

Abstract
The development of SAR around substituted N-piperidinyl indole-based nociceptin opioid receptor (NOP) ligands led to the discovery of a novel series of 2-substituted N-piperidinyl indoles that provide both selective NOP full agonists and bifunctional NOP full agonists–μ opioid (MOP) receptor partial agonists. 2-substituted N-piperidinyl indoles have improved potency at the NOP receptor and are NOP full agonists, compared to our previously reported 3-substituted N-piperidinyl indoles that are selective NOP partial agonists. SAR in this series of 2-substituted N-piperidinyl indoles shows that 2-substitution versus 3-substitution on the indole moiety affects their intrinsic activity and opioid receptor selectivity. Molecular docking of these 2-substituted N-piperidinyl indoles in an active-state NOP homology model and MOP receptor structures provides a rationale for the differences observed in the binding, functional profiles and selectivity of 2-substituted versus 3-substituted N-piperidinyl indoles.
Keywords: nociceptin opioid receptor, mu opioid receptor, N/OFQ, NOP, MOP, nociceptin full agonist, nociceptin partial agonist, bifunctional agonist, structure-based drug design, structure activity relationship, G-protein coupled receptor, orthosteric binding site
1. Introduction1
The nociceptin opioid (NOP) receptor, discovered in 1994,1,2 and previously known as ORL1, is a G-protein-coupled receptor (GPCR) and considered to be the fourth member of the opioid receptor family due to its high homology with the μ (MOP), δ (DOP), and κ (KOP) opioid receptors.3 The endogenous ligand for the NOP receptor is nociceptin/orphinan FQ (N/OFQ),4,5 and it has been shown that the NOP-N/OFQ system is associated with various physiological functions and pathophysiologies–including anxiety, pain and analgesia, Parkinson’s Disease (PD), PD-based dyskinesia, learning, memory, sleep, as well as modulating tolerance development and reward in drug abuse.6-16 Unlike the extensive studies on pharmacology of ligands for the related MOP receptor that produce analgesia and undesired side effects, the pharmacology of the NOP receptor and its ligands remain nebulous due to various factors that influence the pharmacological action of NOP ligands, such as route of administration, type of pain (noxious, inflammatory, neuropathic, etc.) as well as type of species used (rodents vs. non-human primates (NHP)).17,18 Nevertheless, there has been significant drug development of NOP ligands over the past 25 years for both agonists and antagonists, with a few clinical candidates advancing to Phase 1 clinical trials. Yet to date, there are no FDA-approved drugs that target the nociceptin receptor.19
We have investigated the nociceptin receptor as a target for various CNS diseases such as Parkinson’s Disease (PD) and PD-related dyskinesia, pain and substance abuse disorders, and have reported several novel series of small-molecule NOP receptor ligands.20-29 We recently reported a series of 3-substituted N-piperidinyl indoles that showed efficacy in reducing motor deficits in preclinical Parkinson’s disease models. (Figure 1).28 These piperidinylindoles were substituted at the 3-position with various polar functionalities linked via methyl, ethyl, and propyl linkers. SAR of the 3-substituted N-piperidinyl indoles showed that only basic functional groups, such as an amine or guanidine, provided modest improvement in NOP binding affinity and potency compared to the unsubstituted indole. The 3-substituted N-piperidinyl indoles showed modest to high selectivity (20 to 200-fold) for the NOP receptor versus the MOP receptor, and were partial agonists at the NOP receptor.
Figure 1.

Comparative profiles of 3-substituted versus 2-substituted N-piperidinyl indoles.
Recently we published a structure-based SAR study, using an active-state NOP homology model, to dissect how four chemically different NOP ligand scaffolds orient themselves in the orthosteric binding pocket of the NOP receptor.29 Molecular docking of the N-piperidinyl indole series of NOP ligands showed that the modest 3-fold increase in NOP binding affinity of the 3-aminomethyl indole, compared to the unsubstituted indole, is due to an ionic interaction between the positively charged amino group of the indole and two negatively charged amino acids, Glu194 and Glu199, on the NOP extracellular loop 2 (ECL2). Molecular modeling also revealed that a 2-substituted N-piperidinyl indole, (e.g. 2-hydroxymethyl N-piperidinyl indole 1 (n=1), Figure 1), could bind in a different orientation such that the 2-substituted functionality could engage in a hydrogen-bond interaction with polar amino acids on TM7 of the NOP receptor. Indeed, 1 was found to be a NOP full agonist with high binding affinity at NOP–a very different binding and functional efficacy profile than its 3-substituted N-piperidinyl indole analog 2, which was a NOP-selective partial agonist.28 NOP full agonist 1 (AT-312) has been shown to attenuate rewarding effects of morphine, cocaine, and ethanol in the conditioned place preference (CPP) model in rodents in vivo.30,31 Based on these findings, we investigated various polar, ionic, hydrophobic and aromatic substituents with different linker lengths at the 2-position on the indole ring to determine the SAR around binding affinity, functional efficacy, and selectivity versus the other opioid receptors, compared to the 3-substituted indole series of NOP partial agonists. Herein, we report the SAR at the 2-position and the discovery of selective NOP agonists as well as bifunctional NOP–MOP agonist ligands from this series of 2-substituted N-piperidinyl indoles.
2. Results and Discussion
2.1. Chemistry
Synthesis of 2-substituted N-piperidinyl indoles began with the construction of aniline 4 in excellent yield over 3 steps starting from commercially available 2-iodoaniline (3) (Scheme 1).28 Initial attempts to synthesize the indole ring via a one-pot palladium-catalyzed Sonogashira coupling followed by a palladium-catalyzed heterocyclization were unsuccessful (not shown), as it led to decomposition of aniline 4. Instead, a stepwise approach beginning with Sonogashira coupling of aniline 4 with various terminal alkynes such as propargyl alcohol or N-Cbz propargylamine, provided internal alkynes in good yield. Exposure of the latter alkynes to a catalytic amount of Cu(OAc)2 in refluxing toluene, as described by Hiroya,32 readily promoted the intramolecular heterocyclization and provided the desired 2-substituted N-piperidinyl indole compounds 5, 6, 1, 7, and 8 in consistent yield. Smooth oxidation of alcohol 1 using MnO2 provided benzylic aldehyde in nearly quantitative yield, which was then converted to 2-pyrrolidinylmethyl N-piperidinyl indole 9 by exposure to pyrrolidine using standard reductive amination conditions.
Scheme 1. Reagents and conditions.

(a) AcOH, STAB, DCE, rt, 16 h, 96%; (b) TFA, CH2Cl2, 1 h; (c) 4-iPr-cyclohexanone, STAB, AcOH, DCE, rt, 1-2 days, 28% over two steps; (d) alkyne, 2 mol% PdCl2(PPh3)2, 6 mol% Cul, TEA:THF (1:4), rt, 5-7 h, 69-98%; (e) 10-20 mol% Cu(OAc)2, PhMe, reflux, 1-2 h, 49-85%; (f) H2 balloon, 10 wt% Pd/C (10%), 1.5N NH3 in MeOH, rt, 6 h, 75-80%; (g) various electrophiles (Ac2O, ClCO2Et, ClSO2Me), TEA, CH2Cl2, 55-96%; (h) SO2(NH2)2, refluxing dioxane, 56%; (i) Aryl-acid, pyBOP, TEA or iPr2NEt, THF, 20-63%; (j) MnO2, CH2Cl2, 16 h, rt, 98%; (k) amine, STAB, DCE, rt, 16 h, 30%.
Formation of amines 10 and 11 was accomplished by removing the Cbz group with standard hydrogenation conditions. Exposing amine 10 to a variety of electrophiles such as AcCl, ClCO2Et, and ClSO2Me under acylation conditions provided compounds 12, 13, and 14 respectively. Additionally, exposing amine 10 to sulfamide in refluxing dioxane led to sulfamide 14 in 56% yield. Finally, formation of aryl amides 16, 17, and 18 occurred smoothly using amine 10, the corresponding aryl-acids, and pyBOP as the coupling reagent.
2.2. SAR and Molecular interactions of the 2-substituted and 3-substituted N-piperidinyl indoles at the NOP receptor
The 2-substituted N-piperidinyl indoles were tested in vitro for binding affinity at the four opioid receptors subtypes in radioligand competition displacement assays using membranes from human NOP, MOP, KOP, and DOP receptor-transfected Chinese hamster ovary (CHO) cells, as we have described previously.33,34 Binding affinity was calculated as the binding constant, Ki (nM), shown in Table 1. The functional efficacy was determined using the [35S]GTPγS binding assay in membranes prepared from the opioid receptor-transfected CHO cells, using previously described methods.28 Compound potencies are reported as EC50 (nM), and the functional efficacies shown in Table 1 are reported as % stimulation compared to standard full agonists normalized to 100%.
Table 1.
Binding affinities and functional efficacies of 2-substituted N-piperidinyl indoles at human opioid receptors.
| # | Structure | Binding affinities and functional activities of bifunctional NOP-MOP ligands | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Binding Affinities, Ki (nM)a | NOP/MOP selectivity |
[35S]GTPγS Functional Assay (nM)b | ||||||||||||
| NOP | μ (MOP) | δ (DOP) | κ (KOP) | |||||||||||
| NOP | μ (MOP) | δ (DOP) | κ (KOP) | EC50 | % Stim | EC50 | % Stim | EC50 | % Stim | EC50 | % Stim | |||
| N/OFQ | 0.12 ± 0.01 | NT | NT | NT | ---- | 3.6 ± 0.7 | 100 | NT | NT | NT | NT | NT | NT | |

|
9.8 ± 0.86 | 376 ± 37 | 923 ± 413 | 1594 ± 57 | 38.4 | 160 ± 64 | 29.3 ± 9.5 | 232 ± 12.6 | 16.3 ± 7.6 | NT | NT | NT | NT | |
| 19 |

|
3.27 ± 0.30 | 65.3 ± 2.4 | >10K | 1737 ± 172 | 20.0 | 121 ± 52 | 35.9 ± 5.7 | 410 ± 105 | 11.8 ± 2.7 | NT | NT | NT | NT |
| 2 |

|
44.7 ± 9.1 | 716 ± 21 | >10K | >10K | 16.0 | 117 ± 14 | 18.9 ± 0.70 | NT | NT | NT | NT | NT | NT |
| 10 |

|
0.23 ± 0.11 | 0.81 ± 0.78 | 236 ± 43 | 25.21 ± 6.27 | 3.5 | 5.8 ± 3.2 | 100 ± 5 | 24.1 ± 5.6 | 27.9 ± 8.1 | NT | NT | >10K | 9.0 |
| 11 |

