

Summary
Neddylation is a post-translational mechanism that adds a ubiquitin-like protein, namely neural precursor cell expressed developmentally downregulated protein 8 (NEDD8). Here, we show that neddylation in mouse liver is modulated by nutrient availability. Inhibition of neddylation in mouse liver reduces gluconeogenic capacity and the hyperglycemic actions of counter-regulatory hormones. Furthermore, people with type 2 diabetes display elevated hepatic neddylation levels. Mechanistically, fasting or caloric restriction of mice leads to neddylation of phosphoenolpyruvate carboxykinase 1 (PCK1) at three lysine residues—K278, K342, and K387. We find that mutating the three PCK1 lysines that are neddylated reduces their gluconeogenic activity rate. Molecular dynamics simulations show that neddylation of PCK1 could re-position two loops surrounding the catalytic center into an open configuration, rendering the catalytic center more accessible. Our study reveals that neddylation of PCK1 provides a finely tuned mechanism of controlling glucose metabolism by linking whole nutrient availability to metabolic homeostasis.
Keywords: PCK1, glucose metabolism, neddylation, glucagon, fasting, calorie restriction, type 2 diabetes
Graphical abstract
Highlights
-
•
Global neddylation in the liver is modulated by nutrient availability
-
•
Inhibition of hepatic neddylation reduces gluconeogenic capacity
-
•
Neddylation is elevated in the liver of people with type 2 diabetes
-
•
Mutation of lysine residues that can be neddylated worsens PCK1 activity
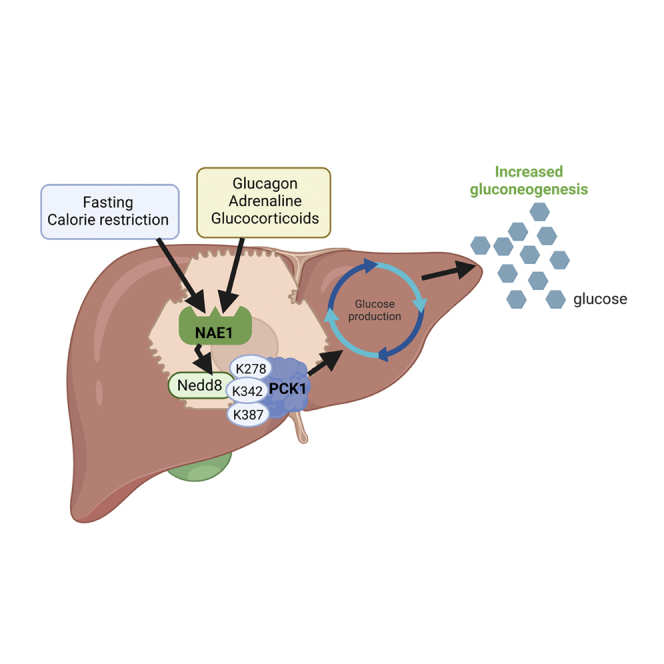
The neddylation of PCK1 at three key lysine residues plays a physiological role in glucose homeostasis by inducing the synthesis of glucose in conditions of nutrient deficiency and by being a common mechanism shared by the counter-regulatory hormones glucagon, adrenaline, and glucocorticoids in the counter-regulatory response to increase glucose availability.
Introduction
Glucose is the main source of cellular energy, and its blood levels are maintained within a narrow range.1 During fasting, the liver plays a critical role in maintaining glucose homeostasis by mobilizing internal nutrient stores and de novo glucose production (gluconeogenesis) from non-carbohydrate precursors.2,3,4 These types of physiological metabolic reprogramming support the body’s energetic demands based on nutrient availability, and alterations in this reprogramming underlies abnormal glucose homeostasis under different pathological conditions, such as cancer and diabetes.4 Gluconeogenesis also contributes to hyperglycemia in type 2 diabetes (T2D).5 This gluconeogenic function is tightly regulated at multiple levels, such as by hormonal secretion, gene transcription, and post-translational modifications (PTMs).6,7,8
The neural precursor cell expressed developmentally downregulated protein 8 (NEDD8) is a ubiquitin-like protein implicated in neddylation, a PTM which consists of the reversible covalent conjugation of NEDD8 to a lysine residue of the substrate protein (predominantly on cullin proteins). This conjugation occurs in a three-step enzymatic process using NEDD8-activating enzyme E1 (NAE1) that initiates the NEDD8 transfer cascade, E2-conjugating enzymes, and E3 ligases.9,10,11,12 Many of the known neddylation substrates have several activities with specific roles in cell cycle progression, cell growth, and survival,9,12,13,14,15 and preclinical results indicate that the inhibition of NEDD8 conjugation might be an effective anti-cancer strategy.16 Indeed, MLN4924 (known as Pevonedistat) is a first-in-class small molecular inhibitor of NAE1 currently in phase I/II clinical trials for patients with myeloid disorders.14,16,17 Despite all these studies, the physiological role of neddylation remains largely unknown, although a pivotal role in liver disease progression from steatosis to hepatocarcinoma had been proposed.
This study shows that hepatic neddylation is regulated by nutrient availability, and the inhibition of neddylation in the liver attenuates hepatic glucose production (HGP) upon fasting or caloric restriction (CR). Mice where hepatic neddylation is disrupted do not exhibit the expected hyperglycemic effect of the counter-regulatory hormones glucagon, adrenaline, and glucocorticoids. In people with T2D, hepatic levels of NAE1 and NEDD8 are increased and positively correlated with fasting glucose. Mechanistically, phosphenolpyruvate carboxykinase (PCK1) was detected to be neddylated at three lysines (K278, K342, and K387) and the ectopic expression of mutant PCK1 was unable to induce glucose production in hepatocytes or mice. Our findings demonstrate that neddylation regulates the gluconeogenic activity of PCK1, an effect of biological relevance on whole-body glucose homeostasis.
Results
Hepatic neddylation is regulated by nutrient availability
Neddylation has been implicated in various aspects of liver physiology, such as β-oxidation18 or insulin signaling,19 as well as in pathophysiological conditions, including non-alcoholic fatty liver disease (NAFLD),20 fibrosis,21 or cholangiocarcinogenesis.22 Given the key role that the liver plays in maintaining glucose homeostasis, we questioned whether neddylation could be involved in the adaptation of glucose production to nutrient availability. Mice that were fasted for 24 h showed increased protein levels of NAE1, NEDD8, and neddylated hepatic cullins (a family of proteins that are neddylated by NEDD8); of note, these fasting-induced levels decreased after refeeding (Figure 1A). In contrast, the ubiquitin E1 enzyme (UBE1), which increases global neddylation following stress conditions,23,24 was unchanged by fasting/refeeding (Figure S1A). Given that circulating glucose is severely affected by energy intake, we fasted mice for 24 h, fed them a one-time supplement of sugar (or not, for the control group), and immediately tested for molecular changes to hepatic proteins. Sugar-fed fasted mice lost the same amount of weight as the non-sugar-fed fasted (control) group; however, their blood glucose levels were similar to those mice fed ad libitum (Figures S1B and S1C), consistent with previous reports.25 Strikingly, we observed that the fasting-induced increases of hepatic protein levels of NAE1, NEDD8, and neddylated cullins were reduced in the sugar-fed group (Figure 1B). Notably, in vitro results reproduced these data as human hepatic THLE2 cells maintained in a medium depleted of nutrients (Krebs-Henseleit-HEPES [KHH]) showed higher levels of NAE1, and the supplementation with glucose reduced its protein levels (Figure S1D). We next tested the effects of leptin, whose levels are markedly reduced upon fasting. Fasting-induced elevation of protein levels of NAE1, NEDD8, and neddylated cullins was blunted when fasted mice were treated with leptin (Figure 1C). In line with this, mice lacking leptin receptors (db/db mice) displayed elevated levels of NEDD8 and neddylated cullins (Figure 1D). Furthermore, we investigated the effect of a CR by feeding mice only 40% of their daily food intake.26 We found that the levels of NAE1 and neddylated cullins, but not of UBE1, were significantly increased compared with mice fed ad libitum (Figures 1E and S1E). Overall, these data suggested that the levels of hepatic neddylation were increased by nutrient deficiency through a leptin-receptor-dependent mechanism.
Figure 1.

Neddylation is activated during nutrient deprivation and is required to maintain blood glucose levels
(A–E) The hepatic protein levels for NAE1, NEDD8, and neddylated-cullins were determined from mice in the following conditions: (A) after (i) ad libitum feeding (as a control), (ii) 24-h fast; or (iii) 24-h fast plus 24-h refeed (n = 4–5 per group); (B) after (i) ad libitum feeding, (ii) 24-h fast; or (iii) 24-h fast followed by a sugar feeding (n = 5 or 8 per group); (C) after (i) ad libitum feeding, (ii) 24-h fast; or (iii) 24-h fast followed by leptin i.p. (n = 5–8 per group); (D) wild-type and db/db mice; and (E) after (i) ad libitum feeding; or (ii) caloric restriction (CR) (n = 8–18 per group).
(F–I) Pyruvate tolerance test (PTT) of (F) wild-type (WT) mice treated with vehicle or the NAE1-inhibitor MLN4924, (G) NAE1+/− mice or their control littermates, (H) Alfp-Cre+/− mice injected with AAV-DIO expressing either GFP or shNAE1 and (I) Alfp-Cre+/− mice injected with AAV-DIO expressing either GFP or shNEDD8.
(J–M) Blood glucose levels after 24-h fasting for (J) WT mice treated with vehicle or MLN4924, (K) NAE1+/− mice and their control littermates, (L) Alfp-Cre+/− mice injected with AAV-DIO expressing either GFP or shNAE1 and (M) Alfp-Cre+/− mice injected with AAV-DIO expressing either GFP or shNEDD8.
(N–Q) Blood glucose levels of mice subjected to 60% CR for (N) WT mice treated with vehicle or MLN4924, (O) NAE1+/− mice or their control littermates, (P) Alfp-Cre+/− mice injected with AAV-DIO expressing either GFP or shNAE1 and (Q) Alfp-Cre+/− mice injected with AAV-DIO expressing either GFP or shNEDD8; n = 5–15 per group.
