

Abstract
Metastatic breast cancer is a prime health concern and leading health burden across the globe. Previous efforts have shown that protein–protein interaction between Metadherin and Staphylococcal nuclease domaincontaining 1 (SND1) promotes initiation of breast cancer, progression, therapy resistance and metastasis. Therefore, small drug molecules that can interrupt the Metadherin and SND1 interaction may be ideal to suppress tumor growth, metastasis and increases chemotherapy sensitivity of triple negative breast cancer. Here, in this study, structure based virtual screening was conducted against the reported active site of SND1 enzyme, which revealed three promising lead molecules from Asinex library. These compounds were; BAS_00381028, BAS_00327287, and BAS_01293454 with binding energy score −10.25 kcal/mol, −9.65 kcal/mol and −9.32 kcal/mol, respectively. Compared to control (5-chloro-2-methoxy-N-([1,2,4]triazolo[1,5-a]pyridin-8-yl)benzene-1-sulfonamide) the lead molecules showed robust hydrophilic and hydrophobic interactions with the enzyme and revealed stable docked conformation in molecular dynamics simulation. During the simulation time, the compounds reported stable dynamics with no obvious fluctuation in binding mode and interactions noticed. The mean root mean square deviation (RMSD) of BAS_00381028, BAS_00327287, and BAS_01293454 complexes were 1.87 Å, 1.75 Å, 1.34 Å, respectively. Furthermore, the MM/GBSA analysis was conduction on the simulation trajectories of complexes that unveiled binding energy score of −19.25 kcal/mol, −27.03 kcal/mol, −34.6 kcal/mol and −29.61 kcal/mol for control, BAS_00381028, BAS_00327287, and BAS_01293454, respectively. In MM/PBSA, the binding energy value of for control, BAS_00381028, BAS_00327287, and BAS_01293454 was −20.45 kcal/mol, −27.89 kcal/mol, −36.41 kcal/mol and –32.01 kcal/mol, respectively. Additionally, the compounds were classified as druglike and have favorable pharmacokinetic properties. The compounds were predicted as promising leads and might be used in experimental investigation to study their anti-SND1 activity.
Keywords: Metastatic breast cancer, Metadherin, Staphylococcal nuclease domaincontaining 1, BAS_00381028, BAS_00327287, BAS_01293454, Molecular dynamics simulation
1. Introduction
Breast cancer develops in the women breast lobules, connective tissues and tubes (Park et al., 2022). The breast cancer is regarded as the most common type of cancer and is grouped into four subtypes; human epidermal growth factor receptor 2 positive, luminal A, luminal B, triple negative breast cancer (Dandamudi et al., 2018, Zeichner et al., 2016). Each of these subtypes has its own metastases, prognosis and treatment methods (Park et al., 2022). In cancer metastasis, the original tumor is transformed into distal secondary tumor and thus represents a hallmark often leads to treatment failure and patient death (Leong et al., 2021). Metastasis is a complex mechanism with multiple cellular actions including primary tumor development, invasion, escaping immune surveillance, and tissue microenvironment regulation (Joyce and Pollard, 2009). The absence of an effective treatment therapy for metastatic cancer in addition to treatment resistance are proved to be two significant hurdles in overcoming metastatic breast cancer mortality (Shen et al., 2022b). The Metadherin protein is reported to show high expression in poor prognosis breast cancer and through its interaction with the staphylococcal nuclease domain-containing 1 (SND1), the protein–protein interaction (PPI) promotes therapy resistance and metastasis (Shen et al., 2022a, Xu et al., 2023). Thus, blocking the PPI interaction between the Metadherin protein and SND1 is considered as a promising therapeutic platform (Shen et al., 2022a, Xu et al., 2023).
The Metadherin protein plays a key role to start tumor initiation, metastasis and chemoresistance (Hu et al., 2009, Wan and Kang, 2013). A recent study showed that knockout of Metadherin gene profoundly affect mammary tumors formation, and similar findings were revealed in studies of whole-body genetic knockout involving Metadherin and astrocyte elevated gene 1 in context of colorectal, liver, lung and prostate cancers (Wan et al., 2014b, Wan et al., 2014a). The Metadherin has been unveiled to play tumor promoting function which is crucially dependent on its interaction with SND1 (Jariwala et al., 2015). The SND1 is characterized as RNA binding protein, and also act transcriptional co-activator (Blanco et al., 2011). As a RNA binding protein, the SND1 function in regulation of RNA stability, ubiquitination, editing and alternate splicing (Jariwala et al., 2015). Both SND1 and Metadherin are reported to share similar functional and clinical importance in promoting chemoresistance and metastasis(Shen et al., 2022b). The SND1 has been regarded as oncogene in different cancers and plays a vital role in cancer cells proliferation, migration, invasion, angiogenesis and metastasis (Chidambaranathan-Reghupaty et al., 2018). Thus, due to integral role of the SND1 in various cancer, makes it a promising therapeutic target.
Previously, two specific inhibitors (C26-A2 and C26-A6) were filtered against SND1 as potential leads. Both the compounds in animal model studies revealed ability to suppressed tumor growth and metastasis. Additionally, the compounds were noticed to increase chemotherapy sensitivity of triple negative breast cancer in preclinical animal models (Shen et al., 2022b). Compared to experimental drug discovery, computer aided drug design methods offer variety of techniques that help in reducing drug research and development time (Macalino et al., 2015). The drug discovery is a complex process, associated with high risks and is costly and long (Van Drie, 2007). Also, the drug discovery often leads to drug failure, thus negatively affecting drug discovery process (Macalino et al., 2015). The computer aided drug design is hence of immense importance in pharmaceutical industry to speed up the drug discovery process (Shaker et al., 2021). Parallelly, recent advancements in DNA microarray technologies are vital in disclosing genes that are involved in disease onset which further provides in-depth information on disease metabolic pathways, disease target and drug toxicity (Talele et al., 2010). Together, the computational methods are of high interest in discovery lead molecules that can strongly bind the target protein and can be subjected to experimental evaluation to prove their biological relevance.
In this work, the authors present the hypothesis that SND1 enzyme inhibition by small drug molecules may block its interaction with Metadherin and thus metastatic breast cancer can be managed. Asinex library was screened against the SND1 crystal enzyme structure with aim to shortlist lead molecules that showed best binding conformation and associated with lowest binding energy score (Maia et al., 2020). Further, intermolecular interactions dynamics were disclosed using molecular dynamics simulation method, which important to show time dependent docked ligands behavior and interaction pattern(Karplus and McCammon, 2002, Shivanika et al., 2020). The intermolecular interactions energies as function of time was determined using MMPB/GBSA analysis (Genheden and Ryde, 2015). The MMPB/GBSA is considered more powerful in term of predicting compounds docked affinity and the predictions are more accurate compared to molecular docking (Ahmad et al., 2018, Wang et al., 2019). To shed light on the filtered lead pharmacokinetics, adsorption, distribution, metabolism, excretion and toxicity.
