

Abstract
Deep learning has led to incredible breakthroughs in areas of research, from self-driving vehicles to solutions, to formal mathematical proofs. In the biomedical sciences, however, the revolutionary results seen in other fields are only now beginning to be realized. Given its public health significance, vaccine research and development efforts, including protein structure prediction, immune repertoire analysis, and phylogenetics are three principal areas in which deep learning is poised to provide key advances. Here, we opine on some of the current challenges with deep learning and how they are being addressed. Despite the nascent stage of deep learning applications in immunological studies, there is ample opportunity to utilize this new technology to address the most challenging and burdensome infectious diseases confronting global populations.
A New Era in Deep Learning
Deep learning has garnered widespread attention in the scientific community and beyond, with groundbreaking results in various domains. The ability of deep learning programs to defeat world champions at games, drive vehicles with human-level performance, and discover novel mathematical proofs[1–4] has spurred intense desire to translate similar results in the biomedical sciences, including immunology and vaccinology. Recent deep learning applications have shown encouraging results in biology for predictive and descriptive tasks[5, 6]. For example, models have been developed to detect cancer from histology images or from genetic information at a higher accuracy than the standard of care[7–9]. To date, the greatest breakthrough of a deep learning model in biology is arguably AlphaFold’s solution to the “protein folding problem”, considered one of the most fundamental and longstanding challenges in biology[10, 11]. As deep learning slowly starts to find its place in biology, it raises the question of what type of impact this area of research may have in contributing to the development of efficacious vaccines (Figure 1), particularly those against viral pathogens, which burden society in the form of continuously evolving circulating strains, and new zoonoses that jump from animal hosts into humans. While antiviral therapies can contribute to disease treatments (e.g. HIV-1), the most effective countermeasure for preventing infectious diseases is the development of highly effective vaccines. To this end, while smallpox and polio eradication campaigns serve as examples of what is possible, developing safe and effective viral vaccines is a difficult and complex process with many more failures than successes. The new era of deep learning and big data suggests that similar potential breakthroughs might be realized in vaccine development for the benefit of public health. To this end, we foresee protein structure prediction, immune repertoire analysis, and phylogenetics as three complementary areas in which deep learning methods will contribute to efforts to advance vaccine research and development.
Figure 1: Deep learning areas of focus in vaccine design.
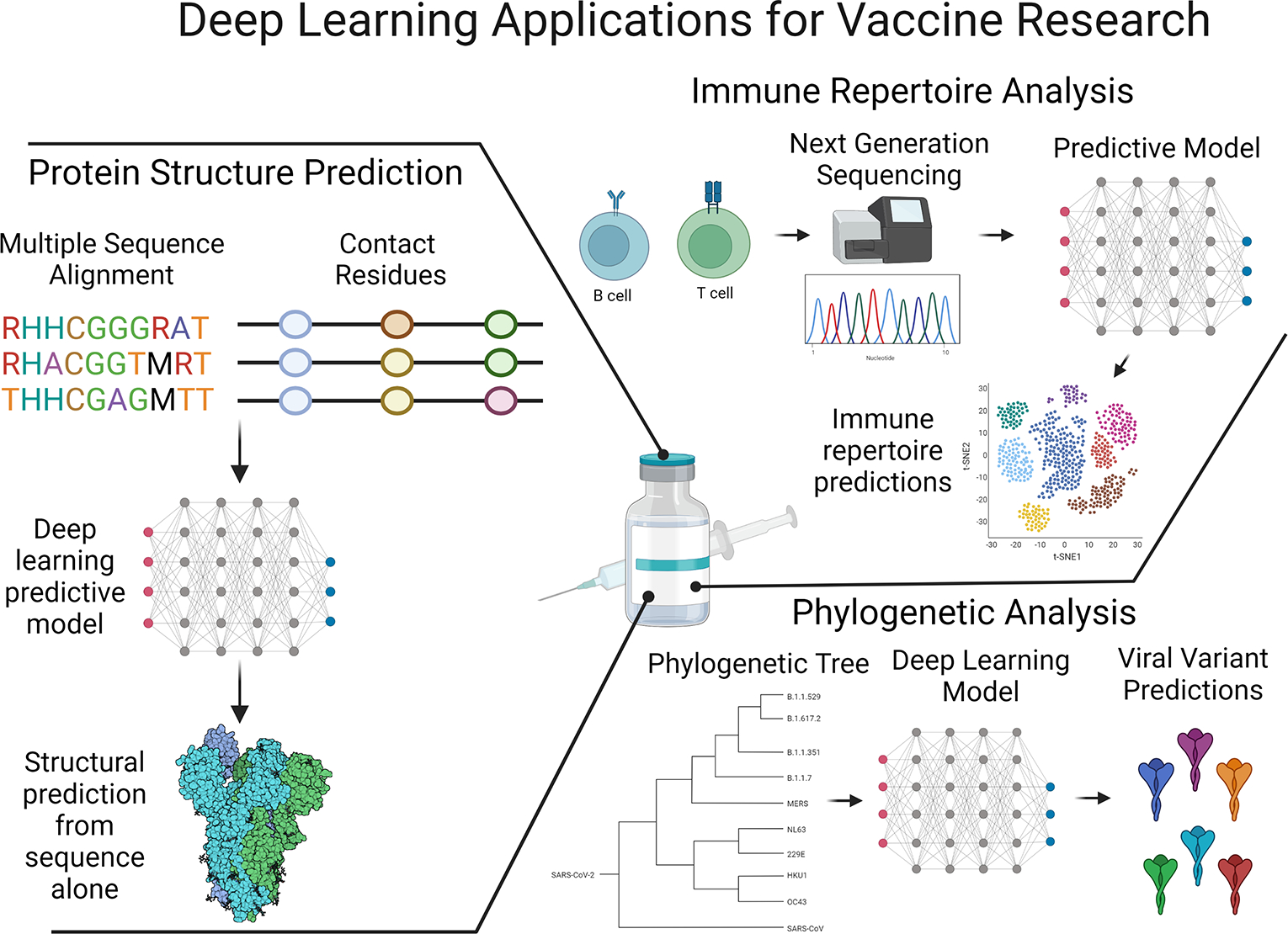
Prediction of protein structures, analysis of antibody and T cell receptor repertoires, and viral phylogenetics are three areas in which deep learning is supporting rapid advances. Deep learning has made the greatest progress so far in structure prediction “solving” the protein folding problem and is now commonly being used to generate antibodies bypassing experimentation steps. Immune repertoire data growth has coincided with deep learning development allowing for prediction of the specificity or disease outcomes of immune responses from sequencing data alone. Phylogenetic analysis of global viral variants can leverage deep learning to better understand mutational patterns and the effect mutations may have on subsequent immune responses as well as pathogen fitness and population susceptibility. This figure was created using BioRender (https://biorender.com/)
Surface level introduction to deep learning
Deep learning is a subset of machine learning that utilizes more complex learning algorithms on large datasets. Although deep learning and classical machine learning (ML) techniques are similar, there are a few key distinctions (Figure 2). At a high level, classical ML and deep learning are computational modeling approaches to take a training data set, learn trends about how data input features relate to outcomes of interest, and create rules to make a prediction (Figure 3). These approaches are validated using a test data set, leading to the determination of the prediction accuracy on “unseen” examples. The overall task of learning how to make correct predictions is the same for both modeling approaches; however, the methods the models use to accomplish this task are different. Deep learning models are modeled after neural networks in which information flows between nodes connected in layers. While the relationships between individual features and prediction outcomes become more abstract at each layer, prediction accuracy can be dramatically better than observed from classical ML approaches. As a result, although deep learning may pose challenges to supporting mechanistic biological insights, these models may more accurately represent true biological complexity, and their improved performance has the potential to lead to new insights in vaccine and therapeutic antibody[12] development, ideally helping to address the most challenging infectious diseases.
