

Abstract
Amplification of EGFR occurs frequently in glioblastoma. Canonically, EGFR is an oncogene with a major role in cancer pathogenesis. A new study posits a tumor suppressive role of EGFR in EGFR-amplified glioblastoma, regulated by ligand abundance. Increased EGFR ligand in EGFR-amplified glioblastoma suppresses invasion by upregulation of BIN3, inhibiting activation of Rho GTPases.
The infiltrating gliomas are highly invasive. Outcome in glioblastoma is a function of grade (histology) rather than stage (extent of disease). The 2021 World Health Organization (WHO) classification system grades gliomas based on increasing degrees of dedifferentiation, anaplasia, and aggressiveness, also incorporating molecular features1. Irrespective of grade, gliomas are highly infiltrative. Glioblastoma, the highest grade and most common primary malignant brain tumor, numbers among the most lethal of all cancers, with median survival of only 12–15 months2. Amplification of the epidermal growth factor receptor (EGFR) occurs in 57.4% of primary glioblastoma patients3. The glioblastoma-specific mutation EGFRvIII (deletion of exons 2–7) is often co-amplified with EGFR in glioblastoma. These cooperate in progression4, triggering either ligand-induced or constitutive EGFR signaling, and activating distinct and mutually exclusive downstream pathways5.
The EGFR is one member of a family of four receptor tyrosine kinases (ERBB1-4/HER1-4). Seven ligands bind to and activate the mammalian EGFR: epidermal growth factor (EGF), transforming growth factor alpha (TGFA), heparin-binding EGF-like growth factor (HB-EGF), betacellulin, amphiregulin, and epiregulin6. EGFR ligands are expressed variably in glioblastoma and differ in binding affinity, kinetics, and function6,7. In this issue of Nature Cell Biology, Guo et al. present evidence that the level of EGFR ligands determines whether EGFR acts as an oncogene or tumor suppressor in the context of EGFR-amplified glioblastoma.
Both small molecule kinase inhibitors and antibodies have been developed against EGFR but have shown little efficacy in glioblastoma patients8. There is an urgent need to better understand why these targeted therapies fail in patients. Hypotheses for this lack of efficacy include: 1) EGFR-targeted therapies fail to penetrate the blood-brain barrier (BBB)9; 2) EGFR-targeted therapies do not effectively inhibit EGFR in the brain10; and 3) Ligand-dependent EGFR signaling drives tumor suppression in EGFR-amplified glioblastomas, a new hypothesis that Guo et al. explore.
Guo et al. hypothesize that EGFR directs two distinct and mutually exclusive pathways (Fig. 1). Canonical ligand-induced EGFR signaling activates downstream pathways including the RAS/RAF/MEK/ERK, the PI3K/AKT, JAK/STAT, and PKC pathways. Non-canonical constitutive EGFR signaling activates not only these canonical pathways but also unique effectors including Src family kinases (SFKs), hepatocyte growth factor receptor (MET), and c-Src7, contributing to migration and tumor progression.
