

Abstract
Commercially available cathinones are drugs of long-term abuse drugs whose pharmacology is fairly well understood. While their psychedelic effects are associated with 5-HT2AR, the enclosed study summarizes efforts to shed light on the pharmacodynamic profiles, not yet known at the receptor level, using molecular docking and three-dimensional quantitative structure–activity relationship (3-D QSAR) studies. The bioactive conformations of cathinones were modeled by AutoDock Vina and were used to build structure-based (SB) 3-D QSAR models using the Open3DQSAR engine. Graphical inspection of the results led to the depiction of a 3-D structure analysis-activity relationship (SAR) scheme that could be used as a guideline for molecular determinants by which any untested cathinone molecule can be predicted as a potential 5-HT2AR binder prior to experimental evaluation. The obtained models, which showed a good agreement with the chemical properties of co-crystallized 5-HT2AR ligands, proved to be valuable for future virtual screening campaigns to recognize unused cathinones and similar compounds, such as 5-HT2AR ligands, minimizing both time and financial resources for the characterization of their psychedelic effects.
Keywords: cathinones, 5-HT2AR, molecular docking, 3-D QSAR
1. Introduction
The aminergic family of G-protein coupled receptors (GPCRs) physiologically respond to hormonal and neurotransmitter stimuli by activating internal signal transduction pathways and cellular responses [1], thus representing a valuable target class for drug discovery [2]. The 5-HT receptors (5-HTRs), as the largest subfamily of GPCRs, are activated by the neurotransmitter and neuromodulator serotonin (i.e., 5-hydroxytryptamine, 5-HT). After being biosynthesized in the intestine from tryptophan, the hormonal behavior of 5-HT in the periphery is associated with mitogenesis and the proliferation of fibroblast cells [3], homeostasis of glucose and lipid metabolism [4], enhanced arterial contraction during cardiac hypertrophy [5,6], bronchoconstriction [7], rhythmic breathing, and circadian rhythms [8], as well as pain, increased appetite, body temperature, etc. [9]. Nevertheless, small amounts of serotonin are biosynthesized in the axons of brain neurons, where 5-HT acts within the CNS (i.e., neocortex and hippocampi of the brain) as a major site for the expression of 5-HT receptors (5-HTRs), regulating mood, social cognition [10], neurogenesis [11], short- and long-term memory [12], spatial memory that enables navigation, sexual behavior, impulsivity, aggression, and migraine attacks [13,14].
At least 14 different serotonin receptors (5-HTRs), divided into seven subgroups (i.e., 5-HT1-7R), constitute the serotonergic system, primarily located on the membranes of either presynaptic or postsynaptic neurons [15]. The studied 5-HT2AR here (Figure 1), together with the 5-HT2BR and 5-HT2CR, is expressed on the postsynaptic membrane and acts as a mediator between the extracellular physiological ligand serotonin and intracellular G-proteins [1,16]. Activation of the 5-HT2AR by 5-HT induces the involvement of the Gαi, Gαq/11, or Gαs proteins and subsequently increases the cellular levels of inositol triphosphate (IP3) and diacylglycerol (DAG) [17]. The 5-HT2AR-mediated pharmacology distinguishes the macromolecule as a drug target in the treatment of Parkinson’s and Alzheimer’s diseases, as well as mental disorders such as schizophrenia, bipolar disorder, depression, anxiety, and insomnia [16]. On the other hand, the 5-HT2AR is also a target for a variety of recreational drugs, mediating the effects of potent psychoactive substances such as lysergic acid diethylamide (LSD) or amphetamines, commonly known as psychedelics or hallucinogens [1,18].
Figure 1.

Left: The crystal structure of 5-HT2AR in complex with LSD (as deposited at PDB, https://www.rcsb.org/, PDB ID: 6HWT [16], accessed on 1 December 2022). LSD is depicted in blue encircled by the transparent sphere, TM1 (residues 69–101) is colored in blue, TM2 (residues 111–137) is depicted in plum, TM3 (residues 114–179) is painted in orange, TM4 (residues 188–217) is colored in lime green, TM5 (residues 231–265) is depicted in dark red, TM6 (residues 315–349) is painted in violet-red, TM7 (residues 353–383) is colored in dark grey, intracellular amphipathic helix H8 (residues 384–399) is depicted in yellow, two intracellular loops ICL1 and ICL2 (residues 102–111 and 179–187) are illustrated in cyan and hot pink, respectively, three extracellular loops ECL1, ECL2, and ECL3 (residues 134–138, 218–230, and 350–352) are labeled in gold, dim gray, and dark slate gray, respectively; Right: The crystal structure of 5-HT2AR immersed into dipalmitoylphosphatidylcholine (DPPC) lipid bilayer (as deposited at OPM, https://opm.phar.umich.edu/, entry: 4282, accessed on 2 March 2023): the DPPCs are illustrated with transparent gray spheres, oxygen atoms from water molecules are colored in red, sodium atoms are presented as pink spheres, and chlorine atoms are described as green spheres. For the clarity of the presentation, hydrogen atoms were omitted.
Although a significant number of ligands co-crystallized with 5-HT2AR were available and deposited in the Protein Data Bank at the start of this investigation (Table 1, 13 ligands found in 14 complexes), only nine of them were homogeneously associated with inhibition constants (i.e., Kis) and thus were insufficient for the development of broad and computationally applicable medicinal chemistry models. Therefore, attention was focused on the literature concerning amphetamines and their β-keto analogs cathinones, as compounds known to exert 5-HT2AR-mediated psychedelic effects and well characterized by corresponding inhibition constants (Table 2), to elucidate their hitherto unknown binding modes and to describe their pharmacodynamic properties using 3D QSAR models. The 5-HT2AR ligands (besides those found co-crystallized within 5-HT2BR, Table 1) were further used either for defying structure-based alignment assessment (SBAA) rules, which were further employed for the generation of bioactive conformations of cathinones, or as an ultimate prediction test set (TSCRY) for external validation of 3-D QSAR models (see further discussion).
As natural products of the khat plant (Catha edulis (Vahl) Forssk. ex Endl.), cathinones were initially structurally optimized for the medical treatment of parkinsonism, obesity, and depression, but are now used as “legal highs” (also known as “bath salts”, “research chemicals not for human consumption”, or “plant food”) [19]. Recently developed DFT-based protocols have successfully reproduced experimental spectral data of selected synthetic cathinones (SCs) and can be further used for the identification/prediction of physicochemical parameters of either known or new SCs, even in forensic applications [20]. SCs primarily target presynaptic plasma membrane transporters for dopamine, norepinephrine, and serotonin (DAT, NAT, and SERT, respectively) [21]. Nevertheless, they increase the concentration of 5-HT in the synaptic space, providing the basis for CNS stimulatory and sympathomimetic effects characterized by increased blood pressure, heart rate, mydriasis, and hyperthermia [22]. As monoamine releasers, they enhance neurotransmitter transmission and increase synaptic concentrations; as monoamine reuptake inhibitors, they prevent the return of neurotransmitters to presynaptic neurons and consequent metabolic degradation while simultaneously activating monoamine receptors within the limbic-corticostriatal pathway [23].
The signaling and neurobehavioral effects of hallucinogens are associated with a 5-HT2AR expression on cortical layer V pyramidal neurons [17,24], suggesting the investigation of SCs as potential ligands for this receptor. Through 5-HT2AR binding, SCs can induce euphoria, increased empathy, sociability, energy, and alertness, but also rhabdomyolysis and autonomic symptoms such as tachycardia, hypertension, hyperthermia, ecstasy, sociability, alertness, empathy, and increased energy [21,22]. However, when taken at higher doses or for longer periods, stimulants can cause many psychiatric symptoms, including anxiety, agitation, and fear, which can progress to psychosis (delusions and hallucinations) and delirium [19,22]. Compared with amphetamines, several SCs, such as α-PPP (9) and MDPV (14) (Table 1), have more severe psychiatric side effects [21].
Since the mechanisms by which SCs exert the above-described effects as 5-HT2AR ligands are not yet fully understood and defined, the present study investigated the putative bioactive conformations and structure-based (SB) pharmacodynamic profiles of SCs (Table 2). The binding modes of the SCs, explored by molecular docking and 3-D QSAR studies were associated with their reported pharmacological profiles, providing a basis for the elucidation of the psychedelic or hallucinogenic effects of the SCs at the molecular level. Derived SB 3-D structure–activity relationships were found in good agreement with various ligands, either reported in the literature or experimentally resolved as 5-HT2AR ligands, for which the obtained results can be used in future virtual screening campaigns to reveal new SCs and similar compounds not yet recognized as 5-HT2AR ligands, minimizing both time and financial resources for the characterization of their potential psychedelic effects.
2. Results and Discussion
2.1. Crystal Dataset Compilation
The experimentally resolved complexes of ligands co-crystallized with either 5-HT2AR or 5-HT2BR (Table 1) were retrieved from the Protein Data Bank (PDB, https://www.rcsb.org/, accessed on 1 December 2022). Fourteen experimental 5-HT2AR/ligand complexes were assembled, including the full 5-HT2AR antagonists (FAs) and antipsychotics risperidone (PDB ID: 6A93) [16], and zotepine (PDB ID: 6A94 [16]), a 5-HT2AR agonist (AG) and psychotic LSD (PDB IDs: 6WGT [25] and 7WC6 [26]), the inverse agonist (IA) 1-methyl-4-[(5~{S})-3-methylsulfanyl-5,6-dihydrobenzo[b][1]benzothiepin-5-yl]piperazine (PDB ID: 6WH4 [25]), the 5-HT2AR partial antagonist (PA) and hallucinogen 25-CN-NBOH (PDB ID: 6WHA [25]), the 5-HT2AR agonist (3R)-3-methyl-5-(1H-pyrrolo[2,3-b]pyridin-3-yl)-1,2,3,6-tetrahydropyridin-1-ium (7RAN [27]), the FAs cariprazine (PDB ID: 7VOD [28]) and aripiprazole (PDB ID: 7VOE [28]), the AGs serotonin (PDB IDs: 7WC4 [26])psilocin (the active metabolite of psilocybin, PDB ID: 7WC5 [26]), lisuride (PDB ID: 7WC7 [26]), and FAs lumateperone (PDB ID: 7WC8 [26]), and the non-hallucinogenic psychedelic analog IHCH-7113 (PDB ID: 7WC9 [26]).
The 5-HT2BR ligands were AGs ergotamine (PDB IDs: 4IB4 [29], 4NC3 [30] and 5TUD [31]), LSD (PDB IDs: 5TVN [31], 7SRR [32] and 7SRS [32]), the agonists N,N-diethyl-N’-[(8α)-6-methyl-9,10-didehydroergolin-8-yl]urea (PDB ID: 6DRX [33]), (8β)-N-[(2S)-1-hydroxybutan-2-yl]-6-methyl-9,10-didehydroergoline-8-carboxamide (PDB ID: 6DRY [33]), (8α)-N-[(2S)-1-hydroxybutan-2-yl]-1,6-dimethyl-9,10-didehydroergoline-8-carboxamide (PDB ID: 6RDZ [33]), as well as FA (1S)-1-[(2-chloro-3,4-dimethoxyphenyl)methyl]-6-methyl-2,3,4,9-tetrahydro-1H-beta-carboline (PDB ID: 6DS0 [33]).
2.2. Literature Datasets Set Compilation
The SCs as 5-HT2AR ligands were retrieved from the literature to compile a training set (TR) (Table 2) [21,34,35,36,37,38,39,40,41,42,43,44,45], whereas the test set (TS) was compiled from listed clinically approved drugs (Table 3) [46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62] with known affinities for 5-HT2AR, but with unknown binding modes for all compounds except 25 (found within the 7VOE protein [28]). For either TR or TS compounds, analysis of the available biological data revealed a homogeneous association with potencies described as pKis (-logKi). Thus, the TR included Cathinone (1) and its derivatives, Flephedrone (2), Mephedrone (3) (the most commonly abused cathinone, non-selective monoamine uptake inhibitor [34,35]), and Methcathinone (4), as likely AGs and psychedelic compounds, as well as FAs and rather antidepressive drugs 4-Bromomethcathinone (5), 3-Bromomethcathinone (6, [21]), 2-Fluoromethcathinone (7), and 2-Trifluoromethoxy-methcathinone (8), also known to act as preferential dopamine active transporter (DAT) and noradrenaline transporter (NAT) inhibitors and α1A adrenoceptor agonists/antagonists [21,34].
The experimentally determined 5-HT2AR IAs/FAs and non-psychostimulants pyrovalerone-based SCs, namely α-PPP (9 [21]), 4-Methyl-α-PPP (10 [21]), 4-Bromo-α-PPP (11), and 3-Bromo-α-PPP (12), were also included in the TR, alongside with Naphyrone (13), MDPV (14), Pyrovalerone (15), and MDPPP (16) as experimentally validated FAs, also known as norepinephrine and dopamine reuptake inhibitors (NRIs and DRIs, respectively) [36,37].
Table 1.
Names, structures, and inhibition constants of co-crystalized 5-HT2AR and 5-HT2BR ligands.
| PDB ID P (Mechanism) a |
Compound’s Structure |
pKi | Ref. | PDB ID P (Mechanism) a |
Compound’s Structure |
pKi | Ref. |
|---|---|---|---|---|---|---|---|
| 5-HT2AR ligand |
7WC7 AG |
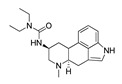
|
8.55 | [26] | |||
|
6A93 FA a |
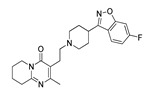
|
8.16 | [16] |
7WC8 AG |
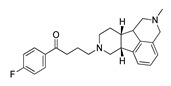
|
9.27 | [26] |
|
6A94 FA |
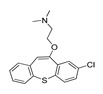
|
7.40 | [16] |
7WC9 AG |
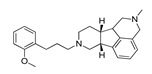
|
NA | [26] |
|
6WGT 7WC6 AG |
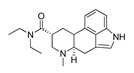
|
8.63 | [16] [26] |
5-HT2BR ligand | |||
|
6WH4 IA |
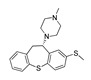
|
9.70 | [25] |
4NC3 4IB4 5TUD AG |
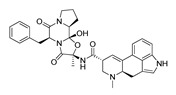
|
8.88 | [29] [29] [31] |
|
6WHA PA |
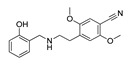
|
9.08 | [25] |
5TVN AG |
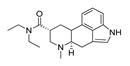
|
NA | [31] |
|
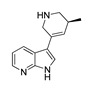
7RAN AG |
|
NA a | [27] |
6DRX AG |
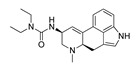
|
8.88 | [33] |
|
7VOD FA |
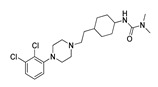
|
7.73 | [28] |
6DRY 7SRQ 7SRS 7SRR AG |
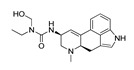
|
9.33 | [33] [32] [32] [32] |
|
7VOE FA |
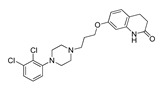
|
8.47 | [28] |
6DRZ AG |
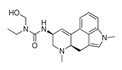
|
10 | [33] |
|
7WC4 AG |
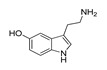
|
7.49 | [26] |
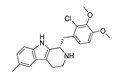
6DS0
FA |
|
NA | [33] |
|
7WC5 AG PA |
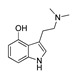
|
NA | [26] | ||||
a Pharmacology: AG—agonist, IA—inverse agonist, PA—partial agonist, FA—full antagonist
Table 2.
