

ABSTRACT
Cleidocranial dysplasia (CCD) is a rare hereditary disease of unknown etiology which was previously known as cleidocranial dysostosis. It usually follows an autosomal dominant mode of transmission with no predilection of genre or ethnic group. It is caused by a mutation of RUNX2, characterized by generalized dysplasia of the bones and teeth. Affected individuals have short stature, atypical facial features, and skeletal anomalies affecting mainly the skull and clavicle. The dental manifestations are mainly delayed exfoliation of the primary teeth and delayed eruption of the permanent teeth, with multiple impacted supernumeraries, and the absence of cellular cementum. The frequency of this disorder is 1 per million individuals. Here we report a rare case of CCD in a 23 year old female patient having most of the characteristic features of this syndrome.
KEYWORDS: Cleidocranial dysplasia, dysostosis, RUNX2
INTRODUCTION
Cleidocranial dysplasia (CCD), also known as Marie and Sainton’s disease; Scheuthauer - Marie - Sainton syndrome; Mutational Dysostosis, is a rare congenital defect of autosomal dominant inheritance affecting both sexes with equal frequency.[1,2]
Cleidocranial dysplasia was first described in a patient with congenital absence of clavicle in 1765 by Martin.[3] In dysostoses, the distribution follows a defect in ectodermal or mesenchyma tissues. Rarely are all bones involved. As derived from Greek, dysplasia refers to an abnormality of development or “ill formed” (Gr. plassein - to form); in pathology, it means an alteration in size, shape, and organization of adult cells.[4]
The etiology though not completely known is thought to be due to a CBFA1 (core binding factor activity 1) gene defect on the short arm of chromosome 6p21. CBFA1 is essential for the differentiation of stem cells into osteoblasts, so any defect in this gene will cause defects in the membraneous and endochondral bone formation. It affects bones of intramembranous origin and endochondral bone formation of long bones, there is also a failure of midline ossification. The involvement of non-membranous bones is also well recognized.[5,6]
The main features of this syndrome are persistent open skull sutures with a bulging calvaria, hypoplasia or aplasia of the clavicle permitting an abnormal ability to appose the shoulders, open fontanelles, wormian bones, a wide pubic symphysis, short middle phalanges of the fifth fingers, and various vertebral and dental abnormalities. The sutures remain open, and the sagittal suture is depressed giving the cranium a flat appearance, sometimes referred to as “Arnold head.” The skull appears brachycephalic with a narrowing of the skull in the anteroposterior direction and an increase in the width of the skull. Dental abnormalities include retained deciduous dentition, delayed eruption or retention of the permanent dentition, multiple supernumerary teeth, crown and root abnormalities, crypt formation around impacted teeth, and a high palate.[7,8]
There is the complete absence of cellular cementum and an increase in the amount of acellular cementum in the roots of the affected teeth.[9,10]
The aim of this article is to illustrate the clinical features, radiological features, and dental abnormalities in a rare case of cleidocranial dysplasia.
CASE PRESENTATION
This case depicts a patient with a hereditary disease and shows all features radiographically and clinically [Figures 1-4]. 23 year old female reported with chief complaint malaligned anterior teeth. On general examination,she was short statured, frontal bossing, nasal bridge flattened, hypoplastic maxilla, drooping shoulders absence of clavicles. Intra Oral examination revealed, over retained deciduous anterior and few posterior teeth. Orthopantomograph revealed , multiple unerupted , permanent teeth , mandibular molars with open apices and in formation stages. All the features of Cleido Cranial dysplasia clinically and radiologically were evident in this patient.
Figure 1.
