

Abstract
A new method to prepare 1,4-oxazinone intermediates was developed based on aza-conjugate addition of β-amino alcohols to electron-deficient alkyne precursors. A tandem intramolecular cycloaddition/cycloreversion reaction sequence was evaluated, leading to the synthesis of the guaipyridine alkaloid natural products rupestine M and L. Starting from (–)-citronellal and thus a known configuration of the C5 stereocenter, a revised absolute configuration of natural rupestine L is suggested based on optical rotation.
Keywords: 1,4-oxazinones; cycloaddition; cycloreversion; pyridine synthesis; domino reaction; Diels–Alder reaction; natural product synthesis
Graphical Abstract

Guaipyridine sesquiterpene alkaloids are a family of nearly 20 structurally related natural products characterized by a 2-methylpyridine fused to a saturated seven-membered ring (Figure 1).1–4 The cycloheptane ring displays variation in substitution and stereochemistry. The C5 methyl group is found in either absolute configuration; the C8 position presents diverse alkyl, alkenyl, acyl, or carboxyl substituents (e.g., 1–6), as well as hydroxyl substitution as shown in rupestine L and M (7, 8). The relative stereochemical relationship between the C5 and C8 substituents may be syn or anti. The guaipyridine natural product family has both a rich chemical history4 and garners current interest. Cananodine (6) has the most recognized biological activity in the family and shows potent and selective anticancer activity against hepatocytes (IC50 0.94 μM, Hep G2 cells).5 In part due to this bioactivity, 6 and other guaipyridines have attracted both synthetic attention6–12 and renewed isolation efforts.2,3,13,14
Figure 1.

Representative guaipyridine alkaloid natural products
We viewed the guaipyridine scaffold as an instructive model to explore our interest in the de novo synthesis of pyridines by domino reaction processes.15,16 We saw an opportunity to advance new chemistry and explore the reactivity and selectivity of an oxazinone precursor in a cycloaddition/cycloreversion sequence. Our synthesis plan highlighted in Scheme 1 targeted rupestine L and M as our entry point to the guaipyridine alkaloid core. We viewed 8 and ent-7 (the structure enantiomeric to rupestine L reported during isolation) as precursors that could potentially be employed in the construction of other congeners in the family. Additionally, we saw an opportunity for synthesis to confirm (or refute, vide infra) the stereochemical assignment of these diastereomeric natural products which had not previously been pursued as synthetic targets.
Scheme 1.

Synthesis plan toward guaipyridine alkaloids
In our synthesis plan, a Diels–Alder reaction of oxazinone 11 and alkyne 10 would give pyridine 13 after cycloreversion and extrusion of CO2 from the intermediate cycloadduct 12. Subsequent annulation to form the seven-membered ring would ensue to deliver the natural product core. General substructure analysis of the requisite oxazinone precursor 11 revealed that both 11 and 9 are at the same oxidation state and thus potential access might be achieved starting from acetylenedicarboxylate 9. Methods to prepare oxazinones have been summarized elsewhere,15 but the synthesis of oxazinone 11 from 9 was not known and would thus represent a new method to rapidly construct functionalized oxazinone precursors. Moreover, oxazinone 11, which possesses a functional group (CO2R) at the position that corresponds to the oxygenated C8 position, could enable construction of target natural products 7 and 8.
To put the plan into practice, 2-amino-1,3-propanediol (15) was combined with dimethyl acetylenedicarboxylate (DMAD, 14) (Scheme 2). Aza-conjugate addition followed by lactonization gave an intermediate hydroxy lactone, which was acylated to give the illustrated product 16. On small scale (<1 g), this compound was purified by chromatography on silica gel (80% yield); on larger scale, we found it more expedient to perform a recrystallization. In this way, a 51% yield (13.7 g) of 16 was afforded in the first crop of crystals.
Scheme 2.