|
0.17 ± 0.02 | 2.78 ± 1.32 | 706 ± 58 | 62.07 ± 15.7 | 16.4 | 10.0 ± 5.9 | 102 ± 2 | 29.7 ± 7.84 | 39.7 ± 9.0 | NT | NT | 1105 ± 50 | 21.9 ± 10.9 |
| 1 |

|
0.34 ± 0.13 | 5.99 ± 0.97 | 129 ± 57 | 73.51 ± 28.28 | 17.6 | 29.9 ± 1.44 | 102 ± 1 | 81.5 ± 16.0 | 24.6 ± 2.37 | 4604 ± 1127 | 38.1 ± 7.1 | >10K | 1.0 |
| 7 |

|
0.93 ± 0.05 | 20.32 ± 5.98 | 154 ± 6 | 51.68 ± 10.33 | 21.8 | 31.4 ± 1.94 | 87.8 ± 3.2 | 72.9 ± 3.33 | 34.9 ± 0.35 | NT | NT | >10K | 6.7 ± 1.5 |
| 12 |

|
1.58 ± 0.7 | 24.32 ± 2.1 | 81.36 ± 0.29 | 176 ± 30 | 15.4 | 50.0 ± 2.9 | 94.1 ± 6.6 | 112 ± 24 | 26.2 ± 5.6 | 135 ± 50 | 45.9 ± 0.3 | NT | NT |
| 13 |

|
2.34 ± 0.55 | 25.35 ± 7.74 | 51.18 ± 4.10 | 408 ± 34 | 10.8 | 17.8 ± 2.9 | 96.7 ± 3.7 | 72.2 ± 9.0 | 43.5 ± 0.5 | 285 ± 10 | 50.8 ± 5.5 | NT | NT |
| 14 |

|
4.14 ± 1.32 | 2.52 ± 0.71 | 139 ± 7 | 177 ± 39 | 0.61 | 22.5 ± 0.74 | 84.0 ± 19.8 | 56.4 ± 12.41 | 37.6 ± 5.7 | 335 ± 63 | 48.8 ± 1.7 | 69.1 ± 40.1 | 36.1 ± 5.5 |
| 15 |

|
0.71 ± 0.21 | 0.66 ± 0.34 | 988c | 20.93 ± 4.78 | 0.93 | 49.2 ± 6.84 | 88.0 ± 7.3 | 65.4 ± 10.25 | 25.8 ± 0.76 | NT | NT | >10K | 2.6 |
| 8 |

|
0.59 ± 0.24 | 38.80 ± 5.88 | 1710c | 32.15 ± 1.54 | 65.8 | 52.3 ± 21.9 | 91.2 ± 8.4 | 38.1 ± 14.78 | 29.8 ± 1.48 | NT | NT | >10K | 1.3 |
| 9 |
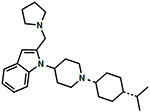
|
1.15 ± 0.09 | 0.58 ± 0.03 | 118 ± 19 | 30.62 ± 9.58 | 0.50 | 7.2 ± 1.4 | 106 ± 4 | 40.0 ± 10.0 | 38.3 ± 5.8 | 934 ± 179 | 42.1 ± 5.4 | 78.2 ± 1.4 | 47.1 ± 0.6 |
| 16 |

|
0.75 ± 0.02 | 5.43 ± 0.22 | 65.74 ± 1.60 | 113 ± 26 | 7.2 | 19.4 ± 5.6 | 85.9 ± 3.8 | 42.9 ± 1.7 | 44.2 ± 3.0 | 195 ± 24 | 45.7 ± 1.7 | >10K | 5.40 ± 0.6 |
| 17 |

|
0.33 ± 0.01 | 0.27 ± 0.08 | 55.2 ± 20.0 | 12.1 ± 2.4 | 0.82 | 9.4 ± 1.9 | 89.9 ± 0.7 | 30.4 ± 1.3 | 37.1 ± 7.4 | 2840 ± 571 | 51.0 ± 3.5 | 1624c | 2.2 |
| 18 |

|
3.36 ± 0.96 | 15.1 ± 5.65 | 63.3 ± 12.4 | 253 ± 30 | 4.5 | 54.3 ± 24.1 | 61.9 ± 2.7 | 60.2 ± 7.5 | 34.5 ± 6.9 | 496 ± 0.3 | 42.1 ± 7.9 | NT | NT |
Binding affinities were determined using radioligand displacement assays performed in membranes of CHO cells stably expressing the human NOP, MOP, KOP and DOP receptors, and their respective radioligands, [3H]N/OFQ–NOP, [3H]U69,593–KOP, [3H]DAMGO-MOP and [3H]DPDPE–DOP receptor. Equilibrium dissociation constants (Ki) were derived from IC50 values using the Cheng–Prusoff equation. Each Ki value represents the arithmetic mean ± SD from at least three independent experiments, each performed in triplicate. NT = not tested.
Compounds with Ki values >100 nM were not tested in functional assays (NT). Functional activity was determined by stimulation of [35S]GTPγS binding to cell membranes, EC50 is the ligand concentration producing half maximal stimulation, % stimulation was obtained as a percentage of stimulation of the standard full agonists, N/OFQ (for NOP), and DAMGO (for MOP), which showed at least 2- to 5-fold stimulation over basal. Results are the means ± SD for at least 3 independent experiments each performed in triplicate.
Performed once.
As reported previously, 3-substituted N-piperidinyl indoles provided NOP partial agonists with high binding affinity and modest to high selectivity for NOP over the MOP receptor (up to 200-fold).28 In this series, 3-aminomethyl-piperidinylindole 19 was a high-affinity NOP partial agonist with a 10-fold higher binding affinity (Ki = 3 nM) than 3-hydroxymethyl-piperidinylindole 2 (Ki = 44 nM), both of which had modest selectivity for NOP over MOP (15-20 fold). However, their 2-substituted regioisomers, 2-aminomethyl-piperidinyl indole 10 and 2-hydroxymethyl 1, show significantly different binding, functional and selectivity profiles at the NOP and MOP receptors compared to their 3-substituted analogs (see Table 1). Compounds 10 and 1 both have subnanomolar binding affinity at the NOP receptor, 0.23 nM and 0.34 nM, respectively. Interestingly, both these 2-substituted indoles show significantly enhanced binding affinity at MOP compared to their 3-substituted analogs. 2-substituted N-piperidinyl indoles also show increased binding affinity at DOP and KOP receptors compared to their 3-substituted analogs (Table 1). However, DOP and KOP binding affinities are still ≥50-fold weaker than the binding affinities at NOP and MOP receptors for 2-substituted N-piperidinyl indoles. The remaining 2-substituted indoles containing diverse functional groups (see Table 1) all show high binding affinity at the NOP receptor (0.5–4 nM).
A key difference observed between the 2-substituted and 3-substituted N-piperidinyl indoles was the full agonist efficacy of the 2-substituted indoles, whereas most of the 3-substituted indoles were NOP partial agonists. For instance, both 2-aminomethyl indole 10 and 2-hydroxymethyl indole 1 were potent NOP full agonists (NOP EC50 = 5.8 and 29.9 nM, NOP % Stim = 100 and 102, respectively), whereas the respective regioisomers at the 3-position, amine 19 and alcohol 2, were NOP partial agonists (NOP EC50 = 121 and 117 nM, NOP % Stim = 35.9 and 18.9, respectively). Other functionalities at the 2-position of the indole including cyclic amines, as well as aliphatic and aryl amides, consistently provided ligands that were full agonists at the NOP receptor (Table 1). One exception was furan 18, which showed partial agonism at the NOP receptor, likely due to the increased steric bulk due to CF3 group on the furan ring.
Another interesting SAR observation from the 2-substituted versus 3-substituted N-piperidinyl indoles is that most 2-substituted analogs had higher binding affinity at the μ opioid receptor compared to the 3-substituted analogs, and that the nature of the 2-substituent influenced the binding affinity of the NOP ligand at the μ opioid receptor, and consequently on the selectivity of the ligand for NOP versus the MOP receptor. 2-hydroxymethylindole 1 has significantly higher affinity for MOP than 3-hydroxymethylindole 2, yet 1 also has subnanomolar affinity at NOP and is ~18-fold selective for NOP over MOP. Similarly, amines 10 and 11 have nanomolar affinity at MOP and hence lower NOP-MOP selectivity of 4-fold and 16-fold, respectively. SAR of the 2-substituents shows that those with an ionic or highly polar character (9, 14, and 15) as well as aryl substituents (16, 17 and 18) tend to have nanomolar affinities at MOP and have bifunctional potencies at NOP and MOP (Table 1).
Molecular docking into a NOP active-state receptor structure was then conducted with the 2- and 3-substituted indoles to investigate whether their docking orientations and molecular interactions could explain the observed higher NOP binding affinities of the 2-substituted indoles compared to their 3-substituted analogs.29,35
When docked into the active-state NOP receptor, the protonated piperidine nitrogen of the 2-substituted N-piperidinyl indoles forms the expected ionic interaction with the carboxylate of Asp1303.32, positioning the N-4-iPr-cyclohexyl moiety into the hydrophobic pocket lined by amino acids Phe2245.47, Trp2766.48, and Met1343.36 (Figure 2A) (Superscript numbers indicate Ballesteros-Weinstein numbering). The top docking pose of 2-aminomethyl 10 orients the phenyl ring of the indole into a minor hydrophobic pocket comprised of residues from transmembrane (TM) helices 2 (Thr1032.56, Leu1042.57), TM-3 (Val1263.28, Ile1273.29) and extracellular loop 2 (ECL2, Cys200). The 2-substituted amine forms an ionic interaction with Glu199 (ECL2), as well as hydrogen bonding interactions with Tyr581.39, Tyr3097.42, and Thr3057.38, the TM7 polar network in NOP known to be involved in receptor activation.35 In contrast, in the preferred (top) docked pose of 3-aminomethyl 19, the 3-aminomethyl substituent forms ionic interactions with Glu194 and Glu199 on ECL2 loop of NOP, but the aromatic indole ring of 19 does not occupy the minor hydrophobic pocket. It appears that this ionic interaction orients the ligand such that the indole ring is rotated approximately 60 degrees toward TM2 (Figure 2a), pulling the indole ring out of the minor hydrophobic pocket. Interestingly, in the binding orientation of most of the 3-substituted N-piperidinyl indoles in the active-state NOP homology model, the indole ring fails to interact with the minor hydrophobic pocket (See Figure 2a).
Figure 2.

