

Abstract
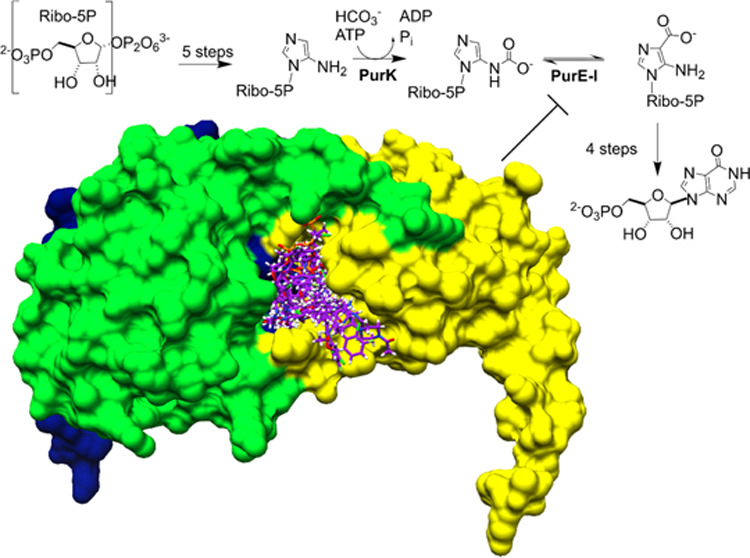
Because purine nucleotides are essential for all life, differences between how microbes and humans metabolize purines can be exploited for the development of antimicrobial therapies. While humans biosynthesize purine nucleotides in a 10-step pathway, most microbes utilize an additional 11th enzymatic activity. The human enzyme, aminoimidazole ribonucleotide (AIR) carboxylase generates the product 4-carboxy-5-aminoimidazole ribonucleotide (CAIR) directly. Most microbes, however, require two separate enzymes, a synthetase (PurK) and a mutase (PurE), and proceed through the intermediate, N5-CAIR. Toward the development of therapeutics that target these differences, we have solved crystal structures of the N5-CAIR mutase of the human pathogens Legionella pneumophila (LpPurE) and Burkholderia cenocepacia (BcPurE) and used a structure-guided approach to identify inhibitors. Analysis of the structures reveals a highly conserved fold and active site architecture. Using this data, and three additional structures of PurE enzymes, we screened a library of FDA-approved compounds in silico and identified a set of 25 candidates for further analysis. Among these, we identified several new PurE inhibitors with micromolar IC50 values. Several of these compounds, including the α1-blocker Alfuzosin, inhibit the microbial PurE enzymes much more effectively than the human homologue. These structures and the newly described PurE inhibitors are valuable tools to aid in further studies of this enzyme and provide a foundation for the development of compounds that target differences between human and microbial purine metabolism.
Introduction
Purine nucleotides are essential for all life, performing vital roles in the transfer of genetic information, energy storage, cell signaling, and as co-enzymes for many types of reactions. In most organisms, nucleotide pools are maintained through the coordinated action of both a de novo biosynthesis and a salvage, or recycling, pathway.1−4 In most higher eukaryotes, including humans, the biosynthesis of inosine monophosphate (IMP) from phosphoribosyl pyrophosphate (PRPP) involves 10 enzymatic steps.1,5 Microbes, however, utilize an 11th enzymatic step to carry out the same overall chemistry.6−8 Unlike aminoimidazole ribonucleotide (AIR) carboxylase, which converts AIR to 4-carboxy-5-aminoimidazole ribonucleotide (CAIR) in humans, most microorganisms, yeast, and plants generate CAIR sequentially by the action of two enzymes, an N5-CAIR synthetase (PurK) and an N5-CAIR mutase (PurE) (Figure 1).
Figure 1.

Role of PurE in purine biosynthesis. In humans, the biosynthesis of IMP is carried out in 10 enzymatic steps, including the conversion of AIR to CAIR by AIR carboxylase (class II PurE). In most microbes, however, AIR is first converted to N5-CAIR by N5-CAIR synthetase, before CAIR is produced by the action of a second enzyme, N5-CAIR mutase (class I PurE).
Because of the central role that purines play in cell growth and viability, the de novo biosynthesis of purine nucleotides has long been targeted by anticancer and antiviral therapies.2,9−13 The distinct chemistry shown in Figure 1 illustrates a potential strategy for selectively disrupting purine metabolism in human pathogens that make use of this alternate biosynthetic route. Support for this approach is provided in studies that demonstrate that genetic inactivation of PurE leads to growth defects and a substantial attenuation of virulence in a number of pathogenic bacteria.14−18 To exploit this potential vulnerability, we set out to elucidate the molecular features of PurE from two pathogenic organisms and use this data as a basis to identify novel PurE inhibitors. Herein, we report the X-ray crystal structures of PurE from B. cenocepacia (BcPurE) and Legionella pneumophila (LpPurE). The latter causes a serious type of pneumonia, called Legionnaires’ disease, while the former is an opportunistic human pathogen that causes lethal infections in immunocompromised patients. Using these crystal structures, we conducted docking studies to screen for potential PurE inhibitors. After testing a subset of these, we identified several with modest activity, from LpPurE and BcPurE enzymes, respectively, that was a partial inhibitor of these enzymes. These data provide a structural framework for further study and describe several new PurE inhibitors that could find use as functional probes or as a basis for drug development.
Results and Discussion
Protein Expression, Data Collection, and Structure Solution
Both BcPurE and LpPurE expressed well and were purified to homogeneity using established methods.19−21 After sparse matrix screening, diffraction-quality crystals of BcPurE were obtained in 0.1 M Bicine, pH 9, and 10% MPD using sitting-drop vapor diffusion, while crystals of LpPurE were obtained in 0.1 M sodium phosphate/potassium phosphate, pH 6.2, 0.2 M sodium chloride, and 10% PEG 8000, also using sitting-drop vapor diffusion. The crystals diffracted well, beyond 2 Å for both, and were indexed in the C2 and I222 space groups for LpPurE and BcPurE, respectively. The BcPurE crystals had a solvent content of 57% with a Matthews coefficient of 2.88 Å3/Da while the LpPurE crystals had 38% solvent and a Matthews coefficient of 1.99 Å3/Da. The structures were solved using molecular replacement and refined to final R/Rfree values of 14.7/17.6 and 14.8/18.2% for BcPurE and LpPurE, respectively. Four chains were present in the asymmetric unit for both BcPurE and LpPurE. The additional four chains, in both cases, are generated by rotation around a twofold axis. A summary of the data collection and refinement statistics is provided in Table 1.
