

Abstract
A “biomarker of aging” is conceptualized as an index of how far an individual has moved along the path from youth to old age. In contrast, an aging rate indicator (ARI) represents a measure of speed, rather than distance, that is, a measure of how rapidly the individual is moving toward the phenotypic changes typical of old age. This essay presents and reviews recent data suggesting common characteristics of slow-aging mice, whether the slowed aging is caused by a mutant allele, the calorie restriction diet, or drugs that slow aging and extend mean and maximal lifespan. Some of the candidate ARIs, shared by nine varieties of slow-aging mice, are physiological changes seen in fat, fat-associated macrophages, muscle, liver, brain, and plasma. Others are molecular measurements, reflecting activity of mTORC1, selective mRNA translation, or each of six MAP kinases in two distinct MAPK cascades in liver, muscle, or kidney. Changes in ARIs are notable in young adult mice after 8 months of drug or diet exposure, are detectable in mutant mice at least as early as 4–6 months of age, and persist until at least 18–22 months. Many of the candidate ARIs are thought to play an influential role in cognition, inflammation, exercise responses, and control of metabolic rate, and are thus plausible as modulators of age-related physiological and neurological illnesses. In principle, screening for drugs that induce alterations in ARIs in normal young adult mice might facilitate the search for preventive medicines that can retard aging and late-life illnesses in mice or in human populations.
Introduction
Researchers interested in the biology of aging, and its potential modification by antiaging drugs, have devoted a substantial amount of community effort to the search for possible biomarkers of aging, conceived as quantifiable traits that can reveal the biological age of an individual animal. The central framework here is that such biomarkers might change monotonically through some relevant portion of adult life, might discriminate younger from older adults, might predict mortality risk at some useful distance from ultimate date of death, and, crucially, might serve, individually or collectively, as surrogate endpoints for studies of putative antiaging diets, drugs, or polymorphic alleles. Like an odometer in a car, biomarkers or weighted combinations of biomarkers might in principle reveal how far along the aging trajectory an individual organism has already proceeded.
Odometers do not reveal the speed at which a car is currently traveling. A low-mileage or high-mileage car might be going quickly or slowly. Speed and distance traveled are independent and unconnected measures of a vehicle’s state.
This essay presents the concept of aging rate indicators (ARIs) as speedometers for aging research. In principle, an ARI is a quantitative trait or measurement, an endpoint, which discriminates slow-aging mice from normal mice; it measures how quickly the aging process is proceeding in an individual organism. (The definition carefully takes no position on whether ARIs might also discriminate fast-aging animals from normal ones1, a topic that will be deferred for another occasion.) An ideal ARI would make this discrimination regardless of the age at which it is measured, at least within the portion of adult life where serious diseases are rare and mortality risk is minimal. The critical feature of an idealized ARI, the critical test of a candidate ARI, is that it should be modulated, in the same direction, by antiaging perturbations, whether the slowed aging and extended lifespan are caused by genetic factors, by dietary intervention, or by lifespan-increasing drugs.
Some ARIs may also change with age, but this is not a necessary or even an expected feature of the proposed definition. For this reason, evidence that a candidate endpoint goes up or down with age, or does not change with age, is irrelevant to the question of whether it can serve as an ARI. ARIs are not the same as biomarkers of aging.
It is helpful to consider the differences between ARIs and the more familiar biomarkers of aging. If a measurement—an estimate of collagen cross-linking, a score of visual acuity or reflex speed, an index combining levels of DNA methylation at several sites, a T cell subset ratio, and so on—is proposed as a biomarker of aging, it is expected to show age-related change in adults, that is, to distinguish young, middle-aged, and older adults. Among older adults, those whose levels of the putative biomarker are closer to the values typically seen in young adults are considered to have a lower biological age, and are inferred to have aged less than age-matched subjects whose biomarker level has diverged more from youthful values. In principle, individuals in older age groups that appear “biologically old” with respect to any one biomarker are expected to show similar elevation across a wide range of biomarkers of aging in multiple physiological, cellular, and biochemical domains. This framework suggests that individuals who appear biologically old, compared to others with the same chronological age, may prove to be at relatively high risk of age-related diseases, and to die at a relatively early age.
In the context of a discussion of ARIs, it is important to see that information about biological age cannot be obtained from young individuals, because these individuals have not yet undergone much aging. If a measurement is to be interpreted as a surrogate of an age-dependent change, it can only be measured in subjects that have undergone age-dependent change, that is, in those that are no longer young. Young adults may well differ from one another in traits that are indeed biomarkers of aging, but these differences must reflect other sources of variance, and not effects of aging, because aging has not yet taken place.
Evaluation of an endpoint as a candidate ARI involves an entirely different set of criteria. Evidence that a candidate ARI changes with age is quite beside the point, because ARIs are taken to be measures of the pace or speed at which aging is currently occurring, and not as indices of how much a given subject has already aged, in the past. In such a framework, estimates of ARIs made in young adults are expected to be highly informative, because these individuals may actually be aging at different rates; for example, one might be on a calorie-restricted (CR) diet, or carry a mutant growth hormone receptor (GHR) allele, or be in a rapamycin (Rapa)-treatment cohort. The key criterion for a putative ARI is that it should be modified, in a consistent direction, by most or all genes, diets, and drugs that are known, on independent evidence, to slow the signs of aging, postpone late-life illnesses, and increase lifespan.
Drugs, diets, and alleles that alter aging will alter both ARIs and biomarkers of aging, but the form and timing of the effects differ. Figure 1A shows some hypothetical biomarker of aging, in this case, one that increases with age, and the expected effects of an antiaging drug initiated at 4 months of age. The measured “biomarker” trait increases more slowly in the drug-treated group than in the control group. Figure 1B shows the effect of a hypothetical antiaging drug on an ARI, in this case, an ARI that is not itself age sensitive and thus could not serve as a biomarker of aging. Treatment with the drug quickly leads to higher levels of the ARI. In the cartoon, the effect of the drug is imagined as instantaneous, although the pace at which ARIs switch to new levels is at present unknown, and may well differ depending on the intervention. Drugs that diminish the level of the ARI would show a similar deflection in a downward direction (not illustrated). Figure 1C,D portrays examples of ARIs that might affect traits that go up (panel C) or down (panel D) with age; in each case, the inference that the trait serves as an ARI does not depend on the direction or speed of age effects, but rather on the ability to discriminate control mice from slow-aging mice at any age after the intervention.
Figure 1.
