

Abstract
Objective
Intraductal papillary mucinous neoplasms (IPMNs) are precursor lesions that can give rise to invasive pancreatic carcinoma. Although approximately 8% of patients with resected pancreatic ductal adenocarcinoma have a co-occurring IPMN, the precise genetic relationship between these two lesions has not been systematically investigated.
Design
We analysed all available patients with co-occurring IPMN and invasive intrapancreatic carcinoma over a 10-year period at a single institution. For each patient, we separately isolated DNA from the carcinoma, adjacent IPMN and distant IPMN and performed targeted next generation sequencing of a panel of pancreatic cancer driver genes. We then used the identified mutations to infer the relatedness of the IPMN and co-occurring invasive carcinoma in each patient.
Results
We analysed co-occurring IPMN and invasive carcinoma from 61 patients with IPMN/ductal adenocarcinoma as well as 13 patients with IPMN/colloid carcinoma and 7 patients with IPMN/carcinoma of the ampullary region. Of the patients with co-occurring IPMN and ductal adenocarcinoma, 51% were likely related. Surprisingly, 18% of co-occurring IPMN and ductal adenocarcinomas were likely independent, suggesting that the carcinoma arose from an independent precursor. By contrast, all colloid carcinomas were likely related to their associated IPMNs. In addition, these analyses showed striking genetic heterogeneity in IPMNs, even with respect to well-characterised driver genes.
Conclusion
This study demonstrates a higher prevalence of likely independent co-occurring IPMN and ductal adenocarcinoma than previously appreciated. These findings have important implications for molecular risk stratification of patients with IPMN.
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is one of the most aggressive human cancers.1 PDAC arises from non-invasive precursor lesions which take several years to transform into invasive carcinoma, providing opportunity for early detection and surgical cure. Intraductal papillary mucinous neoplasms (IPMNs) are the most prevalent cystic precursor lesion in the pancreas.2 Due to the potential for progression to invasive carcinoma, patients with IPMNs are routinely monitored, with surgical intervention recommended for clinical and radiological ‘worrisome features’ and ‘high-risk stigmata’.3 As more IPMNs are discovered incidentally on routine abdominal imaging, optimal surveillance for patients with these lesions is becoming a pressing clinical problem, highlighting the need to balance cancer prevention with overtreatment.4 5
Because of the ability to obtain cyst fluid by fine needle aspiration, pancreatic cysts (including IPMNs) are a promising application of molecular diagnostics.6–8 In recent years, extensive next-generation sequencing analyses have comprehensively characterised the genomic landscape of PDAC, reaffirming the four driver genes altered in the vast majority of tumours (the oncogene KRAS and the tumour suppressor genes TP53, CDKN2A and SMAD4) as well as a much larger number of genes altered at low prevalence.9–13 Additional studies have focused on genetic alterations in histologically distinct precursor lesions, elucidating the timing of specific mutations in pancreatic tumorigenesis.14–16 Next-generation sequencing of cyst fluid DNA to identify key driver gene mutations can reliably distinguish IPMNs from other cystic lesions.17 18 Perhaps more importantly, specific late-occurring driver gene mutations, such as TP53 and SMAD4, may be able to distinguish low-grade from high-grade premalignant lesions, separating patients who benefit from surgical intervention.6 7 This strategy may provide a promising tool for earlier detection of pancreatic carcinoma derived from IPMNs in the future. However, as it relies on mutations identified in the cyst fluid, this approach assumes that the invasive carcinoma arises from the co-occurring IPMN—it may not detect invasive carcinomas that arise from physically adjacent but genetically independent precursor lesions.
Multiple studies have described multifocal neoplasia in the pancreas, particularly in patients with IPMNs.19–21 In pancreata with IPMNs, anatomically separate invasive carcinomas have been termed ‘concomitant’ carcinomas.20 22 23 The traditional definition of IPMN with ‘concomitant’ carcinoma relies on the physical separation of the two lesions by an uninvolved segment of pancreatic parenchyma, assuming that the lack of physical proximity implies independent origin of both lesions.22 However, it is possible that a genetically independent invasive carcinoma could arise in close proximity to an IPMN, making them indistinguishable from IPMN with associated invasive carcinoma by clinical and pathological features alone.
Genetic alterations can be used as molecular tools to assess the relatedness of IPMNs and co-occurring invasive cancers. For example, the identification of discordant KRAS and GNAS mutations in IPMNs and their concomitant invasive carcinomas has been reported and provides evidence for the independent genetic origins of some neoplasms.24 25 An accurate estimation of the proportion of genetically independent PDACs co-occurring with IPMNs is of significant clinical importance; cyst fluid analysis of the nearby IPMN in this situation would likely not identify mutations from the highest risk lesion, as the PDAC would not shed its high-risk mutant DNA molecules into the monitored cyst fluid unless the PDAC invaded the cyst wall.
In this study, we use targeted next-generation sequencing to analyse driver gene mutations in paired neoplastic samples from a cohort of IPMNs with co-occurring PDACs. Using the identified mutations, we determined relatedness and estimated the proportion of these lesions that are genetically independent.
METHODS
We identified all patients who underwent pancreatic resection with a diagnosis of co-occurring IPMN and invasive carcinoma over a 10-year period. We reviewed clinical and pathological data as well as H&E stained slides from each patient to confirm the diagnosis. Neoplastic cells from the invasive carcinoma, IPMN immediately adjacent to the invasive carcinoma (adj-IPMN) and IPMN without invasive carcinoma in the same tissue block (dist-IPMN) were separately isolated from formalin-fixed paraffin-embedded (FFPE) tissue sections by laser capture microdissection (LCM). In all but one case, the adj-IPMN and dist-IPMN samples came from the same grossly defined cyst. Thus, our cohort included only a single case of multifocal IPMN with two pathologically distinct IPMN lesions (IPP12). In addition, three met criteria for IPMN with concomitant carcinoma, with intervening uninvolved pancreas between the two lesions (IPP05, IPP11, IPP48). DNA was extracted from each sample, and DNA of sufficient quantity and quality was obtained from 76 invasive cancers and co-occurring IPMNs, including 56 ductal adenocarcinomas of the pancreas, 13 colloid carcinomas of the pancreas and 7 invasive cancers of the ampullary region (ampullary, distal bile duct and duodenal carcinomas). The cohort for this study initially excluded five patients with IPMN co-occurring with ductal adenocarcinoma that were analysed by whole exome sequencing in a separate study—these cases were selected based on their morphology suggestive of carcinoma arising from an IPMN, and the IPMN and ductal adenocarcinoma shared numerous somatic mutations in each case. These related cases are included in our final estimate of the prevalence of genetically unrelated IPMN/ductal adenocarcinoma in the ‘Discussion’ section and were used to confirm the accuracy of our relatedness assessment. All samples were analysed by targeted next generation sequencing of a custom panel of pancreatic driver genes using Ion AmpliSeq library preparation on an Ion Torrent Personal Genome Machine, and whole exome sequencing was performed on three selected cases by Personal Genome Diagnostics (Baltimore, Maryland, USA).26 Mutations were identified using NextGENe software followed by visual inspection to minimise risk of artifactual calls.27 The relatedness of the lesions from each patient was determined by a categorical algorithm as described in the ‘Results’ section. The validity of this approach was confirmed by an independent statistical model to evaluate the probability of relatedness based on the site-specific mutation distributions in our study cohort. In addition, immunohistochemistry (IHC) for p53 and Smad4 protein was performed on FFPE sections from selected cases. Additional details are provided in the online supplementary methods.