Table 1. Data Collection and Refinement Statistics.
| BcPurE | LpPurE | |
|---|---|---|
| Data Collection | ||
| PDBID | 4GRD | 6O55 |
| beamline | ALS 5.0.1 | APS 21-ID-F |
| detector | ADSC Quantum 315 | Rayonix MX-300 CCD |
| wavelength (Å) | 0.9774 | 0.9787 |
| resolution range (Å) | 48.12–1.85 | 48.34–1.70 |
| space group | I222 | C2 |
| unit cell dimensions | ||
| a,b,c (Å) | 116.51, 117.18, 122.31 | 137.28, 88.56, 52.51 |
| β (°) | 90 | 111.77 |
| no. of unique reflections | 63,895 | 71,473 |
| mean I/sigma(I)aa | 16.28 (3.70) | 16.68 (2.60) |
| completeness (%) | 98.0 (98.3) | 99.5 (99.8) |
| redundancy | 4.22 (3.10) | 3.69 (3.61) |
| Rmerge (%) | 8.9 (51.0) | 5.1 (60.3) |
| CC1/2 | 0.99 (0.80) | 0.99 (0.86) |
| Data Refinement | ||
| total no. of reflections | ||
| test set | 3618 | 2113 |
| Rwork/Rfree (%) | 14.7/17.6 | 14.8/18.2 |
| total no. of atoms | 5696 | 5519 |
| R.M.S.D. | ||
| bonds (Å) | 0.011 | 0.010 |
| angles (°) | 1.351 | 1.250 |
| mean B factor (Å2) | 16.1 | 25.0 |
| Ramachandran plot (%) | ||
| favored | 99.0 | 98.9 |
| allowed | 1.0 | 1.1 |
| outliers | 0 | 0 |
| MolProbity clashscore (%) | 1.0 | 3.0 |
Values in parentheses represent the highest resolution shell.
Structure of Bc and LpPurE Protomer
As observed in the previous solved structures of class I PurE enzymes, the PurE protomer from both Bc and LpPurE is composed of a central domain and a C-terminal α-helix that extends away from the central domain (Figure 2). The central domain is an αβα-sandwich with a 5-stranded parallel β-sheet at the core. A 3D-structure search using the DALI server22 revealed that, in addition to homologous PurE enzymes, this topology is also shared with the human phosphoribosylaminoimidazole carboxylase (PAICS) enzyme (23% sequence identity), and the LarB carboxylase/hydrolase (22% sequence identity) (Table. S1). Superposition of BcPurE and LcPurE illustrates the high degree of structural conservation (RMSD 0.860 Å for protomer), with the only observable differences being small variations in flexible loop regions (Figure 2B).
Figure 2.
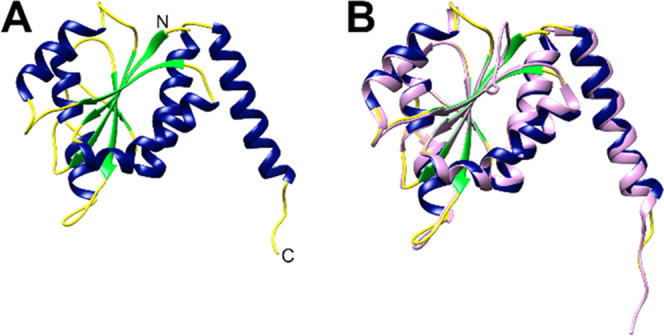
Structure of PurE protomer. The protomers of PurE show the conserved αβα sandwich that is characteristic of class I PurE enzymes. Shown are the protomer from LpPurE (A) with α-helices colored blue, β-strands colored green, and loops colored yellow, and a superposition (B) of the BcPurE protomer (colored in pink) and LpPurE protomer. These two similar PurE proteins (58% sequence identity) show a high level of structural conservation (RMSD = 0.860 Å for protomer) with only subtle differences in some loop regions. The N- and C-termini are labeled with N and C, respectively.
Overall Structure of PurE
The class I PurE enzymes reported to date all have a conserved octameric structure with 422 symmetry.5,23,24 An initial analysis of putative assemblies using PDBePISA25 indicated that both the BcPurE and LpPurE are likely to form octamers. While the size exclusion chromatography data was not conclusive (Figure S1), analysis of multiangle light scattering data (Figure S2) was consistent with an octamer for both proteins (predicted MW of BcPurE = 168 kDa, protomer MW = 19 kDa; predicted MW of LpPurE = 143 kDa; protomer MW = 19 kDa). As expected, the overall structures of BcPurE and LpPurE are octamers of 8 identical protomers (Figure 3). Both LpPureE (Figure 3A) and BcPurE (Figure 3B) have a similar structure and superimpose well with the known structure of the Escherichia coli class I PurE (Figure 3C). While the human PurE homologue, a class II purE, is expressed as a gene fusion with PurC (called PAICS, phosphoribosylaminoimidazole carboxylase-phosphoribosylaminoimidazole succinocarboxamide synthetase), the central PurE octamer is conserved.23 Superposition of Bc and LpPurE with human PurE illustrates that, despite the differences in the chemistry, the overall fold and octameric organization is conserved from bacteria to humans (Figure S3).
Figure 3.

Overall structure of PurE. Both LpPurE (A) and BcPurE (B) are octamers of 8 identical protomers (shown in different colors in (A)). The octameric structure is highly conserved among class I PurE enzymes, as illustrated by the superposition (C) of LpPurE (blue) and BcPurE (pink) with the E. coli PurE (orange, 1D7A). The RMSD for the superposition of LpPurE on E. coli PurE is 0.656 Å, while that for BcPurE on E. coli PurE is 1.160 Å.
Active Site of Bc and LpPurE
Class I PurE enzymes catalyze the reversible conversion of N5-CAIR to CAIR, while Class II PurE enzymes generate CAIR directly from AIR (Figure 1). Because the N5-CAIR intermediate is known to be unstable,26,27 a direct transfer of the intermediate between PurK and PurE has been proposed.28−31 No evidence has yet been found, however, for such a PurK–PurE complex. Mechanistic studies of PurE have identified several residues important for catalysis, including an active site histidine residue (H41 in LpPurE, H50 in BcPurE) and a number of conserved residues that help to position the substrate.26 These residues are conserved in both Lp and BcPurE (Figure S4) and have a similar organization (Figure 4). It is important to note that the active site of PurE resides near an interface of three protomers and, as such, the overall structure of the active site is composed of elements from three distinct PurE chains (Figure 4A). The interactions with the substrate, however, are predominantly with a single chain of PurE (Figure 4B). A superposition of LpPurE and BcPurE (Figure 4C) shows a high degree of structural conservation. The only visible difference is the position of the arginine side chain, which presumably would become more ordered upon binding of the substrate. The active site of the homologous region of the Human class II PurE also shares a high degree of similarity with Lp and BcPurE, with only subtle differences observed in the positions of the active site residues (Figure 5). As observed with the E. coli and other bacterial PurE enzymes, most of the interactions between the substrate (CAIR) and the enzyme are mediated by the protein backbone.24,26 An active site serine hydroxyl (S14 in LpPurE, S23 in BcPurE), and guanidine group from arginine putatively make additional hydrogen bonds with the phosphate of CAIR.