Figure 2. Comparison of common machine learning and deep learning models.
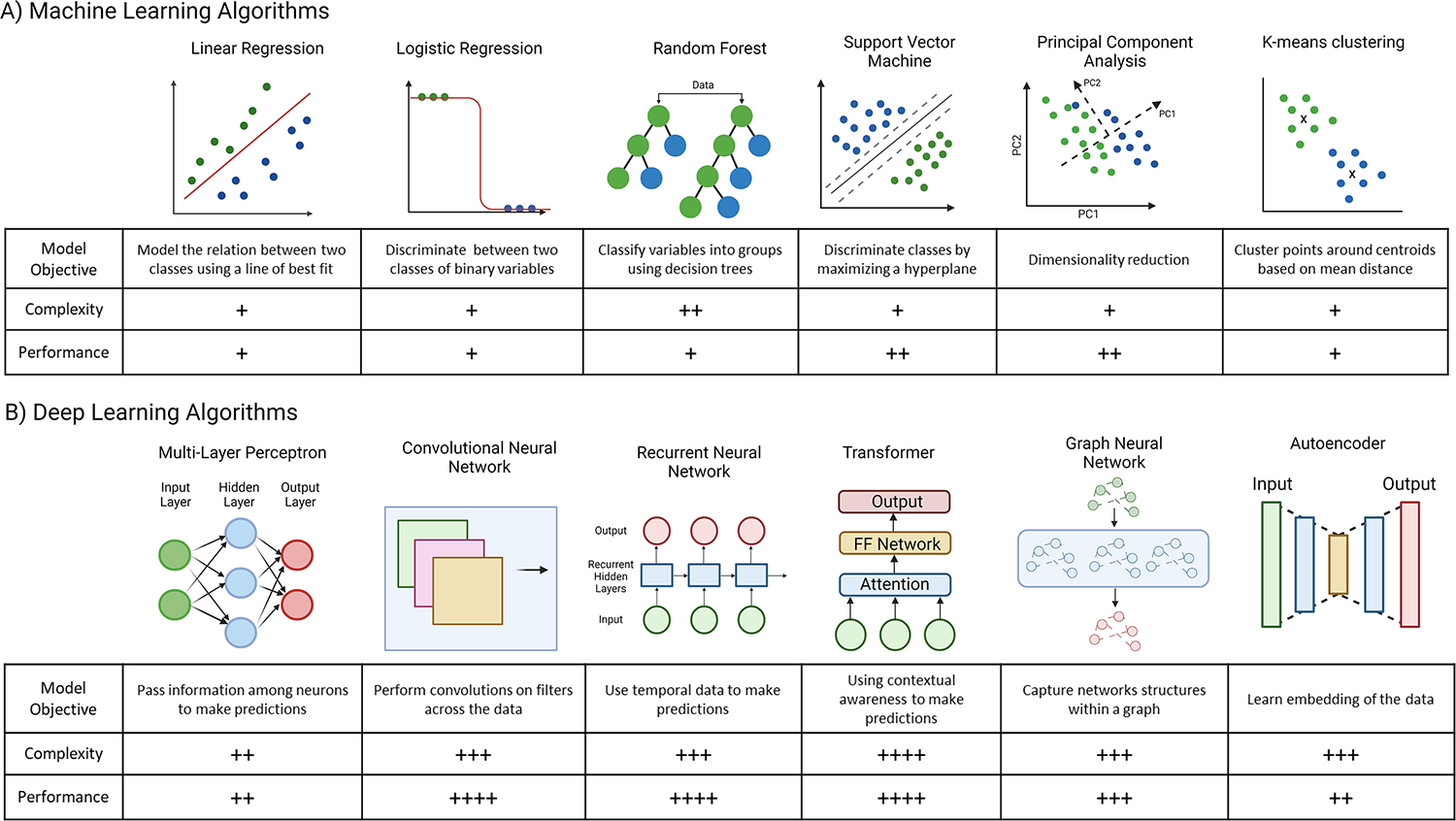
A. Examples of common classical machine learning algorithms. Algorithms are a mix of supervised approaches, such as linear regression, logistic regression, random forest, and support vector machines, in which the models are trained and tested on labelled data, and unsupervised algorithms, such as principal component analysis and K-means, in which the algorithm uses unlabeled data. B. Examples of common deep learning model architectures and associated tasks. Deep learning architectures pass information among nodes within layers to create more abstract data representations that can result in more accurate model predictions. Deep learning models generally have greater performance than machine learning algorithms however are generally more complex to create and are computationally more expensive. This figure was created using BioRender (https://biorender.com/)
Figure 3. Deep Learning Model Workflow.
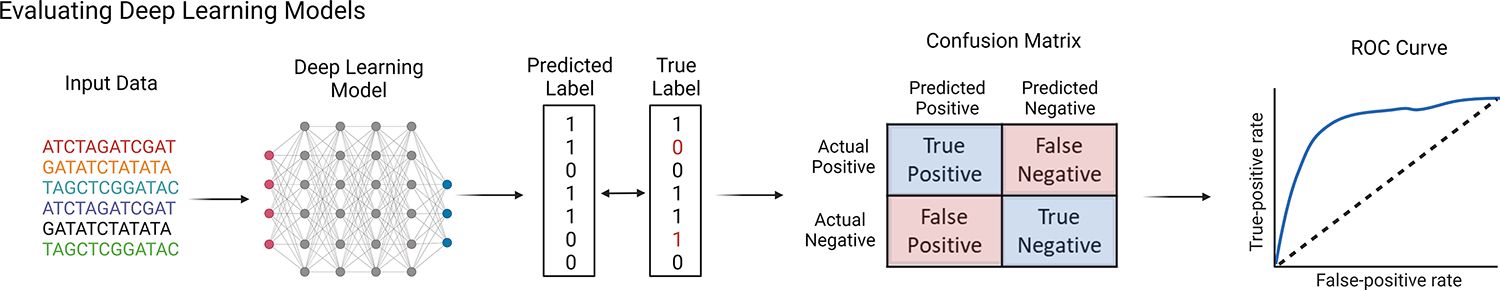
Deep learning models are made using the training data set. Model parameters are refined and tuned until the error is minimized when making predictions in the training set. The model is then tested by making predictions on the test data set, which the model has not seen previously. Standard metrics for classification model evaluation include generation of a confusion matrix which breaks down where the misclassifications happened and a receiver operating characteristic (ROC) curve providing information on how model performance compares to random. This figure was created using BioRender (https://biorender.com/)
Protein structure prediction and immunogen design
In theory, the total surface area of the entire proteome of a pathogen can elicit an adaptive immune response, but in practice, different parts and conformational states of these target antigen surfaces offer differing degrees of protection, as was recently reported for SARS-CoV-2 (responsible for the current COVID-19 pandemic)[13] and has long been appreciated for other viruses, such as HIV-1, and has been referred to as the “neutralizing antibody problem”[14]. Whereas reverse vaccinology sought to employ bioinformatics for antigen selection and has driven great inroads against bacterial pathogens[15], next generation vaccinology approaches for viruses clearly heavily rely on insights from structural biology (Table 1). Knowledge of the three-dimensional structure of relevant immunogens can aid in vaccine development by providing a physical representation above the raw amino acid sequence that can guide studies into directing responses towards certain epitopes or pre-fusion conformational states, and away from others. While for decades the only method of accurately obtaining the structure of a protein was experimentally, deep learning methods have recently predicted structures from amino acid sequences with accuracy equivalent to experimental methods[10, 11, 16–18]. These deep learning models have been evaluated using data from the Critical Assessment of Structure Prediction (CASP)[19], in which models are evaluated by predicting structure from sequence on solved structures that have not been released publicly providing an accuracy benchmark. These new models are able to achieve accuracies on the CASP test structures that are within the same level of variation from true structure as x-ray crystallography and cryo-electron microscopy. Given the importance of structure-based vaccine design against the metastable fusion proteins of viruses, the ability to accurately and rapidly predict structure from sequence alone has the potential to usher in a new era of vaccine discovery.
