

Abstract
PURPOSE
Diagnosis of Mismatch Repair Deficiency (MMRD) is crucial for tumor management and early detection in patients with the cancer predisposition syndrome constitutional mismatch repair deficiency (CMMRD). Current diagnostic tools are cumbersome and inconsistent both in childhood cancers and in determining germline MMRD.
PATIENTS AND METHODS
We developed and analyzed a functional Low-pass Genomic Instability Characterization (LOGIC) assay to detect MMRD. The diagnostic performance of LOGIC was compared with that of current established assays including tumor mutational burden, immunohistochemistry, and the microsatellite instability panel. LOGIC was then applied to various normal tissues of patients with CMMRD with comprehensive clinical data including age of cancer presentation.
RESULTS
Overall, LOGIC was 100% sensitive and specific in detecting MMRD in childhood cancers (N = 376). It was more sensitive than the microsatellite instability panel (14%, P = 4.3 × 10−12), immunohistochemistry (86%, P = 4.6 × 10−3), or tumor mutational burden (80%, P = 9.1 × 10−4). LOGIC was able to distinguish CMMRD from other cancer predisposition syndromes using blood and saliva DNA (P < .0001, n = 277). In normal cells, MMRDness scores differed between tissues (GI > blood > brain), increased over time in the same individual, and revealed genotype-phenotype associations within the mismatch repair genes. Importantly, increased MMRDness score was associated with younger age of first cancer presentation in individuals with CMMRD (P = 2.2 × 10−5).
CONCLUSION
LOGIC was a robust tool for the diagnosis of MMRD in multiple cancer types and in normal tissues. LOGIC may inform therapeutic cancer decisions, provide rapid diagnosis of germline MMRD, and support tailored surveillance for individuals with CMMRD.
INTRODUCTION
Inactivation of the mismatch repair (MMR) and/or polymerase proofreading mechanisms lead to a rapid accumulation of genomic mutations during replication, resulting in hypermutant, treatment-resistant tumors.1,2 At the same time, hypermutation renders these cancers sensitive to immune checkpoint blockade.3 Mismatch Repair Deficiency (MMRD) and/or Polymerase Proofreading Deficiency (PPD) can originate somatically in the tumor itself and is common in multiple cancer types.2,4-6 Their detection in these settings is essential to tailor the therapeutic approach for these patients. Alternatively, germline mutations in either MMR or DNA-polymerase genes lead to cancer predisposition and cancers at younger ages.1-3,7-11 Germline biallelic inactivation of one of the four MMR genes (MSH2, MSH6, MLH1, and PMS2), termed Constitutional Mismatch Repair Deficiency (CMMRD),10,12 results in an extremely aggressive cancer predisposition syndrome in which carriers are affected with cancer in early life and commonly succumb to brain, GI, and hematopoietic malignancies. Both immunotherapy and early cancer detection through a surveillance protocol have recently been shown to improve survival for patients with CMMRD.3,9 Therefore, a robust and affordable tool for MMRD detection in cancer and normal cells can enable both precision cancer therapy and genetic counseling for the patient and their family members.
CONTEXT
Key Objective
Mismatch repair deficiency (MMRD) caused by somatic or germline mutations leads to aggressive cancers that are difficult to treat with conventional chemotherapies. Accurately and efficiently diagnosing somatic and/or germline MMRD is a key challenge in placing patients on life-saving surveillance programs and therapeutic treatments. This study reports the development and, to our knowledge, first use of a new tool, termed low-pass genomic instability characterization (LOGIC), that uses low-pass whole-genome sequencing to detect microsatellite instability and diagnose germline MMRD in a large cohort of pediatric cases.
Knowledge Gathered
LOGIC is an inexpensive method, which achieved 100% diagnostic accuracy in our cohort of patients with genetically confirmed MMRD and revealed important genotype-phenotype associations between different mutations within the mismatch repair genes.
Relevance
LOGIC effectively and rapidly detects MMRD in both the tumor and germline, which can be used to inform important clinical decisions for patient surveillance and treatment.
Diagnosis of MMRD has traditionally relied on established clinical criteria11 and genetic testing, accompanied by functional assays such as immunohistochemistry of the four MMR genes and microsatellite instability (MSI) assays.8 Unfortunately, the clinical criteria are not sufficient to diagnose CMMRD, and both the immunohistochemistry and traditional MSI assays lack robustness, especially when only normal tissues or non-GI cancers are screened.8,13 Furthermore, genetic testing is complicated by multiple variants of uncertain clinical significance (VUS) in the MMR genes and by pseudogenes of PMS2, resulting in difficulty in diagnosing patients, which subsequently delays the implementation of surveillance and therapy.10,14
Recently, several functional genomic assays were developed to detect MMRD and PPD by their characteristic mutational patterns and signatures. Tumor mutational burden (TMB) has been applied to detect hypermutation and the specific MMR-related or polymerase defect–related COSMIC signatures.1,15 However, TMB is not specific to these mechanisms, as many others such as smoking, UV light, and previous chemotherapy result in hypermutation in multiple cancer types.1,16-18 Furthermore, some MMR-deficient cancers do not reach the hypermutation threshold1 and signatures cannot be detected when TMB is low.13
Accumulation of microsatellite indels or instability (MSI) is a unique characteristic of MMR‐deficient cancers and has conventionally been used to diagnose such tumors.19-21 Several MSI-based diagnostic tests exist, such as the Microsatellite Instability Analysis System (MIAS, Promega, WI), which is a five microsatellite loci (MS loci) PCR-based panel widely used to detect MMRD.22 However, these five loci are not sufficient to detect MSI in nonepithelial tissues, such as brain and hematopoietic malignancies, even in the context of CMMRD.13 Larger numbers of microsatellites (24-277 loci) may achieve better sensitivity and specificity.23,24 However, data are still limited regarding the efficacy of these assays in cancers and normal tissues. We previously measured MSI across the genome and used it to characterize unique MS indel signatures that can accurately identify MMRD and PPD in tumors.13 Since the human genome harbors > 23 million microsatellites, we developed a Low-pass genomic instability characterization (LOGIC) assay to test the diagnostic role of low-coverage genome sequencing in cancerous and normal tissues.
PATIENTS AND METHODS
All patients with CMMRD and MMR-proficient (MMRP)/wildtype genotypes (Fig 1) were consented with written consent and registered into the International Replication Repair Consortium and the SickKids Cancer Sequencing Program (KiCS, Toronto, Canada).1,2,13 The study was approved by the Institutional Research Ethics Board at the Hospital for Sick Children (SickKids, Toronto, Canada). CMMRD diagnosis was confirmed by a genetic counselor via established diagnostic criteria8 and by sequencing of the four MMR genes in clinically approved laboratories. MMR-deficient cancers were defined as tumors originating from germline biallelic MMR gene mutations and somatically acquired MMR mutations in the tumor. MMRP tumors and nonmalignant samples lack biallelic mutations in the MMR genes by genetic testing.
FIG 1.

Clinical development of LOGIC—tumors and patients included in the study. Distribution of tumor samples tested using LOGIC (left) and the patients included for germline analysis (right). CMMRD, constitutional mismatch repair deficiency; EPCAM, Epithelial Cellular Adhesion Molecule; LOGIC, low-pass genomic instability characterization; MMR, mismatch repair; NF-1, neurofibromatosis type 1.
High-throughput sequencing, mutation identification, and TMB and MSI signature analysis were performed as described in previous reports.1,2,13 Low-pass genomic analysis was performed using the Illumina Novaseq6000 with 96 samples in a single flow cell at a coverage of 1× per sample. The algorithm for the LOGIC/MMRDness score was described previously.
