

Abstract
Trichoderma genus includes soil-inhabiting fungi that provide important ecosystem services in their interaction with plants and other fungi, as well as biocontrol of fungal plant diseases. A collection of Trichoderma isolates from Sardinia has been previously characterized, but here we selected 113 isolates, representatives of the collection, and characterized their viral components. We carried out high-throughput sequencing of ribosome-depleted total RNA following a bioinformatics pipeline that detects virus-derived RNA-directed RNA polymerases (RdRps) and other conserved viral protein sequences. This pipeline detected seventeen viral RdRps with two of them corresponding to viruses already detected in other regions of the world and the remaining fifteen representing isolates of new putative virus species. Surprisingly, eight of them are from new negative-sense RNA viruses, a first in the genus Trichoderma. Among them is a cogu-like virus, closely related to plant-infecting viruses. Regarding the positive-sense viruses, we report the presence of an ‘ormycovirus’ belonging to a recently characterized group of bisegmented single-stranded RNA viruses with uncertain phylogenetic assignment. Finally, for the first time, we report a bisegmented member of Mononegavirales which infects fungi. The proteins encoded by the second genomic RNA of this virus were used to re-evaluate several viruses in the Penicillimonavirus and Plasmopamonavirus genera, here shown to be bisegmented and encoding a conserved polypeptide that has structural conservation with the nucleocapsid domain of rhabdoviruses.
Keywords: mycovirus, Trichoderma, Mymonaviridae, Negarnaviricota, Bunyavirales, Ormycovirus, evolution, multisementation
Introduction
Trichoderma (kingdom Fungi, division Ascomycota, class Sordariomycetes, order Hypocreales) is a genus of ascomycetous fungi commonly isolated from the soil and rhizosphere. Members of this genus live as saprophytes and play a significant role in the degradation of plant polysaccharides. Currently, more than 450 species of Trichoderma are reported in the literature, among which 382 possess valid nomenclature (Cai et al. 2022). Moreover, most of these validated species (365 out of 382) have been characterized at a molecular level by DNA barcoding for one or more DNA loci (Cai et al. 2022). Beside their widely recognized ecological role, these fungi are also the most common biofungicides and plant growth modifiers in agriculture, with more than 60 per cent of world-registered biofungicides based on fungi belonging to this genus (Abbey et al. 2019). They are sources of enzymes of industrial interest, including those used in the biofuel industry, and they are prolific producers of secondary metabolites, with some having clinical significance (Kredics et al. 2003; Dos Santos and Dos Santos 2023). Other species have been engineered for the heterologous production of important proteins (Zin and Badaluddin 2020). Trichoderma spp. strains are also used for bioremediation, due to their ability to degrade and/or mobilize both organic and inorganic waste compounds, including heavy metals (Tripathi et al. 2013) contributing to valorize agricultural wastes and by-products (Alias et al. 2022). Fungal viruses include viruses infecting and replicating in fungi (kingdom Eumycota) or in oomycetes (kingdom Chromista). They were identified for the first time more than 50 years ago, but description of their diversity increased exponentially in the last 20 years (Ghabrial et al. 2015; Kondo, Botella, and Suzuki 2022). Currently, according to the official ‘Master Species list’ (version 2022_MSL38, published in April 2023) provided by the International Committee on Taxonomy of Viruses (ICTV), viruses infecting fungi are taxonomically classified within twenty-five different officially recognized families. Most of them possess a positive-sense RNA (+RNA) genome and are accommodated within twelve families: Alphaflexiviridae, Deltaflexiviridae, Gammaflexiviridae, Endornaviridae (phylum Kitrinoviricota), Barnaviridae, Fusariviridae, Hadakaviridae, Hypoviridae, Yadokariviridae (phylum Pisuviricota), Botourmiaviridae, Narnaviridae, and Mitoviridae (phylum Lenarviricota). Another considerable amount of fungal viruses possess a double-stranded RNA (dsRNA) genome and belong to eight families: Chrysoviridae, Megabirnaviridae, Quadriviridae, Spinareoviridae, Totiviridae (phylum Duplornaviricota), Curvulaviridae, Partitiviridae (phylum Pisuviricota), and Polymycoviridae (realm Riboviria, phylum unassigned); while just four families accommodate fungal viruses possessing a negative-sense RNA (−RNA) genome, which are as follows: Discoviridae, Mymonaviridae, Phenuiviridae, and Tulasviridae (phylum Negarnaviricota). Finally, just one family named Genomoviridae (phylum Cressdnaviricota) includes viruses having a circular single-stranded DNA genome. Despite the extensive research conducted in the last two decades that has led to the discovery of diverse fungal viruses, most available information concerns phytopathogenic fungi, while relatively little is known on viruses infecting free-living soil fungi, endophytic fungi, and/or epiphytic fungi (Ghabrial et al. 2015; Kondo, Botella, and Suzuki 2022).
Given the ecological relevance and the biotechnological impact of the Trichoderma genus, it is somewhat surprising that their associated mycovirome was only recently investigated, with the first studies in this context mainly identifying fungal viruses possessing a dsRNA genome, some of which have been characterized at a molecular level by providing their complete genome sequence (Chun, Yang, and Kim 2018a, 2018b; Lee et al. 2017; Liu et al. 2019a, 2019b, Wang et al. 2022; Zhang et al. 2018). While three of them were classified as members of the Partitiviridae family (Chun, Yang, and Kim 2018a, 2018b, Wang et al. 2022) and one as a member of the Curvulaviridae family (Liu et al. 2019a), the majority could not be assigned to any officially recognized viral family and hence are listed as unclassified dsRNA. More recently, +RNA viruses have also been identified and characterized in Trichoderma: two belong to the family Hypoviridae (You et al. 2019; Chun et al. 2022) and one to the newly suggested family ‘Ambiguiviridae’ (Gilbert et al. 2019). All these fungal viruses were observed in association with Trichoderma strains collected in the Asian continent (mainly in China or Korea), while, to our knowledge, the mycovirome associated with populations from other continents has never been explored.
In addition, only a few of the above-mentioned viruses were cured from the original strains or transferred to new virus-free isolates by anastomosis, showing the biological effects produced by presence or absence of the virus under examination. Wang and colleagues observed that the presence of Trichoderma harzianum partitivirus 2 (ThPV2) did not produce negative effects on the qualitative biocontrol performance of the fungal host: instead, it showed a moderate but statistically significant improvement in biocontrol activity in experiments with cucumber seeds inoculated with Fusarium oxysporum f. sp. cucumerinum (Wang et al. 2022). In another study, focusing on Trichoderma harzianum partitivirus 1 (ThPV1), inhibition of growth in co-cultured Pleurotus ostreatus and Rhizoctonia solani increased in ThPV1-containing strains compared with ThPV1-cured isogenic strains, and this was associated to a significantly higher β-1,3-glucanase activity, while chitinase activity was not affected (Chun, Yang, and Kim 2018b). Another unexpected result was obtained by You et al. (2019), who observed Trichoderma harzianum hypovirus 1 (ThHV1) in two forms, respectively, present in two different isolates. In one of the two isolates, the virus accumulated with an abundant defective form which is detrimental to the biocontrol properties of two Trichoderma species (You et al. 2019).
The ability of Trichoderma to establish relationships with many organisms in soil and plants makes these fungi an interesting subject to study virus horizontal transfers and virus evolution. For this purpose, we characterized the mycovirome associated with a large subset of Trichoderma spp. isolates (113 isolates) belonging to a wider collection (482 isolates) previously described; fungal strains were isolated from fifteen different soil samples, collected in several habitats, including undisturbed or extensively grazed (EG) grass steppes, forests, and shrub lands on the island of Sardinia (Migheli et al. 2009).
Here, we describe a catalog of new viruses identified in association with the subset of Trichoderma spp. isolates belonging to the collection. We reveal previously undescribed viral clades, expanding our knowledge of virus evolution, and exploring for the first time the mycovirome associated with European populations of Trichoderma species.
Materials and methods
Origin, growth conditions, and harvest of the isolates
The fungal isolates used in this study were part of a previously described collection, hosted at the Department of Plant Protection of the University of Sassari in Sardinia and assembled in 2009 (Migheli et al. 2009). Our subset consisted of 113 isolates of twelve different species belonging to genus Trichoderma, obtained from fifteen different sites comprising both undisturbed and disturbed environments (forest, shrub lands, and undisturbed or extensively grazed grass steppes). Specific details about single isolates are reported in Table S1.
Four fungal plugs obtained from monosporic cultures of each of the 113 Trichoderma isolates were placed on potato dextrose agar (Sigma-Aldrich, St. Louis, MO, USA) medium covered by a cellophane layer and incubated at 26°C for 3 days in the dark. The 4-day-old mycelia were harvested and placed into 1.5 ml Eppendorf tubes for lyophilization along with ten zirconia beads measuring 2 mm in diameter.
Total RNA extraction
The lyophilized mycelia were disrupted by bead-beating (FastPrep-24, MP Biomedicals) and used for total RNA extraction with ‘Spectrum Plant Total RNA’ kit (Sigma-Aldrich, St. Louis, MO, USA) according to the manufacturer’s protocol (Picarelli et al. 2019).
RNA concentration was inferred from ultraviolet (UV) absorbance at a 260-nm wavelength using a NanoDrop Lite Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
Total RNA sequencing and assembly of contigs
Total RNA extracted from the Sardinian collection was sent to Macrogen Inc. (Seoul, Republic of Korea) for sequencing. They were divided into two pools, named 1 and 2, respectively (details on each pool composition are presented in Table S1). Pools were obtained by mixing 2 μg of RNA extracted from each fungal sample. Ribosomal-RNA (rRNA) depletion and complementary DNA (cDNA) libraries were constructed using Illumina TruSeq Stranded Total RNA Gold and sequenced with a NovaSeq 6000 platform. The resulting reads were processed by bioinformatics analysis following a well-established pipeline previously described in detail (Chiapello et al. 2020). Reads from RNA-Seq were first cleaned, in order to remove adapters, artifacts, and short reads using BBMap software (https://sourceforge.net/projects/bbmap/); remaining reads were assembled de novo using Trinity software version 2.9.1 (Haas et al. 2013). Trinity assembly was blasted using DIAMOND software against the sorted viral portion of the NCBI non-redundant (nr) database, and the resulting hits were manually selected and characterized through molecular approaches. The number of reads covering the viral genomes was obtained by mapping the reads from each sequenced library on virus sequences with Bowtie 2, and the number of reads mapping to each segment was retrieved through SAMtools (Li et al. 2009; Langmead and Salzberg 2012).
Since virus identification was performed separately for each library, we compared the results of each one with the other to obtain a list of unique viruses thus reducing redundancy due to contig co-presence in both pools following criteria previously detailed (Chiapello et al. 2020).
ORF prediction and primer design
Starting from the above-mentioned viral contigs, the respective viral open reading frames (ORFs) were predicted using the ORFfinder tool from the National Center for Biotechnology Information (NCBI), selecting the ‘standard’ genetic code for all analyzed contigs. Domain search on each ORF was performed with ‘motif search’ available on the GenomeNet repository (https://www.genome.jp/tools/motif/) with default parameters, along with isoelectric point and molecular weight estimation using the ‘Compute pI/Mw’ tool available on the Expasy Bioinformatics resource portal (https://web.expasy.org/compute_pi/). Only ORFs with a predicted molecular mass of at least 10 kDa were graphically reported in virus genome organizations.
Primer design was performed using NCBI primer BLAST tool (https://www.ncbi.nlm.nih.gov/tools/primer-blast/); results are listed in Table S2. The putative function of the predicted ORF product was suggested by blast analysis, considering the function of the closest proteins in the NCBI database.
cDNA synthesis and real-time reverse-transcription PCR
cDNA synthesis was performed on RNA extracted from each of the 113 isolates present in the Sardinian collection, using the ‘High-Capacity cDNA Reverse Transcription Kit’ (Thermo Fisher Scientific, Waltham, MA, USA) and following the manufacturer’s protocol. Each synthesized cDNA was diluted 1:5 with sterile water for future polymerase chain reaction (PCR) applications.