Docked poses of 2- and 3-substituted piperidinylindoles in the orthosteric binding pocket of the active-state NOP receptor homology model. A. Comparison of 2-aminomethyl indole 10 (magenta) and 3-aminomethyl indole 19 (yellow); B. Comparison of 2-hydroxymethyl indole 1 (blue) and 3-hydroxymethyl indole 2 (red).
In another comparison, 2-hydroxymethyl 1 has subnanomolar NOP binding affinity compared to its 3-substituted analog 2 and is also a full agonist at NOP (Table 1). The preferred docked pose of 1 places its indole ring within the minor hydrophobic pocket, orienting its hydroxyl group to form a hydrogen bond with Tyr3097.42 (Figure 2B). In contrast, the preferred docked pose of 3-position regioisomer 2, positions the indole ring away from the minor hydrophobic pocket while the 3-methylenehydroxy group makes a H-bond interaction with Tyr581.39, (Figure 2b). The higher binding affinity of the 2-substituted indoles and the docking pose comparisons of the 2- and 3-substituted indole pairs 10/19 and 1/2 suggests that interactions with the TM2-TM3 minor hydrophobic pocket and engaging the TM7 polar network (Arg3027.36, Tyr3097.42, and Thr3057.38) are important factors for the higher binding affinity of the 2-substituted indoles. Similar interaction with the TM2-TM3 minor hydrophobic pocket in the MOP receptor has also been shown to be important for the higher intrinsic activity and potency of fentanyl, whose aromatic ring occupies the TM2-TM3 hydrophobic pocket, compared to that of morphine, whose aromatic ring does not interact with the TM2-TM3 pocket in the MOP receptor.36 The importance of the TM2-TM3 hydrophobic pocket interactions for higher NOP ligand intrinsic activity (associated with receptor activation) is suggested from comparing the docking of the 2-(N-pyrrolidinyl)methyl-indole 9 and its 3-substituted analog 20. Unlike most other 3-substituted indoles, 3-(N-pyrrolidinyl)methyl indole 20 showed full agonism at the NOP receptor (EC50 = 97 nM, NOP Emax 88%, Table 1).28 Interestingly, in the preferred docked poses of both the 3- and 2-(N-pyrrolidinyl)methyl indoles, the lipophilic pyrrolidine ring, regardless of position on the indole ring, resides in the minor hydrophobic pocket (Figure 3). Thus, structure-based SAR analysis of the N-piperidinyl indole series at the NOP receptor suggests that ligand occupation of the TM2-TM3 minor hydrophobic pocket improves NOP binding affinity and possibly the intrinsic activity of the ligand.
Figure 3.

