

Abstract
Intercellular signaling drives human development, but there is a paucity of in vitro models that recapitulate important tissue architecture while remaining operationally simple and scalable. We have developed a robust, throughput-compatible microphysiological system to model intercellular signaling including epithelial-mesenchymal interactions that is useful for studying both normal and abnormal orofacial development. We describe the construction and operation of an engineered microplate created using CNC micromilling of 96-well microtiter plates capable of containing up to 20 epithelial-mesenchymal microtissues. A dense 3D mesenchyme is created by embedding cells (O9–1, 3T3) in a biomimetic hydrogel. An epithelial layer is then overlayed on the microtissue by loading cells in engineered microchannels that flank the microtissue. The result in an engineering epithelial-mesenchymal interface that is both on and perpendicular to the imaging plane making it suitable for high-content imaging and analysis. The resulting microtissues and device are compatible with diverse analytical techniques including fluorescent and luminescent cell health and enzymatic reporter assays, gene expression analyses, and protein staining. This tractable model and approach promises to shed light on critical processes in intercellular signaling events in orofacial development and beyond.
Keywords: signaling gradient, paracrine signaling, 3D extracellular matrix, epithelial mesenchymal cross-talk, cleft lip and palate
1. Introduction
Formation of the upper lip and palate requires the orchestrated proliferation and fusion of embryonic facial growth centers and is dependent on paracrine intercellular signaling through multiple pathways including the Sonic Hedgehog (SHH), transforming growth factor-beta (Tgf-β), bone morphogenic protein (BMP), epidermal growth factor (EGF), etc. pathways [1–3]. For example, epithelial ectoderm derived Sonic Hedgehog (SHH) ligand drives orofacial morphogenesis through an induced gradient of SHH dependent transcription in the underlying mesenchyme, which induces proliferation and tissue outgrowth [4,5]. Disruption of the SHH, Tgf-β3, BMP, EGF etc. pathways have been shown to cause orofacial clefts (OFCs) [6,7]. The etiology of most birth defects, including OFCs is complex and is believed to include both genetic and environmental components, which along with the large number of important morphogenetic signaling pathways involved challenges elucidation of the underlying mechanisms [6,7].
Here, we outline our in vitro system that mimics the tissue architecture and signaling interactions that occur during orofacial development to facilitate the study of both normal and abnormal development [8]. Despite some recent advances, current 2D cell cultures techniques are insufficient in providing an accurate 3D morphogenic signaling gradiets. This system, more faithfully recapitulate tissue biology and allows for testing of environmentally relevant drug/chemical exposures, genetic predispositions and/or their interactions to help bridge the in vitro-in vivo gap[9,10]. The devices are constructed using computer numerical control (CNC) micromilling. Unlike many microfluidic/microphysiological systems, these devices are manufactured out of commercially available microplates, providing a familiar platform for new users. The microplate-based format is inherently throughput-compatible enabling low-level chemical and drug screening. This approach also overcomes the problem of hydrophobic molecules being sequestered by the device, which is a problem commonly seen in typical microfluidic devices constructed from polydimethylsiloxane (PDMS) [11].