Figure 1 –
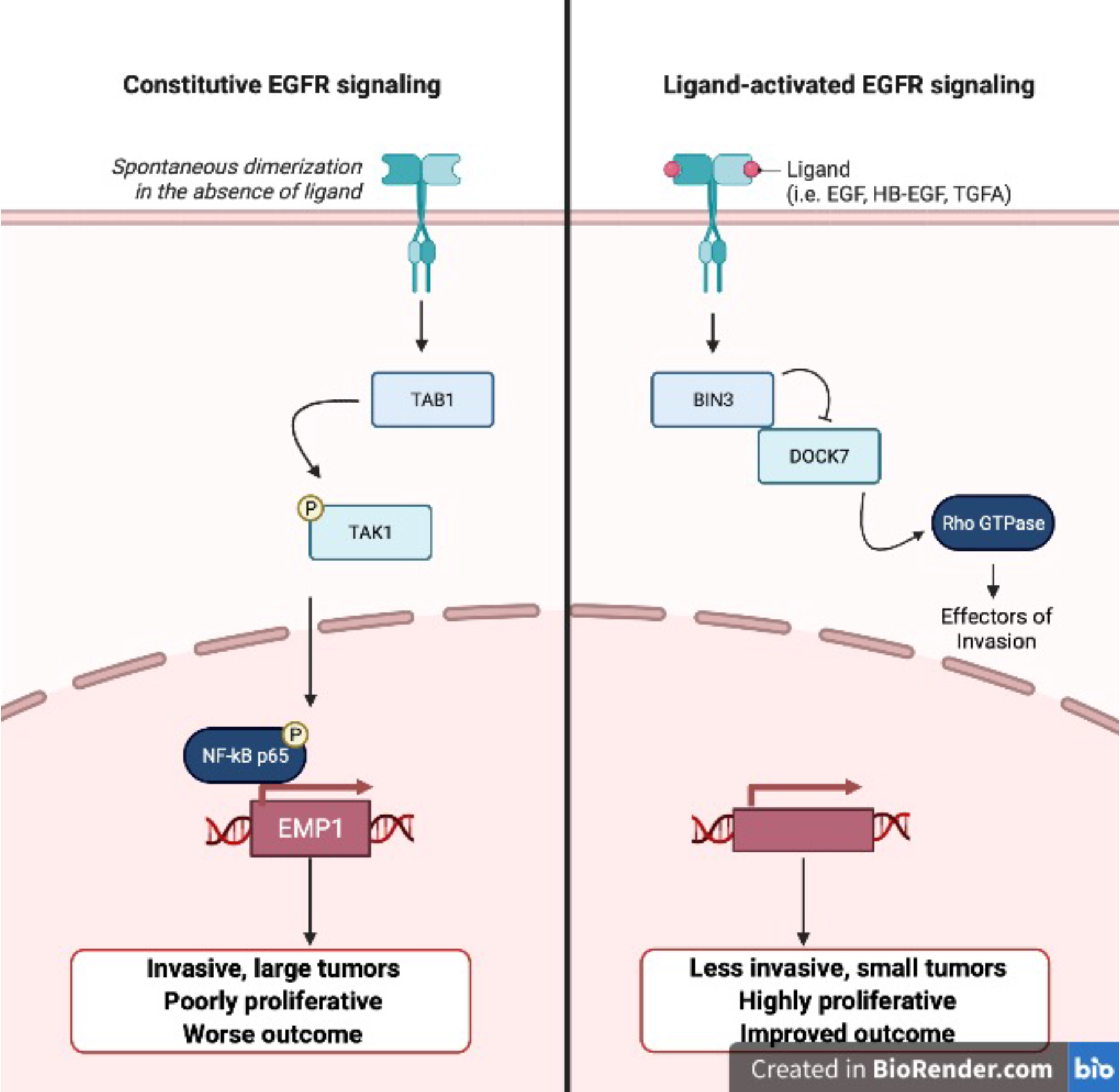
EGFR Signaling Pathways
In a series of RNAi experiments in a panel of PDX lines and primary glioblastoma neurosphere cultures, the authors found that EGFR knockdown without added EGF suppressed invasion. Rescue experiments using a 3’ UTR-targeted siRNA showed that the EGFR knockdown cells became more invasive following overexpression of EGFRWT or EGFRvIII. Cells expressing low levels of EGFR become more invasive when exposed to EGF, but overexpression of EGFR in these lines suppressed invasion in the presence of ligand. Both the abundance of EGFR and of ligand determined whether addition of EGF suppressed or promoted invasion.
Digging into the mechanism, the authors analyzed prior RNA microarray data11 and found that Bridging Integrator 3 (BIN3) was the gene most highly upregulated upon EGF addition. BIN3 functions in membrane and actin dynamics and may suppress cancer by integrating cell polarity signals with regulators of cell cycle arrest and apoptosis12. BIN3 knockdown decreased invasiveness in glioblastoma cells upon exposure to EGF. BIN3 was not upregulated in cells expressing low levels of EGFR, in which EGF induced invasion. Overexpression of BIN3 suppressed invasion, and was not further suppressed by EGF, suggesting BIN3 as a key negative regulator of invasion.
How does BIN3 block invasion in the setting of ligand activated and amplified EGFR? Through a series of siRNA and mass spectrometry experiments for proteins that associate with BIN3, the authors identified dedicator of cytokinesis 7 (DOCK7), a Rac/Cdc42 guanine nucleotide exchange factor, in part due to its role in HGF-induced invasion of glioblastoma cells13. They proposed that BIN3 binding inhibits DOCK7-mediated activation of Rho GTPases, decreasing invasiveness.
In the absence of EGFR amplification, EGFR signaling did not affect levels of BIN3 and instead drove invasion by upregulating epithelial membrane protein 1 (EMP1). The authors had previously validated that EMP1 upregulation was associated with constitutive EGFR signaling, but not ligand-induced EGFR signaling. The authors defined mechanism through a second series of siRNA and mass spectrometry experiments for proteins that bind to EGFR in the absence of EGF. They showed binding of EGFR bound to TGF-beta-activated-kinase 1 (TAB1). TAB1 in turn activated TAK1 and phosphorylated the NF-κB subunit p65, implicating the EGFR-TAB1-TAK1-NF-κB-EMP1 pathway in constitutive EGFR signaling-mediated invasion.
Armed with this mechanistic information, the authors performed a drug screen for compounds that decreased invasiveness in a panel of glioblastoma cell lines. They identified tofacitinib, a brain penetrant Jak1/Jak3 inhibitor. Tofacitinib upregulated BIN3 levels and inhibited invasion via robust activation of EGFR and upregulation of the EGFR ligand HB-EGF. In cells treated with saturating levels of EGF, tofacitinib did not further increase BIN3 levels or inhibit invasion.