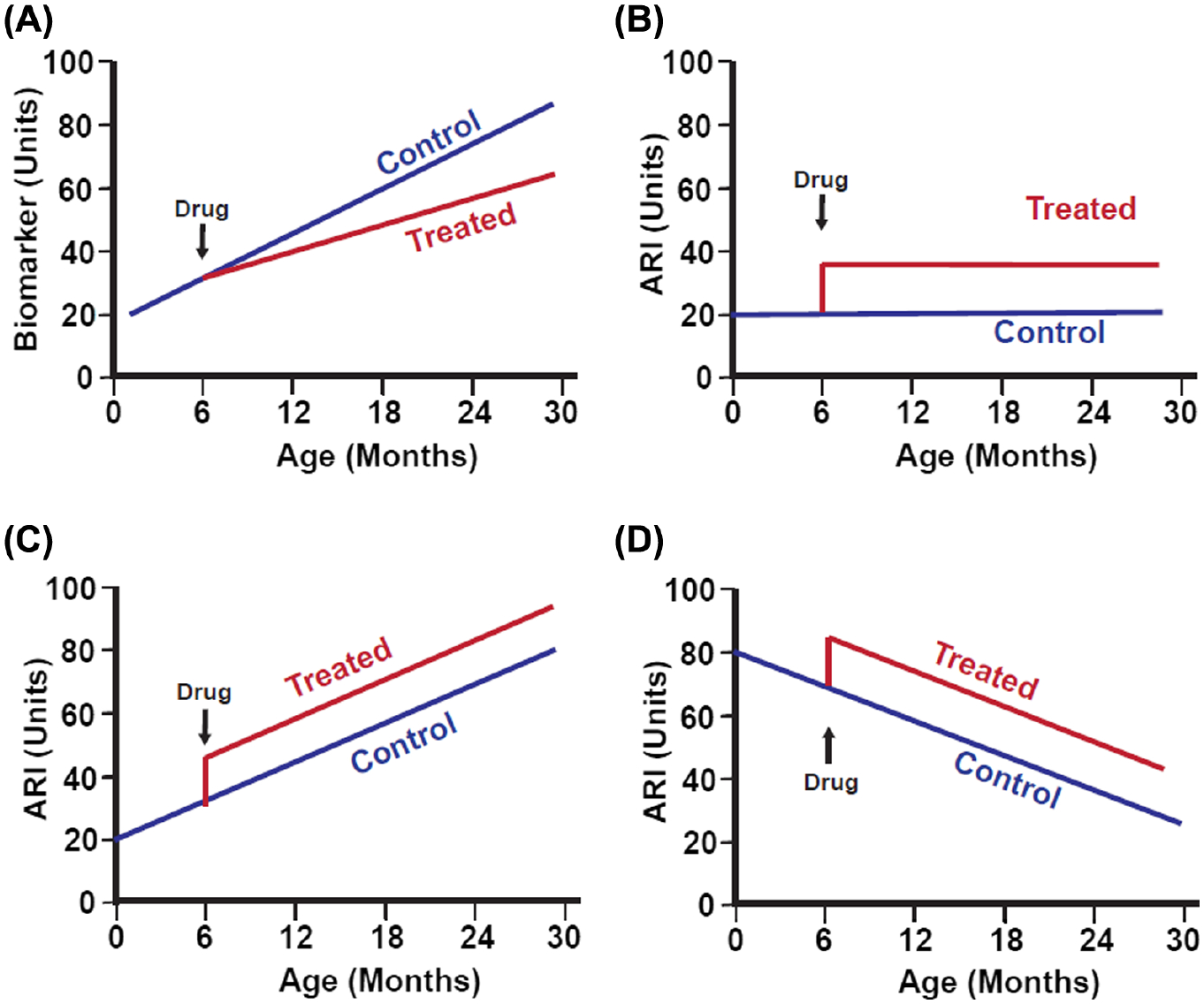
Schematic diagrams showing hypothetical effects of an antiaging drug, initiated at 6 months of age, on (A) a generic biomarker of aging or on three different aging rate indicators (ARIs), (B) one of which does not show age effects, (C) one of which increases with age, and (D) one of which decreases with age. In this hypothetical example, the drug is assumed to induce very rapid changes in the ARIs, but the pace of effect on an ARI may well differ among interventions and must be determined empirically. Classification of a trait as an ARI does not depend on whether it changes with age. In panels (B–D), it is arbitrarily assumed that the hypothetical ARI is elevated in slow-aging mice, but some ARIs are lower in treated than in control animals (such as iNOS and cytokine levels in fat-associated macrophages).
With this conceptual framework, we can now ask some empirical questions. The most important of these would be to ask whether there are indeed any ARIs; in other words, are there traits that change in the same direction in many, highly diverse, models of slower aging? Are there shared mechanisms common to a wide range of antiaging interventions? This question is quite distinct from questions about possible “mechanisms of aging.” It is plausible, for example, that mechanisms of aging, such as oxidative damage to proteins, or death of a specific stem cell type, or oncogenic somatic mutations, or cross-linking of collagen, might lead to aging and its diseases, and yet not be modified, in young adults, by antiaging drugs or mutants or diets.
Evaluation of candidate ARIs was made possible by the evidence, accumulating over the previous 25 y, that the aging process in mice can be slowed, not merely by a CR or methionine-restriction diets2, but also by single-gene mutants3–6 and by antiaging drugs7–11. The foundational idea is that these many, diverse, approaches to delaying death and decelerating many aspects of age-sensitive physiological changes might share one or more common mechanisms. The CR diet, for example, is known to modify expression levels of hundreds of mRNAs and proteins in dozens of cell types and tissues, with effects on hundreds of physiological mediators and circuits of high relevance to health maintenance and disease progression. The same can be said for the growth hormone receptor knockout (GHRKO)5 mutation and for the effects of Rapa7, or acarbose (Aca)8,12, or 17α-estradiol (17aE2)8,13, or canagliflozin (Cana)10,14. So the question then arises: Which, if any, of the many effects of CR diet or Rapa treatment or GHRKO genotype are also seen in each of the other models for delayed aging?
Some recent work has provided encouraging evidence that ARIs may be altered by genes, CR diet, and drugs in many tissues of many varieties of slow-aging mice. Table 1 summarizes several recent publications with relevant data on physiological status of white and brown adipose tissue (WAT and BAT, respectively), fat-associated macrophages, muscle, liver, and brain of Snell dwarf, Ames dwarf, GHRKO, PAPPA knockout (PKO), and CR mice, and mice treated with Rapa, Aca, 17aE2, and Cana.
Table 1.