Table 1:
Structure-based vaccinology for human viruses
| Virus | Structural Modification | Outcome | Ref |
|---|---|---|---|
| RSV | Identification of pre-fusion F protein structure | Allows vaccination with pre-fusion F-containing neutralizing epitopes | [105] |
| Pre-fusion F stabilization | Highly immunogenic responses in vaccines | [106–108] | |
| Epitope focused vaccine design | Proof-of-concept study developing an RSV vaccine for neutralizing epitopes of interest | [109] | |
| SARS-CoV-2 | SARS-CoV-2 spike stabilization | Highly immunogenic vaccines with the S2P and HexaPro stabilizations | [26] |
| SARS-CoV | Identification of SARS-CoV prefusion spike structure | Revealed new epitopes for vaccine design | [34, 110, 111] |
| MERS-CoV | Identification of MERS-CoV prefusion spike structure | Highly immunogenic epitopes for vaccine development | [112, 113] |
| HIV-1 | Stabilization of HIV-1 envelope protein | Generation of BG-SOSIP trimer immunogens capable of eliciting neutralizing antibodies | [27, 114–116] |
| Structure of pre-fusion envelope | Atomic resolution of pre-fusion spike immunogens | [117, 118] | |
| Engineered HIV-1 immunogens | Structural design of germline targeting immunogens | [53, 54, 119, 120] | |
| Structural guided nanoparticle design | Structure based nanoparticle formulations are highly immunogenic for multiple viruses | [38, 121–123] |
The clearest example of the value that structural information can have for vaccines arguably comes from Respiratory Syncytial Virus (RSV)[20, 21]. Specifically, whereas the native F protein of the virus is most frequently presented in its post-fusion conformation, antibody responses with superior neutralizing capacity are associated with recognition of the pre-fusion conformation[22]. These advances in structural knowledge for RSV led to the development of pre-fusion F-based vaccines which have recently shown highly encouraging results in advanced clinical trials (e.g. NCT04785612, NCT03982199, NCT04032093, NCT03334695). These and other studies have demonstrated that the RSV pre-fusion F protein conformation was highly immunogenic and vaccination reduced RSV infection risk compared to placebo [23–25]. Similar observations have been made regarding distinct conformations of HIV-1 and SARS-CoV-2 fusion proteins in structural studies[26, 27] that have informed clinical studies. Indeed, SARS-CoV-2 fusion proteins with structural modifications are the basis of the highly efficacious mRNA vaccines mRNA-1273 and BNT162b2 (NCT04470427, NCT04368728)[28, 29]. Prior efforts to capture metastable proteins in their most vulnerable conformations have sometimes required extensive and iterative exploration of rational modifications or directed evolution[30]. Reliable predictions of structural states from sequence lends naturally to computational alternatives to experimental protein design, which have recently gained traction in diverse design tasks such as designing immunoglobulin scaffolds, protein biosensors, and specific binding proteins using deep learning and sequence alone (Table 2)[31–33].
Table 2:
Recent advances in structure prediction and computational protein design
| Category | Method | Result | Ref |
|---|---|---|---|
| Structure Prediction | AlphaFold | Highly accurate results predicting protein structure from amino acid sequence | [124] |
| AlphaFold-2 | Updated version of AlphaFold that has solved the protein folding problem | [125] | |
| RosettaFold | Similar protein structure prediction as AlphaFold | [126] | |
| ProteinMPNN | Protein backbone sequence design using deep learning | [127] | |
| trRosetta | De novo protein structure prediction using deep neural networks | [128] | |
| RaptorX | Web based server for protein structure prediction from amino acid sequence | [129] | |
| ProGen | Language models can predict protein function from sequence families | [130] | |
| AminoBERT | Structure prediction using a language model | [131] | |
| Pfam | Annotating protein function from amino acid sequence with a deep learning model | [17] | |
| Prediction of protein fitness from evolutionary data | [132] | ||
| Protein Design | Deep learning-based design of zinc finger nucleases for specific DNA binding regions | [133] | |
| Design of IL-2 mimetic protein with reduced toxicity | [134] | ||
| Development of a capsid protein using deep learning | [135] | ||
| De novo design of a chimeric antigen receptor, small molecule regulated, kill switch | [136] | ||
| Computational design of membrane permeable proteins | [137] | ||
| Protein design of axel-rotator-like components | [138] | ||
| Design of proteins binding to specific targets from aa sequence alone | [31] | ||
| Development of nanocage structural proteins | [139] | ||
| Computational design of large multicomponent proteins | [140] | ||
| Rational design of donut-shaped proteins | [141] | ||
| Design of IgG antibodies using multi-state design simulations | [142] | ||
| Design of helical membrane proteins | [143] | ||
| De novo design of a β barrel protein | [144] | ||
Beyond viral antigens themselves, new approaches to present immunogens in defined spatial densities and orientations[34–37] further leverage advances in computation protein design[38]; indeed, approaches to predict attributes of immune responses that may be elicited by these immunogens are advancing. With respect to this latter goal, deep learning approaches appear to be gaining traction. For instance, progress has been made predicting linear B cell epitopes[39–42], in some cases substantially improving sensistivity and specificity, facilitated by growing databases such as the Immune Epitope DataBase (https://www.iedb.org/)[43]. Conformational B cell epitope prediction has been more challenging; however, Graph-based neural networks have recently become more successful at modeling structural space of conformation epitopes by essentially representing interactions in which graph nodes represent atoms and edges represent connections between them[44, 45]. Although substantial room for improvement certainly remains, B cell epitope prediction is expected to continue improving with the combination of larger experimental data sets and novel neural network architectures that might best model the epitope-paratope interactions.
Collectively, each of these aspects of protein structure modeling and prediction advanced by deep learning are in the process of being productively deployed toward vaccine design. At the most advanced end of this spectrum, engineered versions of natural viral proteins are presented, sometimes on designed particles, with the intent of driving recognition of specific epitopes whose recognition might be predicted and result in pathogen neutralization.
Immune repertoire analysis to understand vaccine-induced responses
Whereas the human genome project’s sequencing efforts were widely thought to have provided more information than insight, at least initially, large scale sequencing efforts directed at B and T cell receptors appear to be primed to be effectively coupled to advances in ML in ways that could meaningfully inform vaccine research and development. Given their central positions in building long term immunity after vaccination, B and T cell receptor sequencing has been an intense area of study in the last decade, being revolutionized by next generation sequencing (NGS) technology, resulting, for example, in over 1.5 billion unique human B cell receptor (BCR) sequences available[46]. Coupled to deeper phenotypic characterization, we expect that this rapid expansion in input data can provide rich resources to ML models. To date, deep learning models trained on immune repertoire sequence data have successfully predicted treatment outcomes from immunotherapy[47], as well as infection status or history[48], and infectious disease severity[49].