RESULTs
IPMN lesions co-occurring in pancreata with invasive carcinoma
Within a period of 10 years (from 2006 to 2015), 159 patients diagnosed with invasive pancreatic or periampullary carcinoma and co-occurring IPMN underwent resection at The Johns Hopkins Hospital—112 of these patients had ductal adenocarcinoma, 35 had colloid carcinoma and 12 had a carcinoma of the ampullary region (ampullary, distal bile duct and duodenal carcinomas). In the same period, 1267 patients underwent resection for ductal adenocarcinoma without an associated IPMN; thus, approximately 8% of patients with ductal adenocarcinoma had a co-occurring IPMN. We obtained sufficient high-quality DNA for targeted next generation sequencing analysis from 76 co-occurring IPMN/invasive carcinoma. Of these, 56 were co-occurring IPMN/ductal adenocarcinomas of the pancreas as well as 13 co-occurring IPMN/colloid carcinomas of the pancreas and 7 co-occurring IPMN/carcinomas of the ampullary region. Clinical differences in stage and grade of the carcinoma are readily apparent between the different cohorts, as are differences in the co-occurring IPMNs (table 1 and online supplementary table S1).28 For example, while ductal adenocarcinomas and colloid carcinomas most often occurred in association with high-grade IPMNs, the IPMNs co-occurring with ampullary region carcinoma were typically low-grade, supporting the clinical assumption that ampullary and pancreatic neoplasms are usually independent. Moreover, colloid carcinomas of the pancreas co-occurred with IPMN of intestinal subtype whereas ductal adenocarcinomas were mostly associated with gastric and pancreatobiliary subtypes of IPMNs.
Table 1.
Clinical and pathological features of patients with IPMN and co-occurring carcinoma
| Carcinoma | Total cases (adj-IPMN/dist-IPMN) | Gender | Age | Location carcinoma/adj-IPMN | Location dist-IPMN | Cancer | IPMN | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||||||||||||
| T | N | Grade | Size (cm) | Grade | Type | Subtype | |||||||||||||
|
| |||||||||||||||||||
| Ductal adenocarcinoma | 56 (53/42) | M 63.6% (35) | 70.2 | Head | 62.5% (35) | 36% (20) | T1 | 10.7% (6) | NO | 30.4% (17) | 1 | 3.6% (2) | 2.9 | LGD | 19.6% (11) | BD | 57.4% (27) | Gastric | 61.8% (34) |
| F 36.4% (21) | (+/−10.8) | Neck | 8.9% (5) | 28% (15) | T2 | 39.3% (22) | N1 | 69.6% (39) | 2 | 62.5% (35) | (+/−2.5) | HGD | 80.4% (45) | MD | 29.8% (14) | Intestinal | 7.2% (4) | ||
| Body | 7.1% (4) | 20% (11) | T3 | 46.4% (26) | 3 | 26.8% (15) | Mixed | 12.8% (6) | Pancreatobiliary | 23.6% (13) | |||||||||
| Tail | 14.3% (8) | 12% (6) | T4 | 3.6% (2) | 4 | 3.6% (2) | Oncocytic | 7.2% (4) | |||||||||||
| Multifocal | 5.3% (3) | - | |||||||||||||||||
| Colloid carcinoma | 13 (11/9) | M 76.9% (10) | 62.5 | Head | 92.3% (12) | 50% (6) | T1 | 53.8% (7) | NO | 92.3% (12) | 1 | 61.5% (8) | 4.9 | LGD | 0% (0) | BD | 7.7% (1) | Gastric | 0% (0) |
| F 23.1% (3) | (+/−11.9) | Neck | 0% (0) | 16.7% (2) | T2 | 23.1% (3) | N1 | 7.7% (1) | 2 | 38.5% (5) | (+/−1.94) | HGD | 100% (13) | MD | 69.2% (9) | Intestinal | 100% (13) | ||
| Body | 7.7% (1) | 16.7% (2) | T3 | 23.1% (3) | 3 | 0% (0) | Mixed | 23.1% (3) | Pancreatobiliary | 0% (0) | |||||||||
| Tail | 0% (0) | 16.7% (2) | T4 | 0% (0) | 4 | 0% (0) | Oncocytic | 0% (0) | |||||||||||
| Multifocal | 0% (0) | - | |||||||||||||||||
| Carcinoma of the ampullary region | 7 (6/5) | M 42.8% (3) | 74.1 | Head | 100% (7) | 100% (5) | T1 | 0% (0) | NO | 0% (0) | 1 | 0% (0) | 1.1 | LGD | 71.4% (5) | BD | 71.4% (5) | Gastric | 85.7% (6) |
| F 57.2% (4) | (+/− 8.5) | Neck | 0% (0) | 0% (0) | T2 | 0% (0) | N1 | 100% (7) | 2 | 57.1% (8) | (+/−0.19) | HGD | 28.6% (2) | MD | 14.3% (1) | Intestinal | 0% (0) | ||
| Body | 0% (0) | 0% (0) | T3 | 71.4% (5) | 3 | 42.8% (6) | Mixed | 14.3% (1) | Pancreatobiliary | 14.3% (1) | |||||||||
| Tail | 0% (0) | 0% (0) | T4 | 28.8% (2) | 4 | 0% (0) | Oncocytic | 0% (0) | |||||||||||
| Multifocal | 0% (0) | 0% (0) | |||||||||||||||||
| Significance* | ns | ns | ns | ns | <0.001 | <0.001 | <0.001 | <0.001 | <0.001 | 0.009 | <0.001 | ||||||||
Tests were performed via ANOVA test for continuous variables or Fisher-Exact test for categorical variables.
ANOVA, analysis of variance; BD, branch duct; HGD, high grade dysplasia; IPMN, intraductal papillary mucinous neoplasm; LGD, low grade dysplasia; MD, main duct.
Although our cohort demonstrates that IPMNs and invasive carcinomas frequently occur in the same pancreata, it is not possible to determine relatedness from clinical and pathological features alone. To more accurately determine the genetic relationship between IPMNs and co-occurring invasive carcinomas, we performed targeted next generation sequencing analysis of a panel of pancreatic cancer driver genes for the entire cohort and whole exome sequencing for selected cases. From each case, we separately isolated neoplastic cells from the IPMN and invasive carcinoma using LCM (online supplementary figure S1). We collected IPMN adjacent to the carcinoma (adj-IPMN) and from a separate block not in direct contact with the invasive carcinoma (dist-IPMN) when available.
Molecular landscape of carcinomas and their co-occurring IPMNs
We performed targeted next generation sequencing of IPMN/PDAC driver genes using an IonTorrent Personal Genome Machine to an average coverage depth of >600× (online supplementary table S2). We compared mutations identified in the tumour samples to those in the matched normal tissue from each patient in order to exclude germline variants and report true somatic mutations (online supplementary figure S2). The majority of somatic mutations in the entire cohort (76 carcinoma samples and 95 IPMN samples) were missense mutations (88.1%), while nonsense mutations (5.3%), frameshift insertions/deletions (4.9%), in-frame insertions/deletions (0.9%) and splice site alterations (0.2%) made up only a minor proportion of the identified somatic alterations (online supplementary table S3).