Immunohistochemistry of MMR genes was performed in the clinical pathology laboratory at the Hospital for Sick Children. Microsatellite instability was determined using the MIAS (Promega, WI13). Detailed sequencing and bioinformatic data are available in the Data Supplement (online only).13
Statistical Analysis
All comparisons of MMRDness scores between independent sample groups were performed using the nonparametric Mann-Whitney U-test. Comparisons between groups were performed using two-sided Wilcoxon's rank-sum test and two-sided Fisher's exact test for continuous and categorical variables, respectively. Comparisons of the diagnostic sensitivity of LOGIC and the clinically approved assays for replication repair deficiency diagnosis, including tumor mutational burden, immunohistochemistry, and the microsatellite panel, were performed using McNemar's test. All statistical tests were two-sided, and nominal P values were reported. Goodness of fit between different coverages of sequencing was analyzed using the R2 coefficient of determination test.
RESULTS
LOGIC Accurately Detects MMRD in Malignant Tissues
To test LOGIC's ability to call MMRDness scores as full-coverage WGS, we used a set of MMRP and MMRD cancers with a wide range of TMB and MMRDness scores. As a first step, we bioinformatically downsampled tumor WGS data from 30× to 0.01× coverage and observed comparable MMRDness scores (n = 31; Data Supplement). Next, we performed sequencing at 1× coverage (as compared with 30×) on the same tumors as a pilot cohort and observed high concurrence between the true low-pass sequencing (LOGIC) and downsampled data (n = 31, R2 = 0.96; Data Supplement). We then validated the tool using a discovery cohort of 41 tumors13 and a validation cohort of 174 tumors including paraffin-embedded and frozen tissues (Fig 2A and Data Supplement). LOGIC exhibited complete separation of MMRP and MMRD tumors. To determine a threshold of MMRDness = 0, which separates MMRD from MMRP samples, we performed receiver operator curve and area under the curve analyses using incremental discrimination cutoffs, which yielded an area under the curve value of 1 at the established threshold of MMRDness = 0 (Data Supplement).
FIG 2.
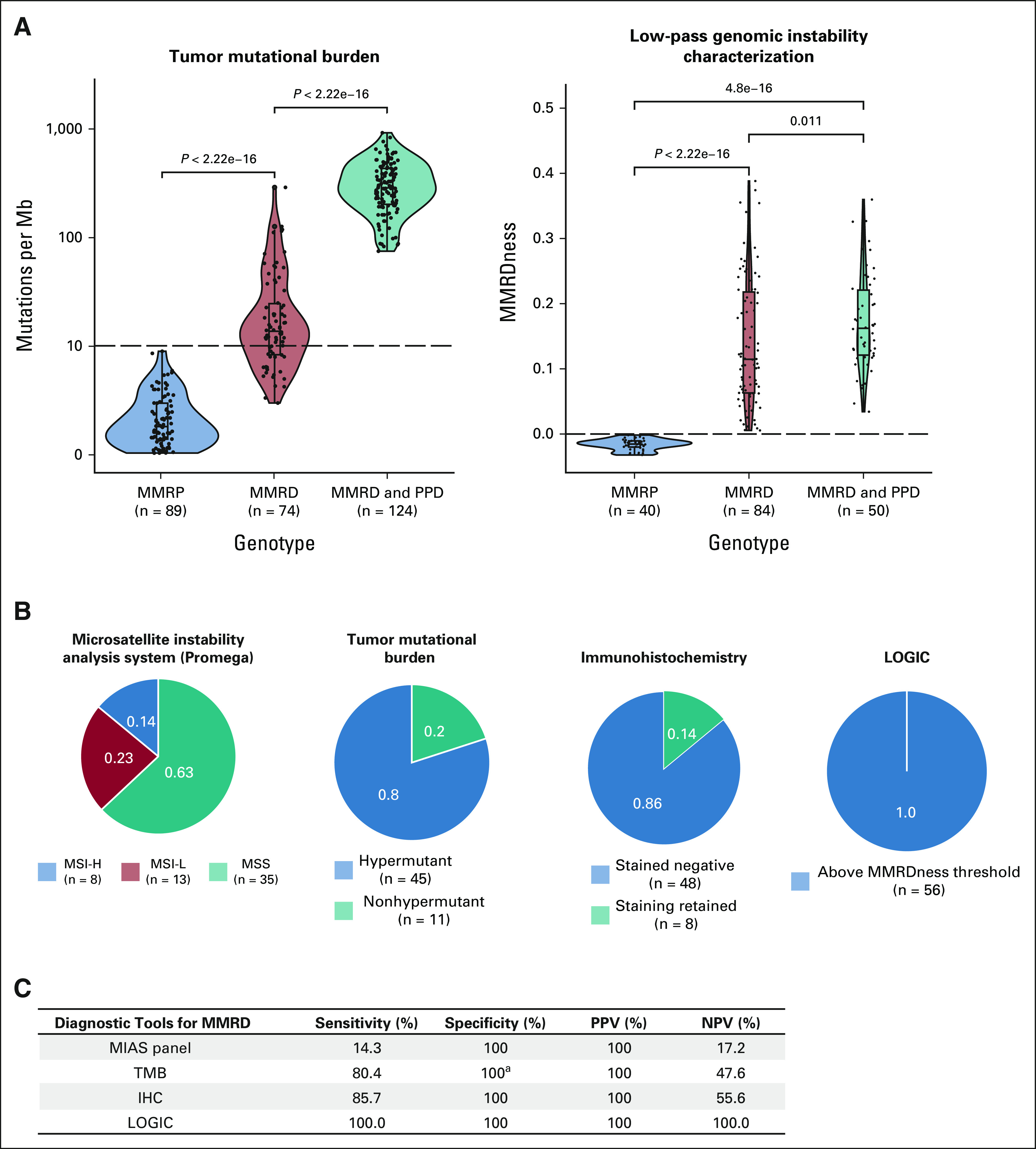
Comparison of current assays in detection of mismatch repair deficiency in childhood cancer. (A) Comparison of TMB with LOGIC in their abilities to detect mismatch repair deficiency in clinically characterized tumors. P values were calculated using the two-sided Mann-Whitney U test. (B) Comparison of the diagnostic ability of immunohistochemistry, the Microsatellite Instability Analysis System (MIAS, Promega, WI) panel, TMB, and LOGIC in mismatch repair–deficient tumors (n = 56). (C) Sensitivity and specificity of the diagnostic tools in detecting MMRD in pediatric cancers. aTMB is not 100% specific in all tumors, especially in adults.7,21-23 LOGIC, low-pass genomic instability characterization; MMRD, mismatch repair deficiency; MMRD, mismatch repair deficient; MMRP, mismatch repair–proficient; MSI-H, microsatellite instability-high; MSI-L, microsatellite instability-low; MSS, microsatellite stable; NPV, negative predictive value; PPD, polymerase proofreading deficiency; PPV, positive predictive value; TMB, tumor mutational burden.
To test the efficacy of LOGIC as a diagnostic tool, we compared MMRDness scores with the current clinically approved tests in a large cohort of well-defined and characterized pediatric MMRP and genetically confirmed MMRD tumors (N = 376; Fig 1 and Data Supplement). We first compared LOGIC with TMB (Fig 2A), which is elevated in MMRD cancers.1-3,13 Although TMB was low in all MMRP (n = 89) childhood tumors and high in all MMRD and PPD cancers (n = 124), 35% (26 of 74) of MMRD-only tumors had a lower TMB than the hypermutation threshold of 10 mutations/Mb (Fig 2A). By contrast, LOGIC displayed 100% specificity and sensitivity in MMRD detection (n = 174, P < .0001; Fig 2A, right panel).
Finally, we performed all functional assays on a subcohort of MMRD tumors (Data Supplement; n = 56).8 LOGIC displayed 100% sensitivity and was superior to all other methods (P < .0133; Data Supplement). By contrast, the MIAS (Promega, WI) was the least sensitive assay (14% MSI-H), whereas TMB and IHC performed better and had sensitivities of 80% and 86%, respectively (Fig 2B and Data Supplement). As expected, all tests were 100% specific and tested negative in MMRP childhood cancers (Fig 2C). Notably, adult cancers are known to have high TMB because of exogenous causes, which leads to lower specificity in MMRD detection.1,16-18 Altogether, LOGIC had 100% positive and negative predictive values in MMRD malignancies.