(ADMET) parameters were evaluated (Van De Waterbeemd and Gifford, 2003). The findings of the current study might be of use to experimentalists to test the filtered compounds in experiments. Furthermore, the study may expedite the drug discovery process against the breast cancer and may provide foundation for discovery of more potent derivates.
2. Methodology
The full methods in step-wise fashion is shown in Fig. 1.
Fig. 1.

The flow of methods starting from the enzyme crystal structure preparation, to ligands library preparation, structure based virtual screening and molecular dynamics simulation. The compounds pharmacokinetics prediction was also part of the study design.
2.1. SND1 enzyme processing
The first step of the methods was to prepare the receptor enzyme. This was accomplished by first retrieving the SND1 crystal structure which is submitted to the protein data bank (PDB) (Berman et al., 2000) under PDB code of 7KNW (https://www.rcsb.org/structure/7KNW). The structure experimental data snapshot is as; X-ray diffraction method was employed for structure determination, resolution value of 2.65 Å, R-value work of 0.264, R-value free of 0.338 and R-value observed of 0.267. The global symmetry of the enzyme is asymmetric and global stoichiometry is monomeric. The enzyme was co-crystalized with 5-chloro-2-methoxy-N-([1,2,4]triazolo[1,5-a]pyridin-8-yl)benzene-1-sulfonamide compound, which was used as a control molecule in this work. The enzyme was processed in UCSF Chimera v1.17 (Kaliappan and Bombay, 2018). The processing was started with addition of missing hydrogen atoms, charge was assigned using Gasteiger method. The energy minimization of the enzyme was completed using two algorithms. First, steepest descent was applied for 2000 cycles with step size of 0.02 Å. This was followed by conjugate gradient for 2500 steps. The energy minimized was then saved as.pdb format for additional processing.
2.2. Ligands library making
For screening purpose, full Asinex library available at https://www.asinex.com/screening-libraries-(all-libraries) was used. The library contains ready-to-use compounds upon request. The library contains different sub-libraries such as BioDesign library, elite library, synergy library, gold and platinum collections. In total, the library harbors 575,302 compounds which have chemical moieties of known pharmacological products. The library has mostly lead-like molecules and proved as to have good ADMET properties. The library was imported into PyRx 0.8 where they were first energy minimized via MM2 force field (Dallakyan and Olson, 2015, Halgren, 1996). The energy minimized compounds were then transformed into.pdbqt form to make them ready for structure based virtual screening.
2.3. Virtual screening of drug library
Afterward, structure based virtual screening of Asinex library was conducted against SND1 crystal structure to identify drug molecules that bind best to the receptor. The screening was performed against active site coordinates by selecting ARG255:NE (Elmaci and Altinoz, 2016). The XYZ coordinates of the atom are; −54.79 Å, 53.50 Å and −51.20 Å, respectively. Each compound in the drug library was docked 100 times to the receptor enzyme. Only, binding conformation of the compound to the enzyme was selected that reported lowest binding energy score as it was predicted as the most stable in terms of binding energy. For the virtual screening, PyRx 0.8 AutoDock Vina 4.2 software was used. The 5-chloro-2-methoxy-N-([1,2,4]triazolo[1,5-a]pyridin-8-yl)benzene-1-sulfonamide was used as a control molecule. To visualize the docked complexes, UCSF Chimera v.1.17 (Kaliappan and Bombay, 2018) and Discovery studio visualizer v2021 (Biovia, 2017) were used.
2.4. Molecular dynamics simulation
As often, the docking predictions are inaccurate, further validation especially through molecular dynamics simulation is highly significant (Alamri et al., 2023, Humayun et al., 2021). In this perspective, molecular dynamics simulations of best docked complexes were performed using AMBER v22 (Case et al., 2022). The preliminary processing of the docked complexes was accomplished using antechamber program (Wang et al., 2001). For enzyme, the FF19SB force field was used while for ligands processing, GAFF2 was considered (Tian et al., 2019, Vassetti et al., 2019). The complexes were submerged into OPC water box by setting padding distance of 12 Å spacing between complex edge and water box boundary (Sengupta et al., 2021). For each complex, the number counter ions (9–11Na+ ions) added were based on the rationale to get a neutral charge system. Next, the docked complexes were energy minimized in a step wise fashion. First, the complexes were passed through 2500 steps of steepest descent algorithm with step size of 0.02 Å. Second, conjugate gradient algorithm was applied for additional 3000 cycles to completely remove steric clashes left in the first phase (Muneer et al., n.d.). The complexes were then gradually heated to 300 K for the time frame of 1000 ps (restrain of 5 kcal/mol- Å2 on carbon alpha atoms). After that, the complexes were equilibrated under NPT ensemble and subjected to production run of 200 ns at 1 atm pressure and 310 K temperature in the presence of NPT ensemble with Berendsen temperature coupling algorithm (Berendsen et al., 1984). The simulation protocol was accomplished using periodic boundary conditions. In the process, long range electrostatic interactions were treated using particle-mesh Ewald protocol (Petersen, 1995). The covalent bonds with hydrogen atoms were constraint via SHAKE algorithm (Kräutler et al., 2001). The temperature of the systems was constraint by Langevin algorithm (Izaguirre et al., 2001). The simulation trajectories were analyzed by CPPTRAJ module (Roe and Cheatham III, 2013). The XMGRACE v5.1 was employed for plotting purposes (Turner, 2005). The simulation frames were visualized using visual molecular dynamics (VMD) v1.93 (Humphrey et al., 1996).
2.5. Intermolecular hydrogen bonds analysis
The number of hydrogen bonds formed between the enzyme active site residues and the compounds during simulation time is important to count as it reflects on the strength of binding (Hubbard and Kamran Haider, 2001). The intermolecular hydrogen bonds analysis was conducted using HBonds plugin of Visual Molecular Dynamics (VMD) v 1.93 software (Gumbart and Luo, 2007, Humphrey et al., 1996). The cut-off distance between the donor and acceptor atoms was set to 3.0 Å and cut-off angle of 20 degrees.