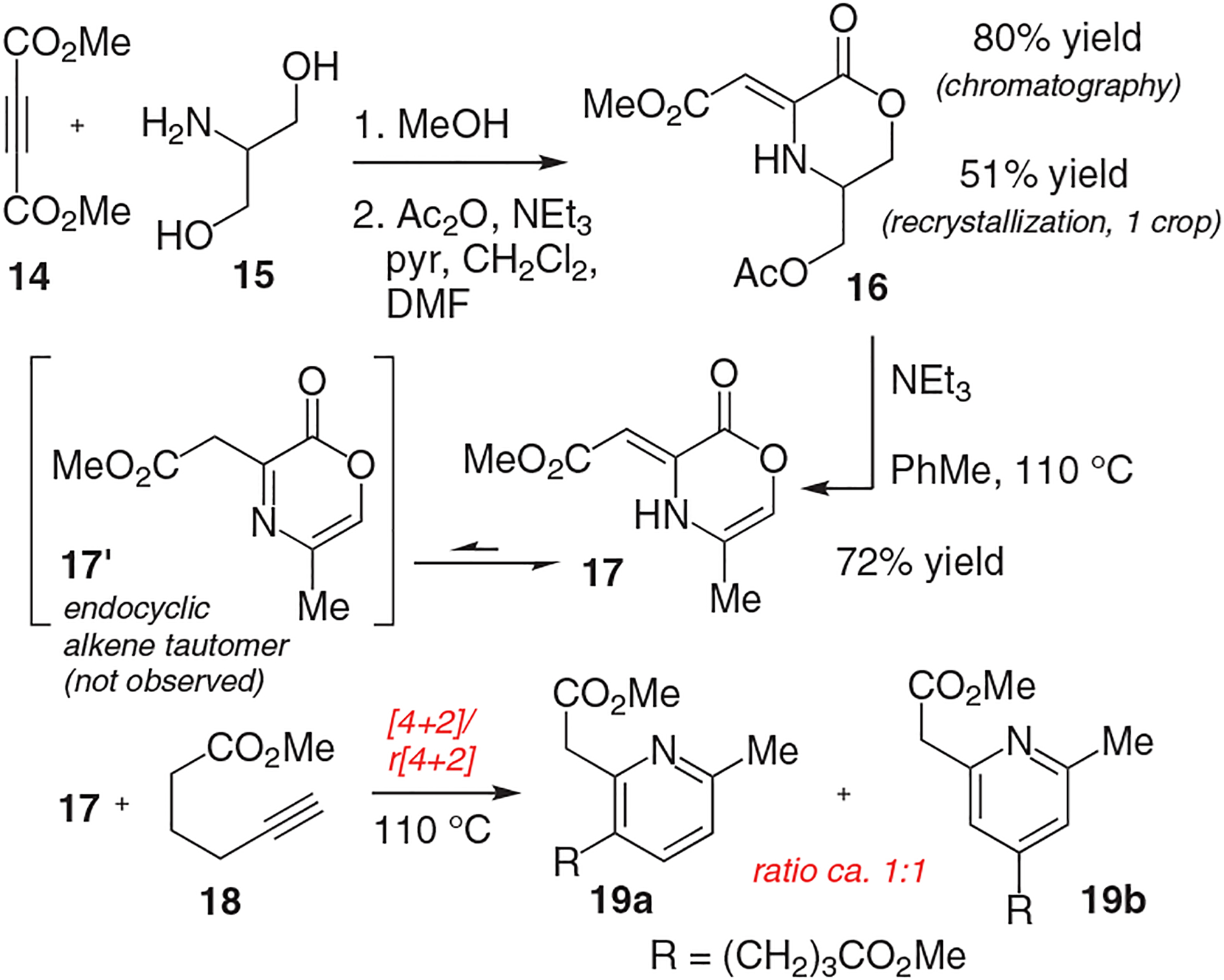
Synthesis of oxazinone precursor and evaluation of the intermolecular [4+2]/retro[4+2] sequence with a model alkyne
Elimination of the acetate in 16 and isomerization to oxazinone 17 was accomplished with NEt3 in toluene at elevated temperatures (110 °C). Spectroscopic analysis of oxazinone 17 revealed that the C3 alkene existed in the exocyclic (vinylogous urethane) form; none of the endocyclic isomer 17′ was apparent. Based on our experience with reactive oxazinone precursors,15 we were unsure if 17 would engage in a productive cycloaddition/cycloreversion sequence. Although somewhat less reactive than other oxazinones we have worked with that reside in endocyclic tautomeric forms primed for cycloaddition, we found that 17 reacted with a model terminal alkyne, 5-hexynoate 18, and gave both the 2,3- and 2,4-pyridine products 19a and 19b (ratio 1:1). Exploration of different reaction conditions including Lewis acid additives failed to improve selection for the desired 2,3-pyridine isomer 19a. In light of these efforts, we elected to pursue a modified synthesis strategy where an analogous intramolecular [4+2] cycloaddition was planned, thereby avoiding selectivity concerns of the intermolecular sequence.
To evaluate the viability of the intramolecular sequence, an achiral precursor, 5-hexynal (20), was prepared17 and reacted with the derived lithium acetylide of methyl propiolate (Scheme 3). The resulting intermediate propargyl alcohol adduct was isolated cleanly but in modest yield (40%) due to competitive degradation during chromatography on silica gel. Oxidation with Dess–Martin periodinane gave the highly sensitive acetylenic ketone 21, which was used directly in the subsequent nucleophilic addition of aminopropanediol 15. The amine addition was selective favoring conjugate addition beta to the more electrophilic ketone in preference to addition beta to the ester moiety. As with addition of 15 to DMAD (Scheme 2), spontaneous lactonization was observed and, as before, we found it more practical to isolate and purify the derived acylation product 22. Overall, product 22 was obtained in 71% yield in three steps from the intermediate propargylic alcohol.