LOGIC can Detect Germline MMRD
Although the MIAS tool (5 MS loci) fails to detect MMRD in individuals with CMMRD who lack replication repair in all cells,13 we reasoned that LOGIC, which uses > 23 million genomic microsatellites, will have sufficient MS indels to detect CMMRD in nonmalignant tissues. Analysis of blood samples from a large cohort (n = 263; Figs 1 and 3) of clinically8 and genetically defined patients revealed high MMRDness scores in CMMRD (n = 87) but not in MMRP individuals or patients with Lynch Syndrome (LS), other pediatric cancer predisposition syndromes such as Li-Fraumeni, and Neurofibromatosis type 1, who often display similar clinical features and cancers with CMMRD.10,23
FIG 3.

LOGIC to detect CMMRD in blood (germline-blood DNA). Comparison of MMRDness scores using low-pass genome sequencing in blood samples from patients with CMMRD and other germline cancer predisposition syndromes. The P value was calculated using the two-sided Mann-Whitney U test. CMMRD, constitutional mismatch repair deficiency; LOGIC, low-pass genomic instability characterization; MMRP, mismatch repair proficient; NF-1, neurofibromatosis type 1; PPD, polymerase proofreading deficiency.
Notably, patients with germline biallelic EPCAM mutations (who only develop MMRD colon cancers) had negative and positive MMRDness in their corresponding blood and colon samples, respectively. Mutations in EPCAM (Epithelial Cellular Adhesion Molecule, OMIM#185535),25 an epithelial cell surface protein upstream of MSH2, cause MMRD and cancer only in EPCAM-expressing cells26 such as GI and genitourinary organs. As blood cells do not express EPCAM, this finding is a proof-of-principle that LOGIC can determine tissue-specific MMRDness, enabling further refinement of differentiating MMRD-related syndromes. LOGIC MMRDness > 0 predicted a diagnosis of CMMRD (P < .0001), whereas none of the demographic variables (Data Supplement) we investigated, including age, sex, regional source, and time of accrual, were associated with the diagnosis (Data Supplement; P > .08).
MMRDness is Organ-Specific and Accumulates Over Time in Normal Tissues
We then examined whether tissues can exhibit different MMRDness scores. We focused on brain, GI, and hematopoietic systems, where tumors typically develop in CMMRD.9 In total, we analyzed 87 blood, 23 normal GI, three normal brain, and five skin biopsy–derived fibroblast samples obtained from individuals with CMMRD undergoing surveillance.9,10 Although all nonmalignant tissues had positive MMRDness scores (Fig 4A), MMR‐deficient GI tissues had significantly higher scores than blood and other tissues (P = 3.8 × 10−8). As this analysis could be confounded by different variants of the MMR genes, we looked at different tissues from the same patient (n = 11; Figs 4B and 4C). Similar patterns were observed where GI tissues had up to six-fold higher MMRDness than blood, whereas blood samples harbored higher scores than other tissues. Paired T-test comparisons between tissues from the same patients also demonstrated this pattern (blood v GI P = .012 and blood v other tissues P = .0054, Data Supplement). Interestingly, MMRDness scores increased from the stomach→duodenum→ileum→colon, within the same patient (Fig 4D).
FIG 4.
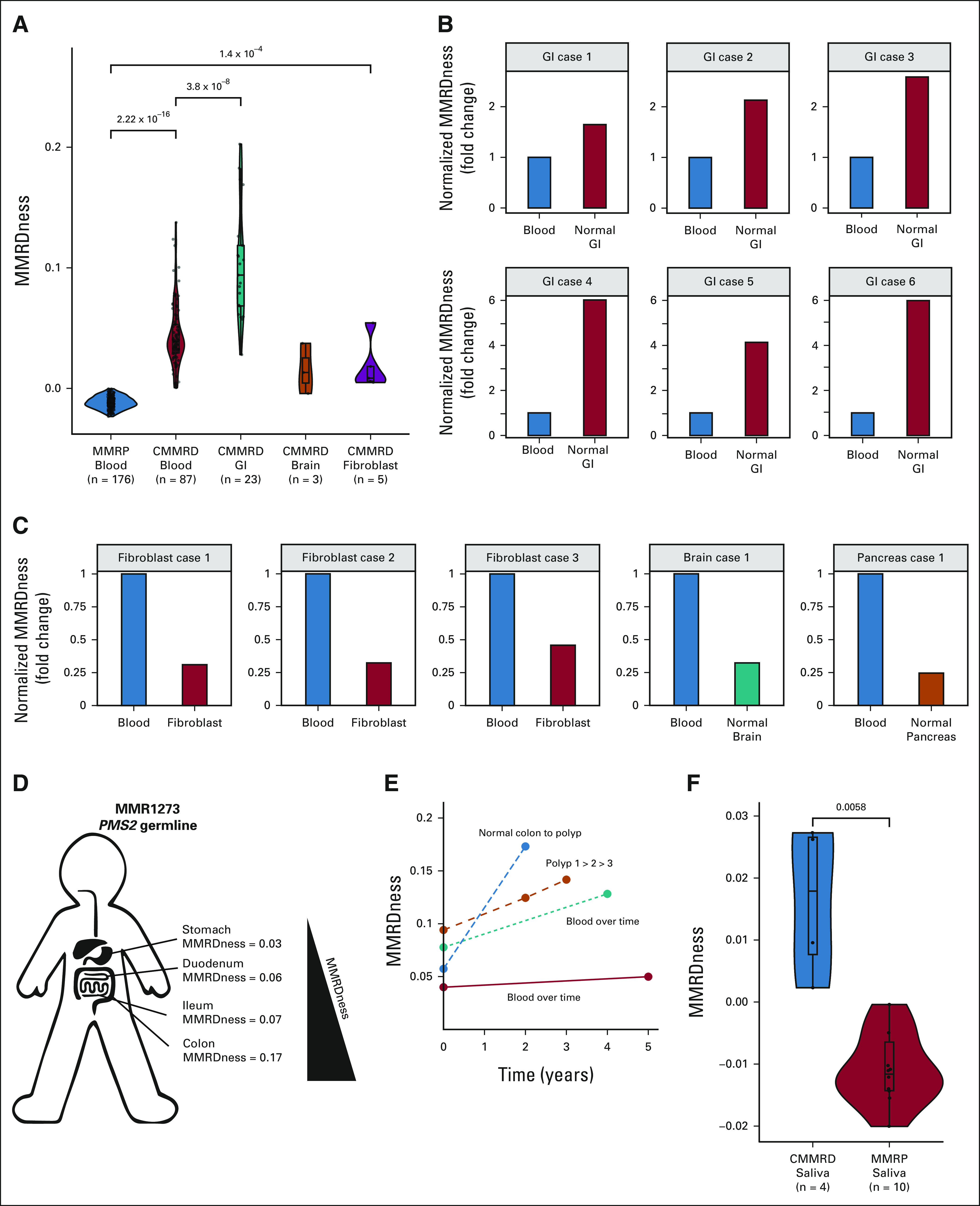
Spatial and temporal analyses of MMRDness in normal tissues. (A) MMRDness scores across different nonmalignant tissues of individuals with CMMRD. (B) MMRDness comparisons between GI and blood from the same patient with CMMRD. Individual scores were normalized to the blood. Cases were anonymized by the label, GI case and an assigned number. (C) MMRDness comparisons of blood and various nonmalignant tissues from the same patient with CMMRD. Individual scores were normalized to the blood. Cases were anonymized by the tissue type and an assigned number. (D) MMRDness of different locations in the GI tract from the same patient with CMMRD taken at the same time. (E) MMRDness scores at different time points (2-5 years apart) in the same tissue of individual patients. (F) MMRDness scores of saliva DNA from patients with CMMRD and MMR-proficient individuals. P values were calculated using the two-sided Mann-Whitney U test. CMMRD, constitutional mismatch repair deficiency; MMR, mismatch repair; MMRD, mismatch repair deficiency; MMRP, mismatch repair proficient.