(R and S) (R) Hepatic glucose production in Alfp-Cre+/− mice injected with AAV-DIO expressing either GFP or shNAE1 upon fasting and (S) caloric restriction; n = 6–10. Expression of glyceraldehyde 3- phosphate dehydrogenase (GAPDH) served as a loading control, and control values were normalized to 100%. Data are presented as mean ± SEM; two-tailed unpaired t test (D–S) and one-way ANOVA followed by Bonferroni post hoc test (A–C) ∗p < 0.05, ∗∗p < 0.01 and ∗∗∗p < 0.001.
Inhibition of hepatic neddylation reduces glucose production
We next aimed to elucidate the physiological role of endogenous hepatic neddylation in glucose metabolism. For this, we used the following four models of neddylation impairment in mice: (1) pharmacological inhibition using MLN4924, a specific, small inhibitor of NAE117; (2) Nae1 heterozygous mice (NAE1+/−) mice27; (3) mice with a Cre-dependent short-hairpin RNA (shRNA)-mediated liver-specific knockdown of NAE1 (associated adenoviruses [AAV]-DIO-shNAE1); and (4) mice with a Cre-dependent shRNA-mediated liver-specific knockdown of NEDD8 (AAV-DIO-shNEDD8). Each of the four animal models was fasted overnight and then subjected to a pyruvate tolerance test (PTT), where pyruvate administration acutely stimulates HGP. The four animal models of disrupted hepatic neddylation showed lower glucose levels compared with control mice, directly reflecting impaired gluconeogenesis since glycogen depots are depleted after an overnight fasting28 (Figures 1F–1I), without differences in glucose tolerance and insulin sensitivity (Figures S2A–S2L). There are two main mechanisms that the liver uses to produce glucose: glycogenolysis and de novo gluconeogenesis. Although glycogenolysis is important during the first hours of fasting, HGP relies exclusively on gluconeogenesis when fasting is prolonged.29 Given our results showing low blood glucose levels during a PTT when hepatic neddylation is inhibited, to further study the specific role of neddylation in hepatic gluconeogenesis and to avoid glycogenolysis contribution to HGP as a confounding factor, in all our further experimental designs, mice were fasted for 24 h or subjected to a 60% CR for 4 days, and physiological conditions where hepatic glycogen depots are depleted.
After a 24-h fast, mice in all models displayed reduced blood glucose levels (Figures 1J–1M). When subjected to CR, none of these models were able to maintain blood glucose at the same level as control mice (Figures 1N–1Q), despite showing a similar course of weight loss during the CR (Figures S2M–S2P). Consistently, fasted (Figure 1R) and calorie-restricted (Figure 1S) hepatic Nae1 knockdown mice exhibited reduced HGP compared with control littermates. We also studied neddylation in the liver of female mice, finding a similar increase in NAE1 protein levels upon fasting (Figure S2Q). In agreement, female mice with a shRNA-mediated knockdown of NAE1 (AAV-DIO-shNAE1) displayed a similar response to male mice, since blood glucose levels after 24 h of fasting and the glucose excursion during a PTT were lower than in control mice, but not after glycerol administration (Figures S2R–S2T). Altogether, these findings demonstrate that neddylation plays an important role in the maintenance of glucose production by the liver in a sex-independent manner.
Inhibition of hepatic neddylation does not modify protein levels of gluconeogenic enzymes or acetyl-CoA
Since inhibition of neddylation in the liver diminished gluconeogenesis, we measured the expression of key gluconeogenic enzymes and acetyl-coenzyme A (CoA) in the liver of two animal models of neddylation impairment: (1) pharmacological inhibition using MLN4924 and (2) mice with a shRNA-mediated knockdown of NAE1. We challenged these mice to 24 h of fasting and CR, and our results indicated that protein levels of pyruvate carboxylase (PC), glucose 6-phosphatase (G6Pase), and phosphoenolpyruvate carboxykinase 1 (PCK1) were increased in the liver of fasted and calorie-restricted mice. However, there were no differences between vehicle- and MLN4924-treated mice or between control and shNAE1-treated mice (Figures S3A, S3B, S3G, and S3H). Similarly, no differences were detected in hepatic levels of acetyl CoA after the inhibition of neddylation (Figures S3A, S3B, S3G, and S3H).
Maintaining glucose homeostasis requires an efficient counter-regulatory response to glucose intake/production. Therefore, we also measured circulating levels of hormones implicated in the glucose counter-regulatory response, namely adrenaline, glucagon, and corticosterone in the models described above. No changes were found between control mice and those animals where neddylation was obstructed (Figures S3C, S3D, S3I, and S3J). Insulin levels were reduced in fasted shNAE1-treated mice (Figure S3D), with no changes in the other experimental models (Figures S3C, S3I, and S3J). Finally, hepatic alanine and total amino acids as well as plasma amino acids were also measured in the two animal models of neddylation impairment described above subjected to 24 h of fasting or CR. These measurements did not detect differences in the liver content of alanine or total amino acids and plasma levels of total amino acids between the two animal models under fasting and CR, except for mice treated with MLN4924 and mice where NAE1 was silenced in the liver and fasted for 24 h, which displayed higher levels of amino acids in the liver (Figures S3E and S3F). As amino acids are important gluconeogenic substrates during fasting and CR, these results indicate that reduced glucose production when hepatic neddylation is disrupted is non-dependent of the availability of amino acids.
Hepatic neddylation regulates the gluconeogenic action of glucagon, adrenaline, and glucocorticoids
Given our results indicating that gluconeogenesis is impaired when hepatic neddylation is inhibited, we wanted to study in detail if neddylation was involved in the glucose counter-regulatory response. To figure this out, we treated our four models of impaired neddylation with glucagon, adrenaline, or glucocorticoids, all of which stimulate gluconeogenesis.30 In normal mice, this administration caused the expected hyperglycemic actions, with upregulated levels of NAE1 and NEDD8 and neddylation of hepatic cullins (Figures S4A–4F). In contrast, neddylation deficiency significantly attenuated the hyperglycemic effects of hormone treatment in each of the four models (Figure 2). Thus, neddylation mediates the action of glucose counter-regulatory hormones and is required for HGP. The levels of gluconeogenic enzymes and acetyl-CoA were similar in mice whose neddylation was pharmacologically inhibited and in mice with a shRNA-mediated knockdown of NAE1 treated with glucagon, adrenaline, or glucocorticoids, except for mice treated with adrenaline and MLN4924, which displayed lower acetyl-CoA (Figures S4G–S4R). This suggests that overall, neddylation does not regulate the protein levels of the gluconeogenic enzymes nor the availability of acetyl-CoA.
Figure 2.

Neddylation is required for the gluconeogenic action of counter-regulatory hormones
(A–D) Blood glucose after intraperitoneal (i.p.) administration of saline or glucagon (200 μg kg−1) for (A) WT mice treated with vehicle or MLN4924, (B) NAE1+/− mice or their control littermates, (C) Alfp-Cre+/− mice injected with AAV-DIO expressing either GFP or shNAE1 and (D) Alfp-Cre+/− mice injected with AAV-DIO expressing either GFP or shNEDD8.
(E–H) Blood glucose levels after i.p. administration of saline or adrenaline (100 μg kg−1) for (E) WT mice treated with vehicle or MLN4924, (F) NAE1+/− and their control littermates, (G) Alfp-Cre+/− mice injected with AAV-DIO expressing either GFP or shNAE1 and (H) Alfp-Cre+/− mice injected with AAV-DIO expressing either GFP or shNEDD8.
(I–L) Blood glucose after i.p. administration of saline or hydrocortisone (20 mg kg−1) for (I) WT mice treated with vehicle or MLN4924, (J) NAE1+/− mice or their control littermates, (K) Alfp-Cre+/− mice injected with AAV-DIO expressing either GFP or shNAE1 and (L) Alfp-Cre+/− mice injected with AAV-DIO expressing either GFP or shNEDD8; n = 3–10 per group. Data are presented as mean ± SEM; one-way ANOVA followed by Bonferroni post hoc test: ∗p < 0.05, ∗∗p < 0.01 and ∗∗∗p < 0.001. # denotes differences among the different animal groups treated with the same glucoregulatory hormone.
Hepatic neddylation is associated with insulin resistance
We next evaluated whether disrupted neddylation could lead to insulin resistance. Insulin inhibits glucose production in the liver, and this action is widely used to measure hepatic insulin function.31 For this, we fed mice with a high-fat diet (HFD) for a short term (4 days), which impaired hepatic insulin sensitivity and stimulated HGP but did not lead to significant changes in body weight, as expected (Figures S4S and S4T).32,33 Strikingly, mice in all four conditions of neddylation inhibition (e.g., pharmacological or genetic) displayed reduced glucose levels during the PTT after 4 days on HFD (Figures 3A–3D). These results indicated that blocking neddylation ameliorated HFD-induced gluconeogenesis. We next performed a gain-of-function experiment by overexpressing NAE1 (AAV-DIO-NAE1) in the liver (livNAE1 mice). These mice maintained the same weight as control mice but displayed an increased capacity to convert pyruvate into glucose with no changes in glucose tolerance or insulin sensitivity compared with control mice (Figures 3E–3J). In addition, livNAE1 mice also exhibited higher glucose levels than control mice during 24 h fasting (Figure 3K) and after acute treatment with insulin (Figure 3L), suggesting that activation of neddylation in liver favors HGP and precludes the hypoglycemic actions of insulin.
Figure 3.

Inhibition of neddylation reduces high-fat diet-induced-glucose levels
(A–K) Pyruvate tolerance test (PTT) from mice fed a HFD (60%) for 4 days, for (A) WT mice treated with vehicle or MLN4924, (B) NAE1+/− mice or their control littermates, (C) Alfp-Cre+/− mice injected with AAV-DIO expressing either GFP or shNAE1 and (D) Alfp-Cre+/− mice injected with AAV-DIO expressing GFP or shNEDD8. NAE1 protein levels detected by immunohistochemistry (E) and western blot (F) in Alfp-Cre+/− mice injected with AAV-DIO-GFP or AAV-DIO-NAE1 to overexpress NAE1. Body weight evolution (G), GTT (H), ITT (I), and PTT (J) for Alfp-Cre+/− mice injected with AAV-DIO expressing GFP or NAE1. Blood glucose levels (K) after 24-h fasting for Alfp-Cre+/− mice injected with AAV-DIO expressing GFP or NAE1.
(L) Blood glucose levels in Alfp-Cre+/− mice injected with AAV-DIO expressing GFP or NAE1 and treated with saline or insulin (0.35 U kg−1); n = 5–15 per group. Expression of GAPDH served as a loading control, and control values were normalized to 100%. Data are presented as mean ± SEM; two-tailed unpaired t test (A–K) and one-way ANOVA followed by Bonferroni post hoc test (L): ∗p < 0.05, ∗∗p < 0.01 and ∗∗∗p < 0.001. # denotes differences among animal groups treated with insulin.