For example, basic studies of how antibody sequences vary among individuals[50, 51], and with disease[52], have started to be paired with immunogen engineering[53] and in vitro assays[54, 55] to build vaccine strategies that aim to generalize the induction of specific antibody responses[56]. With early “natural history” studies[57, 58] maturing toward cohort-sized undertakings[59–61], progress has been made in making inferences of antigen specificity[62–64], positioning deep learning to support more explicit links between antigenic stimuli and resultant responses. Coupled to insights from elegant animal model experiments[65], iterative cycles of vaccination and repertoire sequencing may provide the raw data needed to gain fundamental and quantitative insights into phenomena such as “original antigenic sin”[66](or antigenic imprinting), and might provide a better understanding of how immune history impacts future immune responses at molecular-level resolution.
In the context of the T cell receptor (TCR), previous studies started to uncover features of TCR sequencing datasets that support prediction of epitope specificity[67, 68]. Relative to antibody-antigen complexes, the structural conservation in TCR-peptide-MHC has supported more facile learning of quantifiable descriptive features[47, 69] that can contribute to prediction of specificity or relationships to other biological attributes. Comparisons of TCR repertoires in individuals with progressive and controlled disease have been used in the context of experimental antigen screens to define TCR specificities that are associated with pathogen control, therefore representing promising vaccine targets. For example, in studies of Mycobacterium Tuberculosis, comparison of TCR sequences defined common T cell specificities for peptide-MHC, that represent novel targets for vaccine design[70, 71].
Deep learning has also been used to model antibody-antigen interactions based on data from directed evolution studies of libraries of antibody sequences with changes in antigen binding over rounds of diversification using methods such as error prone PCR to introduce sequence mutations and selection of mutants of interest that have mutations that impact binding affinity or kinetics. Initial applications of deep learning to enrich for antigen binding molecules focused on phage display experiments[72, 73]. Models were used to predict binders, thereby speeding up the experimental process of affinity maturation. Models have also been applied to yeast surface display libraries focused on identifying immunoglobulin CDR3 regions of antibody heavy and light chains with the goal of understanding the impact of sequence mutations[74]. Results from library studies have demonstrated that deep learning models can identify useful patterns in relationships between antibody sequence and structural space, showing that geometric similarity and structural commonalities in CDRs can reflect attributes of antigen recognition[75], particularly over accumulated mutations. Abstracting these efforts toward the analysis of sequence repertoires from the study of immunized and vaccinated individuals, we expect that rapid gains in insights into the specificity and affinity maturation of antibody responses may ensue.
Phylogenetic analysis to understand viral evolution and population susceptibility
Continuous evolution that leads to escape from an immune response within individuals and across populations poses a persistent challenge to the development of vaccines for viral infections. Historically, predicting which viral strains will become dominant relies on modeling that can approximate educated guess work. A leading example is the influenza virus vaccine, which is designed each year based on predictions of which strains will be dominant. Incorrect predictions result in compromised vaccine efficacy and higher burdens of seasonal flu. Modeling whether or when strains may shift host species is even more difficult, but such changes in tropism can be highly consequential, given the vulnerability of naïve populations. Yet, the rapid expansion and exceptional penetrance of SARS-CoV-2 variants of concern demonstrate that viruses can exhibit substantial evolution and point estimates of population susceptibility can vary dramatically, even within a seasonal time scale in populations with a high degree of prior exposure[76–78]. To this end, the insufficiency of the immune system to outpace antigenically variant viruses ranging from common colds to HIV-1 infection, may highlight the value of new approaches to combine advances in repertoire sequencing with insights into viral phylogenies.
Fortunately, advances in technology and global infrastructure have made it easier to sequence viral variants observed in many individuals, raising the prospect of moving well beyond technical enhancements that define sequence phylogenies[79] and toward more functionally informed inferences into the future directions of viral evolution. For example, mutation-resistant amino acid residues and CD8 T cell responses prevalent among individuals able to suppress HIV-1 replication were studied using analysis combining structural data and network theory in order to quantify the structural importance of amino acid mutations on viral evasion from T cell responses. These studies have opened the door to deploying network theory approaches to design novel T cell epitope-based vaccine concepts that are resistant to viral mutation[80].
Similarly, the rapid and thorough study of SARS-CoV-2 and its continued evolution exemplify the push toward data-driven approaches that rely on more comprehensive sequencing and novel functional data streams, combined with ML models. Viral sequence information is now captured globally through robust sampling and sequencing networks[81] and can then be viewed through the lens of deep mutational scanning data[82–84] allowing for investigation of the impact specific amino acid mutations have on antibody and vaccine responses from emerging sequence variants. In these approaches, libraries comprised of thousands to billions of virus or viral antigen sequence variants are screened for phenotypes such as loss of binding to monoclonal or polyclonal antibody pools[85], enhanced infectivity[86] or binding to entry receptors[87], revealing advantageous amino acid mutations in viral sequences.
Integrating data that link attributes of host and pathogen biology from both in vivo and in vitro sources enables estimation of the tendency of different viral sequence variants to escape from contemporaneous antibody responses in the population as well as monoclonal therapies. Thus, these approaches offer opportunities to predict population-level susceptibility and to anticipate the identity of future viral variants of concern[88]. While there is clearly significant utility afforded by classical ML approaches, deep learning models are beginning to be employed on these tasks[89–91]. Given the ambitious plans for serosurveillance[92] across viruses[93], as well as antibody[94] and T cell epitopes[95] by collecting and profiling samples across global populations, and the creation of scientific networks with the capacity for rapid mechanistic and efficacy experiments[96], we envision that the development of models informed by protein structure and predicted interactions, in particular, have the potential to be greatly enhanced by deep learning.
Challenges and considerations
While deep learning can successfully tackle complex biological problems, its application is likely to stretch current immunology datasets and leave gaps in interpretation. Biological datasets are often considerably smaller than those commonly used in deep learning tasks. Immunological datasets often suffer from being “wide”, with a greater number of features than outcomes, are comprised of experimental input data that are inherently noisy due to both technical and biological variability, and are likely to confront the challenges of representing diverse populations that have been popularized by settings such as facial recognition[97]. Understanding how to address experimental and human variability while creating models that are still robust in making predictions is a challenge that is independent of the specific learning task. Moreover, while “interpretable” deep learning models are under development[98–100], the numerous parameters and layered construction inherent to current approaches drive greater and greater abstraction at each level, ultimately resulting in a scenario in which accurate and reliable predictions are made, but insights into mechanisms are obscured. Moreover, computational constraints must be considered; unlike classical ML and other types of statistical analysis, usually, deep learning models cannot run on a personal computer in a reasonable amount of time, and instead require high performance computing clusters with graphics processing units. Indeed, specialized hardware for deep learning is an area of development for several companies and research laboratories, while other groups work on algorithms and data structures that may yield more efficient run times for processing data[101–104]. Fortunately, the excellent predictive performance of deep learning models in various tasks suggests that progress will continue toward these current and future challenges from which vaccine research and development can benefit.
Concluding Remarks
Cutting-edge technological advancements in immunology and data science provide an opportunity for applying new analytical approaches to the development of vaccines, but leave a number of outstanding questions. Specialized domains of structure prediction, immune repertoire analysis, and phylogenetics are current areas of vaccine research on which deep learning is poised to have impact. We anticipate that insights from the application of deep learning on these tasks will offer opportunities to refine the enormous space of molecular possibilities in basic, translational, and clinical trials testing promising vaccine candidates; this can reduce research time and contribute to the development of highly efficacious vaccines for some of the most challenging viruses confronting human populations.