Summary of tissues and mouse models evaluated for candidate aging rate indicators (ARIs).
| Endpoint | Tissue(s) | Seen in These Models |
|---|---|---|
| CIT | Liver, kidney, muscle | Snell, Ames, GHRKO, PKO, Rapa, Aca, 17aE2* |
| MEK1/ERK | Liver, kidney | Rapa, Aca, 17aE2*, Cana* |
| MEK3/p38 | Liver, kidney | Rapa, Aca, 17aE2, Cana |
| mTORCl | Liver, heart, kidney, muscle | Snell, GHRKO, PKO, [CR], Rapa, Aca, 17aE2*. Cana not tested yet. |
| UCP1 | BAT | Snell, Ames, GHRKO, PKO, CR, Aca, 17aE2, Cana; not Rapa |
| UCP1 | Ing-WAT | Snell, Ames, GHRKO, PKO, CR, Aca, 17aE2, Cana; not Rapa |
| UCP1 | Perigonadal WAT | Snell, Ames, GHRKO, PKO, CR, Aca, 17aE2, Cana; not Rapa |
| Ml and M2 | BAT, Ing-WAT, Pg-WAT | Snell, Ames, GHRKO, PKO, CR, Rapa, Aca, 17aE2, Cana |
| FNDC5 | Muscle | Snell, Ames, GHRKO, PKO, CR, Rapa, Aca, 17aE2*, Cana* |
| Irisin | Plasma | Snell, GHRKO, PKO, CR, Rapa, Aca, 17aE2, Cana |
| GPLD1 | Liver, plasma | Snell, Ames, GHRKO, PKO, CR, Rapa, Aca, 17aE2, Cana |
| BDNF | Hippocampus | Snell, Ames, GHRKO, PKO, CR, Rapa, Aca, 17aE2, Cana* |
| DCX | Hippocampus | Snell, Ames, GHRKO, PKO, CR, Rapa, Aca, 17aE2, Cana |
17aE2, 17α-estradiol; Aca, acarbose; BAT, brown adipose tissue; BDNF, brain-derived neurotrophic factor; CIT, cap-independent translation; CR, calorie-restricted; DCX, doublecortin; GHRKO, growth hormone receptor knockout; GPLD1, Glycosylphosphatidylinositol-Specific Phospholipase D1; Ing-WAT, inguinal white adipose tissue; Pg-WAT, perigonadal white adipose tissue; PKO, PAPPA knockout; Rapa, rapamycin.
Sex-specific effect, with ARI changed in male mice but not in females.
Changes in WAT and BAT
The uncoupling protein UCP1, the key factor in thermogenic conversion of fuel to heat energy15,16, has now been shown to increase in BAT, inguinal WAT (Ing-WAT), and intra-perigonadal WAT (Pg-WAT) of Snell dwarf17, Ames dwarf18, PKO19, and GHRKO17 mice. Thus, this shift affects both subcutaneous and intra-abdominal WAT depots. These increased levels of UCP1 protein are accompanied by, and presumably caused by, corresponding changes in the levels of UCP1 mRNA. The effect on UCP1 protein level is seen both at 4–6 months of age (Snell, PKO, and GHRKO) and at 15–18 months (Ames). The WAT depots also show biochemical and morphological changes typical of “browning” of white fat, such as diminished cell size, increased proportion of cytoplasm and mitochondrial number, and expression of surface antigens usually at high levels only in BAT17. Increased UCP1 in BAT, Ing-WAT, and Pg-WAT is also seen in young adult (i.e., 12 months old) UM-HET3 mice exposed for 8 months to the CR diet, or to food containing Aca, 17aE2, or Cana at levels known to increase lifespan18. The only exception to this pattern is that Rapa does not lead to increased UCP1 in WAT, in agreement with previous observations20,21. In contrast, Rapa does increase UCP1 mRNA and protein in BAT-derived pre-adipocytes in vitro22, in good agreement with our data on BAT in mice.
Changes in Fat-Associated Macrophage Populations
BAT and WAT contain two classes of macrophages, the M1 cells that produce inflammatory cytokines, such as tumor necrosis factor α (TNFα), Interleukin 6 (IL6), and monocyte chemoattractant protein 1 (MCP1), and M2 cells that inhibit inflammation23. In general, aging leads to an increase in M1 cells and a decline in M2 cells24. Using arginase 1 (Arg1) protein as an index of M2 cells, and inducible nitric oxide synthase (iNOS) as a marker of M1 macrophages, we demonstrated a decline in M1 and increase in M2 cells in BAT, Ing-WAT, and Pg-WAT in young adult Snell and GHRKO mice17, as well as in Ames dwarf mice at age 18 months18, and in young PKO mice19. In addition, production of mRNA encoding TNFα, IL6, and MCP1 was lower in these slow-aging mutants, in good agreement with the decline in M1 cells. Increases in M2 macrophages and parallel decreases in M1 cells were then also noted in all three fat depots in CR mice, and in mice treated from age 4 to 12 months with Rapa, Aca, 17aE2, or Cana (Li, McPherson, et al., submitted). The responses to Cana and 17aE2 were seen in both male and female mice, even though these two drugs do not increase female lifespan in UM-HET3 mice.
UCP1 and Macrophage Shift Reflects Indirect Effects of the Myokine Irisin and Its Precursor Fibronectin Type III Domain-containing Protein 5 (FNDC5)
To test the idea that these alterations in UCP1 and in fat-associated macrophage populations reflected the effects of hepatic production of insulin-like growth factor 1 (IGF1), a principal mediator of growth hormone (GH) action, we evaluated this set of endpoints in mice with disruption of GHR in liver only (“LKO”); these LKO mice have very low IGF1 levels but are not long-lived25. We found no change in UCP1 protein or mRNA, Arg1 or iNOS protein, or mRNA for MCP1, IL6, or TNFα in any of the three adipose tissues of LKO mice, implying that the effects on fat tissue were not secondary to diminished IGF1 in the GHRKO mice or, presumably, in Snell or Ames dwarf animals17. Next, to see if this suite of changes reflected direct effects on GH on adipocytes, we evaluated mice in which the GH receptor had been disrupted in adipocytes only26. These fat-specific GHRKO mice (“FKO”) are not longer-lived than controls. In these FKO mice, we noted elevated UCP1 in Ing-WAT of males, but not in the other two adipose depots, and saw no changes in any of the three fat depots in females17. All three adipose tissues showed an increase in M1 cells and decline in M2 cells, that is, changes opposite in direction to those seen in global GHRKO mice. Ing-WAT and Pg-WAT also showed higher levels of MCP1 and TNFα mRNA, consistent with the elevation in M1 macrophages, and again opposite in direction to the anti-inflammatory effects in the global GHRKO mice. Thus, with the exception of UCP1 in Ing-WAT of males, the changes in fat in GHRKO mice can apparently not be explained by disruption of GH signals within fat cells per se.
We then evaluated fat tissues of mice in which GHR had been disrupted in skeletal muscle only (MKO mice). MKO females are not long-lived, and survival data for males are ambiguous, with one colony showing no lifespan change and a second, independent colony reporting a small (5%) but significant lifespan increase in males27. We found UCP1 protein to be elevated in both sexes of MKO mice in BAT and in Pg-WAT, and in Ing-WAT of males. M2 macrophages were increased in all three adipose tissues of MKO mice, reaching statistical significance in both sexes except in Ing-WAT of females. M1 macrophages were diminished significant in both sexes of all three adipose tissue depots. TNFα mRNA was diminished in all three varieties of fat, and IL6 and MCP1 mRNA declined in BAT. Thus, nearly all of the effects seen in fat tissue of global GHRKO mice were replicated in mice with GHR disruption limited to muscle.