The authors confirmed the effect of tofacitinib in vivo in multiple orthotopic PDX models, driving ligand expression (or not) by engineering autocrine secretion of TGFA in glioblastoma xenografts. They observed a modest improvement in survival and decreased invasiveness in a model with low levels of EGFR ligand. However, in a model with high levels of EGFR ligand, tofacitinib did not improve survival. Despite the modest survival benefit, confirming the effect of tofacitinib in vivo may offer hope to patients with glioblastoma, a disease with dismal prognosis.
In summary, Guo et al. assert that levels of both EGFR and its ligands determine whether engagement of ligands resulted in a proliferative or invasive phenotype (Fig. 1). Correlative analyses of TCGA data using a combined ligand score for all seven EGFR ligands demonstrated that patients with EGFR-amplified tumors and low expression of EGFR ligands had poorer survival compared to patients with EGFR-amplified tumors and high expression of EGFR ligands. Consistent with this analysis, high levels of EGFR with low levels of ligand promoted larger, more invasive tumors in mouse models. Whereas high levels of both EGFR and ligand resulted in smaller less invasive tumors and improved survival. The authors suggest a model of patient stratification that incorporates levels of EGFR ligands as well as EGFR amplification status to determine the best course of treatment. In patients with EGFR-amplified glioblastoma and high levels of ligands, EGFR-targeted therapy may be counterproductive because EGFR acts as a tumor suppressor in this context. However, in EGFR-amplified GBM with low levels of ligands, tofacitinib may be an efficacious choice.
So where do EGFR inhibitors fit into the therapeutic picture? The authors leave this question largely unresolved, though adaptive responses in vitro led to EGFR inhibitors showing only transient effects on invasion.
In theory, iterating on the current precision-medicine workflow by further stratifying patients represents an opportunity to improve outcomes. However, incorporating levels of EGFR ligands into the current clinical workflow will be challenging. Firstly, how and when will levels of EGFR ligands be assessed during a patient work-up? Secondly, what clinical benefit, if any, would patients derive from tofacitinib treatment if their cancer has already invaded extensively? The authors proposed using tofacitinib prior to surgical resection to shrink the tumor mass. However, given that tofacitinib induced proliferation while suppressing invasion in their studies, this may be a risky approach. Tofacitinib could be utilized later in the disease course to prevent or treat recurrence. However, EGFR amplification status and levels of ligands will likely need to be reassessed at this later stage as tumors could lose EGFR expression or downregulate ligand levels in response to prior treatment.
Guo et al. highlight the complexity of EGFR signaling pathways and identify a potential mechanism underlying the failure of EGFR-targeted therapy in glioblastoma. Whereas a clinical paradigm that incorporates EGFR amplification status as well as levels of EGFR ligands may benefit some patients, curative treatment regimens will likely continue to rely on combination therapy. Each new drug in a therapeutic regimen adds new selective pressure to the system, and glioblastoma has historically proven recalcitrant to treatment, likely due to our incomplete understanding of the complex signaling networks it utilizes to proliferate, invade, and develop resistance to treatment. Importantly, studies here identified and explored a novel tumor suppressive role for EGFR signaling in glioblastoma. Future work, both in the clinical and basic science realms, will be needed to fully understand whether outcomes in patients with EGFR-amplified glioblastoma can be improved by targeting oncogenic functions of EGFR, while sparing potential tumor suppressive roles for EGFR described here.
Acknowledgments
Research in the Weiss lab supported by R01CA221969, U01CA217864, P50CA097257, Cancer Research UK A28592, Evelyn and Mattie Anderson Chair, Panattoni Family, UCSF Jobie Project, Samuel Waxman Cancer Research Foundation.
References
- 1.Louis DN et al. Neuro-Onc (2021) 23(8), 1231–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dirks PB et al. Clin Cancer Res (2020) 26(11), 2457–2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brennan CW et al. Cell (2013) 155, 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fan QW et al. Cancer Cell (2013) 24, 438–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chakraborty et al. Nat Commun (2014) 5, 5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singh B et al. F1000Res (2016) 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hu C et al. Nature (2022) 602, 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.An Z et al. Oncogene (2018) 37(12), 1561–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Vries NA et al. Invest New Drugs (2012); 30(2), 443–449. [DOI] [PubMed] [Google Scholar]
- 10.Barkovich KJ et al. Cancer Discov (2012) 2(5), 450–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramnarain DB et al. Cancr Res (2006) 66, 867–874. [DOI] [PubMed] [Google Scholar]
- 12.Prendergast GC et al. Biochim Biophys Acta (2009) 1796(1), 25–36. [Google Scholar]
- 13.Murray DW et al. Br J Cancer (2014) 110, 1307–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]