

Abstract
Age and diabetes have long been known to induce an oxidative reaction between glucose and collagen, leading to the accumulation of advanced glycation end-products (AGEs) cross-links in collagenous tissues. More recently, AGEs content has been related to loss of bone quality, independent of bone mass, and increased fracture risk with aging and diabetes. Loss of bone quality is mostly attributed to changes in material properties, structural organization, or cellular remodeling. Though all these factors play a role in bone fragility disease, some common recurring patterns can be found between diabetic and age-related bone fragility. The main pattern we will discuss in this viewpoint is the increase of fibrillar collagen stiffness and loss of collagen-induced plasticity with AGE accumulation. This study focused on recent related experimental studies and discusses the correlation between fluorescent AGEs content at the molecular and fibrillar scales, collagen deformation mechanisms at the nanoscale, and resistance to bone fracture at the macroscale.
Keywords: Bone fragility, Advanced glycation end-products, Collagen, Diabetes, Aging
Introduction
Age and type 2 diabetes mellitus (T2DM) are both associated with a drastic increase in fracture risk, in the range of 20% to 900%, independent of bone mass (Hui et al., 1988; Marshall et al., 1996; Bonds et al., 2006; Janghorbani et al., 2007; Vestergaard, 2007; Karim and Bouxsein, 2016; Liang and Chikritzhs, 2016). T2DM disproportionately affects the elderly population (those over 65 years) compared to other adults worldwide, and can lead to increased fragility of bones in older individuals (on U.S. Department of Health and Human Services, 2012; Kirkman et al., 2012; Bradley and Hsueh, 2016; Looker et al., 2016). With the global epidemic of diabetes and the aging of the population, understanding the mechanisms underlying diabetic and age-related bone fragility is urgent.
Since the early 1970s, collagen structural degradation and collagen cross-linking with age and diabetes have been amply documented in different tissues such as skin, joints, arteries, bone, or tendons (Hamlin et al., 1975; Piez, 1968; Traub and Piez, 1971; Robins and Bailey, 1972; Cannon and Davison, 1973; Barnes et al., 1974; Kivirikko, 1974; Tanzer, 1976; Cannon and Davison, 1977; Cerami et al., 1979; Madia et al., 1979). These studies have presented robust evidence that collagen (the most abundant protein in the human body) is modified by non-enzymatic glycation (Maillard reaction) between glucose and proteins and results in advanced glycation end-products (AGEs) cross-links formation. Non-enzymatic AGE cross-links within and in-between collagen molecules are often measured as bulk and comprised of pentosidine, carboxymethyllysine (CML), carboxyethyllysine (CEL), crossline, and vesperlysines (A, B, and C). Due to the long half-life of collagen, AGEs can accumulate with age. This process is quite similar but accelerated in T2DM, explaining why diabetes is often presented as a “fast aging” process (Hamlin et al., 1975; Monnier and Cerami, 1981; Monnier et al., 1984). Thirty years later, studies on bone fragility started to emerge and implicate the accumulation of AGEs in the pathogenesis of diabetes and loss of bone quality due to aging (Catanese et al., 1999; Vashishth et al., 2001; Tang et al., 2007; Vashishth, 2007; Saito and Marumo, 2010; Tang and Vashishth, 2011; Yamagishi, 2011; Zimmermann et al., 2011; Garnero, 2012). This new discovery shifted the research on bone fragility by suggesting new factors affecting bone quality at the collagen level, and new potential therapeutic targets to inhibit AGEs formation (such as blocking the receptor for AGEs [RAGE], for instance). Indeed, loss of bone mass and mineral content could not fully explain osteoporotic fracture with age (Schuit et al., 2004) or address diabetic bone fragility where bone mass is mostly unchanged. In this viewpoint, we discuss here how AGEs accumulation affects bone resistance to fracture (i.e., toughness) and disrupt nanoscale mechanisms of collagen deformation and energy dissipation.
Multiscale Origins of Bone Fragility
The ability of cortical bone to resist fracture originates from its multiscale hierarchical organization (Fig. 1A). Building blocks in bone are organized from the molecular and nanoscopic levels to the macroscopic level. At the nanoscale level, basic building blocks are formed by mineralized collagen fibrils comprising collagen molecules, hydroxyapatite crystals, and water. Mineralized fibrils are aligned, stacked in fibers, and arranged in lamellae, where each lamella layer has a different fiber orientation than its neighbor. At the microscale, concentric lamellae form tube-like structures called osteons, which protect central canals where blood vessels and nerves are running (Fig. 1A). Bone quality, used to characterize bone resistance independently of bone mass, is related to (1) material properties, (2) structure, and (3) remodeling. These three factors of bone quality are impaired with age and T2DM.
FIGURE 1.
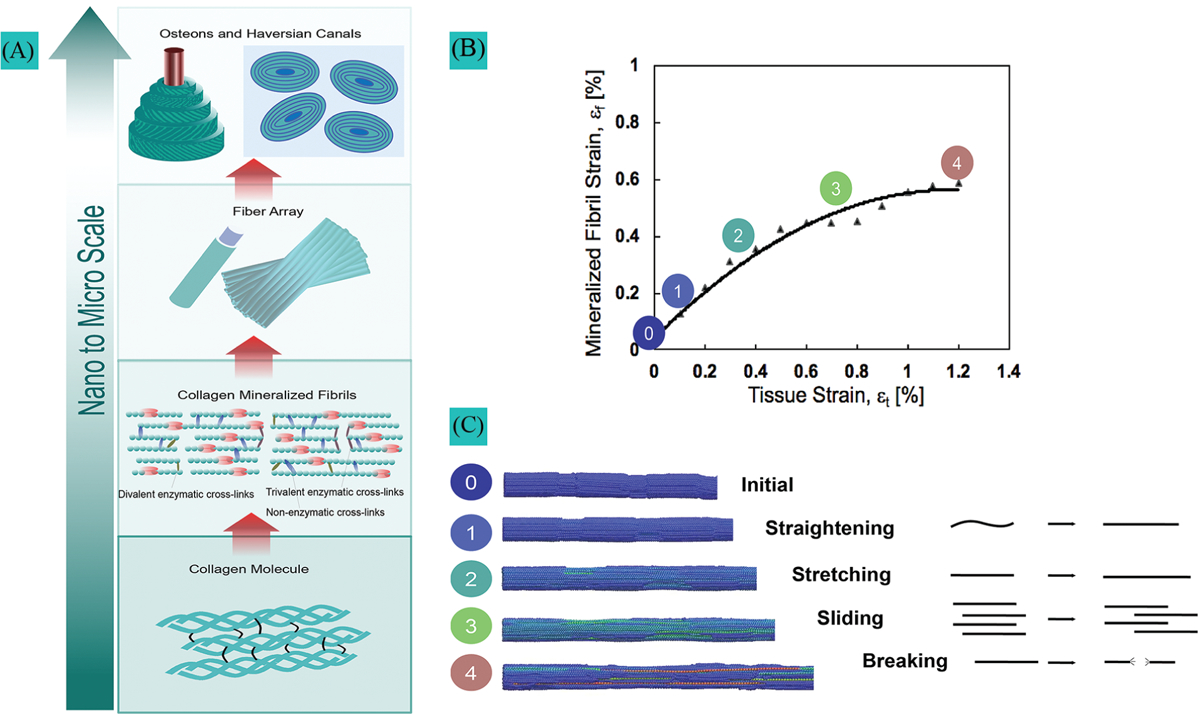
Hierarchical organization in bone and mechanisms of collagen fibril deformation. (A) Hierarchical bone structure with cross-links in the molecular and fibrillar structure of collagen. (B) Collagen fibril deformation measured by small-angle X-ray scattering during rat ulna tensile testing. (C) Collagen loading stages from the initial state to complete failure.