Hepatic neddylation is increased in the liver of people with type 2 diabetes
People with diabetes often have an impaired insulin-dependent suppression of HGP, which contributes to hyperglycemia in the fasted and postprandial states.34 We assessed the levels of neddylation in livers of adults (men and women) with obesity and either normoglycemia or T2D (Table S2). Our data showed higher protein levels of NAE1, NEDD8, and neddylated cullins in the subgroup of people with obesity and T2D vs. levels in people with obesity and normoglycemia (Figure 4A), as well as a positive correlation of the protein levels of NAE1 and NEDD8 with fasting glucose levels and the oral glucose tolerance test (Figures 4B and 4C). Therefore, these findings suggest that changes in hepatic neddylation are associated to hyperglycemia in people with obesity and T2D. When deconvoluted by sex, our data show that protein levels of NAE1 in the liver were equally increased in women and men with obesity and T2D compared with obese women and men with obesity and normoglycemia (Figure 4D). Following a similar trend, protein levels of neddylated cullins and NEDD8 were elevated in both sexes (Figures 4D and 4E).
Figure 4.

Neddylation is elevated in the liver of people with T2D
(A–D) (A) Protein levels of neddylated cullins, free NEDD8 and NAE1 in adults with obesity who were subclassified as having normoglycemia (NG) or type 2 diabetes (T2D) (n = 30 per group).
(B and C) Correlations between the protein levels of NEDD8 and NAE1 and the fasting glucose (mg dL−1) or oral glucose tolerance test (OGTT) (mg dL−1).
(D and E) Protein levels of neddylated cullins, free NEDD8 and NAE1 in (D) men and (E) women with obesity who were subclassified as having normoglycemia (NG) or type 2 diabetes (T2D). Expression of GAPDH served as loading control, and control values were normalized to 100%. Data are presented as mean ± SEM; two-tailed unpaired t test: ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
PCK1 is neddylated upon nutrient deficiency, and this PTM is required for its gluconeogenic activity
Neddylation substrates include not only cullins but also several non-cullin proteins, suggesting that neddylation regulates various cellular processes. For example, neddylation is thought to inhibit the transcriptional activity of the tumor suppressors TP53 and TP7335,36 and to stabilize HuR, thereby supporting the survival of hepatocellular carcinoma and colon cancer cells.37 Furthermore, pharmacological inhibition (using MLN4924) of the neddylation of peroxisome proliferator-activated receptor (PPAR)gamma reduces adipogenesis and lipid droplet formation.20,38,39,40 To elucidate how neddylation affects glucose metabolism, we next delineated the landscape of neddylated proteins under conditions of stimulated gluconeogenesis. For this, we generated transgenic mice that expressed whole-body biotinylated NEDD8 (bionedd8) (Figures S5A and S5B) and subjected them to CR for 4 days or (as a control) an ad libitum diet, where they showed similar body weights and glucose levels to those of wild-type (WT) mice (Figure S5C). Then, we performed biotin pull-down experiments to isolate the hepatic neddylated proteome in the different subgroups. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis using a sequential window acquisition of all theoretical mass spectra (SWATH-MS) approach identified a total of 1,211 proteins. These 1,211 proteins included the proteins present in the eluted (210 proteins that represent 17.3%) and in the flowthrough fractions (Figure 5A). Among the 210 neddylated proteins, not all of them changed their neddylation levels upon nutrient deprivation, but only 61 showed significantly differential neddylated levels after CR. As a positive control for immunoprecipitation of biotin-neddylated proteins, NEDD8 was enriched only in the eluted fraction (but not in the input or flowthrough fractions) (Figure S5D). Functional enrichment analysis of these 61 differentially neddylated proteins showed that gluconeogenesis was overrepresented (Figure 5B); for instance, PCK1, the key limiting enzyme of gluconeogenesis, was present. Correspondingly, we found that PCK1 was enriched in the CR-fed mice compared with the ad libitum-fed control mice.
Figure 5.

PCK1 is neddylated in three lysines after caloric restriction and fasting
(A) Pie chart showing the percentage of neddylated proteins from the total hepatic proteins identified in bionedd8 mice, as determined by LC-MS/MS proteomics (n = 4 or 5).
(B–D) (B) PANTHER functional enrichment analysis of neddylated proteins in liver. Neddylation of PCK1 during a 4-day caloric restriction (of 60%) (C) or 24-h fasting (D), as shown by immunoblot after immunoprecipitation of PCK1.
(E and F) PCK1 and NAE1 protein levels after the immunoprecipitation of PCK1, and NAE1 protein levels relativized to immunoprecipitated PCK1, from the livers of mice fed ad libitum or subjected to a 4-day caloric restriction (of 60%) (E) or 24-h fasting (F); n = 2–4.
(G) Immunoprecipitated PCK1 was subjected to mass spectrometry analysis. Possible modified sites are shown. K∗, modified with GG.
(H) Ribbon representation of M. musculus PCK1 structure with identified neddylation sites colored in dark blue. Loops 95–118 and 460–470 are highlighted in cyan, and the Mn2+ ion, in light green.
(I) Average structures of PCK1 (left, olive) and PCK1-K387-NEDD8 (right, red) after molecular dynamics simulation. Neddylation of PCK1 alters the dynamics and conformation of loops 95–118 and 460–470 (cyan dashed lines). In-depth view of the opening the protein’s crevice upon PCK1 neddylation that makes the Mn2+ ion accessible to the substrate.
To corroborate these results, we immunoprecipitated PCK1 from the liver of the CR-fed mice or the 24-h fasted mice; these experiments revealed that NEDD8 co-precipitated at higher levels with PCK1 in both conditions of nutrient deficiency than in ad libitum conditions (Figures 5C and 5D). Moreover, we also immunoprecipitated PCK1 from the liver of CR-fed mice and 24-h fasted mice treated with vehicle or MLN4924. These experiments revealed that the PCK1-NEDD8-signal was markedly abolished by MLN4924 treatment (Figure S5E). In agreement with our results showing that PCK1 is neddylated upon nutrient deprivation, we also detected a physical and direct interaction by coimmunoprecipitation between PCK1 and NAE1 under these conditions, indicating that PCK1 and NAE1 are more associated under these conditions (Figures 5E and 5F). As a positive control, we have also immunoprecipitated Cullin2, a very well-described neddylated substrate, from the liver of the CR-fed mice and the 24-h fasted mice. We found a marked increase in the intensity of NEDD8-smear in Cullin2 upon nutrient deprivation (Figure S5F) but lighter relative to the observed when PCK1 is immunoprecipitated, indicating that under these physiological conditions, neddylation is directed to specific substrates (such as PCK1).
STRING analysis of the functional interactome after PCK1 immunoprecipitation comprised 1,631 proteins in the 24-h fasting condition and 1,142 in the CR condition, with 968 of them common to both conditions; furthermore, in both fasted and CR mice, a group of proteins involved in neddylation were co-precipitated (indicated in red; Figure S5G).
To identify modification sites in PCK1 that could potentially be neddylation sites, we first immunoprecipitated PCK1 from the liver of mice fed ad libitum or subjected to fasting, and then, we performed LC-MS/MS analysis. Analysis by PEAKS software of the peptide spectra from PCK1 revealed that three PCK1 lysines were potentially neddylated under fasting conditions: K278, K342, and K387 (Figure 5G). To determine how the post-translational neddylation could affect PCK1 activity, we analyzed its effects on the structure, paying particular attention to the neddylated residues. To gain insight into the structural and dynamical consequences of neddylation of PCK1, we performed molecular dynamics simulations of PCK1 that was (1) non-neddylated, (2) mono-neddylated, (3) bi-neddylated, or (4) tri-neddylated (Figure S6). The three lysine residues are distributed across the surface of the enzyme and were all accessible to the solvent (Figure 5H). Trajectory analysis revealed that PCK1 neddylation mainly affected the atomic fluctuations and position of loops 95–118 and 460–470, which surround the enzyme cavity and catalytic center. Mono-neddylation at K387 induced the greatest perturbation of the enzyme cavity, opening the crevice of the protein and allowing the entry of Mn2+ into the catalytic center of PCK1—a critical step for PCK1 activity (Figures 5I and S6). Of note, K387 is the most highly conserved lysine across evolution of the three lysines and is present in all the PCK1 sequences that we analyzed, over a broad range of species, from mycobacteria to mammals (Figure S7A).
Neddylation of PCK1 in residues K278, K342, and K387 is required for its gluconeogenic activity in vitro
Finally, to gain insights into the functional consequences of neddylation of PCK1, we incubated hepatocytes in a medium without nutrients (to create an in vitro fasting condition) and then silenced NAE1 and NEDD8. Fasted cells had increased PCK1 activity; in contrast, NAE1 and NEDD8 inhibition blocked the fasting-induced PCK1 activity, with concomitant lower glucose levels in the medium (Figures 6A–6C and S7B–S7D). These results were also corroborated in vivo: NAE1 inhibition in liver decreased PCK1 activity in CR-fed mice (Figure S7E). In line with these findings as well as with proteomics and modeling, we found that mutating either the three lysine residues of PCK1 at the same time (e.g., K278, K342, and K387) or K387 alone reduced PCK1 activity and glucose levels in the medium (Figures 6D and 6E). Consistently, activity and glucose production could be restored in PCK1-inhibited, nutrient-depleted hepatocytes by overexpressing WT PCK1 but not by overexpressing either the triple PCK1 mutant or the single (K387R) PCK1 mutant (Figures 6D and 6E), highlighting the relevance of neddylation at these lysine residues for the gluconeogenic functions of PCK1. Importantly, protein levels of PCK1 were similarly elevated by WT and PCK1 mutants (Figure 6F), supporting that neddylated PCK1 and not total protein levels were modulating PCK1 activity.
Figure 6.

Neddylation of PCK1 is required for its gluconeogenic function in vitro
(A and B) (A) PCK1 activity and (B) glucose production in AML12 cells maintained in complete medium (CM) or fasted in KHH medium, and transfected with small interfering RNA (siRNA)-scrambled or siRNA NAE1.
(C) Correlation between glucose production and PCK1 activity.