Names, structures, and inhibition constants of cathinones as human 5-HT2AR ligands compiling the TR.
| Name (Number) P (Mechanism) a |
Compound’s Structure |
pKi | Ref. | Name (Number) P (Mechanism) a |
Compound’s Structure |
pKi | Ref. |
|---|---|---|---|---|---|---|---|
| Cathinone and its derivatives |
Naphyrone (13) FA |
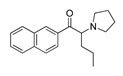
|
4.96 | [34] | |||
|
Cathinone (1) AG a |
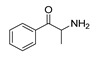
|
6.00 | [34] |
MDPV (14) FA |
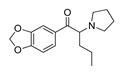
|
4.88 | [34] |
|
Flephedrone (2) AG |
|
6.00 | [34] |
Pyrovalerone (15) FA |
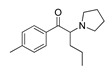
|
4.88 | [34] |
|
Mephedrone (3) AG |
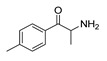
|
5.68 | [34] |
MDPPP (16) FA |
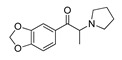
|
4.20 | [21] |
|
Methcathinone (4) AG |
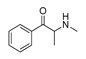
|
5.23 | [34] | Benzo[d][1,3]dioxole-based SCs | |||
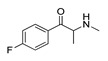
|
4-Bromomethcathinone (5) IA/FA |
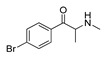
|
5.20 | [21] |
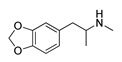
MDBD (17) AG |
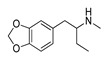
|
5.20 | [34] |
|
3-Bromomethcathinone (6) IA/FA |
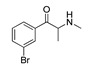
|
5.00 | [21] |
MDMA (18) AG |
|
5.11 | [34] |
|
2-Fluoromethcathinone (7) IA/FA |
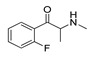
|
5.00 | [21] |
Butylone (19) AG |
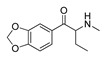
|
4.88 | [34] |
|
2-(Trifluoromethoxy) -methcathinone (8) IA/FA |
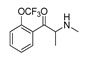
|
5.00 | [21] |
Ethylone (20) AG |
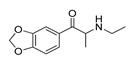
|
4.88 | [34] |
| Pyrovalerone-based SCs |
MDEA (21) AG |
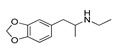
|
4.88 |
MDEA
AG (21) |
|||
|
α-PPP (9) IA/FA |
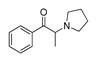
|
5.60 | [21] |
Methylone (22) AG |
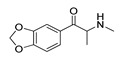
|
4.88 | [34] |
|
4-Methyl-α-PPP (10) IA/FA |
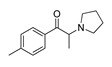
|
5.50 | [21] | SCs ’ precursors | |||
|
4-Bromo-α-PPP (11) IA/FA |
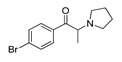
|
5.40 | [21] |
Amphetamine (23) AG |
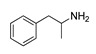
|
4.88 | [34] |
|
3-Bromo-α-PPP (12) IA/FA |
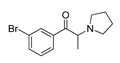
|
5.00 | [21] |
Methamphetamine (24) AG |
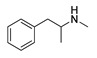
|
4.88 | [34] |
a Pharmacology: AG—agonist, IA—inverse agonist, PA—partial agonist, FA—full antagonist.
Table 3.
Commercial names, structures, and inhibition constants of human 5-HT2AR ligands compiling the TS.
| Name (Number) P (Mechanism) a |
Compound’s Structure |
pKi | Ref. | Name (Number) P (Mechanism) a |
Compound’s Structure |
pKi | Ref. |
|---|---|---|---|---|---|---|---|
|
Aripiprazole (25) FA a |
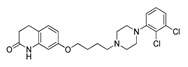
|
8.57 | [46] |
Norfenfluramine (36) AG |
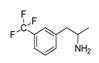
|
6.82 | [47] |
|
BW-723C86 (26) AG |
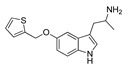
|
7.2 | [48] |
Olanzapine (37) IA |
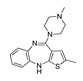
|
8.88 | [49] |
|
Clozapine (27) FA |
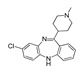
|
8.39 | [50] |
Quentiapine (38) FA |
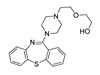
|
6.81 | [50] |
|
CP-809,101 (28) AG |
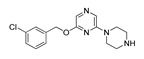
|
8.22 | [51] |
R060-0175 (39) IA |
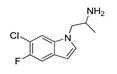
|
7.44 | [48] |
|
Ketanserin (29) FA |
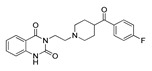
|
9.67 | [52] |
Risperidone (40) IA |
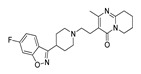
|
9.69 | [50] |
|
Lorcaserin (30) AG |
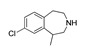
|
6.95 | [53] |
RS-127,455 (41) FA |
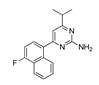
|
6.03 | [48] |
|
MDL-100,907 (31) FA |
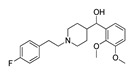
|
8.77 | [54] |
Saprogrelate (42) FA |
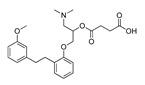
|
8.52 | [55] |
|
Mesulergine (32) FA |
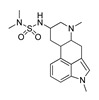
|
7.34 | [56] |
SB-204,741 (43) FA |
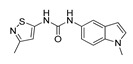
|
5.00 | [57] |
|
Mianserin (33) FA |
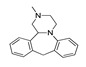
|
8.15 | [58] |
SB-206,553 (44) FA |
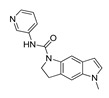
|
5.64 | [59] |
|
Mirtazapine (34) IA |
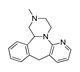
|
7.78 | [60] |
SB-242,084 (45) FA |
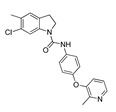
|
6.07 | [61] |
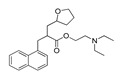
|
Naftidrofuryl (35) IA |
|
6.20 | [55] |
WAY-161,503 (46) AG |
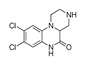
|
7.40 | [62] |
a Pharmacology: AG—agonist, IA—inverse agonist, PA—partial agonist, FA—full antagonist.
Benzo[d][1,3]dioxole-based SCs, MBDB (17), MDMA (ecstasy, 18), Butylone (19), Ethylone (20), MDEA (21), and Methylone (22) were also listed in the TRs as examples of SCs with the ability to directly bind 5-HT2AR most likely as AGs and psychostimulants, resulting in the release of 5-HT. Of these, 18 induces excitation and hallucinogenic-like perceptual changes at higher doses [38,39,40], while 19, 20, and 22 are known as non-selective monoamine uptake inhibitors [33,34,35,36]. The precursors of SCs, amphetamine (23) and methamphetamine (“crystal meth”, 24), likewise considered to be AGs, completed the TR compilation. Binding to 5-HT2AR, 23 and 24 induce behavioral effects opposite to those induced by stimulation of 5-HT1AR (5-HT2AR-mediated depolarization vs. the 5-HT1AR-mediated hyperpolarization) [18,41].
Since the quantitative parameters for psychedelic or hallucinogenic effects, such as the onset of action (OA) and duration of action (DA), are known for only a few compounds (1–8 [35,43,44,45], 13, 14, and 16 [36,37], and 23 and 24 [34], Table S1, Supplementary Materials), it is not possible to derive a robust quantitative psychoactivity relationship model. Therefore, the pKis of the SCs (Table 2) were correlated with the SB bioactive conformations of their 5-HT2ARs using 3-D QSAR models, whose predictive abilities were externally validated by the compiled TS.
2.3. Definition of Optimal Protocol for the Alignment of 5-HT2AR Ligands
In the development of the CoMFA-like 3-D QSAR models, a crucial step is represented by the alignment rules [63]. Therefore, considering the lack of SCs co-crystallized with 5-HT2AR (Table 2), their bioactive conformations were modeled using as much experimentally available structural data as possible on 5-HT2ARs and 5-HT2BRs crystallized in complexes with different ligands (Table 1) [16,25,26,27,28,29,30,31,32,33,64]. Attention was initially focused on lysergic acid diethylamide (LSD), the prototypical human hallucinogen, one of the most potent psychoactive (and recreational) drugs [31], and an experimentally confirmed agonist for both 5-HT2AR (PDB IDs: 6WGT and 7WC6, [25,26] and 5-HT2BR (PDB IDs: 5TVN [31], 6DRX [33], 7SRR [32], and 7SRS [32]) isoforms [64].
Regardless of the active site, LSD showed an almost constant binding conformation with the lowest root mean square deviation (RMSD) (Table 4). Sequence and active site similarities of 5-HT2AR and 5-HT2BR were approximately 40% and 60%, respectively (Figure 2), therefore both targets were used to establish a protocol for the alignment of SCs in the 5-HT2AR. To this end, an alignment evaluation of ligands co-crystallized in either 5-HT2AR (Figure 3a, Table 4) [16,25,26,27,28] or 5-HT2BR (Figure 3b, Table 5) [29,30,32,33] was performed to establish the alignment rules to define the binding modes of TR and TS compounds. The quality of alignment was evaluated by SB 3-D QSAR models in terms of q2 value [65].
Table 4.
The RMSD matrix of either 5-HT2AR or 5-HT2BR proteins bound to LSD.
| 5TVN | 6DRX | 6WGT | 7SRR | 7WC6 | |
|---|---|---|---|---|---|
| 5TVN | 0.000 | 1.012 | 0.731 | 1.154 | 1.077 |
| 6DRX | 1.012 | 0.000 | 0.795 | 1.135 | 1.165 |
| 6WGT | 0.731 | 0.795 | 0.000 | 0.765 | 0.727 |
| 7SRR | 1.154 | 1.135 | 0.765 | 0.000 | 1.195 |
| 7WC6 | 1.077 | 1.165 | 0.765 | 1.195 | 0.000 |
Figure 2.

Sequence alignment between human 5-HT2AR (Uniprot entry P28223) and 5-HT2BR (UniProt entry P41596) performed by Clustal Omega [66]. Special characters meanings: * (Asterix) positions with a single, fully conserved residue; “:” (colon) positions with conservation between amino acid groups of similar properties; “.” (period) positions with conservation between amino acid groups of weakly similar properties.
Figure 3.

The docking assessment of LSD co-crystallized into 5-HT2AR (the 6WGT crystal [16]), EC conformation in pink, ECRD conformation in blue, RCRD conformation in green, ECCD conformation in red, and RCCD conformation in yellow (a); LSD co-crystallized into 5-HT2BR (the 5VNT crystal [31]), EC conformation in pink, ECRD conformation in blue, RCRD conformation in green, ECCD conformation in red, and RCCD conformation in yellow (b).
Table 5.
Structure-based alignment assessment of 5-HT2AR ligands within the RCCD stage.
| Code | AutoDock | Vina | SMINA | DOCK | PLANTS | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| SF a | Vina | Vinardo | ad4 | chemplp | plp | plp95 | ||||
| Randomized Conformation Cross-Docking (RCCD) | ||||||||||
| 6A93 b | 2.784 c | 2.843 | 2.742 | 2.563 | 3.563 | 3.231 | 2.573 | 2.742 | 2.746 | [16] |
| 6A94 | 2.657 | 2.224 | 3.241 | 3.431 | 3.431 | 3.422 | 2.991 | 2.664 | 2.561 | [16] |
| 6WGT | 2.943 | 1.943 | 2.941 | 2.567 | 2.567 | 2.993 | 2.892 | 3.245 | 2.993 | [16] |
| 6WH4 | 2.995 | 1.971 | 2.518 | 2.783 | 2.783 | 2.961 | 2.973 | 2.835 | 3.762 | [25] |
| 6WHA | 3.726 | 2.941 | 3.663 | 3.452 | 3.452 | 2.693 | 2.116 | 2.985 | 3.426 | [25] |
| 7RAN | 3.651 | 2.365 | 2.632 | 2.954 | 3.954 | 3.652 | 2.639 | 2.954 | 3.624 | [27] |
| 7VOD | 3.648 | 1.984 | 3.654 | 3.642 | 3.642 | 2.584 | 2.652 | 2.667 | 3.621 | [28] |
| 7VOE | 6.257 | 1.965 | 2.548 | 2.984 | 2.984 | 2.524 | 2.547 | 2.647 | 2.457 | [28] |
| 7WC4 | 3.658 | 2.695 | 2.458 | 2.658 | 2.658 | 3.632 | 3.698 | 3.965 | 3.621 | [26] |
| 7WC5 | 2.398 | 2.657 | 2.654 | 2.984 | 2.984 | 3.324 | 3.258 | 3.541 | 2.514 | [26] |
| 7WC6 | 3.695 | 3.625 | 3.254 | 3.652 | 3.652 | 2.659 | 2.987 | 2.564 | 2.558 | [26] |
| 7WC7 | 2.698 | 3.954 | 2.698 | 2.874 | 2.874 | 2.584 | 2.898 | 2.842 | 2.774 | [26] |
| 7WC8 | 3.654 | 3.625 | 2.548 | 2.636 | 2.636 | 3.395 | 2.235 | 2.665 | 2.664 | [26] |
| 7WC9 | 3.658 | 3.695 | 2.547 | 2.584 | 2.584 | 2.987 | 2.397 | 2.635 | 2.981 | [26] |
| DA d | 21.43% | 50.00% | 35.71% | 35.71% | 28.57% | 28.57% | 42.85% | 42.85% | 32.14% | |
a Scoring function; b Experimental conformation; c Root-Mean-Square-Deviation measured between the heavy atoms of the ligand’s experimental and the ligand’s re-/cross-aligned conformation; d Docking accuracy.