These results suggested that the adipose tissue changes might be due to a change in GH-dependent production of a myokine with effects on fat tissue. Irisin, a cleavage product of the muscle protein FNDC5, has been suggested28,29 to mediate some of the benefits of exercise on fat tissue metabolism and activity. We, therefore, measured irisin in plasma of our mouse models and found17 a significant elevation in Snell dwarf, GHKRO, and MKO mice, but not in LKO or FKO, in excellent agreement with the pattern of changes in adipocytes and fat-associated macrophage subpopulations. FNDC5, the precursor protein for irisin, showed the same pattern of effects in the muscle tissue itself: elevated levels in Snell, GHRKO, and MKO, but not in LKO or FKO mice. These parallels imply that low GH effects in muscle lead to augmented FNDC5, and, in turn, to lower plasma irisin levels, and then to the observed changes in UCP1 and M1/M2 ratio, with little or no direct involvement of hepatic IGF1 or direct GH effect in fat.
We were also able to demonstrate an increase in muscle FNDC5 in Ames dwarf mice tested at 18 months of age, suggesting that this set of myokine-dependent effects is detectable on a third genetic background, in another vivarium, and in middle-aged mice18. (We did not have access to plasma of these Ames mice and, therefore, could not assess irisin directly.) Similarly, FNDC5 and irisin increases were seen in young adult PKO mice. Thus, this pattern of changes—FNDC5, irisin, UCP1, and macrophage changes—is characteristic of all four of the mutant models for slower aging that were available to us.
FNDC5 and Irisin Are Also Elevated by Diets and Drugs That Extend Mouse Lifespan
Because CR diet, Rapa, Aca, 17aE2, and Cana had been found to alter fat and fat-associated macrophages in young adult, nonmutant, UM-HET3 mice, we also evaluated FNDC5 in skeletal muscle, and irisin in plasma, of all five of these mouse models. We found higher levels of FNDC5 and plasma irisin in all five varieties of mice (Li, McPherson et al., submitted). The effects of 17aE2 and Cana on FNDC5 were seen in males only, consistent with the sex-specific lifespan effect of these two drugs. Plasma irisin levels were elevated by Cana and 17aE2 in both sexes, at odds with the sex-specificity in the FNDC5 data; the basis for this discrepancy is unclear. These data show that elevation of FNDC5/irisin is characteristic of nine varieties of slow-aging mice, including four mutants, the CR diet, and four drugs, and including mice of ages 4–18 months, in two distinct vivaria, and on five genetic backgrounds. It is plausible, but not proven, that the changes in irisin underlie the downstream alterations in fat and in fat-associated macrophages in each of these nine models.
Cap-Independent mRNA Translation in Mutant and Drug-Treated Slow-Aging Mice
Most mRNAs are translated into protein by a process that involves binding of ribosomal subunits to the 5-prime 7-methylguanylate cap structure, but a subset can also be translated when ribosomes bind to 6-methyladenosine (m6A) residues in the untranslated region (UTR) between the cap and the translation start site30, a process called “cap-independent translation” (CIT). We have reported31 that several CIT proteins are elevated in muscle, liver, and kidney of Snell, GHRKO, and PKO mice, in the absence of any increase in the corresponding mRNAs. This subset of proteins includes those involved in DNA repair, mitochondrial biogenesis, and protection of mitochondria from oxidative injury. The same set of mRNA-independent changes are seen in mice treated for 48 hr with the mTORC1 inhibitor Rapa and in the liver of Rapa-treated mice31. A follow-up paper showed that these long-lived mutant mice had elevated levels of two proteins, YTH domain family, member 1 (YTHDF1) and YTHDF2, that can detect m6A signals in the 5-prime UTR and thus promote CIT of selected mRNAs32. The long-lived mutant mice also had elevation of N6-adenosine-methyltransferase 70 kDa subunit 3 (METTL3) and METTL14, which add m6A residues in the UTR. Increases in YTHDF1, YTHDF2, METTL3, and METTL14 likely to lead to the increase in translation of CIT mRNAs seen in these long-lived mutant mice. The third paper in this series33 then showed that production of CIT proteins declines with age, and that this decline is prevented by treating mice with Rapa or Aca. 17aE2 also blocks the age-dependent decline in CIT proteins, but does so only in male mice. Tissues from CR mice have not yet been evaluated for CIT proteins. Thus, an increase in production of CIT proteins is a shared feature seen in four varieties of slow-aging mutant mice and mice treated with any of three antiaging drugs.
Elevation of Glycosylphosphatidylinositol-Specific Phospholipase D1 in Many Varieties of Slow-Aging Mice
Glycosylphosphatidylinositol-specific phospholipase D1 (GPLD1) can hydrolyze inositol phosphate linkages in extracellular proteins attached by glycosylphosphatidlylinositol (GPI) linkages to the cell membrane34. Forced overexpression of GPLD1 in mice leads to enhanced cognition and enhanced neurogenesis, although the mechanism of this effect is unknown, and peripheral GPLD1 does not itself cross the blood-brain barrier35. Physical exercise increases plasma levels of GPLD1 in mice and in humans, and increases in GPLD1 might contribute to beneficial effects of exercise on cognitive function. We have reported increased levels of GPLDL1 protein in liver and plasma of young adult Snell and GHRKO mice36, as well as in 18-month-old Ames dwarf mice18. Consistent with the results in mice overexpressing GPLD1, we found elevations of brain-derived neurotrophic factor (BDNF), which helps maintain CNS function, and of doublecortin (DCX), an indicator of increased neurogenesis, in these slow-aging mutant mice36. The changes in liver production of GPLD1 seem not to reflect a direct effect of GH on liver cells, because they are not seen in mice in which the GH receptor has been disrupted in liver only, and additional studies will be needed to learn which GH-sensitive tissue(s) modulate liver GPLD1 synthesis. GPLD1 levels in plasma were also increased in 12-month-old mice exposed to a CR diet (from age 4 months), and in mice exposed to Rapa, Aca, 17aE2, or Cana to 12 months of age. Although the effects of 17aE2 and Cana on plasma GPLD1 were seen in both sexes, liver GPLD1 protein was affected by these two drugs only in male mice. Effects of CR, Rapa, and Aca on liver GPLD1 were significant independent of sex. The effects of these antiaging drugs on BDNF and DCX in brain are also seen in the CR and drug-treated mice (Li, McPherson et al., submitted). Thus, elevated levels of plasma and liver GPLD1 seem to be shared across multiple models of delayed aging in mice, and may be useful as ARIs.
GPLD1 Protein Is Produced by CIT
The increase in liver GPLD1 protein in Snell and GHRKO mice is not accompanied by a corresponding increase in GPLD1 mRNA36, suggesting the idea that GPLD1 protein levels might be regulated by selective mRNA translation. In support of this idea, we found that liver GPLD1 was elevated in transgenic mice overexpressing YTHDF1, which promotes synthesis of multiple CIT proteins by enabling m6A-dependent translation from sites downstream of the 5-prime cap. When mice were treated briefly (7 days) with 4-EGI1, a drug that blocks cap-dependent translation but increases CIT, GPLD1 levels increased in liver, without any change in GPLD1 mRNA. 4-EGI1 also increased GPLD1 protein, but not its mRNA, by primary mouse fibroblast cells in culture. These three lines of evidence support the idea that GPLD1 protein is regulated, at least in part, by CIT, and that the increases in CIT seen in long-lived mutant, CR, and drug-treated mice may lead to increased plasma GPLD1 and, as a consequence, to increased hippocampal BDNF and DCX.
