

Abstract
B cells are the antibody-producing arm of the adaptive immune system and play a critical role in controlling pathogens. Several groups have now demonstrated the feasibility of using engineered B cells as a therapy, including infectious disease control and gene therapy of serum deficiencies. These studies have largely utilized ex vivo modification of the cells. Direct in vivo engineering would be of utility to the field, particularly in infectious disease control where the infrastructure needs of ex vivo cell modification would make a broad vaccination campaign highly challenging. In this study we demonstrate that engineered adenoviral vectors are capable of efficiently transducing murine and human primary B cells both ex vivo and in vivo. We found that unmodified human adenovirus C5 was capable of infecting B cells in vivo, likely due to interactions between the virus penton base protein and integrins. We further describe vector modification with B cell-specific gene promoters and successfully restrict transgene expression to B cells, resulting in a strong reduction in gene expression from the liver, the main site of human adenovirus C5 infection in vivo.
Keywords: B lymphocytes, B cells, gene delivery, adenovirus, vectors, targeting
Graphical abstract
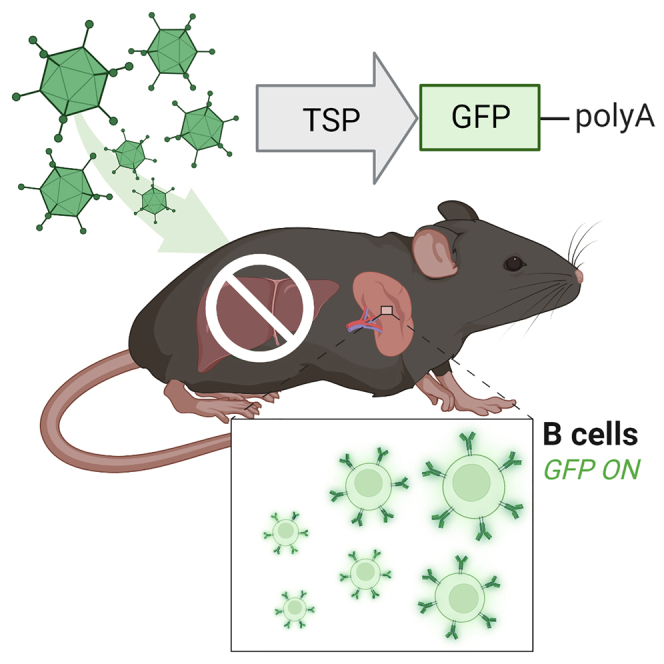
Curiel and colleagues demonstrate that adenoviral vectors could achieve specific B cell gene transfer in vivo in this study. This could lead to direct in vivo genetic modification of B cells by circumventing challenging ex vivo protocols, potentially enabling the development of new B cell therapies.
Introduction
Cellular therapy using genetically modified T lymphocytes (CAR-T cells) has resulted in several clinical successes in the treatment of cancer, with drugs approved for several hematological malignancies.1 These medicines reprogram a patient’s own lymphocytes to target cancer cells, potentially resulting in clearance of the disease. Despite these successes, several challenges remain, including engraftment rejection, limited efficacy in solid tumors, and toxicities such as cytokine release syndrome.2 These difficulties and the potential to expand the reach of cellular medicine have generated interest in using other cell types as therapies. Several alternatives have been explored, including NK cells, non-canonical T cells such as γδ T cells, and others.3
In recent years, B lymphocytes (B cells) have become the subject of active investigation. B cells are part of the adaptive immune system and are defined by the expression of a membrane-tethered antibody, known as the B cell receptor or BCR. Upon binding of the BCR to a cognate antigen in conjunction with various co-stimulatory signals, B cells proliferate and differentiate into memory B cells and plasma cells. These cell populations possess several features that are critical to pathogen control, namely (1) the capacity to be maintained throughout the life of the organism, (2) the ability to secrete large amounts of antibodies, and (3) the capability to rapidly respond to antigen re-exposure via massive proliferation of memory B cells and production of antibodies.4 These same abilities also make B cells highly interesting targets for genetic modification, particularly for control of infectious disease—their highly specialized ability to produce and secrete large amounts of immunoglobulin potentially enables applications beyond the reach of other T cell-based therapies.5 Furthermore, as B cells generally do not directly kill cells, it is conceivable that B cell-based therapies will be subject to fewer toxicities than those utilizing T cells—although an important consideration is the production of anti-host antibodies, which could result in autoimmune disorders.6 In total, these unique features and therapeutic possibilities make B cells a highly attractive target for a new range of medicines. Toward this end, several groups have now demonstrated the use of CRISPR-Cas technology to alter the BCR, replacing the endogenous receptor with an engineered one, potentially resulting in control of previously untreatable illnesses such as HIV.7,8,9,10,11
These studies have largely leveraged ex vivo techniques to achieve BCR modification. Of note, ex vivo engineering typically involves taking a sample from the patient, purifying and culturing the cell of interest, modifying the cells using a vector, and finally returning the cells to the patient.12 While this technique has become the clinical standard for cell therapy, it requires a complex and costly infrastructure. If a cell therapy is to be used for infectious disease control, such as in the case of engineered B cells, deploying ex vivo modification at the scale necessary is likely to be challenging and require specialized technology. In contrast, direct in vivo cell modification involves the delivery of a targeted vector to the cells of interest in the patient. The vector carries the necessary cell engineering machinery (such as the Cas9 protein, guide RNA, and donor template DNA) required for cell modification and generates the desired cell changes in situ. This technique could circumvent the technical and infrastructure challenges involved in ex vivo cell engineering and allow novel medicines to be developed and delivered to populations previously unreachable by cell therapy.13 A few works have explored the utility of this technology for T cell modification, and one group recently demonstrated the use of adeno-associated vectors to generate BCR-modified B cells in vivo.14,15,16,17,18,19
Vectors designed for in vivo gene delivery must meet several stringent requirements, ideally including safety and tolerability, low-to-no gene expression in off-target tissues or cells, and sufficient capacity to deliver the genes required by the therapy. With these goals in mind, we explored the use of adenoviral vectors (Ads) to achieve gene transfer to B cells in vivo. Ads possess several features that make them well suited to meet the criteria for in vivo cell modification, including their large packaging capacity (up to 35 kB), their ability to be rationally engineered to infect targeted cell types, and their proven safety record in the clinic with systemic and local administration.20,21,22,23 This technology has been exploited by our group and others for several applications, including in vivo transduction of hematopoietic stem cells.24,25,26 In addition, the SARS-CoV-2 pandemic demonstrated the ability of the current infrastructure to produce Ads at a global scale at low cost, making them an ideal vector for infectious disease control.27,28 In this study, we therefore genetically engineered Ads for gene transfer to B cells and demonstrated their ability to achieve highly specific gene expression in vivo.