(D–F) (D) PCK1 activity, (E) glucose production, and (F) protein levels of PCK1 in AML12 cells maintained in complete or KHH medium and transfected with empty siRNA-scrambled, siRNA-PCK1, plasmid control, a plasmid encoding wild-type PCK1 or a plasmid encoding triple- or single-mutated PCK1 (pPCK1-K278R-K342R-K387R or pPCK1-K387R).
(G–O) PCK1 activity, glucose production, and protein levels of PCK1 in AML12 cells under the same conditions as before and treated with MLN4924. n = 5–6. Expression of GAPDH served as a loading control, and control values were normalized to 100%. Data are presented as mean ± SEM; two-tailed unpaired t test (B) and one-way ANOVA followed by Bonferroni post hoc test (A and D–O) ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001.
Since modifications other than neddylation can target these lysines and even other potential PCK1 residues could be neddylated and could not be detected, we repeated the experiments treating our different in vitro experimental groups with MLN4924 to study any potential additive or synergic effect, which is critical especially in the absence of assays assessing the neddylation of the PCK1 mutants. As such, hepatocytes were treated with vehicle or MLN4924 under nutrient-depleted conditions (KHH medium for 6 h). The results indicated that, as expected, the silencing of PCK1 reduced protein levels and activity of PCK1 and glucose release in the medium. The rescue of PCK1-WT restored protein levels and activity of PCK1 and consistently increased glucose secretion into the medium (Figures 6G–6I). However, MLN4924 treatment decreased the activity of PCK1-WT, preventing glucose release into the medium (without changes in PCK1 protein levels) (Figures 6G–6I). Following the same experimental procedure, hepatocytes expressing the triple-mutant PCK1 or single-mutant PCK1 (K387R) were also treated with vehicle or MLN4924. The results indicate that the triple-mutant or the single-mutant PCK1 (K387R) were unable to restore PCK1 activity or glucose levels, and thereby, MLN4924 did not cause any effect (Figures 6J–6O). These findings indicate that mutations in the three lysine residues of PCK1 at the same time (K278, K342, and K387) are relevant to maintain the gluconeogenic capacity of PCK1 and that if PCK1 is neddylated in other residues different from K278, K342, and K387, they are not involved in or are not essential for PCK1 gluconeogenic activity since the treatment with MLN4924 in siPCK1 + pPCK1 K278R-K342R-K387R did not have an additive or synergistic effect, namely MLN4924 did not further reduce glucose production or PCK1 activity in those cells.
Neddylation of PCK1 in residues K278, K342, and K387 triggers hepatic glucose production in vivo
To demonstrate the in vivo relevance of neddylated PCK1 in the three residues described above, we disrupted PCK1 in the liver of adult mice by injecting an AAV-Cre virus in the tail vein of PCK1 loxP mice (PCK1 liver knockout [LKO]). Then, in these mice, we injected AAV-DIO-PCK1wt to rescue the expression of hepatic WT PCK1 or an AAV-DIO-PCK1mut virus to overexpress the triple-mutant PCK1 (K278R-K342R-K387R) in the liver. The deletion of PCK1 in the liver of adult mice reduced glucose levels after the administration of pyruvate, whereas the rescue of WT PCK1 normalized glucose levels during the PTT with no changes in glucose tolerance or insulin sensitivity (Figures 7A–7D). We next challenged these mice to fasting for 24 h and found that the rescue of WT PCK1 displayed higher levels of glucose and HGP than mice where hepatic PCK1 was disrupted (Figure 7E).
Figure 7.

Neddylation of PCK1 is required for its gluconeogenic function in vivo
(A–E) (A) PCK1 protein levels, (B) glucose tolerance test (GTT), (C) insulin tolerance test (ITT), (D) pyruvate tolerance test (PTT), (E) 24 h fasting blood glucose levels, and hepatic glucose production in control mice, mice lacking PCK1 in the liver (PCK1 LKO) (PCK1 loxP mice injected with AAV-Cre in the tail vein) and PCK1 LKO injected with AAV-PCK1 wild type (WT).
(F–K) (F) PCK1 protein levels, (G) GTT, (H) ITT, (I) PTT, (J) glycerol tolerance test (GlyTT), (K) 24 h fasting blood glucose levels and hepatic glucose production in control mice, PCK1 LKO, and PCK1 LKO injected with AAV-PCK1 triple mutant (n = 5–10 animals per group). Expression of GAPDH served as a loading control, and control values were normalized to 100%. Data are presented as mean ± SEM; one-way ANOVA followed by Bonferroni post hoc test: ∗p < 0.05 and ∗∗∗p < 0.001. # denotes differences between PCK1 LKO + AAV-PCK1 WT mice and PCK1 LKO + AAV-PCK1-K278R-K342R-K387R.
However, the hepatic expression of the triple-mutant PCK1 in mice where PCK1 was previously knocked down (Figure 7F) did not normalize glucose levels during a PTT with no changes in GTT and ITT (Figures 7G–7I). To further corroborate our results, we also subjected our mice to a glycerol tolerance test, since glycerol bypasses PCK1 activity, as it is incorporated into the gluconeogenic pathway in later steps. As shown in Figure 7J, there were no differences in glucose production after glycerol administration among different groups, highlighting that gluconeogenic capacity of these mice is specifically disrupted at PCK1 level. Finally, when subjected to 24 h of fasting, mice expressing the triple-mutant PCK1 were unable to increase glycemia or HGP, showing similar blood glucose levels as hepatic PCK1 KO mice (Figure 7K).
Discussion
Overall, our findings show that neddylation of PCK1 at three key lysine residues plays a physiological role in glucose homeostasis, both by regulating the synthesis of glucose in conditions of nutrient deficiency as well as by being a common mechanism shared by the counter-regulatory hormones glucagon, adrenaline, and glucocorticoids in the counter-regulatory response to increase glucose availability.
Post-translational modifications are major regulators of cell functions, and their failure results in different human diseases. NEDD8 is a small protein with a broad expression pattern in adult tissues. Neddylation is the reversible covalent conjugation of NEDD8 to a lysine residue of the substrate protein that modifies proteins. The best-characterized function of NEDD8 is the activation of the cullin-RING family of E3 ubiquitin-ligases (CRLs).15,41 However, multiple other proteins have been associated with or modified by NEDD8, including p53 and Mdm2, epidermal growth factor receptors, among others.9,12,15 Thus, NEDDylation, besides CRLs, regulates many biological processes.
In the liver, neddylation has been associated with several diseases. Neddylation is increased in earlier phases of NAFLD by reducing fatty acid oxidation20 in advanced liver fibrosis and hepatocellular carcinoma21,42,43 and in cholangiocarcinoma.22 Our findings indicate that hepatic neddylation is not only upregulated by liver damage but also by energy status. Although energy deficiency (i.e., fasting or CR) increased neddylation, its levels were reversed when energetic requirements were restored (refeeding). The regulation of neddylation levels by nutrient availability was functionally linked to its role in glucose production in the liver. For instance, pharmacological and genetic inhibition of neddylation in mice significantly reduced the gluconeogenic capacity of the animals. These results are in agreement with a recent work indicating that inhibition of neddylation attenuated hyperglycemia in obese mice.19 Although our results point also to an important role of neddylation in modulating glucose levels, the mechanism that we have identified is totally different. Although in the previous study, the inhibition of neddylation enhanced hepatic insulin signaling by delaying CRL-mediated insulin receptor substrate (IRS) turnover, resulting in a reduced hyperglycemia.19 We propose that neddylation of a specific substrate namely PCK1 in three specific lysines (K278, K342, and K387) plays a key role in gluconeogenesis.
Previous work has indicated that PCK1 activity and stability were regulated at the post-translational level by acetylation, phosphorylation, and ubiquitination in response to cell energy input.6,44,45 For instance, PCK1 was acetylated by p300 acetyltransferase under high glucose levels, and conversely, SIRT1 deacetylated PCK1 under low-energy conditions.6 These modifications were associated with different biological functions since acetylated PCK1 displayed anaplerotic activity, whereas deacetylated PCK1 promoted cataplerosis.6 Altogether, earlier studies together with present results indicate that different post-translational modifications of PCK1 play a key role controlling its activity.
An earlier report showed that cytosolic PCK1 does not solely control hepatic gluconeogenesis, and a 90% reduction in PCK1 content showed only a 40% reduction in gluconeogenic flux.46 However, it is important to highlight that this previous study inactivated the gene at embryonic stages, and therefore, compensatory mechanisms could take over the gluconeogenic function. Concerning PCK1 expression in T2D, existing literature shows different data since some have shown that there was no relationship between fasting hyperglycemia and protein expression of PCK1,47 whereas others detected an increased expression of PCK1 in diabetic mice.48 Certainly, the regulation of PCK1 function is quite complex and the varied experimental phenotypes in mice have been extensively discussed.49
Although our results identify the neddylation of PCK1 as a novel mechanism to regulate glucose homeostasis in rodents, a limitation of the present work is that we did not investigate whether the described mutations in PCK1 neddylated sites are also relevant in humans and whether these mutations are related to human T2D. Further genetic studies in people with T2D should elucidate these aspects. Indeed, our work does not exclude that other proteins can also be neddylated upon fasting or CR. Actually, our proteomics approach detected more proteins that were neddylated in those conditions. Therefore, further investigation will be necessary to elucidate the functional role in gluconeogenesis of other neddylated proteins.
Taken together, our results reveal a new mechanism that participates in the finely tuned control of whole-body glucose homeostasis, associating the response of the neddylation of PCK1 and its gluconeogenic activity to nutrient availability.