An increase in MMRDness was observed in blood and GI samples between initial and subsequent time points spanning 2-5 years (Fig 4E). Furthermore, a continuous increase in MMRDness was observed between normal colon (Data Supplement; n = 23), polyps (n = 13, P = .0059) and colon cancers (n = 21, 7.8 × 10−6).
Finally, to further test LOGIC as a screening tool for CMMRD in the International Replication Repair Deficiency Consortium (IRRDC),27 we tested DNA from saliva collected and shipped from 14 individuals in our international centers. Patients with CMMRD had scores within the MMRD range, which were significantly higher than MMRP controls (P = .0058; Fig 4F).
MMRDness Score Is Associated With the Age of Cancer Onset in Individuals With CMMRD
Although all CMMRD blood samples harbored high MS indels, a significant variability in their MMRDness scores was observed (Fig 3). As the range of scores can be related to a genotype-phenotype effect in the MMR genes and therefore the penetrance of the CMMRD phenotype,13,20 we initially examined the seven patients with MMRDness scores that were significantly above the MMR-proficient threshold (P = 7.7 × 10−6; Fig 5A), but in the lower range for MMRD (Data Supplement). Strikingly, in these patients, the median age of initial cancer was much higher (27 years; Data Supplement) than the median age of cancer diagnosis for other individuals with CMMRD (8.8 years, P = 2.9 × 10−5). Of the three patients who had immunohistochemical analysis of their normal tissues, two had residual staining of the mutated proteins (Fig 5B and Data Supplement) and 5 cases retained partial MMR ability as shown by in vitro functional repair assay data28-30 (Data Supplement). These lower-penetrance CMMRD cases carried biallelic variants in PMS2 or MSH6 (Fig 5C), which were compound heterozygous with mixed pathogenicity. More specifically, 6 of 7 cases harbored one known pathogenic driver and another variant, currently classified as VUS. The remaining patient with a homozygous variant was shown to retain 5% repair ability by the in vitro functional repair assay30 (Fig 5C and Data Supplement). LOGIC demonstrated that these are true drivers with attenuated MMR function and not benign variants.
FIG 5.
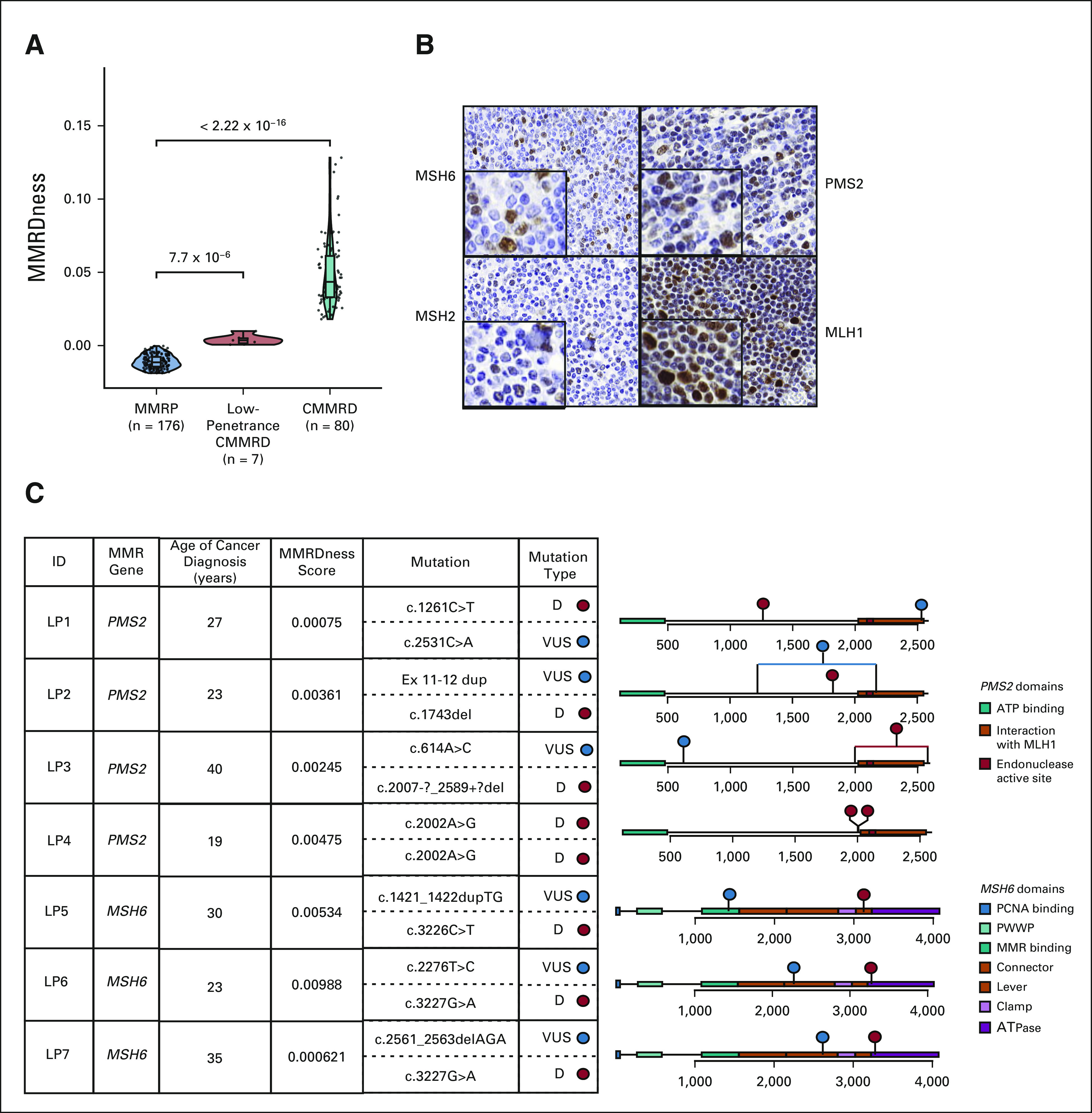
Genotype/phenotype associations using MMRDness scores. (A) Identification of a LP subcohort of CMMRD with lower MMRDness scores than other individuals with CMMRD. P values were calculated using the two-sided Mann-Whitney U test. (B) Partial retention of MMR protein stain in the normal GI tissue of a LP case harboring compound heterozygous PMS2 germline variants. (C) Genotype/phenotype associations of germline mutations in the MMR genes in individuals with LP CMMRD mutations and age of cancer diagnosis. The table on the left indicates the affected MMR gene, age of cancer onset, LOGIC MMRDness scores from blood DNA, and the specific variants and their associated pathogenicity of each case. On the right are lollipop plots with the cDNA of the MMR genes color-labeled with their functional domains. Red lollipops indicate pathogenic or likely pathogenic mutations, and blue represents variants currently classified as VUS. The red line indicates a deletion in the gene. The blue line indicates a duplication in the gene. Patients are anonymized with LP and a number. CMMRD, constitutional mismatch repair deficiency; LOGIC, low-pass genomic instability characterization; LP, low-penetrance; MMR, mismatch repair; MMRD, mismatch repair deficiency; VUS, variants of uncertain clinical significance.
Further comparison of blood MMRDness scores with age of initial cancer in patients with CMMRD revealed a significant negative association (P = 2.2 × 10−5, P = 5.0 × 10−3; Fig 6A) where lower scores were associated with later age of cancer onset. LOGIC analysis of confirmed cases of CMMRD cancer (n = 86; Fig 6B) revealed that none of the individuals with a score of < 0.015 developed cancer before age 18 years.
FIG 6.