Results
Development of transcriptionally targeted Ad vectors
We have developed a “triple targeting” strategy for the generation of highly targeted vectors, which involves (1) fiber modifications to enhance gene transfer to the cell type of interest, (2) the use of a cell-specific promoter, and (3) liver un-targeting of the vector through hexon modifications that ablate binding of the virus to serum factors responsible for liver tropism.29 To initiate development of such a system for B cells, we first screened several vectors developed in our lab including AdRGD, AdPK4, and SAd36 against an Ad5 control for their ability to infect both resting and LPS-activated murine primary B cells (Figure S1A). Standard human adenovirus serotype C5 (Ad5), the most commonly used Ad vector, infects cells by first binding the coxsackie and adenovirus receptor (CAR) through its fiber protein. AdRGD is isogenic to Ad5 but contains an integrin binding peptide in the HI loop domain of the fiber protein that expands vector tropism to both CAR+ and integrin+ cells.30 AdPK4 contains the wild-type Ad5 fiber tail and shaft domains to dock the protein with the virus capsid but swaps the fiber head domain responsible for binding to cells with the corresponding domain from porcine adenovirus 4. This modification has been demonstrated to retarget the virus toward glycans.31 SAd36 is a vector based on chimpanzee adenovirus 36 for which the target receptor has not yet been determined.32 We found that AdRGD and AdPK4 were both capable of enhancing gene transfer into B cells by about 2- to 3-fold compared with the Ad5 control, reaching about 30% eGFP+ cells at higher multiplicities of infection (MOIs). Given that we observed the B cells undergoing roughly one doubling between vector delivery and analysis, we believe these results indicate that we infected a majority of the B cells. We found this acceptable as a starting point and selected the AdRGD backbone for further development due to its well-characterized cell entry biology, ease of upscale, and previous use in the clinic.33
We next selected a series of promoters characterized in the literature as driving strong and/or specific gene expression in B cells or other hematopoietic cells, including the spleen focus forming virus (SFFV) promoter, previously described synthetic promoters EEK and MH based on endogenous human immunoglobulin promoters, and the Epstein-Barr virus Wp1168 promoter34,35,36 (Figure 1A). We generated and attempted to rescue and upscale AdRGD vectors with each promoter driving eGFP from the E1 region of the vector. We were able to purify vectors containing all the described promoters at similar yields to the cytomegalovirus (CMV) promoter control.
Figure 1.

Assessment of promoter-modified vectors in B cells ex vivo
(A) Vector schematic. All vectors were based on first-generation E1/E3 deleted Ads and expressed eGFP from the E1 region. Ad5.CMV is HAdV-C5 with the ubiquitous cytomegalovirus (CMV) promoter. AdRGD contains the RGD4C peptide inserted in the virus fiber knob HI loop region to expand vector tropism toward integrins. A molecular model of this modification was generated using AlphaFold 2.0 and is shown in the inset.37 Model visualization was carried out using UCSF ChimeraX.38 (B) Assessment of promoter-modified vectors in murine B cells ex vivo. Vectors were first screened at 1,000 MOI (left panels), and standout promoters were then assessed at increasing MOIs and vector toxicity was assessed using flow cytometry viability dyes (right panels). For the left panels, six total technical replicates were performed across two separate experiments, except for AdRGD.CMV, which contained five total technical replicates. For the right panel, two replicates were carried out. Statistical analysis was carried out using ordinary one-way ANOVA with Tukey’s multiple comparisons test. (C) Assessment of vectors in human B cells ex vivo. Panels are as described in (B). Two total replicates were performed across two separate experiments. In all cases, eGFP expression is normalized to a PBS mock control. All data are expressed as means ± SD.
We initially assessed these vectors ex vivo in activated primary mouse and human B cells and found that vectors carrying the targeted promoters were all capable of driving reasonable gene expression, often >50% of the CMV control (Figures 1B and 1C). In an initial screen at a single MOI, we found that EEK and MH promoters showed significantly higher numbers of GFP+ cells than the other promoters (other than the AdRGD.CMV control) and the highest mean fluorescence intensity (MFI) in those cells (Figures 1B and 1C, left panels). We selected the EEK vector for further experiments as it trended slightly toward the highest number of eGFP+ cells and screened the vector with the Ad5.CMV and AdRGD.CMV controls at varying MOIs. We obtained similar results across multiple MOIs, although the EEK promoter did appear to show a more limited range of infectivity than the CMV vectors (Figures 1B and 1C, right panels). We confirmed that these trends held true in human B cells across two separate donors. Interestingly, in human B cells a much higher concentration of viral particles was required to achieve similar gene expression. It is unclear if this characteristic is due to an intrinsic difference between the cells or due to differences in the activation and culturing protocols used (LPS in mouse versus CD40 + interleukins + CpG DNA in human). Importantly, no vector showed signs of toxicity in mouse or human cells even at high MOIs, at least based on flow cytometry viability dye binding (Figures 1B and 1C, rightmost panels). Given that EEK appeared to be the standout promoter in both mouse and human cells in terms of the number of GFP+ cells, we developed a fully triple targeted vector based on this backbone, inserting the previously described H5/H3 hexon modification for liver un-targeting.29
We next injected C57BL/6J mice retro-orbitally with 1 × 1011 vp/mouse and harvested major organs (liver, spleen, lungs, heart, and kidneys) 72 h post-injection. One-half of each spleen was assessed for B cell eGFP expression using flow cytometry, while the other half and remaining organs were assessed for tissue gene expression using a fluorimetric plate assay (Figure 2A). In vivo, we were surprised to find that the Ad5.CMV control achieved statistically identical B cell gene expression levels to the targeted AdRGD.CMV vector—in addition, quantitative tissue data showed no difference between either vector in the liver (Figures 2B–2D). As expected given the strong liver tropism of Ad5, both Ad5.CMV and AdRGD.CMV showed strong gene expression in the liver, followed by the spleen, lung, heart, and kidneys (Figure 2D). In contrast, the AdRGD.EEK vector showed significantly reduced gene expression in B cells—a roughly 6-fold decrease compared with AdRGD.CMV (Figures 2B and 2C). However, we observed dramatically reduced eGFP expression in off-target tissues, including near-undetectable expression in the liver, ultimately resulting in a much higher targeting ratio and demonstrating the B cell specificity of this promoter. We also assessed gene expression in T cells via flow cytometry. Although we did detect eGFP+ cells in the AdRGD.EEK group, the MFI was much lower than in control groups (Figure 2B, right). We did not observe any significant differences between the AdRGD.EEK and AdH5/H3RGD.EEK vectors in B cell gene transfer, potentially indicating that the liver sequestration of vector particles does not impact gene transfer in the spleen, at least to B cells (Figure 2C).
Figure 2.

In vivo assessment of promoter-modified vectors
(A) Experimental setup. Six mice per group were injected with 1 × 1011 vp of the vectors indicated in Figure 1A, plus the hexon modified AdH5/H3RGD.EEK vector. Seventy-two hours later, mice were sacrificed and major organs were harvested. One-half of the spleen was processed for flow cytometry scoring of eGFP expression in B and T cells, while the remaining half and heart, lungs, liver, and kidneys were prepared for total tissue eGFP quantification using a fluorometric assay. B cells were scored as CD45+/CD19+/CD3– and T cells were scored as CD45+/CD19–/CD3+. (B) Flow cytometry scoring of eGFP expression in B and T cells after 1 × 1011 vp delivery. All samples were normalized to the PBS control. Left: percent of total eGFP+ cells. Right: mean fluorescent intensity in eGFP+ cells. Data are expressed as means ± SD. Statistical analysis was carried out using ordinary one-way ANOVA with Tukey’s multiple comparisons test. (C) Statistical analysis of B cell (left) and liver (right) eGFP expression compared with PBS using ordinary one-way ANOVA with Tukey’s multiple comparisons test. Both datasets failed the Shapiro-Wilk test for normality of residuals upon analysis, so the data were log transformed for further statistical analyses. Bars represent means. (D) Tissue eGFP expression after 1 × 1011 vp delivery via quantitative assay. Tissues were homogenized in lysis buffer and assessed for eGFP expression using a fluorometric kit. (E) Flow scoring of eGFP expression in B and T cells after 5 × 1010 vp delivery, normalized to PBS. Left: percent of total eGFP+ cells. Right: mean fluorescent intensity in eGFP+ cells. Data are expressed as means ± SD. Statistical analysis was carried out using ordinary one-way ANOVA with Tukey’s multiple comparisons test. (F) Tissue eGFP expression after 5 × 1010 vp delivery. In all flow cytometry panels, eGFP expression is normalized to a PBS mock control.