Real-time PCR was performed using the above-mentioned primers (Table S2) on each cDNA sample guided by mapping results (i.e. in case a certain contig was present in just one RNA pool only isolates belonging to that specific pool were investigated), to associate specific viruses to specific fungal samples.
The PCR was performed in 10 µl using iTaq™ Universal SYBR Green Supermix (BioRad, Hercules, USA), and reaction volumes were loaded in 96-well plates and processed by ‘7500 Fast Real-Time PCR system’ (Thermo Fischer Scientific, Waltham, MA, USA), with thermocycling conditions of 3 min at 95°C, 15 s at 95°C, 30 s at 60°C, for 35 cycles. A dissociation curve analysis was performed at the end of the reverse transcriptase-quantitative PCR (RT-qPCR) protocol to check for non-specific amplification products. Isolates showing a cycle threshold equal or lower than 31 were considered as virus-positives.
Probe synthesis and northern blot analysis
Infected isolates were used to clone viral genomic cDNA fragments of a length between 300 and 400 base pairs (bp) to obtain run-off transcripts for strand-specific hybridization experiments. For this purpose, cDNA was synthetized according to the previously described protocol and then amplified in a PCR using custom-designed primers (Table S2). PCR products were isolated from agarose gel after electrophoresis and purified using the Zymoclean Gel DNA Recovery kit (Zymo Research, Irvine, CA, USA). Purified PCR fragments were ligated in pCR® II Vector—Dual Promoter TA Cloning Kit (Invitrogen-Thermo Fisher Scientific, Waltham, MA, USA) and subsequently used for Escherichia coli transformation on competent cells using the Mix & Go! E. coli Transformation Kit (Zymo Research, Irvine, CA, USA) according to the manufacturer’s protocol. Positive clones with the predicted insert (checked by digestion and subsequent vector sequencing) were used to amplify and purify plasmids; the same plasmids were then linearized and used as templates to synthesize digoxigenin (DIG)-labeled probes. Northern blot analysis was carried out using a glyoxal denaturation system exactly as previously described (Rastgou et al. 2009). DIG-labeling was obtained using ‘DIG RNA Labeling Mix’ (Roche, Basel, CH), while for the following hybridization, ‘DIG Easy Hyb Granules hybridization solution’ (Roche, Basel, CH) was employed. Signal was then detected using ‘Anti-digoxigenin-AP Fab fragments’ (Roche, Basel, CH), ‘CDP-Star’ solution (Roche, Basel, CH), and ‘Blocking Reagent’ powder (Roche, Basel, CH).
ORFan sequences detection and RNA-origin validation
Assembled contigs were submitted to a DIAMOND (v 2.0.8.146—released on March 2021) search of the NCBI nr whole database (version updated on 05 February 2022). All contigs with a homolog were discarded, whereas the remaining ones that were over 1 kb in length, showed an e-value lower than a threshold of 10–10, and encoded a protein of at least 90 amino acids (aa) (around 9 kDa) were kept, defining a preliminary set of contigs coding for ‘ORFan’ protein products. In order to select putative ORFans of viral origin alone, high-throughput sequencing (HTS) reads were mapped on these newly obtained ORFan contigs, keeping in consideration those who mapped on both anti-sense and sense contig sequences, since a typical feature of replicating viruses is the presence of a minus and plus sense genomic template for replication. Further confirmation of their viral origin was later obtained by checking the absence of a DNA amplification product after a PCR run on total nucleic acid obtained from isolates that tested positive for putative ORFan presence after RT-qPCR. To this extent, the OneTaq® DNA Polymerase kit (New England BioLabs Inc.) was used, exploiting the above-mentioned primer pairs and following the manufacturer’s protocol with a 20 µl reaction volume. Amplified bands were then separated by an electrophoretic run on a Tris Acetate EDTA 1X agarose gel (1 per cent V/V) and UV visualized. Positive control for DNA amplification was achieved using internal transcribed spacer sequence (ITS)4 and ITS5 primers (Bruns, Lee, and Taylor 1990).
Virus names assignment
The name of viruses described in this work has been attributed using the following criteria: (i) the first part of the name reflects the source of the virus (fungal species); (ii) the second part of the name identifies the virus taxonomical group of the first blast hit; and (iii) the last part of the name is a progressive number.
Phylogenetic analysis
Genome segments encoding for RNA-directed RNA polymerase (RdRp) proteins from all identified viruses, closest homologs, and representatives of the target phylogenetic clade present in the NCBI database were aligned using the online version of the Clustal Omega software, with default settings, at the European Bioinformatics Institute Web Services (Sievers et al. 2011; Madeira et al. 2019). Subsequent alignment results were first screened using MEGA 11 software (https://www.megasoftware.net/) to evaluate alignment consistency of all viral sequences under analysis: this was achieved checking the proper alignment of subdomain C of the palm domain (including the GDD aminoacidic triad). Then, the same alignment results were submitted to the IQ-TREE web server to produce phylogenetic trees under a maximum likelihood model (Trifinopoulos et al. 2016). The best substitution model was estimated automatically by IQ-TREE with ModelFinder (Kalyaanamoorthy et al. 2017), and ultrafast bootstrap analysis with 1,000 replicates was performed.
In addition, an estimation of the evolutionary distance between the identified viral RdRps and other viral sequences used for the phylogenetic tree production was obtained, using the ‘p-distance’ substitution model for pairwise distance computation in MEGA 11. All ambiguous positions were removed for each sequence pair (the ‘pairwise deletion’ option). Results were then transposed in aa identity, by one’s complement (1 − p) using excel and thus presented as pairwise-identity matrices.
In-silico protein structure prediction and comparison
Protein structure was predicted in-silico using ColabFold v1.5.2 (https://colab.research.google.com/github/sokrypton/ColabFold/blob/main/AlphaFold2.ipynb) (Mirdita et al. 2022). Obtained models were then employed for structural comparison and root mean square deviation (RMSD) estimation using University of California, San Francisco (UCSF) ChimeraX ‘matchmaking’ function, with default parameters. UCSF Chimera X is developed by the Resource for Biocomputing, Visualization, and Informatics at the UCSF, with support from National Institutes of Health R01-GM129325 and the Office of Cyber Infrastructure and Computational Biology, National Institute of Allergy and Infectious Diseases (Pettersen et al. 2021).
Results
Total RNA sequencing, contigs assembly, and RT-qPCR
After the sequencing runs on the entire Trichoderma collection, we gathered 207,195,944 total reads, with 105,787,432 for Pool 1 and 101,408,512 for Pool 2. Trinity assembly generated an initial amount of 314,762 contigs; the subsequent Basic Local Alignment Search Tool (BLAST) search on a custom-prepared viral database produced a total of twenty-six putatively unique viral contigs (Table 1). Among those, seventeen contained the typical conserved motifs of a viral RdRp, implying the likely identification of at least seventeen distinct viruses. In addition, we identified nine other segments predicted to encode capsid proteins or conserved protein products of unknown function and belong to bi- or tri-partite viruses. Finally, four putative viral contigs that did not show any match in nr databases were found, encoding proteins of unknown function (ORFans) without any conservation to existing catalogs through similarity searches. Among these four fragments, three (ORFan1, ORFan2, and ORFan4) possess RNA-only counterparts with no DNA detected corresponding to them using total nucleic acid as template for PCR amplification (see dedicated paragraph). This allowed to exclude that ORFan1, ORFan2, and ORFan4 were transcripts derived from a DNA genome or from endogenization of an RNA virus or that their replication occurred through a DNA intermediate.
Table 1.
List of putative viral contigs obtained by the application of our bioinformatic pipeline to rRNA-depleted total RNA extracted from Trichoderma strains. Columns show: Trinity contig ID, assigned name and abbreviation, segment length, first BLASTx hit organism, a brief description for the putatively encoded protein product of the first BLASTx hit and accession ID of the latter, percentage identity (* when >90 per cent), query coverage, and number of reads mapping for each RNA pool. N.a. stands for ‘not available’.
| TRINITY ID | Assigned name (Abbreviation) | Segment length (bp) | BLASTx first hit | Description | NCBI accession ID | % Identity | Query coverage | Mapped reads (Pool 1–Pool 2) | GenBank ID |
|---|---|---|---|---|---|---|---|---|---|
| TRINITY_DN216_c0_g2_i2 | Trichoderma atroviride partitivirus 1-RNA 1 (TaPV1) | 2,096 | TaPV1 | RdRp | AYQ58321.1 | 98* | 92.1 | 99,206–14,740 | OQ463832 |
| TRINITY_DN216_c0_g1_i2 | Trichoderma atroviride partitivirus 1-RNA 2 (TaPV1-b) | 2,126 | TaPV1 | CP | AYQ58322.1 | 95.3* | 83.7 | 71,166–14,950 | OQ463833 |
| TRINITY_DN1441_c0_g2_i1 | Trichoderma atroviride partitivirus 1-RNA 3 (TaPV1-c)/ORFan1 | 2,003 | n.a. | n.a. | n.a. | n.a. | n.a. | 80,674–23,236 | OQ463834 |
| TRINITY_DN74718_c0_g1_i1 | Trichoderma gamsii alphapartitivirus 1-RNA 1 (TgAPV1) | 1,948 | Medicago sativa alphapartitivirus 2 | RdRp | QBC36014.1 | 64.3 | 91.2 | 6720–68,048 | OQ463835 |