Limitations of the study
Despite our findings clearly showing that NAE1 protein levels are increased in the conditions of nutrient deficiency, the mechanism by which this is modulated remains unknown. Further studies should clarify whether this is a transcriptional of post-transcriptional modulation. Moreover, although our in vitro results were performed in a human hepatocyte cell line, our data do not directly show the role of neddylation in the liver of people with obesity and T2D.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| 1:1000 Rabbit monoclonal NEDD8 | Abcam | Cat# ab81264; RRID: AB_1640720 |
| 1:1000 Rabbit polyclonal Phosphoenolpyruvate Carboxykinase 1 (PCK1) | Abcam | Cat# ab70358; RRID: AB_1925305 |
| 1:1000 Rabbit monoclonal NEDD8-activating enzyme (NAE1) | Cell Signaling | Cat# 14321; RRID: AB_2798448 |
| 1:1000 Rabbit monoclonal Glucose 6-Phosphatase (G6Pase) | Abcam | Cat# ab93857; RRID: AB_10903775 |
| 1:1000 Rabbit monoclonal Pyruvate Carboxylase (PC) | Abcam | Cat# ab126707; RRID: AB_11130771 |
| 1:1000 Mouse monoclonal Cullin-2 | Santa Cruz | Cat# sc-166506; RRID: AB_2230072 |
| 1:5000 Mouse monoclonal Glyceraldehyde 3- phosphate Dehydrogenase (GAPDH) | Merck | Cat# G8795; RRID: AB_1078991 |
| 1:1000 Rabbit polyclonal UBE1 | Cell Signaling | Cat# 4891; RRID: AB_2211438 |
| 1:5000 Mouse monoclonal a-Tubulin | Sigma | Cat# T5168; RRID: AB_477579 |
| 1:5000 Mouse monoclonal β –Actin | Sigma | Cat# A5316; RRID: AB_47674 |
| Chemicals, peptides, and recombinant proteins | ||
| Glucagon | Sigma-Aldrich | #G1774 |
| Adrenaline | ThermoFisher | L04911 |
| Hydrocortisone 21-hemisuccinate | Santa Cruz Biotechnology | sc-250130 |
| Leptin | Sigma-Aldrich | L-4146 |
| Insulin | Actrapid, Novo Nordisk | N/A |
| Sodium pyruvate | Sigma-Aldrich | P2256 |
| glycerol | Sigma-Aldrich | G5516 |
| MLN4924 | MedChemExpress | #HY-70062 |
| 2-Hydroxypropyl)-β-cyclodextrin | Sigma | #H107 |
| [3-3H] glucose | Perkin Elmer | N/A |
| Acetyl-CoA Assay kit | Abcam | ab87546 |
| Adrenaline ELISA KIT | Cusabio | CSB-E08679m-96T |
| Corticosterone ELISA KIT | Enzo Life Sciences | ADI-900-097 |
| Glucagon ELISA KIT | Mercodia | 10-1281-01 |
| Insulin ELISA KIT | Merck Milipore | EZRMI |
| Total Amino Acid Assay Kit | Abbexa | 298965 |
| L-Alanine Assay Kit | Abcam | 83394 |
| si-RNA to NEDD8 | Dharmacon | #4738 |
| si-RNA to NAE1 | Dharmacon | #8883 |
| si-RNA to PCK1 | Dharmacon | #L-006796-00-0005 |
| si-RNA to negative control | Dharmacon | #D-001810-10-05 |
| plasmid wild type PCK1 | Origene | plasmid #RC204758 |
| Plasmid mutated PCK: PCK1K387R | Vector Builder | #VB210901-1404whn |
| Plasmid PCK1K278R-K342R-K387R | Vector Builder | #VB210921-1406frv |
| Lipofectamine 2000 | Invitrogen | #11668-019 |
| High Sensitivity Glucose Assay Kit | Sigma-Aldrich | #MAK181-1KT |
| PCK1 activity kit | Biovision | #K359-100 |
| Deposited data | ||
| Mass spectrometry proteomics data | ProteomeXchange | PRIDE: PXD043054 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact. Ruben Nogueiras. Email: ruben.nogueiras@usc.es
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Animals
Animal experiments were conducted in accordance with the standards approved by the Faculty Animal Committee at the University of Santiago de Compostela, and the experiments were performed in agreement with the Rules of Laboratory Animal Care and International Law on Animal Experimentation. Animals were fed ad libitum with a standard diet (Scientific Animal Food &Engineering, proteins 16%, carbohydrates 60%, and fat 3%) and tap water unless otherwise indicated. The number of animals used in each experiment is indicated in the corresponding figure legend. Wild type (WT) mice, Alfp-Cre, heterozygous NAE1 (C57BL/6 background) and FloxPCK1 were housed in air-conditioned rooms (22–24 °C) under a 12:12 h light/dark cycle. BKS.Cg-+Leprdb/+Lepr+/OlaHsd (db/db) mice were purchased from The Jackson Laboratory (#000642) and were housed in the same air-conditioned rooms as WT mice. Animals were sacrificed and tissues were removed rapidly and immediately frozen in dry ice. Tissues were kept at −80 °C until their analysis.
Generation of bioNEDD8 transgenic mice
In the present paper, it is presented for the first time the transgenic BioNEDD8 mice as a new tool to identify potential direct targets of Neddylation in vivo regulated by the nutritional status. The generation of these animals is based on the previously publication of BioUbiquitin and Bir A mice.50 The pCAG564 vector was subcloned with bioNEDD8 human sequence or BirA using oligos: BirAf(5’-GAGCCGACAAGGTGAGAGTG-3’), BirAr(5’-TTGATTCCGGCTCCGATCAC-3’), bioNEDD8f (5’-GATATCGCTGCGCTGGTC-3’), and bioNEDD8r (5’-CATCGATCCCCAAGAAAACC-3’). Using Quiagen vector purification columns the plasmids were purify and subsequently injected in pronuclear zygotes (C57Bl6xCBA) with 2ng/μl of each. Animals that were positive to the plasmid were used to stablish the mice colony. The Scientific Procedures Act 1986 regulation and the University of Cambridge Ethical Review approved all animal procedures. As the insertion of the vectors in the mice genome was random, all animals used to develop the present work have been genotyped and contain the same numbers of the copies.
Animal experiments
Glucagon administration
Mice were injected with 200 μg kg−1 body weight of glucagon diluted in saline (Sigma-Aldrich, #G1774) intraperitoneally (ip) and after 30 min, blood glucose levels were measured51
Adrenaline administration
Mice were injected with 100 μg kg−1 body weight of L-adrenaline diluted in saline (ThermoFisher, L04911) ip.51
Cortisol administration
Mice were treated with 20 mg kg−1 body weight hydrocortisone 21-hemisuccinate sodium salt diluted in saline (Santa Cruz Biotechnology, sc-250130) ip.51
Leptin administration
WT animals were treated with saline or recombinant leptin (Sigma-Aldrich, L-4146) at a dose of 0.5 mg kg-1 of body weight every 12 h for 24 hours by an intraperitoneal injection (ip). WT mice were divided into three groups: (a) ip saline fed ad libitum chow diet, (b) ip. saline while fasting for a period of 24 h and (c) ip leptin while fasting for a period of 24 h.51
Insulin administration
WT mice were injected with 0.35 U kg−1 body weight of insulin (Actrapid, Novo Nordisk) ip and after 10 and 20 min, blood glucose levels were measured.
Caloric restriction
Five days before initiation of calorie restriction, 8-week-old mice were placed in individual cages and fed the chow diet ad libitum. During these days of acclimation, food intake was monitored to determine the average amount of food consumed daily by each mouse. Thereafter, mice were randomly separated into two groups: one group continued to receive the chow diet ad libitum while the other group was subjected to 60% calorie restriction. Each mouse subjected to calorie restriction was fed at 6 p.m. every day with an amount of food equal to 40% of their daily food intake during the week of acclimation. Body weight and blood glucose were measured daily at 5:30 p.m. before feeding. Finally, mice were sacrificed at 5:30 p.m. (before feeding) on the fourth day of calorie restriction to collect liver and serum for analyses.
Fasting
To study the role of neddylation on glucose production during fating, mice were subjected to a 24 h of fasting.
Glucose, insulin, glycerol, and pyruvate tolerance tests
Basal blood glucose levels were measured after an overnight fast (12 h) for the GTT, GlyTT and PTT, and after 6h for the ITT, with a Glucocard Glucometer (ARKRAY, USA). GTT, ITT, PTT and GlyTT were done after an intraperitoneal injection of either 2 g kg−1 D-glucose (Sigma-Aldrich, G8270), 0.35 U kg−1 insulin (Actrapid, Novo Nordisk), 1.25 g kg−1 sodium pyruvate (Sigma-Aldrich, P2256), or 1 g kg−1 glycerol (Sigma-Aldrich, G5516), and area under the curve (AUC) values were determined.
MLN4924 treatment
To check the effect of the blockade of neddylation, MLN4924 (MedChemExpress, #HY-70062), a NEDD8-activating enzyme (NAE) inhibitor, was injected to WT mice at a dose 60 mg kg−1 body weight subcutaneously 3 days before the experiment. (2-Hydroxypropyl)-β-cyclodextrin (Sigma, #H107) was used as vehicle.
In vivo virogenetic procedures
To achieve a specific effect on the hepatocytes, AAV-DIO Cre-dependent viruses were injected into the tail vein of Afp-Cre mice to downregulate (AAV-DIO-shNAE1 and AAV-DIO-shNEDD8) or upregulate (AAV-DIO-NAE1) neddylation (Vector Builder). As well, AAV-Cre-GFP (Tebubio, ref. 189SL100835) virus was injected into the tail vein of FloxPCK1 mice to generate liver knockout (LKO) mice, and then, PCK1 expression was recovered with the injection of WT PCK1 or K278R-K342R-K387R-PCK1 mutant viruses (Vector Builder). Control mice were injected with GFP (Vector Builder). As such, mice were held in a specific restrainer for intravenous injections: Tailveiner (TV-150, Bioseb, France).
Injections into the tail vein were carried out using a 27 G × 3/8″ (0.40 mm × 10 mm) syringe. Mice were injected with 100 μl of viral vectors diluted in saline. Viruses were injected one month before the beginning of the experiments.
Determination of HGP
HGP measurement was performed in conscious unrestrained catheterized mice. Catheters (BTSIL-025, Instech Laboratories Inc) were surgically implanted under ketamine (100 mg kg−1 body weight)/xylazine (15 mg kg−1 body weight) anesthesia, 7 days prior to the experiment in the right jugular vein and exteriorized above the neck using a vascular access button (VAB62BS/25, Instech Laboratories Inc.). Catheters were washed and flux verified using heparinized saline (NaCl 0.9%, 200 IU/ml heparin). The day of the experiment, mice then received a 120-min infusion of [3-3H] glucose (0.05 μCi/min) (Perkin Elmer). Blood was sampled from the tail vein at 110 and 120 min (10 μl) and treated with 0.3 N ZnSO4 and 0.3 N Ba(OH)2. Glucose concentration was measured, and measurement of [3-3H] glucose was done after overnight drying for the counting of radioactivity in the blood.
Biomolecular analysis
Western blot analysis
Total protein lysates from liver (20 μg) and cells (8 μg) were subjected to SDS-PAGE, electrotransferred onto polyvinylidene difluoride membranes (Millipore) and probed with the indicated antibodies (Table S1).