Structure-Based Alignment Assessment
Either ligands/5-HT2AR (Table 5) or ligands/5-HT2BR (Table 6) experimentally resolved complexes were superimposed using 7WC8 [26,66] as a template, with the lowest resolution of 2.45 Å. For the SB alignment assessments (SBAAs) procedure [67], the best performing molecular docking algorithm/scoring function pair in reproducing the experimentally bound conformations of the ligands was investigated using ligands extracted from either 5-HT2AR (Table 5) or 5-HT2BR (Table 6) complexes (at the same time providing a basis for future cross-pharmacology in a discrete fashion) [67]. The following protocol was established: first, any ligand was subjected to SBAA [67] using free or open-source molecular docking programs: AutoDock [68], AutoDock Vina (hereafter Vina) [69], DOCK [70], SMINA [71], and PLANTS [72], with all available scoring function (SF) variants. As previously described, the SBAA protocol [67] was investigated at four levels of difficulty: experimental or randomized ligand conformation (EC and RC, respectively), re-docking (RD) (ECRD and RCRD, respectively), and cross-docking (CD) (ECCD and RCCD, respectively). While the RMSD data associated with ECRD, RCRD, and ECCD are reported as Supplementary Materials (Tables S2 and S3), the RCCD-related RMSD values, which describe the docking programs’ abilities in reproducing experimental binding modes from initial random ligand conformations into a never-seen protein environment (the most difficult scenario), are reported for either 5-HT2AR or 5-HT2BR as Table 5 and Table 6, respectively.
Table 6.
Structure-based alignment assessment of 5-HT2BR ligands.
| Code | AutoDock | Vina | SMINA | DOCK | PLANTS | Ref. | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| SF a | Vina | Vinardo | ad4 | chemplp | plp | plp95 | ||||
| Randomized Conformation Cross-Docking (RCCD) | ||||||||||
| 4NC3 b | 4.454 c | 2.785 | 2.986 | 2.784 | 2.984 | 4.313 | 3.125 | 2.742 | 3.421 | [29] |
| 4IB4 | 3.431 | 2.895 | 4.124 | 3.984 | 2.634 | 3.453 | 3.784 | 2.563 | 2.741 | [29] |
| 5TUD | 3.625 | 2.748 | 2.642 | 3.614 | 3.569 | 3.695 | 2.539 | 2.597 | 3.625 | [57] |
| 5TVN | 2.895 | 1.993 | 1.963 | 1.943 | 3.964 | 3.241 | 2.431 | 3.254 | 2.346 | [31] |
| 6DRX | 2.674 | 2.864 | 2.424 | 2.435 | 2.874 | 3.254 | 2.964 | 3.541 | 2.758 | [33] |
| 6DRY | 2.895 | 2.998 | 3.894 | 3.784 | 2.695 | 2.451 | 2.531 | 2.431 | 2.321 | [33] |
| 6DRZ | 2.992 | 2.674 | 3.431 | 3.895 | 3.624 | 2.563 | 2.728 | 3.522 | 2.462 | [33] |
| 6DS0 | 2.567 | 1.864 | 2.974 | 2.983 | 2.874 | 2.451 | 2.351 | 2.214 | 3.325 | [33] |
| 7SQR | 2.517 | 2.957 | 2.987 | 2.524 | 2.595 | 3.648 | 2.487 | 2.845 | 2.635 | [32] |
| 7SRS | 2.698 | 1.997 | 1.987 | 1.987 | 2.749 | 2.874 | 2.228 | 2.457 | 3.984 | [32] |
| 7SRR | 3.624 | 2.487 | 2.964 | 1.987 | 2.996 | 3.625 | 2.487 | 2.625 | 3.635 | [32] |
| DA d | 31.82% | 63.64% | 45.45% | 45.45% | 36.36% | 18.18% | 40.91% | 36.36% | 31.82% | |
a Scoring function; b Experimental conformation; c Root-Mean-Square-Deviation measured between the heavy atoms of the ligand’s experimental and the ligand’s re-/cross-aligned conformation; d Docking accuracy.
Regarding the reproduction of co-crystallized ligand conformations in 5-HT2AR (Table 5, Figure 3a), in the ECRD phase (Table S2, Supplementary Materials), all available docking algorithms/SFs pairs, except those implemented in AutoDock and DOCK, showed a high level of docking accuracy (DA) [67,73,74]. Among the most accurate, Vina was found to be the best, with a DA value of 89.28%, while SMINA, with either vina [69], vinardo [75], or ad4 [68] SFs, showed DA ranging from 75.00% (SMINA/vina) to 78.57% (SMINA/vinardo). The PLANTS/plp pair (DA = 82.14%) almost reached the accuracy of Vina, while either PLANTS/chemplp or PLANTS/lp95 gave DA values close to SMINA/vinardo. At the RCRD stage (Table S2, Supplementary Material), Vina maintained a DA value of more than 80%, with a physiological decrease of about 7% compared to ECRD, while each SMINA/SF pair-associated DA was less than 75%. The ability of PLANTS/plp to reposition 5-HT2AR’ ligand conformations showed a 5% decrease in DA, while PLANTS/chemplp remained as accurate as SMINA/vinardo. In the ECCD evaluation phase (Table S2, Supplementary Materials), Vina remained the most accurate (DA equal to 75%), followed by SMINA/vina and SMINA/vinardo (DA ~ 72%), while the SMINA/ad4 and PLANTS pairs, plp and plp95, were 50–60% accurate. At the last and most difficult level, the RCCD stage (Table 5), Vina proved to be the docking program with the highest DA (50%).
As for the docking evaluation for ligands co-crystallized in 5-HT2BR (Table 6, Figure 3b), all docking algorithms/SF pairs showed DAs higher than 50% during the ECRD phase (Table S3, Supplementary Materials). In particular, Vina and SMINA/vina/vinardo showed perfect DAs (100%), followed by SMINA/ad4 (DA higher than 80%), while for AutoDock, DOCK, and PLANTS, the DAs ranged between approximately 59% (DOCK) and 73% (PLANTS/chemplp/plp). In the RCRD assessment (Table S3, Supplementary Materials), Vina retained the highest DA (86.36%), while SMINA was associated with DAs of approximately 68% and 73%. A decreasing trend was also observed for AutoDock, DOCK, and PLANTS (chemplp and plp SFs) with DA values equal to or greater than 50% but not exceeding 65%. At the ECCD level (Table S3, Supplementary Materials), Vina and SMINA/vina/vinardo/ad4 were the only ones able to cross-dock 5-HT2BR ligands with acceptable DA values of 72.73, 63.64, 54.00, and 50.00%, respectively, while AutoDock, DOCK, and all PLANTS SFs suffered from the initial conformation with low DA values. Finally, during the RCCD (Table 4), as in the case of 5-HT2AR, Vina was confirmed as the program that was able to reproduce the experimental binding conformations with the least error (DA = 63.64%).
2.4. SCs’ Binding Mode Analysis into 5-HT2A-DPPC and 3-D QSAR Models Interpretation
Based on the above SB assessments, Vina was selected as an SB tool to investigate the binding modes of either TR or TS molecules. SCs were cross-docked into the 6A93 [16] crystal of 5-HT2AR, previously immersed in the dipalmitoylphosphatidylcholine (DPPC) membrane system (5-HT2AR-DPPC, https://opm.phar.umich.edu/, entry: 4282, accessed on 2 December 2022), and co-aligned with the 5-HT2AR and 5-HT2BR crystals used for SBAAs (Table 3 and Table 4) to elaborate their binding modes, simulating a cell-like environment. TR compounds thus adopted a binding mode similar to that of LSD and were found in the vicinity of extracellular loop 2 (EL2) and the extracellular space, which forms a narrow cleft lined mainly by hydrophobic side chains of residues in transmembrane (TM) helices TM3, TM5, TM6, and TM7, as well as near the laterally extended cavity surrounded by hydrophobic residues of TM3, TM4, TM5, and ECL2, to connect the binding site to the plasma membrane near the lower hydrophobic cleft [16]. None of the compounds occupied the orthosteric region in front of the hydrophobic cleft [16]. Upon deeper analysis, SCs were observed to establish only electrostatic interactions with TM3 Asp155, instead of a hydrogen bond (HB) established by LSD, which has been identified as a crucial interaction for the drug’s psychotic behavior at the 5-HT2AR level [76]. To quantitatively support the observed SB overlap, 3-D QSAR models were built using the TR docked conformations proposed by Vina. The 3-D QSAR models were generated using Open3DQSAR software [77] with multiple probes (Table 7), and the corresponding CoMFA-like maps were analyzed. The final models were obtained with the fractional factorial design (FFD) feature selection applied to the model preoptimized with the variable pretreatment optimization (VPO) protocols by varying the lattice spacing and lattice extension [78].
Table 7.
Statistical results of the best Open3DQSAR models derived after VPO and FFD optimization.
| Probe | Field | GS a | PC b | CO c | Z d | SD e | r 2 f | q2LOO g | q2LSO h | r2YS i | q2YS LOO j | q2YS LSO k |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CR | STE | 1 | 5 | 4 | 0.05 | 0.01 | 0.932 | 0.552 | 0.476 | 0.864 | −0.217 | −0.264 |
| ELE | 1 | 5 | 4 | 0.05 | 0.01 | 0.956 | 0.476 | 0.411 | 0.812 | −0.231 | −0.284 | |
| BOTH | 1 | 5 | 4 | 0.05 | 0.01 | 0.961 | 0.512 | 0.417 | 0.856 | −0.231 | −0.246 | |
| CB | STE | 1 | 4 | 4 | 0.01 | 0.01 | 0.943 | 0.473 | 0.442 | 0.836 | −0.251 | −0.256 |
| ELE | 1 | 4 | 4 | 0.01 | 0.01 | 0.967 | 0.414 | 0.412 | 0.822 | −0.236 | −0.238 | |
| BOTH | 1 | 4 | 4 | 0.01 | 0.01 | 0.934 | 0.486 | 0.446 | 0.821 | −0.245 | −0.217 | |
| NC=O | STE | 1 | 5 | 3 | 0.02 | 0.04 | 0.951 | 0.536 | 0.498 | 0.861 | −0.236 | −0.284 |
| ELE | 1 | 5 | 3 | 0.02 | 0.04 | 0.931 | 0.254 | 0.436 | 0.856 | −0.258 | −0.264 | |
| BOTH | 1 | 5 | 3 | 0.02 | 0.04 | 0.962 | 0.521 | 0.432 | 0.754 | −0.312 | −0.157 | |
| NR | STE | 1 | 4 | 5 | 0.03 | 0.01 | 0.943 | 0.484 | 0.461 | 0.814 | −0.264 | −0.264 |
| ELE | 1 | 4 | 5 | 0.03 | 0.01 | 0.945 | 0.512 | 0.421 | 0.806 | −0.254 | −0.235 | |
| BOTH | 1 | 4 | 5 | 0.03 | 0.01 | 0.952 | 0.504 | 0.412 | 0.825 | −0.147 | −0.135 | |
| O=C | STE | 1 | 3 | 5 | 0.05 | 0.02 | 0.912 | 0.501 | 0.495 | 0.841 | −0.264 | −0.244 |
| ELE | 1 | 3 | 5 | 0.05 | 0.02 | 0.925 | 0.482 | 0.472 | 0.822 | −0.254 | −0.251 | |
| BOTH | 1 | 3 | 5 | 0.05 | 0.02 | 0.976 | 0.498 | 0.426 | 0.734 | −0.137 | −0.146 | |
| OH2 | STE | 1 | 5 | 5 | 0.05 | 0.02 | 0.973 | 0.684 | 0.594 | 0.845 | −0.264 | −0.241 |
| ELE | 1 | 5 | 5 | 0.05 | 0.02 | 0.981 | 0.562 | 0.534 | 0.824 | −0.247 | −0.224 | |
| BOTH | 1 | 5 | 5 | 0.05 | 0.02 | 0.971 | 0.671 | 0.316 | 0.842 | −0.145 | −0.321 |
a Grid spacing (in Å); b Optimal number of principal components/latent variables; c CutOff values ± interval in kcal/mol; d Zeroing values in kcal/mol; e Standard deviation; f Conventional square-correlation coefficient; g Cross-validation correlation coefficient using the leave-one-out method; h Cross-validation correlation coefficient using the leave-some-out (LSO) method with 5 random groups; i Average square correlation coefficient obtained after Y-scrambling process. j Average cross-validation correlation coefficient using the leave-one-out (LOO) method obtained after Y-scrambling process. k Average cross-validation correlation coefficient using the leave-some-out (LSO) method with five random groups obtained after Y-scrambling process.
The best models were obtained with the OH2 probe, with associated q2 values of 0.684, 0.562, and 0.671 with steric (STE), electrostatic (ELE), and both fields (BOTH or STE + ELE), respectively (Table 7, Figure 4). Although the OH2 probe STE model was statistically superior to the model derived with either field (BOTH), for the sake of completeness, the final model interpretation was performed using the OH2 probe-based PLS-coefficients (OH2PLS-coefficients maps) obtained with the BOTH field model. Thus, in a CoMFA-like fashion, the positive STE OH2PLS-coefficients (green isocontours) cover the regions of the molecules where the bulky substituents would increase the activity, while the negative STE OH2PLS-coefficients (shown as yellow isopleths) indicate the regions where the bulky substituents decrease the activity. Positive ELE OH2PLS-coefficients (red polyhedra) indicate regions where positively charged functions and hydrogen bond donors (HBDs) are positively correlated with activity, while negative ELE OH2PLS-coefficients (blue maps) indicate regions where negatively charged functions and hydrogen bond acceptors (HBAs) are positively correlated with potency. Despite the intrinsic limitations of the generated OH2-based STE, ELE, and BOTH models (limited TR chemical diversity), the low error of the cross-validated absolute error of prediction for either TR or TS and TSCRY activities (Tables S4–S6, Supplementary Materials) enabled the model as a tool to predict the activities of yet-untested SCs analogs or similar compounds (see further discussion).
Figure 4.

Experimental versus recalculated (blue circles) and predicted (red circles) pKis for TR using OH2 probe BOTH (STE + ELE) LOO model at 5 PCs (a); OH2 probe BOTH (STE + ELE) LSO model at 5 PCs (b).
2.4.1. The Cathinones’ Benzene Ring and Its Substituents’ Contribution
The benzene ring (Ph) of the SCs can be considered as a first pharmacophoric feature. Thus, the unsubstituted Ph (found in AGs 1, Figure 5a,b; 4, Figure 6a,b; 23, Figure S1a,b, and 24, Figure S2a,b, as well as within an IA 9, Figure 6c,d; Supplementary Materials, Table S2) was placed at the bottom of the hydrophobic cleft. Within the listed SCs, the Ph’s ortho-positions proximal to the β-carbonyl group (β-CO) orientation (see further discussion), according to the positive STE OH2PLS-coefficients maps, formed favorable steric interactions with Ser159, Thr160, and Ser242 side chains, implying that the additional bulkiness would be preferable for the agonistic pharmacology and likely the hallucinogenic effects. In contrast, the yellow STE OH2PLS-coefficients maps emphasized the unfavorable T-shaped (i.e., edge-to-face) van der Waals interactions of the opposite-side o-positions with the benzyl part of Trp336′s indole ring, as well as with Phe340, toward which bulkiness should be reduced, perhaps endowing lower psychostimulation.
Figure 5.

