reporting, by emphasizing the returned benefit to investigators, may be crucial to promoting best practices and compliance. Usability and value of reported data may be maximized by improving back-end data models, increasing the user-friendliness of data input interfaces, and creating novel interfaces for data repositories, like the efforts of AACT and ClinicalTrials.gov modernization. This may include currently unavailable explicit mapping between eligibility criteria, treatments, patient counts, and outcomes at arm level and subgroup level, and reduction of unstructured data fields through standard terminologies and controlled vocabularies. In addition to enabling previously infeasible automated analyses, these changes can facilitate creation of investigator-facing tools for interactive and customizable on demand analyses using the existing body of trial data. Second, there is an unmet need for a central resource (akin to the CBTRUS Reports) that explicitly outlines key assumptions utilized in trial design, including baseline survival and response rates resulting from standard-of-care therapies and realistic effect sizes for evaluating novel therapies in the neuro-oncology context. Accounting for the type of intervention, these guidelines may permit greater optimism for targeted therapies or in cases of rational enrichment of the participant cohort and recommend more modest effect size expectations otherwise. Finally, supporting the increased resources required for both data-hosting and data-reporting institutions to work toward such improvements when allocating funds for clinical research may further encourage compliance and novel utilization of data.
Supplementary Material
Contributor Information
Yeonju Kim, Neuro-Oncology Branch, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA.
Terri S Armstrong, Neuro-Oncology Branch, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA.
Mark R Gilbert, Neuro-Oncology Branch, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA.
Orieta Celiku, Neuro-Oncology Branch, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA.
Funding
This work was supported by the Intramural Research Program of the National Cancer Institute, National Institutes of Health.
Conflict of interest statement
The authors declare no conflicts of interest.
Authorship statement. Study design, data curation, formal analysis, and manuscript writing YK and OC. Contributions to the study design and manuscript writing MRG and TSA. All authors read and approved the manuscript.
Prior abstract presentations of this study, in part. Kim Y, Armstrong TS, Gilbert MR, & Celiku O, EPID-24. TRANSCRIPT: A cenTRAl NERVOUS SYSTEM CANCER CLINICAL tRIals PostmorTem, Neuro-Oncology, Volume 23, Issue Supplement_6, November 2021, Page vi91, https://doi.org/10.1093/neuonc/noab196.357.
REFERENCES
- 1. Wen PY, Weller M, Lee EQ, et al. Glioblastoma in adults: a society for neuro-oncology (SNO) and european society of neuro-oncology (EANO) consensus review on current management and future directions. Neuro Oncol. 2020;22(8):1073–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lapointe S, Perry A, Butowski NA.. Primary brain tumours in adults. Lancet. 2018;392(10145):432–446. [DOI] [PubMed] [Google Scholar]
- 3. Aldape K, Brindle KM, Chesler L, et al. Challenges to curing primary brain tumours. Nat Rev Clin Oncol. 2019;16(8):509–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Vanderbeek AM, Rahman R, Fell G, et al. The clinical trials landscape for glioblastoma: is it adequate to develop new treatments? Neuro Oncol. 2018;20(8):1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grossman SA, Schreck KC, Ballman K, Alexander B.. Point/counterpoint: randomized versus single-arm phase II clinical trials for patients with newly diagnosed glioblastoma. Neuro Oncol. 2017;19(4):469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lee EQ, Weller M, Sul J, et al. Optimizing eligibility criteria and clinical trial conduct to enhance clinical trial participation for primary brain tumor patients. Neuro Oncol. 2020;22(5):noaa015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee EQ, Chukwueke UN, Hervey-Jumper SL, et al. Barriers to accrual and enrollment in brain tumor trials. Neuro Oncol. 2019;21(9):1100–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Armstrong TS, Gilbert MR.. Clinical trial challenges, design considerations, and outcome measures in rare CNS tumors. Neuro Oncol. 2021;23(23 Suppl 5):S30–S38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ostrom QT, Patil N, Cioffi G, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2013–2017. Neuro Oncol. 2020;22(12 Suppl 2):96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Louis DN, Perry A, Reifenberger G, et al. The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131(6):803–820. [DOI] [PubMed] [Google Scholar]
- 11. Definition of rare cancer. NCI Dictionary of Cancer Terms. 2011. https://www.cancer.gov/publications/dictionaries/cancer-terms/def/rare-cancer. Accessed June 14, 2022.
- 12. Bagley SJ, Kothari S, Rahman R, et al. Glioblastoma clinical trials: current landscape and opportunities for improvement. Clin Cancer Res. 2021;28(4):1078CCR–043221-2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Miksad RA, Abernethy AP.. Harnessing the power of real-world evidence (rwe): a checklist to ensure regulatory-grade data quality. Clin Pharmacol Ther. 2018;103(2):202–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. International Clinical Trials Registry Platform (ICTRP). https://www.who.int/clinical-trials-registry-platform. Accessed June 7, 2022.
- 15. FDAAA 801 and the Final Rule. https://clinicaltrials.gov/ct2/manage-recs/fdaaa. Accessed June 7, 2022.
- 16. ICMJE Clinical Trials Recommendations. https://www.icmje.org/recommendations/browse/publishing-and-editorial-issues/clinical-trial-registration.html. Accessed June 7, 2022.