Acetyl-CoA assay kit
Acetyl-CoA levels were quantified from liver (100 mg) and serum (2 ul) extracts using the Acetyl-CoA Assay kit (Abcam, ref. ab87546).
Adrenaline, cortisol, glucagon, and insulin quantification in mice serum
Serum levels of glucorregulatory hormones were quantified using the adrenaline (Cusabio, ref. CSB-E08679m-96T), corticosterone (Enzo Life Sciences, ref. ADI-900-097), glucagon (Mercodia, ref.10-1281-01), and insulin (Merck Milipore, ref. EZRMI) ELISAS kits.
Total amino acids and L-alanine quantification
Total amount of amino acids and L-alanine levels were quantified using Total Amino Acid Assay Kit (Abbexa, ref. 298965) and L-Alanine Assay Kit (Abcam, ref. 83394), respectively, in serum and liver samples following manufacture's indications.
PCK1 immunoprecipitation
Extracts (prepared from 1 mg protein) were examined by immunoblot. For the immunoprecipitation assay, liver extracts were incubated with 1 μg of PCK1 specific antibody (Abcam; #ab70358) coupled to protein-G-Sepharose. After an overnight incubation at 4 °C with agitation, the captured proteins were centrifuged at 10,000g, the supernatants collected, and the beads washed four times in lysis buffer. Beads were boiled for 5 min 95 °C in 10 μl sample buffer. Extracts and immunoprecipitates were examined by SDS–PAGE and blotted with antibodies to the following targets: PCK1 (Abcam; #ab70358 1:1000) and NEDD8 (Abcam; Ab81264 1:1000). The membrane was incubated first with the anti-NEDD8 primary antibody, and then a β-mercaptoethanol based stripping procedure was performed to check for total immunoprecipitated PCK1.
Protein modelling
Mus musculus PCK1 and NEDD8 structures were obtained with AlphaFold prediction algorithm.52 All neddylated protein models were built using the WT protein as a template with UCSF Chimera and the isopeptide bond parameters were obtained by RHF/6-31G∗ computations using GAMESS-US. Hessian matrix in Cartesian coordinates was analyzed by Seminario method, using CartHess2FC module of AMBER. Molecular dynamics (MD) simulations were performed with OpenMM software53 in a CUDA platform with AMBER ff19SB force field, particle mesh Ewald for long-range electrostatic interactions with an Ewald summation cutoff of 10 Å, and constraint of the length of all bonds that involve a hydrogen atom. Each system was solvated with optimal-3-charge, 4-point rigid water model (OPC) water molecules and neutralized with chlorine and sodium counter-ions according to the total charge of the protein. Firstly, each system was subjected to 2500 steps of energy minimization at 298 K. Langevin thermostat was used to control the temperature with a friction coefficient of 1 ps-1 and a step size of 0.002 ps. To evaluate the motions of residues and atoms, each system was subjected to 250 ns MD simulation and snapshots of the trajectories were saved every 1 ps. Finally, the trajectory analysis was performed with the CPPTRAJ module of AMBER.53 Graphic representations and further analysis were made with OriginPro 2019b and molecular graphics with UCFS ChimeraX.
Phylogenetic analysis
Sets of amino acid sequences in FASTA format from both distant and closely related organisms were obtained from Uniprot database. Only sequences reviewed and manually annotated by Swiss-Prot were selected. CLUSTAL multiple sequence alignment was conducted by using MUSCLE v3.8.31, and phylogenetic analysis of the FASTA sequences was carried out by the BIONJ distance algorithm with 100 bootstrap replicates, both approaches were carried out through Seaview Version 5.0.4 software. The resulting phylogenetic tree was customized using the graphic viewer Figtree v.1.4.4.
Cell cultures and transfections
Cells were kept in a 5% CO2 incubator at 37°C and were routinely assessed for mycoplasma. 2.5 × 105 cells were seeded in a six-well plate for all experiments. To knock down the expression of NEDD8 and NAE1 in AML12 cells, cells were transfected with specific small-interference RNA (si-RNA) to NEDD8 (Dharmacon, #4738), to NAE1 (Dharmacon, #8883) and to PCK1 (Dharmacon, #L-006796-00-0005). Non-targeting siRNA was used as negative control (Dharmacon, #D-001810-10-05). The transfection was performed using Dharmafect 1 reagent (Dharmacon, #t-2001-03) following the protocol: 50 pmol of the siRNA diluted in 200μl of optiMEM (Life Technologies, #31985-070) was mixed with 6.5 μl of Dharmafect 1 diluted in 193.5 μl of optiMEM; the mixture was added into each well, resulting in a final volume of 1.5 ml with DMEM:F12 complete medium for 6 hours. 48 hours after the transfection, cells were collected for protein extraction.
To upregulate the expression of PCK1, cells were transfected with a plasmid encoding wild type PCK1 (Origene; plasmid #RC204758), or mutated PCK: PCK1K387R (Vector Builder; #VB210901-1404whn) and PCK1K278R-K342R-K387R (Vector Builder; #VB210921-1406frv). Lipofectamine 2000 (Invitrogen, #11668-019) was used to transfect cells with the following protocol: 4 μl of Lipofectamine 2000 diluted on 150 μl of Opti-MEM mixed with 2.5 μg of DNA diluted on 150 μl of Opti-MEM. This mixture was incubated in a final volume of 1.5 ml of complete medium for 24 hours.
Cell treatments
Cells were starved for 6h in Krebs–Henseleit–HEPES buffer (KHH; composition: 120 mmol l-1 NaCl, 4.7 mmol/l KCl, 2.5 mmol/l CaCl2, 1.2 mmol l-1 MgSO4, 1.2 mmol l-1 KH2PO4, 25 mmol l-1 NaHCO3, 25 mmol/l HEPES pH 7.4.), a fasting medium with neither nutrients nor hormones.
To study the regulation of NAE1 protein expression by glucose in vitro, cell medium from untreated cells were changed to KHH, in the presence or absence of glucose (D-glucose, Sigma-Aldrich, #G8270) 1 mM.
To pharmacologically inhibit neddylation in vitro, a dose of 10 μM of MLN4924 was added to the medium and incubated during 24h.
Glucose measurement in cell culture medium
Cells were incubated in glucogenic medium (KHH medium supplemented with lactate 20 mM and pyruvate 2 mM) for indicated times. Glucose released to the media was quantified using the High Sensitivity Glucose Assay Kit (Sigma-Aldrich, #MAK181-1KT). Results were normalized by the amount of protein.
Phosphoenolpyruvate Carboxykinase Activity assay
PCK1 activity was measured in cell and tissue samples following manufacturer instructions (Biovision, #K359-100). Briefly, samples were homogenized with 200 μl ice cold Assay Buffer and kept for 10 minutes on ice. After centrifuging them for 10 minutes at 10000 x g at 4°C, supernatant is collected, and protein level concentration and PCK activity are measured separately. In this assay, PCK is coupled with a set of enzymes that convert PEP and carbonate into a series of intermediates and hydrogen peroxide, which in turn, reacts with a probe and converted generating a colorimetric signal (OD: 570 nm). The color intensity is directly proportional to the amount of active PCK present in samples. Results were relativized by the amount of protein of each well.
Human cohort
Liver samples were obtained from people with morbid obesity undergoing bariatric surgery (n=60) at the Clínica Universidad de Navarra. Obesity was defined as a BMI≥30 kg/m2 and body fat percentage (BF)≥35%. BMI was calculated as weight in kilograms divided by the square of height in meters, and BF was estimated by air-displacement plethysmography (Bod-Pod®, Life Measurements, Concord, CA, USA). People with obesity were sub-classified into two groups [normoglycaemia (NG) or type 2 diabetes (T2D)] following the criteria of the Expert Committee on the Diagnosis and Classification of Diabetes.54 Inclusion criteria encompassed a complete diagnostic work-up including physical examination, laboratory investigation, ultrasound echography and liver biopsy consistent with the diagnosis of non-alcoholic fatty liver disease (NAFLD) according to the criteria of Kleiner and Brunt by an expert pathologist masked to all the results of the assays.55 Features of steatosis, lobular inflammation and hepatocyte ballooning were combined to obtain NAFLD activity score (NAS) (0-8).55 Exclusion criteria were: a) excess alcohol consumption (≥20 g for women and ≥30 g for men); b) the presence of hepatitis B virus surface antigen or hepatitis C virus antibodies in the absence of a history of vaccination; c) use of drugs linked to NAFLD, including amiodarone, valproate, tamoxifen, methotrexate, corticosteroids or anti-retrovirtals; d) evidence of other specific liver diseases, such as autoimmune liver disease, haemochromatosis, Wilson’s disease or α-1-antitrypsin deficiency. People with type 2 diabetes were not on insulin therapy or medication likely to influence endogenous insulin levels. It has to be stressed that people with type 2 diabetes did not have a long diabetes history (less than 2-3 years or even de novo diagnosis as evidenced from their anamnesis and biochemical determinations). All reported investigations were carried out in accordance with the principles of the Declaration of Helsinki, as revised in 2013, and approved by the Hospital’s Ethical Committee responsible for research (protocol 2021.005). Written informed consent was obtained from all the participants.
Protein quantification by SWATH-MS (Sequential Window Acquisition of all Theoretical Mass Spectra) in pull-down experiments
The analysis was performed as described previously by our group.56 Briefly the method used includes: creation of the spectral library, relative quantification by SWATH acquisition, SWATH data analysis, mass spectrometric analysis by LC-MSMS of PCK1 immunoprecipitation and neddylation site determination, proteomic data analysis. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE [1] partner repository with the dataset identifier PXD043054.
Neddylation site determination
The acquired raw files using a DDA method were analyzed with PEAKS 8 studio software (Bioinformatics Solutions Inc., Waterloo, Canada) using a mouse uniprot database. The peptide mass tolerance was set to 10 ppm and 0.01 Da for MS/MS. Variable modifications included were as follows: Oxidation of M, carbamidomethylation of C, and Glygly of K (neddylation of cysteines) as described previously.24,40,57 For high-confidence peptide identification a FDR (False Discover Rate) of 1% and a minimum of 2 ranked peptides were used for peptide filtering. Functional analysis was performed by FunRich open access software (Functional Enrichment analysis tool) for functional enrichment and interaction network analysis (http://funrich.org/index.html). For statistics, FunRich use hypergeometric test, BH and Bonferroni.
