

Key Points
-
•
Myeloid relapse can originate from various differentiation stages of MLL/AF4+ ALL.
-
•
Dysregulation of epigenetic regulators underpins fundamental lineage reprogramming.
Visual Abstract
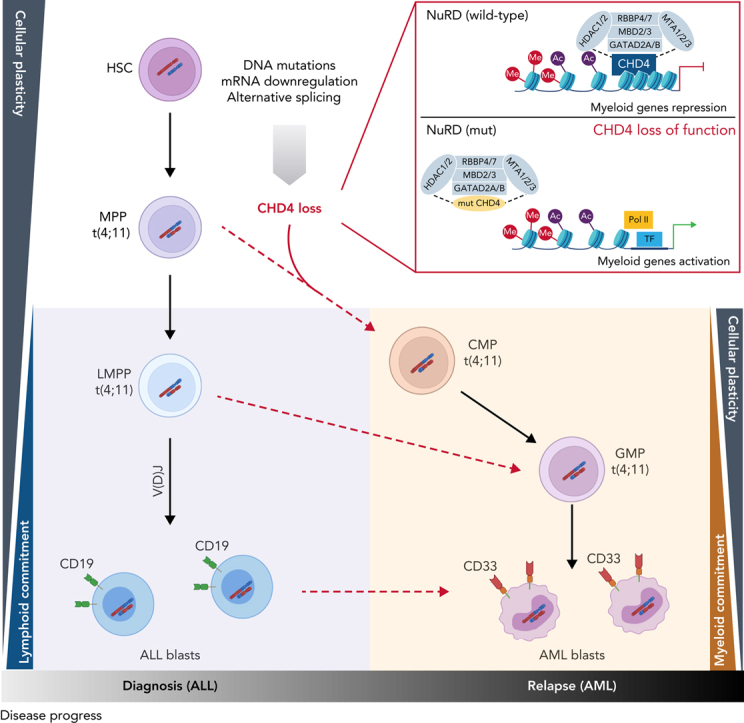
Abstract
The fusion gene MLL/AF4 defines a high-risk subtype of pro-B acute lymphoblastic leukemia. Relapse can be associated with a lineage switch from acute lymphoblastic to acute myeloid leukemia, resulting in poor clinical outcomes caused by resistance to chemotherapies and immunotherapies. In this study, the myeloid relapses shared oncogene fusion breakpoints with their matched lymphoid presentations and originated from various differentiation stages from immature progenitors through to committed B-cell precursors. Lineage switching is linked to substantial changes in chromatin accessibility and rewiring of transcriptional programs, including alternative splicing. These findings indicate that the execution and maintenance of lymphoid lineage differentiation is impaired. The relapsed myeloid phenotype is recurrently associated with the altered expression, splicing, or mutation of chromatin modifiers, including CHD4 coding for the ATPase/helicase of the nucleosome remodelling and deacetylation complex. Perturbation of CHD4 alone or in combination with other mutated epigenetic modifiers induces myeloid gene expression in MLL/AF4+ cell models, indicating that lineage switching in MLL/AF4 leukemia is driven and maintained by disrupted epigenetic regulation.
Introduction
Translocation of mixed lineage leukemia (MLL) with 1 of >130 alternative partner genes is a recurrent cytogenetic finding in both acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL) and is generally associated with a poor prognosis.1,2 Among the most common translocations is t(4;11)(q21;q23), forming the MLL/AF4 (also known as KMT2A/AFF1) fusion gene. Unique among MLL rearrangements (MLLr), MLL/AF4 is almost exclusively associated with pro-B cell ALL and is prototypical of infant ALL, where it carries a very poor prognosis.1 However, despite this general lymphoid presentation, MLL/AF4 leukemias have an intriguing characteristic, that of lineage-switched relapses. Lineage-switched acute leukemias lose their lymphoid-specific features and gain a myeloid phenotype upon relapse.3, 4, 5 Alternatively, MLL/AF4 leukemias may harbor distinct lymphoid and myeloid populations at the same time and thus are classified as mixed-phenotype acute leukemias (MPALs) of the bilineage subtype.6
Lineage plasticity has been associated with the loss of original therapeutic targets.7,8 To understand the molecular basis of lineage promiscuity and switching, we examined a unique cohort of MLL/AF4+ lineage-switched acute leukemia presentation/relapse pairs and MPALs. We demonstrate that disruption of the epigenetic machinery, including the nucleosome remodelling and deacetylation (NuRD) complex, is associated with the loss of lymphoid restriction. Lineage switching is then enacted through redistribution of transcription factor binding and chromatin reorganization. These findings provide novel insight into factors that may prove critical to the effective implementation of lineage-specific, epitope-directed therapies, such as chimeric antigen receptor T-cell (CAR T-cell) or bispecific T-cell–engaging antibody approaches.
Methods
Patient samples and data
Patients were diagnosed by local hematology specialists according to contemporary clinical diagnostic criteria based on morphology and immunophenotypic analysis. All patient samples were collected at the point of diagnosis, remission after treatment, or relapse and were stored with written informed consent for research in 1 of 6 centers (Newcastle Haematology Biobank, Newcastle, United Kingdom; University Hospital Schleswig-Holstein, Kiel, Germany; Dmitry Rogachev National Medical Research Center of Pediatric Hematology, Oncology and Immunology, Moscow, Russia; Haematological Malignancy Diagnostic Service, Leeds, United Kingdom; Princess Maxima Center for Pediatric Oncology, Utrecht, The Netherlands; and Cincinnati Children’s Hospital Medical Center, Cincinnati, OH). Mononuclear cells were isolated from bone marrow or peripheral blood by density centrifugation followed by immediate extraction of DNA or RNA, or cryopreservation in the presence of 10% v/v dimethyl sulfoxide.
Samples were requested and used in accordance with the ethics approvals granted to each of the local/institutional ethics review boards (NRES [National Research Ethics Service] Committee North East, Newcastle & North Tyneside 1, United Kingdom, reference 07/H0906/109 + 5; Medical Faculty Christian-Albrechts University, Kiel, reference A 103/08; Dmitry Rogachev National Medical Research Center, Moscow, Russia, references MB2008: 22.01.2008, MB2015: 22.01.2015, and ALL-REZ-2014: 28.01.2014; Haematological Malignancy Research Network, Yorkshire, United Kingdom, reference 04/Q1205/69; Haematological Malignancy Diagnostic Service, Leeds, United Kingdom, reference 14/WS/0098; Erasmus MC METC (Medisch Ethische Toetsings Commissie), The Netherlands, reference MEC-2016-739; and Institutional Review Board (IRB) of Cincinnati Children’s Hospital, Cincinnati, OH, reference 2010-0658), and in accordance with the Declaration of Helsinki. Each patient and sample was allocated an anonymized reference, and no identifiable information was shared.
Additional methods are described in the supplemental Methods, available on the Blood Web site.
Results
Characterization of MLL/AF4 acute leukemias with lineage switching
We focused on lineage switches that originally presented as ALL and relapsed as AML and MPALs presenting with distinct lymphoid and myeloid populations. Lymphoid and myeloid phenotypes were defined by morphology and by expression of either B lymphoid (CD19, CD22, and CD79A) or myeloid (CD33, CD117/KIT, and CD64/FCGR1A) antigens (Figure 1A; supplemental Table 1). To exclude de novo and therapy-associated AMLs, which are unrelated to the original ALL and do not share the initiating event, the lymphoid and myeloid presentations and relapses had to display identical MLL/AF4 breakpoints as proof of genetic relationship (Figure 1B; supplemental Figure 1, supplemental Table 1). Using these definitions, we collected a cohort of 12 cases of MLL/AF4 ALL comprising 6 infant, 2 pediatric, and 2 adult patients who relapsed with AML, including 1 infant (LS10) who relapsed after B-lineage–directed blinatumomab treatment and 2 infant MLL/AF4 MPALs (supplemental Table 1).