Bone material properties
These properties (e.g., tissue modulus, yield stress/strain, ultimate stress/strain, strain to failure, work-to-fracture, and toughness) are often measured using strength and toughness tests. Strength tests are designed to measure the maximum load a material can withstand before breaking or deforming permanently. Toughness tests, on the other hand, measure the ability of a material to absorb energy before breaking (Ritchie, 2011). Material properties are conferred at the nanoscale by collagen fibrils (100 nm diameter) reinforced by mineral crystals; elastic and pre-yield properties are attributed to the stiffness of the mineral, whereas the plastic, post-yield properties, and intrinsic toughness are attributed to the ability of collagen to deform plastically and prevent crack initiation and growth (Burstein et al., 1975; Nalla et al., 2004; Currey et al., 2007; Launey et al., 2010). Most studies have shown that aging does not significantly impair the elastic modulus of bone (Nyman et al., 2007; Koester et al., 2011; Zimmermann et al., 2011). However, yield strength and ultimate strength are decreased, respectively, by approximately 1% and 2% per decade in human cortical bone for adults older than 30 years of age (Zimmermann et al., 2011; Morgan et al., 2018). Even more significantly, toughness, energy dissipation, and ultimate strain are reduced by approximately 10%–15% every decade (Burstein et al., 1976; Nalla et al., 2004; Nyman et al., 2007; Koester et al., 2011; Zimmermann et al., 2011; Morgan et al., 2018). Studies on material properties in diabetic human bones are not as abundant as those in aging bones, but animal studies show evidence of a significant decrease in post-yield properties, including a reduction of 10%–15% in ultimate strength and 30%–40% in ultimate strain and toughness (Campbell et al., 2016; Acevedo et al., 2018a). Based on comparative models (Nyman et al., 2007; Acevedo et al., 2018a), the reduced post-yield properties and toughness in aging and diabetic bones seem to be mostly attributable to deficits in material properties (conferred at the collagen level) rather than structural deficits.
Bone structure
The structure of bones (microarchitecture and geometry) is also known to influence resistance to fracture and energy dissipation during crack growth (i.e., extrinsic toughening mechanisms). Extrinsic toughening mechanisms act at the microscale level to reduce the driving force that propagates the crack. These mechanisms include crack deflection/ twisting and crack bridging (Ager et al., 2006; Peterlik et al., 2006; Ritchie et al., 2006). When the crack propagates transversely to the osteons, crack deflection occurs around the hypermineralized osteonal boundaries (i.e., cement lines) to dissipate energy (Martin and Burr, 1982; Nalla et al., 2005). Other microstructural features, such as osteocyte lacunae or porosity, can deflect the crack (Currey, 1962). When the crack propagates longitudinally along the osteons (in-between lamellae and fibers), crack bridging leaves uncracked regions to resist fracture along the crack path. Aged bone shows a significant increase in osteon density by 100% to 200% between 30 to 80 years of age and an increase in vascular canal diameters (Busse et al., 2010; Zimmermann et al., 2011; Jast and Jasiuk, 2013), which vastly expand the area of cement lines at which microcracks can form, resulting in both smaller crack bridges and reduced crack deflection (Koester et al., 2011; Zimmermann et al., 2011). Diabetes moderately affects cortical bone geometry, such as a change in moment of inertia due to a different distribution of bone mass (Prisby et al., 2008; Acevedo et al., 2018a). Recent studies (Ay et al., 2020) have found that hyperglycemia increases osteocyte lacunar density and decreases vascular canal diameter. Changes in microstructure are closely tied to cellular remodeling, as the process of cellular remodeling involves the removal and formation of bone tissue, which can alter bone microstructure.
Cellular-regulated remodeling
Bone health is maintained through dynamic remodeling executed by resident cells in this tissue: osteoclasts resorb old bone, followed by osteoblast deposition of bone. The osteocyte, a third bone cell type, directs osteoclast and osteoblast activity and dynamically remodels their local bone matrix in a process called perilacunar remodeling (Qing et al., 2012; Tang et al., 2012). Disruption of remodeling causes an increased risk of fracture due to bone fragility, even with normal bone mass. Age is known to create an imbalance between osteoclasts and osteoblast activity, leading to bone loss, as well as elevated osteoblasts and osteocyte apoptosis. T2DM reduces osteoblast differentiation and osteoblast-induced bone formation while enhancing osteoclast activity and increased bone resorption, which can also lead to bone loss (Lu et al., 2003; Suzuki et al., 2005; Alikhani et al., 2007; Liu et al., 2006; Takizawa et al., 2008). It is also thought that hyperglycemia in T2DM might induce osteocyte apoptosis (Picke et al., 2019). Disruption of osteocyte-mediated remodeling causes distinctive collagen disorganization and a pattern of hypermineralization of the mid-cortical bone matrix (Qing et al., 2012; Tang et al., 2012). In the following discussion, we further explore how the three factors of bone quality are affected by the increase of AGEs with age and diabetes.
Accumulation of Advance Glycation End-Products in Collagen
To understand how AGEs accumulate in collagen, we must first understand how collagen is structured. Collagen is composed of three chains with a repeating sequence of gly-X-Y, where X/Y can be any amino acid but is generally proline or hydroxyproline. During bone development and maturation (before the age of 20 years), enzymatic cross-links are created to stabilize the triple-helix of collagen molecules and connect collagen fibrils. Enzymes, such as lysyl oxidase, prolyl hydroxylase, galactosyltransferase, and matrix metalloproteinase, play a crucial role in regulating the formation and degradation of enzymatic cross-links in bone, which is vital for maintaining optimal bone strength and remodeling. These enzymes work together to balance the formation and breakdown of cross-links, providing stiffness and strength to the bone matrix (Knott and Bailey, 1998). Enzymatic cross-links are first divalent (immature) and then trivalent (mature) cross-links that link collagen molecules head to tail. Increased enzymatic cross-link density and maturity can beneficially increase collagen fibril stiffness and bone stiffness and strength (Oxlund et al., 1995; Bailey et al., 1998; Depalle et al., 2015).