| TRINITY_DN6615_c0_g2_i1 | Trichoderma gamsii alphapartitivirus 1-RNA2 (TgAPV1-b) | 1,823 | Rhizoctonia oryzae-sativae partitivirus 1 | Coat protein | AYV61426.1 | 59.4 | 80.1 | 1916–21,104 | OQ463836 |
| TRINITY_DN906_c12_g1_i1 | Trichoderma gamsii alphapartitivirus 1-RNA3 (TgAPV1-c)/ORFan4 | 1,645 | n.a. | n.a. | n.a. | n.a. | n.a. | 1420–14,466 | OQ463837 |
| TRINITY_DN13792_c0_g1_i1 | Trichoderma gamsii cogu-like virus 1-RNA 1 (TgClV1) | 6,735 | Botrytis cinerea bocivirus 1 | RdRp | QJT73693.1 | 68.6 | 98.1 | 0–1786 | OQ513277 |
| TRINITY_DN109836_c0_g1_i1 | Trichoderma gamsii cogu-like virus 1-RNA 2 (TgClV1-b) | 1,645 | Botrytis cinerea bocivirus 1 | MP | QJT73692.1 | 60.4 | 86.1 | 0–5976 | OQ513278 |
| TRINITY_DN492_c0_g1_i1 | Trichoderma gamsii cogu-like virus 1-RNA 3 (TgClV1-c) | 1,274 | Botrytis cinerea bocivirus 1 | CP | QJT73691.1 | 54.9 | 81.2 | 0–24,140 | OQ513279 |
| TRINITY_DN9647_c0_g1_i1 | Trichoderma gamsii mycobunyavirus 1 (TgMBV1) | 9,319 | Macrophomina phaseolina mycobunyavirus 1 | RdRp | QOE55579.1 | 35.7 | 84.6 | 1270–16,836 | OQ463838 |
| TRINITY_DN6421_c0_g1_i2 | Trichoderma gamsii negative-stranded virus 1 (TgNV1) | 6,902 | Botrytis cinerea negative-stranded RNA virus 6 | RdRp | QJT73694.1 | 37.4 | 97.2 | 0–2662 | OQ463839 |
| TRINITY_DN95_c0_g1_i1 | Trichoderma hamatum dsRNA virus 1 (ThaDSV1) | 9,605 | TaMV1 | RdRp | YP_009553633.1 | 52.4 | 41.8 | 823,514–149,384 | OQ463840 |
| TRINITY_DN71000_c0_g1_i1 | Trichoderma hamatum mycoophiovirus 1 (ThaMOV1) | 7,203 | Plasmopara viticola lesion-associated mycoophiovirus 5 | RdRp | QJX19791.1 | 44.9 | 97.6 | 86,894–12 | OQ463841 |
| TRINITY_DN71583_c0_g1_i1 | Trichoderma harzianum orthocurvulavirus 1—RNA 1 (ThOCV1) | 2,054 | ThOCV1 | RdRp | YP_009553330.1 | 98.3* | 92.2 | 19,962–198 | OQ463842 |
| TRINITY_DN77701_c0_g1_i1 | Trichoderma harzianum orthocurvulavirus 1—RNA 2 (ThOCV1-b) | 1,605 | ThOCV1 | Hypothetical protein | YP_009553331.1 | 99.4* | 58.9 | 11,346–606 | OQ463843 |
| TRINITY_DN23294_c0_g1_i4 | Trichoderma harzianum dsRNA virus 1-RNA 1 (ThDSV1) | 2,147 | Fusarium graminearum dsRNA mycovirus-4 | Putative RdRp | YP_003288790.1 | 66.1 | 90.1 | 1682–958 | OQ463844 |
| TRINITY_DN2883_c0_g1_i1 | Trichoderma harzianum dsRNA virus 1 -RNA 2 (ThDSV1-b) | 941 | Botrytis cinerea mycovirus 5 | Hypothetical protein | QJT73710.1 | 61.4 | 84 | n.a. | OQ463858 |
| TRINITY_DN229_c1_g1_i2 | Trichoderma harzianum dsRNA virus 2 (ThDSV2) | 8,684 | TaMV1 | RdRp | YP_009553633.1 | 51.9 | 46.4 | 36,954–0 | OQ463845 |
| TRINITY_DN72954_c0_g1_i1 | Trichoderma harzianum dsRNA virus 3 (ThDSV3) | 6,346 | Diatom colony-associated dsRNA virus 17 genome type B | RdRp | YP_009551502.1 | 34.3 | 32.1 | 13,128–7266 | OQ463846 |
| TRINITY_DN72949_c0_g1_i1 | Trichoderma harzianum mononegavirus 1 (ThMV1) | 11,124 | PvLaMAV8 | RdRp | QHD64783.1 | 46.3 | 53.3 | 35,102–0 | OQ463847 |
| TRINITY_DN72958_c0_g1_i1 | Trichoderma harzianum mononegavirus 2—RNA 1 (ThMV2) | 6,195 | PvLaMAV8 | RdRp | QHD64783.1 | 45.7 | 92.4 | 28,858–0 | OQ463848 |
| TRINITY_DN72957_c0_g1_i1 | Trichoderma harzianum mononegavirus 2—RNA 2 (ThMV2-b) | 5,031 | Hubei rhabdo-like virus 4 | Hypothetical protein 2 | YP_009336594.1 | 27.2 | 19.1 | 28,380–0 | OQ463849 |
| TRINITY_DN262_c0_g1_i5 | Trichoderma harzianum negative-stranded virus 1 (ThNV1) | 7,061 | Botrytis cinerea negative-stranded RNA virus 6 | RdRp | QJT73694.1 | 38.4 | 95 | 21,072–7028 | OQ463850 |
| TRINITY_DN59_c0_g1_i5 | Trichoderma harzianum negative-stranded virus 2 (ThNV2) | 7,389 | Botrytis cinerea negative-stranded RNA virus 3 | RdRp | QJT73696.1 | 55.3 | 78.4 | 117,984–0 | OQ463851 |
| TRINITY_DN71003_c0_g1_i1 | Trichoderma harzianum partitivirus 3—RNA 1 (ThPV3) | 1,753 | Ustilaginoidea virens partitivirus | RdRp | AGO04402.1 | 86.3 | 92.5 | 9784–27,582 | OQ463852 |
| TRINITY_DN71535_c0_g1_i1 | Trichoderma harzianum partitivirus 3—RNA 2 (ThPV3-b) | 1,563 | Verticillium dahliae partitivirus 1 | Coat protein | YP_009164039.1 | 75.7 | 83.9 | 9990–29,704 | OQ463853 |
| TRINITY_DN76119_c0_g1_i1 | Trichoderma harzianum partitivirus 3—RNA 3 (ThPV3-c) | 1,526 | Aspergillus fumigatus partitivirus 1 | Unknown | CAA7351346.1 | 35.7 | 43.8 | 19,888–30,768 | OQ463854 |
| TRINITY_DN74521_c1_g1_i1 | Trichoderma tomentosum ormycovirus 1-RNA 1 (TtOV1) | 2,645 | Erysiphe lesion-associated ormycovirus 4 | Putative RdRp | USW07208.1 | 53.20 | 90 | 191,226–52,136 | OQ463855 |
| TRINITY_DN72921_c9_g1_i1 | Trichoderma tomentosum ormycovirus 1-RNA2 (TtOV1-b) | 1,893 | Erysiphe lesion-associated ormycovirus 4 | Hypothetical protein | USW07213.1 | 60,8 | 71 | 602,130–164,856 | OQ463856 |
| TRINITY_DN112775_c0_g1_i1 | Trichoderma spirale orphan RNA (ORFan2) | 1,471 | n.a. | n.a. | n.a. | n.a. | n.a. | 0–77,196 | OQ463857 |
Association of each viral contig or ORFan sequence with each fungal isolate was then assessed through a RT-qPCR assay (Table S3, Fig. 1). The total number of different viral contigs in each single isolate was quite variable, ranging from zero to a maximum of five (e.g. Isolate #99). The total number of fungal isolates hosting at least one viral contig were 36 out of the 113 screened isolates: these virus-infected isolates comprised six fungal species belonging to T. harzianum (19 isolates), Trichoderma gamsii (7 isolates), Trichoderma hamatum (6 isolates), Trichoderma tomentosum (2 isolates), Trichoderma samuelsii (Jaklitsch et al. 2013) (1 isolate), and Trichoderma. spirale (1 isolate) (Fig. 1).
Figure 1.
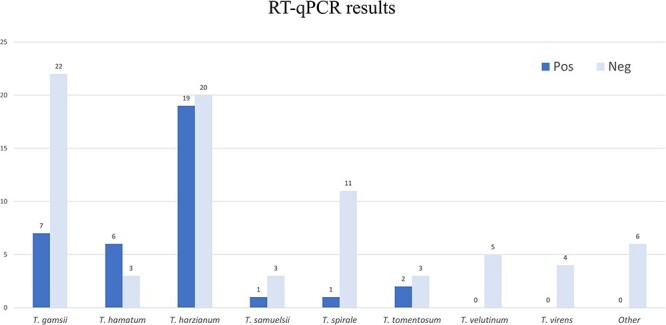
Virus detection by RT-qPCR for the entire Trichoderma collection. Height of the bars represents the number of isolates. The group defined as ‘Other’ includes: Trichoderma koningii (2 isolates), Trichoderma koningiopsis (2 isolates), Trichoderma asperellum (1 isolate), and an anamorph of Hypocrea semiorbis (1 isolate).
For bi- or multi-segmented viruses, when possible, the read count/library has also been used as a guide to identify contigs belonging to the same virus. If this approach resulted in ambiguous associations, a RT-qPCR assay to find strict associations within specific isolates was used (Table S4).
The seventeen putative viral sequences encoding for an RdRp had identity to previously reported viruses ranging from 37.4 to 98.3 per cent. Almost half of these putative viruses were predicted to have negative-sense RNA genomes and to be related to viruses present within the orders Bunyavirales (four novel virus sequences), Mononegavirales (three novel virus sequences), or Serpentovirales (one novel virus sequence). Additionally, another half of the identified viral sequences were predicted to possess a dsRNA genome and were assigned to the Durnavirales order (three novel and two already known sequences) or to the Ghabrivirales order (three novel sequences). Finally, one sequence appeared to belong to a recently proposed group of allegedly positive single-stranded RNA (ssRNA) viruses named ‘ormycoviruses’ (Forgia et al. 2022).
Viruses characterized in the Bunyavirales order
Bunyavirals are mostly enveloped viruses with a genome generally consisting of three ssRNA segments (called L, M, and S) (Ferron et al. 2017). The majority of the families included in this order have invertebrates, vertebrates, or plants as hosts, but recently specific clades infecting fungi have been characterized (Lin et al. 2019; Velasco et al. 2019). Interestingly, in our study, we found only one virus (out of four) related to bunyavirals that possesses a tripartite genome (i.e. Trichoderma gamsii cogu-like virus 1—TgClV1), while the remaining three showed only the presence of one genomic segment (Trichoderma gamsii mycobunyavirus 1—TgMBV1; Trichoderma gamsii negative-sense virus 1—TgNV1; and Trichoderma harzianum negative-sense virus 1—ThNV1). The L segment of these putative bunyavirals includes the complete coding region in a single large ORF coding for the RdRp (Fig. 2). The RdRp-coding nucleotide (nt) sequences range from ≈6.6 kb (TgClV1) to ≈9.3 kb (TgMBV1) and are predicted to encode a protein product ranging from 2200 to 3000 aa. In addition, based on ‘Motif search’ analysis, all these ORF products host a ‘bunyavirus_RdRp’ domain (pfam04196) spanning in an average of 400 aminoacidic residues (Fig. 2, Table S5). In TgMBV1 and ThNV1, an L-protein N-terminal endonuclease domain (pfam15518) was also present on the L segment (Fig. 2).
Figure 2.
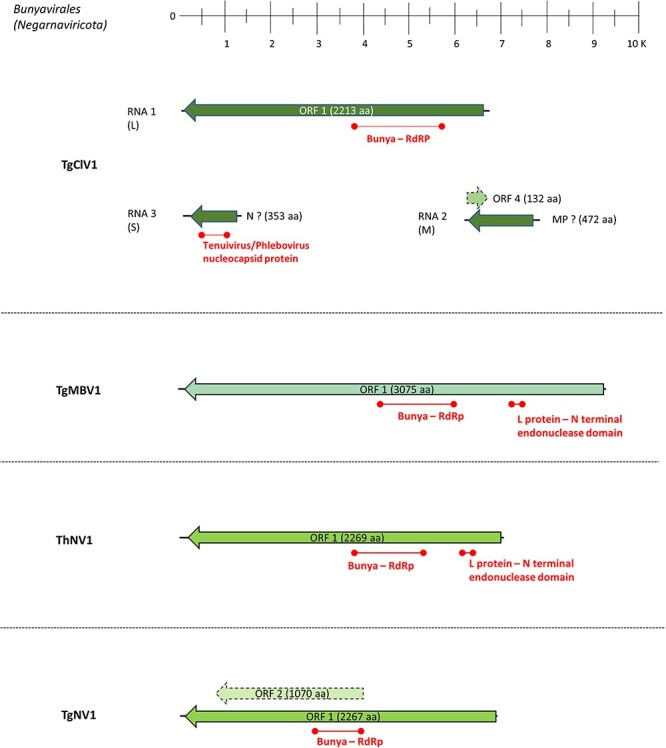
The genome organization of putative viruses belonging to the Bunyavirales order; top ruler indicates length in kb. ORFs which returned at least one BlastP hit are represented with solid lines, while dotted lines represent ORFans. The presence of known protein domains predicted with ‘Motif search’ analysis is highlighted.
For TgClV1, the two other putative genomic segments (RNA2 and RNA3) correspond to putative M and S segments, respectively. The RNA3 segment is around 1300 nt and hosts an ORF encoding for a 353 aa product having its first blast hit with Botrytis cinerea bocivirus 1-capsid protein (CP), although motif search analysis evidenced the additional presence of a Tenuivirus/Phlebovirus CP domain (pfam05733) (Fig. 2). RNA2 is a segment of 1645 nt which putatively codes for a 472 aa protein product, with the latter having a first blast hit with Botrytis cinerea bocivirus 1—movement protein (MP), along with an additional ORFan sequence carried on the positive sense of the segment which encodes for an ORFan protein of 132 aa (ORF4; Fig. 2). Phylogenetic analysis shows that all the viruses assigned to Bunyavirales based on close Blast hits are indeed in the order Bunyavirales, with one clearly belonging to the Phenuiviridae family, while the remaining three clustering with different unclassified Bunyavirales members (Fig. 3).