Docked poses of 2-(N-pyrrolidinyl)methyl indole 9 (gold) and its 3-position regioisomer 20 (cyan) in the active-state NOP receptor.
Notably, 2-substituted indoles with larger aromatic or ethyl carbamate substituents (13, 16, 17 and 18, Table 1), which are high affinity NOP full agonists (Table 1) show preferred docked poses in which their 2-substituents occupy the TM2-TM3 minor hydrophobic pocket, similar to the orientation of 2-(N-pyrrolidinyl)methyl 9 (Figure 3). Analyzing docked poses of the compounds 12, 13, 14, 16 and 17 in the NOP receptor showed that 2-substituents with shorter side-chains like acetamide, methyl sulfonamide and sulfamide (in compounds 12, 14, and 15, respectively), make H-bonds with various residues of the TM7 polar network, whereas 2-substituents with longer side-chains like ethyl carbamate (in 13) and bulkier aromatic side-chains (in 16,17) flip 180° to occupy the minor hydrophobic pocket. It is possible that these preferred conformations by longer and bulkier side-chains avoid steric crowding with the residues of the TM7 polar network and make hydrophobic interactions with the minor hydrophobic pocket. Interestingly, these analogs 13, 16, and 17 have high binding affinity at NOP receptor. An additional H-bond interaction between the oxygen atom of the oxazole ring in compound 17 and backbone NH of Cys200 in ECL2 likely explains the subnanomolar binding affinity of 17 at the NOP receptor. The docked pose of 2-CF3 substituent on the furan ring of compound 18 shows a likely steric clash with Val1263.28 in the minor hydrophobic pocket, and may explain the reduced binding affinity of the compound 18 compared to the 17. Overall, the molecular docking and the Table 1 SAR indicate that occupying the minor hydrophobic pocket consistently provided ligands with high binding affinity at the NOP receptor for the reported ligands. However, the effect of occupancy of the minor hydrophobic pocket on NOP ligand intrinsic activity needs further investigation and will be reported in the future publications.
2.3. SAR and Molecular docking of 2- and 3-substituted N-piperidinyl indoles at the MOP receptor.
Binding affinities at the MOP receptor vary significantly between 2-substituted N-piperidinyl indoles and 3-substituted N-piperidinyl indoles. In general, 2-substituted indoles show higher binding affinity at MOP than most 3-substituted indoles (Table 1).28 However, within the 2-substituted indole series in Table 1, several showed subnanomolar affinity for MOP (e.g. 10, 14, 17) compared to other 2-substituted indoles (e.g. 13, 8). Molecular docking of these indoles into the cryo-EM structure of the MOP receptor was conducted to determine the MOP binding interactions that may affect the binding affinity of the indole series of NOP ligands and hence their selectivity for NOP versus the MOP receptor.36
Interestingly, almost all the 2-substituted indoles in Table 1 showed top docking poses wherein their phenyl (indole) ring occupied the hydrophobic TM2-TM3 pocket of the MOP receptor. It is possible, therefore, that the differences in MOP binding affinity of the various 2-substituted N-piperidinyl indoles (0.3–39 nM) are due to interactions between the 2-substituent and amino acids on TM7 in the orthosteric pocket of the MOP receptor, which are considerably different than the polar amino acids on TM7 in the NOP receptor. Although the subnanomolar binding affinity at the NOP receptor for amines 9 and 10 can be explained given the ionic interactions of the corresponding protonated amines with Asp130 and the ECL2 acidic residues, respectively, the subnanomolar binding affinity at the MOP receptor for these amines, and the relatively low MOP binding affinity for cyclic sulfone 8, are not easily explained, as only hydrogen bonding interactions are observed for these 2-substitutent amines within the MOP receptor. However, despite the high binding affinity at the MOP receptor, these 2-substituted indole amines show only partial agonist efficacy at MOP with modest potencies, while showing significantly higher functional selectivity for the NOP receptor (Table 1).
3. Conclusion
Herein, we report a SAR of 2-substituted N-piperidinyl indoles and the discovery of selective NOP full agonists as well as bifunctional NOP full agonists-MOP partial agonists from this chemical class of NOP ligands. This builds on our earlier work around 3-substituted N-piperidinyl indoles, which provided NOP-selective partial agonists. The SAR showed that the key differences between 2- and 3-substituted indoles were: 1) 2-substituted indoles have higher NOP binding affinities than 3-substituted indoles, and 2) 2-substitution provided NOP full agonists while 3-substitution provided NOP partial agonists. Molecular docking suggests that the increased binding affinity for 2-substituted N-piperidinyl indoles at both NOP and MOP receptors may be due to the ligands occupying a minor hydrophobic pocket, conserved in both receptors, on the extracellular end of the orthosteric binding pockets. In the active-state NOP receptor, effect of the interaction with the minor hydrophobic pocket of either the phenyl (indole) ring or the 2-substituent on ligand intrinsic activity needs further investigation to explain the high potency full agonist efficacy of the 2-substituted N-piperidinylindole-based NOP ligands. In the MOP receptor, 2-substituted indoles consistently docked with the phenyl (indole) ring into the hydrophobic TM2-TM3 pocket but had partial agonist efficacy at MOP. Further efforts to expand the SAR of 2-substituted N-piperidinyl indoles to improve the overall druggability are ongoing and will be reported in due course.
4. Experimental Section
4.1. Molecular modeling of compounds in the active-state NOP receptor homology model and MOP cryo-EM structure
4.1.1. Molecular docking of compounds in the active-state NOP receptor model.
The NOP ligands were docked into an active-state homology model of the NOP receptor we previously reported.35 The details of the model building, loop building, and refinement can be found in our previously published report on homology modeling and molecular dynamics simulation of the NOP receptor.35 As the extracellular loop-2 (ECL2) is an integral part of the binding site, the disulfide bridge between TM3 and the second extracellular loop (ECL2) was included in the homology model. This model was utilized in this study to conduct structure-based SAR analysis of the NOP ligands.
Compounds were docked into the orthosteric site of NOP using Surflex-Dock. The Surflex-Dock protomol is a precomputed molecular representation of an idealized ligand and represents a negative image of the binding site to which putative ligands are aligned. The structure template used for building the active-state NOP homology model did not contain a ligand. Usually, in such a case, it becomes necessary to use available algorithms for finding putative binding pockets. Instead of using such standard site-finding algorithms, we preferred to use the existing knowledge of the NOP binding site from literature mutagenesis studies to locate the orthosteric binding site.37,38 Since its discovery, a number of mutagenesis studies on the NOP receptor have identified cognate differences between NOP and the other opioid receptors, as well as residues important for binding with N/OFQ. These studies over the years have identified amino acids such as Asp1303.32, Thr3057.38, and Val2796.51 to be important for binding of NOP. Hence, for this study, the protomol was constructed using a set of active site residues consisting of Tyr581.39, Asp1303.32, Met1343.36, Val2796.51, Thr3057.38, and Tyr3097.42. To optimize the results further, during the Surflex-Dock docking studies, hydrogen atoms (attached to hydroxyl and thiol) and heteroatoms, whose van der Waals surface distances from the docked ligands were <4 Å in the NOP receptor, were allowed to move to adopt energy-minimized active site conformations of the docked ligands. In addition, the maximum number of starting conformations were kept at four and ring flexibility was also permitted. A maximum of twenty binding poses of each compound were generated. The docked poses were ranked according to the “Total Score” in Sybyl’s Surflex-Dock docking suite and the binding pose with the best score was selected for each compound to compare the binding interactions below.
4.1.2. Molecular docking of compounds in the cryoEM MOP receptor-Gi complex (8EFL)36.
Compounds were docked into the orthosteric site of MOP using Surflex-Dock. The protomol for defining the binding site for the docking studies was generated fron the bound MOP ligand in the structure. Similar to the molecular docking in the NOP receptor, further optimization occurred during the Surflex-Dock docking studies in which hydrogen atoms (attached to hydroxyl and thiol) and heteroatoms whose van der Waals surface distances from the docked ligands <4 Å in the MOP receptor, were allowed to move to adopt energy-minimized conformations of the docked ligands at the active site. In addition, the maximum number of starting conformations for each ligand were kept at four and ring flexibility was also permitted. A maximum of twenty binding poses of each compound were generated and evaluated for possible interactions with the binding site. The docked poses were ranked according to the “Total Score” in Sybyl’s Surflex-Dock docking suite and the binding pose with the best docking score was selected for each compound to compare the binding interactions below.
4.2. Chemistry
4.2.1. General Methods
