

Abstract
Development and function of the heart are crucially controlled by individual genetic factors determining the continuum from health to disease. A prime example of a quantitative pathological phenotype is left ventricular hypoplasia, affecting the left ventricle (LV) and its associated inflow and outflow structures and reducing its size to varying degrees1. Current models suggest polygenic contributions on heart development and disease, but we are just beginning to understand the impact of naturally occurring sequence variations. Here we show how natural genetic variants exert cumulative, environment-sensitive effects on both embryonic heart development as well as adult heart function. We addressed this in embryos of five inbred strains of the Japanese rice fish medaka (Oryzias latipes2 and Oryzias sakaizumii3), analyzing heart rates as a functional proxy to capture combined characteristics of the conduction system and structural morphology. We identified one strain, HO52, with embryonic hypoplastic ventricles, fast heart rates, and significantly impaired cardiac function and overall fitness in adults. We created an interpopulation cross of the divergent HO5 strain to HdrR, which exhibits a morphologically normal heart and comparably slow heart rates, to leverage segregation variance in the second filial generation (F2) for genetic mapping. The correlation of 1192 individually phenotyped F2 embryos (heart rate) with the corresponding whole-genome sequences established a single nucleotide polymorphism-based linkage map. Integrated analysis of parental cardiac transcriptomes highlighted 59 loci containing genes prominently linked to human cardiovascular phenotypes. In vivo knockout models of the top 12 candidate genes demonstrated a causal and differential impact on heart development, ventricular size, and heart rate, evidencing the polygenic nature of heart phenotypes and establishing new loci for genetic diagnostics of human congenital heart disease. The controllable and scalable genetics and experimental validation in inbred vertebrate models complement and expand human association studies to provide novel and unique insights into the role of natural genetic variation in the mechanism of cardiac disease and allow to identify genetic susceptibility in individual human subjects.
Congenital heart disease (CHD) has a global prevalence of approximately 1% of live births and can severely impair health from infancy to adulthood4,5. CHD phenotypes generally have specific morphological and functional hallmarks that can be assessed quantitatively and are typically not binary; instead, they manifest along a continuum that stretches from healthy to pathological forms. This causes a dilemma for both diagnosis and research into the early stages of the disease because normal variability between individuals examined pre- or postnatally can mask quantitative phenotypes. For example, LV hypoplasia is a quantitative phenotype ranging from mildly reduced ventricular size to a diminutive left ventricle observed in hypoplastic left heart syndrome (HLHS)1. While the pathophysiological description of HLHS dates back to 18516, its hereditary nature7 has been a moving target as the genetics are not simple: HLHS is thought to be the outcome of multiple genetic factors that interact in an environmentally sensitive way8,9. As a result, studies of cardiac traits have shifted from single gene analysis to exome- or genome-wide sequencing. Most of these efforts have recovered private mutations (found only in one individual) and thus emphasize the polygenic nature of CHD phenotypes. Due to the limitations posed by a single genome, it has not been possible to assess how an individual’s entire collection of genomic variants drives cardiac phenotypes in processes of health and disease. Identical twins or even better isogenic lines overcome the limitations of a single genomic context and allow incorporating the variability in individual phenotypes and genotypes, environmental factors, genetic relatedness and population stratification10–13.
One step toward resolving these issues is to turn to well-established models of cardiac development and function. Here, we use inbred strains of the teleost medaka14 (Oryzias latipes and Oryzias sakaizumii) to resolve genomic variant complexity underlying quantitative cardiac trait variability. These strains represent fixed states of individually composed natural genetic variants crossed to isogenicity. They allow capturing a snapshot in the spectrum of phenotypic variation and establishing correlations with the underlying genotype by quantitative readouts in a controlled environment. We used the heart rate as proxy for heart function and examined the phenotypic distributions of five highly inbred medaka strains using automated heartbeat detection in a controlled environment. We identify two strains, HO5 and HdrR, that differ most widely in terms of the basal heart rate. With ongoing development, the HO5 strain exhibits a hypoplastic ventricle that severely impairs fitness upon reaching adulthood. We identified 59 significant loci with a maximum association peak on medaka chromosome 3 in a quantitative trait locus mapping based on heart rate and whole-genome sequences of 1192 individually phenotyped embryos of a two-generation segregation population. We refined these loci with differential gene expression analyses in the hearts of the parental strains and teleost-to-human comparisons. In vivo CRISPR-Cas9 and base editor-mediated gene inactivation demonstrated the causal impact of specific genes on the heart rate and morphology of the developing heart.
Heart phenotype contrasts
The heart rate is a product of the properties of the conduction system and structural morphology of the heart, which means it can be used as a readout of the way these properties are connected. We used automated microscopy and image-based heartbeat quantification under controlled physiological conditions15, to study the maximum range of differences between phenotypes in highly homozygous inbred medaka strains/species from southern (HdrR, iCab, HO5; O. latipes16) and northern (Kaga, HNI; O. sakaizumii3 Japan (Fig. 1a). We intentionally selected inbred strains/species of different geographic origins, assuming that genetic differences derived from their founder populations are relevant for cardiac phenotypes. Heart rates build up with developmental time and we addressed the dynamics of embryonic heart rates in four-hour intervals from the onset of heartbeat to the pre-hatching stage (Fig. 1b). The heart rate profiles we obtained revealed a significant and consistent spread between the five inbred strains, with the most prominent contrast between the southern O. latipes strains HO5 (fastest heart rate) and HdrR (slowest heart rate, Fig. 1b).
Fig. 1: Cardiac phenotype contrast in medaka inbred strains.

a, Layout of the automated heartbeat detection in medaka embryos in native environment (28°C) using high-throughput imaging and image-based heart rate quantification. b, Distribution of embryonic heart rates in five inbred strains derived from Southern Japanese medaka populations (HdrR, HO5, iCab) and Northern Japanese populations (HNI, Kaga) across embryonic development starting with the onset of heartbeat. Heart rates of 6–18 embryos per strain were determined every four hours under a 12 h-light/12 h-dark cycle; smoothed line between single (4 h interval) heart rate measurements for each strain (Supplementary Table 1); dotted lines, window of circadian-rhythm-stable heartbeat for comparative analysis (100–104 hours post fertilization, hpf). c, Cardiac morphology in HdrR and HO5 hatchlings; end-systolic frame, scale bar, 50 μm. d, e, Exercise assessment and swim performance of adult fish in a swim tunnel assay. White line in e used for kymograph - note stable swimming behavior in HdrR versus fluctuating HO5 individual. f, Pulsed-wave (pw) doppler of ventricular inflow (atrium-ventricle) and outflow (ventricle-bulbus arteriosus) tracts.
We ensured consistency by measuring heart rates in the time window between 100–104 hpf (dotted rectangle, Fig. 1b), after the completion of cardiovascular development17 and thus avoided the impact of circadian oscillations observed from 3 dpf onwards15.
The differences in embryonic heart rate were substantial, prompting us to examine cardiac morphology at 4 dpf (raised at 28 °C), the time at which the critical stages of cardiac development are complete17. Our analyses revealed that HO5 embryos exhibited an imbalance between the dimensions of the atrium, which was relatively large, and an underdeveloped (hypoplastic) ventricle. This apparent hypoplastic ventricular morphology was associated with a high basal heart rate (Fig. 1c), potentially compensating the reduced ventricular volume. As the unique combination of natural genetic variation fixed in the HO5 genome contributes to its cardiac phenotype, we next assessed the extent to which it influences the function and physical fitness of the heart at adult stages. We subjected adult HO5 and HdrR individuals to a swim tunnel exercise protocol (Fig. 1e). Video monitoring indicated the efficient performance of HdrR; fish assumed stable positions in a defined water flow, requiring minimal fin excursions to generate the necessary swimming speed (Fig. 1f and Supplementary Video 1). In contrast, HO5 individuals (apparently higher BMI) used almost the entire body length to generate forward movement. In addition, they failed to assume stable positions within the constant water flow (cf. kymograph in Fig. 1e). Following swim tunnel exercise, we found that HO5 exhibited significantly reduced velocities in intracardiac blood flow, which are generated by sequential contractions of the atrium and ventricle, as observed using echocardiography, including pulse-waved (pw) Doppler measurements of the ventricle in- and outflow (Fig. 1f). These findings argue for early alterations of physiological cardiac traits that occur in HO5 embryos and severely impact on physical fitness and cardiovascular health in adulthood.