In our cohort of 56 ductal adenocarcinomas, KRAS was mutated in 91% and TP53 in 57% of the invasive carcinomas (online supplementary figure S2). The two other major genetic drivers of ductal adenocarcinoma, CDKN2A and SMAD4, were altered by single nucleotide variants at lower prevalence, 11% and 30%, respectively. However, as these genes are also frequently altered by homozygous deletion, we performed copy number analysis on defined targeted loci which showed deletion of CDKN2A in 7% and SMAD4 in 16% of ductal adenocarcinomas (online supplementary table S3). GNAS and RNF43 mutations were frequently present in ductal adenocarcinomas, at 16% and 23%, respectively, highlighting that at least a subset of these PDACs likely arose from IPMNs.
GNAS mutations occurred in 85% (11 of 13) of colloid carcinomas, which are known to arise almost exclusively from intestinal-type IPMNs (online supplementary figure S2).29 30 TP53 mutations were detected in 46% of colloid carcinomas, but KRAS mutations were found in only 31% of cases. The vast majority (90%) of GNAS mutations in the entire carcinoma cohort (n=76) occurred at the hotspot codon 201, with a surprising enrichment of R201C mutations in colloid carcinomas (73% of GNAS mutations), in contrast to an enrichment of R201H mutations in ductal adenocarcinomas (75% of GNAS mutations) (online supplementary figure S3). Mutations in CDKN2A and SMAD4 were rarely detected in colloid carcinomas.
Our cohort also included seven carcinomas of the ampullary region (ampullary, distal bile duct and duodenal carcinomas) with co-occurring IPMNs. In these cancers, KRAS was the most frequently altered driver with mutation in 71%, while SMAD4 was the only other driver with mutation in more than one of these carcinomas (online supplementary figure S2).
IPMNs co-occurring with ductal adenocarcinomas, colloid carcinomas and ampullary region carcinomas were dissected and separately analysed. IPMNs immediately adjacent to ductal adenocarcinomas (adj-IPMN; n=53) were characterised by prevalent mutations in IPMN-specific drivers such as GNAS (25%) and RNF43 (36%), while they had fewer mutations in CDKN2A (7%), TP53 (34%) and SMAD4 (9%) compared with their neighbouring invasive carcinomas (online supplementary figure S2). IPMNs co-occurring with colloid carcinomas displayed a molecular signature characteristic of intestinal subtype IPMNs, showing a higher prevalence of alterations in GNAS (82%) and a lower prevalence of KRAS mutations (45%). At least one oncogenic hotspot mutation (in KRAS and/or GNAS) was identified in 92% of all IPMNs in our entire cohort of 76 patients (online supplementary table S3).
Validation of somatic mutations and their accuracy in classifying relatedness
In order to determine the relatedness of the IPMN and invasive carcinoma in each patient, we applied two independent analytic approaches. First, we developed a categorical algorithm based on shared mutations between the two lesions. ‘Likely related’ lesions shared more than two hotspot mutations or one mutation other than a hotspot mutation (see details in online supplementary methods), while ‘likely independent’ lesions had no shared mutations. In addition, as GNAS mutations are rare in ductal adenocarcinomas not derived from IPMNs, we considered IPMNs and invasive carcinomas with shared GNAS mutations as ‘likely related’. Lesion pairs with only a shared hotspot KRAS mutation were designated as ‘indeterminate’—the high prevalence of such hotspot mutations in IPMNs as well as ductal adenocarcinomas makes it an unreliable indicator of relatedness, as such a mutation could also occur independently by chance in unrelated lesions.
We confirmed the reliability of this classification in two ways. First, in order to determine the specificity of our ‘likely related’ categorization, we classified the relatedness of IPMNs and carcinomas from different patients in our cohort using the identified mutations (online supplementary figure S4). Of the 3136 total relatedness assessments of lesion pairs from different patients, only 60 (1.9%) were classified as ‘likely related’. Of note, these ‘likely related’ classifications were almost exclusively driven by shared mutations in the GNAS hotspot in codon 201. However, this strategy for evaluating our algorithm likely overestimates ‘false positives’ in our classification of ‘likely related’ lesions. While common GNAS hotspot alterations are likely to be shared by IPMNs and IPMN-associated carcinomas from different patients, such mutations are unlikely to be shared by independent IPMNs and PDACs in the same patient, as such GNAS alterations are rare in PDACs arising outside of IPMNs.10–12 Second, in order to determine the sensitivity of our ‘likely related’ classification, we performed our targeted next generation sequencing (NGS) assay on five pairs of co-occurring IPMN/PDAC with concrete evidence of relatedness based on numerous shared somatic mutations in whole exome sequencing performed for a separate study (online supplementary table S4). All five of these lesion pairs were classified as ‘likely related’ based on our categorical algorithm.
In order to further validate our relatedness assessment, we developed a quantitative statistical model to calculate the probability of the shared mutations in any two samples occurring by chance (online supplementary methods). These probabilities were plotted on heat maps, highlighting that the vast majority of lesion pairs with low probabilities were derived from the same patient (online supplementary figure S5). When comparing our categorical classification to these statistical measures, ‘likely related’ lesions had a mean probability of 0.0017 or <0.2% probability of sharing these mutation profiles by chance. In addition, 85% of cases classified as ‘likely related’ by our qualitative algorithm had a probability of <0.001, further demonstrating the robustness of our classification (online supplementary table S5). Also of note, as all of our ‘likely independent’ cases shared no somatic mutations, all had a probability of >0.9 in this approach, as approximately 9 million synthetic IPMN/PDAC pairs shared no somatic mutations (online supplementary methods). Cases classified as ‘indeterminate’ by our categorical algorithm cases had an average probability value of 0.049, highlighting a higher probability of developing shared mutation profiles by chance and supporting our reluctance to designate relatedness based on shared KRAS hotspot mutations alone.
Finally, in order to determine the frequency of sequencing errors that altered relatedness assessment in our targeted NGS assay, we conducted a validation experiment by resequencing 16 adj-IPMN/PDAC pairs with our assay at a much higher coverage (average coverage of 2,192X in the targeted region). We confirmed that >95% of the somatic mutations which conferred relatedness in these samples were present in the resequenced samples (see online supplementary table S6). In all cases, the relatedness classification was confirmed based on the results of this deep resequencing, underscoring the reliability of our original targeted NGS assay.
Together, these results confirm that our method for evaluating genetic relatedness of these lesions was accurate.
Relatedness of IPMNs to co-occurring carcinomas
Using our qualitative algorithm, 46% of the IPMN/ductal adenocarcinomas were ‘likely related’, further helping to establish IPMNs as precursor lesions to co-occurring ductal adenocarcinomas (figure 1A and online supplementary table S7). Importantly, only a subset of these ‘likely related’ cancers (23%) harboured a GNAS mutation, highlighting the inadequacy of this IPMN-associated gene mutation as a single biomarker to identify IPMN-associated ductal adenocarcinomas. Surprisingly, despite the close anatomic proximity of the two lesions, a sizeable proportion (20%) of IPMNs and co-occurring ductal adenocarcinomas were ‘likely independent’, as they shared no driver gene mutations. The remaining IPMN/ductal adenocarcinoma pairs (34%) were classified as ‘indeterminate’, as their relatedness could not be confidently assigned by mutations in the driver genes in our targeted sequencing assay. Considering the five cases of known relatedness initially excluded from our cohort but later used for validation, we identified the following prevalences of relatedness in the entire cohort—’likely related’: 51%, indeterminate: 31% and likely independent: 18%. Representative cases are presented in table 2, overall proportions in figure 1 and relatedness assessment for individual cases in online supplementary table S7.