Scheme 3.

Intramolecular cycloaddition/cycloreversion pathway toward guaipyridine alkaloids
Using similar conditions to that previously described (NEt3, Δ), the subsequent elimination of acetate from 22 to the Diels–Alder-reactive oxazinone 23 was slower than with the analogous ester-derived substrate 17; higher temperatures were required to promote the reaction at a comparable rate. The intermediate oxazinone 23 was isolable as a minor component under these reaction conditions, but at the temperatures necessary for elimination, intermediate 23 also competitively underwent the [4+2]/retro[4+2] sequence. In practice, heating a solution of 22 for 48 hours with NEt3 in refluxing chlorobenzene (bp 132 °C) gave the bicyclic pyridine 24 in 43% isolated yield. Production of pyridine 24 in this way validated the synthetic sequence with achiral material and gave us confidence to proceed with an analogous chiral variant that could lead to guaipyridine alkaloid structures bearing the C5 methyl group.
Following established chemistry from the Ma18 and Shenvi19 labs, the requisite (S)-4-methylhex-5-ynal was prepared in three steps from (–)-citronellal, a widely available chiral pool reagent. The analogous acetylide addition and oxidation to the acetylenic ketone 26 preceded aza-conjugate addition and lactonization. The sequence was terminated with acylation to provide the domino reaction precursor 27. This sequence (25 to 27) proceeded with little variation from the model route (20 to 22). The key domino sequence comprising acetate elimination, alkene isomerization, cycloaddition, and cycloreversion was observed on heating in chlorobenzene at reflux (132 °C) with NEt3. Compound 27 was thus converted into the guaipyridine core 29 in more favorable efficiency (63% yield) compared with the model route that intercepted achiral material. Finally, reduction of the resulting ketone in 29 with DIBAL-H gave nearly equal amounts (ca. 1:1 as judged by 1H NMR of the unpurified material) of rupestine M (8) and (ent)-7, the enantiomeric structure of rupestine L that was isolated from the natural source. Spectroscopic data for 8 are in good agreement with those provided by the isolation group and the optical rotation of 8 was similar [synthetic [α]D23 −24.8 (c 0.4, MeOH); Lit.3 [α]D20 −38 (c 0.05, MeOH)].
We saw agreement between our 1H and 13C NMR data for the synthetic sample of 7 with the literature.3 We anticipated that the optical rotation of synthetic material would be opposite to that reported by the isolation group; however, our synthetic material 7 showed similar sign and magnitude [synthetic [α]D23 −32.2 (c 0.4, MeOH); Lit.3 [α]D20 −40 (c 0.03, MeOH)]. Because our synthesis started from a known chiral pool reagent [(–)-citronellal], our results suggest a revision of the absolute configuration of rupestine L (7) to that shown in Scheme 3. The assignment of natural rupestine L as (5S,8R) was made based on comparison of calculated and experimental CD spectra.3 Our data from synthetic material support the structural reassignment of rupestine L as (5S,8R). A table comparing NMR spectroscopic data for natural and synthetic rupestine M and L is provided in the Supporting Information.
In summary, a new method to prepare oxazinone precursors was established using electron-deficient alkyne substrates including dimethyl acetylenedicarboxylate and 4-keto ynoate starting materials. Oxazinone materials thus prepared were reactive in both intermolecular and intramolecular manifolds. The latter intramolecular pathway was employed in a domino reaction sequence whereby elimination, alkene isomerization, and [4+2]/retro[4+2] cycloaddition led to the formation of the fused 6/7 guaipyridine alkaloid core. In this way, a synthesis of rupestine M and rupestine L was achieved in eight steps starting from (–)-citronellal. Lastly, we suggest a revision to the absolute stereochemistry proposed during isolation. Our data support revision of rupestine L to the (5S,8R)-configuration.