Engineering the epithelial-mesenchymal interface relies upon the principles of microfluidics to create dense 3D mesenchyme covered with a layer of oral epithelial-mesenchyme organization. Epithelial-mesenchymal microtissues are generated with 09–1 (murine cranial neural crest mesenchymal) or 3T3 Shh-Light2 (murine mesenchymal) cells, and GMSM-K (human fetal oral epithelial) cells. To form microtissues, custom devices are seeded using mesenchymal cells embedded in a hyaluronic acid based (HA)/collagen hydrogel, where HA has known importance in palatogenesis[12]. Surface tension in the mesenchymal/hydrogel matrix causes the liquid to pin at a phase barrier between the adjoining microchannel that polymerizes creating a vertical wall in the microchannel. The epithelium is added by perfusing GMSM-K cells through the microchannels of the device. Laminar flow in the microchannel facilitates the attachment of epithelial cells to the hydrogel surface. This predictably creates a functional 3D epithelial-mesenchymal microtissue perpendicular to and on the plate bottom facilitating microscopic analyses. These microtissues can be used to study the effects of drugs or other environmental factors on paracrine interactions, including gene: environment interactions. To illustrate the broad tractability and analytical versatility of the platform, we detail the use of a mesenchymal Gli-driven luciferase reporter assay to quantify and spatially characterize the gradient of epithelial secreted SHH activity. We also present protocols to extract RNA for gene expression analysis, as well as extraction of the microtissues for standard histopathological processing and H&E staining. Diverse analytical techniques including fluorescent and luminescent cell health and enzymatic reporter assays are also easily implemented. This tractable model and approach are a unique tool to study critical processes in intercellular signaling events in orofacial development and beyond [13,8].
2. Materials
2.1. Device Design and Construction
.19mm clear polystyrene sheet (Goodfellow, Huntingdon, England)
100% isopropyl alcohol
70% ethanol (EtOH)
Acetonitrile (ACN)
Computer aided design (CAD) software (e.g. Solidworks, Fusion 360 etc.) (Solidworks, Dessault Systems, Vélizy-Villacoublay, France)
Computer aided manufacturing (CAM) software (e.g. SprutCAM, MasterCAM, SolidCAM etc.) (SprutCAM America, Waunakee, WI, USA)
Clear 96-well non tissue culture treated plate (Corning Inc., Corning, NY, USA)
Compressed air
CNC Mill with appropriate fixtures and endmills (Tormach Inc., Waunakee, WI, USA)
DI water
Hot plate (Fisher Scientific, Waltham, MA, USA)
Lint free toweling
Razor blade
Ultrasonic bath (Branson Ultrasonics, Sterling Heights, MI, USA)
1000μL non-filtered pipette tips (USA Scientific, Ocala, FL, USA)
200μL filtered pipette tips (USA Scientific, Ocala, FL, USA)
2.2. Device Sterilization and Pre-Culture Treatment
Cell culture hood
P10 Pipetman (Gilson, Middleton, WI, USA)
Glutaraldehyde
Plastic wrap (e.g. Saran)
Polyethylenimine (Sigma Aldrich, St. Louis, MO, USA)
Sterile Water
200μL large orifice non-filtered pipette tips (USA Scientific, Ocala, FL, USA)
2.3. Culturing Cell Lines for Devices
Falcon 100mm cell culture treated plate (Corning Inc., Corning, NY, USA)
GMSM-K GFP human fetal oral epithelial cells
GMSM-K RFP human fetal oral epithelial cells
Matrigel: Growth Factor Reduced, Basement Membrane Matrix, Phenol Red-Free, LDEV-Free (Corning Inc., Corning, NY, USA)
Serological pipette
3T3 Shh-Light2 murine mesenchymal cells
3T3 Shh-Light2 Media: DMEM, 10% FBS,.4mg/mL G418 (Invivogen, San Diego, CA, USA), .15mg/mL zeocin (Invivogen, San Diego, CA, USA) [14]
09-1 murine cranial neural crest mesenchymal cells
DMEM: 1%PenStrep, 10%FBS
2.4. Making 3D HA Cell Suspensions and Device Loading of 09–1 or 3T3 cells
DMEM: 1% FBS, 1% Pen-Strep
EDTA
Hepes
Hystem-C (Sigma-Aldrich, St. Louis, MO, USA)
Kim wipes (Kimberely-Clark, Irving, TX, USA)
P10 Pipetman (Gilson, Middleton, WI, USA)
PBS
Trypsin
2.5. Experimental Procedure for Gene Expression
Chloroform
Confocal microscope (e.g. BD Pathway) (BD Biosciences, San Jose, CA, USA)
1.5mL PCR Tubes
Qiagen MagAttract RNA Universal Tissue Kit (Qiagen, Hilden, Germany)
QIAGEN QIAzol (Qiagen, Hilden, Germany)
2.6. Evaluation of Gli-driven Luciferase
2X dissecting microscope lens
Chemicdoc luminescent imager (Bio-Rad, Hercules, CA, USA)
HEPES
Pherastar plate reader (BMG Labtech, Ortenberg, Germany)
Quantification software (ImageLab, ImageJ, etc)
VivoGlo:2mM luciferin (Promega, Madison, WI, USA)
2.7. H&E Staining
Formalin
Hematoxylin and Eosin (H&E) stain
Histo-wrap 2”x2” (OBEX Industries)
Microtome
Paraffin
3. Methods
All procedures are performed at room temperature unless otherwise stated.