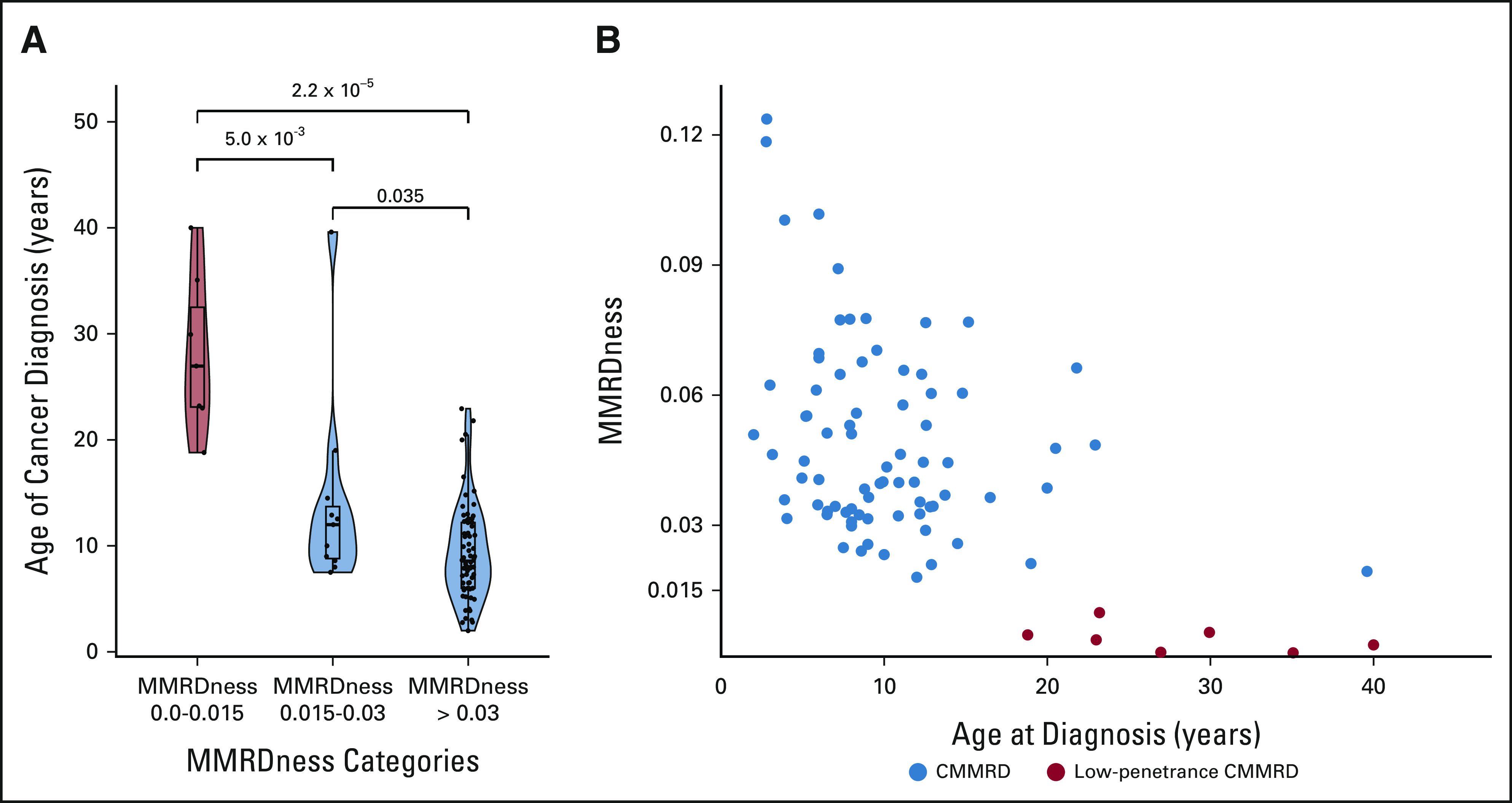
MMRDness score and age of cancer onset in patients with CMMRD. (A) Age of initial cancer diagnosis as a function of the MMRDness score. The box and whisker plots indicate the median age at cancer diagnosis and the interquartile range. MMRDness scores are grouped into low medium and high. The violin plots describe the distribution of patient age at cancer diagnosis in relation to the MMRDness group. P values were calculated using the two-sided Mann-Whitney U test. (B) Association between MMRDness and age of first cancer in each individual patient. Red dots denote patients with low-penetrance CMMRD, whereas blue dots indicate all other individuals with CMMRD. CMMRD, constitutional mismatch repair deficiency; MMRD, mismatch repair deficiency.
DISCUSSION
In this study, we used a well-annotated data set of childhood cancers and patients with cancer predisposition syndromes to provide evidence that low-pass genome analysis can be used to diagnose replication repair deficiency in cancerous and normal tissues. This functional genomic tool performs substantially better than currently used screening assays and is able to quantify the degree of MMRDness to enable risk stratification and precision-based decisions.
Mutational signatures imprinted on the genome by cancer drivers is a growing field of research with major implications for our understanding of cancer.1,4-6,15,31-34 These signatures also have the potential of being implemented into clinical decision making.35 Our data suggest that LOGIC can use MSI signatures to distinguish MMRP from MMRD tumors at low-coverage sequencing. Although it is clear why calling MS indels from > 23 million microsatellites is superior to the five used in the MIAS panel (Promega, WI), LOGIC also has several considerable advantages over other diagnostic assays. For example, immunohistochemistry of MMR proteins is an inexpensive and relatively sensitive assay, but its sensitivity is hampered by the retained expression of some dysfunctional mutant proteins, which still lack the ability to repair mismatches. Technical issues also lead to inconclusive results, especially in CMMRD when normal cells within the tumor biopsy, which are commonly used as internal positive control, stain negative. Genetic testing is generally considered the gold standard for MMRD diagnosis in both tumor and germline tissues. However, in many cases, the genetics community finds it challenging to differentiate true drivers from passenger mutations in MMR genes. This results in a multitude of VUSs, which makes it difficult to diagnose CMMRD and plan clinical management accordingly.
Finally, using TMB and the COSMIC single nucleotide variant (SNV)–based signatures fails in pediatric MMRD cancers where TMB is not sufficiently high (Fig 2). Adding to the lack of sensitivity, TMB is not specific enough to detect MMRD since hypermutation is commonly observed in many adult cancers because of other mechanisms such as UV light, smoking, and other genotoxic factors.1,16-18 Since MS indel accumulation is exclusively dependent on the lack of replication repair, LOGIC provides both superior sensitivity and specificity for MMRD detection in cancerous tissues.
LOGIC's ability to detect MMRD in normal tissues is unique, as a significant accumulation of MS indels is needed to detect such an abnormality. LOGIC is especially advantageous as it enables fast (96 samples analyzed simultaneously), robust, and affordable (< $150 Canadian dollars) detection of germline MMRD. This contrasts with current tools that are cumbersome, reliant on local expertise, expensive, and less sensitive. Importantly, LOGIC is able to distinguish between LS and CMMRD using nonmalignant (germline) tissue DNA, as patients with LS retain MMR function from the unaffected allele. The biallelic loss of MMR in the cells of patients with CMMRD is detected as an increase in the MMRDness score, in comparison with patients with LS. This is critical, given the differences in screening and clinical management between the two syndromes.