Figure 1.

Relatedness of IPMNs and co-occurring carcinomas. Bars show the fraction of likely related (black), likely independent (white) and indeterminate (grey) neoplasms. For each patient the relatedness of three pairs of neoplasms was assayed: carcinoma and adj-IPMN (left), carcinoma and dist-IPMN (middle) and adj-IPMN and dist-IPMN (right). Molecular analysis of three different pancreatic cancer entities, ductal adenocarcinoma (A), colloid carcinoma (B) and carcinoma of the ampullary region (C), revealed a distinct prevalence of relatedness to co-occurring IPMN in each cancer type. adj-/dist-IPMN, adjacent/distant IPMN; IPMN, intraductal papillary mucinous neoplasm.
Table 2.
Somatic mutations in representative cases of cancer and co-occurring IPMN
| Patient ID | Diagnosis | Genetic alterations | shared mutations | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
||||||||
| Carcinoma | adj-IPMN | dist-IPMN | Carcinoma / adj-IPMN | Carcinoma / dist-IPMN | adj-IPMN / dist-IPMN | |||||
|
| ||||||||||
| IPP08 | Ductal adenocarcinoma | KRAS: p.G12V | missense | KRAS: p.G12V | missense | KRAS: p.G12V | missense | KRAS: p.G12V | KRAS: p.G12V | KRAS: p.G12V |
| RNF43: p.G67D | missense | RNF43: p.G67D | missense | GNAS: p.R201H | missense | RNF43: p.G67D | ||||
| TGFBR2: p.P526L | missense | TGFBR2: p.P526L | missense | TGFBR2: p.P526L | ||||||
| ARID1A: p.Q2207X | missense | likely related* <0.0000001† | indeterminate* 0.0403† | indeterminate* 0.0407† | ||||||
| IPP14 | Ductal adenocarcinoma | KRAS: p.G12D | missense | KRAS: p.G12D | missense | KRAS: p.G12D | missense | KRAS: p.G12D | KRAS: p.G12D | KRAS: p.G12D |
| ARID1A: p.N1313S | missense | ARID1A: p.N1313S | missense | KRAS: p.Q61H | missense | ARID1A: p.N1313S | ARID1A: p.N1313S | ARID1A: p.N1313S | ||
| RNF43: p.P686R | missense | RNF43: p.P686R | missense | ARID1A: p.N1313S | missense | RNF43: p.P686R | RNF43: p.P686R | RNF43: p.P686R | ||
| RNF43: p.P686R | missense | likely related* <0.0000001† | likely related* <0.0000001† | likely related* <0.0000001† | ||||||
| IPP22 | Ductal adenocarcinoma | KRAS: p.G12V | missense | KRAS: p.G12V | missense | GNAS: p.R201H | missense | KRAS: p.G12V | GNAS: p.R201H | GNAS: p.R201H |
| GNAS: p.R201H | missense | GNAS: p.R201H | missense | TP53: p.V272L | missense | GNAS: p.R201H | TP53: p.V272L | TP53: p.V272L | ||
| PIK3CA: p.H1047L | missense | PIK3CA: p.H1047L | missense | SMAD4: p.W398X | nonsense | PIK3CA: p.H1047L | SMAD4: p.W398X | SMAD4: p.W398X | ||
| TP53: p.V272L | missense | TP53: p.V272L | missense | TP53: p.V272L | ||||||
| SMAD4: p.W398X | nonsense | SMAD4: p.W398X | nonsense | SMAD4: p.W398X | ||||||
| likely related* <0.0000001† | likely related* 0.00607† | likely related* 0.00679† | ||||||||
| IPP26 | Ductal adenocarcinoma | KRAS: p.G12V | missense | KRAS: p.G12D | missense | KRAS: p.G12D | missense | none | none | KRAS: p.G12D |
| SMAD4: p.R372K | missense | GNAS: p.R201H | missense | GNAS: p.R201H | missense | GNAS: p.R201H | ||||
| ARID1A: p.M1300I | missense | TP53: p.R205Q | likely independent* >0.9† | likely independent* >0.9† | likely related* 0.08 | |||||
| IPP43 | Ductal adenocarcinoma | KRAS: p.G12D | missense | KRAS: p.G12V | missense | KRAS: p.G12V | missense | none | none | KRAS: p.G12V |
| GNAS: p.R201C | missense | TP53: p.R196X | missense | |||||||
| likely independent* >0.9† | likely independent* >0.9† | indeterminate* 0.0407† | ||||||||
| PIK3CA: p.M1004V | missense | SMAD4: p.K70fs | frameshift | |||||||
| IPP54 | Ductal adenocarcinoma | KRAS: p.G12V | missense | KRAS: p.G12A | missense | KRAS: p.G12D | missense | none | none | none |
| TP53: p.V272L | missense | TP53: p.P390Lfs | frameshift | |||||||
| likely independent* 0.9† | likely independent* 0.9† | likely independent* 0.9† | ||||||||
| IPP38 | Ductal adenocarcinoma | KRAS: p.G12V | missense | KRAS: p.G12R | missense | KRAS: p.G12V | missense | KRAS: p.G12V | KRAS: p.G12V | KRAS: p.G12V |
| TP53: p.C275Y | missense | KRAS: p.G12V | missense | |||||||
| SMAD4: p.E235K | missense | ARID1A: p.D385H | missense | |||||||
| indeterminate* 0.0403† | indeterminate* 0.0403† | indeterminate* 0.0407† | ||||||||
| IPC03 | Colloid carcinoma | GNAS: p.R201C T | missense | GNAS: p.R201C | missense | N/A | GNAS: p.R201C | N/A | N/A | |
| TP53: p.G245D | missense | TP53: p.G245D | missense | TP53: p.G245D likely related* | ||||||
| PIK3CA: p.M1004I | missense | |||||||||
| IPC06 | Colloid carcinoma | PIK3CA: p.M1004I GNAS: p.R201C TP53: p.H193R | missense missense missense |
GNAS: p.R201C TP53: p.H193R | missense missense |
GNAS: p.R201C TP53: p.H193R RNF43: p.C511F | missense missense missense |
GNAS: p.R201C TP53: p.H193R |
GNAS: p.R201C TP53: p.H193R |
GNAS: p.R201C TP53: p.H193R |
| TP53: p.H193R | missense | TP53: p.H193R | missense | TP53: p.H193R | missense | TP53: p.H193R | TP53: p.H193R | TP53: p.H193R | ||
| RNF43: p.C511F | missense | |||||||||
| likely related* | likely related* | likely related* | ||||||||
| IPA03 | Carcinoma of the ampullary region | TGFBR2: p.E270K | missense | KRAS: p.G12R | missense | GNAS: c.1018+1G>A | Splice variant | none | none | none |
| GNAS: p.R201C | missense | |||||||||
| likely independent* | likely independent* | likely independent* | ||||||||
| IPA07 | Carcinoma of the ampullary region | KRAS: p.G12D | missense | KRAS: p.G12V | missense | KRAS: p.Q61H | missense | none | none | none |
| CDKN2A: p.G111fs | frameshift | GNAS: p.R201H | missense | |||||||
| likely independent* | likely independent* | likely independent* | ||||||||
Categorical assessment.