Given the differences in results obtained ex vivo and in vivo, we decided to test the remaining promoters in vivo as well. We selected a lower virus dose, 5 × 1010 vp/mouse, hypothesizing that the similarity in gene expression Ad5.CMV and AdRGD.CMV may have been due to saturation of the mouse with the high virus dose. We saw similar results to our previous assay, with Ad5.CMV and AdRGD.CMV showing very similar gene expression profiles in B cells and elsewhere (Figures 2E and 2F). AdRGD.EEK remained the standout promoter-modified vector, again showing a roughly 6-fold decrease in B cell gene expression compared with AdRGD.CMV and little-to-no off-target gene expression, including in T cells, highlighting the exceptional selectivity of this promoter, especially at the lower vector dose. Furthermore, all the promoter-modified vectors showed reduced off-target gene expression, as might be expected given the design criteria for these vectors. Although the EEK promoter did trend toward the highest number of GFP+ B cells, the MH promoter showed similar results with a higher MFI (non-significant), and could also be a useful promoter for future studies.
Analysis of the mechanism of gene transfer to B cells
Intrigued that the AdRGD.CMV and control Ad5.CMV vectors showed comparable in vivo gene transfer to B cells, we hypothesized that Ad-mediated gene transfer to B cells might be driven by virus-cell interactions different than the classic fiber protein-receptor pathway. In this regard, Ad5 (the backbone all our vectors are based on) infection of cells involves a two-step entry mechanism—the virus first binds to cells via high-affinity interactions between the fiber protein and its receptor (CAR). The penton base protein then binds αvβ3 and αvβ5 integrins through an RGD-containing loop, triggering uptake of the virus particle into the cell. We hypothesized that this penton RGD interaction might be responsible for Ad infection of B cells in vivo, explaining the lack of differences between Ad5.CMV and AdRGD.CMV, which carry identical penton base proteins.
To assess this hypothesis, we first pre-treated murine B cells with varying amounts of recombinant Ad5 fiber protein, then infected the cells with Ad5.CMV or AdRGD.CMV (Figure 3A). With Ad5.CMV we observed a modest decrease in gene transfer in the pre-treated groups, dependent on the fiber dose. However, the magnitude of this response was much lower than in A549 cells, which express CAR and bind Ad5 through fiber-receptor interactions (Figure 3B). We did not observe a similar decrease with the AdRGD.CMV vector as expected. We also infected murine B cells and A549 cells with Ad5E3gfpFFc3.luc, which contains a premature stop codon prior to the fiber knob region, resulting in a vector completely incapable of binding cells through CAR. In B cells this vector was nearly as infective as the Ad5.CMV control, but in A549 the vector was almost incapable of gene transfer (Figure 3C). Finally, to determine directly if integrins are responsible for Ad5 gene transfer to B cells, we pre-incubated cells with varying amounts of an integrin blocking peptide, GRGDSP (Figure 3D). We observed a dose-dependent reduction in gene transfer, which was not observed when cells were pre-incubated with GRADSP, a non-binding irrelevant peptide control.
Figure 3.

Analysis of the mechanism of gene transfer to B cells via Ad vectors
(A) Blocking of Ad infection in murine B cells via recombinant fiber. Activated B cells (5 ×105) were resuspended in 400 μL infection medium and incubated with the indicated amounts of recombinant Ad5 fiber for 15 min at room temperature before infection with 1,000 MOI of the indicated vectors. Two technical replicates are summarized. (B) Blocking of Ad infection in A549 cells via recombinant fiber. A549 cells (5 × 105) were seeded in a 12-well plate and incubated overnight. Recombinant fiber was added and cells were infected as above. One technical replicate is shown. Recombinant Ad5 fiber has been described previously.39 (C) Comparison of gene transfer to B cells and A549 cells by a CAR-binding ablated vector. Ad5E3gfpFFc3.luc contains a premature stop codon prior to the fiber knob, resulting in a vector unable to bind CAR. Murine B cells were infected via our standard protocol described in the methods. A549 cells (5 × 104) were seeded in a 24-well plate and infected 24 h later. Two technical replicates are summarized. (D) Blocking of murine B cells with anti-integrin peptide. GRGDSP (Sigma, St. Louis, MO, SCP0157) has been used previously to analyze penton binding to integrins and GRADSP (Sigma, SCP0156) is a non-binding control.40 Activated murine B cells (5 × 105) were resuspended in 25 μL infection medium plus 25 μL of the indicated treatment for 30 min at room temperature before infection. For GRGDSP, two technical replicates are summarized, while for GRADSP one replicate is shown. In (A)–(D) flow cytometry was used to score for eGFP+ cells approximately 48 h after infection. (E) Analysis of integrin expression in eGFP+ B cells after in vivo delivery of 5 × 1010 viral particles of the indicated vectors. Three mice per group were injected with 5 × 1010 vp as described previously. Statistical analysis was carried out using ordinary one-way ANOVA with Tukey’s multiple comparisons test. All data are expressed as means ± SD if applicable and normalized to a PBS mock control.
We also attempted to indirectly analyze the mechanism of gene transfer in vivo using flow cytometry. We assessed B cells for αv and β3 integrin expression after vector delivery and found that these populations in the Ad5.CMV and AdRGD.CMV groups were statistically enriched with eGFP+ cells compared with the overall CD19+ group (Figure 3E). The AdRGD.EEK group did not reach significance but did show a clear trend toward enrichment as well. This effect was especially pronounced in the αv group, suggesting a strong correlation between expression of this receptor and susceptibility to gene transfer from the Ad vectors.
Immunophenotyping of B cells
We next asked if certain B cell subpopulations in the spleen might be more susceptible to Ad gene transfer. We developed a flow cytometry panel to assess marginal zone, follicular, germinal center (GC), and memory B cells, as well as plasmablasts (Figure 4A). We also included a marker for proliferation, hypothesizing that actively growing B cells might be more likely to express the reporter gene. We injected mice retro-orbitally with 5 × 1010 vp as previously and found that, compared with the overall CD19+ group, we observed enriched eGFP+ cells, in order, in the plasmablast, GC, proliferating, and marginal zone groups (Figure 4B). We did not observe any major qualitative differences between Ad5.CMV and AdRGD.CMV in these groups. However, AdRGD.EEK possessed a much higher ratio of eGFP+ cells in the GC and plasmablast groups compared with Ad5.CMV or AdRGD.CMV (Figure 4C). We also compared expression of individual markers in eGFP+ and eGFP– B cells and found that the eGFP+ population was highly enriched for CD95 expression and moderately enriched for GL7 and CD38 expression. This population also showed lower levels of IgD in comparison with the eGFP– group (Figure 4D).
Figure 4.

Immunophenotyping of murine B cells after in vivo vector administration
(A) Gating strategy to define individual B cell populations. All populations were initially gated as singlets/live/CD19+/CD3–. GC, germinal center; FO, follicular; MZ, marginal zone; CD71hi, proliferating; PBs, plasmablasts. (B) eGFP expression in defined cell populations. Statistical analysis was carried out using Ad5.CMV as a representative group using ordinary one-way ANOVA with Tukey’s multiple comparisons test on log transformed data. (C) Fold change in eGFP expressing cells in individual B cell populations compared with all B cells. GC and PB subsets were subjected to analysis via ordinary one-way ANOVA with Tukey’s multiple comparisons test. (D) Representative analysis of cell marker expression in eGFP+ and eGFP– cells. In all cases three mice per group were injected with 5 × 1010 vp as described previously. All data are expressed as means ± SD and normalized to a PBS mock control.