Figure 3.
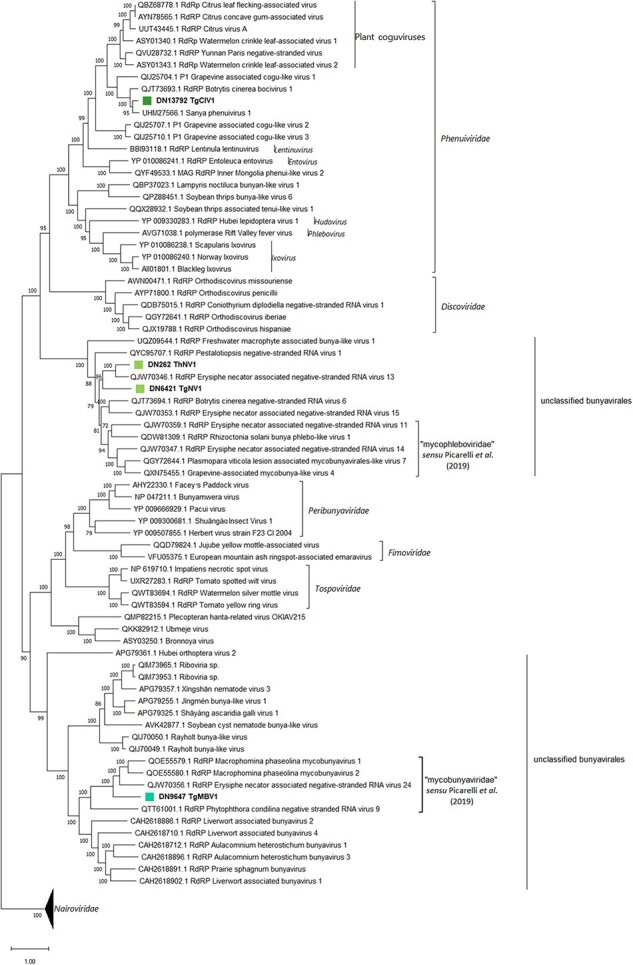
The Bunyavirales phylogenetic tree as computed by IQ-TREE stochastic algorithm, which infers phylogenetic trees by maximum likelihood methodology. Model of substitution: VT + F + I + G4. Consensus tree is constructed from 1,000 bootstrap trees. Log-likelihood of consensus tree: −396,949.8245. The percentage bootstrap values can be found at the nodes. Distinct colors indicate specific viruses in different sub-groups.
TgClV1 clusters within the Phenuiviridae family, in a clade closely related to plant coguviruses. For this reason, the name ‘Trichoderma cogu-like virus 1’ was assigned. With respect to the remaining three viral sequences, two of them (TgNV1 and ThNV1) seem closely related to a clade previously described that, so far, have no specific nucleocapsid (N) or non structural (NS) associated (Picarelli et al. 2019). Finally, according to our phylogenetic analysis, TgMBV1 seems to associate in a distinct clade that includes viruses infecting fungi and oomycetes such as Macrophomina phaseolina negative-sense RNA virus 1 and 2 and Phytophthora condilina negative-stranded RNA virus 9 (Botella and Jung 2021; Picarelli et al. 2019; Wang et al. 2021). Phylogenetic analysis locates this recently proposed group of fungal viruses in a well-defined clade which seems related to viruses infecting liverwort and mosses (Bryophyta) and, to a lesser extent, to nematodes-infecting viruses (Fig. 3). Stronger evidence, which corroborates our taxonomical hypothesis, can also be observed by examining the pairwise-identity matrices (Table S6).
Viruses characterized in the Mononegavirales and Serpentovirales orders
The order Mononegavirales includes negative-sense RNA viruses that are mostly monopartite, with multiple ORFs typically in the same orientation. In our analysis, we have found three viruses putatively belonging to this order, namely Trichoderma harzianum negative-sense virus 2 (ThNV2), Trichoderma harzianum mononegavirus 1 (ThMV1), and Trichoderma harzianum mononegavirus 2 (ThMV2). Initially, just two of the above-mentioned viruses were actually identified as monosegmented, while, surprisingly, for ThMV2, a second RNA segment was detected (see further).
The genome length of these three mononegavirals ranges from 7 to 11 kb, and specifics can be seen in Table S5. All of them host an ORF-encoding RdRp of ≈1,950 aa in length that contains a catalytic domain characteristic of Mononegavirales RdRp L-protein (pfam00946) and messenger RNA (mRNA)-capping region V domain (pfam14318), which is known to host a specific motif, GxxTx(n)HR, that is essential for mRNA cap formation (Fig. 4).
Figure 4.
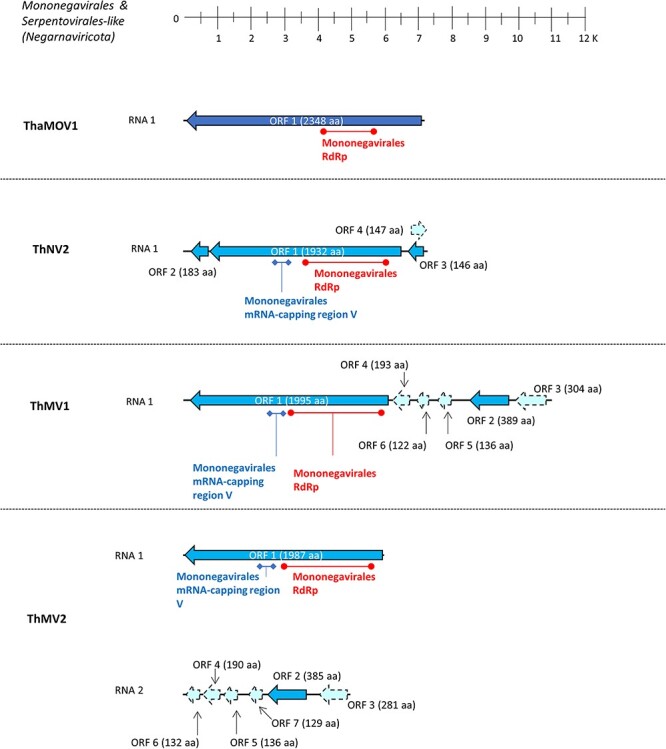
The genome organization of putative viruses belonging to the Mononegavirales and Serpentovirales orders; top ruler indicates length in kb. Solid lines represent ORFs which returned at least one BlastP hit, while dotted lines represent ORFans. The presence of known protein domains predicted with ‘Motif search’ analysis is highlighted.
These three putative viral RdRps had a first blast hit with negative-sense viruses: specifically, ThNV2 had Botrytis cinerea negative-stranded RNA virus 3 as the first hit, while the other two (ThMV1 and ThMV2) both had Plasmopara viticola lesion-associated mononegaambivirus 8 (Chiapello et al. 2020) as the first hit. Other additional ORFs were detected for all three mononegavirals; in fact, ThNV2 hosts two ORFs flanking the RdRp-coding ORF (Fig. 4), and these two small ORFs (ORF2 and ORF3; 552 and 441 nt, respectively) had a first blast hit with hypothetical proteins of unknown function belonging to Fusarium graminearum negative-sense RNA virus 1 and Plasmopara viticola lesion-associated mononegaambivirus 2, respectively. Also, on the sense-strand of ThNV2, an ORFan-coding region was detected (ORF4; Fig. 4). Regarding ThMV1 and ThMV2-RNA2, both possess an ORF coding for a ≈380 aa protein product in the second to last position in the reverse complement genome, which had a first blast hit with Magnaporthe oryzae mymonavirus 1-CP (ThMV1 and ThMV2-RNA2 ORF2; Fig. 4).
Interestingly, a certain degree of synteny could be highlighted between ORFan-coding regions of ThMV1 and ThMV2-RNA2 (dotted lines in Fig. 4): in fact, they all have similar length, orientation, and, to a lesser extent, sequence similarity (Fig. S1, Table S7). After PCR amplification using a primer pair spanning the ThMV2-RNA segment junction, no amplification product coherent with a monopartite genome organization was obtained, confirming the possibility of a bisegmented genome organization for ThMV2. On the other hand, when performing a PCR amplification using primers specific for the same intergenic region on ThMV1, we could obtain and visualize a coherent amplification product. Finally, after northern blot analysis, we indeed confirmed that ThMV1 exists as one unique genomic species of the expected length (11 kb), while ThMV2 exists as a bisegmented genomic species, with two segments of the expected length (6 kb for RNA1 and 5 kb for RNA2) (data not shown).
The last negative-sense RNA virus (Trichoderma hamatum Mycoophiovirus 1 (ThaMOV1)) was hypothesized to belong to the Serpentovirales order, which currently includes only one family (Aspiviridae) and one genus (Ophiovirus) of segmented negative-sense RNA viruses that infect plants. We have identified the presence of one segment coding for an ophio-like viral RdRp in our collection and have not detected any additional segments. This single viral sequence hosts one single ORF of ≈7 kb in length, which codes for a putative protein of 2348 aa which, again, had the characteristic catalytic domain of Mononegavirales RdRp L-protein (pfam00946). It had as its first blast hit Plasmopara viticola lesion-associated mycoophiovirus 5—RdRp, so, for this reason, the sequence was named Trichoderma hamatum Mycoophiovirus 1 (ThaMOV1).
Phylogenetic analysis in this case suggests accommodating ThNV2, ThMV1, and ThMV2 within the Mymonaviridae family (Mononegavirales order). While ThNV2 belongs to the genus Sclerotimonavirus, ThMV1 and ThMV2 appear to be members of the Plasmopamonavirus genus (Fig. 5). ThaMOV1 clearly belongs to a well-defined clade closely related to the plant-infecting viruses in Aspiviridae, which includes several recently characterized fungal viruses that Chiapello and colleagues suggested the taxon name ‘mycoaspiviridae’ for (Chiapello et al. 2020). Pairwise-identity matrices obtained with MEGA11 (Table S8) also corroborated our taxonomical hypothesis.
Figure 5.
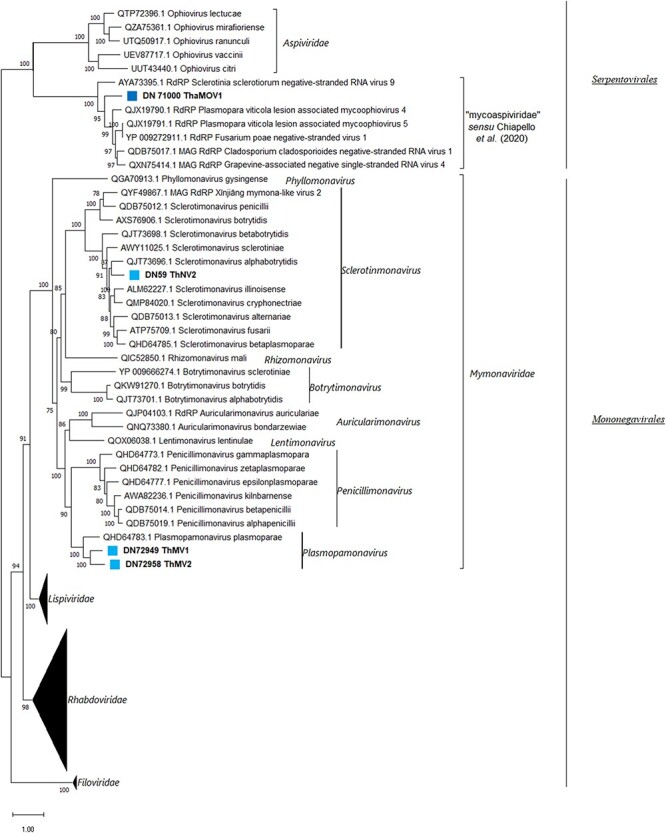
The Mononegavirales and Serpentovirales phylogenetic tree as computed by the IQ-TREE stochastic algorithm, which infers phylogenetic trees by maximum likelihood methodology. Model of substitution: LG + F + I + G4. Consensus tree is constructed from 1,000 bootstrap trees. Log-likelihood of consensus tree: −324,352.0477. The percentage bootstrap values are displayed at the nodes. Distinct colors indicate specific viruses in different sub-groups.