Figure 1.

Characterization of MLL/AF4 lineage-switched cases. (A) Morphological change from lymphoblastic leukemia (left) to acute monoblastic/monocytic leukemia (right) in patient LS01. Bar represents 20 μm. (B) Sanger sequencing of MLL/AF4 and reciprocal AF4/MLL fusions in LS01 presentation ALL (top) and relapsed AML (bottom) identifies a common breakpoint with identical filler sequences in the ALL and AML samples.
Lineage-switched leukemia is associated with transcriptional reprogramming
We hypothesized that lineage switching would be linked with changes in gene expression. Because the changes in transcriptome composition may include altered regulation of both transcription and mRNA maturation,9 we compared gene expression and splicing between lymphoid and myeloid populations from lineage switching and patients with MPAL. Cluster analysis of differential gene expression robustly separated both population types (Figure 2A). We identified 1374 upregulated (adjusted P < .01; log fold change >2) and 1323 downregulated genes in the AML lineage switches and the myeloid populations of patients with MPAL linked to reduced lymphoid and increased myeloid gene expression (Figure 2B, supplemental Table 2). Changed gene expression included the loss of lymphoid genes, such as PAX5, EBF1, CD19, CD20 (MS4A1) and CD22, diminished gene expression of immunoglobulin genes, genes involved in the VDJ recombination (RAG1, RAG2, and DNTT), and gain-of-myeloid genes, including CLEC12A, PRAM1, and CSF3R and members of the CEBP transcription factor family (Figure 2C-D; supplemental Figure 2A-B).10, 11, 12 Moreover, almost 30% of direct bona fide target genes of MLL/AF4 including PROM1, encoding the stem cell markers CD133, IKZF2, and HOXA7 showed lower expression in myeloid cells despite sharing the same MLL/AF4 isotype (supplemental Figure 3A-D, supplemental Table 2).13, 14, 15 These data show that lineage switching also involves differential MLL/AF4-driven gene expression.
Figure 2.

Transcriptional reprogramming in lineage-switched and MPAL cases. (A) Heat map showing the top 100 differentially expressed genes between ALL and AML from 6 lineage-switched (LS01, LS03, LS04, LS05, LS06, and LS10) and 2 MPAL cases, ranked by Wald statistics. (B) Enrichment of myeloid growth and differentiation signature in relapse samples (left) identified by gene set enrichment analyses (GSEA), also pointing to downregulation of genes highly correlating with acute lymphoblastic leukemia (middle and right). GSEAs were performed based on data derived from 6 lineage-switched samples. FDR, false-discovery rate; NES, normalized enrichment score. Differential expression of lineage-specific (C) and immunoglobulin recombination machinery genes (D) in lineage-switched and MPAL cases. Error bars show standard error of the mean for lineage-switched cases and ranges for 2 MPAL cases.
The analysis of RNA isoform compositions showed that lineage switching is associated with altered splicing, comprising changes in intron retention and differential usage of exons and exon-exon linkages (Figure 3A; supplemental Tables 3 and 4). Interestingly, 85% of all differentially used exon-exon linkages were noncanonical and mainly consisted of exon skipping and complex splicing events (Figure 3A-B; supplemental Table 4). Pathway analysis revealed an enrichment of alternatively spliced genes in immune pathways, including antigen processing and membrane trafficking, suggesting that alternative splicing is linked to the change from a lymphoid to a myeloid differentiation state (Figure 3C).
Figure 3.


Alternative splicing in lineage-switched and MPAL cases. (A) Pie charts showing the classification of nondifferential (non-DEEj) and differential (DEEj) exon-exon junctions. Shown are the percentages of splicing events assigned to a particular mode of splicing. A complex splicing event corresponds to several (2 or more) alternative splicing incidents occurring simultaneously in the same sample. (B) Volcano plots demonstrating differential usage of exon-exon junctions in the transcriptome of AML/myeloid vs ALL/lymphoid cells of cases of lineage switching (LS01, LS03, and LS04) or MPAL. The vertical dashed lines represent twofold differences between the AML and ALL cells, and the horizontal dashed line shows the FDR-adjusted q-value threshold of .05 (left). Venn diagrams (right) showing distribution of splice variants identified as significantly changed in AML (or myeloid fraction of patients with MPAL), including DEEjs, differential exon usage (DEU), and retained introns (RI). (C) Enrichment analysis of affected signaling pathways by the DEEjs and DEU in the lineage-switched acute leukemia (LSAL) AML relapse and myeloid compartment of patients with MPAL. Pathway enrichment analysis was performed with g:Profiler (https://biit.cs.ut.ee/gprofiler/gost) at the highest significance threshold, with multiple-testing correction (g:SCS algorithm). (D) Fold log2 change of total transcript levels among genes affected by alternative splicing (left) and of differentially spliced variants in lineage-switched and myeloid compartments of patients with MPAL (right). (E) Representation of impact of alternative splicing on mRNA composition and open reading frames (ORFs) of select genes. Column graphs on the right indicate corresponding fold changes of variant expression between AML (or myeloid) and ALL (or lymphoid) populations in 2 tested lineage-switched cases (LS03 and LS04) and 1 case of MPAL.
Interestingly, lineage switching also affected total expression and the composition of alternatively spliced fusion transcript isoforms for both MLL/AF4 and AF4/MLL. For instance, we detected in relapse tissue from patient LS01 a fusion variant skipping MLL exon 9 (supplemental Figure 3E). In addition, we observed changes in transcription and splicing for genes regulating the chromatin landscape. Several epigenetic regulators, including the polycomb PRC1-like complex component AUTS2 and the SWI/SNF complex component BCL7A were downregulated in myeloid compared with lymphoid cells (Figure 2A). Several other spliceosome and SWI/SNF members were either differentially expressed or spliced. Among all NuRD complex members, only CHD4 demonstrated differential expression, whereas CHD4, CHD3, and HDAC2 were differentially spliced in AML relapse cells or the myeloid subpopulations of MPALs (Figure 3D-E; supplemental Table 4). For instance, CHD4 encoding the ATPase/helicase subunit of the histone-modifying NuRD complex showed a significantly lower expression in AML relapses of patients with lineage switching, but was differentially spliced in patients with MPAL, resulting in premature stops or intron retention, most likely leading to loss of function isoforms.
Reorganization of chromatin accessibility and transcription factor binding site occupancy upon lineage switching
The substantial gene expression changes, including those affecting epigenetic regulators and lineage-determining transcription factors, prompted us to link transcriptional changes to altered genome-wide chromatin accessibility. High-resolution DNase I-hypersensitive site mapping, combined with digital footprinting analysis using the Wellington algorithm,16 uncovered multiple differentially accessible genes, including the hematopoietic surface marker genes CD33 and CD19 and transcription factors (Figure 4A-C; supplemental Figure 4A-B). These alterations occurred both at locations distal and proximal to transcription start sites indicating the involvement of enhancers and promoters (Figure 4D; supplemental Figure 4C). Digital footprinting is now generally accepted to highlight factors that are essential for regulating specific cell fates.17, 18, 19 These analyses showed that changes in chromatin accessibility after lineage switching were linked to an altered pattern of occupancy of transcription factor binding sites (Figure 4E; supplemental Figure 4D), with a loss of occupancy of consensus binding sites for lymphoid transcription factors, including EBF and PAX5, and a corresponding increased occupancy of binding motifs for myeloid factors including C/EBP family members (Figure 4E-F). We also observed a redistribution of footprinted sites for transcription factors controlling both lymphoid and myeloid maturation, such as RUNX, AP-1, and ETS family members, to alternative cognate motifs (Figure 4E; supplemental Figure 4D).20,21 This finding is exemplified by decreased accessibility of a region located 1 kb upstream of the CD19 transcription start site with concomitant loss of EBF binding site occupancy at this element (Figure 4C). In summary, the transition from lymphoid to myeloid immunophenotype is associated with genome-wide alterations in chromatin accessibility and occupancy of transcription factor binding sites.