2.6. Prediction of binding free energies
The binding free energies of docked ligands with the SND1 enzyme were predicted by MMPB/GBSA analysis (Hou et al., 2011, Zhang et al., 2017). This was completed by running MMPBSA.py script available in AMBER v22 software (Miller et al., 2012). The script considered 5000 frames from the trajectories which were picked at regular time interval. The MMPB/GBSA energy was estimated using the following formula;
Both the MM/PBSA and MM/GBSA methods are found to perform similarly. The MM/PBSA employs Poisson-Boltzmann equation to disclose contribution of electrostatic energy to the free energy while the MM/GBSA uses Generalized Born equation which is considered as a faster treatment of former equation (Hou et al., 2011).
2.7. Entropy energy calculations
The entropy energy of complexes was predicted separately by AMBER normal mode analysis (Genheden et al., 2012). For this purpose, only 10 frames were selected from simulation trajectories.
2.8. WaterSwap absolute binding energy estimation
The WaterSwap binding energy of complexes was decoded using Sire WaterSwap method (Woods et al., 2014). This method is more robust and sophisticated than the MMPB/GBSA especially in highlighting the role of water molecules that played a prime role in bridging a ligand molecule to the enzyme key active site residues (Bergström and Larsson, 2018).
2.9. Compounds pharmacokinetics prediction
Predictions about compounds pharmacokinetic which includes absorption, distribution, metabolism, excretion and toxicity (Lombardo et al., 2017). The pharmacokinetic properties of the selected lead compounds were predicted using SWISSADME online server while toxicity of the compounds was unveiled using pkCSM server (Daina et al., 2017, Pires et al., 2015).
3. Results
3.1. Virtual screening process
The structure based virtual screening process was accomplished by screening all the ligands of Asinex library against the SND1 enzyme active pocket. The process screened three compounds; BAS_00381028, BAS_00327287, and BAS_01293454 with binding energy score −10.25 kcal/mol, −9.65 kcal/mol and −9.32 kcal/mol, respectively. The control molecule binding energy score was −9.05 kcal/mol. Chemically, the BAS_00381028, BAS_00327287, and BAS_01293454 is (E)-4-(1-(2-hydroxybenzylidene)hydrazin-1-ium-2-yl)-6-methoxypyrimidine-1,3-diium, 6-(2-ethoxy-2-oxoethyl)-1,3-dihydro-[1,2,5]oxadiazolo[3,4-b]pyrazin-4,7-diium-5-olate and 3-carboxy-1-phenyl-1H-pyrazol-2-ium-5-olate, respectively. The selected compounds structure and binding energy score are given in Table 1. Inspection of the compounds binding mode and interactions were carried out. All the three lead molecules were found to docked at Pocket 2 identified by Shen et al., 2022. Regarding the binding mode, all the leads and control molecule occupied the same binding pocket 2 and gained deep binding inside the pocket. The binding conformation of docked lead structures and control are given in Fig. 2A. The BAS_00381028 (2-hydroxyphenyl)methaniminium) ring was found to engaged with Arg255 residues while 4-amino-6-methoxypyrimidine-1,3-diium chemical moiety of the compound formed a strong hydrogen bond with Ser254. Both these hydrogen bonds were in close distance of < 3 Å and vital for holding and stability of ligand binding mode. The residues Gly282, Ile284, Leu287, His279, Arg36, and Gly92 were found van der Waals interactions with the compound. The Asn281 and Leu256 formed Pi-pi stacked, amide-pi stacked and pi-alkyl interactions with the BAS_00381028. The BAS_00327287 formed three hydrogen bonds each with Arg255, Ile284 and Asn281. The ethyl acetate formed a close distance hydrogen bond with Arg255 (2.68 Å). The chemical moiety 1,3-dihydro-[1,2,5]oxadiazolo[3,4-b]pyrazin-4,7-diium-5-olate produced hydrogen bonds with Ile284 and Asn281 at distance of 2.41 Å and 3.02 Å, respectively. The compound also formed van der Waals interactions with Ile277, His279, Phe251, Asn283, Gly282, and Arg259. Most of the hydrogen bonds interactions of BAS_01293454 came from 3-carboxy-1H-pyrazol-2-ium-5-olate. This chemical moiety was found engaged with Ser254, Arg255 and Arg259. Beside this, the opposite benzene ring formed van der Waals bond with Leu287, Gly282, and His279. The control molecule was seen mostly engaged in hydrophobic interaction and only one hydrogen bonding was reported with Arg255. The chemical interactions of the compounds can be found in Fig. 2B-E.
Table 1.
Virtually screened docked compounds with lowest binding energy score in kcal/mol. The RMSD of all the given solutions is 0.
| Compound | Rank | Structure | Binding Energy |
|---|---|---|---|
| BAS_00381028 | 1 | 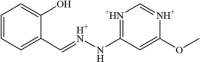 |
−10.25 kcal/mol |
| BAS_00327287 | 2 |  |
−9.65 kcal/mol |
| BAS_01293454 | 3 | 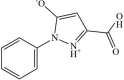 |
−9.32 kcal/mol |
| Control | 4 | 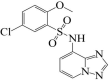 |
−9.05 |
Fig. 2.

Virtual screening of Asinex library. A. Docked binding mode of best binding drug molecules to SND1 enzyme. The SND1 enzyme is cyan ribbon while compounds/control are in different color sticks. The control, BAS_00381028, BAS_00327287, and BAS_01293454 are in red, yellow, green and magenta color, respectively. Further, docked site residues are labeled in zoom. B-E. Intermolecular binding interactions of virtually screened compounds and control with SND1 enzyme active pocket. A. BAS_00381028, B. BAS_00327287, C. BAS_01293454 and D. Control.
3.2. Molecular dynamics simulation
Molecular dynamics simulation studies were conducted on docked complexes in order to reported time dependent behavior of docked ligands (Ahmad et al., 2019, Navid et al., 2021, Wahedi et al., 2020). Among the statistical analyses conducted the most important was root mean square deviation (RMSD) which provide structural insights of docked complexes during simulation time (Alamri et al., 2022, Ehsan et al., 2018). Higher RMSD complies to higher structural changes and vice versa. The mean RMSD of BAS_00381028, BAS_00327287, BAS_01293454 and control was 1.87 Å, 1.75 Å, 1.34 Å and 0.87 Å, respectively (Fig. 3A). All the values indicate high stable structure of the SND1 enzyme in the presence of the ligands during the course of simulation time. Further, this highlights formation of very stable intermolecular docked complex with robust interactions network keeping the ligand stable at the docked site. Next, root mean square fluctuation (RMSF) was calculated for the complexes. The RMSD shed light on the residue’s stability. The mean RMSF of BAS_00381028, BAS_00327287, BAS_01293454 and control was 0.84 Å,0.97 Å, 0.82 Å and 0.97 Å, respectively (Fig. 3B). The RMSF is again support formation of stable complex and the presence of ligand molecules does not affect overall structure stability of the SND1 enzyme. The key residues of the SND1 enzyme involved in hydrogen bonds interaction with the ligands have RMSF value as; control (Aeg255 (1.32 Å) and Ser254 (1.34 Å)), BAS_00381028 (Arg255 (0.78 Å) and Asn281 (0.56 Å)), BAS_00327287 (Ser254 (1.24 Å), Arg255 (1.30 Å) and Arg259 (1.05 Å)), and BAS_01293454 (Arg255 (1.13 Å)). These values are conclusive about the SND1 enzyme residues stability in the presence of ligand molecules. To further study, the strength of binding between the lead molecules and the enzyme, hydrogen bonds analysis was conducted. The hydrogen bonds are critical in determining intermolecular strength. As can be seen in Fig. 3C, each complex reported 2–3 hydrogen bonds in each frame of simulation trajectories. This implies that the findings predicted by the docking and above RMSD, and RMSF are quite similar in demonstrating the strong compounds binding with the enzyme.