Discussion
In this study we developed and characterized Ad vectors for specific gene transfer to B cells in vivo. We leveraged our triple targeting platform as a basis for these vectors, utilizing the RGD fiber modification to enhance infectivity, tissue-specific promoters for specificity, and liver un-targeting. In primary activated murine B cells, we found ex vivo that the fiber modification enhanced gene transfer to B cells, and a variety of B cell-targeted promoters showed good results compared with the CMV control. Importantly, these results crossed to human primary B cells as well, potentially enabling further development of these vectors for clinical use.
In vivo, we found that the lead promoter-modified vector, AdRGD.EEK, showed significantly weaker eGFP expression in all B cells (in terms of number of cells expressing the reporter and MFI) than the control groups, possibly due to its dependence on immunoglobulin production (discussed further below). However, in off-target tissues including the liver we were unable to detect any meaningful eGFP expression, highlighting the selectivity of the promoter. Similar results were obtained for the other tissue-specific promoters used in this study. This selectivity may be especially critical for in vivo cell engineering applications where off-target expression of the gene editing machinery is unacceptable. Although these promoters did yield a relatively low number of B cells expressing the gene of interest, this may be overcome through a boosting strategy as described by other groups whereby vaccination induces B cell expansion, or by further vector engineering strategies to enhance B cell gene transfer, such as the penton modifications suggested below.11,14 Particularly, in a vaccine-induced expansion strategy a very small number of B cells may be required to respond to the antigen and proliferate. The relatively low number of cells expressing the transgene from the EEK promoter might therefore be sufficient for engineering in this context.
Interestingly, we did not find that incorporating the H5/H3 hexon modification increased the number of eGFP+ B cells in vivo, counter to our hypothesis that reducing liver viral particle sequestration might improve splenocyte gene transfer. The mechanism of this finding is unclear—it is possible that this vector accumulated in a different compartment in vivo (such as macrophages) and was not significantly more available to infect B cells. Alternatively, our phenotyping data indicate that expression of the viral transgene is correlated with B cell activation status. It is possible that increased numbers of B cells were infected by the H5/H3 modified vector but did not express eGFP. Interactions between the viral particles and the blood and immune system are highly complex, and this study further highlights the challenges predicting how vector modifications will impact tissue gene transfer.
Similarly, when delivered systemically in vivo, we found that the standard Ad5.CMV vector and the AdRGD.CMV vector achieved the same levels of gene transfer to B cells. This result was surprising to us, given that historical studies have indicated that the murine spleen and B cells express very low or no levels of the receptor used by Ad5, CAR.41,42 We therefore attempted to determine the mechanism of gene transfer to B cells by Ad5 using a variety of methods. We found that gene transfer was only slightly reduced by pre-treatment of B cells with recombinant fiber, and that a vector totally deficient in its ability to bind CAR was nearly as infectious as the control, potentially indicating that the primary mode of gene transfer is fiber independent. In comparison, blocking murine B cells with a synthetic peptide that binds to integrins showed a strong dose-dependent reduction in gene transfer. These data correlate with our finding that eGFP+ B cells in vivo are highly enriched for αv and β3 integrin expression. Furthermore, one study previously reported that penton base mutated vectors lacking the binding RGD motif accumulated much less in the spleen than the control, demonstrating that this motif is critical for splenic gene transfer in vivo.43
In total, these data suggest that Ads infect B cells primarily through the penton base protein in vivo. Since all the vectors used in this study carry an identical penton base protein, this may explain the similarities between Ad5.CMV and AdRGD.CMV B cell gene expression in vivo. We hypothesize that, in the “simple” ex vivo system, the vector can interact with B cells through either the fiber or penton base, explaining the infectivity enhancement from the RGD4C fiber modification and the relatively high basal infectivity of Ad5.CMV. In contrast, in vivo the virus may only be able to interact with B cells through the penton base. Further engineering efforts may therefore focus on penton base engineering to replace the RGD motif with other motifs targeting integrins expressed exclusively and at high levels on B cells. Modification of the penton base with a peptide binding integrin α4β1 (a leukocyte-specific integrin) has already been described, making this a high priority target for further vector engineering.44,45
However, we cannot rule out additional mechanisms influencing gene transfer both in vivo and ex vivo. Although the majority of B cells expressing eGFP in vivo did show high levels of αv integrin, a significant population were negative, indicating that there may be other mechanisms of virus infection occurring—for example, natural IgMs are known to bind Ad5 in naive mice.46 Tissue macrophage populations are believed to then interact with the IgM bound virus through various receptors.47 In particular, CD36 was recently identified as one of the major scavenger receptors involved in sequestration of Ad5 in Kupffer cells via binding to IgM on the virus capsid.48 Intriguingly, it has also been shown that marginal zone B cells express high levels of CD36.49 This finding is in alignment with immunohistochemical imaging of the spleen after virus administration—although we were able to detect rare populations such as GC B cells expressing eGFP using flow cytometry in this study, it is clear that the vast majority of eGFP+ splenocytes are in the marginal zone (Figure S2). This pathway may thus represent another mechanism by which B cells are infected in vivo. Clearly, additional studies are needed to fully explore the mechanism of Ad-mediated gene transfer to B cells in vivo.
We also assessed the phenotype of B cells expressing eGFP after in vivo vector delivery. We found that several populations were enriched for eGFP+ cells compared with the whole B cell group, including plasmablasts, GC B cells, and marginal zone B cells, and that eGFP+ B cells generally appeared to possess an activated phenotype, including strong enrichment of GL7 and CD95 markers. A straightforward hypothesis is that activated and/or rapidly proliferating B cells are more susceptible to either Ad virus uptake or transgene expression (a mechanistic difference that requires further studies directly assessing the presence of Ad genomes to fully elucidate). Such a phenomenon has been shown with adeno-associated vectors, and our own preliminary data indicate that culturing B cells with activating agents strongly enhances gene expression (Figure S1B).7 This finding is important for our future goals of in vivo B cell engineering, as homology-directed repair generally requires the cell to be proliferating in the S/G2 cell-cycle phases.50
However, it is also possible that Ad infection triggers cellular pathways in B cells that lead to an activated phenotype. To assess these hypotheses in the future, we aim to pre-condition mice via immunization prior to vector delivery and assess if the number of eGFP+ B cells increases. We envision that such an experiment will provide insight into how B cell activation status upon vector delivery affects transgene expression. In further work, we also aim to track the development of infected B cells over time in the spleen and bone marrow after vector delivery. The ultimate cell populations likely required for successful gene therapy are memory B cells and plasma cells. It will therefore be important to determine if the eGFP+ GC and plasmablast populations are capable of transitioning into memory and plasma cells. At present we have little data on the persistence of transgene expression in infected B cells due to the challenges in tracking a proliferating and differentiating cell population with our non-integrating Ads. In future experiments we aim to leverage our established Ai9-SauSpyCas9 mouse model, which possesses a genomic switch that activates RFP expression upon successful Cre- or Cas9-mediated edits.51 We expect delivery or either of these gene editors with our Ads will thus enable long-term tracking of infected B cells.
We found few if any qualitative differences between B cell subsets infected by Ad5.CMV or AdRGD.CMV or markers expressed by eGFP+ cells infected by these vectors. However, we did observe clear differences between those groups and the AdRGD.EEK group, which showed a much higher ratio of plasmablast and GC eGFP+ cells compared with all B cells than Ad5 or AdRGD.CMV. The EEK promoter is a synthetic promoter based on the immunoglobulin kappa light chain promoter with enhancer elements. This promoter is likely more dependent on the B cells actively dividing and producing immunoglobulin than the ubiquitous viral CMV promoter, which aligns with its strong expression in the plasmablast and GC groups.