Segregation analysis
To map the loci contributing to these complex phenotypes, we established a mapping population in which we correlated heart rates and whole-genome sequences (WGS). To model a human-relevant modifiable environmental factor of embryonic development, we turned to the well-established heart rate increase in elevated water temperatures15 and measured embryonic heart rates at an incremental ambient temperature ramp (21°C, 28°C, 35°C). For mapping we employed a two-generation segregation design and used single-nucleotide polymorphisms (SNPs) as markers (Fig. 2a). SNP calling in HO5 WGS against the HdrR reference genome established 979,713 differential homozygous SNPs as marker system.
Fig. 2: F2 segregation analysis reveals temperature-sensitive QTLs affecting the heartbeat.

a, Crossing setup used to generate HdrR × HO5 offspring with segregated SNPs in the second generation: isogenic HO5 and HdrR parents are crossed to generate hybrid (heterozygous) F1 generation (grey) with intermediate phenotype, which after incrossing results in F2 individuals with individually segregated SNPs resulting from one cycle of meiotic recombination. Automated embryonic heartbeat analysis at 4 days post fertilization (dpf), performed on all 1260 individual embryos and challenged by increasing temperatures (21 °C, 28 °C, 35 °C). Whole-genome sequencing of measured individuals (1192 genomes) with an effective average coverage of 0.78× allows phenotype-genotype correlation. b, Individual embryonic heart rates of the inbred strains HdrR (slow heart rate, blue) and HO5 (fast heart rate, orange) increase with temperature (21 °C, 28°C and 35°C). The F2 individuals with recombined HdrR × HO5 genomes (green) span the range of parental (F0) heart rates between the two strains with a subgroup of F2 individuals exhibiting heart rate variance beyond the parental extremes (21 and 28 °C); sample sizes (n) for 21°C, 28°C and 35°C: n (HdrR F0) = 35, 35, 35, n (HO5 F0) = 20, 22, 22, n (HdrR × HO5 F2) = 1260, 1260, 1260. c, Minus log10 p values from genome-wide association tests of recombination block genotypes and heart rate measures at different temperatures using a linear mixed model. Chromosomes 3 and 5 hold the most segregated recombination blocks associated with heart rate differences. Twelve selected genes from the loci passing the significance threshold are indicated; red line: 1% false discovery rate (FDR), blue line: 5% FDR, determined by permutation.
We crossed the two isogenic strains with distinctly differentiated cardiac phenotypes to generate a hybrid F1 population with haploid sets of chromosomes from each parental strain and observed heart rates that were intermediate between the two parental strains. We next performed F1 intercrosses and sampled 1260 individuals of the F2 generation with unique inherited genotypes (linkage blocks) to serve as the mapping population (Fig. 2a). We conducted individual heart rate phenotyping of all 1260 F2 embryos and found (with a few outliers showing sporadic arrhythmia) that the phenotypic distribution covered the range between the parental phenotypes at all three temperatures (Fig. 2b). This reflects the differential segregation of multiple alleles that directly impact the heart rate or secondarily due to morphological-functional effects on the heart.
Genome-wide QTL mapping
We raised individually phenotyped F2 embryos in 96-well plates compatible with automated DNA extraction, library preparation, and sequencing. Out of the 1260 phenotyped F2 offspring embryos, we whole-genome sequenced 1192 samples with an average coverage of 0.78×. We used a three-state Hidden Markov Model (HMM) to segment all crossover locations and determine genotype states (AA, AB, BB) based on SNPs homozygous divergent in HO5 (AA) and HdrR (BB). Interestingly, we observed a distortion in the expected Mendelian ratio of 1:2:1 (AA:AB:BB alleles) with an overrepresentation of AA (HO5) alleles suggestive of a potential reference bias. This bias was evident in most F2 offspring samples (Extended Data Fig. 1) and was not restricted to specific regions of the genome. Although it could be possible that certain crossover events between HdrR and HO5 are incompatible, the most parsimonious explanation is a tendency towards homozygous reference calls within the SNP genotype calls used to train the HMM. Having called crossover events and generated a recombination map across all F2 offspring samples independently, we merged the crossover locations and segmented the genome at every breakpoint resulting in a genotype matrix containing AA:AB:BB calls for variable sized blocks. The median genotype block size, once segmented across all F2 cross recombination positions, was 24kb.
Using this genome matrix we then conducted genome-wide association analyses of 101,265 segmented recombination block regions on individual heart rate measures at three different ambient temperatures and for absolute differences in repeated measurements across all temperatures 21°C, 28°C and 35°C (variance phenotype), using a linear mixed model from the Grid_LMM software package18. We found significantly associated quantitative trait loci (QTLs) that were mostly consistent across the three temperatures (Fig. 2c) and different peaks in the test on heart rate variance from 21–35°C (Extended Data Fig. 2). Overall we detected 1385 significant loci across all phenotype tests and performed collapsing down to 59 distinct fine-mapped regions after linkage disequilibrium (LD)-based SNP pruning. The maximum achievable resolution for loci fine mapping was 10kb due to the window size used for recombination block mapping, and the median size across all fine-mapped loci was 115 kb. We were able to fine-map to 10 kb for only 5/59 loci; however, most fine-mapped regions contained small numbers of genes (median of 1 and a range between 0–36 genes per fine-mapped block). Overall we detected similar numbers of fine-mapped loci for the different phenotype measures we tested, with 17, 16, 8, and 17 unique fine-mapped regions for 21°C, 28°C, 35°C and the variance-based phenotype, respectively.
To aid gene prioritization and interpretation of significantly associated loci in the F2 population, we undertook total RNA sequencing of heart tissues in 4 samples from both the HO5 and HdrR strains. After quality control 18,321 genes (75% of known medaka genes) had sufficient coverage to allow differential expression analysis. At a false discovery rate (FDR) of 0.01 and minimum fold change of 2 we detected 1,161 significantly differentially expressed genes between HO5 and HdrR hearts (Extended Data Fig. 3a), which we then used to prioritize genes within significantly associating F2 recombination blocks for subsequent validation experiments. Genes expressed in liver samples of both strains were used as base line reference (Extended Data Fig. 3b). Additionally we assessed the likely impact of SNP calls in HO5 against the HdrR reference using the variant effect predictor (VEP) from Ensembl19. As expected, loss-of-function (LoF) variants were rare within these blocks with a median of 0 variants and maximum of 3 per gene. However using the combination of information it was possible to rank genes and variants more likely to be impactful for the observed phenotypic differences. One case supported by LoF variation is a single premature stop codon variant in the blzf1 gene, and in contrast the rrad gene where no LoF variants in HO5 were observed but a significant differential expression in heart tissues.
Gene enrichments
Across all 59 significantly associated fine mapped regions there were 173 annotated genes in the Oryzias latipes (HdrR) ENSEMBL reference, 28 of the fine mapped regions had no annotated gene within their boundaries (8, 10, 6 and 4 loci with no annotated genes for heart rate phenotypes at 21°C, 28°C, 35°C, and variance respectively), leaving 34 containing 1 or more genes with a median of 3 per fine mapped region. The largest region was found on chromosome 1 associated with the variance phenotype and contained 36 genes, only two of which had any LoF variant called in HO5 against the HdrR reference (Supplementary Table 2, block ID-2). Using the 173 annotated genes, we performed multiple gene enrichment models using the PANTHER overrepresentation test (released 20230705), gProfiler functional profiling and functional annotation clustering using DAVID20–22. Using the overrepresentation test from PANTHER, we found significant associations (FDR P < 0.05) for 72 different biological process GO terms including for “thrombocyte differentiation”, which has a greater than 100-fold enrichment. Both gProfiler and DAVID found a significant enrichment (adjusted P value 4.393×10–10) for the GO term “galactoside binding” with 6 of the 172 genes being involved in the binding of glycoside carbohydrate derivatives, as well as a weaker but significant enrichment (adjusted P value 1.895×10–2) for the KEGG term “Linoleic acid metabolism”. For functional annotation clustering using DAVID with a classification stringency set to Medium using the Benjamini adjustment, there were 21 clusters defined, the strongest of which had an overall enrichment score of 4.56 and included the GO terms “galactoside binding” and “laminin binding”.