Statistics and data analysis
Data are expressed as mean ± standard error mean (SEM). Statistical significance was determined by two-tail Student’s t-test when two groups were compared or ANOVA and post-hoc two-tailed Bonferroni test when more than two groups were compared. P<0.05 was considered significant for all the analysis. Correlations were analyzed by Pearson’s correlation coefficient (r). Data analysis was performed using GaphPad Prism Software Version 8.0 (GraphPad, San Diego, CA) and Microsoft Excel. All the results shown in the manuscript are representative of, at least, two independent experiments with the same result.
Acknowledgments
This work was supported by grants from: FEDER/Ministerio de Ciencia, Innovación y Universidades-Agencia Estatal de Investigación (M.L.M.-C.: PID2020-117116RB-I00; C.D.: BFU2017-87721; M.L.: RTI2018-101840-B-I00; R.N.: PID2021-126096NB-I00 and RED2018-102379-T); Xunta de Galicia (R.N.: 2021-CP085 and 2020-PG0157); Fundación BBVA (to R.N.); Subprograma Retos Colaboración RTC2019-007125-1 (to M.L.M.-C.); Proyectos Investigación en Salud DTS20/00138 (to M.L.M.-C.); Proyectos Investigación en Salud (M.L.M.-C.: DTS20/00138); Fundación Atresmedia (to M.L. and R.N.); and Fundación La Caixa (to M.L., M.L.M.-C., and R.N.). This research also received funding from the European Community’s H2020 Framework Programme (ERC Synergy Grant-2019-WATCH- 810331, to R.N., V.P., and M.S.). The Centro de Investigación Biomédica en Red (CIBER) de Fisiopatología de la Obesidad y Nutrición (CIBERobn) and the Centro de Investigación Biomédica en Red (CIBER) de Enfermedades Hepáticas y Digestivas (CIBERehd) are initiatives of the Instituto de Salud Carlos III (ISCIII) of Spain, which is supported by FEDER funds. We thank MINECO for the Severo Ochoa Excellence Accreditation bioGUNE (SEV-2016-0644) to CIC.
Author contributions
M.J.G.-R., and U.F. conceived the study, designed and conducted experiments, analyzed results, and contributed to the manuscript preparation. T.P., E.N., M.F.F., N.d.S.L., L.R., A.R., M.S.-M., G.P.-M., P.C.-V., C.R., C.V.-D., S.T., R.C., A.W., M.S., V.P., T.C. D, M.L., A.D.-Q., C.D., D.G., G.F., I.D.-M., and S.B.B. contributed to the performance of experiments and to the manuscript preparation. M.L.M.-C. and R.N. conceived the study; designed, coordinated, and supervised experiments; wrote the manuscript; and secured funding. All authors discussed the results, commented on the manuscript before submission, and agreed with the final submitted manuscript.
Declaration of interests
The authors declare no competing interests.
Published: August 3, 2023
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cmet.2023.07.003.
Contributor Information
Maria L. Martinez-Chantar, Email: mlmartinez@cicbiogune.es.
Ruben Nogueiras, Email: ruben.nogueiras@usc.es.
Supplemental information
Data and code availability
-
•
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD043054.
-
•
Uncropped scans of all Western blots and all raw data used to generate graphs are included in Data S1. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
-
•
This paper does not report original code.
References
- 1.Petersen M.C., Vatner D.F., Shulman G.I. Regulation of hepatic glucose metabolism in health and disease. Nat. Rev. Endocrinol. 2017;13:572–587. doi: 10.1038/nrendo.2017.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang T., Hung C.C.Y., Randall D.J. The comparative physiology of food deprivation: from feast to famine. Annu. Rev. Physiol. 2006;68:223–251. doi: 10.1146/annurev.physiol.68.040104.105739. [DOI] [PubMed] [Google Scholar]
- 3.Yoon J.C., Puigserver P., Chen G., Donovan J., Wu Z., Rhee J., Adelmant G., Stafford J., Kahn C.R., Granner D.K., et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. doi: 10.1038/35093050. [DOI] [PubMed] [Google Scholar]
- 4.Roden M., Shulman G.I. The integrative biology of type 2 diabetes. Nature. 2019;576:51–60. doi: 10.1038/s41586-019-1797-8. [DOI] [PubMed] [Google Scholar]
- 5.Zhang X., Yang S., Chen J., Su Z. Unraveling the regulation of hepatic gluconeogenesis. Front. Endocrinol. 2018;9:802. doi: 10.3389/fendo.2018.00802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Latorre-Muro P., Baeza J., Armstrong E.A., Hurtado-Guerrero R., Corzana F., Wu L.E., Sinclair D.A., López-Buesa P., Carrodeguas J.A., Denu J.M. Dynamic acetylation of phosphoenolpyruvate carboxykinase toggles enzyme activity between gluconeogenic and anaplerotic reactions. Mol. Cell. 2018;71:718–732.e9. doi: 10.1016/j.molcel.2018.07.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ruan H.B., Han X., Li M.D., Singh J.P., Qian K., Azarhoush S., Zhao L., Bennett A.M., Samuel V.T., Wu J., et al. O-GlcNAc transferase/host cell factor C1 complex regulates gluconeogenesis by modulating PGC-1α stability. Cell Metab. 2012;16:226–237. doi: 10.1016/j.cmet.2012.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cantó C., Auwerx J. Clking on PGC-1alpha to inhibit gluconeogenesis. Cell Metab. 2010;11:6–7. doi: 10.1016/j.cmet.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Enchev R.I., Schulman B.A., Peter M. Protein neddylation: beyond cullin-RING ligases. Nat. Rev. Mol. Cell Biol. 2015;16:30–44. doi: 10.1038/nrm3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwechheimer C. NEDD8-its role in the regulation of Cullin-RING ligases. Curr. Opin. Plant Biol. 2018;45:112–119. doi: 10.1016/j.pbi.2018.05.017. [DOI] [PubMed] [Google Scholar]
- 11.Brown J.S., Jackson S.P. Ubiquitylation, neddylation and the DNA damage response. Open Biol. 2015;5:150018. doi: 10.1098/rsob.150018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rabut G., Peter M. Function and regulation of protein neddylation. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 2008;9:969–976. doi: 10.1038/embor.2008.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fernández-Ramos D., Martínez-Chantar M.L. NEDDylation in liver cancer: the regulation of the RNA binding protein Hu antigen R. Pancreatology. 2015;15(Suppl):S49–S54. doi: 10.1016/j.pan.2015.03.006. [DOI] [PubMed] [Google Scholar]
- 14.Watson I.R., Irwin M.S., Ohh M. NEDD8 pathways in cancer, Sine Quibus Non. Cancer Cell. 2011;19:168–176. doi: 10.1016/j.ccr.2011.01.002. [DOI] [PubMed] [Google Scholar]
- 15.Nakayama K.I., Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat. Rev. Cancer. 2006;6:369–381. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- 16.Zhou L., Jiang Y., Luo Q., Li L., Jia L. Neddylation: a novel modulator of the tumor microenvironment. Mol. Cancer. 2019;18:77. doi: 10.1186/s12943-019-0979-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Soucy T.A., Smith P.G., Milhollen M.A., Berger A.J., Gavin J.M., Adhikari S., Brownell J.E., Burke K.E., Cardin D.P., Critchley S., et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 18.Rajerison R.M., Faure M., Morel F. Relationship between cell volume and cation content in thick ascending limb of rat kidney. Pflugers Arch. 1988;412:497–502. doi: 10.1007/BF00582538. [DOI] [PubMed] [Google Scholar]
- 19.Chen C., Gu L., Matye D.J., Clayton Y.D., Hasan M.N., Wang Y., Friedman J.E., Li T. Cullin neddylation inhibitor attenuates hyperglycemia by enhancing hepatic insulin signaling through insulin receptor substrate stabilization. Proc. Natl. Acad. Sci. USA. 2022;119 doi: 10.1073/pnas.2111737119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Serrano-Maciá M., Simón J., González-Rellan M.J., Azkargorta M., Goikoetxea-Usandizaga N., Lopitz-Otsoa F., De Urturi D.S., Rodríguez-Agudo R., Lachiondo-Ortega S., Mercado-Gomez M., et al. Neddylation inhibition ameliorates steatosis in NAFLD by boosting hepatic fatty acid oxidation via the DEPTOR-mTOR axis. Mol. Metab. 2021;53:101275. doi: 10.1016/j.molmet.2021.101275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zubiete-Franco I., Fernández-Tussy P., Barbier-Torres L., Simon J., Fernández-Ramos D., Lopitz-Otsoa F., Gutiérrez-de Juan V., de Davalillo S.L., Duce A.M., Iruzubieta P., et al. Deregulated neddylation in liver fibrosis. Hepatology. 2017;65:694–709. doi: 10.1002/hep.28933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Olaizola P., Lee-Law P.Y., Fernandez-Barrena M.G., Alvarez L., Cadamuro M., Azkargorta M., O'Rourke C.J., Caballero-Camino F.J., Olaizola I., Macias R.I.R., et al. Targeting NAE1-mediated protein hyper-NEDDylation halts cholangiocarcinogenesis and impacts on tumor-stroma crosstalk in experimental models. J. Hepatol. 2022;77:177–190. doi: 10.1016/j.jhep.2022.02.007. [DOI] [PubMed] [Google Scholar]
- 23.Abidi N., Xirodimas D.P. Regulation of cancer-related pathways by protein NEDDylation and strategies for the use of NEDD8 inhibitors in the clinic. Endocr. Relat. Cancer. 2015;22:T55–T70. doi: 10.1530/ERC-14-0315. [DOI] [PubMed] [Google Scholar]
- 24.Lobato-Gil S., Heidelberger J.B., Maghames C., Bailly A., Brunello L., Rodriguez M.S., Beli P., Xirodimas D.P. Proteome-wide identification of NEDD8 modification sites reveals distinct proteomes for canonical and atypical NEDDylation. Cell Rep. 2021;34:108635. doi: 10.1016/j.celrep.2020.108635. [DOI] [PubMed] [Google Scholar]
- 25.Levine S., Saltzman A. Feeding sugar overnight maintains metabolic homeostasis in rats and is preferable to overnight starvation. Lab Anim. 2000;34:301–306. doi: 10.1258/002367700780384735. [DOI] [PubMed] [Google Scholar]