Radius of Gyration Analysis
Fig. 3.

Different statistical analysis done on simulation trajectories of docked complexes. A. RMSD, B. RMSF and C. Hydrogen bonds analysis.
Further, to complement the RMSD analysis about structure stability of the SND1 enzyme in the presence of control and lead molecules, radius of gyration (Rg) analysis was performed on the simulation trajectories (Ahmad et al., 2017, Lobanov et al., 2008). This analysis is vital to understand the receptor enzyme compact nature during the simulation. The compact nature of the receptor enzyme illustrates that no major structural and domain changes occurred once the ligands molecules bind to the enzyme. As the RMSD indicates that no vital structure deviations were noticed, similar results were reported (Fig. 4A). The mean Rg of BAS_00381028, BAS_00327287, BAS_01293454 and control was 50.62 Å,50.78 Å, 45.36 Å and 41.69 Å, respectively. The control system was found the most compact while among the lead complexes, BAS_01293454 showed stable equilibrium. Further, solvent accessible surface area (area) analysis for the complexes was performed to investigate the surface area of SND1 enzyme which interact with the solvent molecules. The average value for the systems is; BAS_00381028 (18204 nm2), BAS_00327287 (17845 nm2), BAS_01293454 (17350 nm2) and control (16824 nm2). The plots indicate that no major deviations were noticed after ligand bindings (Fig. 4B).
Fig. 4.

Rg analysis (A) and SASA analysis (B) of the complexes. The unit of Rg is given in Å while that of SASA is in Area (nm2).
3.3. MMPB/GBSA analysis
The MMPB/GBSA analysis was accomplished for selected docked and control complexes. These methods are considered more effective in determining docked ligand binding affinity with the receptor enzyme. The net binding energy of all docked and control complexes were highly negative, demonstrating formation of very strong intermolecular systems and stable complexes. Among the energies produced by the complexes, van der Waals was found as the most dominating force responsible for making systems stable and keeping ligands docked at the docked site. The net van der Waals energy of control, BAS_00381028, BAS_00327287 and BAS_01293454 was −25.20 kcal/mol, −29.34 kcal/mol, −36.54 kcal/mol and –33.67 kcal/mol, respectively. The electrostatic energy of all docked complexes was also unveiled as highly stable. The solvation energy was non-favorable among the estimated energies and contributed non-favorable to overall net energy. The net solvation energy of control, BAS_00381028, BAS_00327287 and BAS_01293454 was 16.97 kcal/mol, 14.36 kcal/mol, 12.17 kcal/mol and 15.08 kcal/mol, respectively in MM-GBSA. Similarly, in MM/PBSA, the net solvation energy was 15.77 kcal/mol, 13.50 kcal/mol, 10.36 kcal/mol, and 12.68 kcal/mol for control, BAS_00381028, BAS_00327287 and BAS_01293454, respectively. More details of the energy terms and values can be found in Table 2.
Table 2.
MMPB/GBSA analysis of docked complexes. The energy unit is in kcal/mol.
| Parameter | Control | BAS_00381028 | BAS_00327287 | BAS_01293454 |
|---|---|---|---|---|
| MM/GBSA | ||||
| Energy van der Waals | −25.20 | −29.34 | −36.54 | −33.67 |
| Energy Electrostatic | −11.02 | −12.05 | −10.23 | −11.02 |
| Total Gas Phase Energy | − 36.22 | − 41.39 | − 46.77 | − 44.69 |
| Total Solvation Energy | 16.97 | 14.36 | 12.17 | 15.08 |
| Net Energy | − 19.25 | − 27.03 | − 34.6 | − 29.61 |
| MM/PBSA | ||||
| Energy van der Waals | −25.20 | −29.34 | −36.54 | −33.67 |
| Energy Electrostatic | −11.02 | −12.05 | −10.23 | −11.02 |
| Total Gas Phase Energy | − 36.22 | − 41.39 | − 46.77 | − 44.69 |
| Total Solvation Energy | 15.77 | 13.50 | 10.36 | 12.68 |
| Net Energy | −20.45 | −27.89 | −36.41 | −32.01 |
3.4. WaterSwap binding energy
The WaterSwap binding free energy of each docked complex and control docked system was determined to re-validate the MMPB/GBSA predictions. The WaterSwap is a more sophisticated method as it considers the contribution of water molecules that bridge the ligands to receptor active pocket residues (Bergström and Larsson, 2018, Woods et al., 2014). The WaterSwap swaps the docked ligand with equal volume of water cluster present at the docked site. The WaterSwap binding energy predicted by different algorithms is given in Fig. 5. Three algorithms were used such as Bennetts, Thermodynamic integration (TI) and free energy perturbation (FEP). The BAS_00327287 and BAS_01293454 were found as the promising lead molecules with very stable binding energy. The Bennetts, TI and FEP binding free energy of BAS_00327287 was –32.05, –32.97 and −31.66, respectively. Similarly, the BAS_01293454 secures binding energy value of –33.41, –33.04 and –33.63 kcal/mol in Bennetts, TI and FEP algorithms, respectively.
Fig. 5.

Bar graph representation of WaterSwap binding energies secured by lead molecules and control molecule. The value of binding energy was estimated in kcal/mol.
3.5. Entropy energy calculation
The AMBER normal mode analysis was performed in order to disclose entropy energy contribution to overall binding energy. The entropy energy contribution of each complex is given in Table 3. Among the docked complexes, BAS_00327287 and BAS_01293454 were found to have favorable entropy energy contribution with value of −5.61 kcal/mol and −5.08 kcal/mol, respectively. These values contribute to stable formation of complexes.
Table 3.