Figure 4.

Chromatin reorganization and differential transcription factor binding underpins lineage switching. (A) DNase I hypersensitive site sequencing identified 13 619 sites with a log2-fold reduction and 12 203 sites with a log2-fold increase after lineage switching to AML. Relative peak heights in the AML sample were plotted against those of the ALL sample. (B) A University of California, Santa Cruz (UCSC) Genome Browser screenshot displaying differential expression at lineage-specific loci (red tracks) accompanied by altered DNase I hypersensitivity (black tracks) proximal to the transcriptional start site (TSS) of CD33. (C) UCSC Genome Browser screenshot for CD19 zoomed in on an ALL-associated DHS with EBF occupation as indicated by high-resolution DHS-seq and Wellington analysis. FP, footprint. (D) Heat maps showing distal DHS regions specific for AML relapse on a genomic scale. Red and green indicate excess of positive and negative strand cuts, respectively, per nucleotide position. Sites are sorted from top to bottom in order of decreasing footprint occupancy score. (E) De novo motif discovery in distal DHSs unique to AML relapse as compared with ALL relapse, as shown in panel D. (F) EBF1 and C/EBP binding motifs demonstrate differential motif density in presentation ALL and relapsed AML. DHS, DNase-hypersensitive site.
The mutational landscape of lineage switching
Next, we examined the mutational landscape of lineage-switched MLL/AF4 leukemias by performing exome sequencing on the entire cohort. In agreement with previously reported mutation rates in MLL-rearranged leukemias, presentation ALLs displayed a relatively quiet mutational landscape, with a median of 25 nonsynonymous somatic single nucleotide variants (SNVs) or insertions/deletions (indels) (supplemental Figure 5A-B; supplemental Table 5).10,22 Most of them were subclonal with <30% of the reads. The group of AML relapses showed a mean of 92 SNVs and indels. However, this increase was related to the more heterogeneous composition of the relapse group: 2 cases (LS07AML and LS08AML) carried mutated DNA polymerase genes resulting in increased mutational burden. We observed this phenotype in only 2 of 10 relapses, arguing against this phenomenon being a general requirement for the lineage switch.
In general, we found only a limited overlap between mutations in ALL presentation and AML relapses (Figure 5A-B; supplemental Table 5). Although ALL mutations were not associated with genes belonging to specific functional pathways, AML-specific mutations were associated with the regulation of transcription and chromatin binding and modification, further emphasizing the notion of transcriptional reprogramming during lineage switching. Most of the subclonal mutations identified in presentation samples were subsequently lost at relapse, indicating alternative subclones as the origin of relapse. This included KRAS and NRAS mutations, which have been shown to confer a worse clinical outcome to infants with MLL-rearranged ALL (Figure 5C).23 Also the MPALs harbored many mutations that were exclusively found in either the lymphoid or myeloid subpopulation indicating the presence of subclones with a lymphoid and myeloid bias (Figure 5A-B). These combined data show that lymphoid and myeloid leukemia phenotypes are associated with distinctive mutation signatures both in lineage switches and in MPALs.
Figure 5.

Molecular characterization of lineage-switched MLL/AF4 leukemias. (A) Whole-exome–sequencing data showing genes recurrently mutated within the analyzed cohort and genes clonally mutated in relapsed cases belonging to the same function protein complexes (eg, DNA polymerases, epigenetic complexes, and transcriptional regulators). Data are presented according to the disease time point/cell lineage and age of the patient. Depicted are major SNVs/indels that were found in >30% of reads and minor SNVs/indels present in <30% reads. (B) Comparison of total mutation load (SNVs and indels) identified in patients at presentation (ALL) and relapse (AML) disease stage or lymphoid and myeloid fraction in MPALs. Listed are common SNVs predicted (by Condel scoring) to have deleterious effects. (C) Evolution of KRAS/NRAS mutation–carrying cells during the lineage-switching process. Clonal vs subclonal mutations were defined based on variant allele frequencies (VAFs) of identified hits at setup cutoff of 30%.
Perturbation of CHD4 and PHF3 disrupts lymphoid development in MLL/AF4-expressing cells
To identify factors contributing to the lineage plasticity in MLL/AF4+ leukemic cells, we compared all genes demonstrating differential expression, alternative splicing or mutation in the AML relapse (Figure 6A). This comparison highlighted 8 genes common to all patients with lineage-switched relapse. One common gene was CHD4, which codes for the ATPase/helicase subunit of the NuRD, a multiprotein transcriptional corepressor complex with both histone deacetylase and adenosine triphosphate (ATP)–dependent chromatin remodelling activity. NuRD is critical for determination of lymphoid lineage by interacting with the transcription factor IKZF1.24, 25, 26 CHD4 shows significantly lower expression in lineage-switched AML when compared with ALL presentation and is differentially spliced in the MPAL cases (Figures 3E and 6B). Finally, although CHD4 mutations have been reported in <1.5% MLL-germline childhood cases of ALL,27 as with the R1068H mutation found in the relapse of patient LS01, these variants commonly affect highly conserved residues in the helicase/ATPase domains and are thought to disrupt its activity (Figure 6C; supplemental Figure 5C).28, 29, 30 In contrast, recurrent mutations in other NuRD complex members have not been described in ALL, and no other NuRD complex member was clonally mutated in our cohort (supplemental Table 5).
Figure 6.

Epigenetic modulatory genes influence lineage-specific expression profiles. (A) Intersection between identified hits of clonal mutations (variant allelic frequency [VAF] >30%), differentially expressed genes and alternatively spliced, differentially used exon-exon junctions (adj. P < .01) in lineage-switched myeloid relapse/myeloid fraction of MPALs, present in the analyzed cohort. (B) Fold change in expression of NuRD complex members (CHD4, MTA1, RBBP4, and MBD3) and PHF3, after lineage-switched relapse (left) and in MPAL cases (right). (C) CHD4 structure; the R1068H mutation (red) is located in the critical helicase domain of CHD4 at a highly conserved residue. ∗Number of positions that have a single, fully conserved residue; colon, conservation between groups of strongly similar properties, scoring >.5 in the Gonnet PAM 250 matrix; period, conservation between groups of weakly similar properties, scoring ≤0.5 in the Gonnet PAM 250 matrix. (D) Identification of regions of differential chromatin accessibility before and after knockdown of CHD4 and PHF3 depicted in red in MLLr SEM cells (left) and non-MLLr 697 cells (right). For all reads, the fold change in ATAC-peak height was calculated relative to the control (shNTC) and ATAC peaks from knockdown cells were plotted according to their fold change vs the control signals. CHD4 ChIP density plots from SEM cells (depicted in blue) were plotted along with the corresponding DNA regions of the shNTC control. Differentially expressed genes associated with changing ATAC peaks (log2FC analyzed vs shNTC) identified in each cellular variant are represented by heat maps included at the right side of each gene (for SEM and 697 cells). (E) UCSC genome browser screenshots representing differential chromatin accessibility (ATAC-seq) and gene expression level (RNA-seq) in the myeloid CEBPA and the lymphoid RAG2 loci after CHD4 and PHF3 knockdown in MLLr SEM cells and non-MLLr 697 cells. ChIP-seq traces representing normal CHD4 occupancy in non-MLLr B-ALL (REH cells), MLLr B-ALL (SEM cells) and MLLr AML cells (MV-4;11) are shown as a reference at the bottom of each screenshot. TSS, transcriptional start site is depicted for each gene. (F) Expression of the lineage-specific cell surface markers CD19 (lymphoid) and CD33 (myeloid) after culture of MLL/Af4-transformed hCD34+ cord blood progenitor cells in lymphoid-permissive conditions. Knockdown of PHF3, CHD4, or a combination disrupts the dominant lymphoid differentiation pattern in nontargeting control (shNTC). (G) PHF3 knockdown is capable of influencing the surface marker expression after a longer incubation period (33 days). CHD4 knockdown impaired cellular survival upon longer in vitro culture (data not shown). ChIP, chromatin immunoprecipitation.