Bisegmented Mymonaviridae members are detected in metatranscriptomic sequences from previous studies using ThMV2 RNA2–encoded proteome as a query
To shed light on the possible bisegmented nature and the evolutionary origin of ThMV2, we used the putative N sequence encoded by ThMV2-RNA2 as query for blast interrogation. This analysis was carried out on some previously described Trinity assemblies shown to host recently characterized mymonavirals that have been previously included in the Penicillimonavirus and Plasmopamonavirus genera and reported as monopartite: some were assemblies from our previous work (Chiapello et al. 2020), while others were newly produced and obtained using published reads (Short Sequence Reads = SSR from 8303984 to 830390) (Nerva et al. 2019). In both cases, reads were originated by high-throughput sequencing (HTS) approaches on metagenomic samples (e.g. plant material showing grapevine downy mildew or esca disease symptoms). In this way, we were able to identify eleven new contigs, undetected in the previous studies, which possessed similar length and genome organization with respect to ThMV-RNA2 (Fig. 6). Moreover, when short reads obtained in the two aforementioned works (Nerva et al. 2019; Chiapello et al. 2020) were mapped onto the eleven newly identified contigs, we found a clear co-distribution of reads, shared with some previously described viral segments belonging to Penicillimonavirus or Plasmopamonavirus genus members. This evidence allows to unequivocally associate each previous RdRp-encoding segment with its newly found RNA2 (Fig. S2). All these second RNAs were therefore associated with their respective first segments, belonging to: Plasmopara viticola lesion-associated mononegaambi virus 1-2-3-4-5-6-7-8-9 (PvLaMAV1–9), Penicillium glabrum negative-sense RNA virus 1 (PgNSV1), and Penicillium adametzioides negative-sense RNA virus 1 (PaNSV1).
Figure 6.

The genome organizations of putative bisegmented members of the Mymonaviridae family; top ruler indicates length in kb. Solid lines represent ORFs which returned at least one BlastP hit, while dotted lines represent ORFans.
RNA2 of the latter viruses ranged from 3,900 to 5,000 nt in length and hosted 4 or 5 ORFs mainly coding for ORFan protein products, with the exception of the one ORF coding for the putative N, in the second to last position of the anti-sense strand (Fig. 6). The degree of conservation evidenced by multiple sequence alignment of the putative N proteins led us to discard our initial hypothesis of a putative N-coding ORF carried in sense orientation on the first RNA of some of these mymonaviruses (Chiapello et al. 2020), instead indicating that the putative N is hosted on these newly identified second segments (Fig. S3).
To further reinforce this last hypothesis that ORF2 of RNA2 is a putative N protein, we compared the structural conservation of these proteins (predicted in-silico), with that of some experimentally confirmed N proteins in the Mononegavirales (Sonchus yellow net virus (SYNV) and Sclerotinia sclerotiorum negative-stranded RNA virus 1 (SsNSRV-1)). The AlphaFold2-predicted N protein structures of ThMV1, ThMV2, PvLaMAV1, PvLaMAV2, SYNV, and SsNSRV-1 were over-imposed by matchmaking using UCSF ChimeraX. Results of the analysis (Fig. 7) allowed the clear recognition of a structurally conserved region of two α-helices that was shared among all the tested N models, spanning from Residue 203 to Residue 240 of SYNV (used as reference structure for matchmaking). Interestingly, this same region (203::240) indeed falls within the ‘Rhabdo_ncap_2’ Rhabdovirus nucleoprotein motif (pfam03216), which is exhibited by SYNV-N in residue position 142::535 after motif search analysis.
Figure 7.
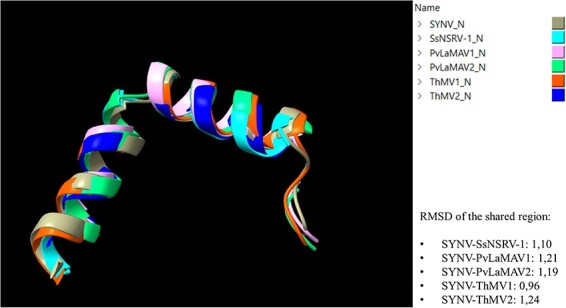
The structural comparison between N models of different mymonaviruses (SsNSRV-1, PvLaMAV1, PvLaMAV2, ThMV1, and ThMV2) and the N model of SYNV. Different colors indicate different N predicted models (legend on top right of the image). RMSD indicates the average distance in Ångström between backbone atoms of different protein structures.
Viruses characterized in the Durnavirales order
Five viral sequences belonging to the Durnavirales order were identified within our collection, among which two were already officially recognized fungal viruses (Trichoderma atroviride partitivirus 1 (TaPV1) and Trichoderma harzianum orthocurvulavirus 1 (ThOCV1)). As for others, three sequences were assigned to the Partitiviridae family, while two were assigned to the Curvulaviridae family. For viruses belonging to Durnavirales and Ghabrivirales orders, a finer description can be found in Supplementary Text, along with genome representations (Figs. S4 and S6), phylogenetic trees (Figs. S5 and S7), and identity matrices (Tables S9 and S10).
Interestingly, a third segment was associated to ThPV3, TaPV1, and Trichoderma gamsii alphapartitivirus 1 (TgAPV1): ThPV3-RNA3 hosts an ORF of 1176 bp which should encode a protein product having 35.6 per cent identity with a protein of unknown function from Aspergillus fumigatus partitivirus 1. The third segment of TaPV1 was instead initially referred to as ORFan1 due to a lack of any sequence conservation with proteins present in the databases and difficulties in associating it to any RdRp-coding viral segments. Nevertheless, RT-qPCR analysis always detected the co-presence of TaPV1 and ORFan1 contigs (Table S3), suggesting that these three segments belonged to the same virus. The same conclusion can be applied for ORFan4 with TgAPV1, which resulted to be the third segment of the latter virus (Table S3). For this reason, we changed the name of ORFan1 to ‘TaPV1-RNA3’ and that of ORFan4 to ‘TgAPV1-RNA3’. With respect to TaPV1-RNA3, the segment displays two ORFan-coding sequences, having opposite orientation, spanning 1605 and 291 bp (ORF3 and ORF6; Fig. S4). Curiously, this is the first time that a third of RNA segment has been associated with TaPV1 (a virus previously characterized by Chun, Yang, and Kim (2018a)). Regarding TgAPV1-RNA3, it is predicted to host an ORFan-coding sequence putatively encoding a 455 aa protein product (Table S5; ORF3; Fig S4). The association of these third partitivirus segments to their specific partitivirus genomes is also supported by the conservation of the terminal sequences (for ORFan results, see further).
Viruses characterized in the Ghabrivirales order
Three viruses were detected that were partially related to officially recognized totiviruses.
Phylogenetic analysis results clearly indicated that these novel viruses are not bona fide totiviruses (member of the genus Totivirus), but, while Trichoderma hamatum dsRNA virus 1 (ThaDSV1) and Trichoderma hamatum dsRNA virus 2 (ThDSV2) seems to belong to the recently suggested family of ‘fusagraviridae’ (Wang et al. 2016), Trichoderma hamatum dsRNA virus 3 (ThDSV3) clusters in a much more distant clade that includes unclassified members of the Totiviridae family (Fig. S7). Pairwise-identity matrices produced with MEGA11 showed for ThDSV2 and ThaDSV1 an identity level of 52.6 and 53.1 per cent, with Trichoderma asperellum dsRNA virus 1 (member of this new suggested family, ‘fusagraviridae’), respectively. On the other hand, ThDSV3 shared a 42.6 per cent identity with Prasiola crispa toti-like virus which is an unclassified putative member of the family Totiviridae (Table S10).
Ormyco-like sequences
The term ‘ormycoviruses’ has been recently coined to describe a novel group of viruses that includes three conserved clades of protein-coding RNA segments of viral origin that are typically associated with a second RNA segment of unknown function (Forgia et al. 2022). Using in-silico structural prediction approaches, we were able to demonstrate a clear structural conservation of these RNA segments with previously characterizedx viral RdRps, however they had very limited protein sequence conservation, which initially prevented their detection through similarity searches. Moreover, no ‘GDD’ catalytic triad was present within these putative RdRps: the most common putative catalytic triads were ‘NDD’ and ‘GDQ’ and, to a lesser extent, ‘SDD’, ‘HDD’, and ‘ADD’ (Forgia et al. 2022).
One alleged new member of this recent ‘ormycovirus’ group, which was named Trichoderma tomentosum ormycovirus 1 (TtOV1), was identified along with a second RNA detected in strict association with the latter and named TtOV1-b (Table S4). TtOV1 segments 1 and 2 were around 2.6 and 1.8 kb of length, respectively, with the first likely encoding a putative 779 aa long RdRp that shows a 53.4 per cent sequence identity with Erysiphe lesion-associated ormycovirus 4 putative RdRp, while the second seems to encode a hypothetical protein of 533 aa that shares 60.8 per cent identity with Erysiphe lesion-associated ormycovirus 4 (Fig. 8A, Table S5). A reliable phylogenetic analysis that includes representatives of the five classes of RNA viruses could not be performed due to the very limited similarity of the ‘ormycoviruses’ group members to those already included in the RNA viruses’ monophyletic tree. Nevertheless, some conservation among ‘ormycoviruses’ was detected through BlastX analysis by querying our ormyco-like contig (data not shown) against the whole nr NCBI protein database; starting from the small subset (twelve sequences) returned by blast search, we constructed a phylogenetic tree and a pairwise-identity matrix (Fig. 8B, Table S11).
Figure 8.
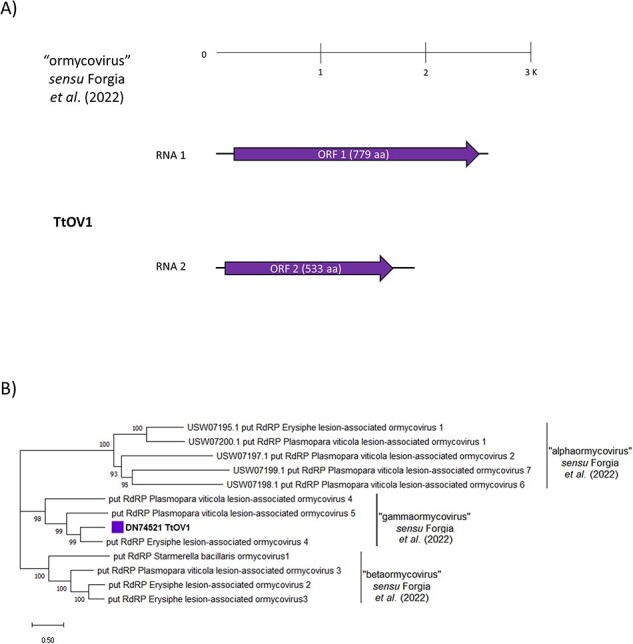
(A) The genome organization of the putative ‘ormycovirus’ TtOMV1; top ruler indicates length in kb. Solid lines represent ORFs which returned at least one BlastP hit, while dotted lines represent ORFans. (B) The ‘Ormycovirus’ phylogenetic tree as computed by the IQ-TREE stochastic algorithm, which infers phylogenetic trees by maximum likelihood. Model of substitution: VT + F + G4. Consensus tree is constructed from 1,000 bootstrap trees. Log-likelihood of consensus tree: −24,861.9845. Percentage bootstrap values are displayed at the nodes. Distinct colors indicate specific viruses in different sub-groups.