Recent studies in humans have looked at the pleiotropic regulatory activities of Galectins in relation to cardiovascular disease (CVD) and their proinflammatory role in the atherosclerotic and plaque formation process23. Here we found 6 Galectin related genes (ENSORLG00000030479, ENSORLG00000026315, ENSORLG00000010700, ENSORLG00000010715, ENSORLG00000010697, ENSORLG00000024256) to be associated to heart rate differences in medaka, associated to the variance in heart rate and found within the same fine mapped region at chromsome 8:13655000–13935000 (Supplementary Table 2, block ID-42). Galectins are promiscuous, with multiple cellular functions and impede arteriogenesis24. Consequently, galectin inhibitors are an attractive target for therapeutics pertaining to remodeling in myocardial infarction25. Thus, associations based on medaka heartbeat dynamics directly surfaced genes with highly relevant translational implications.
In vivo validation
Our genome-wide QTL mapping identified loci that impact both, the cardiac structure and heartbeat, as the heart rate applied in the mapping is a combined result of the properties of the conduction system and the heart’s morphology. To provide evidence for a causal role of the identified loci, we selectively deactivated genes in medaka embryos and investigated the respective impact on heart development, function, and heart rate in vivo. After analyzing the search space (Extended Data Table 1), we narrowed our selection to 12 candidate genes based on their high linkage probabilities, human relevance, and novelty (cf. Methods “Candidate gene selection”): adprhl1, blzf1, btbd1, gpc5a, gse1, irf1, pcdh17, phka2, ptprd, rrad, sec61a1, and zrsr2. Within this set, rrad and adprhl1 have previously been associated with cardiovascular traits, confirming the validity of this approach in identifying candidate genes. To examine potential functional effects more closely, we assessed cardiac-specific phenotypes, including looping defects, pericardial edema, arrhythmia, and aberrant atrial and ventricular size. Targeted inactivation was achieved by CRISPR-Cas9- or base editor-mediated knockout in embryos, and analysis was performed after cardiovascular development was expected to be complete at 4 dpf (28°C). Phenotyping was performed on all embryos devoid of global developmental defects (Fig. 3, Extended Data Fig. 4, Extended Data Table 2).
Fig. 3: Heart phenotypes in knockout models of candidate genes.
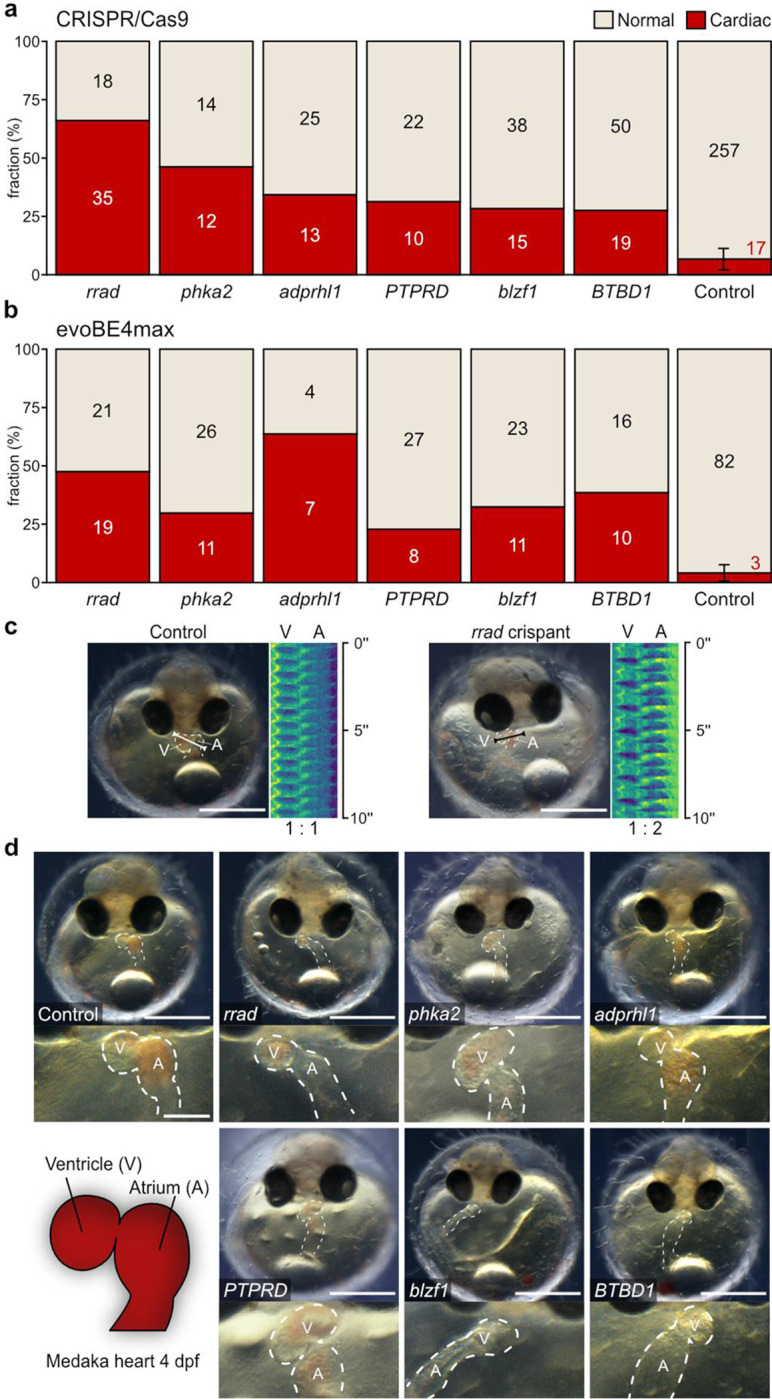
a, Proportion (bars) and counts (values) of cardiac affected and normally developed embryos after CRISPR-Cas9-mediated knockout of indicated candidate genes versus control (mock injection) quantified at 4 days post fertilization (dpf). b, Independent replication using base editing: phenotypic distribution (proportion/bars and counts/values) resulting from targeted gene inactivation mediated by introducing premature termination codons via the cytosine base editor evoBE4max and a set of distinct guide RNAs targeting the same genes as in (a). c, Heart rhythm analysis as depicted by kymographs derived from atrium (A) to ventricle (V) spanning line selection in 10 second time-lapse movies. In contrast to the regular rhythm in the control embryo (left), the representative rrad crispant (right) displays a 2:1 atrioventricular block; cf. Supplementary Video 2. d, Representative cardiac phenotypes of knockout embryos for all six candidate genes targeted with CRISPR-Cas9 and base editor compared to a control embryo at 4 dpf; bright-field overview of the injected specimen (top; scale bar, 500 μm), close-up image of the heart (bottom; scale bar, 125 μm); cf. Supplementary Video 3.
Our targeted inactivation of candidate genes prominently increased the proportion of cardiac phenotypes above the baseline threshold established by injected controls (max. 7% heart phenotypes; Fig. 3a and Extended Data Fig. 4). To underline the specificity of observed phenotypes, we replicated the results of the CRISPR-Cas9 gene interrogations in an independent experimental series, i.e., we introduced premature termination codons (PTCs) with the cytosine base editor evoBE4max in six out of the twelve selected candidate genes (Fig. 3b). Specifically, adprhl1, blzf1, btbd1, phka2, ptprd, and rrad showed high proportions of prominent heart phenotypes in the CRISPR-Cas9 approach. Introducing PTCs in these genes through base editing yielded similar phenotypic profiles.
We then used the automated heart rate assay to assess the specific effects of candidate genes on heart rate in embryos with normally developed hearts (Supplementary Table 3). We found that in eight out of the twelve candidate genes the heartbeat was affected. While six of them showed an increase in heart rate across the three different temperatures tested (Fig. 4), interfering with zrsr2 and gse1, however, resulted in a marked decrease in heart rate (Extended Data Fig. 5).
Fig. 4: Loss-of-function mutations in candidate genes affect the heart rate in medaka embryos.