Figure 4.

Active site of PurE. The active site of PurE enzymes is at the interface of three protomers, as illustrated by the structure of LpPurE with AIR shown in the active site ((A), different protomers shown in cartoon representation and colored in green, yellow, and dark blue; in this figure, the AIR ligand was positioned by superimposing the AIR-bound structure of E. coli PurE, 1D7A, to the structure of LpPurE). Conserved residues (see Figure S4) from two different chains position the ligand and are responsible for the chemisty (B). Other than a flexible arginine side chain (R51), the active site residues of LpPurE and BcPurE (C) superimpose well (BcPurE shown in pink).
Figure 5.

Active site comparison of BcPurE and human PurE. Superposition of BcPurE (yellow) active site residues and human PurE (gray) reveal subtle differences in spatial orientation of some residues (RMSD of 1.581). In this representation, nitrogen atoms are colored blue and oxygen atoms are colored red. There is one distinct difference depicted in the figure where an arginine (R51) in BcPurE is replaced with a lysine(K304) in human PurE.
Steady-State Enzyme Kinetics of LpPurE and BcPurE for CAIR
Due to the instability of the substrate N5-CAIR, it can be difficult to assay Class I PurE enzymes. To determine the kinetics of LpPurE and BcPurE, the activity was assayed in the reverse biosynthetic direction using an established method.7,32 Lp and BcPurE display similar kinetics for the decarboxylation of CAIR, with the L. pneumophila enzyme turning over slightly faster (Figure 6). The kcat/KM values are also similar, with a value of 0.55 s–1 μM–1 for LpPurE, and 0.12 s–1μM–1 for BcPurE. These values are similar to what is observed for E. coli (KM of 23 μM and kcat of 16 s–1).26
Figure 6.

Enzyme kinetics of Bc and LpPurE. Using the decarboxylation of the product, CAIR, the steady-state enzyme kinetics of LpPurE (A) and BcPurE (B) were determined. Both exhibit saturable enzyme activities with similar kinetics. The data are given as the mean ± standard deviation of triplicate measurements.
In Silico Screen to Identify Potential PurE Inhibitors
Because of the differences in how microbes and humans synthesize CAIR, microbial CAIR enzymes are a potential target for the development of novel antibiotics.14−18,33,34 To identify inhibitors of Bc and LpPurE, as a basis for further drug discovery, we first conducted an in silico screen of 1,999 FDA-approved compounds from the ZINC15 database.35 After preparation of the protein structures and library, as described in the Materials and Methods section, we used DOCK6 to dock the compounds and ranked these based on the results. As an internal positive control, we re-docked AIR in the E. coli protein (for which a liganded structure is known—1D7A) to determine if we could recapitulate the crystallography results, and docked this compound to both Lp and BcPurE. The docked ligand superimposed well with that observed in the crystal structure (Figure S6A). Note that, because of the nature of the PurE active site, the protein model used for docking included residues from three chains of the protein. In addition to BcPurE and LpPurE, we repeated the screen using the Bacillus anthracis PurE (1XMP), Francisella tularensis PurE (3OPQ), E. coli PurE (1D7A), and the human PurE (2H31). Overall, the compounds bind broadly around the active site and there do not appear to be any binding hotspots that drive interactions (Figure S6B,F). The resulting grid scores, which are used as a measure of how well compounds bind to the protein, show a good distribution with a mean of −44.87 (Figure S7). We further filtered the results based on molecular weight, cLogP values, and number of rotatable bonds (see the Materials and Methods section). From these data, we selected 25 compounds to further analyze biochemically (Figures 7 and S8, Table 2).
Figure 7.

Some of the structures of the top compounds from screening selected for further analysis. Additional compound structures are provided in Figure S8.
Table 2. DOCK6 Grid Scores and Ranks for Compounds Evaluated in This Study (Figure 6).
| DOCK
grid scorea (rank)b |
||||||
|---|---|---|---|---|---|---|
| compound name | 6O55 | 4GRD | 1XMP | 3OPQ | 2H31 | 1D7A |
| Lapatinib | –71.28 (5) | –75.16 (5) | –66.00 (12) | –68.59 (178) | –55.44 (37) | –70.13 (6) |
| Crizotinib | –69.44 (8) | –70.32 (7) | –49.82 (57) | –48.29 (364) | –47.94 (194) | –69.28 (8) |
| Famotidine | –69.36 (9) | –61.77 (80) | –51.59 (85) | –48.18 (367) | –43.46 (374) | –68.1 (11) |
| Valacyclovir | –67.99 (12) | –63.20 (57) | –52.56 (165) | –55.06 (107) | –46.74 (239) | –68.71 (9) |
| Nefazodone | –68.44 (10) | –70.75 (6) | –57.72 (42) | –59.58 (51) | –49.31 (152) | –64.82 (23) |
| Carvedilol | –68.10 (11) | –62.56 (67) | –56.24 (64) | –61.72 (32) | –51.59 (90) | –66.12 (14) |
| Hydroxychloroquine | –67.42 (14) | –63.76 (42) | –52.66 (161) | –51.23 (224) | –45.04 (306) | –68.31 (10) |
| Avanafil | –66.98 (15) | –63.51 (48) | –61.12 (18) | –62.48 (26) | –51.20 (100) | –65.19 (20) |
| Cangrelorc | –84.17 | –79.05 | –101.34 | –86.60 | –94.07 | –82.75 |
| Ketoconazole | –65.56 (13) | –68.16 (10) | –58.36 (36) | –54.22 (126) | –52.50 (68) | –63.55 (31) |
| Nintedanib | –66.79 (16) | –63.57 (45) | –57.89 (41) | –62.13 (29) | –49.95 (133) | –59.3 (94) |
| Aclidinium | –66.49 (18) | –68.34 (9) | –53.88 (121) | –57.96 (61) | –50.45 (117) | –65.28 (19) |
| Formoterol | –65.50 (22) | –67.42 (15) | –55.52 (76) | –57.09 (797) | –51.64 (89) | –64.7 (24) |
| Nizatidine | –65.29 (23) | –52.74 (343) | –46.83 (397) | –50.35 (266) | –44.81 (318) | –62.8 (33) |
| Valbenazine | –64.27 (29) | –64.07 (39) | –49.85 (256) | –45.65 (705) | –43.06 (395) | –63.95 (27) |
| Afatinib | –64.21 (30) | –66.28 (24) | –55.84 (70) | –49.88 (334) | –47.87 (199) | –60.16 (80) |
| Valganciclovir | –70.32 (6) | –67.87 (11) | –56.29 (63) | –57.40 (72) | –45.26 (292) | –72.56 (5) |
| Sertaconazole | –62.85 (44) | –61.45 (88) | –51.42 (190) | –54.91 (110) | –44.30 (233) | –61.74 (48) |