Thin layer chromatography was performed on Analtech silica gel GF 250 micron TLC plates. The plates were visualized with a 254 nm UV light and iodine. Flash chromatography was carried out on F60 silica gel, 43-60 μm (230-400 mesh), 60 Å (Silicycle SiliaFlash). All solvents and chemicals were purchased from commercial suppliers and used without further purification. All reactions were capped from the atmosphere unless otherwise stated. NMR was recorded on a Varian Mercury Plus NMR (300 MHz) using CDCl3 (7.27 ppm standard) or DMSO-d6 (2.50 ppm standard). Data for 1H NMR were recorded as follows: δ chemical shift (ppm), multiplicity (s, singlet; d, doublet; t, triplet; dd, doublet of doublets; dt, doublet of triplets; q, quartet; dq, doublet of quartets; m, multiplet; br, broad; etc.), coupling constant (Hz), integration. Mass spectra were obtained on a LCQ Fleet Ion Trap LCMS, a micromass ZMD 1000 or PE Sciex API 150EX LCMS using electrospray ionization (ESI), or APCI mode. Elemental analyses were performed by Atlantic Microlabs, Norcross, GA. HRMS analyses were performed by the Mass Spectrometry Service Laboratory, University of Minnesota Department of Chemistry, Minneapolis, MN on a Bruker BioTOF II HRMS using ESI mode. HPLC analysis was performed on a reverse phase Agilent Zorbax SB-Phenyl column (5 μm, 2.1 x150 mm), using a binary gradient of 95:5→5:95 solvent A (95/5 H2O/ACN + 0.1% formic acid): solvent B (5/95 H2O/ACN + 0.1% formic acid) for 12 minutes, at a flow rate of 0.40 mL/min. Eluted peaks were monitored at 254 nm with a Shimadzu SPD-10AVP UV-Vis detector. All final compounds were confirmed to be of ≥95% purity by the HPLC method described above. Purity of final compounds was confirmed by elemental analysis and was within ±0.4% of the theoretical value. Molecular weights of selected compounds were also confirmed by HRMS, as indicated in the experimental section. Based on 1H NMR, HPLC, combustion analysis data and HRMS, all final compounds were ≥95% pure.
4.2.2. Synthesis of compounds
General procedure for the Sonogashira coupling reaction and intramolecular heterocyclization using catalytic Cu(OAc)2.
Iodoaniline 4 (1.00 equiv) was charged into a round bottom flask, followed by the addition of terminal alkyne (2.00-3.00 equiv). A 4:1 mixture of THF:trimethylamine (TEA) (0.30M) was added, and the mixture was stirred at room temperature. PdCl2(PPh3)2 (0.020 equiv) and Cul (0.060 equiv) were weighed out on one weighing paper and added simultaneously to the reaction mixture. The reaction was quickly capped with an argon ballon attached to a three-way adapter. The reaction was purged under light vacuum, and backfilled with argon 3x total. The reaction was then covered with aluminum foil and remained in the dark with stirring. The reaction was monitored by TLC, and was complete after 5–7 hours. The reaction was quenched with water, then EtOAc was added and the biphasic layers stirred for 15 minutes. If a small amount of dark precipitate forms, simply filter over filter paper and rinse with EtOAc. The remaining EtOAc-H2O mixture was transferred to a separating funnel, H2O layer drained, and the remaining EtOAc was washed 5x with H2O to provide an orange-brown EtOAc solution. This solution was washed once with brine, dried with MgSO4, filtered, and concentrated in vacuo to provide a slightly crude oil that underwent immediate flash chromatography using EtOAc:Hexanes:NH4OH (aq.) as eluent to provide the desired internal alkyne product in 80-95% yield.
Next, the internal alkyne (1.00 equiv) was dissolved in toluene (0.10-0.25M) at room temperature, and Cu(OAc)2 (0.10-0.30 equiv) was added. The reaction was heated to reflux and monitored by TLC. Upon completion (30-180 minutes), the reaction was allowed to cool to room temperature, diluted with water and either EtOAc or CH2Cl2, then the organic layer was separated and washed 3x with H2O, brine, dried with MgSO4, filtered, and concentrated in vacuo to provide the crude indole product. The product was purified via flash chromatography using EtOAc:Hexanes:NH4OH (aq.).
Benzyl cis-((1-(1-(-4-isopropylcyclohexyl)piperidin-4-yl)-1H-indol-2-yl)methyl)carbamate (5)
Sonogashira Reaction.
Iodoaniline 420 (7.68 g, 18.0 mmol, 1.00 equiv), N-Cbz-propargylamine (8.51 g, 45.0 mmol, 2.50 equiv), PdCl2(PPh3)2 (253 mg, 0.36 mmol, 0.020 equiv), Cul (206 mg, 1.08 mmol, 0.060 equiv), THF (48 mL), triethylamine (12.0 mL). Rxn time: 7 hours. Product purified by flash chromatography using 25:75:1 EtOAc:Hexanes:NH4OH (aq.) to provide the product as an orange oil (8.51 g, 97% yield). Rf = 0.30 (30:70:3 drops EtOAc:Hexanes:NH4OH (aq.), UV, I2).
Cu(OAc)2 catalyzed heterocyclization.
Internal alkyne (8.50 g, 17.4 mmol, 1.00 equiv), Cu(OAc)2 (950 mg, 5.22 mmol, 0.30 equiv), toluene (70 mL, 0.25M). Refluxed 30 minutes. Crude material was preloaded onto silica gel, and then purified via flash chromatography using 20:80:1→25:75:1 EtOAc:Hexanes:NH4OH (aq) to provide the desired product 5 as light-yellow solid (5.5 g, 65% yield). Rf = 0.40 (30:70:3 drops EtOAc:Hexanes:NH4OH (aq.), UV, I2). 1H NMR (300 MHz, CDCl3) δ 7.65 (d, J = 8.1 Hz, 1H), 7.55 (d, J = 7.5 Hz, 1H), 7.33 (m, 5H), 7.15 (t, J = 8.1 Hz, 1H), 7.06 (t, J = 7.5 Hz, 1H), 6.39 (s, 1H), 5.17 (s, 2H), 4.90 (br, 1H), 4.59 (d, J = 5.7 Hz, 1H), 4.15 (m, 1H), 3.10 (d, J = 10.2 Hz, 2H), 2.57 (dq, J = 12.0, 3.6 Hz, 2H), 2.31 (m, 1H), 2.11 (t, J = 12.0 Hz, 2H), 1.35-1.70 (m, 11H), 1.16 (m, 1H), 0.93 (d, J = 6.9 Hz, 6H).
Benzyl cis-(2-(1-(1-(-4-isopropylcyclohexyl)piperidin-4-yl)-1H-indol-2-yl)ethyl)carbamate (6)
Sonogashira Reaction.
Iodoaniline 420 (5.10 g, 12.0 mmol, 1.00 equiv), benzyl but-3-yn-1-ylcarbamate (4.88 g, 24.0 mmol, 2.00 equiv), PdCl-2(PPh3)2 (168 g, 0.24 mmol, 0.020 equiv), CuI (137 mg, 0.72 mmol, 0.060 equiv), THF (32 mL), triethylamine (8 mL). Rxn time: 6 hours. Product purified by flash chromatography using 30:70:1 EtOAc:Hexanes:NH4OH (aq.) to provide the product as an orange oil (5.1 g, 85% yield). Rf = 0.40 (30:70:3 drops EtOAc:Hexanes:NH4OH (aq.), UV, I2).
Cu(OAc)2 catalyzed heterocyclization.
Internal alkyne (5.10 g, 10.2 mmol, 1.00 equiv), Cu(OAc)2 (278 mg, 1.53 mmol, 0.15 equiv), toluene (41 mL, 0.25M). Refluxed 90 minutes. Crude material purified via flash chromatography using 25:75:1 EtOAc:Hexanes:NH4OH (aq) to provide the desired product 6 as an orange oil (4.32 g, 85% yield). Rf = 0.30 (30:70:3 drops EtOAc:Hexanes:NH4OH (aq.), UV, I2). 1H NMR (300 MHz, CDCl3) δ 7.65 (d, J = 8.1 Hz, 1H), 7.55 (d, J = 7.5 Hz, 1H), 7.33 (m, 5H), 7.15 (t, J = 8.1 Hz, 1H), 7.06 (t, J = 7.5 Hz, 1H), 6.39 (s, 1H), 5.17 (s, 2H), 4.90 (br, 1H), 4.15 (m, 1H), 3.30 (m, 2H), 3.02 (t, J = 4.8 Hz, 2H), 3.10 (d, J = 10.2 Hz, 2H), 2.57 (dq, J = 12.0, 3.6 Hz, 2H), 2.31 (m, 1H), 2.11 (t, J = 12.0 Hz, 2H), 1.38-1.72 (m, 11H), 1.16 (m, 1H), 0.92 (d, J = 6.6 Hz, 6H).
cis-(1-(1-(-4-Isopropylcyclohexyl)piperidin-4-yl)-1H-indol-2-yl)methanol (1).
Sonogashira Reaction.
Iodoaniline 220 (21.0 g, 49.2 mmol, 1.00 equiv), propargyl alcohol (8.60 mL, 148 mmol, 3.00 equiv), PdCl2(PPh3)2 (690 g, 0.984 mmol, 0.020 equiv), CuI (562 mg, 2.95 mmol, 0.060 equiv), THF (131 mL), triethylamine (33 mL). Rxn time: 5 hours. Product purified by flash chromatography using 30:70:1 EtOAc:Hexanes:NH4OH (aq.) to provide the product as a dark glue (14.5 g, 83% yield). Rf = 0.20 (30:70:3 drops EtOAc:Hexanes:NH4OH (aq.), UV, I2).
Cu(OAc)2 catalyzed heterocyclization.
Internal alkyne (14.0 g, 39.5 mmol, 1.00 equiv), Cu(OAc)2 (716 mg, 3.95 mmol, 0.10 equiv), toluene (158 mL, 0.25M). Refluxed 30 minutes. Crude material purified via flash chromatography using 25:75:1→40:60:1→50:50:1 EtOAc:Hexanes:NH4OH (aq) to provide the desired product 1 as a light-yellow solid in a free base form (9.38 g, 67% yield). Rf = 0.25 (30:70:3 drops EtOAc:Hexanes:NH4OH (aq.), UV, I2). 1H NMR (300 MHz, CDCl3) δ 7.69 (d, J = 6.3 Hz, 1H), 7.59 (d, J = 6.3 Hz, 1H), 7.18 (t, J = 6.0 Hz, 1H), 7.08 (t, J = 6.0 Hz, 1H), 4.81 (s, 2H), 4.39 (m, 1H), 3.19 (d, J = 8.7 Hz, 2H), 2.62 (dq, J = 9.3, 2.7, 2H), 2.36 (m, 1H), 2.26 (t, J = 8.7 Hz, 2H), 1.89 (m, 2H), 1.52-1.80 (m, 8H), 1.41 (m, 2H), 1.16 (m, 1H), 0.92 (d, J = 5.1 Hz, 6H) MS(ESI) m/z: 355.6 [M+H]+. The acetate salt of compound 1 (free base) was made by reacting 1 with 3 eq. of acetic acid in DCM. Anal. Calc’d for C23H34N2O·1.00 AcOH: C, 72.4; H, 9.2; N, 6.8; found: C, 72.1; H, 9.2; N, 6.6.
cis-2-(1-(1-(-4-Isopropylcyclohexyl)piperidin-4-yl)-1H-indol-2-yl)ethan-1-ol (7).
Sonogashira Reaction.
Iodoaniline 220 (36.0 g, 84.4 mmol, 1.00 equiv), but-3-yn-1-ol (12.8 mL, 169 mmol, 2.00 equiv), PdCl2(PPh3)2 (1.19 g, 1.69 mmol, 0.020 equiv), CuI (964 mg, 5.06 mmol, 0.060 equiv), THF (224 mL), triethylamine (56 mL). Rxn time: 5-6 hours. Product purified by flash chromatography using 40:60:1→50:50:1→60:40:1→70:30:1 EtOAc:Hexanes:NH4OH (aq.) to provide the product as an orange-brown glue (30.5 g, 98% yield). Rf = 0.40 (50:50:3 drops EtOAc:Hexanes:NH4OH (aq.), UV, I2).
Cu(OAc)2 catalyzed heterocyclization.
Internal alkyne (30.5 g, 82.8 mmol, 1.00 equiv), Cu(OAc)2 (1.50 mg, 8.28 mmol, 0.10 equiv), toluene (800 mL, 0.10M). Refluxed 3 hours. Crude material purified via flash chromatography using 40:60:1→50:50:1→60:40:1 EtOAc:Hexanes:NH4OH (aq) to provide the desired product 7 as a brown solid in a free base form (19.1 g, 63% yield). Rf = 0.30 (40:60:3 drops EtOAc:Hexanes:NH4OH (aq.), UV, I2). 1H NMR (300 MHz, CDCl3) δ 7.66 (d, J = 6.0 Hz, 1H), 7.55 (d, J = 5.7 Hz, 1H), 7.13 (t, J = 5.4 Hz, 1H), 7.07 (t, J = 5.4 Hz, 1H), 6.33 (s, 1H), 4.14 (m, 1H), 3.94 (t, J = 4.8 Hz, 2H), 3.20 (d, J = 8.7 Hz, 2H), 3.09 (t, J = 4.8 Hz, 2H), 2.64 (dq, J = 7.5, 2.7, 2H), 2.36 (m, 1H), 2.22 (t, J = 8.4 Hz, 2H), 1.50-1.86 (m, 10H), 1.42 (m, 2H), 1.18 (m, 1H), 0.92 (d, J = 4.8 Hz, 6H). MS(ESI) m/z: 369.6 [M+H]+. The acetate salt of compound 7 (free base) was made by reacting 7 with 3 eq. of acetic acid in DCM. Anal. Calc’d for C24H36N2O·0.50 AcOH + 0.50 H2O: C, 73.7; H, 9.6; N, 6.9; found: C, 73.8; H, 9.3; N, 6.6.
4-cis-((1-(1-(-4-Isopropylcyclohexyl)piperidin-4-yl)-1H-indol-2-yl)methyl)thiomorpholine 1,1-dioxide (8).
Sonogashira Reaction.
Iodoaniline 220 (1.54 g, 3.61 mmol, 1.00 equiv), 4-(prop-2-yn-1-yl)thiomorphine 1,1-dioxide (1.00 g, 5.77 mmol, 1.60 equiv), PdCl2(PPh3)2 (50.7 mg, 0.0722 mmol, 0.020 equiv), CuI (41.3 mg, 0.217 mmol, 0.060 equiv), THF (9.6 mL), triethylamine (2.40 mL). Rxn time: 7 hours. Product purified by flash chromatography using 50:50:1→60:40:1→70:30:1→80:20:1→90:10:1 EtOAc:Hexanes:NH4OH (aq.) to provide the product as a brown semi-solid (1.18 g, 69% yield). Rf = 0.20 (50:50:3 drops EtOAc:Hexanes:NH4OH (aq.), UV, I2).
Cu(OAc)2 catalyzed heterocyclization.