All reactions were carried out under an atmosphere of nitrogen in flame-dried or oven-dried glassware with magnetic stirring unless otherwise indicated. THF and toluene were degassed with argon and purified by passage through a column of molecular sieves and a bed of activated alumina. Dichloromethane was distilled from CaH2 prior to use. All reagents were used as received. Flash column chromatography was performed using P60 silica gel (230–400 mesh). Analytical TLC was performed on SiliCycle 60 Å glass plates. Visualization was accomplished with UV light, anisaldehyde, ceric ammonium molybdate (CAM), or potassium permanganate, followed by heating. Optical rotations were acquired using a Perkin Elmer 341 polarimeter. IR spectra were recorded using a Digilab FTS 7000 FTIR spectrophotometer. 1H NMR spectra were recorded on a Varian Mercury 400 (400 MHz) spectrometer and are reported in ppm using solvent (CDCl3 at 7.26 ppm) or tetramethylsilane (0.00 ppm) as an internal standard. Proton-decoupled 13C NMR spectra are reported using solvent as an internal standard (CDCl3 at 77.00 ppm). All compounds were judged to be homogeneous (>95% purity) by 1H and 13C NMR spectroscopy unless otherwise noted. Mass spectra data analyses were obtained through positive electrospray ionization (with NaCl) on a Bruker 12 Tesla APEX-Qe FTICR-MS with an Apollo II ion source using an ICR ion trap mass analyzer.
Methyl (Z)-2-(5-(Acetoxymethyl)-2-oxomorpholin-3-ylidene)acetate (16)
A dry flask charged with 2-amino-1,3-propanediol (15; 10.15 g, 111.5 mmol) dissolved in MeOH (250 mL) was flushed with N2 for 10 min. Dimethyl acetylenedicarboxylate (14; 13.7 mL, 111.5 mmol) was added dropwise at rt over 15 min. The reaction was stirred at rt for 1 h and concentrated in vacuo. The resulting residue was resuspended in CH2Cl2 (100 mL) and DMF (5 mL), and cooled to 0 °C. NEt3 (18 mL, 134 mmol) and pyridine (10.8 mL, 134 mmol) were added and the mixture was stirred until solids dissolved (ca. 10 min). Ac2O (12.6 mL, 134 mmol) was added in one portion and the resulting mixture was allowed to warm to rt over 24 h. The reaction mixture was acidified with 1 M HCl (300 mL) and extracted with CH2Cl2 (3 × 50 mL). The combined organic layers were washed with sat. NaHCO3 and brine, dried over Na2SO4, filtered, and concentrated. The resulting residue (29.4 g) was recrystallized by dissolving in hot EtOAc (30 mL), then dilution with hexanes, followed by slow cooling to rt and further at 4 °C overnight. The crystals were collected and washed with a 4:1 mix of hexanes and EtOAc to provide the desired acetate product (13.7 g, 56.3 mmol, 51% yield) as a light pink solid.
IR (ATR): 3302, 1732, 1668, 1608, 1219 cm−1.
1H NMR (400 MHz, CDCl3): δ = 8.49 (broad s, 1 H), 5.69 (s, 1 H), 4.54 (dd, J = 11.0, 3.1 Hz, 1 H), 4.43 (dd, J = 5.1, 1.2 Hz, 1 H), 4.39 (apparent d, J = 5.48 Hz, 2 H), 3.81 (m, 1 H), 3.70 (s, 3 H), 2.10 (s, 3 H).
13C NMR (100 MHz, CDCl3): δ = 170.4, 170.1, 159.8, 143.2, 91.3, 67.9, 62.9, 51.1, 49.3, 20.6.
HRMS (ESI): m/z [M + Na]+ calcd for C10H13NO6Na+: 266.06351; found: 266.06354.
Methyl (Z)-2-(5-Methyl-2-oxo-2H-1,4-oxazin-3(4H)-ylidene)acetate (17)
A dry flask fitted with a condenser and charged with 16 (1.585 g, 6.58 mmol) dissolved in toluene (25 mL) was flushed with N2. NEt3 (3.67 mL, 26.3 mmol) was added and the reaction was heated to reflux (110 °C) using an aluminum block set to an external temperature of 130 °C. After being heated for 24 h, the reaction mixture was cooled to rt, diluted with sat. aq NH4Cl (50 mL), and extracted with EtOAc (3 × 25 mL). The combined organic layers were washed with sat. aq NaHCO3 and brine, dried (Na2SO4), filtered, and concentrated. The resulting mixture was purified by flash column chromatography on silica gel (gradient elution: 5% to 45% EtOAc in hexanes) to afford the desired product 17 (0.872 g, 4.77 mmol, 72% yield) as a yellow solid.
IR (ATR): 3256, 2959, 2852, 1744, 1651, 1629, 1173, 1132 cm−1.
1H NMR (400 MHz, CDCl3): δ = 9.98 (br s, 1 H), 6.22 (t, J = 0.8 Hz, 1 H), 5.81 (s, 1 H), 3.75 (s, 3 H), 1.93 (d, J = 0.8 Hz, 3 H).
13C NMR (100 MHz, CDCl3): δ = 170.4, 156.8, 139.3, 122.0, 118.2, 88.6, 51.2, 14.0.
HRMS (ESI): m/z [M + Na]+ calcd for C8H9NO4Na+: 206.04238; found: 206.04243.