| Vardenafil | –62.65 (48) | –63.42 (54) | –55.55 (75) | –56.42 (85) | –56.24 (32) | –61.4 (55) |
| Indacaterol | –61.97 (53) | –67.83 (12) | –53.40 (132) | –58.39 (57) | –48.83 (164) | –64.94 (22) |
| Sildenafil | –61.71 (56) | –63.18 (58) | –53.88 (122) | –57.29 (77) | –54.28 (50) | –61.22 (58) |
| Isavuconazonium sulfatec | –82.28 | –92.24 | –72.58 | –64.02 | –63.43 | –78.88 |
| Alfuzosin | –61.24 (60) | –65.50 (51) | –53.14 (149) | –55.36 (98) | –45.03 (307) | –61.32 (56) |
| Bosutinib | –62.25 (52) | –64.71 (54) | –53.20 (147) | 53.92 (132) | –53.09 (60) | –61.63 (50) |
| Olodaterol | –63.18 (39) | –63.06 (91) | –51.54 (186) | –54.45 (117) | –49.59 (142) | –62.30 (41) |
Dock Grid energy score—a summation of the electrostatic and van der Waals interactions in kcal/mol.
Rank on list after filtering chemical descriptors (see the Materials and Methodssection for additional details).
Compounds on the list before filtering that had a relatively high grid score when docked against 4GRD and 6O55 but did not fit the filtering criteria.
Analysis of Putative PurE Inhibitors
Using the decarboxylation assay described above, we initially tested the selected compounds at two concentrations (10 and 100 μM) to identify those that reduced activity. In this initial screen, five of the compounds, including Avanafil, Ketoconazole, Afatinib, Indacaterol, and Alfuzosin (Figure 8), showed some degree of inhibition of PurE activity. Further analysis showed that most of these compounds were modest inhibitors of both Bc and LpPurE, with IC50 values ranging from 47 to 282 μM (Figures 9, S9, and S10). In particular, Afatinib, Alfuzosin, and Ketoconazole inhibit LpPurE with IC50 values of 49, 69, and 82 μM, respectively, while only Alfuzosin showed a reasonable inhibition of BcPurE, with an IC50 value of 47 μM. An HPLC-based assay was used as an orthogonal confirmation of the observed inhibition of PurE (Figure S11). Considering how similar in structure the active sites are, the difference in potency for Afatinib and Ketoconazole for LpPurE compared to BcPurE suggests that these inhibitors do not act as competitive inhibitors. Additional studies are needed to fully explore these potential mechanisms of inhibition. These five compounds were also tested using the human protein. While Avanafil, Ketoconazole, and Alfuzosin show little or no inhibition, Afatinib and Indacaterol do inhibit human PAICS (Figure 10). A comparison of the docking results for these compounds suggests that protein-ligand binding is driven primarily by Van der Waals and weak electrostatic interactions, with few hydrogen bonds, similar to what is observed for substrate binding (Figures 11, S12, and S13). In addition, distinct putative binding modes are observed for the inhibitors docked to the human PurE compared to the bacterial protein (Figure S14). While few commonalities exist among the putative interactions observed for these compounds, the two most potent inhibitors, Alfusozin and Afatinib, have terminal furan groups that project into the active sites of both Bc and LpPurE (Figures S12 and S13). Interestingly, the screening library contained two isomers (tautomers of the quinoline ring) of indacaterol. Further analysis of the results showed that the tautomer that ranked lower in the docking study had a similar binding orientation for both LpPurE and BcPurE (Figure S15). In both cases, the quinoline ring sits deep in the active site and a hydrogen bond is made between a conserved aspartate residue of the protein and a hydroxyl group on the indacaterol backbone. Note that, for our docking studies, we limited the search to a sphere of 10 Å radius around the known substrate binding site. As such, we cannot rule out the possibility that some of these inhibitors bind to PurE at locations outside of the active site.
Figure 8.

Twenty-five compounds were chosen, based on the docking study, to test as potential PurE inhibitors. Five of these compounds, including Avanafil, Ketoconazole, Afatinib, Indacaterol, and Alfuzosin, inhibited BcPurE at one or both of the concentrations tested. The data shows the mean ± standard deviation for triplicate experiments.
Figure 9.

Dose–response curves for Afatinib and Alfuzosin. Using the CAIR decarboxylation assay, dose–response curves were determined for the inhibition of LpPurE by Afatinib (A) and Alfuzosin (B). Additional curves for LpPurE and BcPurE are shown in Figures S9 and S10. The hill slope is given, denoted by h.
Figure 10.

Inhibition of human PAICS by top PurE inhibitors. The effect of the putative PurE inhibitors on human PAICS decarboxylation activity was determined at two concentrations, 10 and 100 μM against BcPurE. While Afatinib and Indacaterol inhibit the enzyme to some degree, Avanafil, Ketoconazole, and Alfuzosin showed little to no inhibition. The data shows the mean ± standard deviation for triplicate experiments.
Figure 11.

Putative binding interactions of inhibitors with BcPurE. The position and orientation of the lowest-energy pose of inhibitors Alvanafil (A), Ketoconazole (B), and Indacaterol (C) from the docking results are shown. While these compounds clearly bind at or near the active site, no obvious hydrogen-bonding interactions are observed. The docked structures of Alfuzosin and Afatinib with BcPurE and the top 5 compounds bound to LpPurE are shown in Figures S10 and S11, respectively.
The two new bacterial PurE structures described herein help expand our understanding of the structure and function of this important purine metabolic protein. These data also enabled the structure-guided discovery of several new PurE inhibitors from existing FDA-approved drugs. While more detailed kinetic studies are needed to better understand the mechanism of action and to explore structure–activity relationships, this work describes new tools and insights that will aid in the further development of antibiotics targeting class I PurE enzymes.