GPLD1 is also detectable in the brain, but its levels seem to be controlled independently of liver and plasma GPLD1. Hippocampal GPLD1 is not altered in Snell or GHRKO mice36 or in 18-month-old Ames mice18, or in PKO mice19. Nor does hippocampal GPLD1 protein increase in YTHDF1 transgenic mice36.
These results provide a suggested molecular mechanism for changes in GPLD1, BDNF, and DCX in multiple varieties of slow-aging mice, and imply that further work on how aging and antiaging interventions modulate CIT may be rewarding. They also provide impetus for seeking drugs that might promote CIT in normal young and older mice.
Lifelong Patterns of ARI Can Be Set by Transient, Early, Regulation of GH Levels
Bartke and his colleagues have shown that the long lifespan of Ames dwarf mice can be abrogated by transient GH treatment of very young Ames pups, starting at 2 weeks of age and continuing for approximately 6 weeks of treatment37. Ames mice treated, transiently, with GH not only lose the lifespan benefit of the Ames mutation, they also lose the multiplex stress-resistance characteristic of short-term cultures of skin fibroblasts37. They also lose their resistance to age-dependent increases in hypothalamic inflammation38.
These prior studies motivated a survey of potential ARIs in 18-month-old Ames mice, contrasted with age-matched mice that had been exposed to GH injections ending at 8 weeks of age18. Early exposure to GH blocked the effect of the Ames genotype on BDNF and DCX in hippocampus, on UCP1 in BAT, Ing-WAT, and Pg-WAT, and on M2 (Arg1) macrophages in all three fat depots. A similar effect of GH exposure was seen on M1 (iNOS) macrophages in BAT, with similar but not significant effects in the two WAT depots. Consistent with the changes in UCP1 and in fat-associated macrophages, the Ames-associated increase in muscle FNDC5 was abrogated by the early-life GH treatment procedure. Similarly, the increase in GPLD1 protein in liver and in fat was not seen if the mice had previously been subjected to early life GH treatment. It was not possible to test for irisin or GPLD1 in plasma, because plasma samples were not available to us. Thus, the entire suite of ARI endpoints was blocked, in parallel, by a treatment procedure that interfered with the longevity, stress-resistance, and lowered hypothalamic inflammation that is characteristic of Ames dwarf mice. These observations show that the collection of ARIs can be modified, as a group, by the early life endocrine environment. Lifespan of normal mice can be increased by transient early life exposure to Rapa39, and by crowded litter intervention in which milk availability is limited in the first three weeks of life40, but it is not known if these positive effects are also modulated by GH signals, or if they are accompanied by changes in ARIs.
Diminished Activity of mTORC1
mTOR, the “target of rapamycin” protein kinase, plays a central role in responses to multiple stimuli, including nutritional changes, stresses, and endocrine signals, and influences a very wide range of cellular functions including protein synthesis, cell growth, autophagy, and cell death. mTORC1 function can conveniently be assessed by measuring phosphorylation of two of its substrates: Eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1), a translation initiation factor, and S6-kinase (S6K) or its own substrate ribosomal protein S6. The ratio of phosphorylated substrate protein to the amount of the total protein in a cell lysate is typically interpreted as a surrogate for mTORC1 activity, although it is important to recognize that an increase in this “phosphorylation ratio” might reflect either an increase in the amount of phosphoprotein, or a decrease in the amount of total protein without any change in phosphoprotein levels. Young adult Snell, GHRKO, and PKO mice all show diminished phosphoprotein ratios for S6, S6K, and 4EBP1 in liver, heart, muscle, and kidney, particularly in the non-fasted condition31,41. The effect is apparently not due to GH-induced production of IGF1 in the liver, because no such decline in mTORC1 action is seen in mice, where GHR is disrupted in liver only41.
mTORC1 substrate phosphorylation was also examined in mice exposed to Rapa, Aca, or 17aE233. All the mice were 22 months old at the time of testing, and had been drug-treated from the age of 6 months (“early” onset) or 18 months (“late” onset). Phosphorylation ratios increased with age in males and in females, in liver and in kidney, for both 4EBP1 and S6. For S6, the increase in ratio reflected higher levels of phospho-S6 in the tissues of aged mice, whereas for 4EBP1 the higher ratio reflected a decline in the total amount of 4EBP1 without a change in the amount of phosphorylated 4EPB1. The increase in phosphorylation ratio for the two mTORC1 substrates was not seen in 22-month-old mice that had been treated with Rapa or Aca, regardless of the age of initial drug onset. 17aE2 had the same pattern of effects in male mice, but did not modify mTORC1 substrate phosphorylation in females, consistent with the sex-specific lifespan effect for 17aE2. Thus, a lower phosphorylation ratio of both mTORC1 substrate proteins is shared by three varieties of long-lived mutant mice and mice treated with any of three distinct antiaging drugs. It will be of interest to determine the time course of this drug effect, that is, to learn how long an exposure to these drugs is required to modify mTORC1 substrate phosphorylation.
MAP Kinase Cascades Affected by Three Antiaging Drugs
The pace of mRNA translation is modulated by mTORC1 function, and the specific selection of mRNAs is modulated by enzymes and binding factors involved in CIT. To evaluate signals that are upstream of translation initiation, we evaluated the effects of Rapa, Aca, and 17aE2 on a protein kinase cascade that responds to insulin, GH, and other endocrine stimuli to control phosphorylation of eIF4E, the key cap-dependent initiation and translation factor42, which is also regulated by mTORC1 via 4EPB1. Cell membrane receptors lead to phosphorylation and thus activation of dual specificity mitogen-activated protein kinase kinase 1 (MEK1), which, in turn, phosphorylates extracellular signal-regulated kinases (ERK1/2), which, in turn, phosphorylates MNK1/2, which is responsible for phosphorylation of eIF4E43,44. Aging in UM–HET3 mice led to increases in each of these four phosphorylation steps, that is, an increase in the entire “ERK1/2” kinase cascade. Each step in this cascade was diminished in 22-month-old mice that had been treated with Rapa or Aca, with equal effects seen in early onset and in later onset animals. The same set of effects was produced by 17aE2, but in male mice only. The results on liver tissue were also noted in kidney samples from the same set of mice.
A second, distinct, MAP kinase cascade also increases in liver and kidney of aging mice. This cascade (the “p38” cascade) responds to cytokine stimuli, such as IL6 and TNFα, by phosphorylation of MEK3, its substrate p38, and the p38 target MK243–45. Phosphorylation of MK2 then, in turn, leads to increased production of acute phase proteins such as caspase 6, serum amyloid protein, and hemoxygenase. Each kinase in this sequence has diminished activity in mice treated with Rapa, Aca, or 17aE2, regardless of age of drug onset. All three of the tested acute phase proteins show the same pattern: an increase with age, blocked by each of the three drugs. It is notable that the effects of 17aE2 on the p38 cascade are not sex specific, hinting that the effects on this set of kinases may be less closely associated with the lifespan effect than the drug effects on the ERK1/2 cascade.