Statistical assessment.
adj-/dist-IPMN, adjacent/distant IPMN; IPMN, intraductal papillary mucinous neoplasm; N/A, sample not available.
Because they share only a KRAS hotspot, ‘indeterminate’ cases could be either related or independent. In order to more definitively determine the relatedness of these lesions, we performed whole exome sequencing on three cases classified as ‘indeterminate’ in our targeted NGS assay for which we had sufficient DNA (IPP17, IPP39, IPP41). Surprisingly, the whole exome sequencing data suggest that two of these three co-occurring IPMNs/PDACs are genetically independent, as they share no somatic mutations aside from the KRAS hotspot alteration (online supplementary table S8). In contrast, 21 somatic mutations were shared between the IPMN and PDAC in the other ‘indeterminate’ case, providing strong evidence that this PDAC arose from the co-occurring IPMN.
There was no correlation between IPMN/PDAC relatedness and clinicopathological features including cyst size, grade and histological subtype (online supplementary table S9). Many of the ‘likely independent’ cases were resected because preoperatively their IPMNs met criteria for resection; clinical and radiological features of the IPMN led to resection of 58% of ‘likely independent’ cases compared with 52% of ‘likely related’ cases. Thus, most of the IPMNs in the ‘likely independent’ cases were not merely incidental small cysts adjacent to large ductal adenocarcinomas. Crucially, mutations in driver genes that suggest a high risk for advanced disease (TP53, SMAD4) were not present in the co-occurring IPMN in 9 of 11 (82%) of ‘likely independent’ cases; thus, analysis of mutations in IPMN-derived DNA in cyst fluid would not have identified these patients as high-risk. Survival analysis revealed a significant trend towards improved survival in patients with ‘likely related’ IPMN/ductal adenocarcinomas cases compared with those with ‘likely independent’ lesions (P=0.017; Log-rank test; figure 2).
Figure 2.

Survival analysis of patients with pancreatic cancer based on their molecular relationship with co-occurring IPMNs. Cumulative survival of ductal adenocarcinoma patients with and without co-occurring IPMNs after 5-year follow-up. Colloid carcinomas have significantly better survival than ductal adenocarcinoma without IPMN (P value 0.001; Log-rank test). Patients follow a divergent survival pattern when considering the relatedness of ductal adenocarcinomas with co-occurring IPMNs based on results of the targeted sequencing assay (P value: 0.017; Log-rank test). Ductal adenocarcinomas likely related to IPMNs show improved overall survival (28.8 months), while ductal adenocarcinomas likely independent of IPMNs have poorer survival similar to ductal adenocarcinoma without IPMN (16.2 months). IPMN, intraductal papillary mucinous neoplasm.
In contrast to the observation of unrelatedness of many of the adjacent IPMN/ductal adenocarcinoma cases, we found that all of the colloid carcinomas were ‘likely related’ to the co-occurring IPMN (figure 1B). This result adds to the growing body of evidence that this histological subtype of pancreatic carcinoma develops essentially exclusively in the setting of an intestinal-type IPMN, making IPMN its obligate precursor.29 30 The ampullary region carcinomas with co-occurring IPMNs serve as additional negative controls, as carcinoma of the ampullary region are not known to arise from IPMNs. As expected, all cases of carcinoma of the ampullary region and adj-IPMN were classified as ‘likely independent’ by our assay (figure 1C).
Genetic heterogeneity in IPMNs
Heterogeneity with respect to driver gene mutations has been reported within grossly contiguous IPMNs.8 In order to minimize the likelihood that such heterogeneity resulted in artifactual characterisation of lesions as ‘likely independent’, we analysed an additional section of IPMN from a separate tissue block. In all but one case (IPP12), the two IPMN samples were derived from the same grossly identified cyst. Genetic analysis of matched adj-IPMN and dist-IPMN from the same patient allowed us to describe such genetic heterogeneity within individual IPMNs in more depth.
Examining the relatedness of adj-IPMN and dist-IPMN to the co-occurring invasive carcinoma revealed distinct patterns. The majority of paired IPMN samples (69%) had the same relationship to the co-occurring ductal adenocarcinomas. Of the cases with discordant relationships, the majority (88%) of ductal adenocarcinomas were ‘likely related’ or ‘indeterminate’ to the adj-IPMN and ‘likely independent’ from the dist-IPMN. This suggests that anatomical distance is associated with genetic relatedness; IPMN lesions in close proximity to the invasive carcinoma were more likely to be related than IPMNs at a greater distance.
Comparing the alterations in the two distinct regions of the same IPMN, IPMN foci were ‘likely related’ to one another in 39% of the cases, whereas 28% were ‘likely independent’, highlighting the possibility of polyclonality in individual IPMNs. The relatedness of the remaining paired IPMN samples was ‘indeterminate’ in our assay. Intriguingly, the relatedness of the two IPMN samples varied in our different cancer groups. While 86% of the paired IPMN samples were ‘likely related’ in patients with colloid carcinoma, only 39% of such samples were ‘likely related’ in patients with ductal adenocarcinoma, and all of the paired IPMN samples were ‘likely independent’ in patients with carcinoma of the ampullary region.
Pancreatic ductal neoplasms (including both IPMNs and ductal adenocarcinomas) are typically initiated by oncogenic hotspot mutations. In 56 dissected IPMNs from patients with co-occurring ductal adenocarcinoma, 13 IPMNs (23%) harboured more than one mutation in the same oncogene, including 11 IPMNs with multiple KRAS mutations and 2 IPMNs with multiple GNAS mutations. Up to four different KRAS hotspot mutations in a single sample were detected (online supplementary figure S6). Intriguingly, this may still be an underestimation, as two additional IPMN samples submitted for whole exome sequencing revealed additional oncogenic hotspot mutations due to further sampling of the same blocks to collect additional DNA (online supplementary table S8). Only two ductal adenocarcinomas (4%) had multiple oncogene mutations, in both cases KRAS. Interestingly, the presence of multiple unique mutations in other driver genes in the same neoplasm was an uncommon event. However, even in ‘likely related’ cases, there were many mutations present in only a single sample, reflecting heterogeneity in driver mutations; only 64 of 104 identified somatic mutations were shared among related samples. Taken together, these data show that there is striking genetic heterogeneity within IPMNs, highlighting the challenges of capturing all the genetic alterations in a single tissue sample.
spatial distribution of mutant and wild-type clones
Although we used LCM to obtain samples of IPMN and invasive carcinoma with high neoplastic cellularity, visualisation of mutant and wild-type clones in situ in tissue sections can confirm our targeted sequencing results, particularly in cases in which the IPMN and ductal adenocarcinomas have discordant mutations. Mutations in two of the tumour suppressor genes analysed, TP53 and SMAD4, have well-documented effects on protein expression as assayed by IHC.31 32 To more definitively assess the spatial distribution of mutant and wild-type clones, we performed IHC for Smad4 and p53 protein expression in a subset of our IPMN/ductal adenocarcinoma cases analysed by next generation sequencing. For each protein, we analysed ductal adenocarcinoma and adj-IPMN from five patients who had discordant mutation status between the two lesions (online supplementary table S10).