Materials and Methods
Protein Expression and Purification
Cloning, expression, and purification followed standard protocols as previously described.19−21 The full-length gene for N5-carboxyaminoimidazole ribonucleotide mutase from Burkholderia cenocepacia J2315 (BcPurE; Uniprot B4EA21) encoding amino acids 1–173 was PCR-amplified from gDNA using the primers shown in Table S2. The gene was cloned into the ligation-independent cloning (LIC) vector pBG1861, encoding a noncleavable hexahistidine tag (MAHHHHHH-ORF).20,36 Chemically competent E. coli BL21(DE3)R3 Rosetta cells were transformed with the plasmid DNA. The transformed cells were expression tested, and 2 L of culture was grown using auto-induction media37 in a LEX Bioreactor (Epiphyte Three, Inc.) as previously described.21 The expression clone BuceA.01377.a.B1.GE34990 is available at https://www.ssgcid.org/available-materials/expression-clones/. The full-length gene for phosphoribosylaminoimidazole carboxylase, catalytic subunit PurE, from L. pneumophila Philadelphia 1 (LpPurE; Uniprot Q5ZYZ3) encoding amino acids 1–166 was PCR-amplified from gDNA using the primers shown in Table S3. The gene was cloned into the ligation-independent cloning (LIC) vector pBG1861, encoding a noncleavable hexahistidine tag (MAHHHHHH-ORF).20,36 Chemically competent E. coli BL21(DE3)R3 Rosetta cells were transformed with the plasmid DNA. The transformed cells were expression tested, and 2 L of culture was grown using auto-induction media37 in a LEX Bioreactor (Epiphyte Three Inc.) as previously described.21 The expression clone LepnA.01377.a.B1.GE41630 is available at https://www.ssgcid.org/available-materials/expression-clones/.
His-BcPurE and His-LcPurE were purified in a two-step protocol consisting of an immobilized metal (Ni2+) affinity chromatography (IMAC) step and size exclusion chromatography (SEC). All chromatography runs were performed on an ÄKTApurifier 10 (GE Healthcare) using automated IMAC and SEC programs.19 Thawed bacterial pellets (∼25 g) were lysed by sonication in 200 mL of buffer containing 25 mM HEPES pH 7.0, 500 mM NaCl, 5% glycerol, 0.5% CHAPS, 30 mM imidazole, 10 mM MgCl2, 1 mM TCEP, 250 ug/mL AEBSF, and 0.025% sodium azide. After sonication, the crude lysate was clarified with 20 mL (25 units/μL) of benzonase and incubated while mixing at room temperature for 45 min. The lysate was clarified by centrifugation at 10,000 rev min–1 for 1 h using a Sorvall centrifuge (Thermo Scientific). The clarified supernatant was then passed over a Ni-NTA His-Trap FF 5 mL column (GE Healthcare) which was pre-equilibrated with loading buffer composed of 25 mM HEPES pH 7.0, 500 mM NaCl, 5% glycerol, 30 mM imidazole, 1 mM TCEP, and 0.025% sodium azide. The column was washed with 20 column volumes (CV) of loading buffer and was eluted with loading buffer plus 250 mM imidazole in a linear gradient over 7 CV. Peak fractions were pooled and concentrated to 5 mL. An SEC column (Superdex 75, GE) was equilibrated with a running buffer composed of 20 mM HEPES, pH 7.0, 300 mM NaCl, 5% glycerol, and 1 mM TCEP. The peak fractions were collected and analyzed using SDS page for the protein of interest. For BcPurE, the protein eluted as a single large peak in the molecular mass range of 73 kDa. The peak fraction was pooled and concentrated to 62.8 mg/mL peak using an Amicon purification system (Millipore). Aliquots of 110 μL were flash-frozen in liquid nitrogen and stored at −80 °C until used. For LpPurE, the protein eluted as a single large peak in the molecular mass range ∼85 kDa. The peak fractions were pooled and concentrated to 28.5 mg/mL using an Amicon purification system (Millipore). Aliquots of 110 μL were flash-frozen in liquid nitrogen and stored at −80 °C until use for crystallization.
Size Exclusion Chromatography–Multiangle Light Scattering (SEC-MALS)
To confirm sizing information, SEC was conducted for both BcPurE and LpPurE using a Zenix SEC-300 column (Sepax) running on an Agilent 1200-series HPLC. Both proteins eluted as a single, large peak with predicted MWs of 88 and 121 kDa for LpPurE and BcPurE, respectively (Figure S1). MALS analysis was conducted using a miniDAWN TREOS light scattering instrument at 660 nm equipped with an Optilab T-REX RI detector (Figure S2) in line with the HPLC.
Crystallization, Data Collection, and Structure Solution
Purified His-BcPurE and His-LpPurE were screened for crystallization in 96-well plates against JBScreen JCSG++ HTS (Jena Bioscience) and MCSG1 (Molecular Dimensions) crystal screens. Equal volumes of protein solution (0.4 μL) and precipitant solution were set up at 290 K against reservoir (80 μL) in sitting-drop vapor diffusion format. For His-BcPurE, crystals were obtained in 0.1 M Bicine, pH 9.0, and 10% (V/V) MPD. For His-LpPurE, crystals were obtained in 0.2 M sodium chloride, 0.1 M sodium phosphate/potassium phosphate, pH 6.2, and 10% (W/V) PEG 8000. The crystals were flash-cooled by harvesting them and plunging them directly into liquid nitrogen. For His-BcPurE, crystals were first transferred to well solution supplemented with 20% ethylene glycol. Data were collected at 100 K on beamline 21-ID-F at the Advanced Photon Source, Argonne National Laboratory, or on the 5.0.1 beamline at the Advanced Light Source, Brookhaven National Laboratory, for LpPurE and BcPurE, respectively. Data were integrated with XDS and reduced with XSCALE.38 The structure of BcPurE was solved by molecular replacement using Phaser39 with 1XMP as the starting model. Refinement was carried out using REFMAC540 followed by manual model building in Coot.41 The structure of LpPurE was solved using MoRDa42 with 3OOW as the starting model. Refinement was carried out using phenix.refine43 followed by manual model building in Coot.41 Refinement statistics for both models are given in Table 1.