We therefore hypothesized that CHD4 was vital in maintaining lineage fidelity in MLL/AF4-positive ALL. To test this idea, we performed knockdown experiments in the MLL/AF4-expressing and CD33− ALL cell line SEM, where we included ACAP1, DHX36, NCOA2, PHF3, and PPP1R7 as 5 additional genes with potentially deleterious mutations in patient LS01 (supplemental Figure 6A). Reverse engineering of a mutual gene network from 216 ALL and AML gene expression data sets identified CHD4 and PHF3, a cofactor in RNA Pol II–mediated transcription,31 as the most relevant network components of the mutated genes investigated (PHF3: 21 edges, P = .010; CHD4: 12 edges, P = .0005; supplemental Figure 6B, supplemental Table 6).32,33
Only knockdown of CHD4 and of PHF3 induced robust expression of the myeloid surface marker CD33 with a combined knockdown resulting in an even stronger CD33 expression (supplemental Figures 6A,C). Moreover, knockdown of either CHD4 or PHF3 also increased CD33 levels in RS4;11, another MLL/AF4 ALL cell line, but not in the 2 MLL-germline ALL cell lines 697 and REH (supplemental Figure 6D), indicating that loss of CHD4 or PHF3 may only affect CD33 in the context of MLL/AF4. Finally, the combined knockdown of CHD4 and PHF3 in PDX from diagnostic ALL cells significantly increased the fraction of CD33+ cells from 8% to more than 25% (supplemental Figure 6E). These combined data suggest that CHD4 and PHF3 restrict MLL/AF4+ leukemic cells to a lymphoid phenotype.
To examine the role of additional mutations of chromatin modifiers found in our cohort, we investigated the impact of the PRC1 members PCGF6 and AUTS2, genes with known roles in B-lymphoid malignancy34 and mutated in LS07RAML and LS08RAML (Figure 5A). Although knockdown of AUTS2 did not change the CD33 levels, depletion of PCGF6 increased CD33 surface expression in SEM cells, further supporting the notion of epigenetic factors in regulating lineage determination in ALL (supplemental Figure 6F).
To establish a direct link between CHD4/PHF3 binding to the upregulation of myeloid genes, we investigated the impact of CHD4 or PHF3 perturbation on gene expression and chromatin organization by performing RNA-sequencing (RNA-seq), ATAC-seq, and chromatin immunoprecipitation–seq for CHD4 in SEM cells and the MLL germline cell line 697 (Figure 6D; supplemental Figure 7A-B). In this analysis, we ranked the ATAC-seq and chromatin immunoprecipitation-seq signals according to their fold changes alongside the control patterns, which demonstrated that ATAC-seq analysis of control-treated SEM cells show a very similar pattern to CHD4 binding (Figure 6D), confirming that this factor is a global regulator of chromatin accessibility. Knockdown of both factors caused a shift in the overall chromatin accessibility pattern, as shown by clustering analysis (supplemental Figure 7A-B, bottom) suggesting that the postknockdown cells shifted their cistrome and thus their identity, whereby CHD4 knockdown resulted in a gain of open chromatin sites (Figure 6D, top). The knockdown of PHF3 caused both a loss and a gain of open chromatin sites (Figure 6D, bottom). Gene set enrichment analysis demonstrated a strong correlation of these gene expression changes in SEM cells after knockdown of CHD4 and PHF3 and lineage-switching cases (supplemental Figure 7C-D). However, these changes were particular to MLL/AF4 cells, because in MLL germline 697 cells, CHD4 knockdown-induced changes in chromatin accessibility were not linked to altered gene expression, and knockdown of PHF3 did not affect chromatin accessibility (Figure 6D, right).
Knockdown of CHD4 or PHF3 in SEM cells changed chromatin structure and reduced expression of CD79B, RAG2, VPREB1, and CD22, whereas concomitantly increasing transcription of CEBPA, LYZ, SIRPA, and CD33 (Figure 6E; supplemental Figure 8A-B). However, 697 cells showed neither a change in immunophenotype nor altered expression of these genes, suggesting that CHD4- and PHF3-mediated changes in gene expression correlate with the presence of an MLL fusion gene.
Given that the relapse-initiating cell may arise within an uncommitted, MLL/AF4-translocated HSPC population, we assessed the impact of loss of CHD4 and PHF3 in a human cord blood model that harbors a chimeric MLL/Af4 fusion.35 Knockdown of either CHD4 or PHF3 under lymphoid culture conditions significantly impaired lymphoid differentiation potential, whereas knockdown of both CHD4 and PHF3 disrupted differentiation entirely (Figure 6F-G; supplemental Table 7). Transcriptomic analysis of the sorted populations revealed that CD33+ cells exhibited a metagene expression pattern similar to MLLr AML, whereas the pattern describing CD19+ cells was most similar to MLLr ALL, confirming that changes in surface marker expression were associated with the corresponding changes in the transcriptomic profiles (supplemental Figure 6G).
Taken together, our data show the important role of CHD4 and PHF3 in the epigenetic control of lymphoid lineage maintenance in MLL/AF4+ leukemia. In particular, dysregulation of CHD4/NuRD was mediated by mutation, downregulation of expression, and differential splicing across the entire cohort. These data support a role for these factors in the lineage-determining capacity of MLL/AF4, whereas their loss undermines execution and maintenance of the lymphoid lineage fate.
Clonal evolution of AML relapse
The observed cooperation of CHD4 and PHF3 in the control of lineage determination predicted that both mutations co-occur in the same cell. Furthermore, because both mutations may be necessary for the lineage switch in patient LS01, we hypothesized that they should be detectable in the most immature populations of this AML sample for which we had viable cellular material. We therefore investigated the order of acquisition of these secondary mutations within the structure of the normal hematopoietic hierarchy. Dissecting the relapsed AML sample using cell sorting, we isolated HSC-, MPP-, LMPP- and GMP-like, as well as more differentiated populations, followed by targeted deep sequencing that examined MLL/AF4 and 12 SNVs, including mutated CHD4 and PHF3, that were unique to the relapse sample. The fusion oncogene was found in the multipotential progenitor population (MPP, CD34+CD38−CD45RA−CD90−) and in the lymphoid-primed multipotent progenitorlike population (LMPP; CD34+CD38−CD45RA+), with lymphoid, myeloid, but not megakaryocyte-erythroid potential. (supplemental Figure 9A-B; supplemental Table 8). When examining the presence of the 12 SNVs across the different populations, only PHF3 and CHD4 mutations were present within the purified MPP-like fraction with variant allelic frequency ≥0.3 (Figure 7A, supplemental Table 8). In contrast, LMPP- and GMP-like populations contained all 12 SNVs at high variant allelic frequency. These findings place the CHD4 and PHF3 mutations among the earliest genetic events in this patient during the evolution of lineage-switched relapse. Moreover, they suggest, at least for this patient, an MPP-like or even more immature cell population as the origin of relapse.