After bone maturation, non-enzymatic cross-links are created by an oxidative reaction between glucose and amino acid residues on proteins (e.g., lysine or hydroxylysine in type I collagen). This reaction, called the Maillard reaction (Sroga et al., 2015), forms cross-links among AGEs along the triple helical region of the molecule. Since AGEs are not regulated by enzymes, they can accumulate (Valcourt et al., 2007), especially in aging tissues with low turnover or in the presence of high blood sugar concentrations (like in T2DM) or oxidative stress. The content of AGEs, specifically the content of the well-studied pentosidine, increases with age and T2DM in cortical and trabecular bone (Tang et al., 2007; Karim et al., 2013). Contrary to enzymatic cross-links, the accumulation of AGEs with aging or diabetic cortical bone is associated with increased mechanical fragility, as will be discussed in the following section (Vashishth et al., 2001; Wang et al., 2002; Garnero et al., 2006; Siegmund et al., 2008; Silva et al., 2009; Saito and Marumo, 2010; Tang and Vashishth, 2010; Ionova-Martin et al., 2011; Snedeker and Gautieri, 2014; Acevedo et al., 2018a).
Advance Glycation End-Products Correlate with Loss of Collagen Deformation and Loss of Bone Toughness
The best explanation for the decrease in plastic strain energy in bone fragility is the impairment of collagen deformation by AGEs accumulation (Nyman et al., 2007; Acevedo et al., 2018a). The current state-of-the-art technique for quantifying collagen deformation at the nanoscale is in situ small-angle x-ray scattering (SAXS). The scattering pattern of the collagen fibrils (d-spacing) can be used to measure fibrillar strain, while digital image correlation of bone samples can be used to measure the tissue strain at the macroscale. Only a few studies (Barth et al., 2011; Zimmermann et al., 2011, 2016; Acevedo et al., 2015, 2018a) have gathered data on bone fluorescent AGEs concentration at the molecular and fibrillar levels, on collagen deformation mechanisms at the nanoscale (using SAXS), and bone fracture resistance at the macroscale. These studies investigated the effect of age, diabetes, long-term alendronate treatment, and irradiation, which can affect the collagen structure and increase AGEs content.
A novel finding of these studies was to identify the different stages of fibril deformation in cortical bone and how aging and T2DM affect these mechanisms. The results indicated that factors such as age, diabetes, band high AGEs content did not influence the fibrillar straightening and stretching (stages 1 and 2, Figs. 1B and 1C) that occurs at lower levels of tissue strain, which typically precedes tissue yielding. However, the process of fibrillar sliding (stage 3, Figs. 1B and 1C), which occurs when fibrils decouple during tissue yielding, was systematically impaired with high AGEs content in these studies (Zimmermann et al., 2011; Acevedo et al., 2015, 2018a). Fibrillar sliding was found to be essential to promote plasticity at the nanoscale as the main intrinsic toughening mechanism to resist crack initiation and growth. SAXS results suggest that age (Zimmermann et al., 2011; Barth et al., 2011; Zimmermann et al., 2016), diabetes (Acevedo et al., 2018a), and exposure to x-ray radiation (Barth et al., 2011) limit the ability of collagen fibrils to deform plastically by fibrillar sliding due to the accumulation of AGEs. Aging and diabetes stiffen the fibrils and inhibit fibrillar sliding (Zimmermann et al., 2011; Acevedo et al., 2018a, 2018b). This phenomenon, called collagen stiffening, prevents the collagen in aged- and T2DM-affected bone from sustaining as much tissue deformation without brittle failure. The deficit in collagen ductility must be compensated at higher length scales (micro-scale) via energy dissipation through extrinsic toughening mechanisms such as crack deflection and microdamage accumulation. Evidence revealed that age, diabetes, and the AGEs content correlate with greater microdamage and microcrack accumulation (Schaffler et al., 1995; Diab et al., 2005, 2006; Diab and Vashishth, 2007; Mohsin et al., 2019; Liu et al., 2020).
Cumulatively, these studies show that the increase of AGEs (compared with healthy controls (Barth et al., 2011; Acevedo et al., 2015; Zimmermann et al., 2016; Acevedo et al., 2018a)) or young (Zimmermann et al., 2011) is highly correlated with fibrillar stiffening (r2 = 0.81, Fig. 2A). Decrease in toughness is moderately correlated with fibrillar stiffening (r2 = 0.51, Fig. 2B) and not very correlated with AGEs content (r2 = 0.26, Fig. 2C). This brings evidence that AGEs accumulation directly leads to reduced collagen fibril deformation (reduced plasticity and resistance to fracture). However, collagen deformation itself is not the only contributor to bone toughness, although it is an important one. Structural deficits and remodeling impairment are also key contributors to bone fragility. AGEs are detrimental because they can also affect cells responsible for remodeling and microstructure, as explained in the following section.
FIGURE 2.
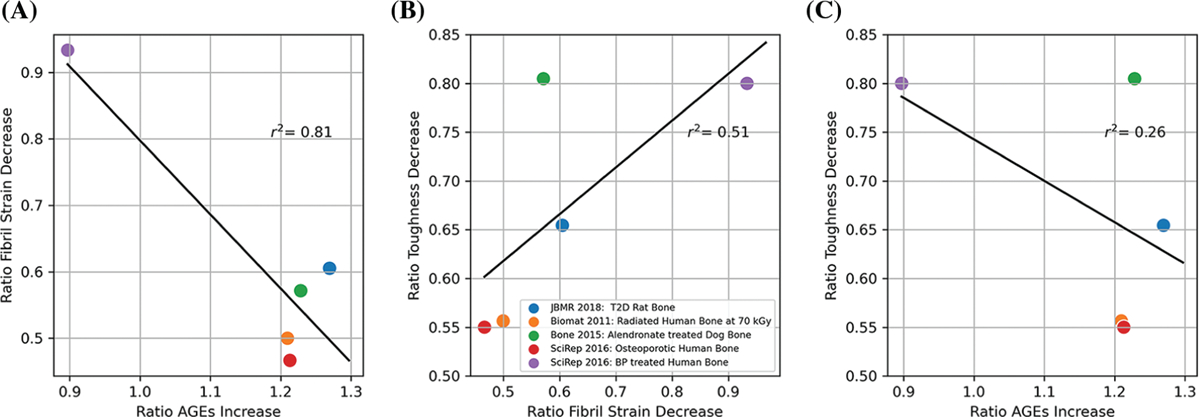
(A) Correlation between change in ultimate fibril strain and advance glycation end-product (AGE) accumulation. (B) Correlation between change in toughness and change in fibril strain. (C) Correlation between the decrease in toughness and AGE accumulation. The ratios were obtained from published values for age (Zimmermann et al., 2011, 2016), diabetes (Acevedo et al., 2018a), bisphosphonate treatment (Acevedo et al., 2015; Zimmermann et al., 2016), and irradiation (Barth et al., 2011).