Methyl 4-Hydroxynona-2,8-diynoate
A dry flask was flushed with N2 and charged with methyl propiolate (0.73 mL, 7.03 mmol) dissolved in THF (16 mL). The reaction mixture was cooled to −78 °C and stirred for 15 min prior to careful dropwise addition of 1 M LiHMDS in hexane (7.03 mL, 7.03 mmol) down the side of the flask over 15 min. The reaction was held at −78 °C for 30 min. A separate flask was charged with hex-5-ynal (20; 614 mg, 6.39 mmol) dissolved in THF (16 mL). The hex-5-ynal solution was added to the propiolate solution dropwise over 15 min via syringe. The reaction was stirred for 30 min at −78 °C and quenched with sat. NH4Cl (75 mL). After warming to rt, the mixture was extracted with Et2O (3 × 20 mL). The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The resulting residue was purified by flash column chromatography on silica gel (gradient elution: 5% to 40% EtOAc in hexanes) to afford the desired propargylic alcohol product (0.455 g, 2.55 mmol, 40% yield) as a yellow oil.
IR (ATR): 3290, 2951, 1712, 1668, 1209, 1037, 636 cm−1.
1H NMR (400 MHz, CDCl3): δ = 4.55 (apparent q, J = 5.9 Hz, 1 H), 3.79 (s, 3 H), 2.27 (td, J = 2.3, 6.7 Hz, 2 H), 1.98 (t, J = 2.7 Hz, 1 H), 1.92 (m, 2 H), 1.73 (t, J = 7.8 Hz, 2 H).
13C NMR (100 MHz, CDCl3): δ = 153.7, 87.7, 83.5, 76.3, 69.0, 61.6, 52.9, 35.6, 23.7, 18.0.
HRMS (ESI): m/z [2 M + Na]+ calcd for [C10H12O3]2Na+: 383.14651; found: 383.14647.
((Z)-6-Oxo-5-(2-oxohept-6-yn-1-ylidene)morpholin-3-yl)methyl Acetate (22)
To a dry flask was added methyl 4-hydroxynona-2,8-diynoate (153 mg, 0.85 mmol) and dissolved in CH2Cl2 (5.5 mL). Dess–Martin periodinane (0.424 g, 1.0 mmol) was added in one lot and the reaction was stirred for 2 h at rt under N2. The reaction mixture was diluted with sat. NaHCO3 (20 mL), filtered, and the filter pad was washed with CH2Cl2 (10 mL). The filtrate was transferred to a separatory funnel, the organic layer was removed, and the aqueous portion was extracted with additional CH2Cl2 (3 × 10 mL). The organic layers were combined, dried over MgSO4, filtered, and concentrated in vacuo. The resulting residue was dissolved in MeOH (4.3 mL). The reaction flask was cooled to −20 °C. In a separate flask, a solution of 2-amino-1,3-propanediol (15; 78 mg, 0.86 mmol) in MeOH (5 mL) was prepared at rt. The aminopropanediol solution was added to the reaction mixture which was stirred with slow warming to 0 °C over 3 h. The reaction mixture was concentrated and the residue resuspended in CH2Cl2 (4.3 mL) and DMF (0.86 mL), and cooled to 0 °C. NEt3 (0.35 mL, 2.58 mmol), pyridine (0.21 mL, 2.58 mmol), and Ac2O (0.24 mL, 2.58 mmol) were added to the reaction mixture. The reaction was allowed to warm to rt over 20 h, diluted with 1 M HCl (20 mL), and extracted with CH2Cl2 (3 × 20 mL). The combined organic layers were washed with sat. NaHCO3 and brine, dried over Na2SO4, filtered, and concentrated. The resulting residue was purified by flash column chromatography on silica gel (gradient elution: 15% to 70% EtOAc in hexanes) to yield compound 22 (168 mg, 0.60 mmol, 71% yield) as a yellow oil.
IR (ATR): 3285, 2951, 1736, 1630, 1580, 1463, 1038, 644 cm−1.
1H NMR (400 MHz, CDCl3): δ = 10.20 (br s, 1 H), 6.11 (s, 1 H), 4.57 (dd, J = 3.5, 11.3 Hz, 1 H), 4.46 (dd, J = 5.2, 11.3 Hz, 1 H), 4.26–4.19 (m, 2 H), 3.84 (m, 1 H), 2.60 (t, J = 7.4 Hz, 2 H), 2.25 (td, J = 2.7, 7.0 Hz, 2 H), 2.12 (s, 3 H), 1.97 (t, J = 2.0 Hz, 1 H), 1.86 (t, J = 7.4 Hz, 2 H).
13C NMR (100 MHz, CDCl3): δ = 201.8, 170.3, 160.0, 142.5, 98.6, 83.6, 69.0, 67.9, 62.8, 46.8, 41.6, 23.6, 20.6, 18.0.