This study forms the basis for our future research toward in vivo B cell engineering for various therapies. In previous work, we demonstrated that Ads could be used to deliver the CRISPR-Cas machinery to the liver in vivo to achieve phenotypic correction of serum deficiencies.52,53 We envision the application of these techniques to B cells may enable in vivo BCR engineering to control infectious diseases, as described by several groups previously.7,8,9,10,11 Alternatively, a single B cell may last the lifetime of an organism if it differentiates into a long-lived plasma cells. If we are able to obtain sufficient numbers of plasma cells expressing the delivered transgene from the episomal Ad backbone, this might be sufficient to correct genetic diseases that require only low levels of corrective protein expression. Our proposed future experiments will help address these feasibilities. We do note that one barrier to the translation of this technology is anti-vector/transgene immune responses triggered by the Ad causing elimination of engineered B cells. While we hope that the restriction of transgene expression to B cells endeavored in this study and further research toward directly altering viral particle tropism will reduce vector immunogenicity, we may also transition our vectors to a high-capacity adenovirus system, which has been demonstrated to achieve long-term gene expression.54,55
In total, we found that Ads were capable of infecting B cells in vivo and could be engineered to restrict gene expression using tissue-specific promoters. Gene transfer did not appear to be dependent on the identity of the fiber protein but was likely rather mediated by the penton base protein and other as-of-yet undefined pathways. The susceptibility of B cells to express of the virus transgene appeared to be influenced by the phenotype of the cells and was strongly correlated with B cell activation markers. Our study may also provide insight into interactions between Ads and the immune system—to our knowledge, this is the first report demonstrating that Ads are able to infect distinct lymphocyte populations after systemic delivery. These data may have implications for other applications such as oncolytics and vaccines.
Materials and methods
Cells
HEK293 (ATCC, Manassas, VA, CRL-1573) and A549 (ATCC CCL-185) cells were grown in Dulbecco’s modified Eagle’s medium/Ham’s F12 1:1 mixture supplemented with L-glutamine, 15 mM HEPES, 10% fetal bovine serum (FBS), and 100 U/mL penicillin-streptomycin (Gibco, Waltham, MA, 11330-032). Cells were grown at 37°C with 5% CO2 under sterile conditions.
Viruses
Vectors developed were all E1/E3 deleted adenoviruses based on the human adenovirus C5 genome (HAdV-C5). HAdV-C5 and AdRGD have been described previously.30,56 Viral genomes were released from plasmids via digestion with PacI (New England Biolabs, Ipswich, MA) or AbsI (SibEnzyme, Russia) and transfected into HEK293 cells for upscale. Viruses were purified and the concentration of viral particles was determined as described previously by our group.51
To facilitate rapid cloning of fiber and promoter-modified vectors, we utilized an Ad backbone with unique restriction sites surrounding the fiber (JC201: pMVP/Ad/5-35-DEST was a gift from Christopher Newgard [Addgene plasmid no. 121847; http://n2t.net/addgene:121847; RRID:Addgene_121847, Watertown, MA]).57 We made several modifications to this backbone to further suit our purposes. We restored the U exon region downstream of the fiber by digesting the backbone with SrfI and SgrDI (New England Biolabs) and inserting the corresponding genomic region with the U exon from pAdEasy1 via NEB HiFi DNA assembly (New England Biolabs), as U exon-deficient vectors have been shown to have reduced yields.56,58
We then replaced the E1 region with a synthetic spacer region flanked by unique SwaI/ClaI restriction sites. We accomplished this swap by cloning the spacer (synthetized by Integrated DNA Technologies, Coralville, IA) into pShuttle (from the pAdEasy system) via NEB HiFi DNA assembly. This plasmid was then linearized via PmeI (New England Biolabs) and swapped into the Ad genome via homologous recombination in BJ5183 bacteria (Agilent, Santa Clara, CA). Digestion of the vector backbone with either enzyme thus released the spacer region and allowed Gibson assembly of desired DNA fragments directly into the vector genome.
We also modified the fiber region by digesting the backbone with BarI (SibEnzyme) and BstBI (New England Biolabs). Standard HAdV-C5 fiber was PCR amplified from pAdEasy1 and assembled into the digested backbone via NEB HiFi DNA assembly.56 The AdRGD fiber was assembled with the backbone in the same manner after amplification from the previously described backbone.30 These new plasmids were termed “prAd5” and “prAdRGD” or plasmid rapid Ad5/AdRGD. To generate AdH5/H3RGD.EEK containing the liver un-targeting hexon modification, the prAdRGD backbone was digested with SfiI (New England Biolabs) and substituted with the modified hexon from a shuttle vector as described previously.29
Promoters
To generate promoter-modified vectors, prAd5 and prAdRGD were digested with SwaI (New England Biolabs) to release the spacer region. DNA fragments encoding each promoter, eGFP and the SV40 polyadenylation tail were then assembled with the vector backbone via NEB HiFi DNA assembly.
The ubiquitous CMV promoter was used as a control and was amplified from the previously described pTrackCMVLuc.56 The SFFV promoter was obtained from pHR-SFFV-dCas9-BFP, a gift from Stanley Qi and Jonathan Weissman (Addgene plasmid no. 46910; http://n2t.net/addgene:46910; RRID:Addgene_46910, Watertown, MA).59 The synthetic promoters EEK and MH have been described previously and were synthesized by Integrated DNA Technologies.35 The Epstein-Barr virus Wp1168 fragment has been described previously and was synthesized in the pUC57m carrier plasmid by GenScript (Piscataway, NJ).36 The promoter was liberated from the carrier by EcoRV digestion and assembled with prAdRGD via NEB HiFi DNA assembly.
Primary B cell culture
Isolation and culture of murine primary B cells has been described previously.11 In brief, spleens were crushed on a 70 μm filter using a syringe plunger and washed through using 1× PBS + 2% FBS (B cell isolation medium [BCIM]). Cells were pelleted via centrifugation at 1,200 rpm or 500 × g for 5 min and resuspended in 3 mL ACK Lysis Buffer (Gibco, A10492-01). After 3 min incubation, 7 mL BCIM was added and cells were pelleted again. Cells were resuspended in 3 mL BCIM and washed through a second 70 μm filter to obtain a single-cell suspension. B cells were purified from this suspension using a magnetic isolation kit according to the manufacturer’s instructions (Miltenyi, Gaithersburg, MD, 130-090-862).
Purified murine B cells were cultured and activated at 37°C for 30 h before virus infections. Unless otherwise indicated, infections were conducted by resuspending 5 × 105 B cells in 40 μL base B cell medium with 5% FBS and without activation. Ten microliters of virus diluted in 1× PBS to the indicated MOI was then added and cells were infected overnight at 37°C, then transferred to 450 μL B cell medium with 10% FBS and 50 μg/mL LPS. Cells were cultured for approximately 48 h after infection then assessed via flow cytometry.
Human primary B cells were obtained from healthy donors. In brief, peripheral blood mononuclear cells (PBMCs) were isolated from LRS chambers and frozen in liquid nitrogen prior to B cell isolation. B cells were then magnetically isolated from 1.5 × 108 PBMCs using a kit according to the manufacturer’s instructions (Stem Cell Technologies, Canada, 17954). B cells were cultured and differentiated according to previously published methods for 72 h before infection.7,60,61 For infections, 2.5 × 105 cells were resuspended in 20 μL StemMACS HSC expansion medium supplemented with 0.2% FBS. Vectors were diluted in PBS so that the appropriate virus number would be delivered in 20 μL. Virus was added to cells and incubated for 3 h at 37°C, then transferred to 1 mL complete B cell medium, and incubated for approximately 48 h before flow cytometry analysis.