Other Effects of Advance Glycation End-products Accumulation on Bone Cell Function and Geometry/ Microstructure
Excessive AGE cross-link accumulation could affect the bone cells’ ability to maintain optimal geometry and microarchitecture via unbalanced matrix resorption/formation and favors bone fragility. AGEs can alter osteoclast differentiation and reduce resorption (Valcourt et al., 2007; Ural et al., 2015), reduce the phenotypic expression of osteoblasts (Katayama et al., 1996), impact the osteocyte mechano-sensitivity (Hie et al., 2011; Tanaka et al., 2015; Yang et al., 2021), trigger cell senescence (Almeida and O’Brien, 2013; Teissier et al., 2022), and stimulate secretion of pro-inflammatory and catabolic factors (Takagi et al., 1997). AGEs also affect the collagen surface and cell-matrix interactions, impairing bone repair (Snedeker and Gautieri, 2014). Therefore, AGE accumulation with diabetes and age may impair cortical bone geometry, microstructure, and damage repair leading to a decreased resistance to fracture. More research is needed to investigate whether and how AGEs can directly impact cortical bone geometry and microstructure with age and diabetes.
Discussion and Conclusion
We showed here that the dramatic reduction in cortical bone toughness and bone quality with age and diabetes is primarily attributable to deficits in material properties, specifically in collagen-related plasticity, and secondly to structural deficits and remodeling impairment. Non-enzymatic cross-linking plays a key role in these three aspects of bone quality (material properties, structure, and remodeling). The studies we combined in this viewpoint consistently show a direct relationship between the accumulation of AGEs and the reduction of fibrillar deformation, which is closely associated with the loss of toughness and plasticity. AGEs are also known to affect the cells responsible for bone formation, resorption, and damage repair leading to structural changes. This contribution of AGEs to toughness via remodeling is not well quantified yet. This knowledge would give evidence that AGEs affect bone quality via different mechanisms and thus are key regulators of bone quality.
Studies have reported many limitations that hinder the ability to draw further conclusions. While AGEs are commonly measured in serum, urine, and the skin, currently, no strong correlation between AGEs in bone and these measurable AGEs has been noted (Kida et al., 2019; Waqas et al., 2020). AGEs content in bones varies greatly from one study to another (Karim and Bouxsein, 2016); the content can be pentosidine (via HPLC) or bulk fluorescent AGEs (combining pentosidine, CML, CEL, crossline, and vesper lysines A, B, and C). However, it is not clear which AGE is the most important or whether pentosidine is the right target to study. Indeed, pentosidine has been the most studied AGEs even though it represents less than 1% of fluorescent AGEs in bones (Dyer et al., 1991); it is only weakly correlated to the total amount of fluorescent AGEs in human cortical and cancellous bone (Karim et al., 2013). Glucosepane can be another promising (non-fluorescent) AGEs to investigate in bones because it has been found to be abundant in the skin of human diabetic patients (Monnier et al., 2014). CML seems to have the potential to be an efficient (non-fluorescent) marker of AGEs (Thomas et al., 2018); these (non-crosslinking) AGE adducts might be associated with the loss of enzymatic cross-links and resistance to fracture (Thomas et al., 2018; Willett et al., 2022). In general, more information about the process of AGEs cross-linking (abundance, binding sites, size, and shape) at the molecular level would help understand the mechanical effects of AGEs on the molecular collagen behavior and identify the most detrimental AGEs to target. Measuring AGEs, such as pentosidine or CML, in skin, urine, or serum may be a potential method to predict bone fragility, but further investigation is needed to confirm the efficacy of the technique.
Researchers have been exploring therapeutic agents that can inhibit the formation of AGEs, block the interaction between AGEs and the RAGE, or break down existing AGEs to restore bone resistance to fracture in diabetic or aged patients (Willett et al., 2022). One of the most promising therapies lately involved pyridoxamine (vitamin B6) (Tanimoto et al., 2007; Nagai et al., 2012; Mascolo and Vernì, 2020). Whether or not current anti-resorptive treatments such as bisphosphonate (used to treat elderly and diabetic patients) have a beneficial or detrimental effect on AGEs accumulation is still under debate. In our combined study (Fig. 2C), we observed conflicting results; long-term bisphosphonate in healthy dogs increased AGEs, whereas bisphosphonate treatment in osteoporotic patients decreased AGEs.
In conclusion, further research is necessary to fully understand the molecular mechanisms by which AGEs affect the molecular functions of collagen in aging and diabetic bone fragility. This knowledge will help us identify whether AGEs quantification and removal have a clinical future in fracture prediction and therapies development to restore the biomechanical properties of bones. Improving our understanding of these mechanisms will be crucial for developing effective strategies for improving bone health in diabetic and aged individuals.
Funding Statement:
The authors were supported by the National Institutes of Health under Award Number 1R21AR077881.
Footnotes
Conflicts of Interest: The authors declare that they have no conflicts of interest to report regarding the present study.