In the p53 IHC assay, three of five cases with discordant TP53 mutation status between IPMN and ductal adenocarcinoma showed the expected expression patterns in both components, with strong and diffuse p53 expression limited to the invasive carcinoma (figure 3). These three cases all had missense TP53 mutations, while the remaining two had nonsense and frameshift mutations, which are not expected to cause the same aberrant p53 expression pattern.32 In the Smad4 IHC assay, three of five cases with discordant SMAD4 mutation status in the IPMN and invasive ductal adenocarcinoma showed the expected expression pattern in both components, with loss of Smad4 expression limited to the invasive ductal adenocarcinoma (figure 3). Overall, these results confirm the discordant mutation status in the IPMN and ductal adenocarcinoma in the majority of cases (online supplementary table S10).
Figure 3.
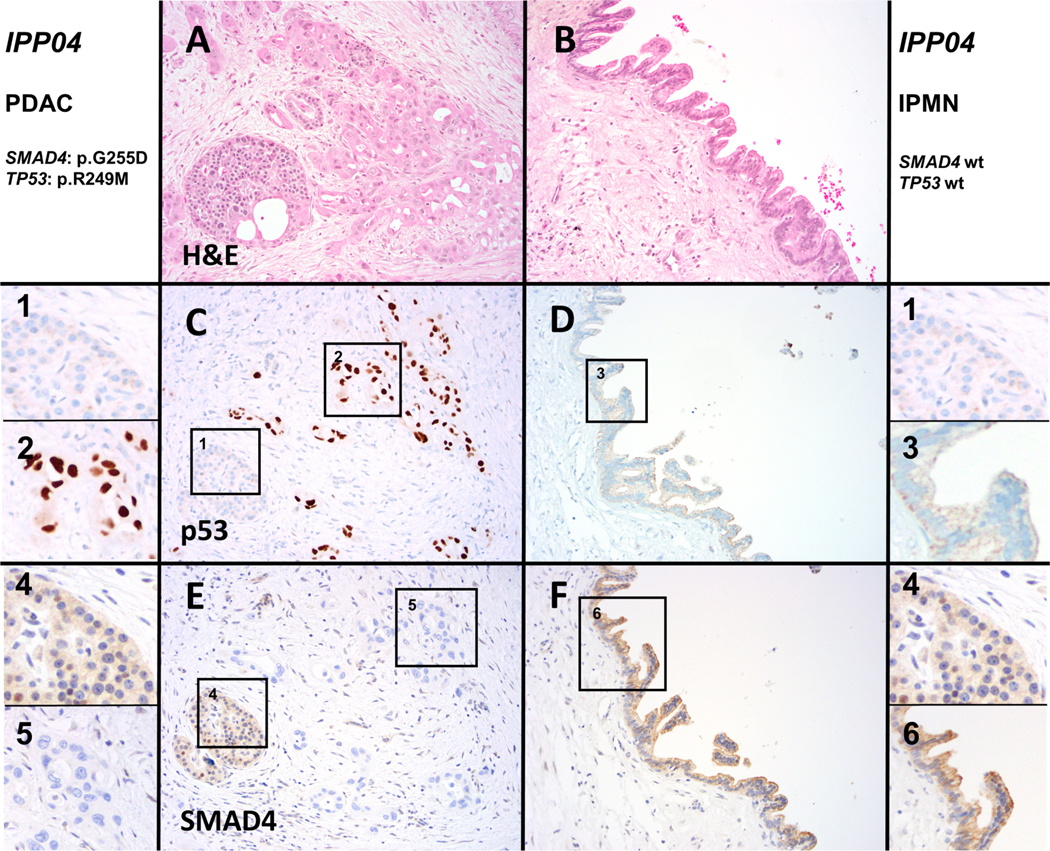
Immunohistochemistry for p53 and Smad4 confirms discordant genotypes in IPMN and co-occurring cancer. A representative H&E stained section shows case IPP04 (20× magnification), which harbours mutations in TP53 and SMAD4 in the ductal adenocarcinoma (A), while the adj-IPMN (B) is wild-type. In the ductal adenocarcinoma (C), strong-diffuse expression of p53 was found in contrast to normal expression in the adj-IPMN (D). Normal islets are shown in the inset as an internal control (1), while (2) shows a high-power view of cancer cells with aberrant p53 expression and (3) shows a high-power view of wild-type p53 expression pattern in the IPMN. Expression of Smad4 was lost in ductal adenocarcinoma (E), while expression was retained in adj-IPMN (F). Normal islets are shown in the inset as an internal control (1), while (2) shows a high-power view of cancer cells with loss of Smad4 expression and (3) shows a high-power view of retained Smad4 expression in the IPMN. adj-/dist-IPMN, adjacent/distant IPMN; IPMN, intraductal papillary mucinous neoplasm; PDAC, pancreatic ductal adenocarcinoma.
DISCUSSION
New diagnostic approaches are needed to more accurately identify patients with high-risk pancreatic lesions while they are still surgically curable.4 33–35 Although imaging findings can stratify the risk of progression of pancreatic cysts, additional approaches are needed to further improve diagnostic sensitivity and specificity. Analysis of molecular alterations in biological samples (including cyst fluid and pancreatic juice) is a promising adjunct to existing clinical and radiological criteria, as alterations in specific genes (such as TP53 and SMAD4) are closely associated with the development of high-risk neoplasia.6 7 14 The identification of mutations in pancreatic cyst fluid is beginning to be applied as a clinical test to preoperatively diagnose and risk-stratify pancreatic cysts.18 36 37 However, this approach assumes that the IPMN is the obligate precursor in cases where an invasive carcinoma develops. The high prevalence of multifocal neoplasia in the pancreas raises the possibility that a carcinoma co-occurring with an IPMN could arise from a genetically independent precursor, such as pancreatic intraepithelial neoplasia.8 15 22
The central objective of this study was to investigate the molecular relationship of co-occurring IPMNs and invasive carcinomas in an unbiased approach. Meticulous LCM to separately isolate DNA from two adjacent neoplasms, followed by next generation sequencing analysis of critical driver genes in pancreatic neoplasia, allowed us to estimate the relatedness in the majority of patients in our cohorts. More than one-sixth (18%) of the co-occurring IPMN/invasive ductal adenocarcinoma pairs appear to be genetically unrelated, meaning they shared no mutations in the assayed genes. Importantly, this is likely an underestimate of prevalence of unrelated lesions. Two of three cases that met clinical criteria for IPMN with ‘concomitant’ carcinoma (IPP05, IPP48) were classified as ‘likely independent’ in our assay, while the third (IP11) was classified as ‘indeterminate’, suggesting that at least a subset of the indeterminate cases also represent independent neoplasms. In addition, analysis of three different ‘indeterminate’ cases (IPP17, IPP39, IPP41) by whole exome sequencing revealed that two were genetically independent, as they did not share any other somatic mutations aside from the KRAS hotspot. This finding has clinical implications, as it suggests that in this subset of patients with IPMN, sampling and analysis of their IPMN cyst fluid would not be evaluating the precursor lesion destined to progress to invasive ductal adenocarcinoma. In the present invasive ductal adenocarcinoma cohort, more than 80% of unrelated IPMNs lacked mutations in high-risk genes (TP53, SMAD4) that are expected to be detected in more than 50% of ductal adenocarcinomas. The implications for cyst fluid analysis should be more directly validated by comparing mutations identified in tissue samples and cyst fluid in future studies. Alternative samples for molecular analysis (such as secretin-stimulated pancreatic juice) may provide a more complete mutation profile of the entire pancreatic duct system and thus may be more able to overcome this issue.6 7 25
Through analysis of two different regions of each IPMN, our study also provides an assessment of genetic heterogeneity in these lesions. First, some IPMNs harboured two or more hotspot mutations in a single oncogene, suggesting presence of multiple clones within the dissected tissue. Intriguingly, the proportion of IPMNs with multiple hotspot mutations was notably more than that found in invasive ductal adenocarcinomas (23% vs 4%), perhaps suggesting a more diverse mixture of clones in lower grade lesions. In addition, many samples displayed discordant hotspot mutations in adj-IPMN and dist-IPMN, which were classified as ‘likely independent’ in one-third of patients. There were also notable differences in IPMN heterogeneity between cancer types. While a majority of the two IPMN samples co-occurring with colloid carcinoma were ‘likely related’, IPMNs that co-occurred with carcinoma of the ampullary region were mostly unrelated. This suggests that the IPMNs that give rise to colloid carcinomas are genetically more homogeneous, while those that co-occur with ampullary region carcinoma are more frequently heterogeneous (table 1).