3.1. Device Design and Construction
Design and model device using 3D modeling software. The device and illustrations of its’ function can be seen in Figure 1.
To machine devices, toolpaths generated using CAM software are provided (G-code is provided for machining this device using a .8mm cylindrical two fluted square endmill).
Secure clear 96-well non tissue treated culture plate to deck of mill using clamps or a vise and check for flatness. The provided code is for use of .8mm cylindrical endmill and will mill the device at a feed rate of 10in/min and a spindle speed of 10000 RPM. The long axis of the plate should align with the y axis of the mill. The code is set so that the origin is the injection port in the center of the plate. The z-axis should be zeroed on a flat portion of the plate bottom (not on the injection port).
Sonicate the machined plate in 100% isopropyl alcohol in the ultra-sonic bath for at least 15 minutes to clean
Remove plate from ultra-sonic bath and rinse thoroughly with DI water then dry with compressed air (see Note 1)
Cut a piece of .19mm clear polystyrene to be bonded to the plate approximately 110×74mm. The piece should be slightly larger than the area of the plate to be bonded.
Spray polystyrene piece with 70% EtOH, rinse with DI water then dry with compressed air
Turn on hot plate and set to 70°C
Place device and polystyrene sheet on the hot plate as in Figure 2 and allow to reach set temperature (see Note 2)
Use a 200μL filtered pipette tip to transfer 35μL of acetonitrile (ACN) to the bonding port (hole) in the upper left corner of the device