References
- Acevedo C, Bale H, Gludovatz B, Wat A, Tang SY et al. (2015). Alendronate treatment alters bone tissues at multiple structural levels in healthy canine cortical bone. Bone 81: 352–363. 10.1016/j.bone.2015.08.002 [DOI] [PubMed] [Google Scholar]
- Acevedo C, Stadelmann VA, Pioletti DP, Alliston T, Ritchie RO (2018a). Fatigue as the missing link between bone fragility and fracture. Nature Biomedical Engineering 2: 62–71. 10.1038/s41551-017-0183-9 [DOI] [PubMed] [Google Scholar]
- Acevedo C, Sylvia M, Schaible E, Graham JL, Stanhope KL et al. (2018b). Contributions of material properties and structure to increased bone fragility for a given bone mass in the UCD-T2DM rat model of type 2 diabetes. Journal of Bone and Mineral Research 33: 1066–1075. 10.1002/jbmr.3393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ager JW, Balooch G, Ritchie R (2006). Fracture, aging, and disease in bone. Journal of Materials Research 21: 1878–1892. 10.1557/jmr.2006.0242 [DOI] [Google Scholar]
- Alikhani M, Alikhani Z, Boyd C, MacLellan CM, Raptis M, Liu R, Pischon N, Trackman PC, Gerstenfeld L, Graves DT (2007). Advanced glycation end products stimulate osteoblast apoptosis via the MAP kinase and cytosolic apoptotic pathways. Bone 40: 345–353. 10.1016/j.bone.2006.09.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida M, O’Brien CA (2013). Basic biology of skeletal aging: Role of stress response pathways. Journals of Gerontology Series A: Biomedical Sciences and Medical Sciences 68: 1197–1208. 10.1093/gerona/glt079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ay B, Parolia K, Liddell RS, Qiu Y, Grasselli G, Cooper DM, Davies JE (2020). Hyper-glycemia compromises rat cortical bone by increasing osteocyte lacunar density and decreasing vascular canal volume. Communications Biology 3: 1–9. 10.1038/s42003-019-0747-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey AJ, Paul RG, Knott L (1998). Mechanisms of maturation and ageing of collagen. Mechanisms of Ageing and Development 106: 1–56. 10.1016/S0047-6374(98)00119-5 [DOI] [PubMed] [Google Scholar]
- Barnes M, Constable B, Morton L, Royce P (1974). Age-related variations in hydroxylation of lysine and proline in collagen. Biochemical Journal 139: 461–468. 10.1042/bj1390461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barth HD, Zimmermann EA, Schaible E, Tang SY, Alliston T, Ritchie RO (2011). Characterization of the effects of X-ray irradiation on the hierarchical structure and mechanical properties of human cortical bone. Biomaterials 32: 8892–8904. 10.1016/j.biomaterials.2011.08.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonds DE, Larson JC, Schwartz AV, Strotmeyer ES, Robbins J, Rodriguez BL, Johnson KC, Margolis KL (2006). Risk of fracture in women with type 2 diabetes: The women’s health initiative observational study. The Journal of Clinical Endocrinology & Metabolism 91: 3404–3410. 10.1210/jc.2006-0614 [DOI] [PubMed] [Google Scholar]
- Bradley D, Hsueh W (2016). Type 2 diabetes in the elderly: Challenges in a unique patient population. Journal of Gerontology & Geriatric Medicine 2: 2–14. 10.23937/2469-5858/1510014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burstein AH, Reilly DT, Martens M (1976). Aging of bone tissue: Mechanical properties. The Journal of Bone and Joint Surgery. American 58: 82–86. 10.2106/00004623-197658010-00015 [DOI] [PubMed] [Google Scholar]
- Burstein AH, Zika J, Heiple K, Klein L (1975). Contribution of collagen and mineral to the elastic-plastic properties of bone. JBJS 57: 956–961. 10.2106/00004623-197557070-00013 [DOI] [PubMed] [Google Scholar]
- Busse B, Hahn M, Schinke T, Püschel K, Duda GN, Amling M (2010). Reorganization of the femoral cortex due to age-, sex-, and endoprosthetic-related effects emphasized by osteonal dimensions and remodeling. Journal of Biomedical Materials Research Part A 92: 1440–1451. [DOI] [PubMed] [Google Scholar]
- Campbell GM, Tiwari S, Picke AK, Hofbauer C, Rauner M, Morlock M, Hofbauer LC, Glüer CC (2016). Effects of insulin therapy on porosity, non-enzymatic glycation and mechanical competence in the bone of rats with type 2 diabetes mellitus. Bone 91: 186–193. 10.1016/j.bone.2016.08.003 [DOI] [PubMed] [Google Scholar]
- Cannon D, Davison P (1973). Cross-linking and aging in rat tendon collagen. Experimental Gerontology 8: 51–62. 10.1016/0531-5565(73)90051-X [DOI] [PubMed] [Google Scholar]
- Cannon DJ, Davison PF (1977). Aging, and crosslinking in mammalian collagen. Experimental Aging Research 3: 87–105. 10.1080/03610737708257091 [DOI] [PubMed] [Google Scholar]
- Catanese J, Bank R, TeKoeppele J, Keaveny T (1999). Increased cross-linking by non-enzymatic glycation reduces the ductility of bone and bone collagen. ASME Bioengineering Division 42: 267–268. [Google Scholar]
- Cerami A, Stevens VJ, Monnier VM (1979). Role of nonenzymatic glycosylation in the development of the sequelae of diabetes mellitus. Metabolism-Clinical and Experimental 28: 431–437. 10.1016/0026-0495(79)90051-9 [DOI] [PubMed] [Google Scholar]
- Currey J (1962). Stress concentrations in bone. Journal of Cell Science 3: 111–133. 10.1242/jcs.s3-103.61.111 [DOI] [Google Scholar]
- Currey JD, Pitchford JW, Baxter PD (2007). Variability of the mechanical properties of bone, and its evolutionary consequences. Journal of the Royal Society Interface 4: 127–135. 10.1098/rsif.2006.0166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depalle B, Qin Z, Shefelbine SJ, Buehler MJ (2015). Influence of cross-link structure, density and mechanical properties in the mesoscale deformation mechanisms of collagen fibrils. Journal of the Mechanical Behavior of Biomedical Materials 52: 1–13. 10.1016/j.jmbbm.2014.07.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diab T, Condon KW, Burr DB, Vashishth D (2006). Age-related change in the damage morphology of human cortical bone and its role in bone fragility. Bone 38: 427–431. 10.1016/j.bone.2005.09.002 [DOI] [PubMed] [Google Scholar]
- Diab T, Sit S, Kim D, Rho J, Vashishth D (2005). Age-dependent fatigue behaviour of human cortical bone. European Journal of Morphology 42: 53–59. 10.1080/09243860500095539 [DOI] [PubMed] [Google Scholar]
- Diab T, Vashishth D (2007). Morphology, localization and accumulation of in vivo microdamage in human cortical bone. Bone 40: 612–618. 10.1016/j.bone.2006.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dyer DG, Blackledge JA, Thorpe SR, Baynes JW (1991). Formation of pentosidine during nonenzymatic browning of proteins by glucose. identification of glucose and other carbohydrates as possible precursors of pentosidine in vivo. Journal of Biological Chemistry 266: 11654–11660. 10.1016/S0021-9258(18)99007-1 [DOI] [PubMed] [Google Scholar]