Flow cytometry
For ex vivo analysis, murine B cells were stained with CD19 (Invitrogen, Waltham, MA, RM7717) and Sytox Red or Fixable Far Red stains for live/dead discrimination (Invitrogen, S34859 and L10120). Human B cells were stained with CD19 (BioLegend, San Diego, CA, 302251) and Fixable Far Red stain.
For in vivo studies, spleens were processed into a single-cell suspension as described above. Splenocytes were incubated with 10 μL Fc Blocking Reagent (Miltenyi 130-092-575) for 10 min prior to staining. All B cell subsets were passed through a Live/CD3–/CD19+ gate prior to further analysis. Germinal center B cells were defined as GL7+/CD95+/IgDlo/CD38lo, marginal zone as IgM+/IgDlo, follicular as IgMlo/IgD+/CD38+, memory as IgDlo/GL7-/CD38+, proliferating as CD71hi, and plasmablasts as CD138+/IgD–. Fluorescence minus one controls were used to determine the gating strategy for B cell subpopulations.
Antibodies used were as follows: CD3 (Invitrogen, 46-0032-82), CD45 (Invitrogen, 47-0451-82), CD19, CD71 (BioLegend, 113813, CD95 (BD Biosciences, Franklin Lakes, NJ, 563646), CD138 (BioLegend, 142515), GL7 (BioLegend, 144607), CD38 (Miltenyi, 130-125-522), IgD (Invitrogen, 56-5993-80), IgM (Invitrogen, 47-5790-82), CD51 (BioLegend, 104105), and CD61 (BioLegend, 104313). Live-dead discrimination was carried out using Sytox Red or Fixable Far Red stains, or Zombie UV (BioLegend, 423107) for integrin-staining experiments.
Animal studies
Male C57Bl/6J mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and housed in a pathogen-free environment. Mice aged 7–10 weeks were injected with 1 × 1011 or 5 × 1010 viral particles retro-orbitally and sacrificed approximately 72 h later via anesthetization with avertin followed by cervical dislocation. In the indicated studies, spleen, lungs, heart, kidney, and liver were harvested. One-half of each spleen was used for flow cytometry analysis as described above, while the remaining half and other organs were frozen in liquid nitrogen. Organs were then thawed and processed for eGFP quantification using a commercial kit according to the manufacturer’s instructions (Cell BioLabs, San Diego, CA, AKR-120). In brief, tissues were homogenized with an Omni Prep 96 in the provided lysis buffer with 10% Proteinase Inhibitor Cocktail added (Sigma, Saint Louis, MO, P8340). Homogenates were then centrifuged to remove debris and supernatants were transferred to 96-well plates for analysis against the included reference protein included in the kit via fluorimetry. All experiments were approved by the Institutional Animal Care and Use Committee of Washington University in St. Louis School of Medicine and were performed in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals. All efforts were made to minimize suffering and the total number of animals used in the study.
Statistical analyses
Specific methods used for data analysis are noted in the corresponding figure legends. In all cases, analysis was carried out using GraphPad Prism 9. A p value of <0.05 was used, and significance is indicated as ∗p < 0.0322, ∗∗p < 0.0021, ∗∗∗p < 0.0002, ∗∗∗∗p < 0.0001; ns, not significant.
Acknowledgments
The authors thank James E. Voss and Mary Tenuta for their assistance setting up primary murine B cell culture. This work was supported by UG3TR002851 and R01CA211096 awarded by the National Institutes of Health to D.T.C., T32HL007088-45 awarded by the National Institutes of Health to Stephen Oh, and P21-04949 awarded by Walking Fish Therapeutics to Z.H.L.
Author contributions
Conceptualization, P.J.R.-B. and D.T.C.; formal analysis, P.J.R.-B.; investigation, P.J.R.-B., S.A.M., A.B.A., R.L., and Z.H.L.; visualization, P.J.R.-B.; methodology, P.J.R.-B., S.A.M., A.B.A., A.J.S., R.L., I.P.D., E.A.K., Z.H.L., R.R., M.S., and K.P.; writing – original draft, P.J.R.-B.; ; writing – review & editing, D.T.C.; resources, I.P.D. and E.A.K.; supervision, D.T.C.
Declaration of interests
R.R. and M.S. are employees of and hold equity in Walking Fish Therapeutics. D.T.C. is a scientific advisor for Walking Fish Therapeutics.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2023.07.004.
Supplemental information
Data availability
All reagents and data described in this study are available upon reasonable request.
References
- 1.CAR T cells: Engineering immune cells to treat cancer - NCI. https://www.cancer.gov/about-cancer/treatment/research/car-t-cells
- 2.Finck A.V., Blanchard T., Roselle C.P., Golinelli G., June C.H. Engineered cellular immunotherapies in cancer and beyond. Nat. Med. 2022;28:678–689. doi: 10.1038/s41591-022-01765-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Patel S., Burga R.A., Powell A.B., Chorvinsky E.A., Hoq N., McCormack S.E., Van Pelt S.N., Hanley P.J., Cruz C.R.Y. Beyond CAR T cells: Other cell-based immunotherapeutic strategies against cancer. Front. Oncol. 2019;9:196. doi: 10.3389/FONC.2019.00196/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jeske A.M., Boucher P., Curiel D.T., Voss J.E. Vector Strategies to Actualize B Cell–Based Gene Therapies. J. Immunol. 2021;207:755–764. doi: 10.4049/JIMMUNOL.2100340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ueda N., Cahen M., Danger Y., Moreaux J., Sirac C., Cogné M. Immunotherapy perspectives in the new era of B-cell editing. Blood Adv. 2021;5:1770–1779. doi: 10.1182/BLOODADVANCES.2020003792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rogers G.L., Cannon P.M. Genome edited B cells: a new frontier in immune cell therapies. Mol. Ther. 2021;29:3192–3204. doi: 10.1016/J.YMTHE.2021.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hung K.L., Meitlis I., Hale M., Chen C.Y., Singh S., Jackson S.W., Miao C.H., Khan I.F., Rawlings D.J., James R.G. Engineering Protein-Secreting Plasma Cells by Homology-Directed Repair in Primary Human B Cells. Mol. Ther. 2018;26:456–467. doi: 10.1016/j.ymthe.2017.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johnson M.J., Laoharawee K., Lahr W.S., Webber B.R., Moriarity B.S. Engineering of Primary Human B cells with CRISPR/Cas9 Targeted Nuclease. Sci. Rep. 2018;8:12144. doi: 10.1038/s41598-018-30358-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ou T., He W., Quinlan B.D., Guo Y., Tran M.H., Karunadharma P., Park H., Davis-Gardner M.E., Yin Y., Zhang X., et al. Reprogramming of the heavy-chain CDR3 regions of a human antibody repertoire. Mol. Ther. 2022;30:184–197. doi: 10.1016/J.YMTHE.2021.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hartweger H., McGuire A.T., Horning M., Taylor J.J., Dosenovic P., Yost D., Gazumyan A., Seaman M.S., Stamatatos L., Jankovic M., Nussenzweig M.C. HIV-specific humoral immune responses by CRISPR/Cas9-edited B cells. J. Exp. Med. 2019;216:1301–1310. doi: 10.1084/JEM.20190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang D., Tran J.T., Olson A., Vollbrecht T., Tenuta M., Guryleva M.V., Fuller R.P., Schiffner T., Abadejos J.R., Couvrette L., et al. Vaccine elicitation of HIV broadly neutralizing antibodies from engineered B cells. Nat. Commun. 2020;11:6360. doi: 10.1038/s41467-020-19650-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X., Rivière I. Clinical manufacturing of CAR T cells: foundation of a promising therapy. Mol. Ther. Oncolytics. 2016;3 doi: 10.1038/MTO.2016.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dunbar C.E., High K.A., Joung J.K., Kohn D.B., Ozawa K., Sadelain M. Gene therapy comes of age. Science. 2018;359:eaan4672. doi: 10.1126/science.aan4672. [DOI] [PubMed] [Google Scholar]