- 17. Cihoric N, Tsikkinis A, Minniti G, et al. Current status and perspectives of interventional clinical trials for glioblastoma—analysis of clinical trials. gov. Radiat Oncol. 2017;12(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. R Core Team. R A Language and Environment for Statistical Computing. R Foundation for Statistical Computing; 2019. https://www.R-project.org [Google Scholar]
- 19. Conway J, Eddelbuettel D, Nishiyama T, Prayaga SK, Tiffin N.. RPostgreSQL: R Interface to the “PostgreSQL” Database System. https://CRAN.R-project.org/package=RPostgreSQL. Accessed July 28, 2021.
- 20. Clinical Trials Transformation Initiative. Aggregate Analysis of ClinicalTrials.gov (AACT). http://www.ctti-clinicaltrials.org. Accessed July 28, 2021.
- 21. National Library of Medicine. ClinicalTrials.gov. https://clinicaltrials.gov. Accessed July 28, 2021.
- 22. The Drug Repurposing Hub. https://clue.io/repurposing. Accessed March 24, 2020.
- 23. Schoenfeld DA. Sample-size formula for the proportional-hazards regression model. Biometrics. 1983;39(2):499–503. [PubMed] [Google Scholar]
- 24. Borkowf CB, Johnson LL, Albert PS.. Chapter 25—Power and sample size calculations. In: Gallin JI, Ognibene FP, Johnson LL, eds. Principles and practice of clinical research (Fourth Edition). Fourth Edition. Cambridge: Academic Press; 2018:359–372. doi: 10.1016/B978-0-12-849905-4.00025-3 [DOI] [Google Scholar]
- 25. Bugin K, Woodcock J.. Trends in COVID-19 therapeutic clinical trials. Nat Rev Drug Discov. 2021;20(4):d41573–021–00037–3. [DOI] [PubMed] [Google Scholar]
- 26. Viechtbauer W. Conducting meta-analyses in R with the metafor package. J Stat Softw. 2010;36(3):1–48. [Google Scholar]
- 27. Champely S. Pwr: basic functions for power analysis. 2020. https://CRAN.R-project.org/package=pwr. Accessed January 13, 2022. [Google Scholar]
- 28. Abola MV, Prasad V, Jena AB.. Association between treatment toxicity and outcomes in oncology clinical trials. Ann Oncol. 2014;25(11):2284–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Han CY, Fitzgerald C, Lee M, et al. Association between toxic effects and survival in patients with cancer and autoimmune disease treated with checkpoint inhibitor immunotherapy. JAMA Oncol. 2022;8(9):1352–1354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gao Z, Chen Y, Cai X, Xu R.. Predict drug permeability to blood–brain-barrier from clinical phenotypes: drug side effects and drug indications. Bioinformatics. 2016;33(6):btw713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wen PY, Macdonald DR, Reardon DA, et al. Updated response assessment criteria for high-grade gliomas: response assessment in neuro-oncology working group. J Clin Oncol. 2010;28(11):1963–1972. [DOI] [PubMed] [Google Scholar]
- 32. Kim Y, Armstrong TS, Gilbert MR, Celiku O.. Accrual and access to neuro-oncology trials in the United States. Neuro Oncol Adv. 2022;4(1):vdac048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Unger JM, Cook E, Tai E, Bleyer A.. The role of clinical trial participation in cancer research: barriers, evidence, and strategies. Am Soc Clin Oncol Educ Book. 2016;35:185–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rahman R, Ventz S, McDunn J, et al. Leveraging external data in the design and analysis of clinical trials in neuro-oncology. Lancet Oncol. 2021;22(10):e456–e465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alexander BM, Ba S, Berger MS, et al. Adaptive global innovative learning environment for glioblastoma: GBM AGILE. Clin Cancer Res. 2018;24(4):737–743. [DOI] [PubMed] [Google Scholar]
- 36. Galanis E, Wu W, Cloughesy T, et al. Phase 2 trial design in neuro-oncology revisited: a report from the RANO group. Lancet Oncol. 2012;13(5):e196–e204. [DOI] [PubMed] [Google Scholar]
- 37. Kaley T, Touat M, Subbiah V, et al. BRAF Inhibition in BRAFV600 -Mutant Gliomas: Results From the VE-BASKET Study. JCO. 2018;36(35):3477–3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mandel JJ, Yust-Katz S, Patel AJ, et al. Inability of positive phase II clinical trials of investigational treatments to subsequently predict positive phase III clinical trials in glioblastoma. Neuro Oncol. 2018;20(1):113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Seruga B, Templeton AJ, Badillo FEV, et al. Under-reporting of harm in clinical trials. Lancet Oncol. 2016;17(5):e209–e219. [DOI] [PubMed] [Google Scholar]
- 40. Common Terminology Criteria for Adverse Events (CTCAE). https://ctep.cancer.gov/protocoldevelopment/electronic_applications/ctc.htm. Accessed June 7, 2022.
- 41. Zwierzyna M, Davies M, Hingorani AD, Hunter J.. Clinical trial design and dissemination: comprehensive analysis of clinicaltrials.gov and PubMed data since 2005. BMJ. 2018;361(8156):k2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.