The structure-based alignment within 5-HT2AR-DPPC and OH2 probe-based PLS-coefficients (positive steric coefficients presented in green, negative steric coefficients depicted in yellow, positive electrostatic coefficients portrayed in red, negative electrostatic coefficients displayed in blue) for 1 (a,b); 2 (c,d); 17 (e,f). For the clarity of presentation, DPPC was omitted.
Figure 6.

The structure-based alignment within 5-HT2AR-DPPC and OH2 probe-based PLS-coefficients (positive steric coefficients presented in green, negative steric coefficients depicted in yellow, positive electrostatic coefficients portrayed in red, negative electrostatic coefficients displayed in blue) for 4 (a,b), 9 (c,d), and 18 (e,f). For the clarity of presentation, DPPC was omitted.
On the other hand, the m-positions on the β-CO side interacted with Ile163, where the negative steric 3-D QSAR maps indicated repulsive interactions with the sec-methyl group of Ile163, while the positive STE OH2PLS-coefficients maps indicated the favorable attraction with the pentanoic acid-based side chain of the residue, as well as with the indole ring of Trp164. The m-carbon atoms were observed in favorable T-shaped steric interactions with the indole ring nitrogen of Trp336 (the green STE OH2PLS-coefficients maps as evidence).
Finally, the p-positions could mainly establish van der Waals contacts with Phe332, Val333, and Phe340 in a T-shaped manner, as well as with Phe243 via parallel displaced interactions, described with either positive or negative steric 3-D QSAR maps. The slightly larger extent of the negative steric 3-D QSAR maps can probably be associated with a repulsive character.
In addition, upon the p-substitution of Ph with a halogen moiety (leading to the agonism of 2, Figure 5c,d; full antagonism of 5, Figure S2c,d, and inverse agonism of 11, Figure S1c,d, Supplementary Materials), the p-halogen likely established induced dipole interactions with Phe332 and the surrounding residues, according to the negative ELE OH2PLS-coefficients maps. The slight decrease in electronegativity (and increase in bulkiness) upon p-Br incorporation, as in 11 or 5, resulted in reduced inhibition constants and likely inverse agonism/full antagonism. Moreover, concerning 2, both 11 and 5 were slightly rotated towards Trp336, overlapping the m-position of the unsubstituted benzene ring. Therefore, according to the positive ELE OH2PLS-coefficients fields, as with HBA, the p-Br was not well tolerated by the indole moiety of Trp336, which probably prefers an HBD at the m-position while interfering with SC; according to the alignment of the positive STE OH2PLS-coefficients contours, certain HBD could be of increased steric hindrance.
The p- to m-halogen position shift, i.e., p-Br to m-Br, as seen in 12 (Figure S3a,b, Supplementary Materials) or 6 (Figure S3e,f, Supplementary Materials), in which the m-Br pointed toward Phe332 and Trp336, likely contributed to the reduced binding potency, full antagonism, and the exertion of antidepressive effects. Nevertheless, the model also indicated that a double m-halogen substitution could contribute to an additional decrease in the binding potency of SC.
A decrease in potency is observed when the halogen position is changed from para to ortho, as seen in FAs and non-psychostimulants/antidepressives 7 (Figure S4a,b, Supplementary Materials) and similarly in 8 (Figure S4e,f, Supplementary Materials). The o-F moiety of 7 established unfavorable steric interactions (negative STE OH2PLS-coefficients contours) with the Trp336 side-chain benzene. At the same time, the positive ELE OH2PLS-coefficients isocontours indicated that an o-HDB within the structure of SC is preferred by Trp336 instead of o-F as HBA. The 8′s o-OCF3 as a bulkier and more electronegative scaffold led to an unwanted steric clash (negative STE OH2PLS-coefficients contours as evidence) with Ile163 and Trp164, although the positive ELE OH2PLS-coefficients/negative ELE OH2PLS-coefficients isocontours alignment implied that more of the HBD character is needed for Trp164 engagement.
Replacement of the p-halogen with a hydrophobic moiety such as the methyl of an AG 3 (Figure S5a,b, Supplementary Materials), as well as IAs/FAs 10 (Figure S5e,f, Supplementary Materials) and 15 (Figure S6a,b, Supplementary Materials), resulted in a ~15° rotation of the central aromatic ring, leading to the overlap of the p-CH3 function with the p-Br or m-Br, respectively. The increased bulkiness at the bottom of the hydrophobic cleft reduced the potency, as verified by the incorporation of 1,3-dioxolane at the m- and p-carbons of the phenyl ring, respectively (i.e., benzo[d][1,3]dioxole formation), as in derivatives 14 (Figure S4c,d, Supplementary Materials) and 16–22 (Figure 5, Figure 6 and Figures S3–S8, Supplementary Materials), or fused benzene as the naphthyl moiety in 13 (Figure S6e,f, Supplementary Materials), contributing to inverse agonism/full antagonism (14 and 15).
2.4.2. The Cathinones’ β-Carbonyl Group Contribution
The orientation of the unsubstituted or substituted Ph within the structures of SCs strongly influenced the behavior of β-CO and vice versa. This group can be considered as a second pharmacophoric feature, as in 1 (Figure 5a,b) and 4 (Figure 6a,b). The β-CO formed a strong HB with the side-chain hydroxyl portion of Thr160 (β-CO----HO-Thr160, O-O distance of 2.636 and 2.464 Å, respectively), characterized by negative ELE OH2PLS-coefficients isocontours, thus contributing to the agonistic pharmacology and psychedelic effects. With the incorporation of p-F (2, Figure 5c,d), the β-CO has shifted away from preventing the optimal β-CO----HO-Thr160 O-O distance toward attractive electrostatic interactions (p-F likely compensated for the lack of HB to maintain the high potency of 2 as an AG and psychostimulant). However, with the introduction of the p-Br, as in 11 and 5 (Figure S1c,d and Figure S2c,d, respectively, Supplementary Materials), the β-CO----HO-Thr160 hydrogen bonding distance was restored due to the ~15° in-plane rotation of CSs concerning 2 (O-O distance of 3.396 and 3.238 Å for 11 and 5, respectively), proving the value for IAs/FAs as well. The p-substitution with a methyl group (3, 10, and 15, Figure S5a,b and Figure S5e,f, and Figure S6a,b, respectively, Supplementary Materials), together with both p- and m-substitution with 1,3-dioxolane or benzene (13, 14, 16–22, and Figure 5, Figure 6 and Figures S2–S6, Supplementary Materials), also forced the β-CO----HO-Thr160 HB formation (O-O distance of 2. 728, 3.360, 2.835, 3.321, 3.229, 2.989, 2.892, 3.119, and 3.217 Å, respectively).
However, the p-Br to m-Br switch, as in 12 (Figure S3a,b, Supplementary Materials) and 6 (Figure S3e,f, Supplementary Materials), or the p-F to o-F switch, as in 7 (Figure S4a,b, Supplementary Materials), all inverse agonists/full antagonists, resulted in the alternative β-CO----HO-Ser159 HB formation (O-O distances of 2.585 Å, 3.245 Å, and 3.396 Å, respectively), for which relatively high potency was retained. The β-CO----HO-Ser159 HB (O-O distance = 2.971 Å) was retained even when the bulkier 8′s o-fluoromethoxy moiety (Figure S6e,f, Supplementary Materials) formed an additional HB with Thr160 (O-O distance = 2.288 Å), also covered by negative ELE OH2PLS-coefficients isocontours, or when Ph was unsubstituted (9, Figure 6c,d, dHB = 2.971 Å). The absence of β-CO (14 and 16–22, and 23 and 24, Figure 5 and Figure 6, and S1–S6, Supplementary Materials) resulted in the lowest potencies, which were partially compensated by the benzo[d][1,3]dioxole (14 and 16–22) or Ph (23–24) alone.
2.4.3. The Cathinones’ Methylene Group and Methylene Group’ Substituents Contribution
Here, we focused on the importance of alkyl-substituted methylene fragments (i.e., -CH2-Rs, either methyl- [-CH2-CH3], ethyl- [-CH2-CH2-CH3 ], or propyl- [-CH2-CH2-CH2-CH3] substituted) located between either substituted/non-substituted Ph-β-CO or benzyl (Bn, in the absence of β-CO) moieties and substituted/non-substituted terminal amines (-NH2) (Table 1). Therefore, the orientations of the -CH2-CH3 fragments were almost conserved and the correct SB correlation to the surrounding groups was difficult to establish. Thus, between either Ph-β-CO----HO-Thr160 HB and -NH2 or -NHCH3 (1, Figure 4a and Figure 5b, and 4, Figure 6a,b, respectively) or Bn and -NH2 or -NHCH3 (23, Figure S1a,b: 24, Figure S2a,b, Supplementary Materials), the -CH2-CH3 was involved in the attractive steric interactions with Val156 (see the STE OH2PLS-coefficients maps) that enhanced the agonistic and hallucinogen effects of compounds. Nevertheless, the -CH2-CH3 contribution was not critical for the SCs, as it was the loss of β-CO that directly affected the potency reduction from the highest (1) to the lowest (23 and 24). The conversion of -NH2 to pyrrolidine (9, Figure 6c,d) directed the substituent methyl group toward favorable interactions with Phe339 and Phe340 (as evidenced by positive STE OH2PLS-coefficients maps), but also contributed to inverse agonist/full antagonist pharmacology. Interestingly, a similar orientation of the -CH2-CH3 moiety (accompanied by a further decrease in potency) was observed with m-Br-Ph-β-CO----HO-Ser159 HB and pyrrolidine (12, Figure S3a,b, Supplementary Materials), as well as with o-OCF3-Ph-β-CO----HO-Ser159 HB and -NHCH3 (8, Figure S4e,f, Supplementary Materials). In addition, both -CH2-CH3 (22, Figure S5c,d; 20, Figure S2e,f, Supplementary Materials) and -CH2-CH2-CH3 (19, Figure S1e,f, Supplementary Materials) indicated that Phe339 and Phe340 were bound by 1,3-dioxolane-Ph-β-CO----Thr160 HB and either -NHCH3 or -NH-Et.
The potency remained relatively high (above the 5 pKi units), with the -CH2-CH3 surrounded by p-X-Ph-β-CO----HO-Thr160 HB and either -NHCH3 (2, X=F, Figure 5c,d; 5, X=Br, Figure S4c,d, Supplementary Materials) or pyrrolidine (11, X=Br, Figure S1c,d, Supplementary Materials), and by p-Me-Ph-β-CO----HO-Thr160 HB and pyrrolidine (3, Figure S3a,b; 10, Figure S5a,b Supplementary Materials), which further directed the targeted functional group towards the T-shaped steric interaction with the indole ring of Trp336, where a larger portion of negative STE OH2PLS-coefficients contours relative to positive ones indicated that further bulkiness increase towards Trp336 would lead to inverse agonism/partial agonism and would thus be detrimental for the psychedelic influence of SC, whereas positive/negative ELE OH2PLS-coefficients maps indicated that methyl group replacement by HBD/HBA would be beneficial for agonistic behavior and psychedelic effects.
In contrast, either m-Br-Ph-β-CO----HO-Ser159 HB (6, Figure S3e,f, Supplementary Materials) or o-F-Ph-β-CO (7, Figure S4a,b, Supplementary Materials) next to -NHCH3 forced the -CH2-CH3 to have unproductive (note negative STE OH2PLS-coefficients contours) van der Waals interactions with Val156 and Ser242, which was reflected in inverse agonism/partial agonism pharmacology. The similar interactions of -CH2-CH3 were observed to be surrounded by 1,3-dioxolane-Ph-β-CO----Thr160 HB and pyrrolidine (16, Figure S6c,d, Supplementary Materials), as well as by 1,3-dioxolane-Bn-and-NHCH3 (18, Figure 6e,f; 21, Figure S3c,d, Supplementary Materials), but not for -CH2-CH2-CH3 (17, Figure 5e,f), which remained bound to Phe340. However, the incorporation of the propyl group was accompanied by either naphthalene-β-CO----HO-Thr160 HB (13, Figure S6e,f, Supplementary Materials), 1,3-dioxolane-Ph-β-CO····HO-Thr160 HB (14, Figure S4c,d, Supplementary Materials), or p-Me-Ph-β-CO····HO-Thr160 HB (15, Figure S4a,b, Supplementary Materials), and pyrrolidine turned the -CH2-CH2-CH2-CH3 towards Trp151 and Val156, defining the boundary for Phe340 steric clash tolerance.
2.4.4. The Cathinones’ Amine Nitrogen Contribution
The affinities of SCs to 5-HT2AR and their psychedelic behavior can also be attributed to their interactions with Asp155 via the terminal primary, secondary, or tertiary nitrogen. Thus, the primary amine, as in AGs 1 (Figure 5a,b), 3 (Figure S5a,b, Supplementary Materials) and 23 (Figure S1a,b, Supplementary Materials), provided a salt bridge with Asp155 instead of HB (d = 4.772 and 5.853 Å, respectively, and the strongest interaction could have contributed to the highest potency of 1), despite being characterized with positive ELE OH2PLS-coefficients maps. The absence of hydrogen bond between the -NH2 and Asp155 is likely the reason for lower psychedelic activities of SCs compared to LSD [25,26,31,32]. Nevertheless, distinct interactions were characterized by negative STE OH2PLS-coefficients isocontours, indicating that the increase in bulkiness at the -NH2 level would reduce the potency of SCs and convert AGs into IAs/FAs, i.e., psychostimulants into non-psychostimulants.
The above hypothesis was (in part) supported by the analysis of the secondary, i.e., either N-methyl substituted, as in 2, 4–8, 17–19, 22, and 24 (Figure 5, Figure 6 and Figures S1–S6, Supplementary Materials), or N-ethyl substituted, as in 20 and 21 (Figure S4e,f and Figure S5c,d, respectively, Supplementary Materials) amines. The listed SCs also formed a salt bridge with Asp155 via the -N-H part (highlighted by positive ELE OH2PLS-coefficients isocontours), strongly conditioned by the alignment of the N-methyl/N-ethyl groups with one of the two subregions bounded by Leu228, Val336, Phe339, Trp336, and Tyr370 (as for 5–7 and 20), or Val156 alone (as for 4, 8, 17–19, 21, 22 and 24), described by simultaneous attractive/repulsive 3-D QSAR maps.
The effect of bulky substituents on the -NH2 was further analyzed, starting from the tertiary nitrogen of SCs found in pyrrolidine, as in 9–16 (all IAs/FAs except 16, Figure 6 and Figure S1–S6, Supplementary Materials), which adopted three different conformations. Favorably attracted by Val156 (as in 9, 10, and 12; the positive STE OH2PLS-coefficients contours as evidence), the tertiary nitrogen provided good potency through moderate electrostatic interactions with Asp155 (distance over 6 Å, positive ELE OH2PLS-coefficients maps as validation). On the other hand, the interactions with Phe339 (observed for 11 and 16) moved the heterocycle away from Asp155 into a negative STE OH2PLS-coefficients cloud, implying a need to maximally reduce bulkiness. Displacement of the heterocycle from Val156 towards Ser242 and Phe340, as in 13–15, contributed more negatively than positively (a larger proportion of negative STE OH2PLS-coefficients maps than positive ones observed) to the potency, with a lack of interactions with Asp155.