- 14.Nahmad A.D., Lazzarotto C.R., Zelikson N., Kustin T., Tenuta M., Huang D., Reuveni I., Nataf D., Raviv Y., Horovitz-Fried M., et al. In vivo engineered B cells secrete high titers of broadly neutralizing anti-HIV antibodies in mice. Nat. Biotechnol. 2022;40:1241–1249. doi: 10.1038/s41587-022-01328-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Michels A., Ho N., Buchholz C.J. Precision medicine: In vivo CAR therapy as a showcase for receptor-targeted vector platforms. Mol. Ther. 2022;30:2401–2415. doi: 10.1016/J.YMTHE.2022.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frank A.M., Braun A.H., Scheib L., Agarwal S., Schneider I.C., Fusil F., Perian S., Sahin U., Thalheimer F.B., Verhoeyen E., Buchholz C.J. Combining T-cell-specific activation and in vivo gene delivery through CD3-targeted lentiviral vectors. Blood Adv. 2020;4:5702–5715. doi: 10.1182/BLOODADVANCES.2020002229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huckaby J.T., Landoni E., Jacobs T.M., Savoldo B., Dotti G., Lai S.K. Bispecific binder redirected lentiviral vector enables in vivo engineering of CAR-T cells. J. Immunother. Cancer. 2021;9 doi: 10.1136/JITC-2021-002737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Agarwal S., Weidner T., Thalheimer F.B., Buchholz C.J. In vivo generated human CAR T cells eradicate tumor cells. Oncoimmunology. 2019;8 doi: 10.1080/2162402X.2019.1671761/SUPPL_FILE/KONI_A_1671761_SM3373.PPTX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pfeiffer A., Thalheimer F.B., Hartmann S., Frank A.M., Bender R.R., Danisch S., Costa C., Wels W.S., Modlich U., Stripecke R., et al. In vivo generation of human CD19-CAR T cells results in B-cell depletion and signs of cytokine release syndrome. EMBO Mol. Med. 2018;10 doi: 10.15252/EMMM.201809158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sheldon D.Y., Yoo D.B.-C., Kmiec D.E., Petrelli D.N., Tiesi D.G., Warner D.S. Review of safety data for Adenovirus 5 (Ad5) as a delivery vector for intratumoral cancer gene therapy. 2023. https://home.liebertpub.com/hum. 10.1089/HUM.2022.228 [DOI] [PubMed]
- 21.Blanchette P., Teodoro J.G. A Renaissance for Oncolytic Adenoviruses? Viruses. 2023;15:358. doi: 10.3390/V15020358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Watanabe M., Nishikawaji Y., Kawakami H., Kosai K.I. Adenovirus Biology, Recombinant Adenovirus, and Adenovirus Usage in Gene Therapy. Viruses. 2021;13:2502. doi: 10.3390/V13122502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boucher P., Cui X., Curiel D.T. Adenoviral vectors for in vivo delivery of CRISPR-Cas gene editors. J. Control Release. 2020;327:788–800. doi: 10.1016/J.JCONREL.2020.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang H., Liu Z., Li C., Gil S., Papayannopoulou T., Doering C.B., Lieber A. High-level protein production in erythroid cells derived from in vivo transduced hematopoietic stem cells. Blood Adv. 2019;3:2883–2894. doi: 10.1182/bloodadvances.2019000706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li C., Mishra A.S., Gil S., Wang M., Georgakopoulou A., Papayannopoulou T., Hawkins R.D., Lieber A. Targeted Integration and High-Level Transgene Expression in AAVS1 Transgenic Mice after In Vivo HSC Transduction with HDAd5/35++ Vectors. Mol. Ther. 2019;27:2195–2212. doi: 10.1016/j.ymthe.2019.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li C., Lieber A. Adenovirus vectors in hematopoietic stem cell genome editing. FEBS Lett. 2019;593:3623–3648. doi: 10.1002/1873-3468.13668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mendonça S.A., Lorincz R., Boucher P., Curiel D.T. Adenoviral vector vaccine platforms in the SARS-CoV-2 pandemic. npj Vaccin. 2021;6:1–14. doi: 10.1038/s41541-021-00356-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coughlan L., Kremer E.J., Shayakhmetov D.M. Adenovirus-based vaccines—a platform for pandemic preparedness against emerging viral pathogens. Mol. Ther. 2022;30:1822–1849. doi: 10.1016/J.YMTHE.2022.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaliberov S.A., Kaliberova L.N., Hong Lu Z., Preuss M.A., Barnes J.A., Stockard C.R., Grizzle W.E., Arbeit J.M., Curiel D.T. Retargeting of gene expression using endothelium specific hexon modified adenoviral vector. Virology. 2013;447:312–325. doi: 10.1016/j.virol.2013.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dmitriev I., Krasnykh V., Miller C.R., Wang M., Kashentseva E., Mikheeva G., Belousova N., Curiel D.T. An Adenovirus Vector with Genetically Modified Fibers Demonstrates Expanded Tropism via Utilization of a Coxsackievirus and Adenovirus Receptor-Independent Cell Entry Mechanism. J. Virol. 1998;72:9706–9713. doi: 10.1128/jvi.72.12.9706-9713.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim J.W., Glasgow J.N., Nakayama M., Ak F., Ugai H., Curiel D.T. An Adenovirus Vector Incorporating Carbohydrate Binding Domains Utilizes Glycans for Gene Transfer. PLoS One. 2013;8 doi: 10.1371/JOURNAL.PONE.0055533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roy S., Medina-Jaszek A., Wilson M.J., Sandhu A., Calcedo R., Lin J., Wilson J.M. Creation of a panel of vectors based on ape adenovirus isolates. J. Gene Med. 2011;13:17–25. doi: 10.1002/JGM.1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lang F.F., Conrad C., Gomez-Manzano C., Yung W.K.A., Sawaya R., Weinberg J.S., Prabhu S.S., Rao G., Fuller G.N., Aldape K.D., et al. Phase I study of DNX-2401 (delta-24-RGD) oncolytic adenovirus: replication and immunotherapeutic effects in recurrent malignant glioma. J. Clin. Oncol. 2018;36:1419–1427. doi: 10.1200/JCO.2017.75.8219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Winiarska M., Nowis D., Firczuk M., Zagozdzon A., Gabrysiak M., Sadowski R., Barankiewicz J., Dwojak M., Golab J. Selection of an optimal promoter for gene transfer in normal B cells. Mol. Med. Rep. 2017;16:3041–3048. doi: 10.3892/MMR.2017.6974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luo X.M., Maarschalk E., O’Connell R.M., Wang P., Yang L., Baltimore D. Engineering human hematopoietic stem/progenitor cells to produce a broadly neutralizing anti-HIV antibody after in vitro maturation to human B lymphocytes. Blood. 2009;113:1422–1431. doi: 10.1182/BLOOD-2008-09-177139. [DOI] [PubMed] [Google Scholar]