a, Validation workflow scheme encompassing zygotic microinjections using a HdrR (myl7::eGFP; myl7::H2A-mCherry) reporter line, high-throughput image acquisition at 4 days post fertilization (dpf), and heart rate quantification in normally developed injected specimens. b,c, Heart rate distributions of morphologically normal crispants and mock-injected control embryos at 4 dpf at 21°C, 28°C, and 35°C with CRISPR-Cas9-mediated (b) and base editor-mediated (c) targeted mutagenesis/silencing of candidate genes. The significance of heart rate differences between the mutant group and its corresponding control was assessed using the Wilcoxon test; *, p<0.05; **, p<0.01; ***, p<0.001 (p-values listed in Extended Data Table 3). Data is visualized as box plots (median+/− interquartile range) and overlaid scatter plots of heart rate measurements; knockouts of adprhl1, btbd1, blzf1, phka2, and rrad have temperature-dependent effects on heart rate.
Taken together, the F2 gene segregation workflow combined with quantitative phenotyping and targeted mutagenesis for validation is a powerful tool to uncover (novel) risk factors for human heart disease and beyond.
Discussion
Human sequencing studies have argued that most congenital heart diseases are polygenic in nature. We resolved the polygenic contribution using highly inbred medaka strains with different cardiac phenotypes and uncovered specific SNPs that contribute to differences in heart rate and disproportionate ventricular morphology, two phenotypes interrelated in human CHD. Analyzing almost 1200 genomes and the corresponding heart rate as readout with predictive power26,27, we identified 59 QTLs at high resolution. 173 of the genes contained in these loci showed a direct link to heart as well as extracardiac organ functions, demonstrating the polygenic architecture of quantitative heart phenotypes.
We targeted a subset of these genes to validate their potential roles in the development of cardiac phenotypes, based on a differential expression analysis in medaka and an orthology-guided examination of genes associated with human heart phenotypes. We carried out CRISPR-Cas9- and base editing-mediated inactivation of these candidates and uncovered genes involved in heart rate and rhythm control and structural development of the heart. Intriguingly, using heart rate metrics for phenotype-genotype mapping, we found genes with dual roles affecting heart muscle mass and rhythm; e.g., rrad combining regulative functions on cardiac muscle strength and electrophysiology. It is known to inhibit cardiac hypertrophy through the CaMKII pathway with implications for heart failure28, and is associated with hypertrophic cardiomyopathy (HCM) phenotype in RRAD-deficient cell line29. RRAD mutations detected in a specific form of familiar arrhythmia (Brugada syndrome) trigger cytoskeleton and electrophysiological abnormalities in iPSC-CMs30. This demonstrates the power of complex cardiac trait analysis in medaka inbred strains to detect human-relevant association signals, guiding genome editing experiments to uncover novel players and potential targets in heart disease.
We noticed a significant decrease in ventricular size in a subset of gene knockouts (rrad, adprhl1, blzf1, and btbd1), and it appears that adprhl1 has a major impact on ventricular dimensions. Adprhl1 is a conserved pseudoenzyme with ADP-ribosylarginine hydrolase and magnesium ion binding activity, likely exclusive to the heart. Transcriptomics revealed very strong cardiac expression (3,109.244 RPM compared to weak (1.175 RPM) in the liver) in medaka. Until now, adprhl1 has been studied functionally only scarcely. Of note, in Xenopus, adprhl1 can localize to the cardiac sarcomeres. While morpholino-mediated knockdown of adprhl1 did not affect early cardiogenesis, it disrupts myofibril assembly in the forming ventricle and leads to small, inert ventricles31. In light of the hypoplastic ventricle observed in the HO5 strain, its location on a strongly associated QTL on chromosome 3 in medaka, and targeted inactivation in vivo, we provide evidence for adprhl1 as a strong contender for controlling myofibril assembly and ventricular outgrowth.
The homozygous fixation of causative genomic variants in viable inbred medaka strains allows not only for modeling twin studies with arbitrary scalability but also for longitudinal investigation to estimate the variants’ effects within a lifespan. Notably, we found an inverse relation of metrics for embryonic heart rate and ventricle in the HO5 strain, which exposes pathological heart phenotype with impaired cardiac function and physical performance in exercise tests, demonstrating the predictive power of genotype-specific embryonic heart rate profiles. Finding such early biosignatures is essential for diagnosing and potentially preventing severe pathophysiologies that become less likely correctable over the long term.
Most cardiac disease phenotypes are quantitative and manifest in a health-disease continuum. Here, we demonstrate that inbred medaka is a powerful resource to partition the spectrum of phenotypes in a strain-specific way. Examining the extremes of a physiological trait range bears the power to reveal the genetic factors likely associated with disease susceptibility. The F2 cross of inbred strains with contrasting phenotypes can help us segregate causal alleles in a way that cannot be achieved with human GWAS studies. However, the results can be transferred into the human context and may shed light on genetic variations that have flown under the radar.
Our study demonstrates that inbred medaka has an important potential for cardiac development, physiology and disease. Our mapping identifies and validates novel heart-related genes. By combining a scalable medaka inbred panel32–34 with highly reproducible, quantitative phenotyping assays (beyond the heart, see accompanying manuscript by Seleit et al.) and advanced crossing designs, we expect to significantly increase the resolution by which candidate causal variants in the coding and non-coding regions of the genome can be mapped and validated.
Methods
Fish maintenance
The wild-type Kaga, HNI, iCab, HO5 and HdrR strain and a fluorescent cardiac reporter line HdrR (myl7::EGFP myl7::H2A-mCherry) were used in this study. All fish stocks were maintained (fish husbandry, permit number 35–9185.64/BH Wittbrodt) and experiments (permit numbers 35–9185.81/G-145/15, 35–9185.81/G-10/17 and 35–9185.81/G-271/20) were performed in accordance with local animal welfare standards (Tierschutzgesetz §11, Abs. 1, Nr. 1) and with European Union animal welfare guidelines35. Fish were kept as described previously36. The fish facility is under the supervision of the local representative of the animal welfare agency.
Automated microscopy and heartbeat detection of medaka embryos
The automated microscopy and heart rate quantification was applied as described previously15. Embryonic heart rate profiles over developmental stages were generated by imaging embryos in 96-well plates every four hours under a 12 h-light-12 h-dark-cycle, i.e., 12 h dark minus imaging time of 20 min/plate/time point; incubation temperature was 28 °C (developmental profiles) and 21°C, 28°C, and 35°C in the screen. For display “smoothed conditional means’’ encoded in ‘geom_smooth()’ using regression method = ‘loess’ (ggplot2) with 95% confidence interval was used (Supplementary Table 1).
All F0, F1 and F2 embryos were imaged as separate batches in 96-well plates each with one row (12 embryos) of Cab embryos as an internal control. For phenotype-genotype correlations in individual F2 embryos, fifteen 96-well plates, each containing two F2 crosses (HdrRf × HO5m F2 and HO5f × HdrRm F2), were imaged and processed under the same conditions.
To exclude positional effects of plate coordinates on heart rate, a full 96-well plate with Cab embryos was recorded at 28°C. Heart rates were normally distributed (P=0.86; Shapiro-Wilk normality test). One-way ANOVA indicated no significant effects of plate row on mean heart rates (degrees of freedom (df) = 7.88; P = 0.61), nor of plate column on mean heart rates (df = 11.84; P = 0.15).
Swim tunnel assay and echocardiography of adult medaka fish
Two months before the experiments, HdrR F96 and HO5 F112 were kept at 22°C water temperature and 14 h/10 h light/dark conditions. Swim tunnel (Loligo Systems) assay was performed with the following flow velocity and settings. Equilibration at 3 cm/s for 20 min, then increase of water flow by 5 cm/s every 5 min; mO2 was scored at each step. Two 10 s videos with 100 fps were recorded at 15 cm/s and 20 cm/s (if applicable) during the interval after a flush. The test was stopped when fish remained 3 s or more at the rear of the chamber. Echocardiography was performed as previously described37 with 150 mg/l tricaine and 8 mg/l metomidate.
HdrR × HO5 intercross and phenotyping of F0, F1, and F2 embryos
For heart rate measurements in F0 (i.e., HO5 F112, HdrR F95, Cab F68), embryos were collected from these crosses: 3 female × 1 male HO5 F111, 3 female × 1 male HdrR F94 and as plate control 3 female × 1 male Cab F67.