Figure 7.

Hematopoietic stem/progenitor populations carry MLL/AF4. (A) Summary of MLL/AF4 positivity and 12 SNVs exclusive for the AML relapse, within different populations analyzed in patient LS01RAML. Circles with solid colors indicate VAF >30%, light dashed circle indicates VAF 5% to 30%. Remaining genes (open circle) represent the 10 other SNVs (of 12 SNVs) which showed the same pattern in the frequency of mutation acquisition (described in supplemental Table 8). (B) Summary of the proposed model of the origin of lineage switched relapse. Evaluation of B-cell receptor repertoires on ALL (presentation) and AML (relapse) lineage switched, and MPAL cases identified 3 different patterns. Pattern 1, with clonotypes on the ALL only. Pattern 2, B-cell receptor-containing clones on ALL and AML, but distinct to each other. Pattern 3, B-cell receptor-containing clones shared between ALL and AML. (C) BCR clone frequencies identified in whole-exome–seq data with application of MiXCR software in all lineage-switched acute leukemia (LSAL) and MPAL cases. VAF, variant allele frequency.
Cellular origin of lineage-switched relapse
To examine whether lineage-switched relapse regularly arises from lymphoid primed or even earlier leukemic populations, we examined whether relapsed AML cells contained and even shared B-cell receptor (BCR) rearrangements with the preceding ALL. To interrogate the developmental stage at which the myeloid relapse arose, we analyzed BCR rearrangements with RNA-seq and whole-exome-seq–derived data.36 All cases of ALL showed classic oligoclonal rearrangements of BCR loci, supporting the lymphoid lineage decision (supplemental Figure 9C, supplemental Table 9). We observed 3 distinct patterns for AML relapses (Figure 7B). Pattern 1 comprises AML cells with no BCR rearrangements, implying the presence of a relapse-initiating cell residing in a primitive precursor population before early DJ recombination. This pattern was seen with patient LS01 and, together with the presence of CHD4 and PHF3 mutations, strongly supports an MPP-like population as a putative origin of relapse (Figure 7A). As a second pattern, we found unrelated BCR rearrangements, which may indicate either aberrant rearrangement in a myeloid cell or relapse initiating from B-lymphoid cells committed to undergo rearrangement, or a transdifferentiated minor ALL clone with an alternative rearrangement (Figure 7C; cases LS03, LS06, LS07, LS08, MPAL1, and MPAL2). Interestingly, this pattern is found in a relapse after blinatumomab treatment (LS10) suggesting that immune escape may occur by direct transdifferentiation (Figure 7C). Pattern 3 shows shared BCR rearrangements between diagnostic and relapse material, which suggests a transdifferentiated myeloid relapse from the major ALL clone (cases LS05 and LS09). These data demonstrate that AML relapses can originate from different stages of lymphoid leukemogenesis.
Discussion
This study describes impaired epigenetic control as being central to the phenomenon of lymphoid-myeloid lineage switching in MLL/AF4 leukemia and demonstrates a heterogeneous cellular origin of relapse. The comparison of BCR rearrangements between matched ALL presentation and AML relapse cases demonstrates that whereas relapse can evolve directly from pro-B–like ALL blast populations, in keeping with the general self-renewal capacity of ALL cells,37 it can alternatively originate within the hematopoietic stem and progenitor cell (HSPC) compartment. Indeed, the identification of MLL/AF4-expressing MPP-like cells shows that lineage-switched relapse can originate from very immature hematopoietic progenitor populations. This finding agrees with recently published data pointing at MPP cells as the origin of MLL/AF4 leukemia38 and is in line with transcriptomic similarities between t(4;11) ALL and Lin-CD34+CD38−CD19− fetal liver cells, again suggesting HSPCs as the cells of origin.23 Furthermore, the identification of MLL/AF4 within HSPC populations is consistent with the recent identification of an early lymphoid progenitor, ELP-like signature specifically in MLL-rearranged ALL.39 Nevertheless, and in agreement with previously published findings for MPALs,6 the data derived from the present cohort strongly support a nonlineage committed progenitor compartment as a source for lineage-switched relapse. However, we cannot exclude additional cells of origin of MLL/AF4 ALL.
Irrespective of the cellular origin of the relapse, lineage switching is associated with a major rewiring of gene regulatory networks. At the level of transcriptional control, the decision for lymphoid development relies, not only on the activation of a lymphoid transcriptional program, but also on the silencing of a default myeloid program.40 That decision is enacted by lymphoid master regulators including EBF1, PAX5, and IKAROS, which represent genes commonly mutated in precursor B-ALL and do not just upregulate B-cell specific genes, but also repress the myeloid program.40, 41, 42, 43, 44 Pax5−/− pro-B cells, which lack lymphoid potential, although capable of erythromyeloid differentiation in vitro, maintain expression of early B-cell transcription factors EBF1 and E2A (TCF3).40 In contrast, we showed that lineage switched MLL/AF4 pro-B leukemic relapse is associated with a significant reduction in expression of these earliest B lymphoid transcription factors, which links to changes in the MLL/AF4 transcriptional program, ultimately establishing a myeloid differentiation fate. Unfortunately, we were not able to directly prove changes in transcription factor binding and associated changes in histone modifications because of the lack of available primary patient samples. However, high-resolution DNase-hypersensitive site–seq clearly demonstrated changes in chromatin accessibility and loss of occupation of the corresponding transcription factor binding sites.
The opposite scenario is observed when myeloid transcription factors are expressed in B-lymphoid cells.45 Overexpression of C/EBPα efficiently reprograms such cells into macrophages by suppressing lymphoid genes. CEBPA is strongly upregulated after CHD4 knockdown (Figure 6E) and is likely to be a driving force behind lineage switching. Taken together, these published and newly presented data confirm that (1) the balance between lymphoid and myeloid transcription factors is instructive for lineage choice, and (2) the downregulation of the myeloid program is essential for the maintenance of lymphoid fate.