2.4.5. Generated 3-D QSAR Models’ Predictive Abilities
The Vina-predicted conformations and OH2 probe-derived BOTH 3-D QSAR models were evaluated for predictivity on TS compounds (Table 3 and Table S3, Supplementary Materials; Figure 7, Figure 8 and Figures S9–S15, Supplementary Materials).
Figure 7.

Experimental versus recalculated (green circles) and predicted (purple circles) pKis for TS using OH2 probe BOTH (STE + ELE) LOO model at 5 PCs (a); OH2 probe BOTH (STE + ELE) LSO model at 5 PCs (b).
Figure 8.

The structure-based alignment within 5-HT2AR-DPPC and OH2 probe-based PLS-coefficients (positive steric coefficients presented in green, negative steric coefficients depicted in yellow, positive electrostatic coefficients portrayed in red, negative electrostatic coefficients displayed in blue) for 25 (a,b); 26 (c,d); 27 (e,f). For the clarity of presentation, DPPC was omitted.
The binding modes for all compounds except 25 (co-crystalized within the 7VOE protein [28]) were modeled for SCs and were associated with experimentally available pKi values. The pKi values of TSs were predicted with an average absolute error of prediction (AAEPs) of 0.61 and 1.29 with the LOO and LSO CVs optimized models, respectively (Table S3 Supplementary Materials), to which the associated predictive q2 (q2pred) values were calculated to be 0.43 and 0.307, respectively, confirming the good predictive capabilities of the model [79]. Similar to the SCs, ten TS compounds, namely 26–28, 30, 32–34, 36, 39, and 41 (Figure 8 and Figures S7–S11d, respectively, Supplementary Materials), filled only the hydrophobic pocket of the 5-HT2AR’s active site and were predicted in potency with the absolute error of prediction (AEP) below 1. Among them, the best predicted one was 33, with errors of prediction (EPs) after LOO and LSO CVs of 0.03 and 0.16 pKi units. The AEP was only 0.095 pKi units, while the worst was 32, associated with LOO and LSO CVs AEPs of 0.37 and 0.97 pKi units, respectively. For the bipartite 5-HT2AR ligands, the AEP was greater than 1. The remaining compounds that were SB aligned throughout both the orthosteric region and the hydrophobic pocket of the receptor’s bipartisan active site. Within the subset, only compound 37 (Figure S10a,b, Supplementary Materials) potency was predicted with tolerable accuracy (LOO and LSO CVs AEPs of 1.2 and 0.94 pKi units, respectively), whereas the worst predicted compound was 29 (Figure S7c,d, Supplementary Materials) (LOO and LSO CVs AEPs of 1.83 and 2.24 pKi units, respectively).
Previously published CoMFA/CoMSIA LB 3-D QSAR models based on dibenzazecines [80], 3-aminoethyl-1-tetralones, piperazines, benzothiazepines, pyrrolobenzazepines [81], and arylpiperazines [82], as well as the LB GRID/GOLPE models of (aminoalkyl)benzo and heterocycloalkanones [83], were also more accurate in predicting the potencies of compounds occupying the hydrophobic pocket. On the other hand, within the molecular docking-based SB GRID/GOLPE 3-D QSAR models generated on either butyrophenones [84], or lozapine, ziprasidone, and ChEMBL-listed analogues [84], the quality of the alignment within either the orthosteric area, the hydrophobic pocket, or both, was, as here, evaluated using the highest q2 [63,85]. However, the universal SB 3-D QSAR model(s) defining the agonism/antagonism on the entire 5-HT2AR active site remains to be generated, perhaps after increasing the number of Ki-associated co-crystallized 5-HT2AR compounds to a minimum of 15 (and thus updating Table 1), using either Open3DQSAR [77], 3-D_QSAutogrid/R [78], or Py_CoMFA [31,86].
2.5. Molecular Determinants for SCs
The pharmacodynamic profile obtained from the 3-D QSAR map analysis indicated some pharmacophoric features that may positively or negatively affect the potency of SCs as 5-HT2AR ligands. Therefore, comprehensive 3-D structure–activity relationship (SAR) rules were derived for the SCs structure–activity relationship model [67] and are shown in Figure 1 as a template (Figure 9). This led to the derivation of a unique SAR as a tool that could be used to drive the virtual screening campaign of new cathinones and similar compounds as 5-HT2AR ligands that could, as AGs, exert psychostimulant properties.
Figure 9.

The SAR model for CSs as 5-HT2AR ligands and agonists is seen through 1.
Thus, the central benzene ring (Ph) of SCs may be either unsubstituted or o-substituted proximal to the β-carbonyl group (β-CO) with bulkier portions to sterically engage Ser159, Thr160, and Ser242, while the o-bulkiness increase towards Trp336 and Phe340 would not be tolerated and HDB portions should be forced. As for the m-position on the β-CO side towards Ile163 and Trp164, Ph should either remain unsubstituted or be heavily substituted by a highly electronegative electron withdrawing/HBA group, while Trp336 on the opposite side would tolerate bulkier moieties with an HBD character. The p-position of Ph before Phe243, Phe332, Val333, and Phe340 could accommodate moieties, providing minimal van der Waals contacts with or substituted by a highly electronegative electron withdrawing/HBA group.
The β-CO pharmacophoric feature is responsible for the formation of HBs with Thr160, mitigated by the presence of either p-electronegative groups/HBA or bulky substituents (or functional groups containing each of these features) at the same position. Nevertheless, the most electronegative functional groups incorporated at the p-position of Ph may prevent the β-CO----HO-Thr160 HB formation. On the other hand, the incorporation of m- or o-electronegative groups/HBA (or corresponding hybrid functional groups) leads to the formation of β-CO----HO-Ser159 HB. Since none of the SCs formed the HB with Asp155 (an interaction that leads to the hallucinogenic effect of LSD [31]), the β-CO or similar β-HBA bioisosteric scaffold remains preferred for SCs to ensure the H-bonding described above, since the absence of β-CO resulted in the lowest potencies.
The -CH2- fragment substituted with a methyl group, located between the β-CO (or Bn) and -NH2 (or -NHCH3), seemed to be the best choice to establish attractive steric interactions with Val156. The substitution of -NH2 with bulkier acyclic and cyclic aliphatic moieties could also direct the -CH2-CH3 moiety to favorable van der Waals interactions with Phe339 and Phe340. The m-HBA/o-HBA-Ph-β-CO····HO-Ser159 HB architecture also supported the alignment of -CH2-CH3 with Phe339 and Phe340 but could also direct the moiety to unfavorable steric hindrance with Ser242. The -CH2-CH3 moiety should not be properly lengthened or branched, as the further increase of van der Waals interactions towards the listed residues could lead to decreased potency. Either the m-HBA-Ph-β-CO----HO-Thr160 HB or p-Me-Ph-β-CO····HO-Thr160 construct could force the -CH2-CH3 toward more unfavorable steric interactions with Trp336, a residue that would be more tolerant of the -CH2-HBD/HBA fraction.
The -NH2 of the SCs should remain unsubstituted to maintain a salt bridge interaction with Asp155 and ensure high potency. Substitution with more voluminous moieties such as N-methyl/N-ethyl could be tolerated but could also result in penalizing van der Waals interactions with Leu228, Val336, Phe339, Trp336, and Tyr370, although not interfering with the -N-H moiety to form the required salt bridge. Finally, the tertiary nitrogen could provide attractive steric interactions with Val156 and interfere with Asp155, again resulting in lower potency.
2.6. External Validation of 3-D QSAR Models on Experimentally Determined 5-HT2AR Ligands
Finally, the suitability of the obtained 3D QSAR model to be used as a tool for virtual screening and potency prediction was evaluated on TSCRY (Table 1, Figure 10). Since the bioactive conformations of the TSCRY compounds were experimentally resolved, a deeper discussion in terms of their potency prediction was included concerning the TS compounds. Thus, the alignments of TSCRY compounds were not as conserved as those of TS compounds, where most of the main cores of TS compounds were aligned with SCs, and for some of the compounds, the remaining parts occupied the orthosteric region. Therefore, the AAEPs of TSCRYs’ pKi values were significantly higher for both LOO and LSO CVs optimized models, 1.12 and 1.49 (Table S5, Supplementary Materials), associated with q2pred of 0.33 and 0.28, respectively, confirming the above-described 3-D QSAR model as a useful tool [79] to discover potential new chemical entities as 5-HT2AR ligands to be studied as broad psychedelic agents.
Figure 10.

Experimental versus recalculated (green circles) and predicted (orange circles) pKis for TSCRY using OH2 probe BOTH (STE + ELE) LOO model at 5 PCs (a); OH2 probe BOTH (STE + ELE) LSO model at 5 PCs (b).
Thus, the best-predicted activities were for the 6A94 (Figure 11a,b), 6WH4 (Figure 11c,d), and 6WHA (Figure 11e,f) ligands that were found deeply buried in the hydrophobic pocket, with AEPs of only 0.72, 0.95, and 1.05 pKi units after the LOO CV, and 0.86, 1.16, and 1.69 pKi units after the LSO CV, respectively. Moreover, given that the 6WH4 crystal is one of the most potent 5-HT2AR ligands known to date (Table 1), predicting its potency with acceptable error was very important for the present 3-D QSAR model, proving the model as capable of recognizing the highly potent ligands bound in a hydrophobic pocket. The main cores of 6WGT (Figure S14a,b, Supplementary Materials), 7WC6 (Figure S14c,d, Supplementary Materials), and 7WC7 (Figure S14e,f, Supplementary Materials) were positioned at the top of the hydrophobic pocket, orthogonal to the SCs, and were within the scope of the 3-D QSAR PLS-coefficients, this time resulting in the LOO CV AEPs of 1.11 (for LSD) and 1.17 (for lisuride) pKi units and LSO CV AEPs of 1.51 (for LSD) and 1.46 (for lisuride) pKi units, respectively.
Figure 11.

The structure-based alignment within 5-HT2AR-DPPC and OH2 probe-based PLS-coefficients (positive steric coefficients presented in green, negative steric coefficients depicted in yellow, positive electrostatic coefficients portrayed in red, negative electrostatic coefficients displayed in blue) for 6A94 (a,b); 6WH4 (c,d); 6WHA (e,f). For the clarity of presentation, DPPC was omitted.
On the other hand, 6A93 (Figure S15a,b, Supplementary Materials), 7VOD (Figure S15c,d, Supplementary Materials), and 7VOE (Figure S15e,f, Supplementary Materials) crystals occupied both the orthosteric and hydrophobic pockets. The main core of 6A93 was superimposed by SCs and thus was the only part of the molecule covered by 3-D QSAR PLS-coefficients, as the rest filled the orthosteric cavity, for which the AEPs according to LOO and LSO CVs were 1.80 and 1.47 pKi units, respectively. In contrast, 7VOD and 7VOE crystals were positioned above the SCs and only the central molecular regions were included in the 3-D QSAR PLS-coefficients, resulting in AEPs of 0.76 and 1.65 pKi units after LOO cross-validation and 1.88 and 1.79 pKi units after LOO cross-validation.
3. Materials and Methods
3.1. Crystal Structures Preparation
All selected 5-HT2AR and 5-HT2BR complexes (Table 1) available from the Protein Data Bank (https://www.rcsb.org/, accessed on 1 December 2022) were loaded into the UCSF Chimera v1.10.1 software [87] and were visually inspected. Complexes were superimposed using 5TVN as a template (the best-resolved complex with a resolution of 1.6 Å) using the MatchMaker module and were then separated into chains using the command line implementation of the Chimera split command. Compared to other chains, chains A were complete with respect to the presence of antagonists and were retained. The antagonists were extracted from each chain A complex, completed by adding hydrogens appropriate for pH 7.4, and the AMBER parameters were calculated by Antechamber using a semi-empirical QM method. The protein parts of the stored monomers were improved by adding hydrogen atoms using the embedded leap module of the Amber 12 suite [88], after which the correct hydrogen atoms, appropriate for pH 7.4, were assigned to each amino acid residue. After preparation, the proteins were fused with the appropriate ligands and the complexes were energy minimized as follows. They were solvated by the Leap module with water molecules (TIP3P model, SOLVATEOCT Chimera command) in a box extending 10 Å in all directions, neutralized with either Na+ or Cl− ions, and refined by a single point minimization using the Sander module of the Amber suite with a maximum of 1000 steps of steepest descent energy minimization and a maximum of 4000 steps of conjugate gradient energy minimization, with an unbound cutoff of 5 Å. Minimized complexes were re-aligned (5TVN as a template), after which all ligands were extracted to compose the SB-aligned TR, ready to be used for the subsequent 3-D QSAR model building.
3.2. Alignment Assessment Rules
Structure-based alignment. Regarding SB alignment, a number of docking programs with all available scoring functions, free or open source for academic use, were evaluated to select the best one to reproduce the binding mode of 5-HT2AR and 5-HT2BR ligands. Namely, the programs were AutoDock [68], AutoDock Vina (hereafter referred to as Vina) [69], SMINA [71], DOCK [70], and PLANTS [72]. The entire SB procedure was evaluated using four levels of difficulty:
Experimental Conformation Re-Docking (ECRD): a procedure in which the experimental conformations (EC) are flexibly docked back into the corresponding protein, evaluating the program for its ability to reproduce the observed bound conformations.
Randomized Conformation Re-Docking (RCRD): a similar assessment to ECRD with the difference that the active site of protein is virtually occupied by conformations initially obtained from computational random optimization of corresponding co-crystallized molecules coordinates and positions. Thus, ligands were initially displaced from the active site and their experimental coordinates were changed by means of assigning new coordinates values: X = 0.000, Y = 0.000, Z = 0.000. Following that, allocated conformations were energy-minimized. Here the programs are evaluated for their ability to find the experimental pose, starting from the randomized minimized conformation.
Experimental Conformation Cross-Docking (ECCD): comparable to ECRD, but the molecular docking was performed on all the TR proteins except the corresponding natives. Here the programs are evaluated to find the ligand binding mode in the active site such as the native one by means of amino acid configuration but are different in terms of amino acids induced-fit conformations, mimicking discrete protein flexibility at the same time.
Randomized Conformation Cross-Docking (RCCD): same as the ECCD but using RCs as starting docking conformations. This is the highest level of difficulty since the program is demanded to dock any given molecule into an ensemble of protein conformations not containing the native one. The outcome is considered as the most important ability of the docking program, as the most accurate scoring function in the RCCD experiment is subsequently applied to any TS molecules whose experimental binding mode is unknown. The related docking accuracy (DA) is a direct function of the program’s probability to find a correct binding mode for an active molecule.