Phenotyping F1 hybrid embryos and generation of F1 hybrid stocks: 3 HO5 F111 females × 1 HdrR F94 male resulting in stock HO5 F111f × HdrR-II F94m F1, and 3 HdrR F94 × 1 HO5 F111m resulting in stock HdrR F94f × HO5 F111m F1. Cab F68 embryos were used as plate control obtained from 3 female × 1 male Cab F67.
The embryonic F2 screen (heart rate) was conducted within two months. Two F2 populations were derived from two separate crosses of each F1 hybrid stock (1) HO5 F111f × HdrR F94m F1 and (2) HdrR F94f × HO5 F111m F1: F2 embryos were collected from stocks (1) and (2), for each of which 3 tanks with 3 females × 1 male were set up (both F2 populations derived each from 12 fish). Cab F68 embryos were used as plate control obtained from 3 female × 1 male Cab F67.
Incubation conditions for F0, F1 embryos: medaka embryos were raised at 28°C with either max. 20 embryos/20 ml medaka hatching medium in 60 mm dishes or with max. 50 embryos/50 ml medaka hatching medium in 90 mm dishes.
Incubation conditions for F2 embryos: 55 embryos were cultured at 28°C in 90 mm dishes with medaka hatching medium; Cab controls: 25 embryos in 9 cm dishes with hatching medium.
Embryo preparation for imaging: Embryos were rolled on sandpaper the afternoon/evening before imaging and placed back to the primary culture dishes with fresh hatching medium.
Heart rate assay (cf. “Automated microscopy and heartbeat detection of medaka embryos”): On the day of imaging, embryos were transferred from hatching medium to ERM before mounting. Individual F2 embryos were loaded with 150 μl ERM in 96-well plates (U-bottom). Approximate imaging times were: Start equilibration at 12:00, imaging 21°C at 12:15 PM, 28°C at 13:15 PM and 35°C at 14:15 PM.
F2 sample preparation and whole genome-sequencing
After imaging of F2 embryos in 96-well plates, ERM was exchanged with medaka hatching medium by pipetting out 140 μl ERM and adding 200 μl medaka hatching medium. Subsequently, 190 μl of the medaka hatching medium was exchanged daily until first embryos hatched. When the majority of embryos had hatched, the remaining unhatched embryos were manually dechorionated. All hatchlings were transferred from the 96-well imaging plate to identical coordinates of a 96-DeepWell plate, which was covered with an adhesive aluminum foil (4titude) and frozen acutely at −80°C until further processing. Samples were processed at Wellcome Sanger Institute (UK).
Library preparation and WGS on a HiSeq X Ten instrument (Illumina) with an average sequencing depth of 0.78×. Library preparation was performed following the standard PCR-free Illumina protocol38. In total, 1 μg was picked from DNA extraction plates within the Sanger sample logistics facility and passed into the Sanger sequencing pipeline from library preparation. Following successful preparation of sequencing libraries, the samples were QCed using Qubit and samples passing the facility quality control threshold were multiplexed sequenced in paired end mode with 150 samples per Illumina X10 flow cell.
Whole genome-sequence analysis and mapping
For the genotyping of recombination blocks in the F2 population, a modified algorithm of the previous version was used (https://github.com/tf2/ViteRbi). The analysis was based on SNPs homozygous divergent in HO5 (AA) and HdrR (BB). Homozygous divergent SNPs were determined by the alignment of high-depth HO5 genome sequencing data against the HdrR-II reference genome. At each homozygous divergent SNP-locus read counts supporting each genotype (A, T, C, G) were extracted from the genome alignments. In a fixed window of 5000 bp, read counts were summed and transformed into proportions of reads supporting the AA genotype. This genotype proportion was the observational input for a three-state Hidden Markov Model employing the Viterbi algorithm to segment all crossover locations and to determine genotype states (AA, AB, BB) in the resulting recombination blocks across all samples. Genome-wide association tests using recombination block genotypes and heart rate measures were performed using a linear mixed model18.
RNA sequencing and transcriptome analysis
Heart and liver samples were dissected from euthanized adult HO5 F112 and HdrR F96 female fish and collected into Qiagen Collection Microtubes (racked, 19560) with cabs (19566), acutely frozen on dry ice and then stored at −80°C. Total RNA purification was performed with QIAsymphony RNA, purifying liver samples with RNA CT 400 and heart samples with RNA FT 400. Samples were prepared for Illumina RNA sequencing using the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina and sequenced on a Hiseq 4000 sequencing platform following the manufacturer’s instructions. All pair-end reads were cleaned up using Fastp v0.20.039, aligned to the medaka HdrR reference genome Ensembl Release 98 (ASM223467v1) using STAR v2.7.3a40 and estimated counts per gene were obtained. Differential expression analysis was performed using DESEQ241 with an FDR cut-off of 0.01 and fold change of 2, 1161 and 1803 significantly differentially expressed genes were obtained for heart and liver samples respectively.
Candidate gene selection
QTLs were prioritized according to their strength of linkage. To filter the search space defined in Supplementary Table 2 we included recombination blocks (QTLs) with SNP-based genotypes significantly (log10 p-value>3) associated with heart rate variation. Only the strongest linkage to heart rate variation is given for each block compared at three temperatures (21°, 28°, and 35°C). Phenotype associations of orthologous human genes supported candidate gene selection relevant to human cardiac disease. Therefore, the human GWAS genome-wide association studies (GWAS) catalog (gwas_catalog_v1.0.2-associations_e104_r2021–10-22.tsv downloaded from https://www.ebi.ac.uk/gwas/docs/file-downloads)42 was scanned for terms indicative of heart rate and morphology associations. Candidate genes were additionally studied with the GeneALaCart batch query processor of the GeneCards suite (https://www.genecards.org)43. If blocks contain multiple genes, candidates were chosen according to defined criteria, primarily novelty and human relevance, positively (+) or negatively (−) ranking a candidate gene (Extended Data Table 1).
sgRNA and crRNAs target site selection
rrad (ENSORLG00000024517) sgRNAs, adprhl1 (ENSORLG00000004693) sgRNAs, PTPRD (ENSORLG00000004685) sgRNAs, phka2 (ENSORLG00000003555) sgRNAs, blzf1 (ENSORLG00000003670) sgRNAs, BTBD1 (ENSORLG00000003434) sgRNAs, zrsr2 (ENSORLG00000003476) sgRNA, pcdh17 (ENSORLG00000004535) sgRNA, SEC61A1 (ENSORLG00000016830) sgRNA, irf1a (ENSORLG00000011716) sgRNA, gse1 (ENSORLG00000009542) sgRNA, and gpc5a (ENSORLG00000026952) sgRNA were designed with CCTop and ACEofBASEs as described previously36,44, on the medaka genome in Ensembl release 101 (Japanese medaka HdrR assembly, Aug 2020), Ensembl release 102 (Japanese medaka HdrR assembly, Nov 2020), Ensembl release 103 (Japanese medaka HdrR assembly, Feb 2021) and Ensembl release 106 (Japanese medaka HdrR assembly, Apr 2022), respectively. The target sites and oligonucleotides selected for sgRNA cloning are listed in Supplementary Information. Cloning of sgRNA templates and in vitro transcription was performed as described previously44. The plasmid DR274 used was a gift from Keith Joung (Addgene 42250). For base editing, locus-specific crRNAs and the tracrRNA backbone were obtained from IDT (custom Alt-R crRNA). crRNA (100 μM) and tracrRNA (100 μM) were diluted in nuclease-free duplex buffer (IDT) to a final concentration of 40 μM and incubated at 95°C for 5 min.
In vitro transcription of mRNA
The plasmid pCS2+(Cas9) and pCS2+(evoBE4max) were linearized using NotI, and mRNA in vitro transcription was performed using the mMESSAGE mMACHINE SP6 or T7 Transcription Kit (Thermo Fisher Scientific) and purified with the RNeasy Mini Kit (Qiagen).