- Garnero P (2012). The contribution of collagen crosslinks to bone strength. BoneKEy Reports 1: 182. 10.1038/bonekey.2012.182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnero P, Borel O, Gineyts E, Duboeuf F, Solberg H, Bouxsein ML, Christiansen C, Delmas PD (2006). Extracellular post-translational modifications of collagen are major determinants of biomechanical properties of fetal bovine cortical bone. Bone 38: 300–309. 10.1016/j.bone.2005.09.014 [DOI] [PubMed] [Google Scholar]
- Hamlin CR, Kohn RR, Luschin JH (1975). Apparent accelerated aging of human collagen in diabetes mellitus. Diabetes 24: 902–904. 10.2337/diab.24.10.902 [DOI] [PubMed] [Google Scholar]
- Hie M, Iitsuka N, Otsuka T, Tsukamoto I (2011). Insulin-dependent diabetes mellitus decreases osteoblastogenesis associated with the inhibition of WNT signaling through increased expression of SOST and DKK1 and inhibition of AKT activation. International Journal of Molecular Medicine 28: 455–462. 10.3892/ijmm.2011.697 [DOI] [PubMed] [Google Scholar]
- Hui SL, Slemenda CW, Johnston CC (1988). Age and bone mass as predictors of fracture in a prospective study. The Journal of Clinical Investigation 81: 1804–1809. 10.1172/JCI113523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ionova-Martin S, Wade J, Tang S, Shahnazari M, Ager J, Lane NE, Yao W, Alliston T, Vaisse C, Ritchie R (2011). Changes in cortical bone response to high-fat diet from adolescence to adulthood in mice. Osteoporosis International 22: 2283–2293. 10.1007/s00198-010-1432-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janghorbani M, van Dam RM, Willett WC, Hu FB (2007). Systematic review of type 1 and type 2 diabetes mellitus and risk of fracture. American Journal of Epidemiology 166: 495–505. 10.1093/aje/kwm106 [DOI] [PubMed] [Google Scholar]
- Jast J, Jasiuk I (2013). Age-related changes in the 3D hierarchical structure of rat tibia cortical bone characterized by high-resolution micro-CT. Journal of Applied Physiology 114: 923–933. 10.1152/japplphysiol.00948.2011 [DOI] [PubMed] [Google Scholar]
- Karim L, Bouxsein ML (2016). Effect of type 2 diabetes-related non-enzymatic glycation on bone biomechanical properties. Bone 82: 21–27. 10.1016/j.bone.2015.07.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim L, Tang SY, Sroga GE, Vashishth D (2013). Differences in non-enzymatic glycation and collagen cross-links between human cortical and cancellous bone. Osteoporosis International 24: 2441–2447. 10.1007/s00198-013-2319-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katayama Y, Akatsu T, Yamamoto M, Kugai N, Nagata N (1996). Role of nonenzymatic glycosylation of type I collagen in diabetic osteopenia. Journal of Bone and Mineral Research 11: 931–937. 10.1002/jbmr.5650110709 [DOI] [PubMed] [Google Scholar]
- Kida Y, Saito M, Shinohara A, Soshi S, Marumo K (2019). Non-invasive skin autofluorescence, blood and urine assays of the advanced glycation end product (AGE) pentosidine as an indirect indicator of age content in human bone. BMC Musculoskeletal Disorders 20: 1–8. 10.1186/s12891-019-3011-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkman MS, Briscoe VJ, Clark N, Florez H, Haas LB et al. (2012). Diabetes in older adults: Consensus report. Journal of the American Geriatrics Society 60: 2342–2356. 10.1111/jgs.12035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kivirikko KI (1974). Biosynthesis of collagen. Connective Tissues:107–121. [Google Scholar]
- Knott L, Bailey A (1998). Collagen cross-links in mineralizing tissues: A review of their chemistry, function, and clinical relevance. Bone 22: 181–187. 10.1016/S8756-3282(97)00279-2 [DOI] [PubMed] [Google Scholar]
- Koester K, Barth H, Ritchie R (2011). Effect of aging on the transverse toughness of human cortical bone: Evaluation by R-curves. Journal of the Mechanical Behavior of Biomedical Materials 4: 1504–1513. 10.1016/j.jmbbm.2011.05.020 [DOI] [PubMed] [Google Scholar]
- Launey ME, Buehler MJ, Ritchie RO (2010). On the mechanistic origins of toughness in bone. Annual Review of Materials Research 40: 25–53. 10.1146/annurev-matsci-070909-104427 [DOI] [Google Scholar]
- Liang W, Chikritzhs T (2016). The effect of age on fracture risk: A population-based cohort study. Journal of Aging Research 2016: 1–5. 10.1155/2016/5071438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu R, Bal HS, Desta T, Krothapalli N, Alyassi M, Luan Q, Graves DT (2006). Diabetes enhances periodontal bone loss through enhanced resorption and diminished bone formation. Journal of Dental Research 85: 510–514. 10.1177/154405910608500606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Li W, Cai J, Yan Z, Shao X, Xie K, Guo XE, Luo E, Jing D (2020). Spatiotemporal characterization of microdamage accumulation and its targeted remodeling mechanisms in diabetic fatigued bone. The FASEB Journal 34: 2579–2594. 10.1096/fj.201902011RR [DOI] [PubMed] [Google Scholar]
- Looker AC, Eberhardt MS, Saydah SH (2016). Diabetes and fracture risk in older U.S. adults. Bone 82: 9–15. 10.1016/j.bone.2014.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Kraut D, Gerstenfeld LC, Graves DT (2003). Diabetes interferes with the bone formation by affecting the expression of transcription factors that regulate osteoblast differentiation. Endocrinology 144: 346–352. 10.1210/en.2002-220072 [DOI] [PubMed] [Google Scholar]
- Madia AM, Rozovski SJ, Kagan HM (1979). Changes in lung lysyl oxidase activity in streptozotocin diabetes and in starvation. Biochimica et Biophysica Acta (BBA)-General Subjects 585: 481–487. 10.1016/0304-4165(79)90181-8 [DOI] [PubMed] [Google Scholar]
- Marshall D, Johnell O, Wedel H (1996). Meta-analysis of how well measures of bone mineral density predict occurrence of osteoporotic fractures. BMJ 312: 1254–1259. 10.1136/bmj.312.7041.1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin RB, Burr DB (1982). A hypothetical mechanism for the stimulation of osteonal remodelling by fatigue damage. Journal of Biomechanics 15: 137–139. 10.1016/S0021-9290(82)80001-8 [DOI] [PubMed] [Google Scholar]