Structure Preparation, Ligand Selection, and Docking
The PurE models used for docking studies were the putative catalytic subunits consisting of three protomers of the protein. This catalytic subunit was derived by first generating symmetry mates for LpPurE (PDBID 6O55), and BcPurE (PDBID 4GRD) using PyMOL (Schrodinger). Then, using the MatchMaker tool in UCSF Chimera,44 each protein was individually aligned to E. coli PurE (PDBID 1D7A).24 Once aligned, the catalytic subunits of E. coli PurE were used to define the catalytic subunits of LpPurE or BcPurE. After alignment, one catalytic unit (three protomers) of LpPurE or BcPurE was retained and the other protomers were deleted. To prepare the reference ligand, the AIR molecule from 1D7A was extracted and prepared by adding hydrogen atoms and charges using the AMBER ff14SB force field (Chimera). The PurE protein models were prepared for docking using well-established DOCK6 protocols.45,46 Briefly, hydrogens and charges were added to the protein using the AMBER ff14SB force field.47 Subsequently, the protein molecular surface was generated by using the dms program, then spheres were generated over the surface of the protein using the sphgen program. Spheres within 10 Å of the reference ligand were then selected. Following sphere generation, docking grids were generated using the grid function of DOCK6.45
Virtual Screening
A virtual screening library of 1,999 FDA-approved, immediate-delivery molecules in MOL2 format was derived from the ZINC15 database.35 Each molecule was flexibly docked to the 6O55 and 4GRD energy grids using DOCK6. After docking, the library was filtered based on several chemical descriptors to remove molecules that were less than 300 Da, had a cLogP value less than −5 and greater than 5, and had greater than 11 rotatable bonds. The final list of 847 docked molecules was rank-ordered based on the DOCK Grid Score. The top 100 molecules were retained, and a subset of 25 commercially available molecules was selected for purchase and tested in a binding assay.
Docking Studies with Human PurE Model
The SAICARS domains (residues 7-261) of human PAICS (PDBID 2H31) were removed and the central octameric PurE domain was aligned to E. coli PurE (1D7A) to define the AIR carboxylase catalytic unit. After alignment, a single catalytic unit (three protomers) was saved in PDB format and all other protomers were deleted. The protein and reference ligand were prepared, and the virtual screening was conducted as described above for Lp and BcPurE.
Synthesis of 4-Carboxy-5-aminoimidazole Ribonucleotide
CAIR was synthesized using a previously established method.48 In brief, lithium hydroxide (2.5 mL of 1 M; 2 mmol) was added to 5-aminoimidazole 4-carboxamide ribonucleotide (AICAR, Sigma) (5 mg; 0.015 mmol) in a 25 mL round-bottom flask. The mixture was stirred overnight under nitrogen at 120° C. The dry reaction mixture was cooled, dissolved in 2 mL of distilled H2O, and pH was adjusted to 7 using 1 N acetic acid. The solution was lyophilized and the resulting residue was further washed five times with 100% ethanol. The remaining white solid (pure CAIR) was dried under vacuum. The product (3.7 mg; 73% yield) was verified by mass spectrometry using TOF-MS (calculated—339.2, observed—338.2 for M–H).
Steady-State Enzyme Kinetics
The kinetics of the PurE enzymes were measured using the reverse direction reaction as previously described.7 Briefly, 10 or 50 nmole (for LpPurE or BcPurE, respectively) of PurE enzyme was added to 5–200 μM CAIR in 100 mM Tris, HCl, pH 7.8, and 150 mM NaCl, and the conversion of CAIR to AIR was followed by the decrease in absorbance at 250 nm. Kinetic parameters were derived by fitting the initial rate data using the Michaelis–Menten equation.
HPLC Analysis
The enzymatic consumption of CAIR was monitored by HPLC using a Zorbax Eclipse Plux C-18 column on an Agilent 1100 HPLC using a previously established method.48 Briefly, a reaction mixture containing Bc or LpPurE, 100 μM CAIR in 100 mM Tris, HCl, pH 7.8, and 150 mM NaCl, and 50 or 100 μM Alfuzosin or Indacaterol was incubated for 15 min, and then the enzyme was removed by spinning for 10 min at 15,000 g in a 3000 Da molecular weight cutoff centrifugal concentrator. The flowthrough was applied to the column to determine the concentration of CAIR remaining in the sample. The separation employed a gradient from 2 to 90% methanol, over 15 min, in a solution of 20 mM potassium phosphate and 10 mM tetrabutylammonium bromide. Under these conditions, CAIR had a retention time of 9 min. Inhibition was confirmed by comparing the concentration of CAIR remaining in inhibitor-treated or DMSO-treated samples.
Inhibitor Screening and IC50 Calculation
Potential inhibitors were first screened using 10 or 50 ng of PurE enzyme and 100 μM CAIR in 100 mM Tris, HCl, pH 7.8, and 150 mM NaCl. Compounds, from 10 mM stocks in DMSO, were initially tested at two concentrations (10 and 100 μM) against BcPurE, and those that showed some level of inhibition at either concentration were further characterized. The initial rate of the PurE reaction was then determined for a series of inhibitor concentrations (from 1 mM or 200 to 0.78 μM). The compounds were first incubated with the enzyme for 15 min, prior to the addition of the substrate to initiate the reaction. The initial rate value was calculated from the curve after a delay of 20 s to avoid fluctuations due to mixing. A total of 10 initial rate measurements were taken for each compound, from 1 μM to 1 mM. In some cases, where indicated, poor solubility prevented the measurement of inhibition above 200 μM. In all cases, triplicate measurements were taken and the mean was reported with the standard deviation shown as the error bar. IC50 values were calculated by fitting a 4-parameter logistic equation to this data. For all inhibitors tested, two sets of parallel control measurements were taken. These included a no-enzyme control, containing both substrate and inhibitor, and a no-substrate control, which contained enzyme and inhibitor only. These were used to determine background levels from absorbance of the inhibitor and to assess whether precipitation of the inhibitor or enzyme contributed to the signal. Note that the vast majority of the measurements were well under 1 absorbance unit (λ = 250 nm) and none exceeded 1.5 A.U.
Data Availability Statement
The coordinate files and experimental data for the two structures described herein are freely available through the protein data bank (www.rcsb.org) with identifiers 6O55 for LpPurE and 4GRD for BcPurE.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.2c00705.