Prospects and Implications
Discovery of validated ARI endpoints would represent progress on two distinct problems: They could provide tools for practical evaluation of putative antiaging drugs, and they could direct attention to the mechanistic causes and consequences of coordinated changes in ARI batteries.
Table 1 provides a succinct summary of the main observations. This list of candidate ARIs includes changes in brown and white adipocytes, two types of fat-associated macrophages, muscle, liver, kidney, and hippocampus. The changes are seen in four varieties of long-lived mutant mice, in CR mice, and in mice treated with four distinct antiaging drugs. Not all of the ARIs have been evaluated in all nine varieties of mice, but those that have been tested have produced significant changes in the same direction (with the exception of Rapa effects on UCP1). Several of the candidate ARIs are detectable in plasma—irisin, GPLD1, and the acute phase proteins— and may thus lend themselves to studies in humans or in species, such as dogs and nonhuman primates, from which blood samples can be taken without harm. ARIs in blood may also useful for designs in which changes are measured in the same individual at different ages or in response to some intervention.
The work done to date sets the stage for a broad set of follow-up studies.
If indeed some or all of these ARIs change rapidly, for example within a few months or weeks, when young mice are exposed to a drug that slows aging and extends lifespan, they might form the basis of a screening process to help identify drugs with particularly high potential to affect aging and healthy lifespan. Many of the ARIs are altered by antiaging mutations in mice as young as 6 months, and by drugs and CR diet in mice as young as 12 months, but more work will be needed to learn about the time course of ARI change in the first weeks or months after drug onset. Of drugs accepted for longevity testing by the ITP, only about 10%–15% lead to a significant lifespan extension; improvement of this hit rate to 25% or higher would increase the speed and lower the cost needed to discover new antiaging drugs.
Many of the ARIs in the current, provisional, test battery have well-known connections to important aspects of health maintenance and disease prevention in humans. If indeed antiaging drugs, genes, and diets do modify thermogenesis, inflammatory tone, neurogenesis, protein synthesis, and patterns of mRNA translation, this suite of coordinated changes can serve as a platform for further investigation of a wide range of diseases in rodent models and in people.
At this point, we have essentially no insight into the basis for the coordinated resetting of this set of putative ARI endpoints. There is evidence that the pattern can be set by alterations in GH and/or IGF1 levels prior to puberty18, and also evidence that drugs and diets can lead to similar shifts when started in 4-month-old mice33,36,42. Several of the drugs that extend lifespan, including Rapa, Aca, and 17aE2, can do so when started at ages of 16–20 months11, and it will be interesting to see if these midlife treatments also modify ARIs, and if so how quickly these shifts occur. At least some of the improvements in physiological fitness are seen in mice exposed to Aca and 17aE2 in late middle age46,47, and changes in CIT, mTORC1, and MAP kinases are also characteristic of mice exposed to antiaging drugs from age 16 months33,42. Thus, there appears to be substantial flexibility in the age of onset of the postulated shift in ARI settings. The evidence that GH exposure in the Ames dwarf model can be detected in old age, that is, 18 or more months after transient exposure to GH, implies that the early-life hormonal environment leads to permanent alterations in multiple cell types18,37,38, but whether the memory of early life GH exposure resides in epigenetic changes in one or many cell types, or changes in proportions of cells within one or more tissues, or some self-reinforcing feedback loop, is so far unclear.
It will be interesting to learn of other mutations that extend mouse lifespan also lead to changes in one, many, or all of the elements of the ARI test battery. We have preliminary evidence (Li, Endicott, Miller, unpublished) that at least some of these shifts are seen in young adults of mice that overexpress phosphatase and tensin homolog (PTEN), shown by others to lead to mouse longevity48. Each new drug or new diet or new mutant that expands the list of long-lived mouse models will provide additional clues to the basis for coordinated modulation of ARIs, whereas examples of slow-aging mice that do not show the changes listed in Table 1 may in themselves open new avenues for investigation of how aging rate is controlled.
Hypotheses about connections among these coordinated changes merit exploration. The evidence that GPLD1, a stimulus of beneficial changes in brain, is itself regulated by CIT36 provides one such link, and the data showing control of adipocyte differentiation and fat-associated macrophage polarization by the myokine FNDC5/irisin provides another17, but the overall configuration of the hypothesized upstream control of the ARI suite is still very fragmentary.
Three of the elements of the ARI collection—irisin, GPLD1, and the set of acute phase proteins modulated by the p38 MAPK cascade—are detectable in plasma, and might serve as a bridge between the mouse studies and tests in human populations. A careful presentation of this topic deserves a review of its own, but a sketch of such a program would include some obvious questions. Do drugs that slow aging in mice, such as Aca and Cana, modify these endpoints in healthy humans? Do agents which are postulated to be beneficial in humans despite a lack of lifespan effect in mice, such as NR or metformin9,13, alter ARIs in people? Do levels of these postulated ARIs predict subsequent major health events in healthy middle-aged or older people, either in a prospective study or in an analysis of health changes in a population for which plasma samples had previously been archived? There are relatively small populations in which muscle or adipose tissue biopsies have been collected in addition to plasma samples, and these valuable tissue banks could also be evaluated to seek links between tissue ARIs and health, or between plasma and tissue indicators.
We have tried here to present both a conceptual advance and the first glimpses of a set of cellular and molecular details of how aging rate might be regulated. The conceptual step lies in the realization that there may be a moderately large set of alterations that are caused by antiaging interventions of multiple kinds, and that affect multiple cell types and tissues. The counter-hypothesis—that is, that the effects on aging of specific genes, specific diets, specific drugs are each mediated by distinct perturbations and pathways—was quite plausible, and many experts remain very skeptical of the notion of a “shared common pathway” of antiaging causality that might link these distinct interventions. These skeptics ought now to reconsider the idea of overlapping—perhaps highly overlapping—pathways shared by many varieties of slow-aging mice. It will be critical for those trying to integrate the idea of shared ARIs into their own ideas about aging and age-dependent diseases to see the clear distinction between “biomarkers of aging,” that is, age-sensitive endpoints that might in theory change more slowly in a slow-aging animals, and “aging rate indicators” that, in theory, reflect the rate of aging without regard to their own alterations as an animal ages. This uncoupling between age and ARI status also has an important practical advantage: The causes and effects of each element of the ARI test kit can be studied in young adult mice, which are far easier to accumulate than older animals.