Figure 4. Immune repertoire and deep learning model analysis.
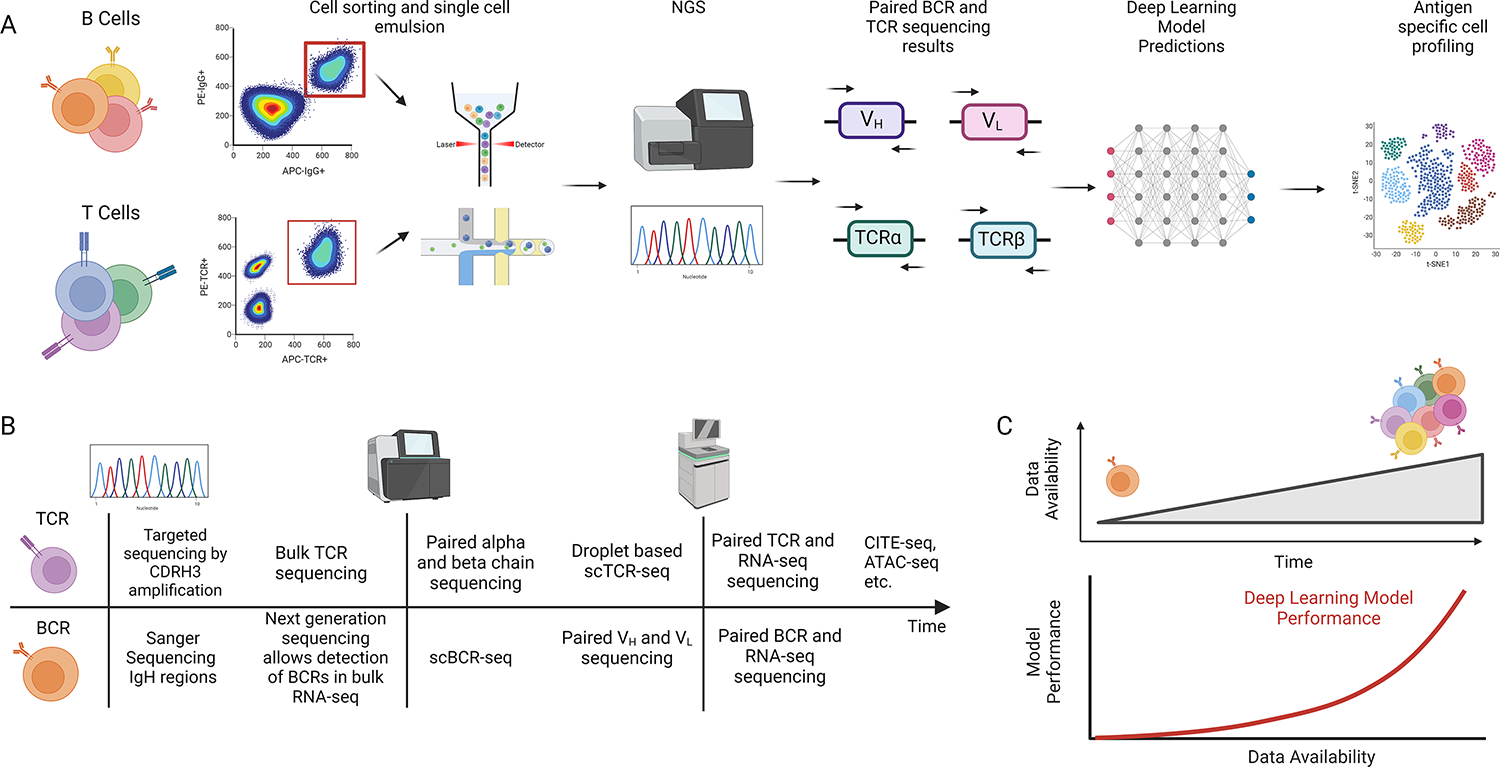
A. Overview of experimental workflow for B and T cell sequencing experiments. After cells are sorted and analyzed on a sequencer, deep learning models can make predictions on various aspects of the immune repertoire. B. Simplified schematic of technology development for sequencing B cells and T cells. C. Over time, sequencing data has continued to accrue with a corresponding growth in deep learning models with improved performance. This figure was created using BioRender (https://biorender.com/)
Acknowledgments
This work was supported in part by NIAID R56AI165448, U19AI145825, and P01AI120756.
Glossary
- Affinity maturation
the process by which antibodies are selected over iterative rounds within germinal centers to select for clones with the greatest binding, which has served as a model for in vitro strategies of protein engineering.
- AlphaFold
protein structure prediction model developed by DeepMind that has solved the protein folding problem by Critical Assessment of protein Structure Prediction (CASP) metrics.
- BCR
B cell receptor, a transmembrane receptor on the surface of B cells that binds antigens
- CASP
Critical Assessment of Structural Predictions, a biannual benchmarking study of novel approaches to predict protein structures that have been solved by experimental methods but not yet deposited in the Protein Data Bank
- Deep learning
a subset of machine learning in which models are built from extracting simpler components into more complicated ones forming many layers that allowing the model to make accurate predictions
- Deep mutational scanning
a set of experimental methods in which a protein or pathogen of interest is diversified in amino acid sequence and then screened in high-throughput fashion to define sequence-function relationships
- Directed evolution
an experimental method in protein engineering that uses sequence variation and selective pressure to iteratively screen and identify variants of a desired phenotype
- Epitope
surface area on an antigen that is recognized by another protein
- Graph-based neural networks
a class of deep learning models that are used when data can be represented by nodes and edges within a graph structure
- Immunogen
an antigen capable of generating an immunological response
- Immune repertoire
the antibodies, BCRs, and TCRs that compose the adaptive immune response observed in an individual
- Layer
a unit of a deep learning model composed of nodes that takes input data, transforms the data with model weights, and applies an activation function to the data
- Neural network
a deep learning model representation that aims to mimic the biological learning process in the brain
- Next generation sequencing
extremely high throughput methods of sequencing that rely on highly parallel processing to determine expression levels and genetic variation in RNA or DNA
- Node
a unit in a deep learning model that is comprised of input connections, weights, and an activation function. Multiple nodes at the same depth comprise a layer
- Machine learning
a set of methods that enable the detection of patterns in data.
- Neutralizing antibody problem
the distinction between antibodies that bind to a given pathogen and those that provide broad anti-pathogen activity
- Next-generation vaccinology
new approaches to vaccine design and research that move beyond empirical approaches
- Original antigenic sin
described more often now as antigenic imprinting, the experimentally supported theory that initial adaptive immune responses to antigen influence characteristics of subsequent exposures to antigenic variants
- Paratope
the part of an antibody that recognizes the respective antigen
- Point estimates
a statistical method that infers information about a population by using a sample statistic
- Predictive model
a model that uses previous data to make a forecast related to unseen data
- Protein folding problem
as initially described, the lack of clarity as to fundamental forces at play in supporting rapid protein folding; as later described, considered the greatest challenge in bioinformatics, the task of computationally predicting the structure of a protein from amino acid sequence alone with the same accuracy as experimental methods
- Reverse vaccinology
a vaccine development approach that uses bioinformatics to systematically identify, prioritize, and then experimentally evaluate the suitability of proteins in the genome of a pathogen as vaccine immunogens.