How can the mutation of global chromatin regulators cause a switch in cell fate? Similar to the Pax5 knockout, loss of IKAROS DNA-binding activity prevents lymphoid differentiation.26 NuRD cooperates directly with IKAROS to repress hematopoietic stem cell self-renewal and myeloid differentiation, permitting early lymphoid development.26,46,47 Lineage switching was associated with heterozygous mutation; reduced expression; or, in the case of 2 MPALs, alternative splicing of CHD4 and other NuRD components. These gene dosage effects are consistent with reports showing that complete loss of CHD4 impairs normal and leukemic proliferation48,49 and myeloid and lymphoid differentiation of HSPCs and causes exhaustion of HSC pools,46 indicating that basal CHD4 expression is essential for maintaining AML. Moreover, a partial inhibition of CHD4 supported induction of pluripotency in iPSCs, whereas a complete deletion eliminated cell proliferation, demonstrating that lowering CHD4 expression may facilitate lineage promiscuitiy.50
Recent studies have identified core NuRD and PRC1 complex members as being direct targets of MLL/AF4 binding.51,52 Moreover, NuRD components including CHD4 were shown to be part of an MLL supercomplex.53 We therefore hypothesize that epigenetic regulator genes are recruited by lineage-specific factors during MLL/AF4 leukemogenesis and mediate fundamental lineage-specific decision-making processes, in this case, the repression of the myeloid lineage program. Multiple routes to their dysregulation may result in escape from this lineage restriction and may be enacted at different stages of hematopoiesis. However, importantly and in keeping with a previous murine study of lineage conversion after CAR T-cell therapy, we did not identify evidence of relapse from a pre-existent myeloid clone.54
Of substantial clinical importance, lineage switching results in the loss of B-cell surface markers (eg, CD19), providing an alternative mechanism for relapse after CAR T-cell or blinatumomab therapy,55,56 in addition to mutations, alternative splicing, and T-cell trogocytosis.57, 58, 59 Whereas these therapies target lineage-specific surface markers, lineage-switched (pre) leukemic progenitor populations escape epitope recognition and provide a potential clonal source for the relapse.60 As recognition of lineage switching after CD19 CAR T-cell therapy grows, 2 recent studies have highlighted the particular vulnerability of patients with MLLr ALL.54,61,62 Given the increasing use of advanced immunological therapies, a detailed understanding of the molecular processes underlying lineage determination and switching will be critical for developing new strategies to avoid this route to clinical relapse. In this report, we highlight an important role of epigenetic regulatory complexes in the context of MLL/AF4 leukemia.
Conflict-of-interest disclosure: Z.K. and J.B. are employees of Illumina, a public company that develops and markets systems for genetic analysis. The remaining authors declare no competing financial interests.
Acknowledgments
The authors thank Jon Coxhead and Raf Hussain (Newcastle University Core Genomics Facility) and Marc van Tuil (Princess Maxima Center Diagnostic department) for development of sequencing strategies; the Newcastle University Flow Cytometry Core Facility and Tomasz Poplonski (Princess Maxima Center Flow Cytometry Core Facility) for assistance with the generation of flow cytometry data and cell sorting strategies; the Newcastle University Bioinformatics Support Unit for helping to develop the analysis approach for sequencing data; and Monique den Boer, Frank van Leeuwen, and Ronald Stam for critically reading the manuscript.
This study was supported by a Cancer Research UK Centre Studentship (C27826/A17312) and Newcastle University Overseas Research Scholarship (R.T.); a CRUK program grant (J.V., O.H.) (C27943/A12788); a Kika programme grant (329) (O.H., J.V.); grants from the North of England Children’s Cancer Research Fund (O.H., J.V., S.B.); Bloodwise grants 12055 and 15005 (O.H.); and a grant from the Kay Kendall Leukaemia Fund (KKL1142) (O.H.). S.B. was supported by an NIHR Academic Clinical Lectureship (CL-2012-01-002), the Sir Bobby Robson Foundation Clinical Fellowship, and a Medical Research Council Clinician Scientist Fellowship (MR/S021590/1). Work in C.B./P.N.C.’s laboratory was funded by a programme grant from Bloodwise (15001). Work in J.M.A.’s laboratory was funded by a programme grant from Bloodwise (13044). E.Z. was supported by a Russian Foundation for Basic Research (RFBR) grant (17-29-06052). Research in the V.V.G. laboratory was supported in part by the Ministry of Education of the Republic of Belarus, grant 3.04.3. Research in A.K.’s laboratory was supported by an Russell Sage Foundation (RSF) grant (20-75-10091). The study made use of data generated by the St Jude Children’s Research Hospital-Washington University Pediatric Cancer Genome Project and the Therapeutically Applicable Research to Generate Effective Treatments (TARGET) initiative, phs000218, managed by the National Cancer Institute (supplemental Methods).
Authorship
Contribution: O.H., S. Bomken, and C.B. conceived the study; O.H., C.B., R.T., K.S., P.M., S. Bomken, A.P., C.M., A.K., Z.K., J.B., V.B., R.M., J.V., J.M.A., and S.L. developed the methodology; S.N., J.Y.H.-K., V.V.G., A.K., D.W., and P.C. were responsible for the software programming; S.N., M.A., J.H.K., V.V.G., A.K., D.W., P.C., P.K., C.B., and O.H. performed the formal analysis; R.T., K.S., P.M., A.P., C.M., P.S.C., H.J.B., S.G.K., A.K., S.A.A., M.R.I., E.K.S., P.E., H.M., A.E., N.M.-S., S.E.F., Y.S., D.P., and P.C. performed the investigations; J.S., F.V., E.Z., A.S., J.C.M., L.J.R., C.E., O.A.H., S. Bailey, R.S., N.M., M.C., V.B., R.M., M.W., C.J.H., C.A.C., D.S., Y.O., M.J.T., P.N.C., J.C.M., M.H., C.B., and O.H. obtained the resources; S.N., D.W., and P.C. were responsible for data curation; S. Bomken, O.H., C.B., R.T., and K.S. wrote the manuscript; O.H., S. Bomken, J.M.A., J.V., and C.B. supervised the study; and O.H., J.V., S. Bomken, C.B., P.N.C., J.M.A., and E.Z. acquired the funding.
Footnotes
∗R.T. and K.S. contributed equally to this study.
†C.B., O.H., and S. Bomken are senior authors.
O.H. would like to dedicate this paper to Fritz Eckstein on the occasion of his 90th birthday.
Exome-sequencing data and genome-sequencing data presented in this article have been deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under project numbers PRJNA547947 and PRJNA547815, respectively. Immunoglobulin and T-cell receptor sequencing data have been deposited in NCBI SRA under project number PRJNA511413. RNA sequencing data and DNase hypersensitivity sequencing data were deposited in Gene Expression Omnibus under accession numbers GSE132396 and GSE130142, respectively. All deposited data will be publically available after publication of the study.
Requests for additional specific data/materials should be made to Olaf Heidenreich (o.t.heidenreich@prinsesmaximacentrum.nl).
The online version of this article contains a data supplement.
There is a Blood Commentary on this article in this issue.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Contributor Information
Olaf Heidenreich, Email: o.t.heidenreich@prinsesmaximacentrum.nl.
Simon Bomken, Email: s.n.bomken@ncl.ac.uk.