Internal alkyne (1.18 g, 2.50 mmol, 1.00 equiv), Cu(OAc)2 (90.8 mg, 0.500 mmol, 0.20 equiv), toluene (12.5 mL, 0.20M). Refluxed 4 hours. Crude material was dissolved in minimal CH2Cl2, and then purified via flash chromatography using 50:50:1 EtOAc:Hexanes:NH4OH (aq) to provide tertiary amine 14 as an off-white solid in a free base form (535 mg, 45% yield). Rf = 0.50 (60:40:3 drops EtOAc:Hexanes:NH4OH (aq.), UV, I2). 1H NMR (300 MHz, DMSO-d6) δ 7.50 (dd, J = 12.0, 7.8 Hz, 1H), 7.08 (t, J = 7.5 Hz, 1H), 6.97 (t, J = 7.5 Hz, 1H), 6.37 (s, 1H), 4.39 (m, 1H), 3.82 (s, 2H), 3.10 (m, 6H), 2.91 (m, 4H), 2.30-2.50 (m, 3H), 2.19 (m, 2H), 1.34-1.82 (m, 11H), 1.17 (m, 1H), 0.89 (d, J = 6.6 Hz, 6H). MS(ESI) m/z: 472.9 [M+H]+. Tartrate salt of 14 (free base) was made using similar procedure as compound 10 to get final compound 14 as a tartrate salt. Anal. Calc’d for C27H36N4O2·0.50 tartaric acid + 0.50 H2O: C, 62.7; H, 8.2; N, 7.6; found: C, 63.0; H, 7.9; N, 7.5.
1-cis-(1-(-4-Isopropylcyclohexyl)piperidin-4-yl)-2-(pyrrolidin-1-ylmethyl)-1H-indole (9).
Alcohol 1 (1.30 g, 3.67 mmol, 1.00 equiv) was >90% dissolved in CH2Cl2 (36.7 mL, 0.10M) at room temperature, and then MnO2 (3.83 g, 44.0 mmol, 12.0 equiv) was added to the reaction. The reaction was capped, and allowed to stir at room temperature overnight. TLC now shows the reaction complete. The reaction was filtered over Celite, and concentrated in vacuo to provide the pure intermediate aldehyde as a white glue (1.27 g, 98%), and was used directly in the following step.
Intermediate aldehyde (87 mg, 0.247 mmol, 1.00 equiv) was charged into a round-bottom flask, and dissolved in 1,2-DCE (2.50 mL, 0.10M). Pyrrolidine (33.0 μL, 0.395 mmol, 1.60 equiv) was then added, followed by glacial AcOH (32.5 μL, 0.568 mmol, 2.30 equiv), and the reaction was allowed to stir for 30 minutes at room temperature, and then sodium triacetoxyborohydride (STAB) (120 mg, 0.568 mmol, 2.30 equiv) was added to the reaction, and the reaction was allowed to stir at room temperature overnight. TLC showed the reaction was complete. The reaction was diluted with EtOAc and water, then the layers were separated, and the EtOAc layer was washed 3x with water, 1x with brine, dried with MgSO4, filtered, and concentrated in vacuo to provide a crude oil that was purified via flash chromatography using 40:60:1 EtOAc:Hexanes:NH4OH (aq.) to provide pyrrolidine 9 as an off-white solid in a free base form (30.3 mg, 30% yield). Rf = 0.20 (30:70:3 drops EtOAc:Hexanes:NH4OH (aq.), UV, I2). 1H NMR (300 MHz, CDCl3) δ 7.66 (d, J = 5.4 Hz, 1H), 7.54 (d, J = 6.9 Hz, 1H), 7.12 (dt, J = 6.0, 1.2, 1H), 7.03 (m, 1H), 6.32 (s, 1H), 4.50 (m, 1H), 3.74 (s, 2H), 3.19 (d, J = 11.7 Hz, 2H), 2.46-2.67 (m, 6H), 2.35 (m, 1H), 2.18 (t, J = 11.7 Hz, 2H), 1.50-1.85 (m, 13H), 1.43 (m, 2H), 1.17 (m, 1H), 0.92 (d, J = 6.6 Hz, 6H). MS(ESI) m/z: 408.5 [M+H]+. Tartrate salt of 9 (free base) was made using similar procedure as compound 10 to get final compound 9 as a tartrate salt. Anal. Calc’d for C27H36N4O2·1.00 tartaric acid + 1.00 Et2O + 1.10 H2O: C, 64.5; H, 9.2; N, 6.45; found: C, 64.2; H, 8.8; N, 6.4.
cis-(1-(1-(-4-Isopropylcyclohexyl)piperidin-4-yl)-1H-indol-2-yl)methanamine (10).
Pure carbamate 3 (14.0 g, 28.7 mmol, 1.00 equiv) was charged into a round-bottom flask, followed by 10% Pd/C (1.40 g, 10 wt%) and a magnetic stirbar. The flask was then cooled in an ice-bath for 5 minutes, and then 1.5N NH3 in MeOH was added dropwise along the flask walls until all of the contents are “wet” with MeOH. Once this has occurred, the ice-bath is removed, the remaining MeOH was added (145 mL total), and the reaction mixture is allowed to warm to room temperature and stir. At this stage, a three-way adapter containing a H2 (g) balloon was attached to the reaction flask, and the reaction was purged under vacuo and then backfilled with H2 (g), repeat 4x total. Initially carbamate 3 is insoluble and the result is a thick grey suspension at the start of the reaction. As the reaction progress, the product is soluble, and thus, the precipitate lightens over time until only black Pd/C was observed. By TLC, the reaction was completed in 4 hours. The reaction was filtered over Celite, rinsed thoroughly with MeOH, and then the MeOH filtrate was concentrated in vacuo to provide to provide amine 10 as a neon glue (9.30 g, 92% yield). This material was salted by adding a solution of tartaric acid (7.90 g, 52.6 mmol, 2.00 equiv) in MeOH to amine 10 (9.30 g, 26.3 mmol, 1.00 equiv) in MeOH (0.10M total). A white precipitate formed immediately, and after 10 minutes, the suspension was cooled in an ice-bath for 20 minutes, filtered, and the white precipitate was washed with cold Et2O 3x and then dried under vacuo to provide a 1.50·tartrate salt of amine 10 (generally 75% yield). Rf = 0.25 (2:98:3 drops MeOH:CH2Cl2:NH4OH (aq.), UV, I2). 1H NMR (300 MHz, DMSO-d6) δ 7.66 (d, J = 8.1 Hz, 1H), 7.54 (d, J = 7.8 Hz, 1H), 7.13 (t, J = 7.2 Hz, 1H), 7.02 (t, J = 7.2 Hz, 1H), 6.51 (s, 1H), 4.40 (m, 1H), 4.28 (s, 2H), 3.97 (s, 3H), 3.28 (m, 2H), 2.50-2.73 (m, 6H), 1.90 (m, 2H), 1.57-1.78 (m, 8H), 1.40 (m, 2H), 1.17 (m, 1H), 0.89 (d, J = 6.6 Hz, 6H). MS(ESI) m/z: 354.6 [M+H]+. Anal. Calc’d for C23H35N3·1.50 tartaric acid + 0.50 H2O: C, 59.3; H, 7.7; N, 7.15; found: C, 59.0; H, 7.7; N, 7.3.
cis-2-(1-(1-(-4-Isopropylcyclohexyl)piperidin-4-yl)-1H-indol-2-yl)ethan-1-amine (11).
Same procedure for Cbz removal as amine 10 to obtain 11 as a free base (75-80% yield). Rf = 0.10 (100:3 drops EtOAc:NH4OH (aq.), UV, I2). 1H NMR (300 MHz, CDCl3) (freebase) δ 7.63 (d, J = 6.4 Hz, 1H), 7.52 (d, J = 5.4 Hz, 1H), 7.01-7.14 (m, 2H), 6.28 (s, 1H), 5.40 (br, 2H), 4.10 (m, 1H), 3.19 (d, J = 12.4 Hz, 2H), 3.04 (m, 2H), 2.92 (m, 2H), 2.63 (dq, J = 12.0, 3.4 Hz, 2H), 2.35 (m, 1H), 2.20 (t, J = 12.0 Hz, 2H), 1.34-1.86 (m, 11H), 1.17 (m, 1H), 0.92 (d, J = 6.6 Hz, 6H). MS(ESI) m/z: 368.5 [M+H]+. Tartrate salt of 11 (free base) was made using similar procedure as compound 10 to get final compound 11 as a tartrate salt. Anal. Calc’d for C24H37N3·1.50 tartaric acid + 1.70 H2O: C, 57.8; H, 8.0; N, 6.7; found: C, 58.0; H, 7.8; N, 6.4.
N-cis-((1-(1-(-4-Isopropylcyclohexyl)piperidin-4-yl)-1H-indol-2-yl)methyl)acetamide (12).
To a solution of amine 10 (98 mg, 0.277 mmol, 1.00 equiv) and trimethylamine (135 μL, 0.776 mmol, 2.80 equiv) in CH2Cl2 (2.80 mL, 0.10M) at room temperature was added acetic anhydride (36.7 μL, 0.388 mmol, 1.40 equiv). The reaction was allowed to stir at room temperature overnight. TLC indicated the reaction was complete. The reaction was diluted with EtOAc and H2O, the layers were separated and the EtOAc layer was washed 2x with water, 1x with brine, dried with MgSO4, filtered, and concentrated in vacuo to provide a yellow-brown solid. The solid was triturated with 100% EtOAc, and washed 3x with cold EtOAc to provide acetamide 12 as an off-white solid in a free base form (64 mg, 58% yield). Rf = 0.40 (100:3 drops EtOAc:NH4OH (aq.), UV, I2). 1H NMR (300 MHz, CDCl3) δ 7.54 (d, J = 7.5 Hz, 1H), 7.18 (t, J = 7.5 Hz, 1H), 7.07 (t, J = 7.5 Hz, 1H), 6.42 (s, 1H), 4.63 (d, J = 2.8 Hz, 2H), 4.20 (m, 1H), 3.30 (m, 1H), 3.17 (m, 2H), 2.65 (m, 2H), 2.08 (m, 3H), 1.35-1.82 (m, 12H), 1.21 (m, 1H), 0.94 (d, J = 3.0 Hz, 6H). MS(ESI) m/z; 396.5 [M+H]+. Acetate salt of compound 12 (free base) was made by reacting 12 with 3.0 eq. of acetic acid in DCM. Anal. Calc’d for C25H37N3O·1.00 AcOH + 0.80 H2O: C, 69.0; H, 9.1; N, 8.9; found: C, 68.8; H, 8.7; N, 8.8.
Ethyl cis-((1-(1-(-4-isopropylcyclohexyl)piperidin-4-yl)-1H-indol-2-yl)methyl)carbamate (13).
To a solution of amine 10 (45 mg, 0.127 mmol, 1.00 equiv) and trimethylamine (35.4 μL, 0.254 mmol, 2.00 equiv) in CH2Cl2 (1.30 mL, 0.10M), cooled in an ice-bath at 0 °C, was added ethyl chloroformate (14.5 μL, 0.152 mmol, 1.20 equiv). The reaction was allowed to warm to room temperature and was monitored by TLC. After 2 hours, TLC showed the reaction was >95% complete. The reaction was diluted with EtOAc and H2O, stirred for several minutes, and then the layers were separated. The EtOAc layer was washed 2x with water, 1x with brine, dried with MgSO4, filtered, and concentrated in vacuo to provide a white solid. This solid was dissolved in minimal CH2Cl2 and loaded onto column and purified via flash chromatography using 10:90:1→15:85:1 EtOAc:Hexanes:NH4OH (aq.) to provide ethyl carbamate 13 as a white solid (52.0 mg, 96% yield). Rf = 0.30 (20:80:3 drops EtOAc:Hexanes:NH4OH (aq.), UV, I2). 1H NMR (300 MHz, CDCl3) δ 7.66 (d, J = 8.1 Hz, 1H), 7.56 (d, J = 7.5 Hz, 1H), 7.16 (t, J = 7.2 Hz, 1H), 7.07 (t, J = 7.2 Hz, 1H), 6.40 (s, 1H), 4.79 (br, 1H), 4.57 (d, J = 5.7 Hz, 2H), 4.18 (m, 3H), 3.18 (d, J = 12.0 Hz, 2H), 2.60 (dq, J = 12.6, 3.6 Hz, 2H), 2.35 (m, 1H), 2.21 (t, J = 12.3 Hz, 2H), 1.36-1.86 (m, 11H), 1.27 (t, J = 7.2 Hz, 3H), 1.18 (m, 1H), 0.92 (d, J = 6.6 Hz, 6H). MS(ESI) m/z: 426.7 [M+H]+. Anal. Calc’d for C26H39N3O2: C, 73.47; H, 9.2; N, 9.9; found: C, 73.4; H, 9.3; N, 9.7.
N-cis-((1-(1-(-4-isopropylcyclohexyl)piperidin-4-yl)-1H-indol-2-yl)methyl)methanesulfonamide (14).
Methanesulfonyl chloride (557 μL, 0.720 mmol, 1.50 equiv) was added to a solution of amine 10 (170 mg, 0.480 mmol, 1.00 equiv) and diisopropylethylamine (251 μL, 1.44 mmol, 3.00 equiv) in CH2Cl2 (4.80 mL, 0.10M) at 0 °C. The reaction was immediately allowed to warm to room temperature upon which the reaction turned yellow. After ca. 1 hour, TLC showed the reaction was >95% complete. The reaction was diluted with EtOAc and water, and the biphasic solution was stirred for 10 minutes. The layers were then separated and the EtOAc layer was washed 2x with water, 1x with brine, dried with MgSO4, filtered, and concentrated in vacuo to provide a crude oil that was purified via flash chromatography using 30:70:1 EtOAc:Hexanes:NH4OH (aq.) to provide sulfonamide 14 as white foam (130 mg, 63%). Rf = 0.20 (30:70:3 drops EtOAc:Hexanes:NH4OH (aq.), UV, I2). 1H NMR (400 MHz, CDCl3) 67.69 (d, J = 8.4 Hz, 1H), 7.58 (d, J = 7.6 Hz, 1H), 7.19 (t, J = 8.4 Hz, 1H), 7.09 (t, J = 7.2 Hz, 1H), 6.47 (s, 1H), 4.51 (s, 2H), 4.48 (br, 1H), 4.27 (m, 1H), 3.20 (d, J = 12.0 Hz, 2H), 2.96 (s, 3H), 2.61 (dq, J = 12.6, 3.6 Hz, 2H), 2.36 (m, 1H), 2.26 (t, J = 12.0 Hz, 2H), 1.87 (d, J = 12.0 Hz, 2H), 1.33-1.78 (m, 7H), 1.42 (m, 2H), 1.18 (m, 1H), 0.92 (d, J = 6.8 Hz, 6H). MS(ESI) m/z: 432.8 [M+H]+. HRMS calculated for C24H37N3O2S (M+H)+ 432.2685, found 432.2674.