- 36.Bell A., Skinner J., Kirby H., Rickinson A. Characterisation of Regulatory Sequences at the Epstein–Barr VirusBamHI W Promoter. Virology. 1998;252:149–161. doi: 10.1006/VIRO.1998.9440. [DOI] [PubMed] [Google Scholar]
- 37.Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pettersen E.F., Goddard T.D., Huang C.C., Meng E.C., Couch G.S., Croll T.I., Morris J.H., Ferrin T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021;30:70–82. doi: 10.1002/PRO.3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krasnykh V., Dmitriev I., Mikheeva G., Miller C.R., Belousova N., Curiel D.T. Characterization of an adenovirus vector containing a heterologous peptide epitope in the HI loop of the fiber knob. J. Virol. 1998;72:1844–1852. doi: 10.1128/JVI.72.3.1844-1852.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wickham T.J., Mathias P., Cheresh D.A., Nemerow G.R. Integrins alpha v beta 3 and alpha v beta 5 promote adenovirus internalization but not virus attachment. Cell. 1993;73:309–319. doi: 10.1016/0092-8674(93)90231-E. [DOI] [PubMed] [Google Scholar]
- 41.Mena I., Perry C.M., Harkins S., Rodriguez F., Gebhard J., Whitton J.L. The Role of B Lymphocytes in Coxsackievirus B3 Infection. Am. J. Pathol. 1999;155:1205–1215. doi: 10.1016/S0002-9440(10)65223-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bergelson J.M., Krithivas A., Celi L., Droguett G., Horwitz M.S., Wickham T., Crowell R.L., Finberg R.W. The Murine CAR Homolog Is a Receptor for Coxsackie B Viruses and Adenoviruses. J. Virol. 1998;72:415–419. doi: 10.1128/JVI.72.1.415-419.1998/ASSET/B316BA2C-D9F4-4E0F-827F-ABAAF201FC2D/ASSETS/GRAPHIC/JV0180552007.JPEG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Di Paolo N.C., Miao E.A., Iwakura Y., Murali-Krishna K., Aderem A., Flavell R.A., Papayannopoulou T., Shayakhmetov D.M. Virus Binding to a Plasma Membrane Receptor Triggers Interleukin-1α-Mediated Proinflammatory Macrophage Response In Vivo. Immunity. 2009;31:110–121. doi: 10.1016/J.IMMUNI.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wickham T.J., Carrion M.E., Kovesdi I. Targeting of adenovirus penton base to new receptors through replacement of its RGD motif with other receptor-specific peptide motifs. Gene Ther. 1995;2:750–756. [PubMed] [Google Scholar]
- 45.Baiula M., Spampinato S., Gentilucci L., Tolomelli A. Novel Ligands Targeting α4β1 Integrin: Therapeutic Applications and Perspectives. Front. Chem. 2019;7:489. doi: 10.3389/FCHEM.2019.00489/BIBTEX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Allen R.J., Byrnes A.P. Interaction of adenovirus with antibodies, complement, and coagulation factors. FEBS Lett. 2019;593:3449–3460. doi: 10.1002/1873-3468.13649. [DOI] [PubMed] [Google Scholar]
- 47.Atasheva S., Yao J., Shayakhmetov D.M. Innate immunity to adenovirus: lessons from mice. FEBS Lett. 2019;593:3461–3483. doi: 10.1002/1873-3468.13696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Atasheva S., Emerson C.C., Yao J., Young C., Stewart P.L., Shayakhmetov D.M. Systemic cancer therapy with engineered adenovirus that evades innate immunity. Sci. Transl. Med. 2020;12:6659. doi: 10.1126/SCITRANSLMED.ABC6659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang P., Li W., Wang Y., Hou L., Xing Y., Qin H., Wang J., Liang Y., Han H. Identification of CD36 as a new surface marker of marginal zone B cells by transcriptomic analysis. Mol. Immunol. 2007;44:332–337. doi: 10.1016/J.MOLIMM.2006.02.030. [DOI] [PubMed] [Google Scholar]
- 50.Lin S., Staahl B.T., Alla R.K., Doudna J.A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife. 2014;3 doi: 10.7554/ELIFE.04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lorincz R., Alvarez A.B., Walkey C.J., Mendonça S.A., Lu Z.H., Martinez A.E., Ljungberg C., Heaney J.D., Lagor W.R., Curiel D.T. In vivo editing of the pan-endothelium by immunity evading simian adenoviral vector. Biomed. Pharmacother. 2023;158 doi: 10.1016/J.BIOPHA.2022.114189. [DOI] [PubMed] [Google Scholar]
- 52.Stephens C.J., Kashentseva E., Everett W., Kaliberova L., Curiel D.T. Targeted in vivo knock-in of human alpha-1-antitrypsin cDNA using adenoviral delivery of CRISPR/Cas9. Gene Ther. 2018;25:139–156. doi: 10.1038/s41434-018-0003-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Stephens C.J., Lauron E.J., Kashentseva E., Lu Z.H., Yokoyama W.M., Curiel D.T. Long-term correction of hemophilia B using adenoviral delivery of CRISPR/Cas9. J. Control Release. 2019;298:128–141. doi: 10.1016/j.jconrel.2019.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ricobaraza A., Gonzalez-Aparicio M., Mora-Jimenez L., Lumbreras S., Hernandez-Alcoceba R. High-Capacity Adenoviral Vectors: Expanding the Scope of Gene Therapy. Int. J. Mol. Sci. 2020;21 doi: 10.3390/IJMS21103643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chuah M.K.L., Schiedner G., Thorrez L., Brown B., Johnston M., Gillijns V., Hertel S., Van Rooijen N., Lillicrap D., Collen D., et al. Therapeutic factor VIII levels and negligible toxicity in mouse and dog models of hemophilia A following gene therapy with high-capacity adenoviral vectors. Blood. 2003;101:1734–1743. doi: 10.1182/BLOOD-2002-03-0823. [DOI] [PubMed] [Google Scholar]
- 56.He T.C., Zhou S., Da Costa L.T., Yu J., Kinzler K.W., Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc. Natl. Acad. Sci. USA. 1998;95:2509–2514. doi: 10.1073/PNAS.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Haldeman J.M., Conway A.E., Arlotto M.E., Slentz D.H., Muoio D.M., Becker T.C., Newgard C.B. Creation of versatile cloning platforms for transgene expression and dCas9-based epigenome editing. Nucleic Acids Res. 2019;47:e23. doi: 10.1093/NAR/GKY1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tollefson A.E., Ying B., Doronin K., Sidor P.D., Wold W.S.M. Identification of a New Human Adenovirus Protein Encoded by a Novel Late l-Strand Transcription Unit. J. Virol. 2007;81:12918–12926. doi: 10.1128/JVI.01531-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gilbert L.A., Larson M.H., Morsut L., Liu Z., Brar G.A., Torres S.E., Stern-Ginossar N., Brandman O., Whitehead E.H., Doudna J.A., et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell. 2013;154:442–451. doi: 10.1016/J.CELL.2013.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Luo B., Zhan Y., Luo M., Dong H., Liu J., Lin Y., Zhang J., Wang G., Verhoeyen E., Zhang Y., Zhang H. Engineering of α-PD-1 antibody-expressing long-lived plasma cells by CRISPR/Cas9-mediated targeted gene integration. Cell Death Dis. 2020;11:973–1018. doi: 10.1038/s41419-020-03187-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cheng R.Y.H., Hung K.L., Zhang T., Stoffers C.M., Ott A.R., Suchland E.R., Camp N.D., Khan I.F., Singh S., Yang Y.J., et al. Ex vivo engineered human plasma cells exhibit robust protein secretion and long-term engraftment in vivo. Nat. Commun. 2022;13:6110–6114. doi: 10.1038/s41467-022-33787-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All reagents and data described in this study are available upon reasonable request.