Entropy energy estimation for docked and control systems. All values are in kcal/mol.
| Complex | Translational | Rotational | Vibrational | ΔS Total |
|---|---|---|---|---|
| Control | 9.64 | 8.34 | 2268.05 | 3.68 |
| BAS_00381028 | 8.52 | 9.67 | 2635.36 | −1.09 |
| BAS_00327287 | 10.20 | 9.34 | 2167.08 | −5.61 |
| BAS_01293454 | 10.58 | 10.25 | 2022.09 | −5.08 |
3.6. Drug-likeness and medicinal chemistry properties
Drug-likeness of selected lead molecules was predicted to get information about whether the compounds have the features to be marketed and branded (Van De Waterbeemd and Gifford, 2003). Druglike molecules have high probability of success and provide good oral bioavailability and absorption thus ensuring high concentration of drug can reach the target site for action (Banerjee et al., 2018). All the three lead molecules and the control were pointed as drug-like by prominent drug rules. Especially, the lead molecules and control were classified to follow Lipinski rule of five, which is a famous rule for describing drug-like molecules (Lipinski, 2004). The BAS_00381028 and BAS_00327287 were reported as non-druglike by Ghose rule. Both these compounds violated the LogP value range set by the Ghose rule (Ahmad et al., 2018). The bioavailability score enables to predict compounds bioavailability of least 10 % in rat. The lead compounds and control molecule were predicted to have better bioavailability. Beside BAS_00381028, other lead molecules were found to have zero alert for pan-assay interference compounds (PAINS) thus highlighting the fact that the compounds only bind to one specific biomolecule and not multiple (Whitty, 2011). Similarly, the compounds were revealed to have good synthetic accessibility score illustrating that these compounds can be easily subjected to chemical synthesis to be used in medicinal chemistry projects. Details of compounds drug-likeness, lead-likeness and medicinal chemistry properties are tabulated in Table 4.
Table 4.
Predicted drug-likeness and medicinal chemistry properties of control and selected lead molecules.
| Compound | Ghose | Veber | Lipinski | Muegge | Egan | Bioavailability score | PAINS | Brenk | Lealikeness | Synthetic accessibility |
|---|---|---|---|---|---|---|---|---|---|---|
| Control | Yes | Yes | Yes | Yes | Yes | 0.55 | 0 alert | 0 alert | Yes | 2.88 |
| BAS_00381028 | No, 1 voilation WLogP < -0.4 | Yes | Yes | Yes | Yes | 0.55 | 1 alert: hzone_phenol_A | 1 alert: imine_1 | No; 1 violation: MW < 250 | 2.84 |
| BAS_00327287 | No; 1 violation: WLOGP < -0.4 | Yes | Yes | Yes | Yes | 0.55 | 0 alert | 0 alert | No; 1 violation: MW < 250 | 3.20 |
| BAS_01293454 | Yes | Yes | Yes | Yes | Yes | 0.56 | 0 alert | 0 alert | No; 1 violation: MW < 250 | 2.08 |
3.7. Physicochemical, lipophilicity and pharmacokinetics properties prediction
Further, lead molecules and control physicochemical, lipophilicity and pharmacokinetic properties were predicted. The lead compounds have molecular weight within accepted range and thus clear as successor of Lipinski rule of five. The topological polar surface area (TPSA) of compounds were reported as good, thus ensuring these compounds might be easily cross the cell membrane (Veber et al., 2002). The compounds were also noticed as good water soluble, ensuring that they might be easily taken orally and high concentration of drugs reach the target site (Veber et al., 2002). Additionally, the compounds revealed good absorption from the gastrointestinal region. In short, the compounds revealed feasible physicochemical, lipophilicity and pharmacokinetic properties which are vital for a successful drug (Table 5).
Table 5.
Physicochemical, lipophilicity and pharmacokinetics properties prediction of lead molecules.
| Compound | Molecular weight | Topological polar surface area | Hydrogen bond acceptors | Hydrogen bond donors | LogP | Water Solubility | Gastrointestinal absorption | Blood brain barrier permeant | CYP1A2 inhibitor | LogKp |
|---|---|---|---|---|---|---|---|---|---|---|
| Control | 338.77 g/mol | 93.97 Å2 | 5 | 1 | 1.82 | Soluble | High | No | No | −7.04 cm/s |
| BAS_00381028 | 247.27 g/mol | 83.74 Å2 | 2 | 5 | 0.99 | Soluble | High | No | No | −5.75 cm/s |
| BAS_00327287 | 227.20 g/mol | 110.93 Å2 | 4 | 4 | 0.03 | Soluble | High | No | No | −6.80 cm/s |
| BAS_01293454 | 204.18 g/mol | 79.43 Å2 | 3 | 2 | 0.03 | Soluble | High | No | No | −6.80 cm/s |
4. Discussion
The strong interaction between Metadherin and SND1 allows breast cancer initiation, progression, metastasis and resistance to treatment (Blanco et al., 2011). Thus, blocking the function of these proteins and resulting interactions could provide an ideal platform for discovery of potent therapy. In particular, small molecules that can interfere with the protein’s interactions would be significant. In this work, BAS_00381028, BAS_00327287, and BAS_01293454 with binding energy score −10.25 kcal/mol, −9.65 kcal/mol and −9.32 kcal/mol, respectively were found as the binders of the SND1 enzyme. The control molecule binding energy score was −9.05 kcal/mol. The compounds binding mode and interactions were found stable throughout the simulation and produced diverse chemical interactions. All the three lead molecules were found to docked at Pocket 2 as reported by Shen et al., 2022 (Shen et al., 2022a). Further, the lead compounds and control occupied the same binding pocket 2 and gained deep binding inside the pocket as the simulation time. Regarding, structural stability of complex, the mean RMSD of BAS_00381028, BAS_00327287, BAS_01293454 and control was 1.87 Å, 1.75 Å, 1.34 Å and 0.87 Å, respectively. These values affirm that the SND1 enzyme in the presence of the selected leads showed considerable structure stability and no obvious conformational changes were noticed. This further explain the good stability of intermolecular docked complexes.