The alignment fitness was quantified through the evaluation of RMSD values and the subsequent docking accuracy (DA) values. As previously reported [31], DA can be used to test how the docking or alignment algorithms, respectively, are capable of predicting a ligand pose as close as possible to the experimentally observed and can be calculated by the following equation:
| xA = frmsd ≤ a + 0.5 (frmsd ≤ b − frmsd ≤ a) | (1) |
In particular, xA is equal to DA in the case of docking accuracy, whereas frmsd ≤ a and frmsd ≤ b represent the fraction of aligned ligands, showing an RMSD value less than 2 Å or equal to 2 Å (a coefficient) and less than 3 Å or equal to 3Å (b coefficient), respectively. The widely accepted standard is that the correctly docked/aligned conformations are those displaying an RMSD value lower than 2 Å on all heavy atoms from the crystallographic structure of the ligand conformation, as found in the inhibitor–enzyme complex. Structures with RMSD between 2 and 3 Å are considered partially docked/aligned, whereas those with a RMSD higher than 3 were mis-docked and were thus not considered in the DA calculation.
3.2.1. AutoDock Settings
For all ligands, the rigid root and rotatable bonds were defined using AutoDockTools. The docking was performed with AutoDock 4.2 by applying the cuboid docking grid coordinates, provided as follows: the xyz coordinates (in Ångströms) for the computation were Xmin/Xmax = −1.805/30.447, Ymin/Ymax = −12.113/9.482, Zmin/Zmax = 48.159/60.462. The coordinate setup was performed in a manner to embrace the minimized inhibitor, spanning 10 Å in all three dimensions. The Lamarckian Genetic Algorithm was used to generate orientations or conformations of ligands within the binding site. The procedure of the global optimization started with a sample of 200 randomly positioned individuals, a maximum of 1.0 × 106 energy evaluations, and a maximum of 27,000 generations. A total of 100 runs were performed with RMS Cluster Tolerance of 0.5 Å.
3.2.2. Vina Settings
The docking simulations were carried out with an energy range of 10 kcal/mol and exhaustiveness of 100 with RMS Cluster Tolerance of 0.5 Å, using the identical grid as for AutoDock4.2. The output comprised 20 different conformations for every receptor considered.
3.2.3. Smina Settings
As Smina is AutoDock Vina fork, an identical setup was used as for Vina for either vina [69], vinardo [75], or ad4 [68] scoring functions.
3.2.4. DOCK Settings
During the docking simulations with the DOCK program, the proteins were considered to be rigid while the inhibitors were regarded as flexible and were subjected to energy minimization. The solvent-accessible surface of each enzyme without hydrogen atoms was calculated using the DMS program [89], using a probe radius of 1.4 Å. The orientation of ligands was described using the SPHGEN and SPHERE_SELECTOR modules. A box around the binding site was constructed with the accessory module SHOWBOX. The steric and electrostatic environment of the pocket was evaluated with the program Grid using a 0.3 Å of grid spacing. Selected spheres were within 8 Å from ligand heavy atoms of the crystal structure, and for computing, the energy grids an 8 Å box margin and 6–9 VDW exponents were used.
3.2.5. PLANTS Settings
The docking site was limited inside a 12 Å radius sphere, centered in the mass center of the crystallized ligand. Docking was performed by default settings using three different scoring functions: chemplp, plp, and plp95.
3.3. Generation of the TR and TS Designed Compounds
TR, TS, and designed compounds were modelled by applying the Chemaxon’s msketch module [90] through molecular mechanic optimization, upon which the hydrogen atoms appropriate to pH 7.4 were assigned. Upon the generation of structures, compounds were uploaded into previously described SB to obtain the bioactive conformations.
3.4. Retrieval of 5-HT2AR-Lipid Bilayer Complex
The crystal structure of 5-hydroxytryptamine receptor 2A in complex with risperidone, deposited at PDB under the code: 6A93, was found modeled in dipalmitoylphosphatidylcholine (DPPC) bilayer system and deposited as entry: 4282 at Orientations of Proteins in Membranes (OPM) database (https://opm.phar.umich.edu/, accessed on 2 December 2022). Upon retrieval, the 5-HT2AR-DPPC complex was subjected to the same preparation protocol with UCSF Chimera, as described in Section 3.1.
3.5. Structure Alignment of LSD, TR, TS, and Designed Compounds within 5-HT2AR
The same relative coordinates were assigned to the 5-HT2AR- DPPC system as for 5-HT2BR, using the UCSF Chimera’s MatchMaker module. Afterwards, either LSD, TR, TS, or designed compounds were docked into 5-HT2AR-DPPC complex using the best performing algorithm/scoring function.
3.6. 3-D QSAR Models Generation
The herein Vina-based TR was submitted to Open3DQSAR procedure to generate partial least squares (PLS)-based 3-D QSAR models using eight probes, namely CR (alkyl carbon), CB (aromatic carbon), NC=O (amide nitrogen), NR (amine nitrogen), O=C (carbonyl oxygen), OH2 (oxygen in water), HNCO (amide hydrogen), and ELE (electrostatic field). The 3D QSAR models were built for each probe using a maximum of 5 principal components. Initially generated molecular interaction fields (MIFs) and PLS 3-D QSAR models were first optimized on 1 Å grid spacing using the standard pretreatment protocol (energy cutoff of ± 5 Kcal/mol, zeroing = 0.01 Kcal/mol, and minimum standard deviation = 0.05) and by storing the corresponding standard (r2) and cross-validated (q2) correlation coefficients. A second stage of optimization was achieved by a Variable Pretreatment Optimization (VPO) procedure using Leave-One-Out (LOO) and Leave-Some-Out (LSO) cross-validation while monitoring q2. Full pretreatment of the data derived from the MIFs calculations was performed by exploring the combinations of cutoff values from −5 to 5 kcal/mol with intervals between the cutoffs equal to 1, zero values from −0.005 to 0.05 kcal/mol with an interval of 0.005, and standard deviation values from −0.01 to 0.1 with an interval of 0.01. The final 3-D QSAR models (Table 7) were obtained by the fractional factorial design (FFD) feature selection procedure. The internal validation (robustness) of the 3-D QSAR models was evaluated by classical cross-validation (CV) techniques (LOO and LSO), while the lack of random correlation was evaluated by CV and Y-scrambling combination (Table 7). The best 3D QSAR models were used to predict the activity of either co-crystalized TR compounds (Table 1) or SB-aligned TS compounds (Table 3).
4. Conclusions
The enclosed manuscript summarizes the efforts to define the pharmacodynamics of SCs available in the literature [21,34] as 5-HT2AR ligands, namely agonists (i.e., psychedelic abusers) and IAs/PAs (i.e., non-psyshostimulants). The bioactive conformations of SCs were obtained using the AutoDock Vina [69] after exhaustive SB alignment evaluation on co-crystallized 5-HT2AR and 5-HT2BR ligands, while the crucial interactions leading to the inhibition constants and possible psychedelic effects for AGs were described by a statistically robust CoMFA-like 3-D QSAR model, as generated by Open3DQSAR [77], starting from the OH2 probe, using both steric and electrostatic fields. The obtained PLS-coefficients maps were summarized in a 3-D SAR model (i.e., molecular determinants), a useful tool for virtual screening campaigns in the search for new SCs with potential psychedelic effects with an etiology in the agonism of 5-HT2AR. The applicability of the OH2 probe 3-D QSAR model as a predictive engine was evaluated on co-crystallized 5-HT2AR ligands, showing satisfactory predictive properties, considering that none of the experimentally resolved 5-HT2AR ligands were structural homologs of SCs.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/molecules28176236/s1, Table S1. Onsets and durations of actions of SCs 5-HT2AR ligands compiling the TR. Table S2. Structure-based alignment assessment of 5-HT2AR ligands. Table S3. Structure-based alignment assessment of 5-HT2BR ligands. Table S4. Best Open3DQSAR Models’ (OH2 model at PC5) Predictive Abilities for TR. TableS5. Best Open3DQSAR Models’ (OH2 model at PC5) Predictive Abilities for TS. TableS6. Best Open3DQSAR Models’ (OH2 model at PC5) Predictive Abilities for TSCRY. Figure S1. The structure-based alignment within 5-HT2AR-DPPC and OH2 probe-based PLS-coefficients (positive steric coefficients presented in green, negative steric coefficients depicted in yellow, positive electrostatic coefficients portrayed in red, negative electrostatic coefficients displayed in blue) for 23 (a,b); 11 (c,d); 19 (e,f). For the clarity of presentation, DPPC was omitted. Figure S2. The structure-based alignment within 5-HT2AR-DPPC and OH2 probe-based PLS-coefficients (positive steric coefficients presented in green, negative steric coefficients depicted in yellow, positive electrostatic coefficients portrayed in red, negative electrostatic coefficients displayed in blue) for 24 (a,b); 5 (c,d); 20 (e,f). For the clarity of presentation, DPPC was omitted. Figure S3. The structure-based alignment within 5-HT2AR-DPPC and OH2 probe-based PLS-coefficients (positive steric coefficients presented in green, negative steric coefficients depicted in yellow, positive electrostatic coefficients portrayed in red, negative electrostatic coefficients displayed in blue) for 12 (a,b); 21 (c,d); 6 (e,f). For the clarity of presentation, DPPC was omitted. Figure S4. The structure-based alignment within 5-HT2AR-DPPC and OH2 probe-based PLS-coefficients (positive steric coefficients presented in green, negative steric coefficients depicted in yellow, positive electrostatic coefficients portrayed in red, negative electrostatic coefficients displayed in blue) for 7 (a,b); 14 (c,d); 8 (e,f). For the clarity of presentation, DPPC was omitted. Figure S5. The structure-based alignment within 5-HT2AR-DPPC and OH2 probe-based PLS-coefficients (positive steric coefficients presented in green, negative steric coefficients depicted in yellow, positive electrostatic coefficients portrayed in red, negative electrostatic coefficients displayed in blue) for 3 (a,b); 22 (c,d); 10 (e,f). For the clarity of presentation, DPPC was omitted. Figure S6. The structure-based alignment within 5-HT2AR-DPPC and OH2 probe-based PLS-coefficients (positive steric coefficients presented in green, negative steric coefficients depicted in yellow, positive electrostatic coefficients portrayed in red, negative electrostatic coefficients displayed in blue) for 15 (a,b); 16 (c,d); 13 (e,f). For the clarity of presentation, DPPC was omitted. Figure S7. The structure-based alignment within 5-HT2AR-DPPC and OH2 probe-based PLS-coefficients (positive steric coefficients presented in green, negative steric coefficients depicted in yellow, positive electrostatic coefficients portrayed in red, negative electrostatic coefficients displayed in blue) for 28 (a,b); 29 (c,d); 30 (e,f). For the clarity of presentation, DPPC was omitted. Figure S8. The structure-based alignment within 5-HT2AR-DPPC and OH2 probe-based PLS-coefficients (positive steric coefficients presented in green, negative steric coefficients depicted in yellow, positive electrostatic coefficients portrayed in red, negative electrostatic coefficients displayed in blue) for 31 (a,b); 32 (c,d); 33 (e,f). For the clarity of presentation, DPPC was omitted. Figure S9. The structure-based alignment within 5-HT2AR-DPPC and OH2 probe-based PLS-coefficients (positive steric coefficients presented in green, negative steric coefficients depicted in yellow, positive electrostatic coefficients portrayed in red, negative electrostatic coefficients displayed in blue) for 34 (a,b); 35 (c,d); 36 (e,f). For the clarity of presentation, DPPC was omitted. Figure S10. The structure-based alignment within 5-HT2AR-DPPC and OH2 probe-based PLS-coefficients (positive steric coefficients presented in green, negative steric coefficients depicted in yellow, positive electrostatic coefficients portrayed in red, negative electrostatic coefficients displayed in blue) for 37 (a,b); 38 (c,d); 39 (e,f). For the clarity of presentation, DPPC was omitted. Figure S11. The structure-based alignment within 5-HT2AR-DPPC and OH2 probe-based PLS-coefficients (positive steric coefficients presented in green, negative steric coefficients depicted in yellow, positive electrostatic coefficients portrayed in red, negative electrostatic coefficients displayed in blue) for 40 (a,b); 41 (c,d); 42 (e,f). For the clarity of presentation, DPPC was omitted. Figure S12. The structure-based alignment within 5-HT2AR-DPPC and OH2 probe-based PLS-coefficients (positive steric coefficients presented in green, negative steric coefficients depicted in yellow, positive electrostatic coefficients portrayed in red, negative electrostatic coefficients displayed in blue) for 43 (a,b); 44 (c,d). For the clarity of presentation, DPPC was omitted. Figure S13. The structure-based alignment within 5-HT2AR-DPPC and OH2 probe-based PLS-coefficients (positive steric coefficients presented in green, negative steric coefficients depicted in yellow, positive electrostatic coefficients portrayed in red, negative electrostatic coefficients displayed in blue) for 45 (a,b); 46 (c,d). For the clarity of presentation, DPPC was omitted. Figure S14. The structure-based alignment within 5-HT2AR-DPPC and OH2 probe-based PLS-coefficients (positive steric coefficients presented in green, negative steric coefficients depicted in yellow, positive electrostatic coefficients portrayed in red, negative electrostatic coefficients displayed in blue) for 6WGT (a,b); 7WC6 (c,d); 7WC7 (e,f). For the clarity of presentation, DPPC was omitted. Figure S15. The structure-based alignment within 5-HT2AR-DPPC and OH2 probe-based PLS-coefficients (positive steric coefficients presented in green, negative steric coefficients depicted in yellow, positive electrostatic coefficients portrayed in red, negative electrostatic coefficients displayed in blue) for 6A93 (a,b); 7VOD (c,d); 7VOE (e,f). For the clarity of presentation, DPPC was omitted. Figure S16. The structure-based alignment within 5-HT2AR-DPPC and OH2 probe-based PLS-coefficients (positive steric coefficients presented in green, negative steric coefficients depicted in yellow, positive electrostatic coefficients portrayed in red, negative electrostatic coefficients displayed in blue) for 7WC4 (a,b); 7RAN (c,d); 7WC5 (e,f). For the clarity of presentation, DPPC was omitted. Figure S17. The structure-based alignment within 5-HT2AR-DPPC and OH2 probe-based PLS-coefficients (positive steric coefficients presented in green, negative steric coefficients depicted in yellow, positive electrostatic coefficients portrayed in red, negative electrostatic coefficients displayed in blue) for 7WC9 (a,b); 7WC8 (c,d). For the clarity of presentation, DPPC was omitted.