According to our results, TtOV1 is closely related to Erysiphe lesion-associated ormycovirus 4 (53.8 per cent identity, see Table S11) and clearly clusters within the proposed ‘gammaormycovirus’ sub-group (Fig. 8B). Further confirmation of these results came from a multiple sequence alignment performed between this small subset of sequences (Fig. S8). TtOV1 shows the conservation of the D residue in motif A and the two G residues in motif B which are the characteristics of ‘ormycoviruses’ in general, along with the catalytic triad ‘GDQ’ in motif C, which is known to be unique for ‘gammaormycoviruses’ (Forgia et al. 2022).
ORFan sequences
Four ORFans, named ORFan 1, 2, 3, and 4, were identified within the collection. Only those having a putative viral origin (the absence of amplification from total nucleic acid sample without reverse transcription, see Fig. S9) were further considered, leaving only ORFan1, ORFan2, and ORFan4. Neither of the three could be located in any known viral taxonomical group, and no evidence of an RdRp domain was detected within them. ORFan1 was found in association with TaPV1 contigs, thus leading us to postulate a possible role as the third segment of TaPV1 genome. The ORFan1 sequence has a good degree of conservation present with the 5′ and 3′ ends of both TaPV1-RNA1 and -RNA2 which may be a confirmation of this hypothesis (Fig. S10); nevertheless, the ORFan nature of the protein product putatively encoded by the segment remains unsolved. On the other hand, ORFan2 did not show any association with other viral contigs (Table S3), and it was only detected within one unique isolate (Number #45) of T. spirale. ORFan2 consisted of a sequence around 1.5 kb in length that hosts three putative ORFan-coding sequences, each of which circa 260 nt long and did not return any results after ‘Motif search’ analysis (Fig. S11, Table S5). Finally, ORFan4 could be a third segment associated to TgAPV1: it was found in strict association with TgAPV1 contigs exclusively, and, additionally, we could find some conservation on the 5′ end of this ORFan sequence with TgAPV1-RNA1 and RNA2 (Fig. S12).
Discussion
Trichoderma spp. are ubiquitous fungi that, as specialized saprotrophs, are able to colonize almost all environments (e.g. agricultural, forest, mountain, and grassland), contributing to soil and carbon mineralization. Moreover, some species are known for their potential value as biocontrol agents due to their highly competitive mycoparasitic behavior and their ability to improve plant health and protection: so, they are broadly appreciated for agricultural applications (Mukherjee et al. 2013). In recent years, viruses of fungi have attracted increasing attention due to their effects on their hosts, but those infecting Trichoderma spp. have not been the subject of extensive studies. A few available research studies suggest that successful application of Trichoderma could depend, on the long term, on the presence or absence of specific viruses which, in turn, affect their mycoparasitic or antifungal activity (Chun, Yang, and Kim 2018b; You et al. 2019).
In this work, we investigated the virome associated with a wide and diverse Trichoderma spp. collection to shed light on the diversity and distribution of fungal viruses, possibly paving the way for potential future biotechnological or agricultural applications. To this extent, we chose to exploit a HTS approach on the rRNA-depleted total RNA fraction obtained from our fungal collection. This method confirmed its capability to characterize fungal viromes of a vast collection of fungi, at low cost, allowing us to identify seventeen viral genomes and a total of twenty-five viral segments (none of them endogenized in the host genome) associated to 36 out of the 113 initial different Trichoderma isolates evaluated. Among these thirty-six isolates, the majority belonged to the T. harzianum species complex, which was the most represented group within the original collection, described in 2009 (Migheli et al. 2009). It is worth mentioning that five of the putative viruses we have identified (TgMBV1, ThNV1, TgNV1, ThaMOV1, and TtOV1) do not currently associate with a known CP and are not directly derived phylogenetically from ancestors coated by a capsid, implying that they are not to be considered ‘viruses’ according to the current definition.
Interestingly, among the total number of isolates described in the previous study (Migheli et al. 2009), the majority were positively identified as pan-European and/or pan-global Trichoderma species from sections Trichoderma and Pachybasium, with only one isolate representing a new, undescribed species belonging to the harzianum–catoptron clade (Migheli et al. 2009). Moreover, ITS analysis revealed only one potentially endemic ITS1 allele of T. hamatum, while all other species exhibited genotypes that were already present in Eurasia or in other continents (Migheli et al. 2009). Evidence pointed out to a significative decline for native Trichoderma-endemic populations of Sardinia, which were probably replaced by widespread invasion of species from Eurasia, Africa, and/or the Pacific Basin (Migheli et al. 2009). The same isolates sampled in Sardinia were also tested for their biological control properties on a Rhizoctonia solani/cotton model pathosystem, with a high proportion of the tested isolates (mainly belonging to T. gamsii) demonstrating remarkable antagonistic properties, leading to an almost complete control of the disease on artificially inoculated cotton seedlings (Scherm et al. 2009).
With respect to the geographical distribution of viral sequences, the highest number of virus-infected Trichoderma spp. was found in soils coded F1, EG2, EG5, and EG6, which correspond either to forest (F1) or EG (pasture) land (EG2, EG5, and EG6) (Migheli et al. 2009). No correlation could be found between viral distribution and abiotic factors, such as soil properties (carbon availability), altitude, climatic conditions, and ecosystem disturbance.
This is, to our knowledge, one of the few wide-ranging studies regarding the Trichoderma mycovirome present in the literature in terms of the number of isolates and diversity of species screened: the others include those conducted by Jom-in and Akarapisan (2009, 156 isolates screened); Liu et al. (2019b, 155 strains screened); and Yun et al. (2016, 315 strains screened). While the majority of studies on the Trichoderma virome focused on one or a few fungal strains always belonging to Asian populations of the fungus (Wu et al. 2020), the possibility to explore such a diverse collection in an European site allowed us to characterize both double-stranded and single-stranded viral strains (8 and 8, respectively) belonging to three of the major phyla constituting the RNA viral kingdom of Orthornavirae, thus further increasing our general knowledge with respect to the Trichoderma virome. This is particularly true for negative-sense viruses; in fact, to our knowledge, no negative-sense virus has been reported up to now in Trichoderma. Among the large diversity of viruses identified here, the absence of members of the Lenarviricota phylum (e.g. mitovirids, botourmiavirids, and narnavirids) is noteworthy, as they are generally the most represented ones within ascomycetes (Mu et al. 2018; Picarelli et al. 2019; Chiapello et al. 2020).
A third ORFan segment associated with TaPV1
Among the seventeen virus strains identified in our study, ThOCV1 and TaPV1 belonged to previously described species within the families Curvulaviridae and Partitiviridae, respectively (Chun, Yang, and Kim 2018a; Liu et al. 2019a).
With respect to TaPV1, Chun and colleagues published the complete bisegmented genome of TaPV1 after total RNA extraction starting from one unique isolate of Trichoderma atroviride (NFCF394) collected on substrates showing green mold symptoms in a Korean shiitake farm (Chun, Yang, and Kim 2018a). Curiously, in our analysis, we have identified a previously unreported third segment (TaPV1-RNA 3); this third segment was detected in all the five isolates carrying both TaPV1-RNA1 and RNA2 (Fig. S4, Table S5).
Interestingly, more examples of multi-segmented partitivirds (i.e. Partitiviridae members possessing more than the two canonical genomic segments) are starting to accumulate in the literature (Kim, Choi, and Lee 2005; Vainio et al. 2018; Jiang et al. 2019). Kuroki Misa and colleagues recently deposited the nt sequences of a partitivirus showing up to eight dsRNA segments (NCBI accessions from LC763222 to LC763246 and from LC763268 to LC763275), identified from four different Aspergillus flavus isolates. Moreover, additional satellite RNA segments have been found in association with helper viruses in the Partitiviridae (Shah, Kotta-Loizou, and Coutts 2015; Jiang et al. 2022), with the former being capable of reducing both pathogenicity and genomic accumulation of the respective helper virus (Jiang et al. 2022).
The presence of this virus in three distinct species from our collection (T. harzianum, T. tomentosum, and T. hamatum) and in T. atroviride from the Korean region of Gyeonggi-do (Chun, Yang, and Kim 2018a) led us to postulate that TaPV1 could possess an uncommon ability to overcome the species-specific barrier, potentially expanding its host range to several Trichoderma spp. Moreover, the possibility of detecting this virus within fungal isolates collected in both Asia (Korea) and Europe could suggest a long co-evolution history with the genus Trichoderma.
New negative-sense RNA viruses
Negative-sense viruses are a relatively recent discovery in fungi, and only a few of them have been characterized so far through virus purification and whole genome characterization (Liu et al. 2014).
In the Bunyavirales order, we characterized four novel viruses, but only one could be clearly assigned to an officially recognized family (TgClV1, Phenuiviridae), while TgMBV1 could accommodate within a recently well-defined clade of viruses (Picarelli et al. 2019). With respect to the remaining ThNV1 and TgNV1, they appear to belong to a distinct sister clade in the Bunyavirales order separate from taxonomically established families. We hypothesize that ThNV1 and TgNV1 (as well as a third virus from powdery mildew of grapevine lesion, still unpublished) should be accommodated in a new family as soon as more members are unveiled. Pairwise amino acid sequence comparison confirms the phylogenetic analysis (Table S6).
One of the most interesting Bunyavirales members found in our study is TgClV1, a tri-segmented virus. Each segment hosts a single ORF coding for a putative Bunya-RdRp, a putative N, and a hypothetical protein with identity to the putative MP of Botrytis cinerea bocivirus 1 (BcBV1), a recently described negative ssRNA related to cogu-like viruses. In their study, Ruiz-Padilla and colleagues describe BcBV1 as a virus closely related to plant coguviruses, and, after phylogenetic analysis using both the RdRp and the N protein, they placed BcBV1 in the same clade as the plant coguviruses (Ruiz-Padilla et al. 2021). Moreover, alignment of the hypothetical BcBV1 protein with the core domain of the 30K viral MP of Laurel Lake virus, Grapevine-associated cogu-like virus 2, and Grapevine-associated cogu-like virus 3 (all currently assigned to Laulavirus genus) showed high conservation in this region which led the authors to suggest that this hypothetical protein could be an ancient MP, that is probably not functional in BcBV1 (Ruiz-Padilla et al. 2021). Since fungal viruses do not typically possess MPs, likely because fungal hyphae have cytoplasmic continuity, Padilla and colleagues postulate a possible cross-kingdom event where an ancient plant virus, coinfecting a plant host with B. cinerea, was transferred from the plant host to the fungus (Ruiz-Padilla et al. 2021). It would be of great interest to collect more reliable experimental evidence and shed light on the possible functional role for these hypothetical proteins of BcBV1 and TgClV1.
The fact that the majority of bunyavirals characterized in this study (apart from TgClV1) did not possess any additional segment encoding for an N protein is not surprising since numerous examples can be found in the literature (Donaire, Pagán, and Ayllón 2016; Marzano and Domier 2016; Chiapello et al. 2020; Botella and Jung 2021). Besides the obvious reasons linked to the intrinsic limitation of homology-based detection methods, the absence of N-coding segments could also be explained by the fact that all these bunya-like viruses possess a modified version of their RdRp that does not need any N nor the common RNPs formation, to achieve a successful replication.
Regarding viruses assigned to the Mononegavirales order, our phylogenetic analysis clearly accommodates ThNV2 within the Sclerotimonavirus genus (Mymonaviridae family), while ThMV1 and ThMV2 fall within the Plasmopamonavirus genus (Mymonaviridae family). ICTV taxonomy rules for members of Mymonaviridae (proposal code: 2020.004F) define the demarcation threshold level at 80 per cent and at 32 per cent of aa sequence identity of the L-protein for the species and for the genus level, respectively. Consequently, we can clearly state that ThNV2 represents a novel species of Sclerotimonavirus, while ThMV1 and ThMV2 are novel species of Plasmopamonavirus.
Finally, for the only identified member of the Serpentovirales order (ThaMOV1), we are confident about its accommodation within the recently suggested ‘mycoaspiviridae’ group, described by Chiapello et al. (2020).