HRMS (ESI): m/z [M + Na]+ calcd for C14H18NO5: 280.11795; found: 280.11848.
2-Methyl-5,6,7,9-tetrahydro-8H-cyclohepta[b]pyridin-8-one (24)
A dry 3-dram vial was charged with 22 (147 mg, 0.526 mmol) dissolved in PhCl (10.5 mL; 0.05 M). NEt3 (0.29 mL, 2.105 mmol) was added and the vial was evacuated, backfilled with N2, and sealed with a Teflon cap. The reaction was heated to 132 °C for 48 h, cooled to rt, and concentrated. The resulting residue was purified by flash column chromatography on silica gel (gradient elution: 10% to 80% EtOAc in hexanes). The resulting pyridine 24 (39 mg, 0.225 mmol, 43% yield) was obtained as a yellow oil.
IR (ATR): 2936, 1742, 1705, 1634, 1576, 1435, 1039 cm−1.
1H NMR (400 MHz, CDCl3): δ = 7.34 (d, J = 7.8 Hz, 1 H), 7.00 (d, J = 7.8 Hz, 1 H), 3.98 (s, 2 H), 2.88 (t, J = 6.3 Hz, 2 H), 2.57 (t, J = 7.0 Hz, 2 H), 2.45 (s, 3 H), 1.97 (m, 2 H).
13C NMR (100 MHz, CDCl3): δ = 208.0, 156.3, 153.4, 137.2, 131.9, 122.0, 53.1, 42.7, 31.1, 25.9, 23.9.
HRMS (ESI): m/z [2 M + Na]+ calcd for [C11H13NO]2Na+: 373.18865; found: 373.18918.
Methyl (7S)-4-Hydroxy-7-methylnona-2,8-diynoate
A dry flask was charged with methyl propiolate (0.76 mL, 8.5 mmol) and flushed with N2. After the propiolate was dissolved in THF (30 mL), the reaction vessel was cooled to −78 °C and stirred for 15 min prior to careful dropwise addition of 1.37 M n-BuLi in hexane (6.16 mL, 8.5 mmol) dropwise down the side of the flask over 15 min. The reaction was stirred an additional 10 min prior to dropwise addition of a solution of (S)-4-methylhex-5-ynal19 (0.75 g, 6.8 mmol) in THF (10 mL) over 10 min. The reaction was stirred for 15 min at −78 °C, then quenched with sat. NH4Cl (10 mL), warmed to rt, diluted with H2O (50 mL), and extracted with Et2O (3 × 20 mL). The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The resulting residue was purified by flash column chromatography on silica gel (gradient elution: 10% to 30% EtOAc in hexanes) to afford methyl (7S)-4-hydroxy-7-methylnona-2,8-diynoate (0.61 g, 3.1 mmol, 46% yield) as a yellow oil.
[α]D23 +17.4 (c 2.25, CH2Cl2).
Rf = 0.23 (20% EtOAc in hexanes; KMnO4).
IR (ATR): 3400, 2955, 2874, 1713, 1435, 1250, 1020, 635 cm−1.
1H NMR (400 MHz, CDCl3): δ = 4.55 (t, J = 1.4 Hz, 1 H), 3.79 (s, 3 H), 2.48 (m, 1 H), 2.07 (s, 1 H), 1.99 (m, 1 H), 1.89 (m, 1 H), 1.62 (m, 2 H), 1.21 (m, 3 H).
13C NMR (100 MHz, CDCl3): δ = 153.8, 153.7, 88.0, 87.8, 87.7, 83.7, 69.0, 61.9, 61.8, 61.7, 52.8, 34.5, 31.9, 31.7, 25.4, 25.3, 20.9.
HRMS (ESI): m/z [M + Na]+ calcd for C11H14O3Na+: 217.08352; found: 217.08384.