N-cis-((1-(1-(-4-isopropylcyclohexyl)piperidin-4-yl)-1H-indol-2-yl)methyl) sulfamide (15).
Amine 10 (364 mg, 1.03 mmol, 1.00 equiv) and sulfamide (198 mg, 2.06 mmol, 2.00 equiv) were charged into a round-bottom flask. Dioxane (6.9 mL, 0.14M) was added and the light suspension was heated to reflux and monitored by TLC. After ca. 3.5 hours, the reaction was complete. The reaction was allowed to cool to room temperature, concentrated in vacuo, and the crude material was dissolved in minimal 5:95 MeOH:CH2Cl2 and purified via flash chromatography using 4:96:1→5:95:1→6:94:1 MeOH:CH2Cl2:NH4OH (aq.) to provide sulfamide 15 as a glassy, soft solid in a free base form (251 mg, 56% yield). Rf = 0.20 (5:95:3 drops MeOH:CH2Cl2:NH4OH (aq.), UV, I2). 1H NMR (300 MHz, CDCl3) δ 7.69 (m, 1H), 7.54 (d, J = 8.1 Hz, 1H), 7.20 (t, J = 7.5 Hz, 1H), 7.07 (t, J = 7.5 Hz, 1H), 6.47 (s, 1H), 4.8 (br, 2H), 4.48 (s, 2H), 4.30 (br, 1H), 3.17 (d, J = 10.5 Hz, 2H), 2.30-2.71 (m, 5H), 1.56-1.92 (m, 10H), 1.41 (m, 2H), 1.19 (m, 1H), 0.92 (d, J = 6.6 Hz, 6H). MS(ESI) m/z: 433.7 [M+H]+. Tartrate salt of 15 (free base) was made using similar procedure as compound 10 to get final compound 15 as a tartrate salt. Anal. Calc’d for C23H36N4O2S·1.50 tartaric acid: C, 53.0; H, 6.9; N, 8.5; found: C, 53.3; H, 7.3; N, 8.7.
N-cis-((1-(1-(-4-isopropylcyclohexyl)piperidin-4-yl)-1H-indol-2-yl)methyl)nicotinamide (16).
To a solution of 3-nicotinic acid (16.7 mg, 0.136 mmol, 1.00 equiv) in DMF (250 μL) at room temperature, was added pyBOP (70.8 mg, 0.136 mmol, 1.00 equiv) followed by diisopropylethylamine (28.4 μL, 0.163 mmol, 1.20 equiv). The reaction was stirred for 5 minutes, then a solution of amine 10 (48.0 mg, 0.136 mmol, 1.00 equiv) in DMF (150 μL, 50 μL rinse) was added and the reaction was allowed to stir at room temperature overnight. TLC showed the reaction was complete. The reaction was diluted with EtOAc and water, the layers were separated, and the EtOAc layer was wash 3x with water, 1x with brine, dried with MgSO4, filtered, and concentrated in vacuo to provide a crude material that was purified via flash chromatography using 50:50:1→70:30:1 EtOAc:Hexanes:NH4OH (aq.) to provide nicotinic amide 16 as a white solid in a free base form (39.0 mg, 63% yield). Rf = 0.20 (50:50:3 drops EtOAc:Hexanes:NH4OH (aq.), UV, I2). 1H NMR (300 MHz, CDCl3) δ 8.96 (d, J = 1.5 Hz, 1H), 8.75 (dd, J = 4.8, 1.5 Hz, 1H), 8.10 (dt, J = 7.8, 2.1 Hz, 1H), 7.68 (d, J = 8.4 Hz, 1H), 7.58 (d, J = 7.5 Hz, 1H), 7.40 (dd, J = 4.8, 0.9 Hz, 1H), 7.18 (m, 1H), 7.09 (m, 1H), 6.49 (s, 1H), 6.27 (br, 1H), 4.89 (d, J = 5.7 Hz, 2H), 4.24 (m, 1H), 3.14 (d, J = 11.1 Hz, 2H), 2.60 (dq, J = 11.1, 2.4 Hz, 2H), 2.31 (m, 1H), 2.18 (t, J = 12.0 Hz, 2H), 1.37-1.82 (m, 11H), 1.18 (m, 1H), 0.91 (d, J = 6.6 Hz, 6H). MS(ESI) m/z: 459.7 [M+H]+. Anal. Calc’d for C29H38N4O°0.50 Et2O: C, 75.1; H, 8.7; N, 11.3; found: C, 75.0; H, 8.4; N, 11.0.
N-cis-((1-(1-(-4-isopropylcyclohexyl)piperidin-4-yl)-1H-indol-2-yl)methyl)oxazole-5-carboxamide (17).
To a room temperature solution containing oxazole-5-carboxylic acid (232 mg, 2.05 mmol, 1.00 equiv) in DMF (4.5 mL), was added pyBOP (1.07 g, 2.05 mmol, 1.00 equiv), followed by triethylamine (343 μL, 2.46 mmol, 1.20 equiv). The reaction was allowed to stir for 5 minutes, and then amine 10 (725 mg, 2.05 mmol, 1.00 equiv) was added to the reaction, and the reaction was allowed to stir overnight at room temperature. TLC indicated the reaction was complete, and the reaction was diluted with EtOAc and water. The layers were separated, and the EtOAc layer was washed 3x with water, 1x with brine, dried with MgSO4, filtered, and concentrated in vacuo to provide a crude solid that was triturated with MeOH to provide oxazole 17 as a free base (204 mg, 22% yield). Rf = 0.30 (40:60:3 drops EtOAc:Hexanes:NH4OH (aq.), UV, I2). 1H NMR (300 MHz, CDCl3) δ 7.89 (s, 1H), 7.80 (s, 1H), 7.58 (d, J = 7.8 Hz, 1H), 7.20 (t, J = 7.8 Hz, 1H), 7.10 (t, J = 7.5 Hz, 1H), 6.50 (s, 1H), 4.84 (d, J = 5.7 Hz, 2H), 4.24 (m, 1H), 3.18 (m, 2H), 2.60 (m, 1H), 2.30 (m, 1H), 1.37-1.84 (m, 15H), 1.19 (m, 1H), 0.91 (d, J = 6.9 Hz, 6H). MS(ESI) m/z: 449.6 [M+H]+. Tartrate salt of 17 (free base) was made using similar procedure as compound 10 to get final compound 17 as a tartrate salt. Anal. Calc’d for C27H36N4O2·1.00 tartaric acid + 1.00 H2O: C, 60.4; H, 7.2; N, 9.1; found: C, 60.4; H, 7.3; N, 8.7.
N-cis-((1-(1-(-4-isopropylcyclohexyl)piperidin-4-yl)-1H-indol-2-yl)methyl)-5-(trifluoromethyl)furan-2-carboxamide (18).
To a room temperature solution containing 5-(trifluoromethyl)furan-2-carboxylic acid (370 mg, 2.05 mmol, 1.00 equiv) in DMF (5.0 mL), was added pyBOP (1.07 g, 2.05 mmol, 1.00 equiv), followed by triethylamine (343 μL, 2.46 mmol, 1.20 equiv). The reaction was allowed to stir for 5 minutes, and then amine 10 (725 mg, 2.05 mmol, 1.00 equiv) was added to the reaction, and the reaction was allowed to stir overnight at room temperature. TLC indicated the reaction was 80-90% complete, and the reaction was diluted with EtOAc and water. The layers were separated, and the EtOAc layer was washed 3x with water, 1x with brine, dried with MgSO4, filtered, and concentrated in vacuo to provide a crude oil that was purified via flash chromatography using 40:60:0.5:0.5→50:50:0.5:0.5→60:40:0.5:0.5 EtOAc:Hexanes:MeOH:NH4OH (aq.) to provide furan 18 in a free base form (210 mg, 20% yield). Rf = 0.25 (50:50:3 drops EtOAc:Hexanes:NH4OH (aq.), UV, I2). 1H NMR (300 MHz, CDCl3) δ 7.57 (m, 1H), 7.05-7.27 (m, 4H), 6.94 (m, 1H), 6.53 (s, 1H), 4.84 (d, J = 6.0 Hz, 2H), 4.69 (m, 1H), 3.20 (m, 2H), 2.80 (m, 2H), 2.60 (m, 2H), 2.04 (m, 1H), 1.36-1.85 (m, 11H), 1.20 (m, 1H), 0.92 (d, J = 6.6 Hz, 6H). MS(ESI) m/z: 516.7 [M+H]+. Tartrate salt of 18 (free base) was made using similar procedure as compound 10 to get final compound 18 as a tartrate salt. Anal. Calc’d for C27H36N4O2·1.00 tartaric acid + 0.50 H2O: C, 58.8; H, 6.4; N, 6.2; found: C, 58.9; H, 6.5; N, 5.9.
4.3. In Vitro Pharmacology
The in vitro pharmacological characterization of binding affinities and functional activity of the test compounds at all four opioid receptors was conducted in-house in membranes from stably transfected Chinese hamster ovary (CHO) cells containing the human opioid receptors, using methods we have described previously.28,33
4.3.1. Cell Culture
Human NOP, mu, delta, and kappa opioid receptors were individually expressed in Chinese hamster ovary (CHO) cells stably transfected with the human receptor cDNA, as we have described previously.33,34 The hORL, hDOR, and hKOR-FLAG19 cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) with 10% fetal bovine serum, in the presence of 0.4 mg/ml G418 and 0.5% penicillin/streptomycin, in 150-mm tissue culture dishes. The hMOR cells were grown in 50% F12/DMEM with 10% fetal bovine serum, in the presence of 0.4 mg/ml G418 and 0.5% penicillin/streptomycin.
4.3.2. Membrane preparation
The cell lines were grown to confluency, then harvested for membrane preparation. The membranes were prepared in 50 mM Tris buffer (pH 7.4). Cells are scraped and centrifuged at 500 × g for 12 mins. The cell pellet was homogenized in 50 mM Tris with a Fisher Scientific PowerGen 125 rotor-stator type homogenizer, centrifuged at 20,000 × g for 25 mins, washed and recentrifuged once more at 20,000 × g for 25 mins, and aliquoted at a concentration of 3 mg/ml protein per vial and stored in a −80 °C freezer till further use. For the hORL-CHO membranes, the aliquots contained 2 mg/ml protein.
4.3.3. Receptor Binding
Test compounds were dissolved at 10 mM stock in 100% DMSO and diluted with 50 nMTris buffer pH 7.4. The assay was performed in a 96-well polystyrene plate with triplicates of six concentrations of each test compound (10μM – 0.1 nM), adding 100 μl of compound and 100 μl of tritiated ligands [3H]DAMGO (51.0 Ci/mmole, Kd 0.59 nM for MOP), [3H]DPDPE (42.0 Ci/mmole, Kd 1.11 nM for DOP), [3H]U69593 (41.7 Ci/mmole, Kd 1.05 nM for KOP), and [3H]N/OFQ (130 Ci/mmole, Kd 0.12 nM for NOP). Nonspecific binding was determined using 1.0 μM of the unlabeled version of each of the radioligands respectively. Assays were initiated by addition of 800 μl of membrane per well (approx.3 mg membrane homogenate per plate). Samples were incubated for 60 min at 25°C in a total volume of 1.0 ml. In NOP receptor experiments, 1 mg/ml BSA was added to the compound dilution buffer. The incubation was terminated by rapid filtration through 0.05% PEI-soaked glass fiber filter mats (GF/C Filtermat A, Perkin-Elmer) on a Tomtec Mach III cell harvester and washed 5 times with 0.5 ml of ice-cold 50 nM Tris-HCl, pH 7.4 buffer. The filters were dried overnight and soaked with scintillation cocktail before counting on a Wallac Beta plate 1205 liquid scintillation counter. Radioactivity was determined as counts per minutes (cpm). IC50 values were obtained using at least six concentrations of test compound, and calculated using GraphPad/Prism (ISI, San Diego, CA). Ki values were determined by the method of Cheng and Prusoff Binding affinities reported are from at least three independent experiments conducted in triplicate. Statistical analysis was conducted using GraphPad Prism.
4.3. [35S]GTPγS binding Assay
The functional efficacy of compounds was determined by their ability to stimulate [35S]GTPγS binding to cell membranes and compared to that of standard agonists. For this binding assay, membrane pellets prepared as described above were resuspended in Buffer A, containing 20 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), 10 mM MgCl2, and 100 mM NaCl, pH 7.4. Membrane prepared as described was incubated with [35S]GTPγS (150,000 cpm/well), guanosine diphosphate (GDP) (10 mM), and the test compound, in a total volume of 1 ml for 60 minutes at 25 °C. Samples were filtered over Filtermat A and counted as described for the binding assays. A dose response curve with a prototypical full agonist at the respective receptor was included in each experiment. Typically, the standard full agonists (N/OFQ for NOP, DAMGO for MOP, U-69593 for KOP, and DPDPE for DOP) showed at least 2-fold to 5-fold stimulation over basal. The EC50 values were determined from at least six concentrations of test compound and calculated using GraphPad Prism 6 (ISI, San Diego, CA). The % stimulation by the test compound was calculated as the percentage of stimulation by the standard full agonist (Table 1).