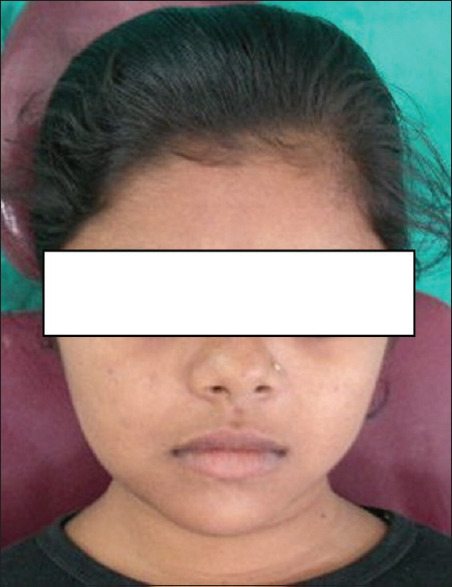
Frontal profile of the patient
Figure 4.
Hand and wrist radiograph
Figure 2.
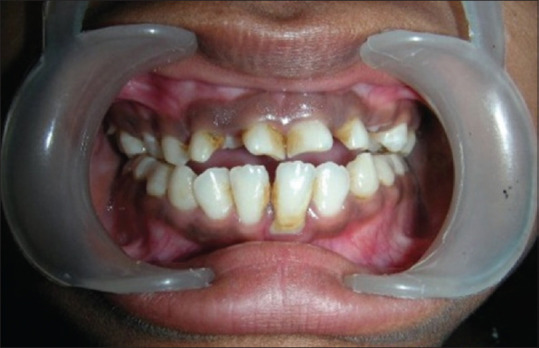
Intraoral image
Figure 3.
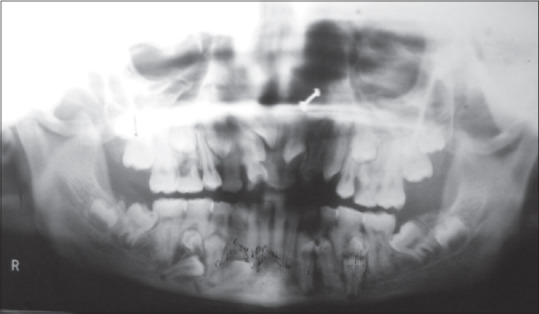
Orthopantomograph of the patient
DISCUSSION
Cleidocranial dysplasia is a dominant, inherited autosomal bone disorder with a wide range of expressivities, primarily affecting bones undergoing intramembranous ossification.
The appearance is generally pathognomonic. Affected individuals are usually short, with males averaging 156.6 to 168.8 cm and females 144.6 to 148.5 cm. The skull is brachycephalic, with pronounced frontal and parietal bossing, and the maxilla and zygomas are hypoplastic; thus, the face appears small. The nose is broad at the base, with the bridge depressed. There is hypertelorism. The neck appears long, the shoulders are narrow and droop markedly the occipital bone is above the inion. Secondary centers of ossification appear in the suture lines (metopic), and many wormian bones are formed. In extreme cases, the parietal bones are not present at birth. The cranial base has a short sagittal diameter. The Mandibular length is increased and the maxilla is short vertically. The foramen magnum, which is large, often exhibits defects in the posterior wall. Paranasal sinuses and mastoids are often underdeveloped or absent.[11]
The clavicles are the first bone to ossify and/or are commonly affected, being either hypoplastic or aplastic. Complete absence of the clavicles occurs in about 10% of cases and usually, only the acromial end is absent. When there is unilateral absence, it is usually in the right clavicle. Complete or partial absence of clavicular calcification, with associated muscle defects, results in hypermobility of the shoulders, allowing for variable levels of approximation in an anterior plane. Other bones also may be affected, including the long bones, vertebral column, pelvis, and bones of the hands and feet. Hemivertebrae and posterior wedging of the thoracic vertebrae may contribute to the development of kyphoscoliosis.[12,13]
Delayed eruption of permanent teeth is seen. It is known that the extraction of deciduous teeth does not promote the eruption of permanent teeth. Rushton and others studied teeth microscopically and observed that roots lacked a layer of cellular cementum. Greater than normal bone density of the jaws might inhibit tooth eruption. Rushton and Hitchin attributed the noneruption of teeth to the failure of the bone to resorb.[3] Usually, the first molars and the mandibular incisors erupt normally. The delayed eruption of the maxillary central incisors is usually the cause for seeking treatment.[11]
CONCLUSION
Cleidocranial dysplasia is a rare congenital disorder of the growth and development of the bones. Early diagnosis allows proper orientation to the treatment and offers a better life quality. A holistic approach takes care of the aspects, including the primary pathology and the psychological aspects. As Cleidocranial dysplasia manifests with a variety of oral manifestations, knowledge of the clinical and radiological features of this disorder is necessary for dental professionals.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Shafer W. G., Hine M. K., Levy B. M. Patologia bucal. Rio de Janeiro, Interamericana, 1979. 1ª. ed. Available from:https://books.google.co.in/books?id=Spk0V6TrCggC&newbks=1&newbks_redir=0&printsec=frontcover&redir_esc=y#v=onepage&q&f=false .