Author Contributions
Conceptualization, N.T., R.R. and M.M.; methodology, N.T., R.R. and M.M.; validation, M.V., E.K., V.R., B.A., S.L.M., M.B., R.F. and E.P., formal analysis, M.V., E.K., V.R., B.A., S.L.M., M.B., R.F. and E.P.; investigation, all authors; resources, all authors.; a curation, all authors; writing—original draft preparation, N.T.; writing—review and editing, R.R. and M.M.; supervision, R.R. and M.M. All authors have read and agreed to the published version of the manuscript.
Informed Consent Statement
Not applicable.
Data Availability Statement
Complexes herein used to derive 3-D QSAR models and perform SB alignments assessments are available at Protein Data Bank (https://www.rcsb.org/). Structures of other training set and test set compounds are retrieved from literature. The obtained 3-D QSAR models are available upon request from Milan Mladenović (E-mail: milan.mladenovic@pmf.kg.ac.rs).
Conflicts of Interest
The authors declare no conflict of interest.
Sample Availability
Not applicable.
Funding Statement
This work was supported by the Serbian Ministry of Science, Technological Development, and Innovation (Agreement Nos. 451-03-47/2023-01/200122 and 451-03-47/2023-01/200378), and supported by two grants from Progetti di Ricerca di Università 2015, Sapienza Università di Roma (C26A15RT82 and C26A15J3BB).
Footnotes
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content.
References
- 1.Nichols D.E., Nichols C.D. Serotonin Receptors. Chem. Rev. 2008;108:1614–1641. doi: 10.1021/cr078224o. [DOI] [PubMed] [Google Scholar]
- 2.Hauser A.S., Attwood M.M., Rask-Andersen M., Schiöth H.B., Gloriam D.E. Trends in GPCR Drug Discovery: New Agents, Targets and Indications. Nat. Rev. Drug Discov. 2017;16:829–842. doi: 10.1038/nrd.2017.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Welsh D.J., Harnett M., MacLean M., Peacock A.J. Proliferation and Signaling in Fibroblasts: Role of 5-hydroxytryptamine2A Receptor and Transporter. Am. J. Respir. Crit. Care Med. 2004;170:252–259. doi: 10.1164/rccm.200302-264OC. [DOI] [PubMed] [Google Scholar]
- 4.El-Merahbi R., Loffler M., Mayer A., Sumara G. The Roles of Peripheral Serotonin in Metabolic Homeostasis. FEBS Lett. 2015;589:1728–1734. doi: 10.1016/j.febslet.2015.05.054. [DOI] [PubMed] [Google Scholar]
- 5.Matsumoto T., Kobayashi T., Ishida K., Taguchi K., Kamata K. Enhancement of Mesenteric Artery Contraction to 5-HT Depends on Rho Kinase and Src Kinase Pathways in the Ob/Ob Mouse Model of Type 2 Diabetes. Br. J. Pharmacol. 2010;160:1092–1104. doi: 10.1111/j.1476-5381.2010.00753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vindis C., D’Angelo R., Mucher E., Nègre-Salvayre A., Parini A., Mialet-Perez J. Essential Role of TRPC1 Channels in Cardiomyoblasts Hypertrophy Mediated by 5-HT2A Serotonin Receptors. Biochem. Biophys. Res. Commun. 2010;391:979–983. doi: 10.1016/j.bbrc.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 7.Hershenson M.B., Chao T.S., Abe M.K., Gomes I., Kelleher M.D., Solway J., Rosner M.R. Histamine Antagonizes Serotonin and Growth Factor-Induced Mitogen-Activated Protein Kinase Activation in Bovine Tracheal Smooth Muscle Cells. J. Biol. Chem. 1995;270:19908–19913. doi: 10.1074/jbc.270.34.19908. [DOI] [PubMed] [Google Scholar]
- 8.Manzke T., Niebert M., Koch U.R., Caley A., Vogelgesang S., Hülsmann S., Ponimaskin E., Müller U., Smart T.G., Harvey R.J., et al. Serotonin Receptor 1A-Modulated Phosphorylation of Glycine Receptor A3 Controls Breathing in Mice. J. Clin. Investig. 2010;120:4118–4128. doi: 10.1172/JCI43029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Berumen L.C., Rodríguez A., Miledi R., García-Alcocer G. Serotonin Receptors in Hippocampus. Sci. World J. 2012;2012:823493. doi: 10.1100/2012/823493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jenkins T.A., Nguyen J.C.D., Polglaze K.E., Bertrand P.P. Influence of Tryptophan and Serotonin on Mood and Cognition with a Possible Role of the Gut-Brain Axis. Nutrients. 2016;8:56. doi: 10.3390/nu8010056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alenina N., Klempin F. The Role of Serotonin in Adult Hippocampal Neurogenesis. Behav. Brain Res. 2015;277:49–57. doi: 10.1016/j.bbr.2014.07.038. [DOI] [PubMed] [Google Scholar]
- 12.Nikoui V., Javadi-Paydar M., Salehi M., Behestani S., Dehpour A.-R. Protective Effects of Lithium on Sumatriptan-Induced Memory Impairment in Mice. Acta Med. Iran. 2016;54:226–232. [PubMed] [Google Scholar]
- 13.Mann J.J. Role of the Serotonergic System in the Pathogenesis of Major Depression and Suicidal Behavior. Neuropsychopharmacology. 1999;21:99–105. doi: 10.1038/sj.npp.1395364. [DOI] [PubMed] [Google Scholar]
- 14.López-Giménez J.F., González-Maeso J. Hallucinogens and Serotonin 5-HT2A Receptor-Mediated Signaling Pathways. In: Halberstadt A.L., Vollenweider F.X., Nichols D.E., editors. Behavioral Neurobiology of Psychedelic Drugs. Volume 36. Springer; Berlin/Heidelberg, Germany: 2017. pp. 45–73. Current Topics in Behavioral Neurosciences. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Švob Štrac D., Pivac N., Mück-Šeler D. The Serotonergic System and Cognitive Function. Transl. Neurosci. 2016;7:35–49. doi: 10.1515/tnsci-2016-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kimura K.T., Asada H., Inoue A., Kadji F.M.N., Im D., Mori C., Arakawa T., Hirata K., Nomura Y., Nomura N., et al. Structures of the 5-HT2A Receptor in Complex with the Antipsychotics Risperidone and Zotepine. Nat. Struct. Mol. Biol. 2019;26:121–128. doi: 10.1038/s41594-018-0180-z. [DOI] [PubMed] [Google Scholar]
- 17.González-Maeso J., Weisstaub N.V., Zhou M., Chan P., Ivic L., Ang R., Lira A., Bradley-Moore M., Ge Y., Zhou Q., et al. Hallucinogens Recruit Specific Cortical 5-HT2A Receptor-Mediated Signaling Pathways to Affect Behavior. Neuron. 2007;53:439–452. doi: 10.1016/j.neuron.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 18.Nichols D.E. Hallucinogens. Pharmacol. Ther. 2004;101:131–181. doi: 10.1016/j.pharmthera.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 19.Penders T.M., Gestring R.E., Vilensky D.A. Excited Delirium Following Use of Synthetic Cathinones (Bath Salts) Gen. Hosp. Psychiatry. 2012;34:647–650. doi: 10.1016/j.genhosppsych.2012.06.005. [DOI] [PubMed] [Google Scholar]
- 20.Vujović M., Ragavendran V., Arsić B., Kostić E., Mladenović M. DFT Calculations as an Efficient Tool for Prediction of Raman and Infra-Red Spectra and Activities of Newly Synthesized Cathinones. Open Chem. 2020;18:185–195. doi: 10.1515/chem-2020-0021. [DOI] [Google Scholar]
- 21.Chen Y., Blough B.E., Murnane K.S., Canal C.E. The Synthetic Cathinone Psychostimulant A-PPP Antagonizes Serotonin 5-HT2A Receptors: In Vitro and in Vivo Evidence. Drug Test. Anal. 2019;11:990–998. doi: 10.1002/dta.2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weinstein A.M., Rosca P., Fattore L., London E.D. Synthetic Cathinone and Cannabinoid Designer Drugs Pose a Major Risk for Public Health. Front. Psychiatry. 2017;8:156. doi: 10.3389/fpsyt.2017.00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Koob G.F. Drugs of Abuse: Anatomy, Pharmacology and Function of Reward Pathways. Trends Pharmacol. Sci. 1992;13:177–184. doi: 10.1016/0165-6147(92)90060-j. [DOI] [PubMed] [Google Scholar]
- 24.Jakab R.L., Goldman-Rakic P.S. 5-Hydroxytryptamine2A Serotonin Receptors in the Primate Cerebral Cortex: Possible Site of Action of Hallucinogenic and Antipsychotic Drugs in Pyramidal Cell Apical Dendrites. Proc. Natl. Acad. Sci. USA. 1998;95:735–740. doi: 10.1073/pnas.95.2.735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim K., Che T., Panova O., DiBerto J.F., Lyu J., Krumm B.E., Wacker D., Robertson M.J., Seven A.B., Nichols D.E., et al. Structure of a Hallucinogen-Activated Gq-Coupled 5-HT2A Serotonin Receptor. Cell. 2020;182:1574–1588.e19. doi: 10.1016/j.cell.2020.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cao D., Yu J., Wang H., Luo Z., Liu X., He L., Qi J., Fan L., Tang L., Chen Z., et al. Structure-Based Discovery of Nonhallucinogenic Psychedelic Analogs. Science. 2022;375:403–411. doi: 10.1126/science.abl8615. [DOI] [PubMed] [Google Scholar]
- 27.Kaplan A.L., Confair D.N., Kim K., Barros-Álvarez X., Rodriguiz R.M., Yang Y., Kweon O.S., Che T., McCorvy J.D., Kamber D.N., et al. Bespoke Library Docking for 5-HT2A Receptor Agonists with Antidepressant Activity. Nature. 2022;610:582–591. doi: 10.1038/s41586-022-05258-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Z., Fan L., Wang H., Yu J., Lu D., Qi J., Nie F., Luo Z., Liu Z., Cheng J., et al. Structure-Based Design of a Novel Third-Generation Antipsychotic Drug Lead with Potential Antidepressant Properties. Nat. Neurosci. 2022;25:39–49. doi: 10.1038/s41593-021-00971-w. [DOI] [PubMed] [Google Scholar]
- 29.Wacker D., Wang C., Katritch V., Han G.W., Huang X.-P., Vardy E., McCorvy J.D., Jiang Y., Chu M., Siu F.Y., et al. Structural Features for Functional Selectivity at Serotonin Receptors. Science. 2013;340:615–619. doi: 10.1126/science.1232808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu W., Wacker D., Gati C., Han G.W., James D., Wang D., Nelson G., Weierstall U., Katritch V., Barty A., et al. Serial Femtosecond Crystallography of G Protein–Coupled Receptors. Science. 2013;342:1521–1524. doi: 10.1126/science.1244142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wacker D., Wang S., McCorvy J.D., Betz R.M., Venkatakrishnan A.J., Levit A., Lansu K., Schools Z.L., Che T., Nichols D.E., et al. Crystal Structure of an LSD-Bound Human Serotonin Receptor. Cell. 2017;168:377–389.e12. doi: 10.1016/j.cell.2016.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cao C., Barros-Álvarez X., Zhang S., Kim K., Dämgen M.A., Panova O., Suomivuori C.-M., Fay J.F., Zhong X., Krumm B.E., et al. Signaling Snapshots of a Serotonin Receptor Activated by the Prototypical Psychedelic LSD. Neuron. 2022;110:3154–3167.e7. doi: 10.1016/j.neuron.2022.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McCorvy J.D., Wacker D., Wang S., Agegnehu B., Liu J., Lansu K., Tribo A.R., Olsen R.H.J., Che T., Jin J., et al. Structural Determinants of 5-HT 2B Receptor Activation and Biased Agonism. Nat. Struct. Mol. Biol. 2018;25:787–796. doi: 10.1038/s41594-018-0116-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simmler L.D., Buser T.A., Donzelli M., Schramm Y., Dieu L.-H., Huwyler J., Chaboz S., Hoener M.C., Liechti M.E. Pharmacological Characterization of Designer Cathinones In Vitro. Br. J. Pharmacol. 2013;168:458–470. doi: 10.1111/j.1476-5381.2012.02145.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Elliott S., Evans J. A 3-Year Review of New Psychoactive Substances in Casework. Forensic Sci. Int. 2014;243:55–60. doi: 10.1016/j.forsciint.2014.04.017. [DOI] [PubMed] [Google Scholar]
- 36.AHC Media, LLC . Pediatric Trauma Care II: A Clinical Reference for Physicians and Nurses Caring for the Acutely Injured Child. AHC Media, LLC; Atlanta, GA, USA: 2014. [Google Scholar]
- 37.Drug Enforcement Administration, Diversion Control Division, Drug & Chemical Evaluation Section, 3,4-Methylenedioxypyrovalerone (MDPV) (Street Names: “Bath Salts,” “Ivory Wave,” “Plant Fertilizer,” “Vanilla Sky,” “Energy-1”) [(accessed on 3 December 2022)];2019 Available online: https://deadiversion.usdoj.gov/drug_chem_info/mdpv.pdf.