Protein structure comparison from DALI Protein Server (Table S1); cloning, expression, and purification details for BcPurE (Table S2); cloning, expression, and purification details for LpPurE (Table S3); size exclusion chromatogram of LpPurE and BcPurE (Figure S1); molecular weight prediction of LpPurE and BcPurE using multiangle light scattering (Figure S2); structure-based alignment of LpPurE and human PurE (Figure S3); multiple sequence alignment of PurE enzymes (Figure S4); active site comparison of BcPurE and human PurE (Figure S5); PurE enzymes with docked compounds (Figure S6); distribution of grid scores (Figure S7); dose–response curves for LpPurE (Figure S8); dose–response curves for BcPurE (Figure S9); putative binding interaction of Alfuzosin and Afatinib with BcPurE (Figure S10); HPLC assay confirming inhibition (Figure S11); putative binding interactions of Alfuzosin and Afatinib (Figure S12); putative binding interactions with LpPurE (Figure S13); comparison of ligand binding conformations (Figure S14); and alternative binding pose of indacaterol (Figure S15) (PDF)
This work was supported in part by the National Institutes of General Medical Sciences, and the National Institute of Allergy and Infectious Diseases, of the National Institutes of Health, Department of Health and Human Services, under grant number R35GM124898 (JBF) and contract number under Contract Nos. 75N93022C00036, HHSN272201700059C, HHSN272201200025C, and HHSN272200700057C (SSGCID). The funding agencies did not play a role in the conception, implementation, or analysis of the research described.
The authors declare no competing financial interest.
Supplementary Material
References
- Hartman S. C.; Buchanan J. M. Nucleic acids, purines, pyrimidines (nucleotide synthesis). Annu. Rev. Biochem. 1959, 28, 365–410. 10.1146/annurev.bi.28.070159.002053. [DOI] [PubMed] [Google Scholar]
- Lane A. N.; Fan T. W. Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res. 2015, 43, 2466–2485. 10.1093/nar/gkv047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaoka T.; Yano M.; Kondo M.; Sasaki H.; Hino S.; Katashima R.; Moritani M.; Itakura M. Feedback inhibition of amidophosphoribosyltransferase regulates the rate of cell growth via purine nucleotide, DNA, and protein syntheses. J. Biol. Chem. 2001, 276, 21285–21291. 10.1074/jbc.M011103200. [DOI] [PubMed] [Google Scholar]
- Natsumeda Y.; Prajda N.; Donohue J. P.; Glover J. L.; Weber G. Enzymic capacities of purine de Novo and salvage pathways for nucleotide synthesis in normal and neoplastic tissues. Cancer Res. 1984, 44, 2475–2479. [PubMed] [Google Scholar]
- Zhang Y.; Morar M.; Ealick S. E. Structural biology of the purine biosynthetic pathway. Cell. Mol. Life Sci. 2008, 65, 3699–3724. 10.1007/s00018-008-8295-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestine S. M.; Poon S. W.; Mueller E. J.; Stubbe J.; Davisson V. J. Reactions catalyzed by 5-aminoimidazole ribonucleotide carboxylases from Escherichia coli and Gallus gallus: a case for divergent catalytic mechanisms. Biochemistry 1994, 33, 11927–11934. 10.1021/bi00205a031. [DOI] [PubMed] [Google Scholar]
- Meyer E.; Leonard N. J.; Bhat B.; Stubbe J.; Smith J. M. Purification and characterization of the purE, purK, and purC gene products: identification of a previously unrecognized energy requirement in the purine biosynthetic pathway. Biochemistry 1992, 31, 5022–5032. 10.1021/bi00136a016. [DOI] [PubMed] [Google Scholar]
- Thoden J. B.; Kappock T. J.; Stubbe J.; Holden H. M. Three-dimensional structure of N5-carboxyaminoimidazole ribonucleotide synthetase: a member of the ATP grasp protein superfamily. Biochemistry 1999, 38, 15480–15492. 10.1021/bi991618s. [DOI] [PubMed] [Google Scholar]
- Elion G. B. The purine path to chemotherapy. Science 1989, 244, 41–47. 10.1126/science.2649979. [DOI] [PubMed] [Google Scholar]
- Jordheim L. P.; Durantel D.; Zoulim F.; Dumontet C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discovery 2013, 12, 447–464. 10.1038/nrd4010. [DOI] [PubMed] [Google Scholar]
- Parker W. B. Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chem. Rev. 2009, 109, 2880–2893. 10.1021/cr900028p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden M. G. Targeting cancer metabolism: a therapeutic window opens. Nat. Rev. Drug Discovery 2011, 10, 671–684. 10.1038/nrd3504. [DOI] [PubMed] [Google Scholar]
- Yates M. K.; Seley-Radtke K. L. The evolution of antiviral nucleoside analogues: A review for chemists and non-chemists. Part II: Complex modifications to the nucleoside scaffold. Antiviral Res. 2019, 162, 5–21. 10.1016/j.antiviral.2018.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford R. M.; Van De Verg L.; Yuan L.; Hadfield T. L.; Warren R. L.; Drazek E. S.; Houng H. H.; Hammack C.; Sasala K.; Polsinelli T.; Thompson J.; Hoover D. L. Deletion of purE attenuates Brucella melitensis infection in mice. Infect. Immun. 1996, 64, 2188–2192. 10.1128/iai.64.6.2188-2192.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flashner Y.; Mamroud E.; Tidhar A.; Ber R.; Aftalion M.; Gur D.; Lazar S.; Zvi A.; Bino T.; Ariel N.; Velan B.; Shafferman A.; Cohen S. Generation of Yersinia pestis attenuated strains by signature-tagged mutagenesis in search of novel vaccine candidates. Infect. Immun. 2004, 72, 908–915. 10.1128/IAI.72.2.908-915.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirsch D. R.; Whitney R. R. Pathogenicity of Candida albicans auxotrophic mutants in experimental infections. Infect. Immun. 1991, 59, 3297–3300. 10.1128/iai.59.9.3297-3300.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polissi A.; Pontiggia A.; Feger G.; Altieri M.; Mottl H.; Ferrari L.; Simon D. Large-scale identification of virulence genes from Streptococcus pneumoniae. Infect. Immun. 1998, 66, 5620–5629. 10.1128/IAI.66.12.5620-5629.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samant S.; Lee H.; Ghassemi M.; Chen J.; Cook J. L.; Mankin A. S.; Neyfakh A. A. Nucleotide biosynthesis is critical for growth of bacteria in human blood. PLoS Pathog. 2008, 4, e37 10.1371/journal.ppat.0040037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan C. M.; Bhandari J.; Napuli A. J.; Leibly D. J.; Choi R.; Kelley A.; Van Voorhis W. C.; Edwards T. E.; Stewart L. J. High-throughput protein production and purification at the Seattle Structural Genomics Center for Infectious Disease. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 2011, 67, 1010–1014. 