- Structure-based vaccine design
a vaccine development approach that leverages structural biology in the selection or engineering of candidate immunogens
- TCR
T cell receptor, a T cell surface receptor that recognizes peptides presented by MHC molecules
- Test data
the portion of the data set not previously seen by the model that is used to measure performance after training
- Training data
the portion of the data set a model uses to learn to make predictions for a given task
References
- 1.Silver D et al. (2017) Mastering the game of Go without human knowledge. Nature 550 (7676), 354–359. [DOI] [PubMed] [Google Scholar]
- 2.Schrittwieser J et al. (2020) Mastering Atari, Go, chess and shogi by planning with a learned model. Nature 588 (7839), 604–609. [DOI] [PubMed] [Google Scholar]
- 3.Silver D et al. (2018) A general reinforcement learning algorithm that masters chess, shogi, and Go through self-play. Science 362 (6419), 1140–1144. [DOI] [PubMed] [Google Scholar]
- 4.Davies A et al. (2021) Advancing mathematics by guiding human intuition with AI. Nature 600 (7887), 70–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schmidt B and Hildebrandt A (2021) Deep learning in next-generation sequencing. Drug Discov Today 26 (1), 173–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alharbi WS and Rashid M (2022) A review of deep learning applications in human genomics using next-generation sequencing data. Human Genomics 16 (1), 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elmarakeby HA et al. (2021) Biologically informed deep neural network for prostate cancer discovery. Nature 598 (7880), 348–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu MY et al. (2021) AI-based pathology predicts origins for cancers of unknown primary. Nature 594 (7861), 106–110. [DOI] [PubMed] [Google Scholar]
- 9.McKinney SM et al. (2020) International evaluation of an AI system for breast cancer screening. Nature 577 (7788), 89–94. [DOI] [PubMed] [Google Scholar]
- 10.Jumper J et al. (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596 (7873), 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Senior AW et al. (2020) Improved protein structure prediction using potentials from deep learning. Nature 577 (7792), 706–710. [DOI] [PubMed] [Google Scholar]
- 12.Wilman W et al. (2022) Machine-designed biotherapeutics: opportunities, feasibility and advantages of deep learning in computational antibody discovery. Brief Bioinform 23 (4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bowen JE et al. (2022) SARS-CoV-2 spike conformation determines plasma neutralizing activity elicited by a wide panel of human vaccines. Sci Immunol 7 (78), eadf1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burton DR et al. (2004) HIV vaccine design and the neutralizing antibody problem. Nat Immunol 5 (3), 233–6. [DOI] [PubMed] [Google Scholar]
- 15.Seib KL et al. (2012) Developing vaccines in the era of genomics: a decade of reverse vaccinology. Clin Microbiol Infect 18 Suppl 5, 109–16. [DOI] [PubMed] [Google Scholar]
- 16.Baek M et al. (2021) Accurate prediction of protein structures and interactions using a three-track neural network. Science 373 (6557), 871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bileschi ML et al. (2022) Using deep learning to annotate the protein universe. Nature Biotechnology 40 (6), 932–937. [DOI] [PubMed] [Google Scholar]
- 18.AlQuraishi M (2019) End-to-End Differentiable Learning of Protein Structure. Cell Systems 8 (4), 292–301.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moult J et al. (1995) A large-scale experiment to assess protein structure prediction methods. Proteins 23 (3), ii–v. [DOI] [PubMed] [Google Scholar]
- 20.Graham BS et al. (2019) Structure-Based Vaccine Antigen Design. Annual Review of Medicine 70 (1), 91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crank MC et al. (2019) A proof of concept for structure-based vaccine design targeting RSV in humans. Science 365 (6452), 505–509. [DOI] [PubMed] [Google Scholar]
- 22.Ngwuta JO et al. (2015) Prefusion F-specific antibodies determine the magnitude of RSV neutralizing activity in human sera. Sci Transl Med 7 (309), 309ra162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Falsey AR et al. (2023) Efficacy and Safety of an Ad26.RSV.preF-RSV preF Protein Vaccine in Older Adults. N Engl J Med 388 (7), 609–620. [DOI] [PubMed] [Google Scholar]
- 24.Papi A et al. (2023) Respiratory Syncytial Virus Prefusion F Protein Vaccine in Older Adults. N Engl J Med 388 (7), 595–608. [DOI] [PubMed] [Google Scholar]
- 25.Schmoele-Thoma B et al. (2022) Vaccine Efficacy in Adults in a Respiratory Syncytial Virus Challenge Study. N Engl J Med 386 (25), 2377–2386. [DOI] [PubMed] [Google Scholar]
- 26.Hsieh CL et al. (2020) Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 369 (6510), 1501–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sanders RW et al. (2013) A next-generation cleaved, soluble HIV-1 Env trimer, BG505 SOSIP.664 gp140, expresses multiple epitopes for broadly neutralizing but not non-neutralizing antibodies. PLoS Pathog 9 (9), e1003618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baden LR et al. (2021) Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N Engl J Med 384 (5), 403–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Polack FP et al. (2020) Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N Engl J Med 383 (27), 2603–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Graham BS et al. (2019) Structure-Based Vaccine Antigen Design. Annu Rev Med 70, 91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cao L et al. (2022) Design of protein-binding proteins from the target structure alone. Nature 605 (7910), 551–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chidyausiku TM et al. (2022) De novo design of immunoglobulin-like domains. Nat Commun 13 (1), 5661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Quijano-Rubio A et al. (2021) De novo design of modular and tunable protein biosensors. Nature 591 (7850), 482–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walls AC et al. (2020) Elicitation of Potent Neutralizing Antibody Responses by Designed Protein Nanoparticle Vaccines for SARS-CoV-2. Cell 183 (5), 1367–1382.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cohen AA et al. (2021) Mosaic nanoparticles elicit cross-reactive immune responses to zoonotic coronaviruses in mice. Science 371 (6530), 735–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cohen AA et al. (2022) Mosaic RBD nanoparticles protect against challenge by diverse sarbecoviruses in animal models. Science 377 (6606), eabq0839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kanekiyo M et al. (2019) Mosaic nanoparticle display of diverse influenza virus hemagglutinins elicits broad B cell responses. Nat Immunol 20 (3), 362–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.King NP et al. (2014) Accurate design of co-assembling multi-component protein nanomaterials. Nature 510 (7503), 103–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Collatz M et al. (2020) EpiDope: a deep neural network for linear B-cell epitope prediction. Bioinformatics 37 (4), 448–455. [DOI] [PubMed] [Google Scholar]
- 40.Liu T et al. (2020) Deep learning methods improve linear B-cell epitope prediction. BioData Mining 13 (1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saha S and Raghava GPS (2006) Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins: Structure, Function, and Bioinformatics 65 (1), 40–48. [DOI] [PubMed] [Google Scholar]
- 42.Sher G et al. (2017) DRREP: deep ridge regressed epitope predictor. BMC Genomics 18 (Suppl 6), 676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vita R et al. (2019) The Immune Epitope Database (IEDB): 2018 update. Nucleic Acids Res 47 (D1), D339–D343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiménez J et al. (2017) DeepSite: protein-binding site predictor using 3D-convolutional neural networks. Bioinformatics 33 (19), 3036–3042. [DOI] [PubMed] [Google Scholar]
- 45.Pittala S and Bailey-Kellogg C (2020) Learning context-aware structural representations to predict antigen and antibody binding interfaces. Bioinformatics 36 (13), 3996–4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Olsen TH et al. (2022) Observed Antibody Space: A diverse database of cleaned, annotated, and translated unpaired and paired antibody sequences. Protein Sci 31 (1), 141–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sidhom J-W et al. (2022) Deep learning reveals predictive sequence concepts within immune repertoires to immunotherapy. Science Advances 8 (37), eabq5089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen Y et al. (2022) A Deep Learning Model for Accurate Diagnosis of Infection Using Antibody Repertoires. The Journal of Immunology 208 (12), 2675–2685. [DOI] [PubMed] [Google Scholar]