The first bisegmented Mononegavirales infecting fungi
Within the order Mononegavirales, members possessing a bisegmented genome are quite rare and mainly belong to the Dichorhavirus and Varicosavirus genera (Rhabdoviridae family Betarhabdovirinae subfamily) and are transmitted by false spider mites (Dichorhavirus) or chytridiomycetes (Varicosavirus) (Kondo et al. 2006; Ramos-González et al. 2017).
In this study, we present strong evidence for the presence of bipartite viruses also within the Mymonaviridae family, specifically within the Plasmopamonavirus and Penicillimonavirus genera. Curiously, among these newly identified bisegmented viruses, two sub-groups could be observed, with the first sub-group (PvLaMAV1, PvLaMAV3, PaNSV1, and PgNSV1) hosting five ORFs on their RNA2, and a second sub-group including viruses possessing only four ORFs on the same segment (PvLaMAV2, 4, 5, 6, 7, and 9). For the majority of viruses present in this second sub-group, an additional ORF is carried instead in sense orientation on RNA1 and, despite all having a similar length, they do not show a strong degree of aa sequence conservation when compared to RNA2-ORF5 of the first sub-group. The remaining PvLaMAV8 and ThMV2 constitute a third sub-group which carries six ORFs on their second segment. Interestingly, the basal branch of this group consists of the monosegmented ThMV1, therefore giving us indirect evidence of a relatively recent transition from monosegmented to bisegmented genome organization inside the Mononegavirales.
Our collection of RNA2 from different mymonavirids allowed us to perform an in-silico structural analysis that identified a conserved N domain that could not be found by similarity searches.
Besides clear evidence of sequence conservation between ORFs encoding the putative N among all these bisegmented viruses (ORF2 on RNA2), it is interesting that sequence conservation can also be found when comparing ORF1 (carried in the last position on the 3′-end of RNA 2) and ORF3 (in the third to last position on 3′-end) only within the three above-mentioned RNA2 sub-groups (i.e. those hosting 4, 5, or 6 ORFs on their second segment).
In general terms, multipartite genomes have been considered less efficient in terms of transmission efficiency (cell to cell and from host to host) with respect to monopartite genomes, due to the fact that the probability of infection for each of their multiple virus particles is lower when compared with their monopartite counterparts and one or more segments could be lost during transmission (Sicard et al. 2013; Lucía-Sanz and Manrubia 2017). On the other hand, the hypothesis that multi-segmentation might be advantageous since it allows for the rapid tuning of gene expression has recently been gaining acceptance (Sicard et al. 2013, 2016), particularly after the introduction of model simulations for evaluating the effect of selective pressures applied on the genome formula (the set of genome–segment frequencies for all genome) of multipartite viruses (Zwart and Elena 2020). A recent study further supports the hypothesis that a possible advantage of having a partitioned genome is the increased ability to quickly modulate gene expression in highly variable environments, which require rapid adaptation responses. In fact, researchers propose a scenario where the availability of a broad range of well-adapted hosts across multiple environments could contribute to the evolution of multipartite viruses (Zwart and Elena 2020). This last hypothesis finds further support when considering the evidence reported here of a bisegmented mymonavirus (ThMV2) infecting a Trichoderma host, a ubiquitous opportunistic fungus known to possess exceptional adaptation capabilities.
New dsRNA viruses
Viruses with a dsRNA genome are currently grouped into two main lineages, due to the recent taxonomical revision of RNA viruses: the Duplopiviricetes class (phylum Pisuviricota) and the Duplornaviricota phylum (Koonin et al. 2020) meaning that dsRNA viruses are not monophyletic. We have identified a total of six novel dsRNA viruses, three belonging to the Duplopiviricetes class (Pisuviricota) and three to the Chrymotiviricetes class (Duplornaviricota).
In the first case, after phylogenetic analysis and pairwise sequence alignment, the three dsRNA viruses TgAPV1, ThPV3, and ThDSV1 clustered within the Alphapartitivirus, Gammapartitivirus, and Orthocurvulavirus genera, respectively. TgAPV1 and ThPV3 fulfill the genus and species demarcation criteria currently adopted for the Partitiviridae family (Vainio et al. 2018), and for ThDSV1, our phylogenetic analysis clearly assigns it to the Orthocurvulavirus genus (Curvulaviridae family) since an 85 per cent RdRp aa identity is defined as the species demarcation criteria in the Orthocurvulavirus approved taxonomy proposal (number: 2020.002F).
With respect to the other group of dsRNA viruses (Chrymotiviricetes class), we have identified three novel members of the Ghabrivirales order named ThaDSV1, ThDSV2, and ThDSV3. Despite the fact that they could not be indicated as bona fide Totivirus members, our phylogenetic analysis located the latter of the three (ThDSV3) in a previously reported unrecognized clade of totiviruses (unclassified totiviruses—clade 1) (Chiapello et al. 2020; Botella and Jung 2021). Noticeably, within the latter clade, we observed two distinct sub-groups, with a clear difference in host range: one accommodating viruses mainly infecting sea-mosses and green algae and the other including invertebrate- or oomycete-infecting viruses. ThDSV3 seems to constitute an additional third sub-group which, pending further evidence, could eventually be recognized as a distinct group of viruses with specific hosts if new members infecting fungi are discovered. Moreover, this virus was also the most common within our collection, and it was found in association with eight isolates, belonging to three different Trichoderma spp.: T. harzianum (4 isolates), T. gamsii (3 isolates), and T. samuelsii (1 isolate). These results suggest that, besides TaPV1, ThDSV3 could also accommodate a wide spectrum of hosts, likely due to its capability of inter- and intra-species transmission and could be quite frequent within Sardinian populations of host fungi.
ThDSV2 and ThaDSV1 are, to some extent, closely related to members of the Megabirnaviridae and Chrysoviridae families (Fig. S7), yet they constitute a distinct and well-supported clade for which the recognition as novel family has been proposed a few years ago, under the name ‘fusagraviridae’ (Wang et al. 2016). According to the researchers, ‘fusagravirids’ can be easily distinguished from other known viral families on account of the length of their monopartite genomes (8112∼9518 bp), their characteristic genomic structure containing a putative Programmed-1 Ribosomal Frameshifting (–1 PRF) translational recoding mechanism (allowed by the presence of a shifty heptameric sequence, typically ‘GGAAAAC’), a long 5′- untranslated region (UTR) (865–1,310 bp), a relatively short 3′-UTR (7–131 bp), and the presence of ‘Phytoreo_S7’ and RdRp domains (Wang et al. 2016). Most of these features can also be found in our ThaDSV1 and ThDSV2 genomes since both have a similar genome length, a putative –1 PRF motif, immediately before the first ORF stop codon (‘GGAAAAC’ at nt 5436–5442, UAG at 5445 for ThaDSV1; ‘GGAAAAC’ at nt 4512–4518, UAG at 4521 for ThDSV2), a short 3′-UTR (27 and 21 bp, respectively) and the ‘Viral RdRp domain’ (pfam02123) on the second ORF of the genome (Wang et al. 2016). The long 5′-UTR could be found only in ThaDSV1 (900 bp), while in ThDSV2, it was extremely short (33 bp). However, some unclassified fungal viruses closely related to the ‘fusagraviridae’ group, such as Diplodia scrobiculata RNA virus 1 and Trichoderma asperellum dsRNA virus 1 (TaMV1) also contain a short 5′-UTR of 29 and 85 bp, respectively. Thus, ThDSV2 may potentially represent further evidence for the establishment of a new putative genus within the ‘fusagraviridae’ suggested family (Wang et al. 2016; Lee et al. 2017). Finally, even though no ‘Phytoreo_S7’ (pfam07236) domain was found on the RdRp-coding ORFs of ThDSV2 and ThaDSV1 after motif search analysis, multiple alignment of RdRp aminoacid sequences from different ‘fusagraviridae’ members highlighted a high degree of conservation within the region hosting the ‘Phytoreo_S7’ domain (Fig. S13). In conclusion, we are confident that both ThaDSV1 and ThDSV2 could be considered new representative members of this tentatively proposed family ‘fusagraviridae’.
Conclusions
Results gathered from this work are just an initial step toward the understanding of the intricate mycovirome associated with Trichoderma. Nevertheless, they could contribute to further knowledge acquisition from both a plain biological standpoint and an agronomical and biotechnological application perspective.
This will potentially lead to the identification of novel Trichoderma-based bio-control agents and eventually a better understanding of the ecology of both Trichoderma communities and their associated virome, within natural or artificial ecosystems.
Supplementary Material
Acknowledgements
The authors thank Anthony Taylor from Pennsylvania State University for reading and editing the manuscript.
Contributor Information
Saul Pagnoni, Department of Agricultural and Environmental Sciences—Production, Landscape, Agroenergy, University of Milan, via Celoria 2, Milan 20133, Italy.
Safa Oufensou, Department of Agricultural Sciences and NRD—Desertification Research Center, University of Sassari, Viale Italia 39a, Sassari, Sardegna 07100, Italy.
Virgilio Balmas, Department of Agricultural Sciences and NRD—Desertification Research Center, University of Sassari, Viale Italia 39a, Sassari, Sardegna 07100, Italy.
Daniela Bulgari, Department of Molecular and Translational Medicine, University of Brescia, Viale Europa 11, Brescia 25123, Italy.
Emanuela Gobbi, Department of Molecular and Translational Medicine, University of Brescia, Viale Europa 11, Brescia 25123, Italy.
Marco Forgia, Institute for Sustainable Plant Protection, National Research Council of Italy, Strada delle Cacce, 73, Torino 10135, Italy.
Quirico Migheli, Department of Agricultural Sciences and NRD—Desertification Research Center, University of Sassari, Viale Italia 39a, Sassari, Sardegna 07100, Italy.
Massimo Turina, Institute for Sustainable Plant Protection, National Research Council of Italy, Strada delle Cacce, 73, Torino 10135, Italy.
Supplementary data
Supplementary data are available at Virus Evolution online.
Data availability
All the raw reads generated have been deposited in the Sequence Read Archive: Bioproject PRJNA936709; Biosamples SAMN33361767 and SAMN33361768; and accessions SRR23531244 and SRR23531245.
Conflict of interest:
None declared.