- 2.Neville B. W., Damm D. D, Allen C. M., Bouquot JE. Patologia oral e maxilofacial. 2ª. ed Rio de Janeiro, Guanabara Koogan, 2004. Avialable from:https://www.elsevier.com/books/oral-and-maxillofacial-pathology/neville/978-1-4557-7052-6 .
- 3.Patil PP, Barpande SR, Bhavthankar JD, Humbe JG. Cleidocranial Dysplasia:A Clinico-radiographic Spectrum with Differential Diagnosis. J Orthop Case Rep. 2015;5:21–4. doi: 10.13107/jocr.2250-0685.264. doi:10.13107/jocr.2250-0685.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marie P., Sainton P. Sur la dysostose cleido-cranienne herediataire, Rev neurol. 6:835, 1898 On hereditary cleido-cranial dysostosis. Clin Orthop Relat Res. 1968;58:5–7. PMID:4875295. [PubMed] [Google Scholar]
- 5.Mundlos S. Cleidocranial dysplasia:Clinical and molecular genetics. J Med Genet. 1999;36:177–82. [PMC free article] [PubMed] [Google Scholar]
- 6.Counts AL, Rohrer MD, Prasad H, Bolen P. An assessment of root cementum in cleidocranial dysplasia. Angle Orthod. 2001;71:293–8. doi: 10.1043/0003-3219(2001)071<0293:AAORCI>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 7.López BSG, Solalinde CO, Ito TK, Carrillo EL, Solalinde EO. Cleidocranial dysplasia:Report of a family. J Oral Sci. 2004;46:259–66. doi: 10.2334/josnusd.46.259. [DOI] [PubMed] [Google Scholar]
- 8.Hemalatha R, Balasubramaniam MR. Cleidocranial dysplasia:A case report. J Indian Soc Pedod Prev Dent. 2008;26:40–3. doi: 10.4103/0970-4388.40322. [DOI] [PubMed] [Google Scholar]
- 9.Counts AL, Rohrer MD, Prasad H, Bolen P. An assessment of root cementum in cleidocranial dysplasia. Angle Orthod. 2001;71:293–8. doi: 10.1043/0003-3219(2001)071<0293:AAORCI>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 10.Fukuta Y, Totsuka M, Fukuta Y, Takeda Y, Yoshida Y, Niitsu J, et al. Histological and analytical studies of a tooth in a patient with cleidocranial dysostosis. J Oral Sci. 2001;43:85–9. doi: 10.2334/josnusd.43.85. [DOI] [PubMed] [Google Scholar]
- 11.Gorlin RJ, Cohen MM, Hennekam . Syndromes of head and neck. 4th ed. Oxford University Press, Inc; New York: pp. 306–310. Available from:https://www.academia.edu/37214951/Gorlin_Syndromes_of_the_Head_and_Neck . [Google Scholar]
- 12.Verma P, Verma KG, Gupta SD. Cleidocranial dysplasia:A dilemma in diagnosis? Arch Orofac Sci. 2010;5:61–4. [Google Scholar]
- 13.Pereira CM, Alves VF, Gasparetto PF, Souza RCN, de Lucena Botelho T. Cleidocranial dysplasia associated with the dentigerous cyst:Review of the literature and report of the clinical case. J Health Sci Inst. 2010;28:137–9. [Google Scholar]