Use a 1000μL non-filtered pipette tip attached to vacuum to aspirate the excess ACN from the adjacent corners followed by the initial well and any surrounding channels (see Note 3)
Repeat step 10–11 for the remaining 3 corners
Using a 200μL filtered pipette tip transfer 25μL of ACN to a well
Aspirate the ACN out using a 1000μL non-filtered pipette tip attached to vacuum
Repeat for each well on the plate
Remove the plate from the hot plate and allow to cool to room temperature
Use a handheld razor blade to trim the excess polystyrene from around the perimeter of the device
Figure 1:
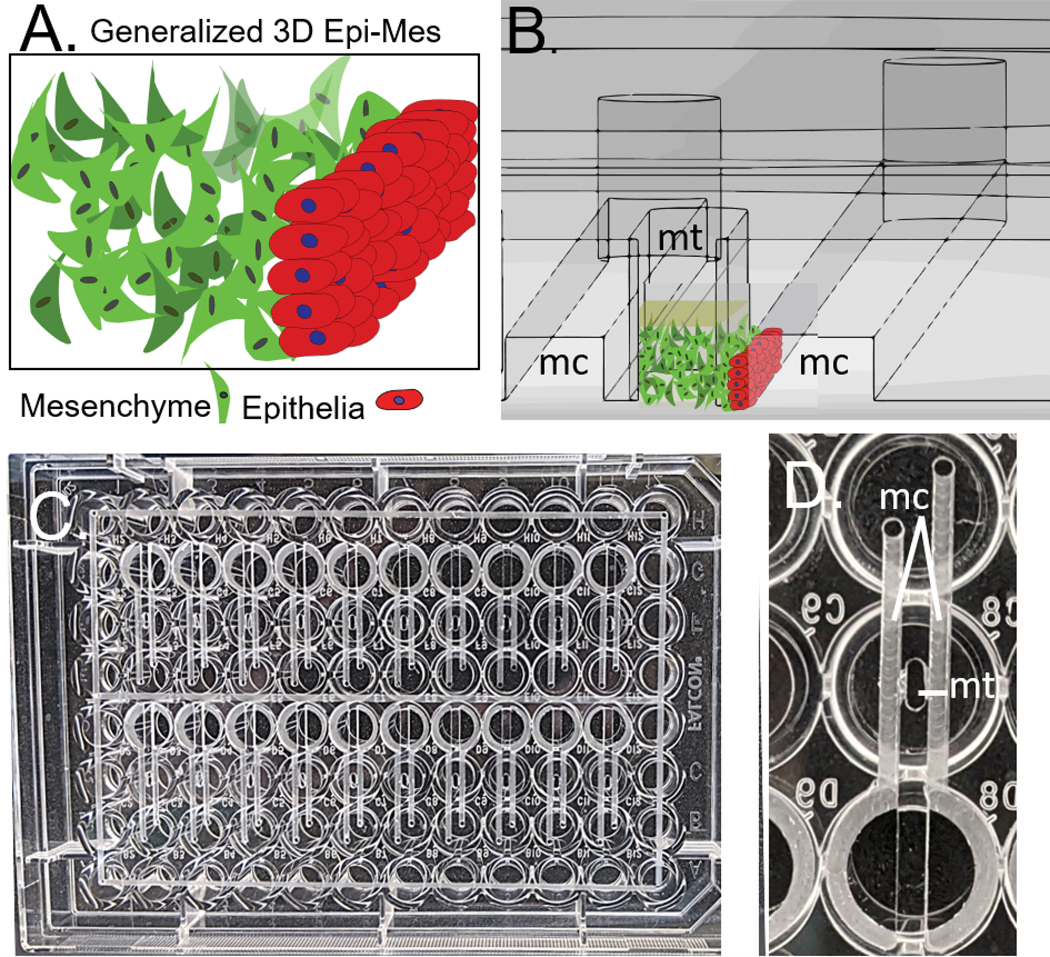
A) Generalized 3D orofacial mesenchyme/epithelia interface B) Cross section of CAD model of the device, flanking microchannels (mc) can be seen on either side of the microtissue well (mt). Mesenchyme/ECM and epithelial cells are illustrated on the cross section to show the function of the device. C) Devices (20) machined into a standard 96 well plate, perimeter channels facilitate bonding. D) Single device as viewed from the bottom of the plate integrates 3 wells.
Figure 2:

A) Schematic of bonding setup. B) Side view of bonding setup. Solvent (ACN) is added to bond the devices at 70˚C. Excess solvent must be quickly removed to avoid etching C) Bottom view of bonded device.
3.2. Device Sterilization and Preparation for Culture.
Place device into UV irradiator and cover the main part of the plate with plastic wrap separate from the lid [15].