Computational modeling and chemoinformatic studies are proved vital in identifying potential lead molecules against therapeutic targets. In one study, high throughput screening of synthesized compounds was conducted against α-glucosidase enzyme. The study findings proposed 63 compounds as active inhibitors of the enzyme with IC50 value ranges from 3.2 μM to 50.0 μM. Further shortlisting was done that surfaced compound 25 (which was an oxadiazole derivates) as a potent inhibitor with IC50 value of 3.23 ± 0.8 μM. The kinetic studies of the compound reported that both Vmax and Km values change with the inhibitor concentration suggesting un-competitive inhibition model. The molecular docking unveiled that the compound formed conventional hydrogen bonds with acidic and basic amino acid residues (Ali et al., 2023). In another work, a series of 2,3-disubstitutedbenzofuran derivatives were synthesized and virtual screened against amyloid beta peptide. Most of the compounds in particular 8a and 8 g showed better binding affinity for Aβ fibrils. Further, it was noticed that the compounds showed 50 % to 67 % reduction in Aβ aggregation growth (Radwan et al., 2023). The computational techniques are quite useful in modern drug discovery as they not only speed up the process but proved vital in identify good lead molecules. For example, the computational drug discovery methods are used successfully in marketing oxymorphone for opioid analgesic, captopril for high blood pressure, dorzolamide for glaucoma and ocular hypertension, zanamivir for influenza A and influenza B, and saquinavir for HIV AIDS (Talele et al., 2010, Van Drie, 2007). The study findings also noticed that the compounds reported good drug-likeness, solubility, gastrointestinal absorption and no alert for PAINS. All these parameters make the compounds to be useful in experimental testing’s.
5. Conclusions
In this work, three drug molecules (BAS_00381028, BAS_00327287, and BAS_01293454) from Asinex library were found to show robust binding interactions and stable binding conformation with the SND1 enzyme. The compounds reported several closed distance hydrophilic and hydrophobic bonds and remained in stable conformation as the simulation time proceeds. Further, binding free energies were noticed at very low demonstrating formation of strong intermolecular bindings. The compounds were further demonstrated to behave favorable pharmacokinetics and are drug-like with no chemical toxicity. These findings suggested to be subjected to experimental studies to confirm their anti-SND2 activity and their ability to stop breast cancer metastasis.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Peer review under responsibility of King Saud University.
References
- Ahmad S., Raza S., Uddin R., Azam S.S. Binding mode analysis, dynamic simulation and binding free energy calculations of the MurF ligase from Acinetobacter baumannii. J. Mol. Graph. Model. 2017;77:72–85. doi: 10.1016/j.jmgm.2017.07.024. [DOI] [PubMed] [Google Scholar]
- Ahmad S., Raza S., Uddin R., Azam S.S. Comparative subtractive proteomics based ranking for antibiotic targets against the dirtiest superbug: Acinetobacter baumannii. J. Mol. Graph. Model. 2018;82:74–92. doi: 10.1016/j.jmgm.2018.04.005. [DOI] [PubMed] [Google Scholar]
- Ahmad S., Raza S., Abro A., Liedl K.R., Azam S.S. Toward novel inhibitors against KdsB: a highly specific and selective broad-spectrum bacterial enzyme. J. Biomol. Struct. Dyn. 2019;37:1326–1345. doi: 10.1080/07391102.2018.1459318. [DOI] [PubMed] [Google Scholar]
- Alamri, M.A., Mirza, M.U., Adeel, M.M., Ashfaq, U.A., Tahir ul Qamar, M., Shahid, F., Ahmad, S., Alatawi, E.A., Albalawi, G.M., Allemailem, K.S., 2022. Structural Elucidation of Rift Valley Fever Virus L Protein towards the Discovery of Its Potential Inhibitors. Pharmaceuticals 15, 659. [DOI] [PMC free article] [PubMed]
- Alamri, M.A., Tariq, M.H., Tahir ul Qamar, M., Alabbas, A.B., Alqahtani, S.M., Ahmad, S., 2023. Discovery of potential phytochemicals as inhibitors of TcdB, a major virulence factors of Clostridioides difficile. J. Biomol. Struct. Dyn. 1–9. [DOI] [PubMed]
- Ali M., Malik K., Zaidi A., Farooq U., Bukhari S.M., Majeed Z., Mahnashi M.H., Nawazish S., Abdulwahab A., Alshaibari K.S. In-vitro high-throughput library screening—Kinetics and molecular docking studies of potent inhibitors of $α$-glucosidase. PLoS One. 2023;18:e0286159. doi: 10.1371/journal.pone.0286159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee P., Eckert A.O., Schrey A.K., Preissner R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018;46 doi: 10.1093/nar/gky318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berendsen H.J.C., van Postma J.P.M., van Gunsteren W.F., DiNola A., Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- Bergström C.A.S., Larsson P. Computational prediction of drug solubility in water-based systems: qualitative and quantitative approaches used in the current drug discovery and development setting. Int. J. Pharm. 2018;540:185–193. doi: 10.1016/j.ijpharm.2018.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E. The protein data bank. Nucleic Acids Res. 2000;28:235–242. doi: 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biovia D.S. San Diego; CA, USA: 2017. Discovery studio visualizer. [Google Scholar]
- Blanco M.A., Alečković M., Hua Y., Li T., Wei Y., Xu Z., Cristea I.M., Kang Y. Identification of staphylococcal nuclease domain-containing 1 (SND1) as a Metadherin-interacting protein with metastasis-promoting functions. J. Biol. Chem. 2011;286:19982–19992. doi: 10.1074/jbc.M111.240077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case, D.A., Duke, R.E., Walker, R.C., Skrynnikov, N.R., Cheatham III, T.E., Mikhailovskii, O., Simmerling, C., Xue, Y., Roitberg, A., Izmailov, S.A., others, 2022. AMBER 22 Reference Manual.
- Chidambaranathan-Reghupaty S., Mendoza R., Fisher P.B., Sarkar D. The multifaceted oncogene SND1 in cancer: focus on hepatocellular carcinoma. Hepatoma Res. 2018;4 doi: 10.20517/2394-5079.2018.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daina A., Michielin O., Zoete V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017;7:1–13. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallakyan S., Olson A.J. Small-molecule library screening by docking with PyRx. Chem. Biol. Springer. 2015:243–250. doi: 10.1007/978-1-4939-2269-7_19. [DOI] [PubMed] [Google Scholar]
- Dandamudi A., Tommie J., Nommsen-Rivers L., Couch S. Dietary patterns and breast cancer risk: a systematic review. Anticancer Res. 2018;38:3209–3222. doi: 10.21873/anticanres.12586. [DOI] [PubMed] [Google Scholar]
- Ehsan N., Ahmad S., Navid A., Azam S.S. Identification of potential antibiotic targets in the proteome of multi-drug resistant Proteus mirabilis. Meta Gene. 2018;18:167–173. [Google Scholar]
- Elmaci I., Altinoz M.A. A metabolic inhibitory cocktail for grave cancers: metformin, pioglitazone and lithium combination in treatment of pancreatic cancer and glioblastoma multiforme. Biochem. Genet. 2016;54:573–618. doi: 10.1007/s10528-016-9754-9. [DOI] [PubMed] [Google Scholar]
- Genheden S., Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 2015;10:449–461. doi: 10.1517/17460441.2015.1032936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genheden S., Kuhn O., Mikulskis P., Hoffmann D., Ryde U. The normal-mode entropy in the MM/GBSA method: effect of system truncation, buffer region, and dielectric constant. J. Chem. Inf. Model. 2012;52:2079–2088. doi: 10.1021/ci3001919. [DOI] [PubMed] [Google Scholar]
- Gumbart, J.C., Luo, D., 2007. HBonds plugin, version 1.2. There is no Corresp. Rec. this Ref. Sch.