Microinjections
Microinjections were performed in HdrR (myl7::EGFP, myl7::H2A-mCherry) zygotes. The Cas9 injection solution contained 150 ng/μl Cas9 mRNA,10 ng/μl sgRNA and 10 ng/μl GFP mRNA as injection tracer. The base editor injection solution contained 150 ng/μl evoBE4max mRNA, 4 pmol sgRNA and 10 ng/μl GFP mRNA as injection tracer. Control injections were performed with 10 ng/μl GFP mRNA as injection tracer. Injected embryos were maintained at 28°C in medaka embryo rearing medium (ERM, 17 mM NaCl, 40 mM KCl, 0.27 mM CaCl2, 0.66 mM MgSO4, 17 mM Hepes) and selected for GFP expression 7 hours post injection. Phenotypes were assessed 4 days post fertilization.
Image acquisition and phenotyping of knockout models
Embryo morphology and heart dynamics were documented with a Nikon SMZ18 equipped with Nikon DS-Ri1 and DS-Fi2 cameras. Embryos were mounted in EMR into injection molds (1.5 % (w/v) agarose in ERM). Heart rate quantification of CRISPR-Cas9 and evoBE4max-mediated knockout embryos and control embryos were obtained using an ACQUIFER Imaging Machine and the HeartBeat software as described previously15,45. To acquire the heart rate, fluorescent image sequences have been captured for 10 sec with 24 fps at 21°C, 28°C, and 35°C.
Genotyping
Single phenotypic, non-phenotypic and control embryos were lysed in DNA extraction buffer (0.4 M Tris/HCl pH 8.0, 0.15 M NaCl, 0.1% SDS, 5 mM EDTA pH 8.0; 1 mg/ml proteinase K) at 60°C overnight. Samples were diluted 1:2 with nuclease-free water and the proteinase K was heat inactivated afterwards for 20min. Precipitation of genomic DNA was done in 300 mM sodium acetate and 3x vol. absolute ethanol at 20,000 × g at 4°C, followed by resuspension in TE buffer (10 mM Tris pH 8.0, 1 mM EDTA in RNAse-free water). The target loci were PCR amplified in 30 PCR cycles using Q5 High-Fidelity DNA Polymerase (New England Biolabs), locus-specific primer pairs (Supplementary Information) and 1 μl precipitated DNA sample. PCR products were purified after agarose gel electrophoresis using the Monarch DNA Gel Extraction Kit (New England Biolabs) and submitted for Sanger sequencing to Eurofins Genomics. Base editing results were analyzed with EditR46.
Data analysis and statistics
Data visualization and statistical analysis were performed using R47 (R Core Team, 2016).
Extended Data
Extended Data Fig. 1: Genotype proportions of the parental lines HdrR and HO5 in the sequenced F2 population.

a, Proportions of homozygous HO5 genotype (AA), homozygous HdrR genotype (BB) or heterozygous genotype (AB). b, Genotypes and recombination block distribution for the F2 samples (rows) across the 24 chromosomes (columns). AA = black, BB = green, AB = red.
Extended Data Fig. 2: Variance phenotype.

Manhattan plot showing −log10 p values from the linear mixed model using the variance phenotype (mean absolute difference between repeated measurements on the same embryo across the 3 different temperatures).
Extended Data Fig. 3: Differentially expressed genes in HdrR versus HO5 heart and liver samples.

a, b, Volcano plots of HdrR and HO5 heart (a) and liver (b) comparative transcriptomics, −log10(adj-pvalue) compared with log2 Fold change.
Extended Data Fig. 4: Heart phenotypes in CRISPR-Cas9 knockout models of candidate genes.
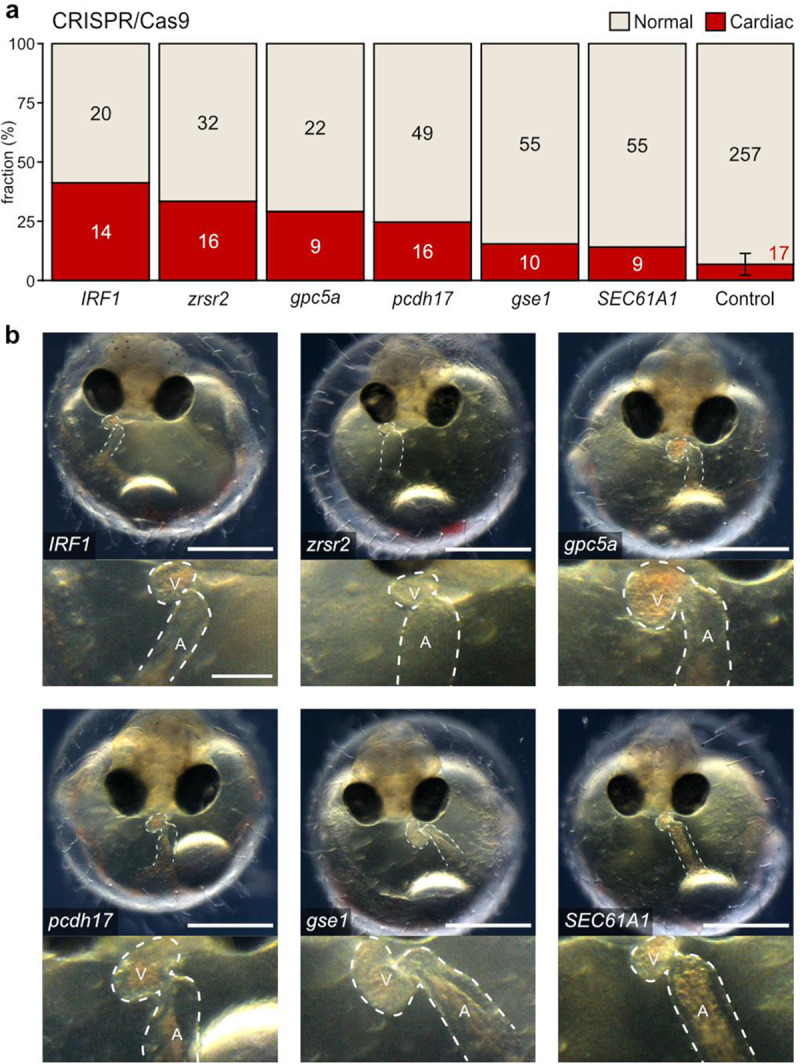
a, Proportion (bars) and counts (values) of cardiac affected and normally developed embryos after CRISPR-Cas9-mediated knockout of indicated candidate genes versus control (mock injection) quantified at 4 days post fertilization (dpf). b, Cardiac phenotypes of knockout embryos for the six candidate genes targeted with CRISPR-Cas9 at 4 dpf; bright-field overview of the injected specimen (top; scale bar, 500 μm), close-up image of the heart (bottom; scale bar, 125 μm). cf. Supplementary Video 4.
Extended Data Fig. 5: Loss-of-function candidate gene mutations affecting the heart rate level of medaka embryos.

Heart rate distributions of morphologically normal CRISPR-Cas9-mediated gene loss-of-function embryos and mock-injected control embryos at 4 dpf at 21°C, 28°C, and 35°C. The significance of heart rate differences between the mutant group and its corresponding control was tested with the Wilcoxon test; *, p<0.05, **, p<0.01, ***, p<0.001 (p-values for all comparison groups are listed in Extended Data Table 3). Data is visualized as box plots (median+/− interquartile range) and overlaid scatter plots of heart rate measurements; zrsr2 mutants and gse1 mutants show a temperature-dependent significant decrease in heart rate, in contrast, gpc5a mutants show a significant increase in heart rate at 35°C.
Extended Data Table 1:
Workflow of candidate gene prioritization.
| Ranking criterion | Database | Positive (+) or negative (–) influence on gene rank |
|---|---|---|
| (1) Differential expression in the heart | Transcriptomes, this study | + |
| (2) Presence of a paralog in medaka (Oryzias latipes) | Ensembl | - |
| (3) Known cardiac phenotype in medaka (Oryzias latipes) | Ensembl | |
| (4) Presence of a high confidence ortholog in zebrafish (Danio rerio) | Ensembl | + |
| (5) Known cardiac phenotype in zebrafish | Ensembl, ZFIN | - |
| (6) Known cardiac phenotype in mouse or rat | Ensembl, MGI/JAX | - |
| (7) Presence of high confidence ortholog in human | Ensembl | + |
| (8) Cardiovascular > ubiquitous expression | Ensembl, NCBI | + |
| (9) (a) Presence of ortholog or (b) variant(s) associations in human | Ensembl | + |
Extended Data Table 2: Number (n) of injected and phenotyped embryos at 4 dpf.