UV irradiate for 15 minutes
Transfer device to sterilized culture hood
Add 3μL of polyethylenimine to each well of the device using a P10 Pipetman
Wait 10 minutes
Aspirate the 3μL of polyethylenimine from each well using a 200μL large orifice non-filtered pipette tip attached to vacuum
Add 3μL of glutaraldehyde to each well of the device using a Gilson P10 Pipetman
Wait 30 minutes
Aspirate the 3μL of glutaraldehyde from each well using a 200μL large orifice non-filtered pipette tip attached to vacuum
Wash each well with 5μL of DI water using the P10 Pipetman
Repeat step 10 for a total of 3 washes with DI water (see Note 4)
Allow device to dry in the cell culture hood
3.3. Culture and Preparation of 09–1, GMSM-K, and 3T3 Shh-Light2 Cells for Seeding in Devices
Prepare Matrigel per manufacturer instructions and keep on ice at all times during use
Add 300μL of Matrigel to 10mL DMEM: 10%, 1% Pen-Strep media using a pipette
Transfer the dilution from step 1 into a Falcon 100mm cell culture treated plate using a serological pipette
Incubate plate at room temperature for 60 minutes in sterile cell culture hood
Plate 09–1 cells on Matrigel coated culture plates in DMEM: 10% FBS, 1% Pen-Strep
Culture cell line 24 hours at 37°C and 5% CO2 prior to trypsinizing for use in 3D co-culture devices
Plate GMSM-K GFP or GMSM-K RFP in 10mL of DMEM: 10%FBS, 1% Pen-Strep (see Note 5)
Culture GMSM-K cell line for 48 hours at 37°C and 5% CO2 prior to trypsinizing for use in 3D co-culture devices
Plate 3T3 Shh-Light2 cells in 10mL of 3T3 Shh-Light2 media
Culture 3T3 Shh-Light2 cells for 48 hours at 37°C and 5% CO2 prior to trypsinizing for use in 3D co-culture devices
3.4. Making 3D Hyaluronic Acid Cell Suspensions and Device Loading of 09–1 or 3T3 cells
Trypsinize 09–1 or 3T3 cells with 0.25% trypsin and EDTA for 3 minutes at 37°C
Add approximately 1mL of 1:1 dilution of DMEM: 10% FBS to neutralize trypsin reaction
Prepare a 100,000 cells/μL solution of either 09–1 or 3T3 cells
Prepare hyaluronic acid (HA) as per manufacturer instructions
Mix Hystem-C (HA and collagen gel solution) 1:1 with the cell solution prepared in step 3
Transfer 1.75 −3μL of HA:cell solution to device wells using a P10 Pipetman
Let device sit for 45 minutes at room temperature to allow for polymerization
Add 100μL of DMEM: 1% FBS, 1% Pen-Strep media to the center well of all devices on top of the hydrogel
Fill outer wells with 100uL H2O to act as sacrificial fluid and prevent evaporation in the devices.
Prepare a solution of 4,000 GMSM-K cells/μL
Load 10μL of the GMSM-K cell solution into the side channel of each well. If desired 5μL of the GMSM-K cell solution can be added to the center well directly on top of the mesenchyme. (see Note 6)
Wait for 30 minutes then flush channels and wells with media to remove unattached cells
Add 100–150μL of DMEM: 1% FBS, 1% Pen-Strep to the center well (see Note 7) a fully loaded device can be seen in Figure 3.
Aspirate the perfused media from the half-moon reservoirs in the bottom wells daily
Change media every 1–2 days and dose daily for 3 days
Figure 3:

Device loading. A) An empty device is seen on the left and a dye-loaded device is on the right. B) Mesenchyme/epithelia microtissue. C) Fluorescent staining/imaging in situ in the left image GMSM-K RFP can be seen next to non-RFP GMSM-K. In the image on the right, GMSM-K GFP-SHH are shown next to standard GMSM-K.
3.5. Experimental Procedure for Gene Expression
Load device as in 3.4
Image plate every 24 hours using a microscope to monitor cell growth and well collapse
After 48 hours of culture aspirate excess media and agarose gel from all center wells to expose the 09–1 cells
Add 100μL of QIAGEN QIAzol to the center well of a device then remove and add the QIAGEN QIAzol to a second center well (see Note 8).