Supplementary Material
Acknowledgements
This work was supported by grants from the National Institutes on Drug Abuse R01DA027811, R44DA042465 and UG3DA048371 to NTZ
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interest
The authors declare that they have no competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Abbreviations: PD, Parkinson’s disease; CPP, conditioned place preference; GPCR, G-protein coupled receptor; NOP, nociceptin opioid; MOP, μ opioid; DOP, δ opioid; KOP, κ opioid; CNS, central nervous system; NHP, non-human primate; TM, transmembrane; ECL2, extracellular loop 2; SAR structure-activity relationship.
References
- 1.Bunzow JR, Saez C, Mortrud M, Bouvier C, Williams JT, Low M, et al. Molecular cloning and tissue distribution of a putative member of the rat opioid receptor gene family that is not a mu, delta or kappa opioid receptor type. FEBS Lett. 1994, 347(2-3), 284–288. [DOI] [PubMed] [Google Scholar]
- 2.Mollereau C, Parmentier M, Mailleux P, Butour JL, Moisand C, Chalon P, et al. ORL1, a novel member of the opioid receptor family. Cloning, functional expression and localization. FEBS Lett. 1994, 341(1), 33–38. [DOI] [PubMed] [Google Scholar]
- 3.Fukuda K, Kato S, Mori K, Nishi M, Takeshima H, Iwabe N, et al. cDNA cloning and regional distribution of a novel member of the opioid receptor family. FEBS Lett. 1994, 343(1), 42–46. [DOI] [PubMed] [Google Scholar]
- 4.Meunier JC, Mollereau C, Toll L, Suaudeau C, Moisand C, Alvinerie P, et al. Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature. 1995, 377(6549), 532–535. [DOI] [PubMed] [Google Scholar]
- 5.Reinscheid RK, Nothacker HP, Bourson A, Ardati A, Henningsen RA, Bunzow JR, et al. Orphanin FQ: a neuropeptide that activates an opioidlike G proteicoupled receptor. Science. 1995, 270(5237), 792–794. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y, Fan Y, Liu J, Mestek A, Tian M, Kozak CA, et al. Molecular cloning, tissue distribution and chromosomal localization of a novel member of the opioid receptor gene family. FEBS Lett. 1994, 347(2-3), 279–83. [DOI] [PubMed] [Google Scholar]
- 7.Narita M, Mizoguchi H, Oji DE, Dun NJ, Hwang BH, Nagase H, et al. Identification of the G-proteicoupled ORL1 receptor in the mouse spinal cord by [35S]-GTPgammaS binding and immunohistochemistry. Br J Pharmacol. 1999, 128(6), 1300–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sandin J, Georgieva J, Schott PA, Ogren SO, Terenius L. Nociceptin/orphanin FQ microinjected into hippocampus impairs spatial learning in rats. Eur. J. Neurosci 1997, 9(1), 194–197. [DOI] [PubMed] [Google Scholar]
- 9.Andero R. Nociceptin and the nociceptin receptor in learning and memory. Prog Neuro-Psychopharmacol Biol Psychiatry. 2014, 62, 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jenck F, Moreau JL, Martin JR, Kilpatrick GJ, Reinscheid RK, Monsma FJ Jr, et al. Orphanin FQ acts as an anxiolytic to attenuate behavioral responses to stress. Proc Natl Acad Sci USA. 1997, 94(26), 14854–14858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gavioli EC, de Medeiros IU, Monteiro MC, Calo G, Romao PR. Nociceptin/orphanin FQ-NOP receptor system in inflammatory and immune-mediated diseases. Vitam Horm. 2014, 97, 241–266. [DOI] [PubMed] [Google Scholar]
- 12.Kiguchi N, Ding H, Kishioka S, Ko MC. Nociceptin/Orphanin FQ Peptide Receptor-Related Ligands as Novel Analgesics. Curr Top Med Chem. 2020, 20(31), 2878–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mercatelli D, Pisano CA, Novello S, Morari M. NOP receptor ligands and Parkinson's disease. Handb Exp Pharmacol. 2019, 254, 213–232. [DOI] [PubMed] [Google Scholar]
- 14.Ciccocioppo R, Borruto AM, Domi A, Teshima K, Cannella N, Weiss F. NOP-Related Mechanisms in Substance Use Disorders. Handb Exp Pharmacol. 2019, 254, 187–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mouledous L. The nociceptin/orphanin FQ system and the regulation of memory. Handb Exp Pharmacol. 2019, 254, 259–78. [DOI] [PubMed] [Google Scholar]
- 16.Kiguchi N, Ding H, Ko MC. Therapeutic potentials of NOP and MOP receptor coactivation for the treatment of pain and opioid abuse. J. Neurosci. Res 2022, 100, 191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daibani AE, Che T. Spotlight on Nociceptin/Orphanin FQ Receptor in the Treatment of Pain. Molecules. 2022, 27(3), 595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coluzzi F, Rullo L, Scerpa MS et al. Current and Future Therapeutic Options in Pain Management: Multi-mechanistic Opioids Involving Both MOR and NOP Receptor Activation. CNS Drugs 2022, 36, 617–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zaveri NT. Nociceptin Opioid Receptor (NOP) as a Therapeutic Target: Progress in Translation from Preclinical Research to Clinical Utility. J. Med. Chem 2016, 59(14), 7011–7028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Toll L, Khroyan TV, Polgar WE, et al. Comparison of the Antinociceptive and Antirewarding Profiles of Novel Bifunctional Nociceptin Receptor/mu-Opioid Receptor Ligands: Implications for Therapeutic Applications. J. Pharmacol. Exp. Ther 2009, 331(3), 954–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Olsen C, Zaveri NT, Yasuda D, et al. Structure-activity relationships of nociceptin receptor (NOP) ligands and the design of bifunctional NOP/mu opioid receptor-targeted ligands. Research and Development of Opioid-Related Ligands. 2013, 1131, 145–160. [Google Scholar]
- 22.Journigan VB, Polgar WE, Khroyan TV, Zaveri NT. Designing bifunctional NOP receptor–mu opioid receptor ligands from NOP-receptor selective scaffolds. Part II. Bioorg. Med. Chem 2014, 22(8), 2508–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Toll L, Bruchas MR, Calo’ G, Cox BM, Zaveri NT. Nociceptin/Orphinan FQ Receptor Structure, Signaling, Ligands, Functions, and Interactions with Opioid Systems. Pharmacol. Rev 2016, 68(2), 419–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferrari F, Cerlesi MC, Malfacini D, et al. In vitro functional characterization of novel nociceptin/orphinan FQ receptor agonists in recombinant and native preparations. Eur. J. Pharmacol 2016, 793, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ding H, Kiguchi N, Yasuda D, Daga PR et al. A bifunctional nociceptin and mu opioid receptor agonist is analgesic without opioid side effects in nonhuman primates. Sci. Transl. Med 2018, 10(456). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arcuri L, Novello S, Frassineti M et al. Anti-Parkinsonian and anti-dyskenetic profiles of two novel potent and selective nociceptin/orphanin FQ receptor agonists. Br. J. Pharmacol 2018, 175(5), 782–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zaver NT, Meyer ME. NOP-Targeted Nonpeptide Ligands. The Nociceptin/Orphinan FQ Peptide Receptor. Handb. Exp. Pharmacol 2019, 254. [DOI] [PubMed] [Google Scholar]
- 28.Kamakolanu UG, Meyer ME, Yasuda D, Polgar WE, Marti M, Mercatelli D, et al. Discovery and structure–activity relationships of nociceptin receptor partial agonists that afford system ablation in Parkinson’s disease models. J. Med. Chem 2020, 63, 2688–2704. [DOI] [PubMed] [Google Scholar]
- 29.Meyer ME, Doshi A, Yasuda D, Zaveri NT. Structure-based SAR in the design of selective or bifunctional nociceptin (NOP) receptor agonists. AAPS J. 2021, 23, 68. [DOI] [PubMed] [Google Scholar]
- 30.Zaveri NT, Marquez PV, Meyer ME, Polgar WE, Hamid A, Lutfy K. A novel and selective nociceptin receptor (NOP) agonist (1-(1-((cis)-4-isopropylcyclohexyl) piperidi4-yl)-1Hindol-2-yl)methanol (1) decreases acquisition of ethanol-induced conditioned place preference in mice. Alcohol Clin Exp Res. 2018, 42, 461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zaveri NT, Marquez PV, Meyer ME, Hamid A, Lutfy K. The nociceptin Receptor (NOP) agonist 1 blocks acquisition of morphine- and cocaine-induced conditioned place preference in mice. Front Psychiatry. 2018, 9, 638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hiroya K, Itoh S, Sakamoto T. Development of an efficient procedure for indole ring synthesis from 2-ethynylaniline derivatives catalyzed by Cu(II) Salts and its application to natural product synthesis. J. Org. Chem 2004, 69, 1126–1136. [DOI] [PubMed] [Google Scholar]
- 33.Toll L, Khroyan TV, Polgar WE et al. Comparison of the antinociceptive and antirewarding profiles of novel bifunctional nociceptin receptor/mu-opioid receptor ligands: implications for therapeutic applications. J. Pharmacol. Exp. Ther 2009, 331, 954–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zaveri NT, Polgar WE, Olsen CM et al. Characterization of opiates, neuroleptics, and synthetic analogs at ORL1 and opioid receptors. Eur. J. Pharmacol 2001, 428, 29–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Daga PR, Zaveri NT. Homology modeling and molecular dynamics siumulations of the active state of the nociceptin receptor reveal new insights into agonist binding and activation. Proteins: Struct., Funct., Genet 2012, 80, 1948–1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhuang Y, Wang Y, He B et al. Molecular recognition of morphine and fentanyl by the human μ-opioid receptor. Cell 2022, 185(23), 4361–4365. [DOI] [PubMed] [Google Scholar]
- 37.Meng F, Taylor LP, Hoversten MT, Ueda Y, Ardati A, et al. Moving from the orphinan FQ receptor to an opioid receptor using four point mutations. J Biol. Chem 1996, 271(50), 32016–32020. [DOI] [PubMed] [Google Scholar]
- 38.Meng F, Ueda Y, Hoversten MT, Taylor LP, Reinscheid RK, et al. Creating a functional opioid alkaloid binding site in the orphinan FQ receptor through site-directed mutagenesis. Mol. Pharmacol 1998, 53(4), 772–777. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.