Overall, the results of our study support the hypothesis of field cancerisation, in which the entire pancreas of some patients is at increased risk for ductal neoplasia. We identify a sizeable proportion of patients in our cohort with two likely independent ductal neoplasms in a small area of the pancreas, highlighting the importance of multifocal neoplasia. In addition, many of the IPMNs analysed had multiple mutations in initiating driver genes, raising the possibility of polyclonality within one IPMN. However, our data do not provide any insight into the underlying mechanism of field cancerisation in the pancreas, though they do suggest that it is not mediated by somatic mutations in well-characterised driver genes. Of note, our study did not comprehensively address large chromosomal alterations as potential genetic drivers in pancreatic cancer precursor lesions. Although these alterations are less useful for determination of relatedness, recent studies suggest that they play an important role in driving pancreatic tumourigenesis, at least in a subset of patients.38 Thus, they remain candidates for the alterations underlying field cancerisation in the pancreas, and future studies could incorporate such alterations into studies of precursor lesions to better address this hypothesis.
Some limitations of our study have to be considered. First, we were not able to confidently determine relatedness in a subset of our IPMN/ductal adenocarcinoma pairs; as demonstrated, more comprehensive sequencing such as whole exome sequencing can help to determine relatedness in these individuals. In addition, it is possible that ‘likely independent’ ductal adenocarcinomas arose out of a subclone of the co-occurring IPMNs that was not sampled or was too small to be detected by our approach. Low neoplastic cellularity and clonal heterogeneity may decrease the sensitivity of molecular analyses in these cases—we address this in several ways. First, we performed LCM of every IPMN and PDAC to enrich neoplastic cellularity as much as possible. Second, we performed deep targeted sequencing (with an average coverage of >600X), improving our ability to detect rare subclones. Third, we sampled the IPMN immediately adjacent to the ductal adenocarcinoma (and thus most likely to be related) as well as a separate section to account for genetic heterogeneity. In addition, we performed validation of our mutation calls in a subset of samples at very high coverage (>2000×) to estimate the risk of misclassification of relatedness in the larger cohort—we confirmed >95% of the mutations conferring relatedness in the original samples. Finally, the early driver mutations should be present in high proportion in both components of related lesions, making this explanation far less likely than true genetic independence.
Collectively, our molecular analysis revealed an unexpected proportion of co-occurring IPMNs and invasive ductal adenocarcinomas that likely arose from genetically distinct precursor lesions in the pancreas. We also show that virtually all colloid carcinomas arise from their associated intestinal-type IPMNs and that ampullary region carcinomas (distal common bile duct, ampullary and duodenal carcinoma) do not arise from co-occurring IPMNs.
Molecular analysis of cyst fluid is a promising technique for risk stratification in patients with IPMN undergoing surveillance. However, we demonstrate that at least one-sixth of invasive ductal adenocarcinomas co-occurring with IPMNs are ‘likely independent’ and that more than 80% of those cases would not have identified high-risk alterations in analysis of IPMN-derived cyst fluid. This highlights a potential limitation of cyst fluid analysis and suggests a need for molecular approaches that more completely sample DNA from the entire pancreas.
Supplementary Material
What is already known on this subject?
Intraductal papillary mucinous neoplasms (IPMNs) are precursor lesions that can give rise to invasive pancreatic ductal adenocarcinoma. Multifocal neoplasia is common in the pancreas, particularly pancreata with IPMN, raising the possibility that a subset of co-occurring IPMNs/carcinomas are independent.
What are the new findings?
Targeted next generation sequencing of co-occurring IPMN and ductal adenocarcinoma samples revealed that more than one-sixth of these lesions are likely independent, suggesting that the carcinoma is derived from a precursor lesion independent of the co-occurring IPMN. Most of the IPMNs that were likely independent from their co-occurring carcinoma lacked mutations in driver genes suggestive of a high risk of malignant transformation.
How might it on impact clinical practice in the foreseeable future?
These results highlight a possible limitation of relying solely on cyst fluid molecular analysis to assess risk in the entire pancreas, as this fluid would not contain high-risk mutations from independent precursor lesions or carcinomas. Other biospecimens that more completely sample the entire pancreas may have improved sensitivity in detecting these likely independent neoplasms.
Funding
The authors acknowledge the following sources of support: NIH/NCI P50 CA62924; NIH/NIDDK K08 DK107781; Sol Goldman Pancreatic Cancer Research Center; DFG-German Research Foundation; Buffone Family Gastrointestinal Cancer Research Fund; Kaya Tuncer Career Development Award in Gastrointestinal Cancer Prevention; AGA-Bernard Lee Schwartz Foundation Research Scholar Award in Pancreatic Cancer; Sidney Kimmel Foundation for Cancer Research Kimmel Scholar Award; AACR-Incyte Corporation Career Development Award for Pancreatic Cancer Research; Rolfe Pancreatic Cancer Foundation; Joseph C Monastra Foundation; The Gerald O Mann Charitable Foundation (Harriet and Allan Wulfstat, Trustees); Dutch Digestive Foundation (CDG 14-02); The Lisa Waller Hayes Foundation; The Nijbakker-Morra Foundation.
Footnotes
Competing interests LDW is a paid consultant for Personal Genome Diagnostics. The other authors declare no competing interests.
Ethics approval Johns Hopkins Hospital Institutional Review Board.
Provenance and peer review Not commissioned; externally peer reviewed.