Isolate the lysis from these 2 wells to create a single sample in a 1.5 mL microfuge tube. Repeat step 4 for all remaining wells
Add 25μL of chloroform to each lysis mixture and shake vigorously
Place samples at room temperature for 2 minutes
Centrifuge samples at 12000 x g for 15 minutes at 4°C
Transfer aqueous phase of samples to new PCR tubes (see Note 9)
Continue RNA extraction using a Qiagen MagAttract RNA Universal Tissue M48 Kit standard protocol
Use the Applied Biosystems High-Capacity cDNA Reverse Transcription Kit standard protocol to perform reverse transcription reactions
Perform qRT-PCR using a Roche Life Science LightCycler 480 Probes Master Protocol
3.6. Evaluation of Gli-driven Luciferase
For 3D luciferase activity assays, include 2mM VivoGlo luciferin and 25mM HEPES in culture media used in 3.4 (see Note 10)
Use a Biorad Chemicdoc luminescent imager or similar (CCD based maintains spatial data to monitor gradients) or Pherastar plate reader (rapid quantification of activity, dwell time = 2 seconds/well) to measure luminescence. Readings should be obtained prior to dosing as well as after the dosing period to enable normalization of any background luminescence. Sample images can be seen in Figure 4.
Place a 2X dissecting microscope lens between the camera and the device to obtain magnified images of the SHH-mediated gene expression gradient
Perform quantification using appropriate image analysis software
Figure 4:

Luminescent imaging of microtissues. A) Microtissues composed of 3T3 cells embedded in hyaluronic acid overlayed with GMSM-K RFP expressing cells top The blue indicates SHH pathway activity. B) i. Fluorescent image of a single microtissue with overlay on both sides and the top. The gradient of SHH can be seen along either edge where the tissue is flanked by epithelium ii. Fluorescent image of a single microtissue with overlay on the left side only.
3.8. H&E Staining
Fix microtissues in 10% formalin for 24 hours
Use a razor blade to remove the bonded polystyrene sheet from each device
Use a sharp probe to extract the microtissue from the device
Embed the microtissue in paraffin
Section the tissue at 5μm
Stain tissue with Hematoxylin and Eosin (H&E) via standard protocols.
Image tissue
Notes
Tweezers may be used to help clean the device during rinsing with DI water. Plastic chips from machining can buildup and become lodged in the fine features of devices.
There are two main ways to setup the plate for bonding. The skirt of the plate can be removed through machining to allow for a flat surface for bonding. A piece of metal that fits within the skirt while providing adequate coverage of the device is another option. A layer of thin fabric between the metal and polystyrene sheet can help reduce scratching. The fabric would be placed on the metal, followed by the sheet and plate. Leaning on the plate while adding the ACN provides downward force to aid in bonding. Use of a fume hood or other method of providing air flow while bonding is recommended while working with ACN. We also tried .12 and .17mm polystyrene sheet and were able to successfully bond both thicknesses.
The ACN needs to be aspirated immediately after pipetting into the device to avoid prolonged contact and subsequent etching, proper chemical safety precautions including use of a chemical fume hood and charcoal canister in the vacuum line (to capture solvent vapor and prevent damage and potential fire at the pump) must be strictly adhered to.
Polyethylenimine and glutaraldehyde will be toxic to cultures if thorough washes with DI water are not completed
Both the epithelial and mesenchymal cell lines were engineered for in situ visualization. The 09–1 cells express RFP and the GMSM-K cells express GFP or RFP. GMSM-K RFP show no little basal SHH secretion and GMSM-K GFP-SHH cells overexpress SHH[16].
The GMSM-K cells can be loaded into one or both channels of each device. 5μL of the GMSM-K cell solution can also be loaded into the center well, directly on top of the mesenchyme. This can help to increase signal for screening but prohibits visualization of gradients.
Adding media to the center well creates a hydraulic head that induces gravity driven perfusion of the created microtissue. Composition of the ECM determines rate of flow.
The reagent used in this step contains phenol which is incompatible with polystyrene. This step must be carried out quickly to avoid problems due to this incompatibility.
From top to bottom the phases in the PCR tube following centrifuging are: aqueous, interphase, and organic.
Persistent exposure to 2mM luciferin added to culture media did not show any adverse cytotoxic effects nor reduction in luminescent signal in presence of SHH ligand in our cultures, but this may be cell-type specific. We used in vivo grade luciferin.