Differentiating true pathogenic variants from VUS in individuals carrying mutations in the replication repair genes is still a major challenge, as clinical and family history is still lacking for most variants. Indeed, many germline mutations in the MMR and polymerase genes are probably not drivers and are not the cause of the corresponding childhood cancer. Recent reports described germline pathogenic/likely pathogenic variants in the MMR and polymerase genes in children with cancer. However, many were not the true driver mutations,36,37 highlighting the importance of a robust functional assay such as LOGIC, which can quantify the strength of the MMR mutation, may be used as an adjunct functional test to supplement genetic testing to determine the pathogenic nature of individual variants, and can aid in variant reclassification, as previously performed in other cancer predisposition genes.38
The ability of LOGIC to further quantify the extent of microsatellite instability in normal tissues has several important biologic and clinical implications. First, the differences in MMRDness observed between tissues are intriguing. As previously described, in contrast to SNVs, which reach a threshold,2,13 MS indels accumulate with each cell division.13 Therefore, the high MMRDness in normal GI and blood samples can explain the high prevalence of GI and hematologic malignancies in individuals with CMMRD. Furthermore, the lower MMRDness in the normal brain can explain why most of these cancers acquire secondary somatic polymerase mutations during the initial steps of carcinogenesis,1,2 enabling the explosive and unique SNV and MS indel accumulation. Higher MMRDness may be related to the higher cell renewal in the GI system, which results in more polymerase slippage. Furthermore, even within the same anatomic system (Fig 4D), different rates of cellular turnover may exist, providing a potential explanation for the higher prevalence of colon cancers when compared with other areas throughout the GI tract. Nevertheless, further experiments are required to confirm this hypothesis. Clinically, since MMRDness in nonmalignant colon tissues is up to 6 times higher than that in the blood, our data suggest that it is possible to use GI MMRDness scores for the diagnosis of CMMRD for patients where the blood and fibroblast biopsy results are ambiguous.
Second, LOGIC can quantify the extent of MMRDness, revealing genotype-phenotype correlations in MMRD. Specifically, LOGIC identified MMR gene variants, which may confer lower mutagenicity than the known pathogenic variants associated with CMMRD. These lower-penetrance CMMRD cases harbored variants, which were previously reported in multiple studies21,28,29,39-41 that described patients with CMMRD diagnosed with their first cancer in adulthood (Data Supplement). Some individuals carrying one lower-penetrance variant did not develop cancer, even up to age 67 years,42 reflecting the low penetrance of these mutations in cancer development. The lower-penetrance mutations described in the PMS2 gene were located outside the functional domains, which can explain the residual MMR function43 and less aggressive phenotype (Data Supplement). However, additional factors interacting at the genomic and proteomic level may contribute to the age of cancer onset in CMMRD. These include additional modifier genes, variance in the rates of cell division between individuals, and external genotoxic stimuli. The complexity of multiple factors contributing to individual MMRDness scores may indeed change the true threshold of cancer risk over time, especially as more patients with low-penetrance CMMRDs are diagnosed clinically and using newer tools. Nevertheless, the identification of a threshold value on the basis of our index cohort and future studies using LOGIC can indeed add a new dimension to our understanding and the risk stratification for cancer onset among families associated with replication repair deficiency.
Third, quantification of MMRDness enables stratification of patients with CMMRD into risk groups, which may allow us to tailor the appropriate age of cancer surveillance for each individual. Most surveillance protocols developed for LS44 and BRCA-associated syndromes45 rely on general clinical risk factors to determine the age to initiate screening for each cancer type, regardless of the function of the specific variant. In syndromes such as Li‐Fraumeni syndrome and neurofibromatosis type 1, where cancer penetrance is high, all patients are expected to undergo surveillance at any age.46 The MMRDness score (Fig 6B) presents a novel option where a functional genomic tool can determine the risk of cancer even for patients with similar genotypes and where cancer penetrance is extremely high. If our pilot data, which reveal an increase in MMRDness over time in four patients with CMMRD (Fig 4E), are validated on a larger set, periodic blood tests may assist with clinical decisions, such as when to initiate surveillance for each patient.
In summary, this study reveals that LOGIC is a sensitive and specific method to measure RRD in both tumor and normal tissues with superior performance to currently used diagnostic assays. This rapid, robust, and economical functional genomic tool can be used in most laboratories worldwide and adds an important dimension to cancer management in the era of precision medicine.
Sumedha Sudhaman
Employment: Strand Life Sciences, Natera Inc
Stock and Other Ownership Interests: Natera Inc
Research Funding: Natera Inc
Travel, Accommodations, Expenses: Natera Inc
Patrick Tomboc
Honoraria: UniCare Health Plan
Consulting or Advisory Role: UniCare Health Plan
Jeffrey Knipstein
Employment: PRA Health Sciences, Servier Pharmaceuticals
Consulting or Advisory Role: Atheneum Partners
Kami Wolfe Schneider
Other Relationship: Journal of Genetic Counseling
Robert McWilliams
Stock and Other Ownership Interests: Zentalis
Consulting or Advisory Role: Zentalis.
Research Funding: NewLink Genetics (Inst), Merck (Inst), Bristol-Myers Squibb (Inst), GlaxoSmithKline/Tesaro (Inst)
Lindsay Peterson
Honoraria: Global Reach Health
Sara Rhode
Honoraria: Myriad Genetics
Speakers' Bureau: Myriad Genetics
Travel, Accommodations, Expenses: Myriad Genetics
An Van Damme
Consulting or Advisory Role: Bayer
Research Funding: Johnson & Johnson
Travel, Accommodations, Expenses: Sobi, Roche
M. Stephen Meyn
Stock and Other Ownership Interests: PhenoTips/Gene42
Patents, Royalties, Other Intellectual Property: I am a coinventor on a patent application for PhenoTips, which is software for phenotyping patients with genetic disorders
Rebecca Auer
Research Funding: Qu Biologics (Inst)
Patents, Royalties, Other Intellectual Property: US201662372406P: Oncolytic rhabdovirus expressing IL12, U.S. Provisional Patent Application No. 63/165,539 filed March 24, 2021, and titled Perioperative Innate Immune Priming in Cancer Therapy
Uncompensated Relationships: Immodulon Therapeutics
Brandie Leach
Employment: Invitae
Stock and Other Ownership Interests: Invitae
Consulting or Advisory Role: Invitae
Carol Burke
Consulting or Advisory Role: Freenome, Johnson & Johnson/Janssen, Johnson & Johnson/Janssen
Research Funding: Emtora Biosciences (Inst), Freenome (Inst), Janssen (Inst)
David Malkin
Consulting or Advisory Role: Bayer
Eric Bouffet
Consulting or Advisory Role: Novartis
Research Funding: Roche (Inst)
Cynthia Hawkins
Consulting or Advisory Role: Bayer
Patents, Royalties, Other Intellectual Property: IP for low-grade glioma and sarcoma fusion panels and medulloblastoma subgrouping panel
Gad Getz
Stock and Other Ownership Interests: Scorpion Therapeutics
Honoraria: Society for Neuro-Oncology, Society of Tumor Oncology, MD Anderson Cancer Center
Consulting or Advisory Role: Scorpion Therapeutics
Research Funding: IBM (Inst), Pharmacyclics (Inst), Leidos Biomedical Research/NCI (Inst)
Patents, Royalties, Other Intellectual Property: Royalties from licensing tools, patents related to bioinformatic tools (Inst), patent related to MSIDETECT (Inst), patent related to MSIDETECT (Inst)
Yosef E. Maruvka
Stock and Other Ownership Interests: Illumina (I)
Consulting or Advisory Role: Foresee Genomics
Speakers' Bureau: AstraZeneca
Patents, Royalties, Other Intellectual Property: Detection of MSI tumors
No other potential conflicts of interest were reported.