- Mascolo E, Vernì F (2020). Vitamin B6 and diabetes: Relationship and molecular mechanisms. International Journal of Molecular Sciences 21: 3669. 10.3390/ijms21103669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohsin S, Kaimala S, AlTamimi EKY, Tariq S, Adeghate E (2019). In vivo labeling of bone microdamage in an animal model of type 1 diabetes mellitus. Scientific Reports 9: 1–12. 10.1038/s41598-019-53487-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnier VM, Cerami A (1981). Nonenzymatic browning in vivo: Possible process for aging of long-lived proteins. Science 211: 491–493. 10.1126/science.6779377 [DOI] [PubMed] [Google Scholar]
- Monnier VM, Kohn RR, Cerami A (1984). Accelerated age-related browning of human collagen in diabetes mellitus. Proceedings of the National Academy of Sciences 81: 583–587. 10.1073/pnas.81.2.583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnier VM, Sun W, Sell DR, Fan X, Nemet I, Genuth S (2014). Glucosepane: A poorly understood advanced glycation end product of growing importance for diabetes and its complications. Clinical Chemistry and Laboratory Medicine 52: 21–32. 10.1515/cclm-2013-0174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan EF, Unnikrisnan GU, Hussein AI (2018). Bone mechanical properties in healthy and diseased states. Annual Review of Biomedical Engineering 20: 119–143. 10.1146/annurev-bioeng-062117-121139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai R, Murray DB, Metz TO, Baynes JW (2012). Chelation: A fundamental mechanism of action of age inhibitors, age breakers, and other inhibitors of diabetes complications. Diabetes 61: 549–559. 10.2337/db11-1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalla RK, Kruzic JJ, Kinney JH, Ritchie RO (2004). Effect of aging on the toughness of human cortical bone: Evaluation by R-curves. Bone 35: 1240–1246. 10.1016/j.bone.2004.07.016 [DOI] [PubMed] [Google Scholar]
- Nalla R, Stölken J, Kinney J, Ritchie R (2005). Fracture in human cortical bone: Local fracture criteria and toughening mechanisms. Journal of Biomechanics 38: 1517–1525. 10.1016/j.jbiomech.2004.07.010 [DOI] [PubMed] [Google Scholar]
- Nyman JS, Roy A, Tyler JH, Acuna RL, Gayle HJ, Wang X (2007). Age-related factors affecting the post yield energy dissipation of human cortical bone. Journal of Orthopaedic Research 25: 646–655. 10.1002/jor.20337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxlund H, Barckman M, Ørtoft G, Andreassen T (1995). Reduced concentrations of collagen crosslinks are associated with reduced strength of bone. Bone 17: S365–S371. 10.1016/8756-3282(95)00328-B [DOI] [PubMed] [Google Scholar]
- Peterlik H, Roschger P, Klaushofer K, Fratzl P (2006). From brittle to ductile fracture of bone. Nature Materials 5: 52–55. 10.1038/nmat1545 [DOI] [PubMed] [Google Scholar]
- Picke A-K, Campbell G, Napoli N, Hofbauer LC, Rauner M (2019). Update on the impact of type 2 diabetes mellitus on bone metabolism and material properties. Endocrine Connections 8: R55–R70. 10.1530/EC-18-0456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piez KA (1968). Cross-linking of collagen and elastin. Annual Review of Biochemistry 37: 547–570. 10.1146/annurev.bi.37.070168.002555 [DOI] [PubMed] [Google Scholar]
- Prisby RD, Swift JM, Bloomfield SA, Hogan HA, Delp MD (2008). Altered bone mass, geometry and mechanical properties during the development and progression of type 2 diabetes in the Zucker diabetic fatty rat. Journal of Endocrinology 199: 379–388. 10.1677/JOE-08-0046 [DOI] [PubMed] [Google Scholar]
- Qing H, Ardeshirpour L, Divieti Pajevic P, Dusevich V, Jähn K, Kato S, Wysolmerski J, Bonewald LF (2012). Demonstration of osteocytic perilacunar/canalicular remodeling in mice during lactation. Journal of Bone and Mineral Research 27: 1018–1029. 10.1002/jbmr.1567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie RO (2011). The conflicts between strength and toughness. Nature Materials 10: 817–822. 10.1038/nmat3115 [DOI] [PubMed] [Google Scholar]
- Ritchie R, Nalla R, Kruzic J, Ager J III, Balooch G, Kinney J (2006). Fracture and ageing in bone: Toughness and structural characterization. Strain 42: 225–232. 10.1111/j.1475-1305.2006.00282.x [DOI] [Google Scholar]
- Robins S, Bailey A (1972). Age-related changes in collagen: The identification of reducible lysine-carbohydrate condensation products. Biochemical and Biophysical Research Communications 48: 76–84. 10.1016/0006-291X(72)90346-4 [DOI] [PubMed] [Google Scholar]
- Saito M, Marumo K (2010). Collagen cross-links as a determinant of bone quality: A possible explanation for bone fragility in aging, osteoporosis, and diabetes mellitus. Osteoporosis International 21: 195–214. 10.1007/s00198-009-1066-z [DOI] [PubMed] [Google Scholar]
- Schaffler M, Choi K, Milgrom C (1995). Aging and matrix microdamage accumulation in human compact bone. Bone 17: 521–525. 10.1016/8756-3282(95)00370-3 [DOI] [PubMed] [Google Scholar]
- Schuit S, Van der Klift M, Weel A, de Laet C, Burger H, Seeman E, Hofman A, Uitterlinden A, Van Leeuwen J, Pols H (2004). Fracture incidence and association with bone mineral density in elderly men and women: The Rotterdam study. Bone 34: 195–202. 10.1016/j.bone.2003.10.001 [DOI] [PubMed] [Google Scholar]
- Siegmund T, Allen MR, Burr DB (2008). Failure of mineralized collagen fibrils: Modeling the role of collagen cross-linking. Journal of Biomechanics 41: 1427–1435. 10.1016/j.jbiomech.2008.02.017 [DOI] [PubMed] [Google Scholar]
- Silva MJ, Brodt MD, Lynch MA, McKenzie JA, Tanouye KM, Nyman JS, Wang X (2009). Type 1 diabetes in young rats leads to progressive trabecular bone loss, cessation of cortical bone growth, and diminished whole bone strength and fatigue life. Journal of Bone and Mineral Research 24: 1618–1627. 10.1359/jbmr.090316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snedeker JG, Gautieri A (2014). The role of collagen crosslinks in ageing and diabetes-the good, the bad, and the ugly. Muscle Ligaments and Tendons Journal 4: 303–308. 10.32098/mltj.03.2014.07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sroga GE, Siddula A, Vashishth D (2015). Glycation of human cortical and cancellous bone captures differences in the formation of maillard reaction products between glucose and ribose. PLoS One 10: e0117240. 10.1371/journal.pone.0117240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K, Kurose T, Takizawa M, Maruyama M, Ushikawa K, Kikuyama M, Sugimoto C, Seino Y, Nagamatsu S, Ishida H (2005). Osteoclastic function is accelerated in male patients with type 2 diabetes mellitus: The preventive role of osteoclastogenesis inhibitory factor/osteoprotegerin (OCIF/OPG) on the decrease of bone mineral density. Diabetes Research and Clinical Practice 68: 117–125. 10.1016/j.diabres.2004.08.006 [DOI] [PubMed] [Google Scholar]