- Halgren T.A. Merck molecular force field. J. Comput. Chem. 1996;17:490–519. doi: 10.1002/(SICI)1096-987X(199604)17:5/6<520::AID-JCC2>3.0.CO;2-W. [DOI] [Google Scholar]
- Hou, T., Wang, J., Li, Y., Wang, W., 2011. Assessing the Performance of the MM_PBSA and MM_GBSA Methods. 1. The Accuracy.pdf 69–82. [DOI] [PMC free article] [PubMed]
- Hu G., Wei Y., Kang Y. The multifaceted role of MTDH/AEG-1 in cancer progression. Clin. Cancer Res. 2009;15:5615–5620. doi: 10.1158/1078-0432.CCR-09-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard, R.E., Kamran Haider, M., 2001. Hydrogen bonds in proteins: role and strength. e LS.
- Humayun, F., Khan, A., Ahmad, S., Yuchen, W., Wei, G., Nizam-Uddin, N., Hussain, Z., Khan, W., Zaman, N., Rizwan, M., others, 2021. Abrogation of SARS-CoV-2 interaction with host (NRP1) Neuropilin-1 receptor through high-affinity marine natural compounds to curtail the infectivity: A structural-dynamics data. Comput. Biol. Med. 104714. [DOI] [PMC free article] [PubMed]
- Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Izaguirre J.A., Catarello D.P., Wozniak J.M., Skeel R.D. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001;114:2090–2098. [Google Scholar]
- Jariwala N., Rajasekaran D., Srivastava J., Gredler R., Akiel M.A., Robertson C.L., Emdad L., Fisher P.B., Sarkar D. Role of the staphylococcal nuclease and tudor domain containing 1 in oncogenesis. Int. J. Oncol. 2015;46:465–473. doi: 10.3892/ijo.2014.2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce J.A., Pollard J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer. 2009;9:239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaliappan, S., Bombay, I.I.T., 2018. UCSF Chimera-Overview.
- Karplus M., McCammon J.A. Molecular dynamics simulations of biomolecules. Nat. Struct. Mol. Biol. 2002;9:646. doi: 10.1038/nsb0902-646. [DOI] [PubMed] [Google Scholar]
- Kräutler V., Van Gunsteren W.F., Hünenberger P.H. A fast SHAKE algorithm to solve distance constraint equations for small molecules in molecular dynamics simulations. J. Comput. Chem. 2001;22:501–508. [Google Scholar]
- Leong, S.P., Naxerova, K., Keller, L., Pantel, K., Witte, M., 2021. Molecular mechanisms of cancer metastasis via the lymphatic versus the blood vessels. Clin. \& Exp. Metastasis 1–21. [DOI] [PMC free article] [PubMed]
- Lipinski C.A. Lead- and drug-like compounds: The rule-of-five revolution. Drug Discov. Today Technol. 2004;1:337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Lobanov M.Y., Bogatyreva N.S., Galzitskaya O.V. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008;42:623–628. [PubMed] [Google Scholar]
- Lombardo F., Desai P.V., Arimoto R., Desino K.E., Fischer H., Keefer C.E., Petersson C., Winiwarter S., Broccatelli F. In silico absorption, distribution, metabolism, excretion, and pharmacokinetics (ADME-PK): Utility and best practices. An industry perspective from the international consortium for innovation through quality in pharmaceutical development: Miniperspective. J. Med. Chem. 2017;60:9097–9113. doi: 10.1021/acs.jmedchem.7b00487. [DOI] [PubMed] [Google Scholar]
- Macalino S.J.Y., Gosu V., Hong S., Choi S. Role of computer-aided drug design in modern drug discovery. Arch. Pharm. Res. 2015;38:1686–1701. doi: 10.1007/s12272-015-0640-5. [DOI] [PubMed] [Google Scholar]
- Maia, E.H.B., Assis, L.C., de Oliveira, T.A., da Silva, A.M., Taranto, A.G., 2020. Structure-based virtual screening: From classical to artificial intelligence. Front. Chem. 8. [DOI] [PMC free article] [PubMed]
- Miller B.R., McGee T.D., Swails J.M., Homeyer N., Gohlke H., Roitberg A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012;8:3314–3321. doi: 10.1021/ct300418h. [DOI] [PubMed] [Google Scholar]
- Muneer, I., Ahmad, Sajjad, Naz, A., Abbasi, S.W., Alblihy, A., Aloliqi, A.A., Alkhayl, F.F., Alrumaihi, F., Ahmad, Sarfraz, El Bakri, Y., n.d. Discovery of Novel Inhibitors from Medicinal Plants for V-Domain Ig Suppressor of T-Cell Activation (VISTA). Front. Mol. Biosci. 951. [DOI] [PMC free article] [PubMed]
- Navid A., Ahmad S., Sajjad R., Raza S., Azam S.S. Structure based in silico screening revealed a potent Acinetobacter baumannii Ftsz inhibitor from Asinex antibacterial library. IEEE/ACM Trans. Comput. Biol. Bioinforma. 2021;19:3008–3018. doi: 10.1109/TCBB.2021.3103899. [DOI] [PubMed] [Google Scholar]
- Park M., Kim D., Ko S., Kim A., Mo K., Yoon H. Breast cancer metastasis: Mechanisms and therapeutic implications. Int. J. Mol. Sci. 2022;23:6806. doi: 10.3390/ijms23126806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen H.G. Accuracy and efficiency of the particle mesh Ewald method. J. Chem. Phys. 1995;103:3668–3679. [Google Scholar]
- Pires D.E.V., Blundell T.L., Ascher D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015;58:4066–4072. doi: 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radwan A.A., Alanazi F.K., Raish M. Design and synthesis of multi-functional small-molecule based inhibitors of amyloid-$β$ aggregation: Molecular modeling and in vitro evaluation. PLoS One. 2023;18:e0286195. doi: 10.1371/journal.pone.0286195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roe D.R., Cheatham T.E., III PTRAJ and CPPTRAJ: software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 2013;9:3084–3095. doi: 10.1021/ct400341p. [DOI] [PubMed] [Google Scholar]