Surviving embryos were grouped into three categories: normal, cardiac affected and global phenotypes.
| Gene | sgRNA/crRNA | n injected | n normal | n cardiac | n global | n dead | Editor |
|---|---|---|---|---|---|---|---|
| rrad | rrad_T1 sgRNA | 93 | 18 | 35 | 20 | 20 | CRISPR_Cas9 |
| adprhl 1 | adprhl1_T1 sgRNA | 81 | 25 | 13 | 17 | 26 | CRISPR_Cas9 |
| BTBD1 | BTBD1_T1 sgRNA | 100 | 50 | 19 | 21 | 10 | CRISPR_Cas9 |
| blzfl | blzf1_T1 sgRNA | 84 | 38 | 15 | 14 | 17 | CRISPR_Cas9 |
| PTPRD | PTPRD_T1 sgRNA | 54 | 22 | 10 | 8 | 14 | CRISPR_Cas9 |
| phka2 | phka2_T1 sgRNA | 62 | 14 | 12 | 7 | 29 | CRISPR_Cas9 |
| pcdh17 | pcdh17_T1 sgRNA | 92 | 49 | 16 | 12 | 15 | CRISPR_Cas9 |
| IRF1 | IRF1a_T1 sgRNA | 83 | 20 | 14 | 27 | 22 | CRISPR_Cas9 |
| gpc5a | gpc5a_T1 sgRNA | 72 | 22 | 9 | 25 | 16 | CRISPR_Cas9 |
| gsel | gse1_T1 sgRNA | 79 | 55 | 10 | 4 | 10 | CRISPR_Cas9 |
| SEC61A1 | SEC61A1_T1 sgRNA | 81 | 55 | 9 | 8 | 9 | CRISPR_Cas9 |
| zrsr2 | zrsr2_T1 sgRNA | 114 | 32 | 16 | 43 | 23 | CRISPR_Cas9 |
| rrad | rrad_T2 crRNA | 75 | 21 | 19 | 15 | 20 | evoBE4max |
| adprhl1 | adprhl1_T2 crRNA | 68 | 4 | 7 | 14 | 43 | evoBE4max |
| BTBD1 | btbd1_T2 crRNA | 53 | 16 | 10 | 10 | 17 | evoBE4max |
| blzf1 | blzf1_T2 crRNA | 68 | 23 | 11 | 13 | 21 | evoBE4max |
| PTPRD | ptprd_T2 crRNA | 54 | 27 | 8 | 0 | 19 | evoBE4max |
| phka2 | phka2_T2_crRNA | 50 | 26 | 11 | 0 | 13 | evoBE4max |
| mock_ctrll | NA | 41 | 36 | 1 | 0 | 4 | NA_Cas9_ctrl |
| mock_ctrl2 | NA | 49 | 44 | 3 | 0 | 2 | NA_Cas9_ctrl |
| mock_ctrl3 | NA | 46 | 42 | 0 | 2 | 2 | NA_Cas9_ctrl |
| mock_ctrl4 | NA | 49 | 44 | 4 | 1 | 0 | NA_Cas9_ctrl |
| mock_ctrl5 | NA | 48 | 42 | 3 | 2 | 1 | NA_Cas9_ctrl |
| mock_ctrl6 | NA | 30 | 21 | 4 | 1 | 4 | NA_Cas9_ctrl |
| mock_ctrl7 | NA | 40 | 28 | 2 | 2 | 8 | NA_Cas9_ctrl |
| mock_ctrl1 | NA | 31 | 27 | 1 | 1 | 2 | NA_BE_ctrl |
| mock_ctrl2 | NA | 46 | 34 | 0 | 3 | 9 | NA_BE_ctrl |
| mock_ctrl3 | NA | 28 | 21 | 2 | 0 | 5 | NA_BE_ctrl |
Extended Data Table 3: Significance levels of heart rate differences at 21°C, 28°C., and 35°C.
P values were assessed with the Wilcoxon test comparing the heart rate of the mutant embryos to the heart rate of the respective mock-injected control embryos.
| Gene_group_editor | Phenotype | p value | Temperature |
|---|---|---|---|
| adprhl1_Cas9 | normal | 0.049 | 21 °C |
| adprhl1_Cas9 | normal | 0.137 | 28 °C |
| adprhl1_Cas9 | normal | 0.250 | 35 °C |
| adprhl1_evoBE4max | normal | 0.129 | 21 °C |
| adprhl1_evoBE4max | normal | 0.042 | 28 °C |
| adprhl1_evoBE4max | normal | 0.953 | 35 °C |
| blzf1_Cas9 | normal | 0.741 | 21 °C |
| blzf1_Cas9 | normal | 0.984 | 28 °C |
| blzf1_Cas9 | normal | 0.104 | 35 °C |
| blzf1_evoBE4max | normal | 0.009 | 21 °C |
| blzf1_evoBE4max | normal | 0.000 | 28 °C |
| blzf1_evoBE4max | normal | 0.015 | 35 °C |
| btbd1_Cas9 | normal | 0.009 | 21 °C |
| btbd1_Cas9 | normal | 0.033 | 28 °C |
| btbd1_Cas9 | normal | 0.269 | 35 °C |
| btbd1_evoBE4max | normal | 0.002 | 21 °C |
| btbd1_evoBE4max | normal | 0.038 | 28 °C |
| btbd1_evoBE4max | normal | 0.032 | 35 °C |
| gpc5a_Cas9 | normal | 0.053 | 21 °C |
| gpc5a_Cas9 | normal | 0.452 | 28 °C |
| gpc5a_Cas9 | normal | 0.018 | 35 °C |
| gse1_Cas9 | normal | 0.039 | 21 °C |
| gse1_Cas9 | normal | 0.779 | 28 °C |
| gse1_Cas9 | normal | 0.455 | 35 °C |
| IRF1_Cas9 | normal | 0.057 | 21 °C |
| IRF1_Cas9 | normal | 0.374 | 28 °C |
| IRF1_Cas9 | normal | 0.079 | 35 °C |
| pcdh17_Cas9 | normal | 0.710 | 21 °C |
| pcdh17_Cas9 | normal | 0.889 | 28 °C |
| pcdh17_Cas9 | normal | 0.605 | 35 °C |
| phka2_Cas9 | normal | 0.301 | 21 °C |
| phka2_Cas9 | normal | 0.242 | 28 °C |
| phka2_Cas9 | normal | 0.010 | 35 °C |
| phka2_evoBE4max | normal | 0.383 | 21 °C |
| phka2_evoBE4max | normal | 0.465 | 28 °C |
| phka2_evoBE4max | normal | 0.258 | 35 °C |
| PTPRD_Cas9 | normal | 0.852 | 21 °C |
| PTPRD_Cas9 | normal | 0.844 | 28 °C |
| PTPRD_Cas9 | normal | 0.140 | 35 °C |
| PTPRD_evoBE4max | normal | 0.607 | 21 °C |
| PTPRD_evoBE4max | normal | 0.105 | 28 °C |
| PTPRD_evoBE4max | normal | 0.160 | 35 °C |
| rrad_Cas9 | normal | 0.001 | 21 °C |
| rrad_Cas9 | normal | 0.022 | 28 °C |
| rrad_Cas9 | normal | 0.007 | 35 °C |
| rrad_evoBE4max | normal | 0.058 | 21 °C |
| rrad_evoBE4max | normal | 0.113 | 28 °C |
| rrad_evoBE4max | normal | 0.004 | 35 °C |
| SEC61A1_Cas9 | normal | 0.195 | 21 °C |
| SEC61A1_Cas9 | normal | 0.469 | 28 °C |
| SEC61A1_Cas9 | normal | 0.366 | 35 °C |
| zrsr2_Cas9 | normal | 0.008 | 21 °C |
| zrsr2_Cas9 | normal | 0.135 | 28 °C |
| zrsr2_Cas9 | normal | 0.014 | 35 °C |
Supplementary Material
Acknowledgements
We thank the Wittbrodt and Birney labs for constructive support of the work and the manuscript. Thanks to R. Hodge for critically reading and commenting on the manuscript. We thank J. Gehrig (ACQUIFER, Bruker) for technical support in high-throughput imaging and benchmarking algorithms. We also thank T. Kellner, B. Wittbrodt, and R. Müller for excellent technical support as well as, E. Leist, M. Majewski, A. Saraceno and S. Erny for fish support.