- 49.Schultheiß C et al. (2020) Next-Generation Sequencing of T and B Cell Receptor Repertoires from COVID-19 Patients Showed Signatures Associated with Severity of Disease. Immunity 53 (2), 442–455.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Briney B et al. (2019) Commonality despite exceptional diversity in the baseline human antibody repertoire. Nature 566 (7744), 393–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Soto C et al. (2019) High frequency of shared clonotypes in human B cell receptor repertoires. Nature 566 (7744), 398–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bashford-Rogers RJM et al. (2019) Analysis of the B cell receptor repertoire in six immune-mediated diseases. Nature 574 (7776), 122–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jardine J et al. (2013) Rational HIV immunogen design to target specific germline B cell receptors. Science 340 (6133), 711–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jardine JG et al. (2016) HIV-1 broadly neutralizing antibody precursor B cells revealed by germline-targeting immunogen. Science 351 (6280), 1458–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Havenar-Daughton C et al. (2018) The human naive B cell repertoire contains distinct subclasses for a germline-targeting HIV-1 vaccine immunogen. Sci Transl Med 10 (448). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leggat DJ et al. (2022) Vaccination induces HIV broadly neutralizing antibody precursors in humans. Science 378 (6623), eadd6502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Doria-Rose NA et al. (2014) Developmental pathway for potent V1V2-directed HIV-neutralizing antibodies. Nature 509 (7498), 55–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liao HX et al. (2013) Co-evolution of a broadly neutralizing HIV-1 antibody and founder virus. Nature 496 (7446), 469–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ju B et al. (2020) Human neutralizing antibodies elicited by SARS-CoV-2 infection. Nature 584 (7819), 115–119. [DOI] [PubMed] [Google Scholar]
- 60.Yang X et al. (2021) Large-scale analysis of 2,152 Ig-seq datasets reveals key features of B cell biology and the antibody repertoire. Cell Reports 35 (6), 109110. [DOI] [PubMed] [Google Scholar]
- 61.Isacchini G et al. (2021) Deep generative selection models of T and B cell receptor repertoires with soNNia. Proceedings of the National Academy of Sciences 118 (14), e2023141118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Richardson E et al. (2021) A computational method for immune repertoire mining that identifies novel binders from different clonotypes, demonstrated by identifying anti-pertussis toxoid antibodies. MAbs 13 (1), 1869406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schneider C et al. (2022) DLAB: deep learning methods for structure-based virtual screening of antibodies. Bioinformatics 38 (2), 377–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Akbar R et al. (2021) A compact vocabulary of paratope-epitope interactions enables predictability of antibody-antigen binding. Cell Rep 34 (11), 108856. [DOI] [PubMed] [Google Scholar]
- 65.Schiepers A et al. (2023) Molecular fate-mapping of serum antibody responses to repeat immunization. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Francis TJ (1960) On the Doctrine of Original Antigenic Sin. Proceedings of the American Philosophical Society 104 (6), 572–578. [Google Scholar]
- 67.Dash P et al. (2017) Quantifiable predictive features define epitope-specific T cell receptor repertoires. Nature 547 (7661), 89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Glanville J et al. (2017) Identifying specificity groups in the T cell receptor repertoire. Nature 547 (7661), 94–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sidhom J-W et al. (2021) DeepTCR is a deep learning framework for revealing sequence concepts within T-cell repertoires. Nature Communications 12 (1), 1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Musvosvi M et al. (2023) T cell receptor repertoires associated with control and disease progression following Mycobacterium tuberculosis infection. Nat Med 29 (1), 258–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Huang H et al. (2020) Analyzing the Mycobacterium tuberculosis immune response by T-cell receptor clustering with GLIPH2 and genome-wide antigen screening. Nature Biotechnology 38 (10), 1194–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liu G et al. (2020) Antibody complementarity determining region design using high-capacity machine learning. Bioinformatics 36 (7), 2126–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Saka K et al. (2021) Antibody design using LSTM based deep generative model from phage display library for affinity maturation. Sci Rep 11 (1), 5852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lim YW et al. (2022) Predicting antibody binders and generating synthetic antibodies using deep learning. MAbs 14 (1), 2069075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Raybould MIJ et al. (2021) Public Baseline and shared response structures support the theory of antibody repertoire functional commonality. PLoS Comput Biol 17 (3), e1008781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nicholson G et al. (2022) Improving local prevalence estimates of SARS-CoV-2 infections using a causal debiasing framework. Nat Microbiol 7 (1), 97–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nikiforuk AM et al. (2022) Simple approximation of sample size for precise estimates of SARS-CoV-2 infection from point-seroprevalence studies. Public Health 212, 7–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Irons NJ and Raftery AE (2021) Estimating SARS-CoV-2 infections from deaths, confirmed cases, tests, and random surveys. Proc Natl Acad Sci U S A 118 (31). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fioravanti D et al. (2018) Phylogenetic convolutional neural networks in metagenomics. BMC Bioinformatics 19 (2), 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gaiha GD et al. (2019) Structural topology defines protective CD8(+) T cell epitopes in the HIV proteome. Science 364 (6439), 480–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Obermeyer F et al. (2022) Analysis of 6.4 million SARS-CoV-2 genomes identifies mutations associated with fitness. Science 376 (6599), 1327–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Greaney AJ et al. (2021) Comprehensive mapping of mutations in the SARS-CoV-2 receptor-binding domain that affect recognition by polyclonal human plasma antibodies. Cell Host & Microbe 29 (3), 463–476.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Greaney AJ et al. (2021) Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition. Cell Host & Microbe 29 (1), 44–57.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Starr TN et al. (2020) Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding. Cell 182 (5), 1295–1310.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yu TC et al. (2022) A biophysical model of viral escape from polyclonal antibodies. Virus Evol 8 (2), veac110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee JM et al. (2018) Deep mutational scanning of hemagglutinin helps predict evolutionary fates of human H3N2 influenza variants. Proc Natl Acad Sci U S A 115 (35), E8276–E8285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zahradnik J et al. (2021) SARS-CoV-2 variant prediction and antiviral drug design are enabled by RBD in vitro evolution. Nat Microbiol 6 (9), 1188–1198. [DOI] [PubMed] [Google Scholar]
- 88.Thadani NN et al. (2022) Learning from pre-pandemic data to forecast viral antibody escape. bioRxiv. [Google Scholar]
- 89.Voznica J et al. (2022) Deep learning from phylogenies to uncover the epidemiological dynamics of outbreaks. Nature Communications 13 (1), 3896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Taft JM et al. (2022) Deep mutational learning predicts ACE2 binding and antibody escape to combinatorial mutations in the SARS-CoV-2 receptor-binding domain. Cell 185 (21), 4008–4022 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hie B et al. (2021) Learning the language of viral evolution and escape. Science 371 (6526), 284–288. [DOI] [PubMed] [Google Scholar]
- 92.Metcalf CJ et al. (2016) Use of serological surveys to generate key insights into the changing global landscape of infectious disease. Lancet 388 (10045), 728–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Carroll D et al. (2018) The Global Virome Project. Science 359 (6378), 872–874. [DOI] [PubMed] [Google Scholar]