- 38.Zawilska J.B. Synthetic Cathinones: Novel Addictive and Stimulatory Psychoactive Substances. Springer; Berlin/Heidelberg, Germany: 2018. [Google Scholar]
- 39.Freye E. Pharmacology and Abuse of Cocaine, Amphetamines, Ecstasy and Related Designer Drugs: A Comprehensive Review on Their Mode of Action, Treatment of Abuse and Intoxication. Springer Science & Business Media; Berlin/Heidelberg, Germany: 2009. [Google Scholar]
- 40.Fattore L., Weinstein A. Novel Psychoactive Drugs. Frontiers Media SA; Lausanne, Switzerland: 2019. [Google Scholar]
- 41.Gatch M.B., Forster M.J., Janowsky A., Eshleman A.J. Abuse Liability Profile of Three Substituted Tryptamines. J. Pharmacol. Exp. Ther. 2011;338:280–289. doi: 10.1124/jpet.111.179705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Janicak P.G., Marder S.R., Pavuluri M.N. Principles and Practice of Psychopharmacotherapy. Lippincott Williams & Wilkins; Philadelphia, PA, USA: 2011. [Google Scholar]
- 43.Karch S.B. Cathinone Neurotoxicity (“The 3Ms”) Curr. Neuropharmacol. 2015;13:21–25. doi: 10.2174/1570159X13666141210225009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Prosser J.M., Nelson L.S. The Toxicology of Bath Salts: A Review of Synthetic Cathinones. J. Med. Toxicol. 2012;8:33–42. doi: 10.1007/s13181-011-0193-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Young R., Glennon R.A. Discriminative Stimulus Effects of S(-)-Methcathinone (CAT): A Potent Stimulant Drug of Abuse. Psychopharmacology. 1998;140:250–256. doi: 10.1007/s002130050765. [DOI] [PubMed] [Google Scholar]
- 46.Shapiro D.A., Renock S., Arrington E., Chiodo L.A., Liu L.-X., Sibley D.R., Roth B.L., Mailman R. Aripiprazole, A Novel Atypical Antipsychotic Drug with a Unique and Robust Pharmacology. Neuropsychopharmacology. 2003;28:1400–1411. doi: 10.1038/sj.npp.1300203. [DOI] [PubMed] [Google Scholar]
- 47.Giovanni G.D., Matteo V.D., Esposito E. Serotonin-Dopamine Interaction: Experimental Evidence and Therapeutic Relevance. Elsevier; Amsterdam, The Netherlands: 2008. [DOI] [PubMed] [Google Scholar]
- 48.Knight A.R., Misra A., Quirk K., Benwell K., Revell D., Kennett G., Bickerdike M. Pharmacological Characterisation of the Agonist Radioligand Binding Site of 5-HT2A, 5-HT2B and 5-HT2C Receptors. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2004;370:114–123. doi: 10.1007/s00210-004-0951-4. [DOI] [PubMed] [Google Scholar]
- 49.Bymaster F.P., Calligaro D.O., Falcone J.F., Marsh R.D., Moore N.A., Tye N.C., Seeman P., Wong D.T. Radioreceptor Binding Profile of the Atypical Antipsychotic Olanzapine. Neuropsychopharmacology. 1996;14:87–96. doi: 10.1016/0893-133X(94)00129-N. [DOI] [PubMed] [Google Scholar]
- 50.Seeman P. Clozapine, a Fast-Off-D2 Antipsychotic. ACS Chem. Neurosci. 2013;5:24–29. doi: 10.1021/cn400189s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Canal C.E., Booth R.G., Morgan D. Support for 5-HT2C Receptor Functional Selectivity in Vivo Utilizing Structurally Diverse, Selective 5-HT2C Receptor Ligands and the 2,5-Dimethoxy-4-Iodoamphetamine Elicited Head-Twitch Response Model. Neuropharmacology. 2013;70:112–121. doi: 10.1016/j.neuropharm.2013.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Roth B.L. The Serotonin Receptors: From Molecular Pharmacology to Human Therapeutics. Springer Science & Business Media; Berlin/Heidelberg, Germany: 2008. [Google Scholar]
- 53.Meltzer H.Y., Roth B.L. Lorcaserin and Pimavanserin: Emerging Selectivity of Serotonin Receptor Subtype–Targeted Drugs. J. Clin. Investig. 2013;123:4986–4991. doi: 10.1172/JCI70678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Johnson M.P., Siegel B.W., Carr A.A. [3H]MDL 100,907: A Novel Selective 5-HT2A Receptor Ligand. Naunyn Schmiedeberg’s Arch. Pharmacol. 1996;354:205–209. doi: 10.1007/BF00178722. [DOI] [PubMed] [Google Scholar]
- 55.Aly S.A.R., Hossain M., Bhuiyan M.A., Nakamura T., Nagatomo T. Assessment of Binding Affinity to 5-Hydroxytryptamine 2A (5-HT2A) Receptor and Inverse Agonist Activity of Naftidrofuryl: Comparison With Those of Sarpogrelate. J. Pharmacol. Sci. 2009;110:445–450. doi: 10.1254/jphs.09124FP. [DOI] [PubMed] [Google Scholar]
- 56.Closse A. [3H]Mesulergine, a Selective Ligand for Serotonin-2 Receptors. Life Sci. 1983;32:2485–2495. doi: 10.1016/0024-3205(83)90375-2. [DOI] [PubMed] [Google Scholar]
- 57.Bonhaus D.W., Bach C., DeSouza A., Salazar F.H., Matsuoka B.D., Zuppan P., Chan H.W., Eglen R.M. The Pharmacology and Distribution of Human 5-Hydroxytryptamine2B (5-HT2B) Receptor Gene Products: Comparison with 5-HT2A and 5-HT2C Receptors. Br. J. Pharmacol. 1995;115:622–628. doi: 10.1111/j.1476-5381.1995.tb14977.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Newton R.A., Elliott J.M. Mianserin-Induced down-Regulation of Human 5-Hydroxytryptamine2A and 5-Hydroxytryptamine2C Receptors Stably Expressed in the Human Neuroblastoma Cell Line SH-SY5Y. J. Neurochem. 1997;69:1031–1038. doi: 10.1046/j.1471-4159.1997.69031031.x. [DOI] [PubMed] [Google Scholar]
- 59.Kennett G.A., Wood M.D., Bright F., Cilia J., Piper D.C., Gager T., Thomas D., Baxter G.S., Forbes I.T., Ham P., et al. In Vitro and in Vivo Profile of SB 206553, a Potent 5-HT2C/5-HT2B Receptor Antagonist with Anxiolytic-like Properties. Br. J. Pharmacol. 1996;117:427–434. doi: 10.1111/j.1476-5381.1996.tb15208.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fernández J., Alonso J.M., Andrés J.I., Cid J.M., Díaz A., Iturrino L., Gil P., Megens A., Sipido V.K., Trabanco A.A. Discovery of New Tetracyclic Tetrahydrofuran Derivatives as Potential Broad-Spectrum Psychotropic Agents. J. Med. Chem. 2005;48:1709–1712. doi: 10.1021/jm049632c. [DOI] [PubMed] [Google Scholar]
- 61.Kennett G.A., Wood M.D., Bright F., Trail B., Riley G., Holland V., Avenell K.Y., Stean T., Upton N., Bromidge S., et al. SB 242084, a Selective and Brain Penetrant 5-HT2C Receptor Antagonist. Neuropharmacology. 1997;36:609–620. doi: 10.1016/S0028-3908(97)00038-5. [DOI] [PubMed] [Google Scholar]
- 62.Rosenzweig-Lipson S., Zhang J., Mazandarani H., Harrison B.L., Sabb A., Sabalski J., Stack G., Welmaker G., Barrett J.E., Dunlop J. Antiobesity-like Effects of the 5-HT2C Receptor Agonist WAY-161503. Brain Res. 2006;1073–1074:240–251. doi: 10.1016/j.brainres.2005.12.052. [DOI] [PubMed] [Google Scholar]
- 63.Ragno R., Esposito V., Di Mario M., Masiello S., Viscovo M., Cramer R.D. Teaching and Learning Computational Drug Design: Student Investigations of 3D Quantitative Structure–Activity Relationships through Web Applications. J. Chem. Educ. 2020;97:1922–1930. doi: 10.1021/acs.jchemed.0c00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Titeler M., Lyon R.A., Glennon R.A. Radioligand Binding Evidence Implicates the Brain 5-HT2 Receptor as a Site of Action for LSD and Phenylisopropylamine Hallucinogens. Psychopharmacology. 1988;94:213–216. doi: 10.1007/BF00176847. [DOI] [PubMed] [Google Scholar]
- 65.Ballante F., Musmuca I., Marshall G.R., Ragno R. Comprehensive Model of Wild-Type and Mutant HIV-1 Reverse Transciptases. J. Comput. Aided Mol. Des. 2012;26:907–919. doi: 10.1007/s10822-012-9586-6. [DOI] [PubMed] [Google Scholar]
- 66.Sievers F., Wilm A., Dineen D., Gibson T.J., Karplus K., Li W., Lopez R., McWilliam H., Remmert M., Söding J., et al. Fast, Scalable Generation of High-Quality Protein Multiple Sequence Alignments Using Clustal Omega. Mol. Syst. Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mladenović M., Patsilinakos A., Pirolli A., Sabatino M., Ragno R. Understanding the Molecular Determinant of Reversible Human Monoamine Oxidase B Inhibitors Containing 2H-Chromen-2-One Core: Structure-Based and Ligand-Based Derived Three-Dimensional Quantitative Structure-Activity Relationships Predictive Models. J. Chem. Inf. Model. 2017;57:787–814. doi: 10.1021/acs.jcim.6b00608. [DOI] [PubMed] [Google Scholar]
- 68.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Trott O., Olson A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization and Multithreading. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lang P.T., Brozell S.R., Mukherjee S., Pettersen E.F., Meng E.C., Thomas V., Rizzo R.C., Case D.A., James T.L., Kuntz I.D. DOCK 6: Combining Techniques to Model RNA-Small Molecule Complexes. RNA. 2009;15:1219–1230. doi: 10.1261/rna.1563609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Koes D.R., Baumgartner M.P., Camacho C.J. Lessons Learned in Empirical Scoring with Smina from the CSAR 2011 Benchmarking Exercise. J. Chem. Inf. Model. 2013;53:1893–1904. doi: 10.1021/ci300604z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Korb O., Stützle T., Exner T.E. PLANTS: Application of Ant Colony Optimization to Structure-Based Drug Design. In: Dorigo M., Gambardella L.M., Birattari M., Martinoli A., Poli R., Stützle T., editors. Ant Colony Optimization and Swarm Intelligence. Springer; Berlin/Heidelberg, Germany: 2006. pp. 247–258. [Google Scholar]
- 73.Mihović N., Tomašević N., Matić S., Mitrović M.M., Kostić D.A., Sabatino M., Antonini L., Ragno R., Mladenović M. Human Estrogen Receptor α Antagonists. Part 1: 3-D QSAR-Driven Rational Design of Innovative Coumarin-Related Antiestrogens as Breast Cancer Suppressants through Structure-Based and Ligand-Based Studies. J. Chem. Inf. Model. 2021;61:5028–5053. doi: 10.1021/acs.jcim.1c00530. [DOI] [PubMed] [Google Scholar]
- 74.Kurtanović N., Tomašević N., Matić S., Proia E., Sabatino M., Antonini L., Mladenović M., Ragno R. Human Estrogen Receptor Alpha Antagonists, Part 3: 3-D Pharmacophore and 3-D QSAR Guided Brefeldin A Hit-to-Lead Optimization toward New Breast Cancer Suppressants. Molecules. 2022;27:2823. doi: 10.3390/molecules27092823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Quiroga R., Villarreal M.A. Vinardo: A Scoring Function Based on Autodock Vina Improves Scoring, Docking, and Virtual Screening. PLoS ONE. 2016;11:e0155183. doi: 10.1371/journal.pone.0155183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kurrasch-Orbaugh D.M., Watts V.J., Barker E.L., Nichols D.E. Serotonin 5-Hydroxytryptamine 2A Receptor-Coupled Phospholipase C and Phospholipase A2 Signaling Pathways Have Different Receptor Reserves. J. Pharmacol. Exp. Ther. 2003;304:229–237. doi: 10.1124/jpet.102.042184. [DOI] [PubMed] [Google Scholar]
- 77.Tosco P., Balle T. Open3DQSAR: A New Open-Source Software Aimed at High-Throughput Chemometric Analysis of Molecular Interaction Fields. J. Mol. Model. 2011;17:201–208. doi: 10.1007/s00894-010-0684-x. [DOI] [PubMed] [Google Scholar]
- 78.Ballante F., Ragno R. 3-D QSAutogrid/R: An Alternative Procedure to Build 3-D QSAR Models. Methodologies and Applications. J. Chem. Inf. Model. 2012;52:1674–1685. doi: 10.1021/ci300123x. [DOI] [PubMed] [Google Scholar]
- 79.Tropsha A. Best Practices for QSAR Model Development, Validation, and Exploitation. Mol. Inf. 2010;29:476–488. doi: 10.1002/minf.201000061. [DOI] [PubMed] [Google Scholar]
- 80.Hamacher A., Weigt M., Wiese M., Hoefgen B., Lehmann J., Kassack M.U. Dibenzazecine Compounds with a Novel Dopamine/5HT2A Receptor Profile and 3D-QSAR Analysis. BMC Pharmacol. 2006;6:11. doi: 10.1186/1471-2210-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Avram S., Duda-Seiman D., Borcan F., Wolschann P. QSAR-CoMSIA Applied to Antipsychotic Drugs with Their Dopamine D2 and Serotonine 5HT2A Membrane Receptors. J. Serb. Chem. Soc. 2011;76:263–281. doi: 10.2298/JSC100806022A. [DOI] [Google Scholar]
- 82.Lin F., Li F., Wang C., Wang J., Yang Y., Yang L., Li Y. Mechanism Exploration of Arylpiperazine Derivatives Targeting the 5-HT2A Receptor by In Silico Methods. Molecules. 2017;22:1064. doi: 10.3390/molecules22071064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brea J., Rodrigo J., Carrieri A., Sanz F., Cadavid M.I., Enguix M.J., Villazón M., Mengod G., Caro Y., Masaguer C.F., et al. New Serotonin 5-HT2A, 5-HT2B, and 5-HT2C Receptor Antagonists: Synthesis, Pharmacology, 3D-QSAR, and Molecular Modeling of (Aminoalkyl)Benzo and Heterocycloalkanones. J. Med. Chem. 2002;45:54–71. doi: 10.1021/jm011014y. [DOI] [PubMed] [Google Scholar]
- 84.Dezi C., Brea J., Alvarado M., Raviña E., Masaguer C.F., Loza M.I., Sanz F., Pastor M. Multistructure 3D-QSAR Studies on a Series of Conformationally Constrained Butyrophenones Docked Into a New Homology Model of the 5-HT2A Receptor. J. Med. Chem. 2007;50:3242–3255. doi: 10.1021/jm070277a. [DOI] [PubMed] [Google Scholar]
- 85.Radan M., Ruzić D., Antonijević M., Djikić T., Nikolić K. In Silico Identification of Novel 5-HT2A Antagonists Supported with Ligand- and Target-Based Drug Design Methodologies. J. Biomol. Struc. Dyn. 2021;39:1819–1837. doi: 10.1080/07391102.2020.1738961. [DOI] [PubMed] [Google Scholar]
- 86.Ragno R. Www.3d-Qsar.Com: A Web Portal That Brings 3-D QSAR to All Electronic Devices—The Py-CoMFA Web Application as Tool to Build Models from Pre-Aligned Datasets. J. Comput.-Aided Mol. Des. 2019;33:855–864. doi: 10.1007/s10822-019-00231-x. [DOI] [PubMed] [Google Scholar]
- 87.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera—A Visualization System for Exploratory Research and Analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 88.Case D.A., Darden T.A., Cheatham T.E., III, Simmerling C.L., Wang J., Duke R.E., Luo R., Walker R.C., Zhang W., Merz K.M., et al. AMBER 12. University of California; San Francisco, CA, USA: 2012. [Google Scholar]
- 89.Distributed MS . A Version of the Solvent-Accessible Surface Computation Program. UCSF Computer Graphics Laboratory; San Francisco, CA, USA: 2009. [Google Scholar]
- 90.Marvin Beans. ChemAxon; Budapest, Hungary: 2015. 15.4.27.0. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Complexes herein used to derive 3-D QSAR models and perform SB alignments assessments are available at Protein Data Bank (https://www.rcsb.org/). Structures of other training set and test set compounds are retrieved from literature. The obtained 3-D QSAR models are available upon request from Milan Mladenović (E-mail: milan.mladenovic@pmf.kg.ac.rs).