J.G. was supported by a Research Center for Molecular Medicine (HRCMM) Career Development Fellowship, the MD/PhD program of the Medical Faculty Heidelberg, the Deutsche Herzstiftung e.V. (S/02/17), and by an Add-On Fellowship for Interdisciplinary Science of Joachim Herz Stiftung. This work was supported by the DZHK (German Centre for Cardiovascular Research, number 81X2500189) to J.W. and J.G., the European Research Council Synergy Grant IndiGene (number 810172) and the NIH (National Institutes of Health, number R01ES029917) to E.B. and J.W..
Footnotes
Competing interest declaration
No competing interests declared.
Main references
- 1.Kearney D. L. The Pathological Spectrum of Left-Ventricular Hypoplasia. Semin. Cardiothorac. Vasc. Anesthesia 17, 105–116 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Hyodo-Taguchi Y. Inbred strains of the medaka, Oryzias latipes. The Fish Biology Jounral MEDAKA 8, 11–14 (1996). [Google Scholar]
- 3.Asai T., Senou H. & Hosoya K. Oryzias sakaizumii, a new ricefish from northern Japan (Teleostei: Adrianichthyidae). Ichthyological Exploration of Freshwaters 22, 289–299 (2011). [Google Scholar]
- 4.Linde D. van der et al. Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J. Am. Coll. Cardiol. 58, 2241–2247 (2011). [DOI] [PubMed] [Google Scholar]
- 5.Bom T. van der et al. The changing epidemiology of congenital heart disease. Nat. Rev. Cardiol. 8, 50–60 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Gehrmann J., Krasemann T., Kehl H. G. & Vogt J. Hypoplastic Left-Heart Syndrome The First Description of the Pathophysiology in 1851; Translation of a Publication by Dr. Bardeleben From Giessen, Germany. Chest 120, 1368–1371 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Hinton R. B. et al. Hypoplastic Left Heart Syndrome Is Heritable. J. Am. Coll. Cardiol. 50, 1590–1595 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Parker L. E. & Landstrom A. P. Genetic Etiology of Left-Sided Obstructive Heart Lesions: A Story in Development. J. Am. Hear. Assoc. 10, e019006 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Krane M. et al. Sequential Defects in Cardiac Lineage Commitment and Maturation Cause Hypoplastic Left Heart Syndrome. Circulation 144, 1409–1428 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Homsy J. et al. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science 350, 1262–1266 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sifrim A. et al. Distinct genetic architectures for syndromic and nonsyndromic congenital heart defects identified by exome sequencing. Nat. Genet. 48, 1060–1065 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin S. C. et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat. Genet. 49, 1593–1601 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Page D. J. et al. Whole Exome Sequencing Reveals the Major Genetic Contributors to Nonsyndromic Tetralogy of Fallot. Circ. Res. 124, 553–563 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wittbrodt J., Shima A. & Schartl M. Medaka--a model organism from the far East. Nat. Rev. Genet. 3, 53–64 (2002). [DOI] [PubMed] [Google Scholar]
- 15.Gierten J. et al. Automated high-throughput heartbeat quantification in medaka and zebrafish embryos under physiological conditions. Sci Rep 10, 2046–12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Naruse K., Hori H., Shimizu N., Kohara Y. & Takeda H. Medaka genomics: a bridge between mutant phenotype and gene function. Mech. Dev. 121, 619–628 (2004). [DOI] [PubMed] [Google Scholar]
- 17.Iwamatsu T. Stages of normal development in the medaka Oryzias latipes. Mech. Dev. 121, 605–618 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Runcie D. E. & Crawford L. Fast and flexible linear mixed models for genome-wide genetics. PLoS Genet. 15, e1007978 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McLaren W. et al. The Ensembl Variant Effect Predictor. Genome Biol. 17, 122 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mi H. et al. Protocol Update for large-scale genome and gene function analysis with the PANTHER classification system (v.14.0). Nat. Protoc. 14, 703–721 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Raudvere U. et al. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 47, W191–W198 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sherman B. T. et al. DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 50, W216–W221 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blanda V., Bracale U. M., Taranto M. D. D. & Fortunato G. Galectin-3 in Cardiovascular Diseases. Int. J. Mol. Sci. 21, 9232 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laan A. M. van der et al. Galectin-2 expression is dependent on the rs7291467 polymorphism and acts as an inhibitor of arteriogenesis. Eur. Hear. J. 33, 1076–1084 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Hollander M. R. et al. Stimulation of Collateral Vessel Growth by Inhibition of Galectin 2 in Mice Using a Single-Domain Llama-Derived Antibody. J. Am. Hear. Assoc.: Cardiovasc. Cerebrovasc. Dis. 8, e012806 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoed M. den et al. Identification of heart rate-associated loci and their effects on cardiac conduction and rhythm disorders. Nat. Genet. 45, 621–631 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eppinga R. N. et al. Identification of genomic loci associated with resting heart rate and shared genetic predictors with all-cause mortality. Nat. Genet. 48, 1557–1563 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Chang L. et al. Rad GTPase Deficiency Leads to Cardiac Hypertrophy. Circulation 116, 2976–2983 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Y. et al. RAD-Deficient Human Cardiomyocytes Develop Hypertrophic Cardiomyopathy Phenotypes Due to Calcium Dysregulation. Front. Cell Dev. Biol. 8, 585879 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Belbachir N. et al. RRAD mutation causes electrical and cytoskeletal defects in cardiomyocytes derived from a familial case of Brugada syndrome. Eur Heart J 40, 3081–3094 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith S. J. et al. The cardiac-restricted protein ADP-ribosylhydrolase-like 1 is essential for heart chamber outgrowth and acts on muscle actin filament assembly. Dev. Biol. 416, 373–388 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spivakov M. et al. Genomic and phenotypic characterization of a wild medaka population: towards the establishment of an isogenic population genetic resource in fish. G3 (Bethesda) 4, 433–445 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fitzgerald T. et al. The Medaka Inbred Kiyosu-Karlsruhe (MIKK) panel. Genome Biol. 23, 59 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leger A. et al. Genomic variations and epigenomic landscape of the Medaka Inbred Kiyosu-Karlsruhe (MIKK) panel. Genome Biol. 23, 58 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods references
- 35.Bert B. et al. Considerations for a European animal welfare standard to evaluate adverse phenotypes in teleost fish. EMBO J. 35, 1151–1154 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cornean A. et al. Precise in vivo functional analysis of DNA variants with base editing using ACEofBASEs target prediction. Elife 11, e72124 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hein S. J. et al. Advanced echocardiography in adult zebrafish reveals delayed recovery of heart function after myocardial cryoinjury. PLoS ONE 10, e0122665 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Head S. R. et al. Library construction for next-generation sequencing: Overviews and challenges. BioTechniques 56, 61–77 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen S., Zhou Y., Chen Y. & Gu J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dobin A. et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Love M. I., Huber W. & Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sollis E. et al. The NHGRI-EBI GWAS Catalog: knowledgebase and deposition resource. Nucleic Acids Res. 51, D977–D985 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stelzer G. et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 54, 1.30.1–1.30.33 (2016). [DOI] [PubMed] [Google Scholar]
- 44.Stemmer M., Thumberger T., Keyer M. D. S., Wittbrodt J. & Mateo J. L. CCTop: An Intuitive, Flexible and Reliable CRISPR/Cas9 Target Prediction Tool. PLoS ONE 10, e0124633 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hammouda O. T. et al. In vivo identification and validation of novel potential predictors for human cardiovascular diseases. PLoS ONE 16, e0261572 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kluesner M. G. et al. EditR: A Method to Quantify Base Editing from Sanger Sequencing. Crispr J 1, 239–250 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.R Core Team. R: A Language and Environment for Statistical Computing. https://www.R-project.org/ (2016).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.