- Sengupta A., Li Z., Song L.F., Li P., Merz K.M., Jr Parameterization of monovalent ions for the OPC3, OPC, TIP3P-FB, and TIP4P-FB water models. J. Chem. Inf. Model. 2021;61:869–880. doi: 10.1021/acs.jcim.0c01390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaker, B., Ahmad, S., Lee, J., Jung, C., Na, D., 2021. In silico methods and tools for drug discovery. Comput. Biol. Med. 104851. [DOI] [PubMed]
- Shen M., Smith H.A., Wei Y., Jiang Y.-Z., Zhao S., Wang N., Rowicki M., Tang Y., Hang X., Wu S., et al. Pharmacological disruption of the MTDH–SND1 complex enhances tumor antigen presentation and synergizes with anti-PD-1 therapy in metastatic breast cancer. Nat. cancer. 2022;3:60–74. doi: 10.1038/s43018-021-00280-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen M., Wei Y., Kim H., Wan L., Jiang Y.-Z., Hang X., Raba M., Remiszewski S., Rowicki M., Wu C.-G., et al. Small-molecule inhibitors that disrupt the MTDH–SND1 complex suppress breast cancer progression and metastasis. Nat. Cancer. 2022;3:43–59. doi: 10.1038/s43018-021-00279-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shivanika, C., Kumar, D., Ragunathan, V., Tiwari, P., Sumitha, A., others, 2020. Molecular docking, validation, dynamics simulations, and pharmacokinetic prediction of natural compounds against the SARS-CoV-2 main-protease. J. Biomol. Struct. Dyn. 1. [DOI] [PMC free article] [PubMed]
- Talele T.T., Khedkar S.A., Rigby A.C. Successful applications of computer aided drug discovery: moving drugs from concept to the clinic. Curr. Top. Med. Chem. 2010;10:127–141. doi: 10.2174/156802610790232251. [DOI] [PubMed] [Google Scholar]
- Tian C., Kasavajhala K., Belfon K.A.A., Raguette L., Huang H., Migues A.N., Bickel J., Wang Y., Pincay J., Wu Q., et al. ff19SB: Amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J. Chem. Theory Comput. 2019;16:528–552. doi: 10.1021/acs.jctc.9b00591. [DOI] [PubMed] [Google Scholar]
- Turner, P.J., 2005. XMGRACE, Version 5.1. 19. Cent. Coast. Land-Margin Res. Oregon Grad. Inst. Sci. Technol. Beaverton, OR.
- Van De Waterbeemd H., Gifford E. ADMET in silico modelling: towards prediction paradise? Nat. Rev. Drug Discov. 2003;2:192–204. doi: 10.1038/nrd1032. [DOI] [PubMed] [Google Scholar]
- Van Drie J.H. Computer-aided drug design: the next 20 years. J. Comput. Aided Mol. Des. 2007;21:591–601. doi: 10.1007/s10822-007-9142-y. [DOI] [PubMed] [Google Scholar]
- Vassetti D., Pagliai M., Procacci P. Assessment of GAFF2 and OPLS-AA general force fields in combination with the water models TIP3P, SPCE, and OPC3 for the solvation free energy of druglike organic molecules. J. Chem. Theory Comput. 2019;15:1983–1995. doi: 10.1021/acs.jctc.8b01039. [DOI] [PubMed] [Google Scholar]
- Veber D.F., Johnson S.R., Cheng H.Y., Smith B.R., Ward K.W., Kopple K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- Wahedi H.M., Ahmad S., Abbasi S.W. Stilbene-based natural compounds as promising drug candidates against COVID-19. J. Biomol. Struct. Dyn. 2020:1–10. doi: 10.1080/07391102.2020.1762743. [DOI] [PubMed] [Google Scholar]
- Wan L., Kang Y. Pleiotropic roles of AEG-1/MTDH/LYRIC in breast cancer. Adv. Cancer Res. 2013;120:113–134. doi: 10.1016/B978-0-12-401676-7.00004-8. [DOI] [PubMed] [Google Scholar]
- Wan L., Hu G., Wei Y., Yuan M., Bronson R.T., Yang Q., Siddiqui J., Pienta K.J., Kang Y. Genetic ablation of metadherin inhibits autochthonous prostate cancer progression and metastasis. Cancer Res. 2014;74:5336–5347. doi: 10.1158/0008-5472.CAN-14-1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan L., Lu X., Yuan S., Wei Y., Guo F., Shen M., Yuan M., Chakrabarti R., Hua Y., Smith H.A., et al. MTDH-SND1 interaction is crucial for expansion and activity of tumor-initiating cells in diverse oncogene-and carcinogen-induced mammary tumors. Cancer Cell. 2014;26:92–105. doi: 10.1016/j.ccr.2014.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E., Sun H., Wang J., Wang Z., Liu H., Zhang J.Z.H., Hou T. End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem. Rev. 2019;119:9478–9508. doi: 10.1021/acs.chemrev.9b00055. [DOI] [PubMed] [Google Scholar]
- Wang J., Wang W., Kollman P.A., Case D.A. Antechamber: an accessory software package for molecular mechanical calculations. J. Am. Chem. Soc. 2001;222:U403. [Google Scholar]
- Whitty A. Growing PAINS in academic drug discovery. Future Med. Chem. 2011;3:797–801. doi: 10.4155/fmc.11.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods C.J., Malaisree M., Michel J., Long B., McIntosh-Smith S., Mulholland A.J. Rapid decomposition and visualisation of protein-ligand binding free energies by residue and by water. Faraday Discuss. 2014;169:477–499. doi: 10.1039/c3fd00125c. [DOI] [PubMed] [Google Scholar]
- Xu, Y., Guo, X., Yan, D., Dang, X., Guo, L., Jia, T., Wang, Q., 2023. Molecular Dynamics Simulation-Driven Focused Virtual Screening and Experimental Validation of Inhibitors for MTDH-SND1 Protein--Protein Interaction. J. Chem. Inf. Model. [DOI] [PubMed]
- Zeichner, S.B., Terawaki, H., Gogineni, K., 2016. A review of systemic treatment in metastatic triple-negative breast cancer. Breast cancer basic Clin. Res. 10, BCBCR--S32783. [DOI] [PMC free article] [PubMed]
- Zhang X., Perez-Sanchez H., Lightstone C.F. A comprehensive docking and MM/GBSA rescoring study of ligand recognition upon binding antithrombin. Curr. Top. Med. Chem. 2017;17:1631–1639. doi: 10.2174/1568026616666161117112604. [DOI] [PMC free article] [PubMed] [Google Scholar]