Supplementary Material
References
- 1.Meyer C, Burmeister T, Gröger D, et al. The MLL recombinome of acute leukemias in 2017. Leukemia. 2018;32(2):273–284. doi: 10.1038/leu.2017.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moorman AV, Ensor HM, Richards SM, et al. Prognostic effect of chromosomal abnormalities in childhood B-cell precursor acute lymphoblastic leukaemia: results from the UK Medical Research Council ALL97/99 randomised trial. Lancet Oncol. 2010;11(5):429–438. doi: 10.1016/S1470-2045(10)70066-8. [DOI] [PubMed] [Google Scholar]
- 3.Germano G, Pigazzi M, del Giudice L, et al. Two consecutive immunophenotypic switches in a child with MLL-rearranged acute lymphoblastic leukemia. Haematologica. 2006;91(5 suppl):ECR09. [PubMed] [Google Scholar]
- 4.Jiang JG, Roman E, Nandula SV, Murty VV, Bhagat G, Alobeid B. Congenital MLL-positive B-cell acute lymphoblastic leukemia (B-ALL) switched lineage at relapse to acute myelocytic leukemia (AML) with persistent t(4;11) and t(1;6) translocations and JH gene rearrangement. Leuk Lymphoma. 2005;46(8):1223–1227. doi: 10.1080/10428190500086055. [DOI] [PubMed] [Google Scholar]
- 5.Rossi JG, Bernasconi AR, Alonso CN, et al. Lineage switch in childhood acute leukemia: an unusual event with poor outcome. Am J Hematol. 2012;87(9):890–897. doi: 10.1002/ajh.23266. [DOI] [PubMed] [Google Scholar]
- 6.Alexander TB, Gu Z, Iacobucci I, et al. The genetic basis and cell of origin of mixed phenotype acute leukaemia. Nature. 2018;562(7727):373–379. doi: 10.1038/s41586-018-0436-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Le Magnen C, Shen MM, Abate-Shen C. Lineage plasticity in cancer progression and treatment. Annu Rev Cancer Biol. 2018;2(1):271–289. doi: 10.1146/annurev-cancerbio-030617-050224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quintanal-Villalonga Á., Chan JM, Yu HA, et al. Lineage plasticity in cancer: a shared pathway of therapeutic resistance. Nat Rev Clin Oncol. 2020;17(6):360–371. doi: 10.1038/s41571-020-0340-z. [published correction appears in Nat Rev Clin Oncol. 2020;17(6):382] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen L, Kostadima M, Martens JHA, et al. Transcriptional diversity during lineage commitment of human blood progenitors. Science. 2014;345(6204) doi: 10.1126/science.1251033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andersson AK, Ma J, Wang J, et al. St. Jude Children’s Research Hospital–Washington University Pediatric Cancer Genome Project. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat Genet. 2015;47(4):330–337. doi: 10.1038/ng.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Novershtern N, Subramanian A, Lawton LN, et al. Densely interconnected transcriptional circuits control cell states in human hematopoiesis. Cell. 2011;144(2):296–309. doi: 10.1016/j.cell.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zangrando A, Dell’orto MC, Te Kronnie G, Basso G. MLL rearrangements in pediatric acute lymphoblastic and myeloblastic leukemias: MLL specific and lineage specific signatures. BMC Med Genomics. 2009;2(1):36. doi: 10.1186/1755-8794-2-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gessner A, Thomas M, Castro PG, et al. Leukemic fusion genes MLL/AF4 and AML1/MTG8 support leukemic self-renewal by controlling expression of the telomerase subunit TERT. Leukemia. 2010;24(10):1751–1759. doi: 10.1038/leu.2010.155. [DOI] [PubMed] [Google Scholar]
- 14.Somervaille TC, Matheny CJ, Spencer GJ, et al. Hierarchical maintenance of MLL myeloid leukemia stem cells employs a transcriptional program shared with embryonic rather than adult stem cells. Cell Stem Cell. 2009;4(2):129–140. doi: 10.1016/j.stem.2008.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilkinson AC, Ballabio E, Geng H, et al. RUNX1 is a key target in t(4;11) leukemias that contributes to gene activation through an AF4-MLL complex interaction. Cell Rep. 2013;3(1):116–127. doi: 10.1016/j.celrep.2012.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Piper J, Elze MC, Cauchy P, Cockerill PN, Bonifer C, Ott S. Wellington: a novel method for the accurate identification of digital genomic footprints from DNase-seq data. Nucleic Acids Res. 2013;41(21) doi: 10.1093/nar/gkt850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonifer C, Cockerill PN. Chromatin structure profiling identifies crucial regulators of tumor maintenance. Trends Cancer. 2015;1(3):157–160. doi: 10.1016/j.trecan.2015.10.003. [DOI] [PubMed] [Google Scholar]
- 18.Kreher S, Bouhlel MA, Cauchy P, et al. Mapping of transcription factor motifs in active chromatin identifies IRF5 as key regulator in classical Hodgkin lymphoma. Proc Natl Acad Sci USA. 2014;111(42):E4513–E4522. doi: 10.1073/pnas.1406985111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Howell ED, Yzaguirre AD, Gao P, et al. Efficient hemogenic endothelial cell specification by RUNX1 is dependent on baseline chromatin accessibility of RUNX1-regulated TGFβ target genes. Genes Dev. 2021;35(21-22):1475–1489. doi: 10.1101/gad.348738.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hohaus S, Petrovick MS, Voso MT, Sun Z, Zhang DE, Tenen DGPU. PU.1 (Spi-1) and C/EBP alpha regulate expression of the granulocyte-macrophage colony-stimulating factor receptor alpha gene. Mol Cell Biol. 1995;15(10):5830–5845. doi: 10.1128/mcb.15.10.5830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leddin M, Perrod C, Hoogenkamp M, et al. Two distinct auto-regulatory loops operate at the PU.1 locus in B cells and myeloid cells. Blood. 2011;117(10):2827–2838. doi: 10.1182/blood-2010-08-302976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dobbins SE, Sherborne AL, Ma YP, et al. The silent mutational landscape of infant MLL-AF4 pro-B acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2013;52(10):954–960. doi: 10.1002/gcc.22090. [DOI] [PubMed] [Google Scholar]
- 23.Agraz-Doblas A, Bueno C, Bashford-Rogers R, et al. Unraveling the cellular origin and clinical prognostic markers of infant B-cell acute lymphoblastic leukemia using genome-wide analysis. Haematologica. 2019;104(6):1176–1188. doi: 10.3324/haematol.2018.206375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arends T, Dege C, Bortnick A, et al. CHD4 is essential for transcriptional repression and lineage progression in B lymphopoiesis. Proc Natl Acad Sci USA. 2019;116(22):10927–10936. doi: 10.1073/pnas.1821301116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ng SY, Yoshida T, Zhang J, Georgopoulos K. Genome-wide lineage-specific transcriptional networks underscore Ikaros-dependent lymphoid priming in hematopoietic stem cells. Immunity. 2009;30(4):493–507. doi: 10.1016/j.immuni.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang J, Jackson AF, Naito T, et al. Harnessing of the nucleosome-remodeling-deacetylase complex controls lymphocyte development and prevents leukemogenesis. Nat Immunol. 2011;13(1):86–94. doi: 10.1038/ni.2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma X, Liu Y, Liu Y, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. 2018;555(7696):371–376. doi: 10.1038/nature25795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.González-Pérez A, López-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet. 2011;88(4):440–449. doi: 10.1016/j.ajhg.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sifrim A, Hitz MP, Wilsdon A, et al. Deciphering Developmental Disorders Study Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat Genet. 2016;48(9):1060–1065. doi: 10.1038/ng.3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Novillo A, Fernández-Santander A, Gaibar M, et al. Role of chromodomain-helicase-DNA-binding protein 4 (CHD4) in breast cancer. Front Oncol. 2021;11 doi: 10.3389/fonc.2021.633233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Appel LM, Franke V, Bruno M, et al. PHF3 regulates neuronal gene expression through the Pol II CTD reader domain SPOC. Nat Commun. 2021;12(1):6078. doi: 10.1038/s41467-021-26360-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kang H, Chen IM, Wilson CS, et al. Gene expression classifiers for relapse-free survival and minimal residual disease improve risk classification and outcome prediction in pediatric B-precursor acute lymphoblastic leukemia. Blood. 2010;115(7):1394–1405. doi: 10.1182/blood-2009-05-218560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gentles AJ, Plevritis SK, Majeti R, Alizadeh AA. Association of a leukemic stem cell gene expression signature with clinical outcomes in acute myeloid leukemia. JAMA. 2010;304(24):2706–2715. doi: 10.1001/jama.2010.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ferreira BI, García JF, Suela J, et al. Comparative genome profiling across subtypes of low-grade B-cell lymphoma identifies type-specific and common aberrations that target genes with a role in B-cell neoplasia. Haematologica. 2008;93(5):670–679. doi: 10.3324/haematol.12221. [DOI] [PubMed] [Google Scholar]