- Takagi M, Kasayama S, Yamamoto T, Motomura T, Hashimoto K, Yamamoto H, Sato B, Okada S, Kishimoto T (1997). Advanced glycation endproducts stimulate interleukin-6 production by human bone-derived cells. Journal of Bone and Mineral Research 12: 439–446. 10.1359/jbmr.1997.12.3.439 [DOI] [PubMed] [Google Scholar]
- Takizawa M, Suzuki K, Matsubayashi T, Kikuyama M, Suzuki H et al. (2008). Increased bone resorption may play a crucial role in the occurrence of osteopenia in patients with type 2 diabetes: Possible involvement of accelerated polyol pathway in its pathogenesis. Diabetes Research and Clinical Practice 82: 119–126. 10.1016/j.diabres.2008.07.008 [DOI] [PubMed] [Google Scholar]
- Tanaka K-i, Yamaguchi T, Kanazawa I, Sugimoto T (2015). Effects of high glucose and advanced glycation end products on the expressions of sclerostin and RANKL as well as apoptosis in osteocyte-like MLO-Y4-A2 cells. Biochemical and Biophysical Research Communications 461: 193–199. 10.1016/j.bbrc.2015.02.091 [DOI] [PubMed] [Google Scholar]
- Tang SY, Herber RP, Ho SP, Alliston T (2012). Matrix metalloproteinase-13 is required for osteocytic perilacunar remodeling and maintains bone fracture resistance. Journal of Bone and Mineral Research 27: 1936–1950. 10.1002/jbmr.1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, Vashishth D (2010). Non-enzymatic glycation alters microdamage formation in human cancellous bone. Bone 46: 148–154. 10.1016/j.bone.2009.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, Vashishth D (2011). The relative contributions of non-enzymatic glycation and cortical porosity on the fracture toughness of aging bone. Journal of Biomechanics 44: 330–336. 10.1016/j.jbiomech.2010.10.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang S, Zeenath U, Vashishth D (2007). Effects of non-enzymatic glycation on cancellous bone fragility. Bone 40: 1144–1151. 10.1016/j.bone.2006.12.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanimoto M, Gohda T, Kaneko S, Hagiwara S, Murakoshi M et al. (2007). Effect of pyridoxamine (K-163), an inhibitor of advancedglycation end products, on type 2 diabetic nephropathy in KK-Ay/TAa mice. Metabolism-Clinical and Experimental 56: 160–167. 10.1016/j.metabol.2006.08.026 [DOI] [PubMed] [Google Scholar]
- Tanzer ML (1976). Cross-linking. In: Biochemistry of Collagen, pp. 137–162. Berlin: Springer. [Google Scholar]
- Teissier T, Temkin V, Pollak RD, Cox LS (2022). Crosstalk between senescent bone cells and the bone tissue microenvironment influences bone fragility during chronological age and in diabetes. Frontiers in Physiology 228: 105. 10.3389/fphys.2022.812157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CJ, Cleland TP, Sroga GE, Vashishth D (2018). Accumulation of carboxymethyl-lysine(CML) in human cortical bone. Bone 110: 128–133. 10.1016/j.bone.2018.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub W, Piez KA (1971). The chemistry and structure of collagen. Advances in Protein Chemistry 25: 243–352. 10.1016/S0065-3233(08)60281-8 [DOI] [PubMed] [Google Scholar]
- U.S. Department of Health and Human Services. (2012). A profile of older Americans: 2012. U.S. Government Printing Office. [Google Scholar]
- Ural A, Janeiro C, Karim L, Diab T, Vashishth D (2015). Association between non-enzymatic glycation, resorption, and microdamage in human tibial cortices. Osteoporosis International 26: 865–873. 10.1007/s00198-014-2938-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valcourt U, Merle B, Gineyts E, Viguet-Carrin S, Delmas PD, Garnero P (2007). Non-enzymatic glycation of bone collagen modifies osteoclastic activity and differentiation. Journal of Biological Chemistry 282: 5691–5703. 10.1074/jbc.M610536200 [DOI] [PubMed] [Google Scholar]
- Vashishth D (2007). The role of the collagen matrix in skeletal fragility. Current Osteoporosis Reports 5: 62–66. 10.1007/s11914-007-0004-2 [DOI] [PubMed] [Google Scholar]
- Vashishth D, Gibson G, Khoury J, Schaffler M, Kimura J, Fyhrie DP (2001). Influence of nonenzymatic glycation on biomechanical properties of cortical bone. Bone 28: 195–201. 10.1016/S8756-3282(00)00434-8 [DOI] [PubMed] [Google Scholar]
- Vestergaard P (2007). Discrepancies in bone mineral density and fracture risk in patients with type 1 and type 2 diabetes—A meta-analysis. Osteoporosis International 18: 427–444. 10.1007/s00198-006-0253-4 [DOI] [PubMed] [Google Scholar]
- Wang X, Shen X, Li X, Agrawal CM (2002). Age-related changes in the collagen network and toughness of bone. Bone 31: 1–7. 10.1016/S8756-3282(01)00697-4 [DOI] [PubMed] [Google Scholar]
- Waqas K, Chen J, Koromani F, Trajanoska K, van der Eerden BC, Uitterlinden AG, Rivadeneira F, Zillikens MC (2020). Skin autofluorescence, a noninvasive biomarker for advanced glycation end-products, is associated with prevalent vertebral and major osteoporotic fractures: The Rotterdam study. Journal of Bone and Mineral Research 35: 1904–1913. 10.1002/jbmr.4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willett TL, Voziyan P, Nyman JS (2022). Causative or associative: A critical review of the role of advanced glycation end-products in bone fragility. Bone 1635: 116485. 10.1016/j.bone.2022.116485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi SI (2011). Role of advanced glycation end products (AGEs) in osteoporosis in diabetes. Current Drug Targets 12: 2096–2102. 10.2174/138945011798829456 [DOI] [PubMed] [Google Scholar]
- Yang X, Liu CJ, Wang ZZ, Ding D, Shi JW, Wu XT, Sun LW, Fan YB (2021). Effects of advanced glycation end products on osteocytes mechanosensitivity. Biochemical and Biophysical Research Communications 568: 151–157. 10.1016/j.bbrc.2021.06.074 [DOI] [PubMed] [Google Scholar]
- Zimmermann EA, Schaible E, Bale H, Barth HD, Tang SY, Reichert P, Busse B, Alliston T, Ager JW III, Ritchie RO (2011). Age-related changes in the plasticity and toughness of human cortical bone at multiple length scales. Proceedings of the National Academy of Sciences 108: 14416–14421. 10.1073/pnas.1107966108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann EA, Schaible E, Gludovatz B, Schmidt FN, Riedel C et al. (2016). Intrinsic mechanical behavior of femoral cortical bone in young, osteoporotic and bisphosphonate-treated individuals in low-and high energy fracture conditions. Scientific Reports 6: 1–12. 10.1038/srep21072 [DOI] [PMC free article] [PubMed] [Google Scholar]