See accompanying editorial on page 727
SUPPORT
The SickKids Cancer Sequencing (KiCS) program was supported by the Garron Family Cancer Centre with funds from the SickKids Foundation. This research was supported by Meagan's Walk (MW-2014-10), b.r.a.i.n.child Canada, LivWise, SickKids Foundation donors—Harry and Agnieszka Hall, the Zane Cohen Center donors—The Mullin Family and Friends, the Canadian Institutes for Health Research (CIHR) grant (PJT-156006), the CIHR Joint Canada-Israel Health Research Program (MOP—137899), a Stand Up to Cancer (SU2C)—Bristol Myers Squibb Catalyst Research (SU2C-AACR-CT07-17) grant, a Genome Applications Partnership Program (GAPP) grant from Genome Canada, a COG NCORP Research Base Administrative Supplement Request: Landscape of somatic and inherited replication repair deficiency toward a childhood cancer vaccine (3UG1CA189955-08S2), CCS/CIHR/BC Spark Grants: Novel Technology Applications in Cancer Prevention and Early Detection (SPARK-21, 707089—funded by both CCS (Canadian Cancer Society) and BC (Brain Canada)), and St Baldrick's Foundation International Scholar Award (with generous support from the Team Campbell Foundation; Grant No.: 697257, A.D.). Stand Up To Cancer is a program of the Entertainment Industry Foundation. Research grants are administered by the American Association for Cancer Research, the scientific partner of SU2C.
J.C. and L.N. contributed equally to this work. Y.E.M. and U.T. are senior authors.
DATA SHARING STATEMENT
All next-generation sequencing data in this report have been deposited in the European Genome Phenome Archive, and the accession number will be available before publication. Public data sets used include the SickKids KiCS program (https://kicsprogram.com/). Further information and/or requests for data will be fulfilled by the corresponding author, Uri Tabori (uri.tabori@sickkids.ca).
AUTHOR CONTRIBUTIONS
Conception and design: Jiil Chung, Logine Negm, Vanessa Bianchi, Lucie Stengs, Martin Komosa, Vanan Magimairajan Issai, Eric Bouffet, Carol Durno, Gad Getz, Yosef E. Maruvka, Uri Tabori
Financial support: Anirban Das, Uri Tabori
Administrative support: Lucie Stengs, Uri Tabori
Provision of study materials or patients: Lucie Stengs, Jodi Lees, David Samuel, Roula Farah, Jeffrey Knipstein, Kami Wolfe Schneider, Shayna Zelcer, Alexandra Zorzi, Robert McWilliams, William D Foulkes, Raymond Bedgood, Isabelle Scheers, Vanan Magimairajan Issai, Rebecca Auer, Carol Burke, Anita Villani, Uri Tabori
Collection and assembly of data: Jiil Chung, Logine Negm, Lucie Stengs, Sumedha Sudhaman, Melyssa Aronson, Ledia Brunga, Melissa Edwards, Scott Davidson, Jodi Lees, Patrick Tomboc, David Samuel, Roula Farah, Anne Bendel, Jeffrey Knipstein, Kami Wolfe Schneider, Agnes Reschke, Shayna Zelcer, Alexandra Zorzi, William D Foulkes, Raymond Bedgood, Lindsay Peterson, Sara Rhode, An Van Damme, Isabelle Scheers, Sharon Gardner, Gabriel Robbins, Vanan Magimairajan Issai, M Stephen Meyn, Rebecca Auer, Brandie Leach, Carol Burke, Anita Villani, Eric Bouffet, Annie Huang, Carol Durno, Cynthia Hawkins, Uri Tabori
Data analysis and interpretation: Jiil Chung, Logine Negm, Anirban Das, Zhihui Amy Liu, Victoria Forster, Scott Davidson, Alexandra Zorzi, Robert McWilliams, Lindsay Peterson, Vanan Magimairajan Issai, David Malkin, Eric Bouffet, Michael D. Taylor, Carol Durno, Adam Shlien, Cynthia Hawkins, Gad Getz, Yosef E. Maruvka, Uri Tabori
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Genomic Microsatellite Signatures Identify Germline Mismatch Repair Deficiency and Risk of Cancer Onset
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated unless otherwise noted. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/authors/author-center.
Open Payments is a public database containing information reported by companies about payments made to US-licensed physicians (Open Payments).
Sumedha Sudhaman
Employment: Strand Life Sciences, Natera Inc
Stock and Other Ownership Interests: Natera Inc
Research Funding: Natera Inc
Travel, Accommodations, Expenses: Natera Inc
Patrick Tomboc
Honoraria: UniCare Health Plan
Consulting or Advisory Role: UniCare Health Plan
Jeffrey Knipstein
Employment: PRA Health Sciences, Servier Pharmaceuticals
Consulting or Advisory Role: Atheneum Partners
Kami Wolfe Schneider
Other Relationship: Journal of Genetic Counseling
Robert McWilliams
Stock and Other Ownership Interests: Zentalis
Consulting or Advisory Role: Zentalis.
Research Funding: NewLink Genetics (Inst), Merck (Inst), Bristol-Myers Squibb (Inst), GlaxoSmithKline/Tesaro (Inst)
Lindsay Peterson
Honoraria: Global Reach Health
Sara Rhode
Honoraria: Myriad Genetics
Speakers' Bureau: Myriad Genetics
Travel, Accommodations, Expenses: Myriad Genetics
An Van Damme
Consulting or Advisory Role: Bayer
Research Funding: Johnson & Johnson
Travel, Accommodations, Expenses: Sobi, Roche
M. Stephen Meyn
Stock and Other Ownership Interests: PhenoTips/Gene42
Patents, Royalties, Other Intellectual Property: I am a coinventor on a patent application for PhenoTips, which is software for phenotyping patients with genetic disorders
Rebecca Auer
Research Funding: Qu Biologics (Inst)
Patents, Royalties, Other Intellectual Property: US201662372406P: Oncolytic rhabdovirus expressing IL12, U.S. Provisional Patent Application No. 63/165,539 filed March 24, 2021, and titled Perioperative Innate Immune Priming in Cancer Therapy
Uncompensated Relationships: Immodulon Therapeutics
Brandie Leach
Employment: Invitae
Stock and Other Ownership Interests: Invitae
Consulting or Advisory Role: Invitae
Carol Burke
Consulting or Advisory Role: Freenome, Johnson & Johnson/Janssen, Johnson & Johnson/Janssen
Research Funding: Emtora Biosciences (Inst), Freenome (Inst), Janssen (Inst)
David Malkin
Consulting or Advisory Role: Bayer
Eric Bouffet
Consulting or Advisory Role: Novartis
Research Funding: Roche (Inst)
Cynthia Hawkins
Consulting or Advisory Role: Bayer
Patents, Royalties, Other Intellectual Property: IP for low-grade glioma and sarcoma fusion panels and medulloblastoma subgrouping panel
Gad Getz
Stock and Other Ownership Interests: Scorpion Therapeutics
Honoraria: Society for Neuro-Oncology, Society of Tumor Oncology, MD Anderson Cancer Center
Consulting or Advisory Role: Scorpion Therapeutics
Research Funding: IBM (Inst), Pharmacyclics (Inst), Leidos Biomedical Research/NCI (Inst)
Patents, Royalties, Other Intellectual Property: Royalties from licensing tools, patents related to bioinformatic tools (Inst), patent related to MSIDETECT (Inst), patent related to MSIDETECT (Inst)
Yosef E. Maruvka
Stock and Other Ownership Interests: Illumina (I)
Consulting or Advisory Role: Foresee Genomics
Speakers' Bureau: AstraZeneca
Patents, Royalties, Other Intellectual Property: Detection of MSI tumors
No other potential conflicts of interest were reported.