5. References
- 1.Bush JO, Jiang R (2012) Palatogenesis: morphogenetic and molecular mechanisms of secondary palate development. Development 139 (2):231–243. doi: 10.1242/dev.067082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jiang R, Bush JO, Lidral AC (2006) Development of the upper lip: morphogenetic and molecular mechanisms. Dev Dyn 235 (5):1152–1166. doi: 10.1002/dvdy.20646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lan Y, Xu J, Jiang R (2015) Cellular and Molecular Mechanisms of Palatogenesis. Curr Top Dev Biol 115:59–84. doi: 10.1016/bs.ctdb.2015.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kurosaka H (2015) The Roles of Hedgehog Signaling in Upper Lip Formation. Biomed Res Int 2015:901041. doi: 10.1155/2015/901041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lan Y, Jiang R (2009) Sonic hedgehog signaling regulates reciprocal epithelial-mesenchymal interactions controlling palatal outgrowth. Development 136 (8):1387–1396. doi: 10.1242/dev.028167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Heyne GW, Melberg CG, Doroodchi P, Parins KF, Kietzman HW, Everson JL, Ansen-Wilson LJ, Lipinski RJ (2015) Definition of critical periods for Hedgehog pathway antagonist-induced holoprosencephaly, cleft lip, and cleft palate. PLoS One 10 (3):e0120517. doi: 10.1371/journal.pone.0120517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lipinski RJ, Bushman W (2010) Identification of Hedgehog signaling inhibitors with relevant human exposure by small molecule screening. Toxicol In Vitro 24 (5):1404–1409. doi: 10.1016/j.tiv.2010.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson BP, Vitek RA, Morgan MM, Fink DM, Beames TG, Geiger PG, Beebe DJ, Lipinski RJ (2021) A Microphysiological Approach to Evaluate Effectors of Intercellular Hedgehog Signaling in Development. Front Cell Dev Biol 9:621442. doi: 10.3389/fcell.2021.621442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beames TG, Lipinski RJ (2020) Gene-environment interactions: aligning birth defects research with complex etiology. Development 147 (21). doi: 10.1242/dev.191064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knudsen TB, Klieforth B, Slikker W, Jr. (2017) Programming microphysiological systems for children’s health protection. Exp Biol Med (Maywood) 242 (16):1586–1592. doi: 10.1177/1535370217717697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Regehr KJ, Domenech M, Koepsel JT, Carver KC, Ellison-Zelski SJ, Murphy WL, Schuler LA, Alarid ET, Beebe DJ (2009) Biological implications of polydimethylsiloxane-based microfluidic cell culture. Lab Chip 9 (15):2132–2139. doi: 10.1039/b903043c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferguson MW (1988) Palate development. Development 103 Suppl:41–60 [DOI] [PubMed] [Google Scholar]
- 13.Kaushik G, Gupta K, Harms V, Torr E, Evans J, Johnson HJ, Soref C, Acevedo-Acevedo S, Antosiewicz-Bourget J, Mamott D, Uhl P, Johnson BP, Palecek SP, Beebe DJ, Thomson JA, Daly WT, Murphy WL (2020) Engineered Perineural Vascular Plexus for Modeling Developmental Toxicity. Adv Healthc Mater 9 (16):e2000825. doi: 10.1002/adhm.202000825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen JK, Taipale J, Young KE, Maiti T, Beachy PA (2002) Small molecule modulation of Smoothened activity. Proc Natl Acad Sci U S A 99 (22):14071–14076. doi: 10.1073/pnas.182542899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharma A An Ultraviolet-Sterilization Protocol for Microtitre Plates. In, 2012.
- 16.Ishii M, Arias AC, Liu L, Chen YB, Bronner ME, Maxson RE (2012) A stable cranial neural crest cell line from mouse. Stem Cells Dev 21 (17):3069–3080. doi: 10.1089/scd.2012.0155 [DOI] [PMC free article] [PubMed] [Google Scholar]