- 26.Zhao T.J., Liang G., Li R.L., Xie X., Sleeman M.W., Murphy A.J., Valenzuela D.M., Yancopoulos G.D., Goldstein J.L., Brown M.S. Ghrelin O-acyltransferase (GOAT) is essential for growth hormone-mediated survival of calorie-restricted mice. Proc. Natl. Acad. Sci. USA. 2010;107:7467–7472. doi: 10.1073/pnas.1002271107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Delgado T.C., Barbier-Torres L., Zubiete-Franco I., Lopitz-Otsoa F., Varela-Rey M., Fernández-Ramos D., Martínez-Chantar M.L. Neddylation, a novel paradigm in liver cancer. Transl. Gastroenterol. Hepatol. 2018;3:37. doi: 10.21037/tgh.2018.06.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Perry R.J., Zhang D., Guerra M.T., Brill A.L., Goedeke L., Nasiri A.R., Rabin-Court A., Wang Y., Peng L., Dufour S., et al. Glucagon stimulates gluconeogenesis by INSP3R1-mediated hepatic lipolysis. Nature. 2020;579:279–283. doi: 10.1038/s41586-020-2074-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Perry R.J., Wang Y., Cline G.W., Rabin-Court A., Song J.D., Dufour S., Zhang X.M., Petersen K.F., Shulman G.I. Leptin mediates a glucose-fatty acid cycle to maintain glucose homeostasis in starvation. Cell. 2018;172:234–248.e17. doi: 10.1016/j.cell.2017.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cryer P.E. Glucose counterregulation in man. Diabetes. 1981;30:261–264. doi: 10.2337/diab.30.3.261. [DOI] [PubMed] [Google Scholar]
- 31.Edgerton D.S., Scott M., Farmer B., Williams P.E., Madsen P., Kjeldsen T., Brand C.L., Fledelius C., Nishimura E., Cherrington A.D. Targeting insulin to the liver corrects defects in glucose metabolism caused by peripheral insulin delivery. JCI Insight. 2019;5 doi: 10.1172/jci.insight.126974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schneeberger M., Gómez-Valadés A.G., Altirriba J., Sebastián D., Ramírez S., Garcia A., Esteban Y., Drougard A., Ferrés-Coy A., Bortolozzi A., et al. Reduced alpha-MSH underlies hypothalamic ER-stress-induced hepatic gluconeogenesis. Cell Rep. 2015;12:361–370. doi: 10.1016/j.celrep.2015.06.041. [DOI] [PubMed] [Google Scholar]
- 33.Jin E.S., Beddow S.A., Malloy C.R., Samuel V.T. Hepatic glucose production pathways after three days of a high-fat diet. Metabolism. 2013;62:152–162. doi: 10.1016/j.metabol.2012.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Magnusson I., Rothman D.L., Katz L.D., Shulman R.G., Shulman G.I. Increased rate of gluconeogenesis in type II diabetes mellitus. A 13C nuclear magnetic resonance study. J. Clin. Invest. 1992;90:1323–1327. doi: 10.1172/JCI115997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xirodimas D.P., Saville M.K., Bourdon J.C., Hay R.T., Lane D.P. Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell. 2004;118:83–97. doi: 10.1016/j.cell.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 36.Watson I.R., Blanch A., Lin D.C., Ohh M., Irwin M.S. Mdm2-mediated NEDD8 modification of TAp73 regulates its transactivation function. J. Biol. Chem. 2006;281:34096–34103. doi: 10.1074/jbc.M603654200. [DOI] [PubMed] [Google Scholar]
- 37.Embade N., Fernández-Ramos D., Varela-Rey M., Beraza N., Sini M., Gutiérrez de Juan V., Woodhoo A., Martínez-López N., Rodríguez-Iruretagoyena B., Bustamante F.J., et al. Murine double minute 2 regulates Hu antigen R stability in human liver and colon cancer through NEDDylation. Hepatology. 2012;55:1237–1248. doi: 10.1002/hep.24795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ju U.I., Jeong D.W., Seo J., Park J.B., Park J.W., Suh K.S., Kim J.B., Chun Y.S. Neddylation of sterol regulatory element-binding protein 1c is a potential therapeutic target for nonalcoholic fatty liver treatment. Cell Death Dis. 2020;11:283. doi: 10.1038/s41419-020-2472-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park H.S., Ju U.I., Park J.W., Song J.Y., Shin D.H., Lee K.H., Jeong L.S., Yu J., Lee H.W., Cho J.Y., et al. PPARgamma neddylation essential for adipogenesis is a potential target for treating obesity. Cell Death Differ. 2016;23:1296–1311. doi: 10.1038/cdd.2016.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang X., Zhang Y.L., Qiu G., Pian L., Guo L., Cao H., Liu J., Zhao Y., Li X., Xu Z., et al. Hepatic neddylation targets and stabilizes electron transfer flavoproteins to facilitate fatty acid beta-oxidation. Proc. Natl. Acad. Sci. USA. 2020;117:2473–2483. doi: 10.1073/pnas.1910765117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kumar S., Yoshida Y., Noda M. Cloning of a cDNA which encodes a novel ubiquitin-like protein. Biochem. Biophys. Res. Commun. 1993;195:393–399. doi: 10.1006/bbrc.1993.2056. [DOI] [PubMed] [Google Scholar]
- 42.Barbier-Torres L., Delgado T.C., García-Rodríguez J.L., Zubiete-Franco I., Fernández-Ramos D., Buqué X., Cano A., Gutiérrez-de Juan V., Fernández-Domínguez I., Lopitz-Otsoa F., et al. Stabilization of LKB1 and Akt by neddylation regulates energy metabolism in liver cancer. Oncotarget. 2015;6:2509–2523. doi: 10.18632/oncotarget.3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kumar D., Das M., Sauceda C., Ellies L.G., Kuo K., Parwal P., Kaur M., Jih L., Bandyopadhyay G.K., Burton D., et al. Degradation of splicing factor SRSF3 contributes to progressive liver disease. J. Clin. Invest. 2019;129:4477–4491. doi: 10.1172/JCI127374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin Y.Y., Lu J.Y., Zhang J., Walter W., Dang W., Wan J., Tao S.C., Qian J., Zhao Y., Boeke J.D., et al. Protein acetylation microarray reveals that NuA4 controls key metabolic target regulating gluconeogenesis. Cell. 2009;136:1073–1084. doi: 10.1016/j.cell.2009.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grimsrud P.A., Carson J.J., Hebert A.S., Hubler S.L., Niemi N.M., Bailey D.J., Jochem A., Stapleton D.S., Keller M.P., Westphall M.S., et al. A quantitative map of the liver mitochondrial phosphoproteome reveals posttranslational control of ketogenesis. Cell Metab. 2012;16:672–683. doi: 10.1016/j.cmet.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Burgess S.C., He T., Yan Z., Lindner J., Sherry A.D., Malloy C.R., Browning J.D., Magnuson M.A. Cytosolic phosphoenolpyruvate carboxykinase does not solely control the rate of hepatic gluconeogenesis in the intact mouse liver. Cell Metab. 2007;5:313–320. doi: 10.1016/j.cmet.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Samuel V.T., Beddow S.A., Iwasaki T., Zhang X.M., Chu X., Still C.D., Gerhard G.S., Shulman G.I. Fasting hyperglycemia is not associated with increased expression of PEPCK or G6Pc in patients with type 2 diabetes. Proc. Natl. Acad. Sci. USA. 2009;106:12121–12126. doi: 10.1073/pnas.0812547106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qiao A., Zhou J., Xu S., Ma W., Boriboun C., Kim T., Yan B., Deng J., Yang L., Zhang E., et al. Sam68 promotes hepatic gluconeogenesis via CRTC2. Nat. Commun. 2021;12:3340. doi: 10.1038/s41467-021-23624-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Beale E.G., Harvey B.J., Forest C. PCK1 and PCK2 as candidate diabetes and obesity genes. Cell Biochem. Biophys. 2007;48:89–95. doi: 10.1007/s12013-007-0025-6. [DOI] [PubMed] [Google Scholar]
- 50.Lectez B., Migotti R., Lee S.Y., Ramirez J., Beraza N., Mansfield B., Sutherland J.D., Martinez-Chantar M.L., Dittmar G., Mayor U. Ubiquitin profiling in liver using a transgenic mouse with biotinylated ubiquitin. J. Proteome Res. 2014;13:3016–3026. doi: 10.1021/pr5001913. [DOI] [PubMed] [Google Scholar]
- 51.Gonzalez-Rellan M.J., Fondevila M.F., Fernandez U., Rodríguez A., Varela-Rey M., Veyrat-Durebex C., Seoane S., Bernardo G., Lopitz-Otsoa F., Fernández-Ramos D., et al. O-GlcNAcylated p53 in the liver modulates hepatic glucose production. Nat. Commun. 2021;12:5068. doi: 10.1038/s41467-021-25390-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eastman P., Swails J., Chodera J.D., McGibbon R.T., Zhao Y., Beauchamp K.A., Wang L.P., Simmonett A.C., Harrigan M.P., Stern C.D., et al. OpenMM 7: rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017;13:e1005659. doi: 10.1371/journal.pcbi.1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.American Diabetes Association (2) Classification and diagnosis of diabetes. Diabetes Care. 2015;38(Suppl 1):S8–S16. doi: 10.2337/dc15-S005. [DOI] [PubMed] [Google Scholar]
- 55.Kleiner D.E., Brunt E.M., Van Natta M., Behling C., Contos M.J., Cummings O.W., Ferrell L.D., Liu Y.C., Torbenson M.S., Unalp-Arida A., et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 56.Chantada-Vázquez M.D.P., García Vence M., Serna A., Núñez C., Bravo S.B. SWATH-MS protocols in human diseases. Methods Mol. Biol. 2021;2259:105–141. doi: 10.1007/978-1-0716-1178-4_7. [DOI] [PubMed] [Google Scholar]
- 57.Vogl A.M., Phu L., Becerra R., Giusti S.A., Verschueren E., Hinkle T.B., Bordenave M.D., Adrian M., Heidersbach A., Yankilevich P., et al. Global site-specific neddylation profiling reveals that NEDDylated cofilin regulates actin dynamics. Nat. Struct. Mol. Biol. 2020;27:210–220. doi: 10.1038/s41594-019-0370-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD043054.
-
•
Uncropped scans of all Western blots and all raw data used to generate graphs are included in Data S1. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
-
•
This paper does not report original code.