The work reviewed here also gives an initial glimpse into some of the specific cellular and molecular changes that deserve more intense scrutiny, including changes in cellular differentiation, cellular composition of complex tissues, intercellular communication (muscle to fat; liver to brain; etc.), kinase control of translation and inflammatory status, selective mRNA translation, and other pathways implicated in the endpoints listed in Table 1. It is clear that the work done so far is just scratching the surface: The observation, for example, that altered macrophage polarity is seen in all nine tested slow-aging models is just a first step in working out both the basis for this shift, its possible implications for macrophage differentiation in other organs and tissues, and its possible effects on a wide range of age-dependent diseases, neoplastic and degenerative.
The biogerontology community has invested many years of work to the search, so far only modestly successful, for biomarkers of aging that can be used as surrogate endpoints in tests for effects of drugs or polymorphic markers or dietary factors on the rate of aging. As work on predictive biomarkers continues, we propose that parallel investigations on ARIs, the “speedometers” of aging rate, are likely to provide additional and complementary levers for exploration of the control and consequences of aging.
Acknowledgments
The work described in this review was supported by National Institutes of Health (NIH) grants AG023122, AG024824, and AG022303, and by the Glenn Foundation for Aging Research. The authors thank Scott Leiser, Scott Pletcher, and David Lombard for comments on a draft of this article.
References
- 1.Miller RA (2004). ‘Accelerated aging’: A primrose path to insight? Aging Cell 3(2), 47–51. doi: 10.1111/j.1474-9728.2004.00081.x. [DOI] [PubMed] [Google Scholar]
- 2.Orentreich N, Matias JR, DeFelice A, & Zimmerman JA (1993). Low methionine ingestion by rats extends life span. J. Nutr 123, 269–274. doi: 10.1093/jn/123.2.269. [DOI] [PubMed] [Google Scholar]
- 3.Brown-Borg HM, Borg KE, Meliska CJ, & Bartke A (1996). Dwarf mice and the ageing process. Nature 384(6604), 33–33. doi: 10.1038/384033a0. [DOI] [PubMed] [Google Scholar]
- 4.Flurkey K, Papaconstantinou J, Miller RA, & Harrison DE (2001). Lifespan extension and delayed immune and collagen aging in mutant mice with defects in growth hormone production. Proc. Natl. Acad. Sci. U. S. A 98(12), 6736–6741. doi: 10.1073/pnas.111158898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coschigano KT, Clemmons D, Bellush LL, & Kopchick JJ (2000) Assessment of growth parameters and life span of GHR/BP gene-disrupted mice. Endocrinology 141(7), 2608–2613. doi: 10.1210/endo.141.7.7586. [DOI] [PubMed] [Google Scholar]
- 6.Conover CA, & Bale LK (2007). Loss of pregnancy-associated plasma protein A extends lifespan in mice. Aging Cell 6(5), 727–729. doi: 10.1111/j.1474-9726.2007.00328.x. [DOI] [PubMed] [Google Scholar]
- 7.Harrison DE, Strong R, Sharp ZD, … Fernandez E, & Miller RA (2009). Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460(7253), 392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Harrison DE, Strong R, Allison DB, … Wilkinson JE, & Miller RA (2014). Acarbose, 17-alpha-estradiol, and nordihydroguaiaretic acid extend mouse lifespan preferentially in males. Aging Cell 13(2), 273–282. doi: 10.1111/acel.12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strong R, Miller RA, Antebi A, … Nadon NL, & Harrison DE (2016). Longer lifespan in male mice treated with a weakly estrogenic agonist, an antioxidant, an α-glucosidase inhibitor or a Nrf2-inducer. Aging Cell 15(5), 872–884. doi: 10.1111/acel.12496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miller RA, Harrison DE, Allison DB, … Lombard DB, & Strong R (2020). Canagliflozin extends life span in genetically heterogeneous male but not female mice. JCI Insight 5(21), e140019. doi: 10.1172/jci.insight.140019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Macchiarini F, Miller RA, Strong R, Rosenthal N, & Harrison DE (2021). NIA Interventions Testing Program: A collaborative approach for investigating interventions to promote healthy aging. In Musi N & Hornsby PJ (Eds.), Handbook of the biology of aging. London (UK): Academic Press, 219–235. [Google Scholar]
- 12.Harrison DE, Strong R, Alavez S, … Wilkinson JE, & Miller RA (2019). Acarbose improves health and lifespan in aging HET3 mice. Aging Cell 18(2), e12898. doi: 10.1111/acel.12898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harrison DE, Strong R, Reifsnyder P, … Bogue M, & Miller RA (2021). 17-a-estradiol late in life extends lifespan in aging UM-HET3 male mice; nicotinamide riboside and three other drugs do not affect lifespan in either sex. Aging Cell 20(5), e13328. doi: 10.1111/acel.13328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Snyder JM, Casey KM, Galecki A, … Miller RA, & Ladiges W (2023). Canagliflozin retards age-related lesions in heart, kidney, liver, and adrenal gland in genetically heterogenous male mice. Geroscience 45(1), 385–397. doi: 10.1007/s11357-022-00641-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Townsend KL, & Tseng YH (2014). Brown fat fuel utilization and thermogenesis. Trends Endocrinol. Metab 25(4), 168–177. doi: 10.1016/j.tem.2013.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loyd C, & Obici S (2014). Brown fat fuel use and regulation of energy homeostasis. Curr. Opin. Clin. Nutr. Metab. Care 17(4), 368–372. doi: 10.1097/MCO.0000000000000063. [DOI] [PubMed] [Google Scholar]
- 17.Li X, Frazier JA, Spahiu E, McPherson M, & Miller RA (2020). Muscle-dependent regulation of adipose tissue function in long-lived growth hormone-mutant mice. Aging (Albany NY) 12(10), 8766–8789. doi: 10.18632/aging.103380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li X, McPherson M, Hager M, Fang Y, Bartke A, & Miller RA (2022). Transient early life growth hormone exposure permanently alters brain, muscle, liver, macrophage, and adipocyte status in long-lived Ames dwarf mice. FASEB J. 36(7), e22394. doi: 10.1096/fj.202200143R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li X, Hager M, McPherson M, … Lombard D, & Miller RA (2022). Recapitulation of anti-aging phenotypes by global, but not by musclespecific, deletion of PAPP-A in mice. Geroscience 45(2), 931–948. doi: 10.1007/s11357-022-00692-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tran CM, Mukherjee S, Ye L, … Seale P, & Baur JA (2016). Rapamycin blocks induction of the thermogenic program in white adipose tissue. Diabetes 65(4), 927–941. doi: 10.2337/db15-0502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guilherme A, Yenilmez B, Bedard AH, … Collins S, & Czech MP (2020). Control of adipocyte thermogenesis and lipogenesis through beta3-adrenergic and thyroid hormone signal integration. Cell Rep. 31(5), 107598. doi: 10.1016/j.celrep.2020.107598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huwatibieke B, Yin W, Liu L, … Zhang W, & Li Y (2021). Mammalian target of rapamycin signaling pathway regulates mitochondrial quality control of brown adipocytes in mice. Front Physiol. 