- 35.Lin S, Luo RT, Ptasinska A, et al. Instructive role of MLL-fusion proteins revealed by a model of t(4;11) pro-B acute lymphoblastic leukemia. Cancer Cell. 2016;30(5):737–749. doi: 10.1016/j.ccell.2016.10.008. [DOI] [PubMed] [Google Scholar]
- 36.Bolotin DA, Poslavsky S, Davydov AN, et al. Antigen receptor repertoire profiling from RNA-seq data. Nat Biotechnol. 2017;35(10):908–911. doi: 10.1038/nbt.3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.le Viseur C, Hotfilder M, Bomken S, et al. In childhood acute lymphoblastic leukemia, blasts at different stages of immunophenotypic maturation have stem cell properties. Cancer Cell. 2008;14(1):47–58. doi: 10.1016/j.ccr.2008.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malouf C, Ottersbach K. The fetal liver lymphoid-primed multipotent progenitor provides the prerequisites for the initiation of t(4;11) MLL-AF4 infant leukemia. Haematologica. 2018;103(12):e571–e574. doi: 10.3324/haematol.2018.191718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khabirova E, Jardine L, Coorens THH, et al. Single-cell transcriptomics reveals a distinct developmental state of KMT2A-rearranged infant B-cell acute lymphoblastic leukemia. Nat Med. 2022;28(4):743–751. doi: 10.1038/s41591-022-01720-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nutt SL, Heavey B, Rolink AG, Busslinger M. Commitment to the B-lymphoid lineage depends on the transcription factor Pax5. Nature. 1999;401(6753):556–562. doi: 10.1038/44076. [DOI] [PubMed] [Google Scholar]
- 41.Mullighan CG, Kennedy A, Zhou X, et al. Pediatric acute myeloid leukemia with NPM1 mutations is characterized by a gene expression profile with dysregulated HOX gene expression distinct from MLL-rearranged leukemias. Leukemia. 2007;21(9):2000–2009. doi: 10.1038/sj.leu.2404808. [DOI] [PubMed] [Google Scholar]
- 42.Boer JM, van der Veer A, Rizopoulos D, et al. Prognostic value of rare IKZF1 deletion in childhood B-cell precursor acute lymphoblastic leukemia: an international collaborative study. Leukemia. 2016;30(1):32–38. doi: 10.1038/leu.2015.199. [DOI] [PubMed] [Google Scholar]
- 43.Witkowski MT, Hu Y, Roberts KG, et al. Conserved IKAROS-regulated genes associated with B-progenitor acute lymphoblastic leukemia outcome. J Exp Med. 2017;214(3):773–791. doi: 10.1084/jem.20160048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pongubala JM, Northrup DL, Lancki DW, et al. Transcription factor EBF restricts alternative lineage options and promotes B cell fate commitment independently of Pax5. Nat Immunol. 2008;9(2):203–215. doi: 10.1038/ni1555. [DOI] [PubMed] [Google Scholar]
- 45.van Oevelen C, Collombet S, Vicent G, et al. C/EBPα activates pre-existing and de novo macrophage enhancers during induced pre-B cell transdifferentiation and myelopoiesis. Stem Cell Reports. 2015;5(2):232–247. doi: 10.1016/j.stemcr.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yoshida T, Hazan I, Zhang J, et al. The role of the chromatin remodeler Mi-2beta in hematopoietic stem cell self-renewal and multilineage differentiation. Genes Dev. 2008;22(9):1174–1189. doi: 10.1101/gad.1642808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu X, Chu CS, Fang T, et al. MTA2/NuRD regulates B cell development and cooperates with OCA-B in controlling the pre-B to immature B cell transition. Cell Rep. 2019;28(2):472–485. doi: 10.1016/j.celrep.2019.06.029. e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sperlazza J, Rahmani M, Beckta J, et al. Depletion of the chromatin remodeler CHD4 sensitizes AML blasts to genotoxic agents and reduces tumor formation. Blood. 2015;126(12):1462–1472. doi: 10.1182/blood-2015-03-631606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Heshmati Y, Türköz G, Harisankar A, et al. The chromatin-remodeling factor CHD4 is required for maintenance of childhood acute myeloid leukemia. Haematologica. 2018;103(7):1169–1181. doi: 10.3324/haematol.2017.183970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mor N, Rais Y, Sheban D, et al. Neutralizing Gatad2a-Chd4-Mbd3/NuRD complex facilitates deterministic induction of naive pluripotency. Cell Stem Cell. 2018;23(3):412–425. doi: 10.1016/j.stem.2018.07.004. e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kerry J, Godfrey L, Repapi E, et al. MLL-AF4 spreading identifies binding sites that are distinct from super-enhancers and that govern sensitivity to DOT1L inhibition in leukemia. Cell Rep. 2017;18(2):482–495. doi: 10.1016/j.celrep.2016.12.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harman JR, Thorne R, Jamilly M, et al. A KMT2A-AFF1 gene regulatory network highlights the role of core transcription factors and reveals the regulatory logic of key downstream target genes. Genome Res. 2021;31(7):1159–1173. doi: 10.1101/gr.268490.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakamura T, Mori T, Tada S, et al. ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell. 2002;10(5):1119–1128. doi: 10.1016/s1097-2765(02)00740-2. [DOI] [PubMed] [Google Scholar]
- 54.Jacoby E, Nguyen SM, Fountaine TJ, et al. CD19 CAR immune pressure induces B-precursor acute lymphoblastic leukaemia lineage switch exposing inherent leukaemic plasticity. Nat Commun. 2016;7(1) doi: 10.1038/ncomms12320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gardner R, Wu D, Cherian S, et al. Acquisition of a CD19-negative myeloid phenotype allows immune escape of MLL-rearranged B-ALL from CD19 CAR-T-cell therapy. Blood. 2016;127(20):2406–2410. doi: 10.1182/blood-2015-08-665547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rayes A, McMasters RL, O’Brien MM. Lineage switch in MLL-rearranged infant leukemia following CD19-directed therapy. Pediatr Blood Cancer. 2016;63(6):1113–1115. doi: 10.1002/pbc.25953. [DOI] [PubMed] [Google Scholar]
- 57.Sotillo E, Barrett DM, Black KL, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov. 2015;5(12):1282–1295. doi: 10.1158/2159-8290.CD-15-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rabilloud T, Potier D, Pankaew S, Nozais M, Loosveld M, Payet-Bornet D. Single-cell profiling identifies pre-existing CD19-negative subclones in a B-ALL patient with CD19-negative relapse after CAR-T therapy. Nat Commun. 2021;12(1):865. doi: 10.1038/s41467-021-21168-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hamieh M, Dobrin A, Cabriolu A, et al. CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature. 2019;568(7750):112–116. doi: 10.1038/s41586-019-1054-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liao W, Kohler ME, Fry T, Ernst P. Does lineage plasticity enable escape from CAR-T cell therapy? Lessons from MLL-r leukemia. Exp Hematol. 2021;100:1–11. doi: 10.1016/j.exphem.2021.07.002. [published correction appears in Exp Hematol. 2021;103:73-74.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lamble AJ, Myers RM, Taraseviciute A, et al. KMT2A rearrangements are associated with lineage switch following CD19 targeting CAR T-cell therapy. Blood. 2021;138(suppl_1) [abstract] Abstract 256. [Google Scholar]
- 62.Leahy AB, Devine KJ, Li Y, et al. Impact of high-risk cytogenetics on outcomes for children and young adults receiving CD19-directed CAR T-cell therapy. Blood. 2022;139(14):2173–2185. doi: 10.1182/blood.2021012727. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.