REFERENCES
- 1.Campbell BB, Light N, Fabrizio D, et al. : Comprehensive analysis of hypermutation in human cancer. Cell 171:1042-1056, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shlien A, Campbell BB, de Borja R, et al. : Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat Genet 47:257-262, 2015 [DOI] [PubMed] [Google Scholar]
- 3.Bouffet E, Larouche V, Campbell BB, et al. : Immune checkpoint inhibition for hypermutant glioblastoma multiforme resulting from germline biallelic mismatch repair deficiency. J Clin Oncol 34:2206-2211, 2016 [DOI] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Network : Comprehensive molecular characterization of human colon and rectal cancer. Nature 487:330-337, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kandoth C, Kandoth C, Schultz N, et al. : Integrated genomic characterization of endometrial carcinoma. Nature 497:67-73, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maruvka YE, Mouw KW, Karlic R, et al. : Analysis of somatic microsatellite indels identifies driver events in human tumors. Nat Biotechnol 35:951-959, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Briggs S, Tomlinson I: Germline and somatic polymerase ε and δ mutations define a new class of hypermutated colorectal and endometrial cancers. J Pathol 230:148-153, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Aronson M, Colas C, Shuen A, et al. : Diagnostic criteria for constitutional mismatch repair deficiency (CMMRD): Recommendations from the international consensus working group. J Med Genet 59:318-327, 2022 [DOI] [PubMed] [Google Scholar]
- 9.Durno C, Ercan AB, Bianchi V, et al. : Survival benefit for individuals with constitutional mismatch repair deficiency undergoing surveillance. J Clin Oncol 39:2779-2790, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tabori U, Hansford JR, Achatz MI, et al. : Clinical management and tumor surveillance recommendations of inherited mismatch repair deficiency in childhood. Clin Cancer Res 23:e32-e37, 2017 [DOI] [PubMed] [Google Scholar]
- 11.Wimmer K, Kratz CP, Vasen HFA, et al. : Diagnostic criteria for constitutional mismatch repair deficiency syndrome: Suggestions of the European consortium 'care for CMMRD' (C4CMMRD). J Med Genet 51:355-365, 2014 [DOI] [PubMed] [Google Scholar]
- 12.Wimmer K, Etzler J: Constitutional mismatch repair-deficiency syndrome: Have we so far seen only the tip of an iceberg? Hum Genet 124:105-122, 2008 [DOI] [PubMed] [Google Scholar]
- 13.Chung J, Maruvka YE, Sudhaman S, et al. : DNA polymerase and mismatch repair exert distinct microsatellite instability signatures in normal and malignant human cells. Cancer Discov 11:1176-1191, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Durno CA, Sherman PM, Aronson M, et al. : Phenotypic and genotypic characterisation of biallelic mismatch repair deficiency (BMMR-D) syndrome. Eur J Cancer 51:977-983, 2015 [DOI] [PubMed] [Google Scholar]
- 15.Alexandrov LB, Kim J, Haradhvala NJ, et al. : The repertoire of mutational signatures in human cancer. Nature 578:94-101, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roberts SA, Gordenin DA: Hypermutation in human cancer genomes: Footprints and mechanisms. Nat Rev Cancer 14:786-800, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. : Signatures of mutational processes in human cancer. Nature 500:415-421, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lawrence MS, Stojanov P, Polak P, et al. : Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499:214-218, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bakry D, Aronson M, Durno C, et al. : Genetic and clinical determinants of constitutional mismatch repair deficiency syndrome: Report from the constitutional mismatch repair deficiency consortium. Eur J Cancer 50:987-996, 2014 [DOI] [PubMed] [Google Scholar]
- 20.Boland CR, Goel A: Microsatellite instability in colorectal cancer. Gastroenterology 138:2073-2087, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lavoine N, Colas C, Muleris M, et al. : Constitutional mismatch repair deficiency syndrome: Clinical description in a French cohort. J Med Genet 52:770-778, 2015 [DOI] [PubMed] [Google Scholar]
- 22.Murphy KM, Zhang S, Geiger T, et al. : Comparison of the microsatellite instability analysis system and the Bethesda panel for the determination of microsatellite instability in colorectal cancers. J Mol Diagn 8:305-311, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gallon R, Muhlegger B, Wenzel SS, et al. : A sensitive and scalable microsatellite instability assay to diagnose constitutional mismatch repair deficiency by sequencing of peripheral blood leukocytes. Hum Mutat 40:649-655, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.González-Acosta M, Marin F, Puliafito B, et al. : High-sensitivity microsatellite instability assessment for the detection of mismatch repair defects in normal tissue of biallelic germline mismatch repair mutation carriers. J Med Genet 57:269-273, 2020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Munz M, Baeuerle PA, Gires O: The emerging role of EpCAM in cancer and stem cell signaling. Cancer Res 69:5627-5629, 2009 [DOI] [PubMed] [Google Scholar]
- 26.Tutlewska K, Lubinski J, Kurzawski G: Germline deletions in the EPCAM gene as a cause of Lynch syndrome—Literature review. Hered Cancer Clin Pract 11:9, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.IRRDC . https://replicationrepair.ca/
- 28.Biswas K, Couillard M, Cavallone L, et al. : A novel mouse model of PMS2 founder mutation that causes mismatch repair defect due to aberrant splicing. Cell Death Dis 12:838, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Drost M, Koppejan H, de Wind N: Inactivation of DNA mismatch repair by variants of uncertain significance in the PMS2 gene. Hum Mutat 34:1477-1480, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shuen AY, Lanni S, Panigrahi GB, et al. : Functional repair assay for the diagnosis of constitutional mismatch repair deficiency from non-neoplastic tissue. J Clin Oncol 37:461-470, 2019 [DOI] [PubMed] [Google Scholar]
- 31.Stephens PJ, Tarpey PS, Davies H, et al. : The landscape of cancer genes and mutational processes in breast cancer. Nature 486:400-404, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chalmers ZR, Connelly CF, Fabrizio D, et al. : Analysis of 100, 000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med 9:34, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lawrence MS, Stojanov P, Mermel CH, et al. : Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505:495-501, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yaeger R, Chatila WK, Lipsyc MD, et al. : Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell 33:125-136, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Das A, Sudhaman S, Morgenstern D, et al. : Genomic predictors of response to PD-1 inhibition in children with germline DNA replication repair deficiency. Nat Med 28:125-135, 2022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang J, Walsh MF, Wu G, et al. : Germline mutations in predisposition genes in pediatric cancer. N Engl J Med 373:2336-2346, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuhlen M, Taeubner J, Brozou T, et al. : Family-based germline sequencing in children with cancer. Oncogene 38:1367-1380, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Richardson ME, Hu C, Lee KY, et al. : Strong functional data for pathogenicity or neutrality classify BRCA2 DNA-binding-domain variants of uncertain significance. Am J Hum Genet 108:458-468, 2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Senter L, Clendenning M, Sotamaa K, et al. : The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations. Gastroenterology 135:419-428, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guerrini-Rousseau L, Varlet P, Colas C, et al. : Constitutional mismatch repair deficiency–associated brain tumors: Report from the European C4CMMRD consortium. Neurooncol Adv 1:vdz033, 2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jasperson KW, Samowitz WS, Burt RW: Constitutional mismatch repair-deficiency syndrome presenting as colonic adenomatous polyposis: Clues from the skin. Clin Genet 80:394-397, 2011 [DOI] [PubMed] [Google Scholar]
- 42.Bougeard G, Olivier-Faivre L, Baert-Desurmont S, et al. : Diversity of the clinical presentation of the MMR gene biallelic mutations. Fam Cancer 13:131-135, 2014 [DOI] [PubMed] [Google Scholar]
- 43.Obmolova G, Ban C, Hsieh P, Yang W: Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature 407:703-710, 2000 [DOI] [PubMed] [Google Scholar]
- 44.Vasen HFA, Moslein G, Alonso A, et al. : Guidelines for the clinical management of Lynch syndrome (hereditary non-polyposis cancer). J Med Genet 44:353-362, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elezaby M, Lees B, Maturen KE, et al. : BRCA mutation carriers: Breast and ovarian cancer screening guidelines and imaging considerations. Radiology 291:554-569, 2019 [DOI] [PubMed] [Google Scholar]
- 46.Kratz CP, Achatz MI, Brugieres L, et al. : Cancer screening recommendations for individuals with Li-Fraumeni syndrome. Clin Cancer Res 23:e38-e45, 2017 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All next-generation sequencing data in this report have been deposited in the European Genome Phenome Archive, and the accession number will be available before publication. Public data sets used include the SickKids KiCS program (https://kicsprogram.com/). Further information and/or requests for data will be fulfilled by the corresponding author, Uri Tabori (uri.tabori@sickkids.ca).