10.1107/S1744309111018367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi R.; Kelley A.; Leibly D.; Hewitt S. N.; Napuli A.; Van Voorhis W. Immobilized metal-affinity chromatography protein-recovery screening is predictive of crystallographic structure success. Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun. 2011, 67, 998–1005. 10.1107/S1744309111017374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serbzhinskiy D. A.; Clifton M. C.; Sankaran B.; Staker B. L.; Edwards T. E.; Myler P. J. Structure of an ADP-ribosylation factor, ARF1, from Entamoeba histolytica bound to Mg(2+)-GDP. Acta Crystallogr., Sect. F: Struct. Biol. Commun. 2015, 71, 594–599. 10.1107/S2053230X15004677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm L. Dali server: structural unification of protein families. Nucleic Acids Res. 2022, 50, W210–W215. 10.1093/nar/gkac387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S. X.; Tong Y. P.; Xie X. C.; Wang Q. H.; Zhou H. N.; Han Y.; Zhang Z. Y.; Gao W.; Li S. G.; Zhang X. C.; Bi R. C. Octameric structure of the human bifunctional enzyme PAICS in purine biosynthesis. J. Mol. Biol. 2007, 366, 1603–1614. 10.1016/j.jmb.2006.12.027. [DOI] [PubMed] [Google Scholar]
- Mathews I. I.; Kappock T. J.; Stubbe J.; Ealick S. E. Crystal structure of Escherichia coli PurE, an unusual mutase in the purine biosynthetic pathway. Structure 1999, 7, 1395–1406. 10.1016/S0969-2126(00)80029-5. [DOI] [PubMed] [Google Scholar]
- Krissinel E.; Henrick K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. 10.1016/j.jmb.2007.05.022. [DOI] [PubMed] [Google Scholar]
- Hoskins A. A.; Morar M.; Kappock T. J.; Mathews I. I.; Zaugg J. B.; Barder T. E.; Peng P.; Okamoto A.; Ealick S. E.; Stubbe J. N5-CAIR mutase: role of a CO2 binding site and substrate movement in catalysis. Biochemistry 2007, 46, 2842–2855. 10.1021/bi602436g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller E. J.; Meyer E.; Rudolph J.; Davisson V. J.; Stubbe J. N5-carboxyaminoimidazole ribonucleotide: evidence for a new intermediate and two new enzymatic activities in the de novo purine biosynthetic pathway of Escherichia coli. Biochemistry 1994, 33, 2269–2278. 10.1021/bi00174a038. [DOI] [PubMed] [Google Scholar]
- Ovádi J. Physiological significance of metabolic channelling. J. Theor. Biol. 1991, 152, 1–22. 10.1016/S0022-5193(05)80500-4. [DOI] [PubMed] [Google Scholar]
- Rudolph J.; Stubbe J. Investigation of the mechanism of phosphoribosylamine transfer from glutamine phosphoribosylpyrophosphate amidotransferase to glycinamide ribonucleotide synthetase. Biochemistry 1995, 34, 2241–2250. 10.1021/bi00007a019. [DOI] [PubMed] [Google Scholar]
- Schendel F. J.; Cheng Y. S.; Otvos J. D.; Wehrli S.; Stubbe J. Characterization and chemical properties of phosphoribosylamine, an unstable intermediate in the de novo purine biosynthetic pathway. Biochemistry 1988, 27, 2614–2623. 10.1021/bi00407a052. [DOI] [PubMed] [Google Scholar]
- Wang W.; Kappock T. J.; Stubbe J.; Ealick S. E. X-ray crystal structure of glycinamide ribonucleotide synthetase from Escherichia coli. Biochemistry 1998, 37, 15647–15662. 10.1021/bi981405n. [DOI] [PubMed] [Google Scholar]
- Tranchimand S.; Starks C. M.; Mathews I. I.; Hockings S. C.; Kappock T. J. Treponema denticola PurE Is a bacterial AIR carboxylase. Biochemistry 2011, 50, 4623–4637. 10.1021/bi102033a. [DOI] [PubMed] [Google Scholar]
- Lei H.; Jones C.; Zhu T.; Patel K.; Wolf N. M.; Fung L. W.; Lee H.; Johnson M. E. Identification of B. anthracis N(5)-carboxyaminoimidazole ribonucleotide mutase (PurE) active site binding compounds via fragment library screening. Bioorg. Med. Chem. 2016, 24, 596–605. 10.1016/j.bmc.2015.12.029. [DOI] [PubMed] [Google Scholar]
- Streeter C. C.; Lin Q.; Firestine S. M. Isatins Inhibit N(5)-CAIR Synthetase by a Substrate Depletion Mechanism. Biochemistry 2019, 58, 2260–2268. 10.1021/acs.biochem.8b00939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin J. J.; Sterling T.; Mysinger M. M.; Bolstad E. S.; Coleman R. G. ZINC: a free tool to discover chemistry for biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. 10.1021/ci3001277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aslanidis C.; de Jong P. J. Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res. 1990, 18, 6069–6074. 10.1093/nar/18.20.6069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier F. W. Protein production by auto-induction in high density shaking cultures. Protein Expression Purif. 2005, 41, 207–234. 10.1016/j.pep.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Kabsch W. XDS Program Package. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 125–132. 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Read R. J. Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2001, 57, 1373–1382. 10.1107/S0907444901012471. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1997, 53, 240–255. 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Vagin A.; Lebedev A. MoRDa, an automatic molecular replacement pipeline. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, s19. 10.1107/S2053273315099672. [DOI] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Echols N.; Headd J. J.; Hung L. W.; Jain S.; Kapral G. J.; Grosse Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R. D.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. The Phenix software for automated determination of macromolecular structures. Methods 2011, 55, 94–106. 10.1016/j.ymeth.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Meng E. C.; Couch G. S.; Croll T. I.; Morris J. H.; Ferrin T. E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. 10.1002/pro.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen W. J.; Balius T. E.; Mukherjee S.; Brozell S. R.; Moustakas D. T.; Lang P. T.; Case D. A.; Kuntz I. D.; Rizzo R. C. DOCK 6: Impact of new features and current docking performance. J. Comput. Chem. 2015, 36, 1132–1156. 10.1002/jcc.23905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S.; Balius T. E.; Rizzo R. C. Docking validation resources: protein family and ligand flexibility experiments. J. Chem. Inf. Model. 2010, 50, 1986–2000. 10.1021/ci1001982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehl R. A.; Begley T. P. Synthesis of 32P-labeled intermediates on the purine biosynthetic pathway. J. Labelled Compd. Radiopharm. 2002, 45, 1097–1102. 10.1002/jlcr.627. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The coordinate files and experimental data for the two structures described herein are freely available through the protein data bank (www.rcsb.org) with identifiers 6O55 for LpPurE and 4GRD for BcPurE.