References
- Abbey J. A. et al. (2019) ‘Biofungicides as Alternative to Synthetic Fungicide Control of Grey Mould (Botrytis Cinerea) – Prospects and Challenges’, Biocontrol Science and Technology, 29: 207–28. [Google Scholar]
- Alias C., Bulgari D., and Gobbi E. (2022) ‘It Works! Organic-Waste-Assisted Trichoderma spp. Solid-State Fermentation on Agricultural Digestate’, Microorganisms, 10: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botella L., and Jung T. (2021) ‘Multiple Viral Infections Detected in Phytophthora condilina by Total and Small RNA Sequencing’, Viruses, 13: 620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns T. D., Lee S. B., and Taylor J. W. (1990) ‘Amplification and direct sequencing of fungal ribosomal RNA Genes for phylogenetics’, in: M. A. Innis, D. H. Gelfand, J. J. Sninsky and T. J. White (eds) PCR Protocols: A Guide to Methods and Applications, pp. 315–22. London UK: Academic Press. [Google Scholar]
- Cai F. et al. (2022) The Current State of Trichoderma Taxonomy and Species Identification‘’, in: N. Amaresan, A. Sankaranarayanan and I. S. Druzinina (eds) Advances in Trichoderma Biology for Agricultural Applications, pp. 3–35. Cham (CH): Springer Nature. [Google Scholar]
- Chiapello M. et al. (2020) ‘Analysis of the Virome Associated to Grapevine Downy Mildew Lesions Reveals New Mycovirus Lineages’, Virus Evolution, 6: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun J. et al. (2022) ‘Molecular Characteristics of a Novel Hypovirus from Trichoderma harzianum’, Archives of Virology, 167: 233–8. [DOI] [PubMed] [Google Scholar]
- Chun J., Yang H.-E.-E., and Kim D.-H.-H. (2018a) ‘Identification and Molecular Characterization of a Novel Partitivirus from Trichoderma atroviride NFCF394’, Viruses, 10: 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun J., Yang H.-E., and Kim D.-H. (2018b) ‘Identification of a Novel Partitivirus of Trichoderma harzianum NFCF319 and Evidence for the Related Antifungal Activity’, Frontiers in Plant Science, 9: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaire L., Pagán I., and Ayllón M. A. (2016) ‘Characterization of Botrytis cinerea Negative-stranded RNA Virus 1, a New Mycovirus Related to Plant Viruses, and a Reconstruction of Host Pattern Evolution in Negative-sense ssRNA Viruses’, Virology, 499: 212–8. [DOI] [PubMed] [Google Scholar]
- Dos Santos U. R., and Dos Santos J. L. (2023) ‘Trichoderma after Crossing Kingdoms: Infections in Human Populations’, Journal of Toxicology and Environmental Health, Part B, 26: 97–126. [DOI] [PubMed] [Google Scholar]
- Ferron F. et al. (2017) ‘Transcription and Replication Mechanisms of Bunyaviridae and Arenaviridae L Proteins’, Virus Research, 234: 118–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forgia M. et al. (2022) ‘Three New Clades of Putative Viral RNA-dependent RNA Polymerases with Rare or Unique Catalytic Triads Discovered in Libraries of ORFans from Powdery Mildews and the Yeast of Oenological Interest Starmerella bacillaris’, Virus Evolution, 8: veac038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghabrial S. A. et al. (2015) ‘50-Plus Years of Fungal Viruses’, Virology, 479–480: 356–68. [DOI] [PubMed] [Google Scholar]
- Gilbert K. B. et al. (2019) ‘Hiding in Plain Sight: New Virus Genomes Discovered via a Systematic Analysis of Fungal Public Transcriptomes’, PLoS One, 14: 1–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas B. J. et al. (2013) ‘De Novo Transcript Sequence Reconstruction from RNA-seq Using the Trinity Platform for Reference Generation and Analysis’, Nature Protocols, 8: 1494–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaklitsch W. M. et al. (2013) ‘Disentangling the Trichoderma viridescens Complex’, Persoonia: Molecular Phylogeny and Evolution of Fungi, 31: 112–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y. et al. (2019) ‘Molecular Characterization of a Debilitation-associated Partitivirus Infecting the Pathogenic Fungus Aspergillus flavus’, Frontiers in Microbiology, 10: 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2022) ‘A Satellite dsRNA Attenuates the Induction of Helper Virus-Mediated Symptoms in Aspergillus flavus’, Frontiers in Microbiology, 13: 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jom-in S., and Akarapisan A. (2009) ‘Characterization of Double-stranded RNA in Trichoderma spp. Isolates in Chiang Mai Province’, Journal of Agricultural Technology, 5: 261–70. [Google Scholar]
- Kalyaanamoorthy S. et al. (2017) ‘ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates’, Nature Methods, 14: 587–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J. W., Choi E. Y., and Lee J. I. (2005) ‘Genome Organization and Expression of the Penicillium stoloniferum Virus F’, Virus Genes, 31: 175–83. [DOI] [PubMed] [Google Scholar]
- Kondo H. et al. (2006) ‘Orchid Fleck Virus Is a Rhabdovirus with an Unusual Bipartite Genome’, Journal of General Virology, 87: 2413–21. [DOI] [PubMed] [Google Scholar]
- Kondo H., Botella L., and Suzuki N. (2022) ‘Mycovirus Diversity and Evolution Revealed/Inferred from Recent Studies’, Annual Review of Phytopathology, 60: 307–36. [DOI] [PubMed] [Google Scholar]
- Koonin E. V. et al. (2020) ‘Global Organization and Proposed Megataxonomy of the Virus World’, Microbiology and Molecular Biology Reviews, 84: e00061–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kredics L. et al. (2003) ‘Clinical Importance of the Genus Trichoderma’, Acta Microbiologica et Immunologica Hungarica, 50: 105–17. [DOI] [PubMed] [Google Scholar]
- Langmead B., and Salzberg S. L. (2012) ‘Fast Gapped-read Alignment with Bowtie 2’, Nature Methods, 9: 357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. H. et al. (2017) ‘Characterization of A Novel dsRNA Mycovirus of Trichoderma atroviride NFCF028’, Archives of Virology, 162: 1073–7. [DOI] [PubMed] [Google Scholar]
- Li H. et al. (2009) ‘The Sequence Alignment/Map Format and SAMtools’, Bioinformatics, 25: 2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.-H. et al. (2019) ‘Two Novel Fungal Negative-strand RNA Viruses Related to Mymonaviruses and Phenuiviruses in the Shiitake Mushroom (Lentinula edodes)’, Virology, 533: 125–36. [DOI] [PubMed] [Google Scholar]
- Liu L. et al. (2014) ‘Fungal Negative-stranded RNA Virus that Is Related to Bornaviruses and Nyaviruses’, Proceedings of the National Academy of Sciences, 111: 12205–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. et al. (2019a) ‘Complete Nucleotide Sequence of a Novel Mycovirus from Trichoderma harzianum in China’, Archives of Virology, 164: 1213–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. et al. (2019b) ‘A Novel Double-stranded RNA Mycovirus Isolated from Trichoderma harzianum’, Virology Journal, 16: 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucía-Sanz A., and Manrubia S. (2017) ‘Multipartite Viruses: Adaptive Trick or Evolutionary Treat?’, Npj Systems Biology and Applications, 3: 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeira F. et al. (2019) ‘The EMBL-EBI Search and Sequence Analysis Tools APIs in 2019’, Nucleic Acids Research, 47: W636–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzano S.-Y. L., and Domier L. L. (2016) ‘Novel Mycoviruses Discovered from Metatranscriptomics Survey of Soybean Phyllosphere Phytobiomes’, Virus Research, 213: 332–42. [DOI] [PubMed] [Google Scholar]
- Migheli Q. et al. (2009) ‘Soils of a Mediterranean Hot Spot of Biodiversity and Endemism (Sardinia, Tyrrhenian Islands) are Inhabited by pan-European, Invasive Species of Hypocrea/Trichoderma’, Environmental Microbiology, 11: 35–46. [DOI] [PubMed] [Google Scholar]
- Mirdita M. et al. (2022) ‘ColabFold: Making Protein Folding Accessible to All’, Nature Methods, 19: 679–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu F. et al. (2018) ‘Virome Characterization of a Collection of S. sclerotiorum from Australia’, Frontiers in Microbiology, 8: 2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee P. K. et al. (2013) in: Mukherjee Prasun K, Horwitz Benjamin, Singh Uma Shnkar, Mukherjee Mala and Schmoll Monika (eds) Trichoderma: Biology and Applications, pp. 230–3. Wallingford UK: CABI. [Google Scholar]
- Nerva L. et al. (2019) ‘Isolation, Molecular Characterization and Virome Analysis of Culturable Wood Fungal Endophytes in Esca Symptomatic and Asymptomatic Grapevine Plants’, Environmental Microbiology, 21: 2886–904. [DOI] [PubMed] [Google Scholar]
- Pettersen E. F. et al. (2021) ‘UCSF ChimeraX: Structure Visualization for Researchers, Educators, and Developers’, Protein Science, 30: 70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picarelli M. A. et al. (2019) ‘Extreme Diversity of Mycoviruses Present in Isolates of Rhizoctonia solani AG2-2 LP from Zoysia japonica from Brazil’, Frontiers in Cellular and Infection Microbiology, 9: 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-González P. L. et al. (2017) ‘Citrus Leprosis Virus N: A New Dichorhavirus Causing Citrus Leprosis Disease’, Phytopathology, 107: 963–76. [DOI] [PubMed] [Google Scholar]
- Rastgou M. et al. (2009) ‘Molecular Characterization of the Plant Virus Genus Ourmiavirus and Evidence of Inter-kingdom Reassortment of Viral Genome Segments as Its Possible Route of Origin’, Journal of General Virology, 90: 2525–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Padilla A. et al. (2021) ‘Novel Mycoviruses Discovered in the Mycovirome of a Necrotrophic Fungus’, MBio, 12: e03705–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherm B. et al. (2009) ‘Identification of Potential Marker Genes for Trichoderma harzianum Strains with High Antagonistic Potential against Rhizoctonia solani by a Rapid Subtraction Hybridization Approach’ Current Genetics, 55: 81–91. [DOI] [PubMed] [Google Scholar]
- Shah U. A., Kotta-Loizou I., and Coutts R. H. A. (2015) ‘Sequence Determination of a Satellite RNA Isolated from Aspergillus foetidus’, Archives of Virology, 160: 883–5. [DOI] [PubMed] [Google Scholar]
- Sicard A. et al. (2016) ‘The Strange Lifestyle of Multipartite Viruses’, PLoS Pathogens, 12: 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ——— et al. (2013) ‘Gene Copy Number Is Differentially Regulated in a Multipartite Virus’, Nature Communications, 4: 1–8. [DOI] [PubMed] [Google Scholar]
- Sievers F. et al. (2011) ‘Fast, Scalable Generation of High-quality Protein Multiple Sequence Alignments Using Clustal Omega’, Molecular Systems Biology, 7: 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trifinopoulos J. et al. (2016) ‘W-IQ-TREE: A Fast Online Phylogenetic Tool for Maximum Likelihood Analysis’, Nucleic Acids Research, 44: W232–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi P. et al. (2013) ‘Trichoderma: A Potential Bioremediator for Environmental Clean Up’, Clean Technologies and Environmental Policy, 15: 541–50. [Google Scholar]
- Vainio E. J. et al. (2018) ‘ICTV Virus Taxonomy Profile: Partitiviridae’, Journal of General Virology, 99: 17–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velasco L. et al. (2019) ‘Viromes in Xylariaceae fungi infecting avocado in Spain’, Virology, 532: 11–21. [DOI] [PubMed] [Google Scholar]
- Wang L. et al. (2016) ‘Two Novel Relative Double-stranded RNA Mycoviruses Infecting Fusarium poae Strain SX63’, International Journal of Molecular Sciences, 17: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. et al. (2021) ‘Divergent RNA Viruses in Macrophomina phaseolina Exhibit Potential as Virocontrol Agents’, Virus Evolution, 7: 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R. et al. (2022) ‘The Newly Identified Trichoderma harzianum Partitivirus (ThPV2) Does Not Diminish Spore Production and Biocontrol Activity of Its Host’, Viruses, 14: 1532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu B. et al. (2020) ‘The Insight of Mycovirus from Trichoderma spp.’, Agricultural Research & Technology, 24: 1–4. [Google Scholar]
- You J. et al. (2019) ‘Defective RNA of a Novel Mycovirus with High Transmissibility Detrimental to Biocontrol Properties of Trichoderma spp.’, Microorganisms, 7: 507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun S.-H. et al. (2016) ‘Incidence of Diverse dsRNA Mycoviruses in Trichoderma spp. Causing Green Mold Disease of Shiitake Lentinula edodes’, (R. Boden, Ed.)FEMS Microbiology Letters, 363: fnw220. [DOI] [PubMed] [Google Scholar]
- Zhang T. et al. (2018) ‘Molecular Characterization of a Novel Double-stranded RNA Mycovirus of Trichoderma asperellum Strain JLM45-3’, Archives of Virology, 163: 3433–7. [DOI] [PubMed] [Google Scholar]
- Zin N. A., and Badaluddin N. A. (2020) ‘Biological Functions of Trichoderma spp. for Agriculture Applications’, Annals of Agricultural Sciences, 65: 168–78 [Google Scholar]
- Zwart M. P., and Elena S. F. (2020) ‘Modeling Multipartite Virus Evolution: The Genome Formula Facilitates Rapid Adaptation to Heterogeneous Environments’, Virus Evolution, 6: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the raw reads generated have been deposited in the Sequence Read Archive: Bioproject PRJNA936709; Biosamples SAMN33361767 and SAMN33361768; and accessions SRR23531244 and SRR23531245.