((Z)-5-((S)-5-Methyl-2-oxohept-6-yn-1-ylidene)-6-oxomorpholin-3-yl)methyl Acetate (27)
A dry flask was charged with methyl (7S)-4-hydroxy-7-methylnona-2,8-diynoate (166 mg, 0.85 mmol) dissolved in CH2Cl2 (5.0 mL). Dess–Martin periodinane (0.424 g, 1.0 mmol) was added in one lot and the reaction was stirred for 2 h at rt under N2. The reaction mixture was diluted with sat. NaHCO3 (20 mL), filtered, and the filter pad was washed with CH2Cl2 (10 mL). The filtrate was transferred to a separatory funnel, the organic layer was removed, and the aqueous portion was extracted with additional CH2Cl2 (3 × 10 mL). The organic layers were combined, dried over MgSO4, filtered, and concentrated in vacuo. The resulting residue was dissolved in MeOH (5 mL) and cooled to −20 °C. In a separate flask, aminopropanediol 15 (77 mg, 0.85 mmol) was dissolved in MeOH (1.2 mL) at rt. The aminopropanediol solution was added dropwise to the reaction mixture. After being stirred at −20 °C for 2 h, the reaction mixture was concentrated, the resulting residue was resuspended in CH2Cl2 (4 mL) and DMF (1 mL), and the reaction vessel was cooled to 0 °C. NEt3 (0.34 mL, 2.43 mmol), pyridine (0.20 mL, 2.43 mmol), and Ac2O (0.23 mL, 2.43 mmol) were added. The reaction was allowed to warm to rt over 19 h before dilution with 1 M HCl (10 mL) and extraction with CH2Cl2 (3 × 5 mL). The combined organic layers were washed with sat. NaHCO3 and brine, dried over Na2SO4, filtered, and concentrated in vacuo. The resulting residue was purified by flash column chromatography on silica gel (gradient elution: 15% to 70% EtOAc in hexanes) to yield compound 27 (186 mg, 0.66 mmol, 77% yield) as a yellow gum.
[α]D23 +16.5 (c 2.3, CH2Cl2).
Rf = 0.38 (40% EtOAc in hexanes; CAM).
IR (ATR): 3285, 2951, 1736, 1630, 1038, 644, 604 cm−1.
1H NMR (400 MHz, CDCl3): δ = 10.19 (s, 1 H), 6.12 (s, 1 H), 4.57 (dd, J = 3.5, 11.3 Hz, 1 H), 4.45 (dd, J = 1.2, 5.0 Hz, 1 H), 4.26–4.19 (m, 2 H), 3.84 (m, 1 H), 2.68–2.60 (m, 2 H), 2.50–2.48 (m, 1 H), 2.11 (s, 3 H), 2.07 (d, J = 6.0 Hz, 1 H), 1.81 (m, 1 H), 1.70 (m, 1 H), 1.21 (d, J = 7.0 Hz, 3 H).
13C NMR (100 MHz, CDCl3): δ = 202.1, 170.3, 160.1, 142.5, 98.7, 88.0, 69.0, 67.9, 62.8, 46.8, 40.7, 31.6, 25.3, 20.9, 20.6.
HRMS (ESI): m/z [M + Na]+ calcd for C15H19NO5Na+: 316.11554; found: 316.11555.
(S)-2,5-Dimethyl-5,6,7,9-tetrahydro-8H-cyclohepta[b]pyridin-8-one (29)
A dry 25-mL flask was charged with 27 (75 mg, 0.25 mmol) dissolved in PhCl (4.0 mL; 0.05 M). NEt3 (0.14 mL, 1.0 mmol) was added, the flask was fitted with a condenser, and the reaction vessel was evacuated and backfilled with N2. The reaction was heated to reflux (132 °C) using an aluminum block. After 48 h, the reaction was cooled to rt, concentrated, and the resulting residue was purified by flash column chromatography on silica gel (gradient elution: 15% to 80% EtOAc in hexanes) to afford pyridine 29 (30 mg, 0.16 mmol, 63% yield) as a yellow oil.
[α]D23 +16.6 (c 1.3, CH2Cl2).
Rf = 0.35 (40% EtOAc in hexanes; KMnO4).
IR (film): 2963, 1742, 1709, 1663, 1221, 1035, 656 cm−1.
1H NMR (400 MHz, CDCl3): δ = 7.47 (d, J = 8.2 Hz, 1 H), 7.09 (d, J = 7.8 Hz, 1 H), 4.05 (d, J = 18.4 Hz, 1 H), 3.85 (d, J = 18.0 Hz, 1 H), 3.10 (m, 1 H), 2.53 (s, 3 H), 2.49 (dd, J = 3.1, 7.4 Hz, 1 H), 2.38 (ddd, J = 2.7, 3.9, 12.1 Hz, 1 H), 2.15 (m, 1 H), 1.61 (m, 1 H), 1.39 (d, J = 6.7 Hz, 3 H).
13C NMR (100 MHz, CDCl3): δ = 209.3, 155.7, 153.9, 135.0, 133.4, 122.2, 52.5, 40.9, 34.0, 33.1, 23.8, 18.9.
HRMS (ESI): m/z [2 M + Na]+ calcd for [C12H15NO]2Na+: 401.21995; found: 401.22052.