- 94.Xu GJ et al. (2015) Viral immunology. Comprehensive serological profiling of human populations using a synthetic human virome. Science 348 (6239), aaa0698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kula T et al. (2019) T-Scan: A Genome-wide Method for the Systematic Discovery of T Cell Epitopes. Cell 178 (4), 1016–1028 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.DeGrace MM et al. (2022) Defining the risk of SARS-CoV-2 variants on immune protection. Nature 605 (7911), 640–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Merler M et al. (2019) Diversity in Faces. arXiv arXiv:1901.10436. [Google Scholar]
- 98.Figueroa Barraza J et al. (2021) Towards Interpretable Deep Learning: A Feature Selection Framework for Prognostics and Health Management Using Deep Neural Networks. Sensors (Basel) 21 (17). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li X et al. (2022) Interpretable Deep Learning: Interpretation, Interpretability, Trustworthiness, and Beyond. arXiv arXiv: 2103.10689. [Google Scholar]
- 100.Wang H et al. (2023) Interpretable Deep Learning Methods for Multiview Learning. arXiv arXiv:2302.07930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Wu H et al. (2022) Efficient Neural Network Analysis with Sum-of-Infeasibilities. arXiv arXiv:2203.11201. [Google Scholar]
- 102.Gholami A et al. (2021) A Survey of Quantization Methods for Efficient Neural Network Inference. arXiv arXiv:2103.13630. [Google Scholar]
- 103.Zhou X et al. (2021) Efficient Neural Network Training via Forward and Backward Propagation Sparsification. arXiv arXiv:2111.05685. [Google Scholar]
- 104.Hu H et al. (2016) Network Trimming: A Data-Driven Neuron Pruning Approach towards Efficient Deep Architectures. arXiv arXiv:1607.03250. [Google Scholar]
- 105.McLellan JS et al. (2013) Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science 340 (6136), 1113–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Joyce MG et al. (2016) Iterative structure-based improvement of a fusion-glycoprotein vaccine against RSV. Nat Struct Mol Biol 23 (9), 811–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Krarup A et al. (2015) A highly stable prefusion RSV F vaccine derived from structural analysis of the fusion mechanism. Nat Commun 6, 8143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Stewart-Jones GB et al. (2015) A Cysteine Zipper Stabilizes a Pre-Fusion F Glycoprotein Vaccine for Respiratory Syncytial Virus. PLoS One 10 (6), e0128779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Correia BE et al. (2014) Proof of principle for epitope-focused vaccine design. Nature 507 (7491), 201–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kirchdoerfer RN et al. (2016) Pre-fusion structure of a human coronavirus spike protein. Nature 531 (7592), 118–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Walls AC et al. (2016) Cryo-electron microscopy structure of a coronavirus spike glycoprotein trimer. Nature 531 (7592), 114–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pallesen J et al. (2017) Immunogenicity and structures of a rationally designed prefusion MERS-CoV spike antigen. Proc Natl Acad Sci U S A 114 (35), E7348–E7357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yuan Y et al. (2017) Cryo-EM structures of MERS-CoV and SARS-CoV spike glycoproteins reveal the dynamic receptor binding domains. Nat Commun 8, 15092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Binley JM et al. (2000) A recombinant human immunodeficiency virus type 1 envelope glycoprotein complex stabilized by an intermolecular disulfide bond between the gp120 and gp41 subunits is an antigenic mimic of the trimeric virion-associated structure. J Virol 74 (2), 627–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Klasse PJ et al. (2013) Influences on trimerization and aggregation of soluble, cleaved HIV-1 SOSIP envelope glycoprotein. J Virol 87 (17), 9873–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sanders RW et al. (2002) Stabilization of the soluble, cleaved, trimeric form of the envelope glycoprotein complex of human immunodeficiency virus type 1. J Virol 76 (17), 8875–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Julien JP et al. (2013) Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science 342 (6165), 1477–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lyumkis D et al. (2013) Cryo-EM structure of a fully glycosylated soluble cleaved HIV-1 envelope trimer. Science 342 (6165), 1484–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jardine JG et al. (2015) HIV-1 VACCINES. Priming a broadly neutralizing antibody response to HIV-1 using a germline-targeting immunogen. Science 349 (6244), 156–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Wu X et al. (2010) Rational design of envelope identifies broadly neutralizing human monoclonal antibodies to HIV-1. Science 329 (5993), 856–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Burkhard P and Lanar DE (2015) Malaria vaccine based on self-assembling protein nanoparticles. Expert Rev Vaccines 14 (12), 1525–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.King NP et al. (2012) Computational design of self-assembling protein nanomaterials with atomic level accuracy. Science 336 (6085), 1171–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yassine HM et al. (2015) Hemagglutinin-stem nanoparticles generate heterosubtypic influenza protection. Nat Med 21 (9), 1065–70. [DOI] [PubMed] [Google Scholar]
- 124.Senior AW et al. (2020) Improved protein structure prediction using potentials from deep learning. Nature 577 (7792), 706–710. [DOI] [PubMed] [Google Scholar]
- 125.Jumper J et al. (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596 (7873), 583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Baek M et al. (2021) Accurate prediction of protein structures and interactions using a three-track neural network. Science 373 (6557), 871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Dauparas J et al. (2022) Robust deep learning-based protein sequence design using ProteinMPNN. Science 378 (6615), 49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Anishchenko I et al. (2021) De novo protein design by deep network hallucination. Nature 600 (7889), 547–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kallberg M et al. (2012) Template-based protein structure modeling using the RaptorX web server. Nat Protoc 7 (8), 1511–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Madani A et al. (2023) Large language models generate functional protein sequences across diverse families. Nat Biotechnol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chowdhury R et al. (2022) Single-sequence protein structure prediction using a language model and deep learning. Nat Biotechnol 40 (11), 1617–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hsu C et al. (2022) Learning protein fitness models from evolutionary and assay-labeled data. Nat Biotechnol 40 (7), 1114–1122. [DOI] [PubMed] [Google Scholar]
- 133.Ichikawa DM et al. (2023) A universal deep-learning model for zinc finger design enables transcription factor reprogramming. Nat Biotechnol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Quijano-Rubio A et al. (2022) A split, conditionally active mimetic of IL-2 reduces the toxicity of systemic cytokine therapy. Nat Biotechnol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Bryant DH et al. (2021) Deep diversification of an AAV capsid protein by machine learning. Nat Biotechnol 39 (6), 691–696. [DOI] [PubMed] [Google Scholar]
- 136.Giordano-Attianese G et al. (2020) A computationally designed chimeric antigen receptor provides a small-molecule safety switch for T-cell therapy. Nat Biotechnol 38 (4), 426–432. [DOI] [PubMed] [Google Scholar]
- 137.Bhardwaj G et al. (2022) Accurate de novo design of membrane-traversing macrocycles. Cell 185 (19), 3520–3532 e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Courbet A et al. (2022) Computational design of mechanically coupled axle-rotor protein assemblies. Science 376 (6591), 383–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Divine R et al. (2021) Designed proteins assemble antibodies into modular nanocages. Science 372 (6537). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Bale JB et al. (2016) Accurate design of megadalton-scale two-component icosahedral protein complexes. Science 353 (6297), 389–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Doyle L et al. (2015) Rational design of alpha-helical tandem repeat proteins with closed architectures. Nature 528 (7583), 585–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lewis SM et al. (2014) Generation of bispecific IgG antibodies by structure-based design of an orthogonal Fab interface. Nat Biotechnol 32 (2), 191–8. [DOI] [PubMed] [Google Scholar]
- 143.Mravic M et al. (2019) Packing of apolar side chains enables accurate design of highly stable membrane proteins. Science 363 (6434), 1418–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Dou J et al. (2018) De novo design of a fluorescence-activating beta-barrel. Nature 561 (7724), 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]