REFERENCEs
- 1.American Cancer Society. Cancer Facts & Figures. Georgia, USA: American Cancer Society, 2017. [Google Scholar]
- 2.Valsangkar NP, Morales-Oyarvide V, Thayer SP, et al. 851 resected cystic tumors of the pancreas: a 33-year experience at the Massachusetts General Hospital. Surgery 2012;152:S4–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tanaka M, Fernández-Del Castillo C, Kamisawa T, et al. Revisions of international consensus Fukuoka guidelines for the management of IPMN of the pancreas. Pancreatology 2017;17:738–53. [DOI] [PubMed] [Google Scholar]
- 4.Crippa S, Pergolini I, Rubini C, et al. Risk of misdiagnosis and overtreatment in patients with main pancreatic duct dilatation and suspected combined/main-duct intraductal papillary mucinous neoplasms. Surgery 2016;159:1041–9. [DOI] [PubMed] [Google Scholar]
- 5.Vege SS, Ziring B, Jain R, et al. American gastroenterological association institute guideline on the diagnosis and management of asymptomatic neoplastic pancreatic cysts. Gastroenterology 2015;148:819–22. [DOI] [PubMed] [Google Scholar]
- 6.Yu J, Sadakari Y, Shindo K, et al. Digital next-generation sequencing identifies low-abundance mutations in pancreatic juice samples collected from the duodenum of patients with pancreatic cancer and intraductal papillary mucinous neoplasms. Gut 2017;66:1677–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kanda M, Sadakari Y, Borges M, et al. Mutant TP53 in duodenal samples of pancreatic juice from patients with pancreatic cancer or high-grade dysplasia. Clin Gastroenterol Hepatol 2013;11:719–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu J, Matthaei H, Maitra A, et al. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci Transl Med 2011;3:92ra66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017;32:185–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bailey P, Chang DK, Nones K, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016;531:47–52. [DOI] [PubMed] [Google Scholar]
- 11.Witkiewicz AK, McMillan EA, Balaji U, et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 2015;6:6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012;491:399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008;321:1801–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hosoda W, Chianchiano P, Griffin JF, et al. Genetic analyses of isolated high-grade pancreatic intraepithelial neoplasia (HG-PanIN) reveal paucity of alterations in TP53 and SMAD4. J Pathol 2017;242:16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amato E, Molin MD, Mafficini A, et al. Targeted next-generation sequencing of cancer genes dissects the molecular profiles of intraductal papillary neoplasms of the pancreas. J Pathol 2014;233:217–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Murphy SJ, Hart SN, Lima JF, et al. Genetic alterations associated with progression from pancreatic intraepithelial neoplasia to invasive pancreatic tumor. Gastroenterology 2013;145:1098–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu J, Jiao Y, Dal Molin M, et al. Whole-exome sequencing of neoplastic cysts of the pancreas reveals recurrent mutations in components of ubiquitin-dependent pathways. Proc Natl Acad Sci U S A 2011;108:21188–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Springer S, Wang Y, Dal Molin M, et al. A combination of molecular markers and clinical features improve the classification of pancreatic cysts. Gastroenterology 2015;149:1501–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raman SP, Kawamoto S, Blackford A, et al. Histopathologic findings of multifocal pancreatic intraductal papillary mucinous neoplasms on CT. AJR Am J Roentgenol 2013;200:563–9. [DOI] [PubMed] [Google Scholar]
- 20.Pea A, Yu J, Rezaee N, et al. Targeted DNA sequencing reveals patterns of local progression in the pancreatic remnant following resection of Intraductal Papillary Mucinous Neoplasm (IPMN) of the Pancreas. Ann Surg 2017;266:133–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tamura K, Ohtsuka T, Matsunaga T, et al. Assessment of clonality of multisegmental main duct intraductal papillary mucinous neoplasms of the pancreas based on GNAS mutation analysis. Surgery 2015;157:277–84. [DOI] [PubMed] [Google Scholar]
- 22.Yamaguchi K, Kanemitsu S, Hatori T, et al. Pancreatic ductal adenocarcinoma derived from IPMN and pancreatic ductal adenocarcinoma concomitant with IPMN. Pancreas 2011;40:571–80. [DOI] [PubMed] [Google Scholar]
- 23.Ingkakul T, Sadakari Y, Ienaga J, et al. Predictors of the presence of concomitant invasive ductal carcinoma in intraductal papillary mucinous neoplasm of the pancreas. Ann Surg 2010;251:70–5. [DOI] [PubMed] [Google Scholar]
- 24.Tamura K, Ohtsuka T, Date K, et al. Distinction of Invasive Carcinoma Derived From Intraductal Papillary Mucinous Neoplasms From Concomitant Ductal Adenocarcinoma of the Pancreas Using Molecular Biomarkers. Pancreas 2016;45:826–35. [DOI] [PubMed] [Google Scholar]
- 25.Ideno N, Ohtsuka T, Matsunaga T, et al. Clinical significance of GNAS mutation in intraductal papillary mucinous neoplasm of the pancreas with concomitant pancreatic ductal adenocarcinoma. Pancreas 2015;44:311–20. [DOI] [PubMed] [Google Scholar]
- 26.Jones S, Anagnostou V, Lytle K, et al. Personalized genomic analyses for cancer mutation discovery and interpretation. Sci Transl Med 2015;7:283ra53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCall CM, Mosier S, Thiess M, et al. False positives in multiplex PCR-based next-generation sequencing have unique signatures. J Mol Diagn 2014;16:541–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Basturk O, Hong SM, Wood LD, et al. A Revised Classification System and Recommendations From the Baltimore Consensus Meeting for Neoplastic Precursor Lesions in the Pancreas. Am J Surg Pathol 2015;39:1730–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seidel G, Zahurak M, Iacobuzio-Donahue C, et al. Almost all infiltrating colloid carcinomas of the pancreas and periampullary region arise from in situ papillary neoplasms: a study of 39 cases. Am J Surg Pathol 2002;26:56–63. [DOI] [PubMed] [Google Scholar]
- 30.Mino-Kenudson M, Fernández-del Castillo C, Baba Y, et al. Prognosis of invasive intraductal papillary mucinous neoplasm depends on histological and precursor epithelial subtypes. Gut 2011;60:1712–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilentz RE, Su GH, Dai JL, et al. Immunohistochemical labeling for dpc4 mirrors genetic status in pancreatic adenocarcinomas : a new marker of DPC4 inactivation. Am J Pathol 2000;156:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yemelyanova A, Vang R, Kshirsagar M, et al. Immunohistochemical staining patterns of p53 can serve as a surrogate marker for TP53 mutations in ovarian carcinoma: an immunohistochemical and nucleotide sequencing analysis. Mod Pathol 2011;24:1248–53. [DOI] [PubMed] [Google Scholar]
- 33.Schnelldorfer T, Ware AL, Sarr MG, et al. Long-term survival after pancreatoduodenectomy for pancreatic adenocarcinoma: is cure possible? Ann Surg 2008;247:456–62. [DOI] [PubMed] [Google Scholar]
- 34.Fatima J, Schnelldorfer T, Barton J, et al. Pancreatoduodenectomy for ductal adenocarcinoma: implications of positive margin on survival. Arch Surg 2010;145:167–72. [DOI] [PubMed] [Google Scholar]
- 35.Dal Molin M, Zhang M, de Wilde RF, et al. Very Long-term Survival Following Resection for Pancreatic Cancer Is Not Explained by Commonly Mutated Genes: Results of Whole-Exome Sequencing Analysis. Clin Cancer Res 2015;21:1944–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nikiforova MN, Khalid A, Fasanella KE, et al. Integration of KRAS testing in the diagnosis of pancreatic cystic lesions: a clinical experience of 618 pancreatic cysts. Mod Pathol 2013;26:1478–87. [DOI] [PubMed] [Google Scholar]
- 37.Al-Haddad M, DeWitt J, Sherman S, et al. Performance characteristics of molecular (DNA) analysis for the diagnosis of mucinous pancreatic cysts. Gastrointest Endosc 2014;79:79–87. [DOI] [PubMed] [Google Scholar]
- 38.Notta F, Chan-Seng-Yue M, Lemire M, et al. A renewed model of pancreatic cancer evolution based on genomic rearrangement patterns. Nature 2016;538:378–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.