12, 638352. doi: 10.3389/fphys.2021.638352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kohno K, Koya-Miyata S, Harashima A, … Ushio S, & Iwaki K (2021). Inflammatory M1-like macrophages polarized by NK-4 undergo enhanced phenotypic switching to an anti-inflammatory M2-like phenotype upon co-culture with apoptotic cells. J. Inflamm. (Lond.) 18(1), 2. doi: 10.1186/s12950-020-00267-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Costantini A, Viola N, Berretta A, … Olivieri F, & Bonafè M (2018). Age-related M1/M2 phenotype changes in circulating monocytes from healthy/unhealthy individuals. Aging (Albany NY) 10(6), 1268–1280. doi: 10.18632/aging.101465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.List EO, Berryman DE, Funk K, … Bartke A, & Kopchick JJ (2014). Liver-specific GH receptor gene-disrupted (LiGHRKO) mice have decreased endocrine IGF-I, increased local IGF-I, and altered body size, body composition, and adipokine profiles. Endocrinology 155(5), 1793–1805. doi: 10.1210/en.2013-2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.List EO, Berryman DE, Funk K, … Bartke A, & Kopchick JJ (2013). The role of GH in adipose tissue: Lessons from adipose-specific GH receptor gene-disrupted mice. Mol. Endocrinol 27(3), 524–535. doi: 10.1210/me.2012-1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.List EO, Berryman DE, Ikeno Y, … Miller RA, & Kopchick JJ (2015). Removal of growth hormone receptor (GHR) in muscle of male mice replicates some of the health benefits seen in global GHR−/− mice. Aging (Albany NY) 7(7), 500–512. doi: 10.18632/aging.100766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bostrom P, Wu J, Jedrychowski MP, … Gygi SP, & Spiegelman BM (2012). A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 481(7382), 463–468. doi: 10.1038/nature10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Norheim F, Langleite TM, Hjorth M, … Jensen J, & Drevon CA (2014). The effects of acute and chronic exercise on PGC-1alpha, irisin and browning of subcutaneous adipose tissue in humans. FEBS J. 281 (3), 739–749. doi: 10.1111/febs.12619. [DOI] [PubMed] [Google Scholar]
- 30.Lacerda R, Menezes J, & Romao L (2017). More than just scanning: the importance of cap-independent mRNA translation initiation for cellular stress response and cancer. Cell. Mol. Life. Sci 74(9), 1659–1680. doi: 10.1007/s00018-016-2428-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dominick G, Bowman J, Li X, Miller RA, & Garcia GG (2017). mTOR regulates the expression of DNA damage response enzymes in long-lived Snell dwarf, GHRKO, and PAPPA-KO mice. Aging Cell 16(1), 52–60. doi: 10.1111/acel.12525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ozkurede U, Kala R, Johnson C, Shen Z, Miller RA, & Garcia GG (2019). Cap-independent mRNA translation is upregulated in long-lived endocrine mutant mice. J. Mol. Endocrinol 63(2), 123–138. doi: 10.1530/JME-19-0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shen Z, Hinson A, Miller RA, & Garcia GG (2021). Cap-independent translation: A shared mechanism for lifespan extension by rapamycin, acarbose, and 17α-estradiol. Aging Cell 20(5), e13345. doi: 10.1111/acel.13345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fujihara Y, & Ikawa M (2016). GPI-AP release in cellular, developmental, and reproductive biology. J. Lipid. Res 57(4), 538–545. doi: 10.1194/jlr.R063032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horowitz AM, Fan X, Bieri G, … Williams KE, & Villeda SA (2020). Blood factors transfer beneficial effects of exercise on neurogenesis and cognition to the aged brain. Science 369(6500), 167–173. doi: 10.1126/science.aaw2622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li X, Shi X, McPherson M, Hager M, Garcia GG, & Miller RA (2022). Cap-independent translation of GPLD1 enhances markers of brain health in long-lived mutant and drug-treated mice. Aging Cell 21(9), e13685. doi: 10.1111/acel.13685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Panici JA, Harper JM, Miller RA, Bartke A, Spong A, & Masternak MM (2010). Early life growth hormone treatment shortens longevity and decreases cellular stress resistance in long-lived mutant mice. FASEB J. 24(12), 5073–5079. doi: 10.1096/fj.10-163253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sadagurski M, Landeryou T, Cady G, … Bartke A, & Miller RA (2015). Growth hormone modulates hypothalamic inflammation in long-lived pituitary dwarf mice. Aging Cell 14(6), 1045–1054. doi: 10.1111/acel.12382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shindyapina AV, Cho Y, Kaya A, … Peshkin L, & Gladyshev VN (2022). Rapamycin treatment during development extends life span and health span of male mice and Daphnia magna. Sci. Adv 8(37), eabo5482. doi: 10.1126/sciadv.abo5482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun L, Sadighi Akha AA, Miller RA, & Harper JM (2009). Life-span extension in mice by preweaning food restriction and by methionine restriction in middle age. J. Gerontol. A Biol. Sci. Med. Sci 64(7), 711–722. doi: 10.1093/gerona/glp051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dominick G, Berryman DE, List EO, … Miller RA, & Garcia GG (2015). Regulation of mTOR activity in Snell dwarf and GH receptor gene-disrupted mice. Endocrinology 156(2), 565–575. doi: 10.1210/en.2014-1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wink L, Miller RA, & Garcia GG (2022). Rapamycin, Acarbose and 17α-estradiol share common mechanisms regulating the MAPK pathways involved in intracellular signaling and inflammation. Immun. Ageing 19(1), 8. doi: 10.1186/s12979-022-00264-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Joshi S, & Platanias LC (2014). Mnk kinase pathway: Cellular functions and biological outcomes. World J. Biol. Chem 5(3), 321–333. doi: 10.4331/wjbc.v5.i3.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morrison DK (2012). MAP kinase pathways. Cold Spring Harb. Perspect. Biol 4(11), a011254. doi: 10.1101/cshperspect.a011254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Canovas B, & Nebreda AR (2021). Diversity and versatility of p38 kinase signalling in health and disease. Nat. Rev. Mol. Cell Biol 22(5), 346–366. doi: 10.1038/s41580-020-00322-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garratt M, Leander D, Pifer K, … Brooks SV, & Miller RA (2019). 17-α estradiol ameliorates age-associated sarcopenia and improves late-life physical function in male mice but not in females or castrated males. Aging Cell 18(2), e12920. doi: 10.1111/acel.12920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Herrera JJ, Louzon S, Pifer K, … Miller RA, & Garratt M (2020). Acarbose has sex-dependent and -independent effects on age-related physical function, cardiac health, and lipid biology. JCI Insight 5(21), e137474. doi: 10.1172/jci.insight.137474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ortega-Molina A, Efeyan A, Lopez-Guadamillas E, … Bischoff JR, & Serrano M (2012). Pten positively regulates brown adipose function, energy expenditure, and longevity. Cell Metab. 15(3), 382–394. doi: 10.1016/j.cmet.2012.02.001. [DOI] [PubMed] [Google Scholar]