Rupestine M (8) and Rupestine L (7)
Pyridine 29 (20 mg, 0.106 mmol) was added to a 3-dram vial, dissolved in toluene (1.5 mL), and cooled to −78 °C. A solution of DIBAL-H in toluene (0.12 mL, 25% w/v) was added dropwise and the reaction was stirred at −78 °C for 40 min. The reaction was quenched with sat. aq Rochelle salt solution (2 mL) and stirred overnight. The resulting clear biphasic solution was diluted with H2O (5 mL), transferred to a separatory funnel, and extracted with CH2Cl2 (3 × 5 mL). The combined organic layers were dried over Na2SO4, filtered, and concentrated in vacuo. 1H NMR analysis of the unpurified mixture revealed the presence of 8 and 7 in nearly equal amounts (ratio 55:45). The mixture was purified by flash column chromatography on silica gel (gradient elution: 0% to 5% MeOH in EtOAc) to afford 8 (8 mg, 40% yield) and 7 (5 mg, 27% yield), both as a light yellow oils. NMR spectroscopic data for 8 and 7 agree with literature values3 (see the Supporting Information for more detail and a table comparing natural and synthetic material).
New data for 8 (rupestine M):
[α]D23 −24.8 (c 0.4, MeOH) [Lit.3 [α]D20 −38 (c 0.05, MeOH)].
Rf = 0.30 (5% MeOH in EtOAc; UV, KMnO4).
New data for 7 (rupestine L, revised structure):
[α]D23 −32.2 (c 0.4, MeOH) [Lit.3 [α]D20 −40 (c 0.03, MeOH)].
Rf = 0.35 (5% MeOH in EtOAc; UV, KMnO4).
Supplementary Material
Acknowledgment
In this memorial issue, David A. Evans (1941-2022) is acknowledged both for his direct support during the PhD studies of J.R.S. (2001-2006) and his indirect support and continued influence that endures to the present.
Funding Information
This work was supported by the National Institutes of Health (National Institute of General Medical Sciences, R15GM107702 to J.R.S.).
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supporting Information
Supporting information for this article is available online at https://doi.org/10.1055/s-0042-1751413.
References
- (1).Su Z; Wu H; Yang Y; Aisa HA; Slukhan U; Aripova SJ Sep. Sci 2008, 31, 2161. [DOI] [PubMed] [Google Scholar]
- (2).Su Z; Wu H-K; He F; Slukhan U; Aisa HA Helv. Chim. Acta 2010, 93, 33. [Google Scholar]
- (3).He F; Nugroho AE; Wong CP; Hirasawa Y; Shirota O; Morita H; Aisa HA Chem. Pharm. Bull 2012, 60, 213. [DOI] [PubMed] [Google Scholar]
- (4).Büchi G; Goldman IM; Mayo DW J. Am. Chem. Soc 1966, 88, 3109. [Google Scholar]
- (5).Hsieh T-J; Chang F-R; Chia Y-C; Chen C-Y; Chiu H-F; Wu Y-CJ Nat. Prod 2001, 64, 616. [DOI] [PubMed] [Google Scholar]
- (6).Craig D; Henry GD Eur. J. Org. Chem 2006, 3558. [Google Scholar]
- (7).Shelton PMM; Grosslight SM; Mulligan BJ; Spargo HV; Saad SS; Vyvyan JR Tetrahedron 2020, 76, 131500. [Google Scholar]
- (8).Shelton P; Ligon TJ; Dell (née Meyer) JM; Yarbrough L; Vyvyan JR Tetrahedron Lett. 2017, 58, 3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Starchman ES; Marshall MS; Vyvyan JR Tetrahedron Lett. 2020, 61, 151837. [Google Scholar]
- (10).Aibibula P; Yusuf A; Zhao J; Wang B; Huang G; Aisa HA Chem. Pap 2021, 75, 5599. [Google Scholar]
- (11).Yusuf A; Zhao J; Wang B; Aibibula P; Aisa HA; Huang GR Soc. Open Sci 2018, 5, 172037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zhang C; Wang B; Aibibula P; Zhao J; Aisa HA Org. Biomol. Chem 2021, 19, 7081. [DOI] [PubMed] [Google Scholar]
- (13).Luo X; Wu R; Han X; Tang X; Wang Q; Li P; Li G RSC Adv. 2022, 12, 2662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Clery RA; Cason JRL; Zelenay VJ Agric. Food Chem 2016, 64, 4566. [DOI] [PubMed] [Google Scholar]
- (15).Carrillo Vallejo NA; Scheerer JR J. Org. Chem 2021, 86, 5863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Williamson JB; Smith ER; Scheerer JR Synlett 2017, 28, 1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Margrey KA; Chinn AJ; Laws SW; Pike RD; Scheerer JR Org. Lett 2012, 14, 2458. [DOI] [PubMed] [Google Scholar]
- (18).Zhou Q; Chen X; Ma D Angew. Chem. Int. Ed 2010, 49, 3513. [DOI] [PubMed] [Google Scholar]
- (19).Lu H-H; Martinez MD; Shenvi RA Nat. Chem 2015, 7, 604. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.